
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
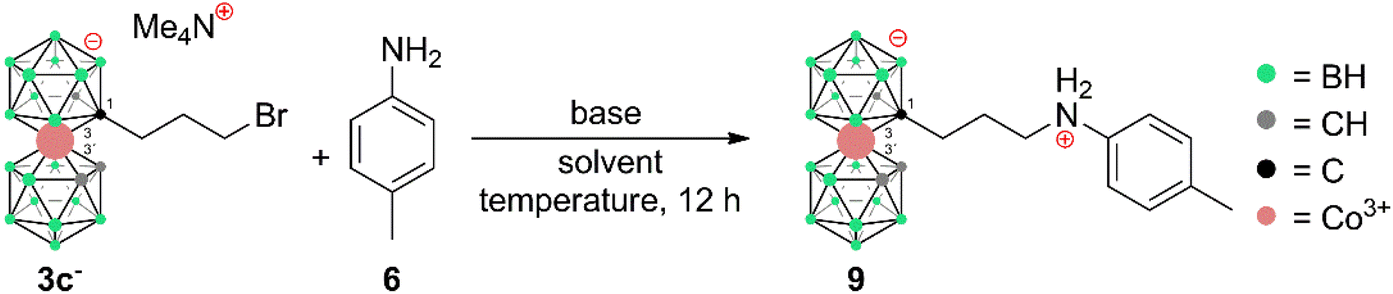
Synthetic routes to carbon substituted cobalt bis(dicarbollide) alkyl halides and aromatic amines along with closely related irregular pathways†
Jan
Nekvinda
 *a,
Dmytro
Bavol
a,
Miroslava
Litecká
a,
Ece Zeynep
Tüzün
a,
Michal
Dušek
b and
Bohumír
Grüner
a
*a,
Dmytro
Bavol
a,
Miroslava
Litecká
a,
Ece Zeynep
Tüzün
a,
Michal
Dušek
b and
Bohumír
Grüner
a
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, Hlavní 1001, Husinec-Řež 25068, Czech Republic. E-mail: nekvinda@iic.cas.cz
bInstitute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, Prague 8, 182 21, Czech Republic
First published on 21st February 2024
Abstract
Carbon substituted cobalt bis(dicarbollide) alkyl halides [(1-X-(CH2)n-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]Me4N (X = Br, I; n = 1–3) are prepared in high yields (>90%) from their corresponding alcohols without side skeletal substitutions. These species offer access to the synthesis of aromatic cobalt bis(dicarbollide) amines, however only for particular terminal halogen substitution, the propylene pendant arm, and under appropriately controlled reaction conditions. Thus, the compounds substituted at cage carbon atoms with a propylene linker and terminal aromatic amine groups could be prepared. In other cases, numerous irregular reaction pathways occur, undoubtedly as a consequence of the bulky anionic boron cage in close proximity to the reaction site. Among them, an unusual intramolecular hydroboration forming rigidified carbon-to-boron bridged isomeric anions with an asymmetric structure that correspond to formulae [(1,8′-μ-C2H4)-(1,2-C2B9H10)(1′,2′-C2B9H10)-3,3′-Co]− and [(1,7′-μ-C2H4)-(1,2-C2B9H10)(1′,2′-C2B9H10)-3,3′-Co]− is described herein and the former isomer is structurally characterized. This product with a restrained geometry is widely accessible through nucleophile and/or thermally induced decomposition of (pseudo)halides attached to the cage via an ethylene linker. Surprisingly enough, also doubly bridged isomeric species [(1,8-μ-C2H4-1,2-C2B9H9)2-3,3′-Co]− and [(1,7-μ-C2H4-1,2-C2B9H9)2-3,3′-Co]− are available in good yield using these methods. Furthermore, other more typical side reactions are discussed, i.e. nucleophilic reactions of propyl halides with Me3N formed apparently by disproportionation of Me4N+ at higher temperatures or with pyridine used as a base.
Introduction
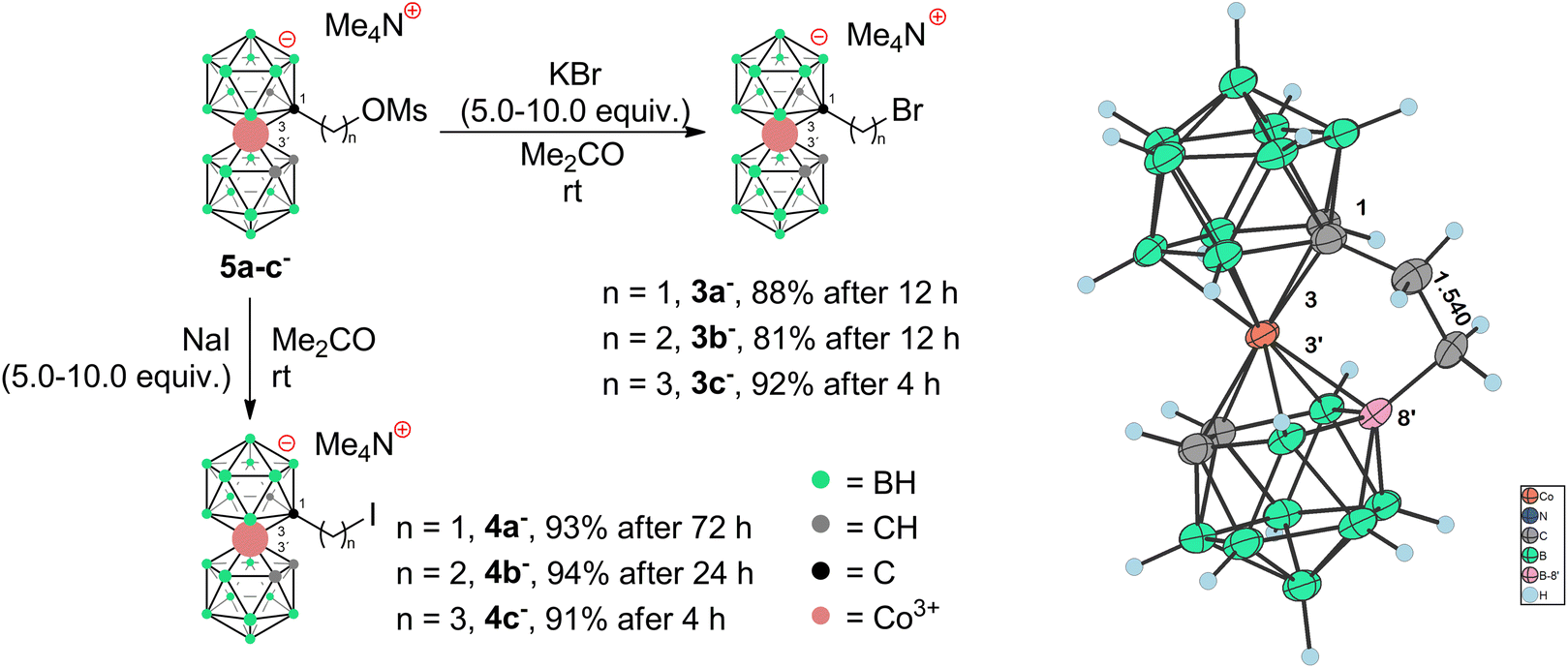
The cobalt bis(dicarbollide)(1-) ion,1 often abbreviated as COSAN, with the empirical formula [(1,2-C2B9H11)2-3,3′-Co]−, has strong amphiphilic character and 3D aromaticity.2–5 For these reasons, it has attracted the attention of chemists interested in innovative solutions to a variety of challenges in areas such as medicinal and physical chemistry, and it is the subject of several review articles and book chapters.2–6 Thus, the COSAN(1-) ion and its derivatives have been investigated in the context of, for example, boron neutron capture therapy,7,8 HIV protease inhibitors,9,10 carbonic anhydrase inhibitors,11,12 or antibacterial agents,13–19 extraction agents of radionuclides from used radioactive fuel,20–23 or photochemistry.24,25 This wide potential feeds a demand for more versatile and variable synthetic methods to yield COSAN derivatives. Two main sites for the substitution of COSAN ions can be distinguished – the hydridic site at the B8 and B8′ vertices and the acidic site at the carbon vertices C1/C2 and C1′/C2′.6 Regarding boron-site substitutions, Plešek et al. disclosed in their pioneering work a dioxane ring-cleavage reaction that opened up a generic route to a series of COSAN derivatives.26 Direct halogenation27–30 and other substitutions at the B8 and B8′ positions have also been studied.31–33 Regarding substitutions at the acidic carbon vertices, Grüner et al. published the direct alkylation of COSAN with either formaldehyde or constrained cyclic ethers to make hydroxy derivatives versatile reagents for further derivatization.33 This work also described that the corresponding disubstitution leads to the formation of two main stereoisomers, which correspond to racemic- and meso-forms. Later works described the preparation of COSAN derivatives with diverse functionalities such as carboxylic acids and amides,34 azides,35 nitriles,36 and primary or secondary amines,37 but, significantly to this contribution, not with aromatic amines. Aromatic amines, e.g., anilines or toluidines, are significantly less nucleophilic than their aliphatic counterparts and therefore the formation of C(sp3)–N(Ar) bonds often requires the use of transition-metal catalysis38–40 or various harsh conditions.41,42 According to our considerations, for certain biological applications, such as the design of antibacterials and antifungals, the presence of a lowly basic aniline fragment in the structure should bring noticeable benefits. Here, we present synthetic routes leading to C-substituted alkyl halides that proved useful in substitution reactions with anilines to produce the corresponding aromatic amino derivatives of COSAN. We also discuss, herein, the influence of reaction conditions and unusual reaction pathways that were observed when applying widely known organic reactions to the chemistry of COSAN ions.Results and discussion
To prepare COSAN derivatives with aromatic amines 7–11 of general formula [(1-Ar-NH2-(CH2)3-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]0 we initially turned to the previously described method dealing with the activation of hydroxy derivatives of the COSAN anion using methanesulfonate esters.37 As we described earlier,37 the SN2-type reaction of Ms esters with primary and secondary amines proceeds well under relatively mild conditions using K2CO3 as a base in the temperature range of 40 to 50 °C. At higher temperatures, the Ms esters begin to decompose. This SN2-type reaction proceeds well with primary amines, under mild conditions, using K2CO3 or an excess of amine as a base in the temperature range of 40 to 50 °C. However, attempting similar conditions with less nucleophilic aromatic amines did not yield positive results. Even increasing the reaction temperature to 100 °C, changing the solvent to MeCN, PhMe, or THF, or experimenting with various bases like K2CO3, Cs2CO3, NaOtBu, KOtBu, or NaH only resulted in the breakdown of the starting material without the formation of the desired products. As a result, we explored alternative leaving groups in the hope of improving reactivity. In the initial trials we pursued the preparation of triflate esters43,44 [(1-(F3C-SO2)-O-(CH2)n-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]− (2a–c−, n = 1–3); however, these proved extremely labile and despite the observation of full consumption of the starting material we were unable to isolate expected products. Instead, we started to observe decomposition products that have appeared repeatedly in other experiments (vide infra). We only observed the formation of product 2c− for the propyl triflate derivative, but we were unable to isolate it using silica gel flash chromatography. Therefore, we moved to the preparation of alkyl bromides of formulae [(1-Br-(CH2)n-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]Me4N (3a–c−, n = 1–3) and corresponding iodides [(1-I-(CH2)n-1,2-C2B9H10)(1,2- C2B9H11)-3,3′-Co]Me4N (4a–c−, n = 1–3). These C-bound alkyl halides were not reported previously. The alkyl bromides of the COSAN ion 3a–c− can be easily prepared directly from the known hydroxy derivatives of the formula [(1-HO-(CH2)n-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]Me3NH (1a–c−, n = 1–3) (Scheme 1) provided that these are reacted with PBr3 in dry acetonitrile. This protocol, known from organic synthesis,45 provided the desired products in a short reaction time and without the formation of any side (halogenated) products, which were observed when using HBr. Surprisingly, all three bromo derivatives 3a–c− are stable enough to undergo a purification process on a silica gel column. Alternatively, the reaction starting from the known methanesulfonate (Ms) esters of the general formula [(1-CH3SO2-O-(CH2)n-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]Me4N (5a–c−)37 with KBr in acetone (Scheme 2) was tested in parallel. Although this reaction proceeded smoothly, it did require a longer reaction time, resulted in slightly lower conversion, and made an extra step necessary for the preparation of the respective esters, making this method less convenient for the synthesis of the bromides 3a–c−. However, the latter reaction conditions did prove advantageous for the synthesis of the respective iodides (Scheme 2), starting from methanesulfonate esters 5a–c− and NaI. In this case, the direct synthesis from hydroxy derivatives 1a–c− and TMSI as a source of iodide ions, adapted from organic chemistry,46 caused difficulties. Complications surfaced particularly in the case of compounds comprising methylene and ethylene linkers, methyl (1a−) and ethyl (1b−), where the reaction rates were too sluggish. This method (Scheme 1) proved to give satisfactory conversion only for the propyl iodide derivative 4c−, when the reaction reached almost completion. However, even then the product could not be sufficiently purified from the remaining starting reagents and organic/inorganic side products. In addition, we observed that the iodo derivatives 4a–c− are water labile species that readily decompose on silica gel. Therefore, an alternative synthesis method that would give an even better conversion, which would allow for bypassing the chromatographic purification step, was needed. Thus, an alternative method, consisting of substitution of the methanesulfonate ester with an iodide anion in acetone (Scheme 2), was developed and proved superior. Under these conditions, the propyl iodide 4c− formed smoothly in 2 hours as could be seen from the monitoring of the reaction mixture by ESI-MS. In the case of compounds 4a,b−, having the reactive centre closer to the cage, lower reaction rates were still observed. Ethyl derivative 4b− required a 5-fold excess of NaI and a prolonged reaction time of 12 hours. The methyl derivative 4a− required 72 hours of reaction time and a 10-fold excess of NaI (Fig. 1). In both cases, the reaction went to completion when heated in a sealed tube. The synthesis of COSAN alkyl halides 3a–c− and 4a–c− allowed for the preparation of a series of carbon-bound anilines 7–11, as the long-time anticipated building blocks useful in the synthesis of designed compounds with biological activity. As we described earlier,37 the SN2-type reaction of Ms esters with primary and secondary amines proceeds well under relatively mild conditions using K2CO3 as a base in a temperature range from 40 to 50 °C. At higher temperatures, the Ms esters begin to decompose. Consequently, the respective reactions with anilines proved feasible only with the use of the alkylbromides and alkyliodides described here, however, with severe limitations for the shorter methylene and ethylene linkers.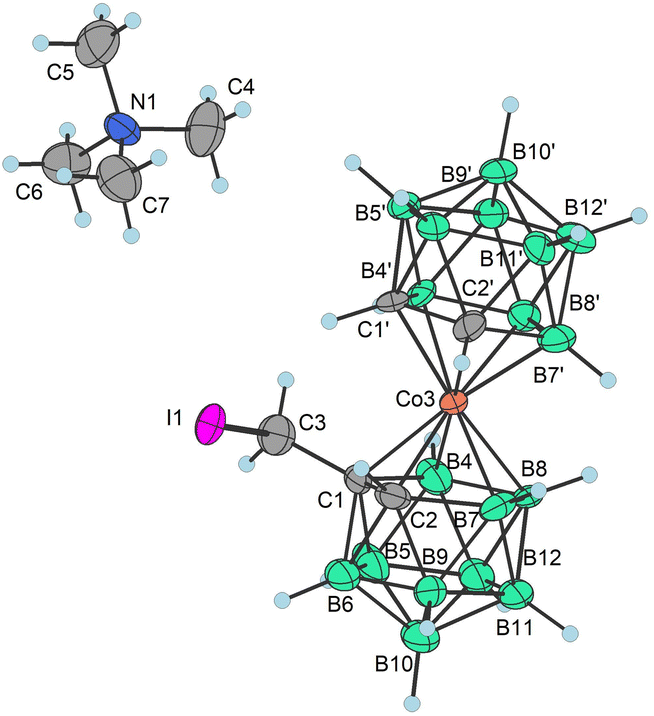 | ||
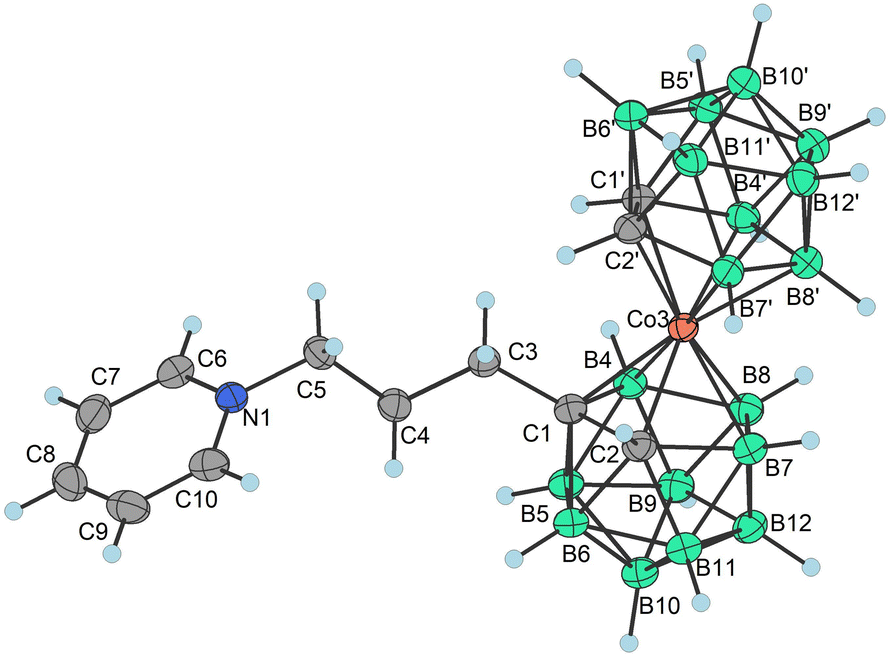
| Fig. 1 XRD structure of Me4N4a− (CCDC 2246096,† ellipsoids drawn at the 50% probability level). Selected interatomic distances [Å] and angles [°]: Co3–B4 2.101(12), Co3–B7 2.109(11), Co3–C1′ 2.045(9), Co3–B8 2.117(11), Co3–C1 2.083(9), Co3–C2 2.069(8), Co3–B8′ 2.085(11), Co3–B7′ 2.102(12), Co3–C2′ 2.058(9), Co3–B4′ 2.099(10), B4–Co3–B7 87.4(6), B4–Co3–B8 51.0(5), B4–Co3–B7′ 132.2(5), B7–Co3–B8 51.5(5), C1′–Co3–B4 130.7(5), C1′–Co3–B7 132.7(5), C1′–Co3–B8 172.8(4), C1′–Co3–B8 172.8(4), C1′–Co3–C1 101.7(4), C1′–Co3–C2 102.9(4), C1′–Co3–B8′ 82.5(4), C1′–Co3–B7′ 83.4(4), C1′–Co3–C2′ 47.3(4), C1′–Co3–B4′ 48.1(4), C1–Co3–B4 48.4(4), C1–Co3–B7 83.1(5), C1–Co3–B8 84.2(4), and C1–Co3–B8′ 135.9(4). | ||
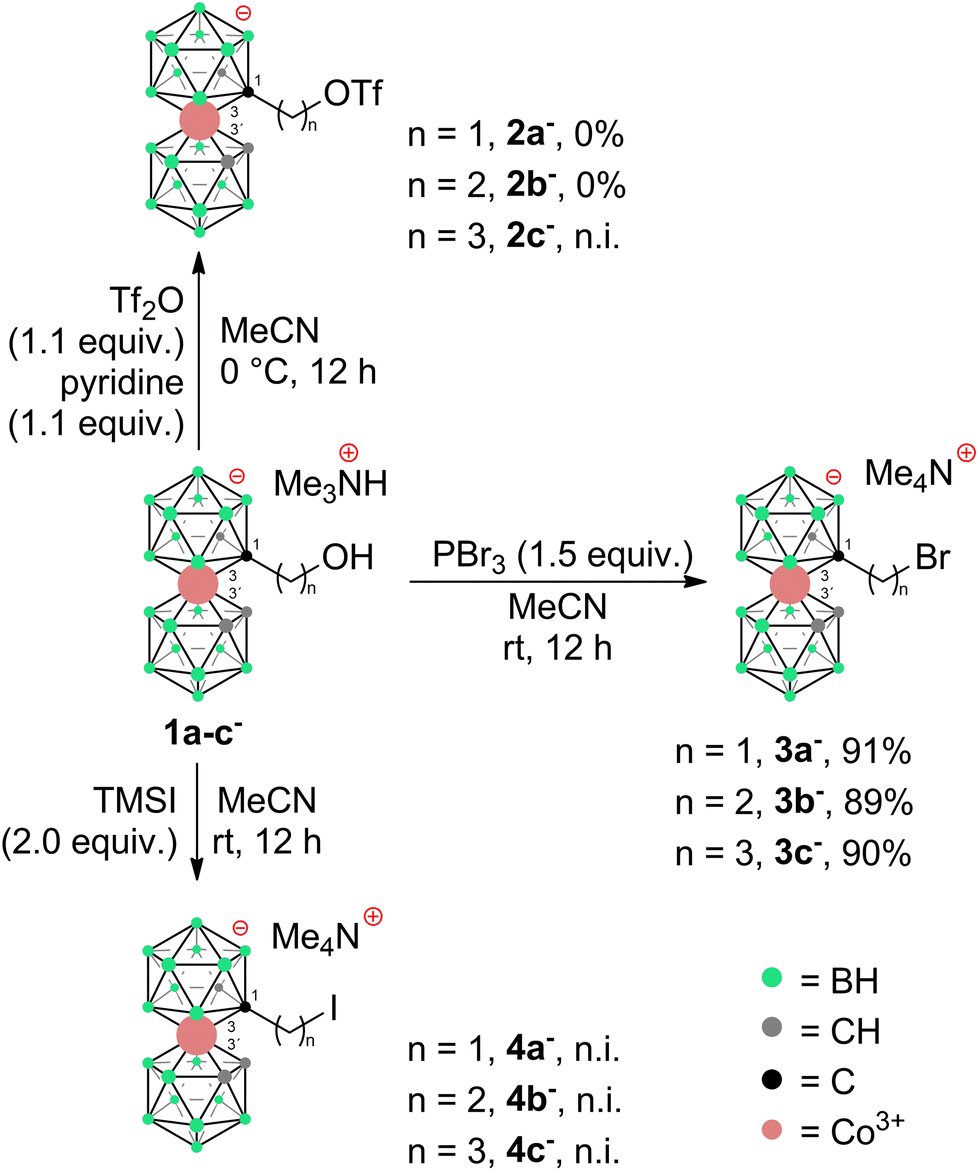 | ||
| Scheme 1 Preparation of (pseudo)halides from alkyl hydroxy COSANs 1a–c−. n.i. = not isolated. | ||
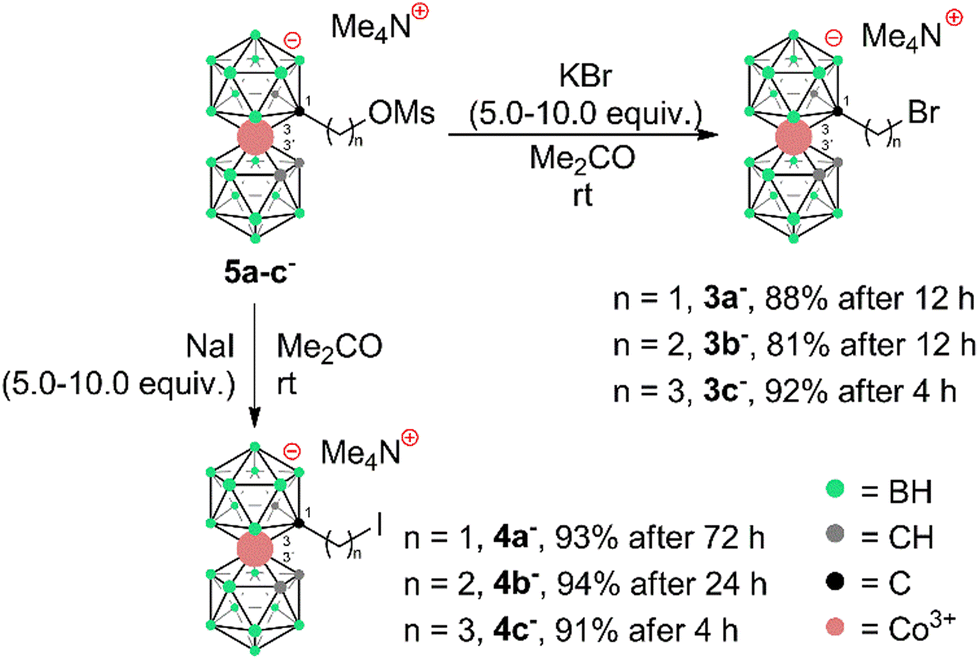 | ||
| Scheme 2 Preparation of alkyl halides 3a–c− and 4a–c− from methanesulfonate esters 5a–c−. | ||
The influence of reaction conditions on the yields of the model aniline product was tested using propylbromide 3c− and 1-amino-4-Me-benzene (p-toluidine) (6) and the data are summarized in Table 1. It can be seen that the most efficient conditions comprise 2.0 equivalents of KOtBu and aniline 6 in PhMe at 100 °C (entry 14). The entries 1–5 clearly indicate that the use of low-polar toluene as the solvent favours the formation of the expected product with the formula [(1-(1-NH2-C6H4-4-CH3)-(CH2)3-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]0 (9). Entries 6–13 reflect the effect of various bases at different temperatures, and it can be seen that the use of a strong base such as NaOtBu, KOtBu, or Cs2CO3 improves the yields; on the other hand, an increase of the temperature to 100 °C apparently plays a more important role than the particular type of base. Data in entries 13 to 15 show that increasing the ratio of 6 over two equivalents had a little effect on the resulting yield. It should be noted that the conditions were also tested using the respective iodide 4c−. However, in this case, the reactions proceed at the expense of the formation of larger quantities of decomposition products, which results in a decrease of isolatable yields (entry 16). The optimized reaction conditions were employed in the synthesis of a wider series of model arylamine derivatives (7–11) with an amine group attached to the carbon atom of the cage with a propyl pendant chain. The schematic structures are shown in Scheme 3. From the data presented in this scheme, it may be seen that the yields correlate well with the electron density at the nitrogen atom of the particular substituted aniline and are thus the highest for 4-methoxy- and 2,3,4-trimethoxy-nitroaniline and the lowest for 4-nitroaniline.
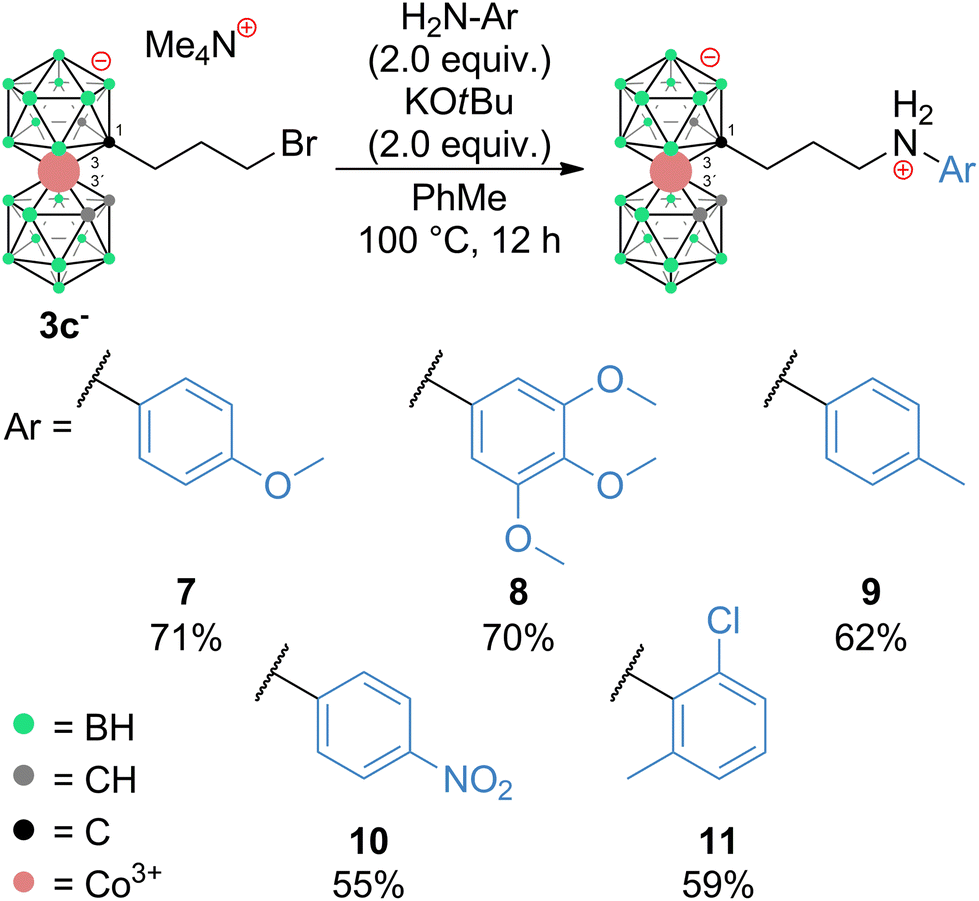 | ||
| Scheme 3 Preparation of zwitterionic aromatic amines 7–11. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | 6 (equiv.) | Basea | Solvent | Temp. (°C) | Yieldb (%) |
| a The same equivalent as that of compound 6. b Isolated yield after flash chromatography. c 6 was treated with NaH before the addition of 3c in PhMe. d Derivative 4c was used, n.r. = no reaction, trace = observed in ESI-MS but in minimum abundance and was not isolated, and time = 12 h until the complete conversion of the starting material. | |||||
| 1 | 1.5 | K2CO3 | MeCN | 60 | n.r. |
| 2 | 1.5 | K2CO3 | MeCN | 80 | Trace |
| 3 | 1.5 | K2CO3 | THF | 80 | Trace |
| 4 | 1.5 | K2CO3 | 1,4-Dioxane | 80 | Trace |
| 5 | 1.5 | K2CO3 | PhMe | 80 | 10 |
| 6 | 1.5 | Cs2CO3 | PhMe | 80 | 21 |
| 7 | 1.5 | KOtBu | PhMe | 80 | 20 |
| 8 | 1.5 | NaOtBu | PhMe | 80 | 15 |
| 9 | 1.5 | NaHc | PhMe | 80 | n.r. |
| 10 | 1.5 | K2CO3 | PhMe | 100 | 35 |
| 11 | 1.5 | Cs2CO3 | PhMe | 100 | 48 |
| 12 | 1.5 | NaOtBu | PhMe | 100 | 45 |
| 13 | 1.5 | KOtBu | PhMe | 100 | 49 |
| 14 | 2.0 | KOtBu | PhMe | 100 | 62 |
| 15 | 2.5 | KOtBu | PhMe | 100 | 63 |
| 16d | 2.0 | KOtBu | PhMe | 100 | 45 |
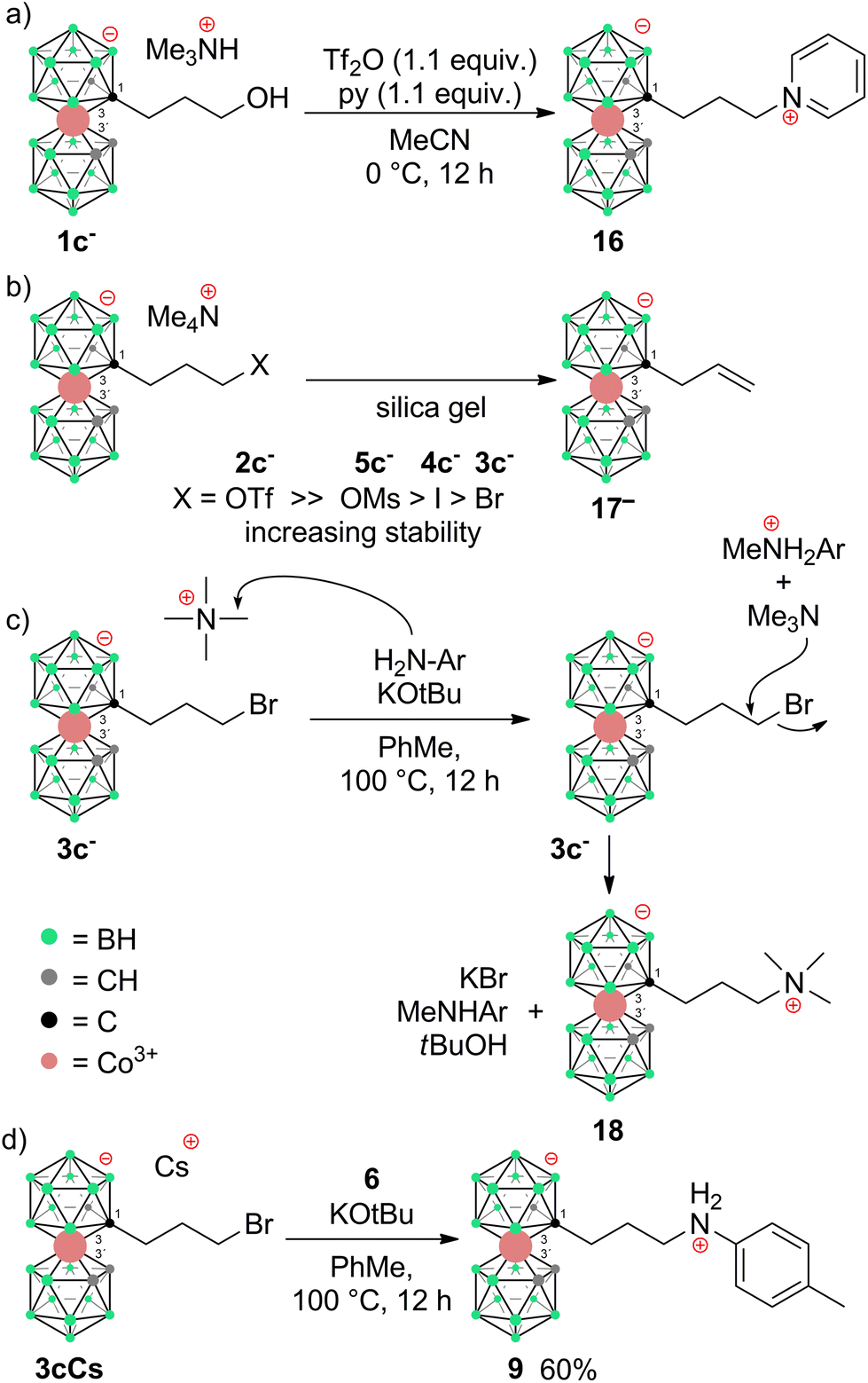
The newly developed reaction conditions were further applied in trials to employ methyl (3a−, 4a−) and ethyl (3b−, 4b−) halide derivatives to synthesize the corresponding anilines with a shorter connector to the cage. However, these attempts did not provide the expected products. Instead, different types of reactions were observed, which could be elucidated only for ethyl halides. In this particular case, high-yield formation of a main product was observed, which remained identical regardless of the type of terminal halogen moiety in the starting compound. This compound forms in two isomeric forms when the solid (pseudo)halide is heated in solution or in the solid state. This unconventional type of substitution contains a diatomic ethylene bridge asymmetrically located between carbon (C1) and boron B(7′) or B(8′) positions and corresponds to the formulae [(1,7′-μ-C2H4)-(1,2-C2B9H10)(1′,2′-C2B9H10)-3,3′-Co]Me4N (15a−) and [(1,8′-μ-C2H4)-(1,2-C2B9H10)(1′,2′-C2B9H10)-3,3′-Co]Me4N (15b−). Both isomers were purified by column chromatography on silica gel and then separated by flash chromatography on a semi-preparative Büchi RP WP column (see the ESI†). Such a type of cage substitution, that interconnects carbon and boron atoms, is unique and has not been previously reported. These products apparently form via an intramolecular hydroboration mechanism outlined in Fig. 2. We suggest that, due to the electronic demands of the boron cluster and the lability of halide or pseudohalide leaving groups, the ethyl chain of transient species {12−} is prone to the dissociation of the terminal halide of the pseudohalide group via an elimination mechanism. A proton in the beta position in the resulting electroneutral intermediate {13} is then either abstracted by a nucleophile, i.e. free halide, methanesulfonate, or triflate anions, or simply released by thermal treatment, resulting in the formation of an unsaturated bond present in another assumed transient species {14−}. The reaction then proceeds as hydroboration of the double bond by the most hydridic Bδ+(8′)–Hδ− hydrogen atom that results in an intramolecular cyclization reaction providing the five membered cyclic moiety (considering also the involvement of a Co(III) central atom) present in the final isolatable product 15b−. On the other hand, the substitution proceeds also on the Bδ+(7′)–Hδ− bond that is in the vicinal position to one carbon atom and has a comparatively lower electron density. The direct substitution of this boron position has never been described before in the literature. The reason could be that most of the reactions on the B(8) site proceed via Electrophile Induced Nucleophilic Substitution (EINS), which is different from the mechanism outlined here.
 | ||
| Fig. 2 Left: the proposed mechanism for the formation of Me4N15a,b. Right: the X-ray structure of Me4N15b plotted for t = 0; for crystallographic parameters, selected interatomic distances and angles see Table S1 in the ESI.† | ||
All new compounds were unequivocally characterized by NMR, HRMS, and, in the case of the Me4N15b species, single-crystal X-ray crystallography. The crystal structure of Me4N15b is incommensurately modulated, with q-vector 0.4584(2) b* and superspace symmetry P21/n(0β0)00. The q-vector is necessary to index satellite reflections in the diffraction pattern, for which three Miller indices cannot be described because the q-vector components are not simple rational numbers. With reflections indexed by more than four indices, the modulated structure must be calculated in a (3 + 1)-dimensional superspace, with a four-dimensional unit cell and each atom described by a modulation function. The crystal structure is then obtained as a three-dimensional section through the superspace, where the t coordinate defines the position of the section. By changing t in the <0,1) interval, we can obtain all possible configurations of the modulated structure. More details about modulated structures can be found in ref. 47. The ion 15b− is rigid and asymmetric due to the presence of the bridge. The asymmetric part of the incommensurately modulated structure of 15b− is shown in Fig. 2 for t = 0. The crystal structure of 15b− was refined with Jana2020![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48 as two independent rigid bodies [(C2B9H10)2−] and free Co atoms and the bridging C2H4. We found that the molecule was harmonically modulated with large modulation amplitudes of about 0.6 Å and, despite modulations, the whole molecule was rigid, with the C22a–C12a–B91a–B81a torsion angle lying between 151.4° and 154.4°, and the dihedral (C11a C21a B71a B91a B81a)–(C12a C22a B92a B82a B72a) angle lies between 3.2° and 4.6°. The bond length between carbon atoms in the ethylene bridging unit corresponds to 1.535 Å, which indicates the presence of a simple bond. The large torsion angle corresponds rather to a transoid-conformation of the ligand planes. As has been discussed previously in the literature,49 this conformation is rare, and in fact, could be seen only in solid-state structures of the caesium salt of the parent ion and compounds substituted with heavier halogens or heterosubstituted with Ph and iodine. In the latter case, the formation of H–Hal or B–H–Ar bonds is apparently responsible for transoid-arrangement.50,51 The conformations of all other substituted derivatives reported in CCDC correspond to cisoid or gauche-arrangement. Therefore, the high torsion angle observed in this structure is unique between the substituted derivatives of the COSAN ion, and in particular when considering bridge structures with a restrained geometry.2 In turn, the dihedral angle between (C11a C21a B71a B91a B81a) and (C12a C22a B92a B82a B72a) is small, with a mean value of 3.9°, which is in line with the diatomic character of the bridge.
48 as two independent rigid bodies [(C2B9H10)2−] and free Co atoms and the bridging C2H4. We found that the molecule was harmonically modulated with large modulation amplitudes of about 0.6 Å and, despite modulations, the whole molecule was rigid, with the C22a–C12a–B91a–B81a torsion angle lying between 151.4° and 154.4°, and the dihedral (C11a C21a B71a B91a B81a)–(C12a C22a B92a B82a B72a) angle lies between 3.2° and 4.6°. The bond length between carbon atoms in the ethylene bridging unit corresponds to 1.535 Å, which indicates the presence of a simple bond. The large torsion angle corresponds rather to a transoid-conformation of the ligand planes. As has been discussed previously in the literature,49 this conformation is rare, and in fact, could be seen only in solid-state structures of the caesium salt of the parent ion and compounds substituted with heavier halogens or heterosubstituted with Ph and iodine. In the latter case, the formation of H–Hal or B–H–Ar bonds is apparently responsible for transoid-arrangement.50,51 The conformations of all other substituted derivatives reported in CCDC correspond to cisoid or gauche-arrangement. Therefore, the high torsion angle observed in this structure is unique between the substituted derivatives of the COSAN ion, and in particular when considering bridge structures with a restrained geometry.2 In turn, the dihedral angle between (C11a C21a B71a B91a B81a) and (C12a C22a B92a B82a B72a) is small, with a mean value of 3.9°, which is in line with the diatomic character of the bridge.
The Me4N+ cation (TMA) molecule was refined as a rigid body flipping between two major configurations following the crenel modulation function.52 Moreover, each position is accompanied by another rotated minor occupied position following the same crenel function. The difference between disordered and modulated structures consists of the presence of satellites. Disordered structures contain statistically distributed variants of configurations, which do not possess translation periodicity and, therefore, cannot give rise to additional Bragg peaks. Modulated structures contain variants of configurations, which exhibit translation periodicity in the superspace, giving rise to additional Bragg peaks – satellites. The crystal structure of Me4N15b is a rare example of an incommensurately modulated disordered structure because the major and minor variants of the TMA configuration cannot be separated (this is the disorder) while their flipped positions possess the translational periodicity in the superspace and are described by the modulation function.
The extraordinarily strong modulation amplitudes in the crystal structure of Me4N15b can be explained by missing hydrogen bonds. Fig. S2 (see the ESI†) shows the TMA molecule surrounded by the four nearest [1,8′-μ-CH2CH2-(1,2-C2B9H10)2-3,3′-Co]− anions plotted for two t values. We can see that TMA flips, probably when some neighbouring cages come too close, causing H–H repulsion.
It should be noted that COSAN ions bridged between carbon atoms by alkyl chain groups are known from the literature; all of which had been prepared by tedious indirect methods of cobalt insertion into modified dicarbollide ions.2,53 Only a solitary report on a similar compound prepared via direct substitution can be found in the literature that also supports the proposed mechanism.54 In this case, an unsaturated symmetrically located ethylene bridge that interconnects two boron (B8,8′) positions formed under Pd catalysed reaction between the trimethylsilyl alkyne and the B(8)–I derivative of the COSAN anion in the presence of CuI and DIPEA. Also here, the hydroboration mechanism is considered as being essential for the observed intramolecular cyclization along with the cooperative action of B–I and B–H sites of the boron cage.
The second isomer 15a− did not crystallize well and could be characterized only by spectroscopic methods. However, its structure should parallel the pattern observed in the disubstituted species (see in the paragraph below). Additional support for the proposed mechanism could follow from later observations showing that compound 15− is also a common product of thermal degradation (>60 °C) of the methanesulfonate ester 5b− either in solution or in the solid state. Indeed, the synthesis of the solid ester proved finally as the most convenient method for the preparation of the isomeric bridged products 15a,b−. The bridged product(s) could be observed by HRMS during the esterification of ethyl hydroxy derivative 5c− using Tf2O in the presence of pyridine. Compound 15− also formed during the purification processes of alkyl triflate 2c− and iodo derivatives 4b− on silica gel. Therefore, for an ethylene connector, all these conditions easily result in the molecule being asymmetrically substituted with an ethylene bridge.
Surprisingly enough, this intramolecular cyclization has almost not appeared while reacting, heating or purifying propylbromo derivative 3b−. Indeed, the derivative 3b− is stable enough to withstand flash chromatography on silica gel and can also be smoothly precipitated from water as its Me4N+ salt. However, it should be noted that this salt must be rapidly isolated by filtration and dried in a vacuum. Thus, considering the length of the alkyl pendant arm, the reaction pathways are significantly altered going from the propyl to ethyl chain. This can be reasonably ascribed to the proximal effect of the COSAN ion. This is shown here in the simplest model of (pseudo)halide terminal groups. We believe, such substantial alterations in the reaction pathway, depending on the length of the pendant arm, reflect the unique properties of the boron cluster ions. Apparently, the energetically favourable formation of a five membered cycle contributes to the formation of a stable bridging arrangement. As follows from the observations of several minor products, a tendency to undergo unexpected substitution or elimination pathways was also observed for the corresponding propyl halides. The first isolated product corresponds to the expected pathway, i.e. the SN2-type reaction product is the propylpyridinium derivative [(1-(C6H5N)-(CH2)3-1,2-C2B9H10)(1′,2′-C2B9H11)-3,3′-Co]016 formed spontaneously in small quantities in reactions of 2c− when pyridine was used as a base. This compound could be isolated and characterized by crystallography (Scheme 4a; X-ray structure in Fig. 3). Also, we observed a mass m/z = 364, which corresponds to the product of elimination of the terminal halide groups and it can be formulated as an allyl derivative {17−} (Scheme 4b). Again, this ion proved to be extremely unstable and underwent a variety of subsequent reactions. However, we could not confirm the final hydroboration step and formation of a triatomic bridge in this case since the exact masses of {17−} and the bridge product are the same. We could get some evidence about bridge formation from a combination of analytical HPLC and MS, which showed several peaks of the same mass. However, the mixture of products was richer in this case, which precluded the isolation of pure compounds. This product appeared repeatedly during every experiment using propyl derivatives 2c− to 4c−, and Ms ester 5c−, and could be observed in HRMS of the reaction mixtures.
 | ||
| Scheme 4 (a) Zwitterionic pyridinium molecule 16, (b) expected transient elimination product {17−}, (c) the disproportionation of the TMA cation and the formation of 18, and (d) reaction of the 3cCs salt tested as an alternative. | ||
 | ||
| Fig. 3 Molecular structure of 16 (CCDC 2246097,† ellipsoids drawn at the 50% probability level). Selected interatomic distances [Å] and angles [°]: Co3–B4 2.064(3), Co3–B7 2.100(3), Co3–C1′ 2.064(3), Co3–B8 2.091(3), Co3–C1 2.108(2), Co3–C2 2.083(3), Co3–B8′ 2.113(3), Co3–B7′ 2.094(3), Co3–C2′ 2.051(3), Co3–B4′ 2.118(3), B4–Co3–B7 85.16(12), B4–Co3–B8 49.43(11), B4–Co3–B7′ 173.50(12), B7–Co3–B8 51.13(12), C2–Co3–C1 47.55(10), B7–Co3–C1 84.49(11), B7′–Co3–C1 133.56(11), B8–Co3–C1 84.03(11), B4–Co3–B8′ 127.92(12), B4–Co3–B4′ 96.25(12), C2–Co3–B7′ 94.44(12), C2–Co3–B8′ 131.07(11), C2–Co3–B4′ 178.52(11), C2–Co3–B7 50.57(12), C2–Co3–B8 86.31(13), B7′–Co3–B8′ 50.81(13), B7′–Co3–B4′ 86.97(13), B7′–Co3–B7 88.44(13), B8′–Co3–B4′ 50.31(12), B7–Co3–C1 84.49(11), B7–Co3–B8′ 90.29(12), B7–Co3–B4′ 130.01(12), B8–Co3–B7′ 124.89(12), B8–Co3–B8′ 88.95(13), and B8–Co3–B4′ 93.26(13). | ||
Apparently, the species {17−} formed as a thermal decomposition product of 2c−. It was also observed during the chromatographic separation of 4c− and 5c− on silica gel as a side product that results upon thermal degradation of these compounds at temperatures higher than 60 °C. Based on the lowest relative abundance of the {17−} derivative observed in the reaction mixtures of propylbromide 3c− we conclude that the latter derivative is the most stable and suitable compound for alkylation of its amine groups. This also correlates with the leaving group's ability to be released from the C–Br bond.55,56 Another side product, which we were able to isolate and characterize is the zwitterionic compound of the formula [(1-(CH3)3N-(CH2)3-1,2-C2B9H10)(1,2-C2B9H11)-3,3′-Co]0 (18). This compound was fully characterized including by XRD. We suppose that during the reaction, the TMA counter ion disproportionates thermally with anilines57 or undergoes Hofmann elimination of tertiary amines producing Me3N which is more basic and nucleophilic than the aromatic amines (Scheme 4c; X-ray structure in Fig. 4).58 Therefore, we have tried the preparation of 9 under alternative reaction conditions using the Cs+ salt of 3c− as a starting material, see the ESI.† The exchange of the cation prevented the formation of 18; however, the overall yield of 9 was not improved, and the formation of 17− (Scheme 4d) could be still seen. Suitable single crystals of 16 and 18 were obtained and the compounds were characterized using single crystal XRD analysis (see Fig. 4 and 5, and the ESI† for structural refinement and crystallographic data). The molecular structures of 16 and 18 unambiguously confirmed the particular type of substitution and the zwitterionic character of both compounds. As we found later, the thermal decomposition of C(1), C(1′) disubstituted ethyl methanesulfonyl ester, rac-[(1,1′-(MsOC2H4)2-1,2-C2B9H10)2-3,3′-Co]Me4N (19)6,9,37 at 90 °C for 48 h provides high yields of an isomeric mixture consisting of doubly bridged compounds [(1,7-μ-C2H4-1,2-C2B9H9)2-3,3′-Co]Me4N (Me4N20a) and [(1,8-μ-C2H4-1,2-C2B9H9)2-3,3′-Co]Me4N (Me4N20b) (see Scheme 5 and Fig. 5 and 6).
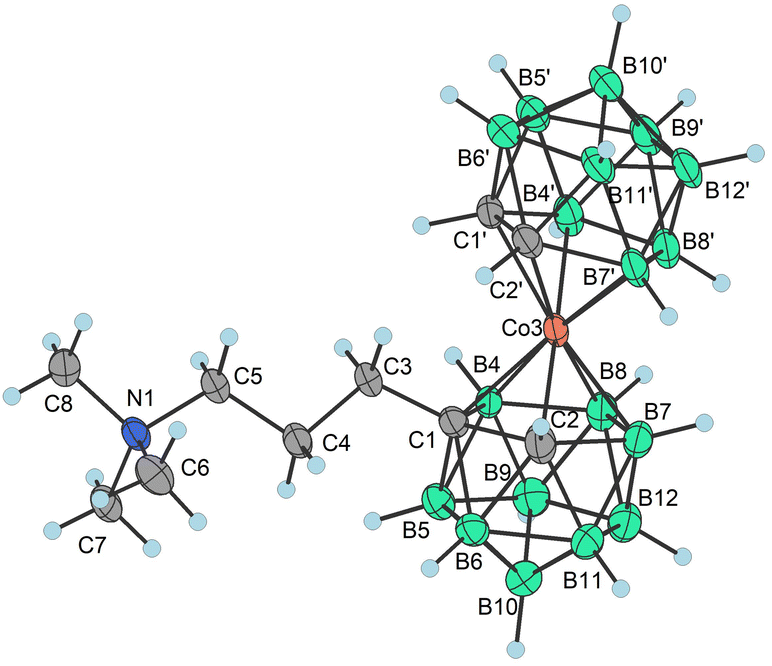 | ||
| Fig. 4 Molecular structure of 18 (CCDC 2246098,† ellipsoids drawn at the 50% probability level). Selected interatomic distances [Å] and angles [°]: Co3–C1′ 2.0499(16), Co3–C1 2.1067(16), Co3–C2′ 2.0736(16), Co3–C2 2.0572(17), Co3–B4′ 2.0914(18), Co3–B8′ 2.1230(19), Co3–B4 2.0905(19), Co3–B7′ 2.1085(19), Co3–B7′ 2.1085(19), Co3–B1 2.0998(19), Co3–B7 2.0854(19), C2′–Co3–B1 170.40(7), C2′–Co3–B7 135.79(7), C2– Co3–C1 46.67(6), C2–Co3–C2′ 104.93(7), C2–Co3–B4′ 171.75(7), C2–Co3–B8′ 125.79(7), C2–Co3–B4 82.35(7), C2–Co3–B7′95.70(7), C2–Co3–B1 84.67(7), C2–Co3–B7 49.43(7), B4′–Co3–C1 135.89(7), B4′–Co3–B8′ 50.91(7), B4′–Co3–B7′ 86.89(7), B4′–Co3–B187.81(8), B4–Co3–C1 47.73(7), B4–Co3–B4′ 95.53(8), B4–Co3–B8′ 133.36(7), B4–Co3–B7′ 175.99(7), B4–Co3–B1 50.88(7), B7′–Co3–B8′ 50.53(7), B1–Co3–C1 84.48(7), B1–Co3–B8′ 91.53(7), B1–Co3–B7′132.57(7), B7–Co3–C1 84.34(7), B7–Co3–B4′ 122.64(8), B7–Co3–B8′ 87.49(7), B7–Co3–B4 87.10(8), B7–Co3–B7′ 94.30(7), and B7–Co3–B1 51.21(7). | ||
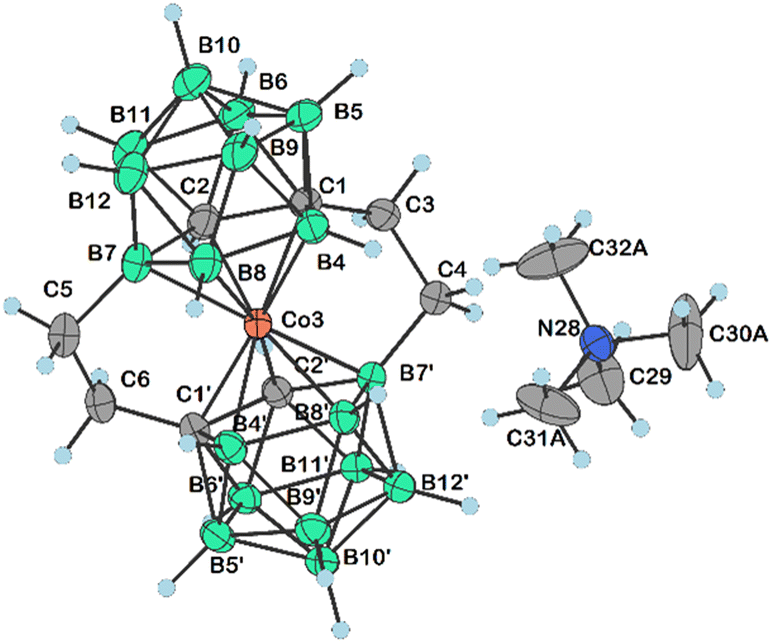 | ||
| Fig. 5 Molecular structure of 20a− (CCDC 2313358,† ellipsoids drawn at the 50% probability level). The CH2Cl2 solvent molecule has been omitted for clarity. Selected interatomic distances [Å] and angles [°]: C4–C3 1.532(3), C6–C5 1.538(3), C1–C3 1.528(3), C2–C1 1.666(3), C2′–C1′ 1.659(3), C1′–C6 1.530(3), B7–C5 1.604(3), C2–B7 1.718(3), B6′–C1′ 1.739(3), C4–B7′ 1.590(3), C1–B6 1.737(3), Co3–C2′ 2.022(2), Co3–C2 2.030(2), Co3–C1 2.037(2), Co3–B4′ 2.089(2), Co3–B7 2.081(2), Co3–C1′ 2.039(2), Co3–B42.076(2), Co3–B8′ 2.119(2), Co3–B7′ 2.078(2), Co3–B8 2.101(2), B7–B11, 1.811(4), B7–B12 1.786(4), B7–B8 1.812(3), C1′–B5′ 1.705(3), B11′–B7′ 1.802(3), B12′–B7′ 1.775(3), B8′–B7′ 1.808(3), C3–C1–C2 120.10(17), C1–C3–C4 112.39(17), C6–C5–B7 111.04(17), C3–C4–B7′ 110.18(16), C1–Co3–B4′ 167.46(9), C1–Co3–B7 86.25(9), C1–Co3–C1′ 142.61(8), C3–C1–Co3 108.09(13), B7′–Co3–B8 143.56(9), B8–Co3–B8′ 102.03(10), C1′–C2′–Co3 66.46(10), C1′–C2′–B7′ 112.63(15), B11′–C2′–B7′ 63.96(13), C1–C2–B7 112.58(16), C5–B7–C2 119.74(18), C5–B7–B11 122.89(18), C5–B7–B12 129.47(18), C5–B7–B8 125.47(18), B6–C2–B7 115.99(17), C1′–B6′–B10′ 105.71(16), C1′–B6′–B11′ 105.08(15), C2–C1–B4 109.48(15), C3–C1–B5, 119.70(17), C3–C1–B4 122.19(17), C3–C1–B6 115.93(17), C1′–B4′–B8′ 106.83(16), C2–B7–B8 104.15(16), C5–B7–Co3 106.05(15), B11–B7–Co3 116.05(14), B11–B7–B8 107.05(18), C2′–C1′–Co3 65.33(10), C2′–C1′–B4′ 109.46(15), C6–C1′–Co3 108.27(14), C6–C1′–C2′ 119.40(16), C6–C1′–B6′ 114.83(16), C6–C1′–B4′ 123.11(17), C6–C1′–B5′ 120.21(17), C1′–C6–C5 112.07(17), C1–B4–Co3 64.31(10), B8–B4–Co3 65.29(11), B4′–B8′–B7′ 106.63(15), C2–B6–C1 57.67(12), C2′–B7′–Co3, 63.52(10), C2′–B7′–B8′ 104.33(15), C4–B7′–Co3 106.78(14), C4–B7′–C2′ 120.76(17), C4–B7′–B11′ 122.04(17), C4–B7′–B12′ 127.90(17), C4–B7′–B8′ 125.12(17), B8′–B7′–Co3 65.65(10), B7–B8–Co3 63.78(11), and B4–B8–B7 106.86(17). | ||
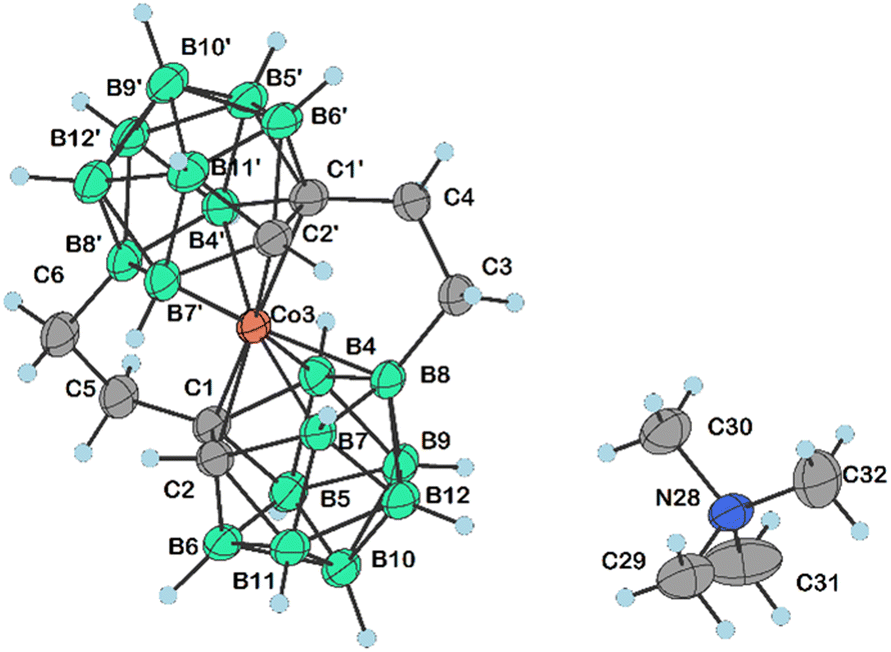 | ||
| Fig. 6 Molecular structure of 20b− (CCDC 2313381,† ellipsoids drawn at the 50% probability level). Selected interatomic distances [Å] and angles [°]: C4–C3 1.541(4), C6–C5 1.556(4), C1–C5 1.536(3), C4–C1′ 1.535(4), C1–C2 1.645(4), C1′–C2′ 1.646(4), Co3–C1 2.026(2), Co3–C1′ 2.024(3), Co3–B7 2.088(3), Co3–B4 2.076(3), Co3–B4′ 2.082(3), Co3–B8 2.106(3), Co3–B7′ 2.080(3), C2′–B7′ 1.709(4), C1–B4 1.736(4), C1–B6 1.745(4), C1–B5 1.723(4), C1′–B4′ 1.733(4), C1′–B6′ 1.742(4), C1′–B5′ 1.708(4), C6–B8′ 1.610(4), B7–B8 1.803(5), B4–B8 1.807(4), C3–B8 1.613(4), B8–B12 1.807(4), B8–B9 1.797(4), B8′–B12′ 1.811(4), B8′–B9′ 1.805(4), C1–C5–C6 113.1(2), C5–C1–C2 119.5(2), C1′–C4–C3 113.1(2), C4–C1′–C2′ 119.8(2), C4–C3–B8 110.9(2), C5–C1–B4 123.4(2), C5–C1–B6 115.4(2), C5–C1–B5 120.1(2), C1–C2–B7 112.6(2), C1–C2–B11 112.8(2), C2–C1–B4 109.51(19), C2–C1–B5 108.6(2), C5–C1–Co3 109.04(17), B4–C1–B6 112.0(2), B6–C1–Co3 122.11(17), B5–C1–Co3 122.28(17), C2′–C1′–B4′ 109.4(2), C2′–C1′–B5′ 108.9(2), C4–C1′–Co3 109.46(17), C4–C1′–B4′ 123.6(2), C4–C1′–B6′ 114.7(2), C4–C1′–B5′ 119.3(2), B6′–C1′–Co3 122.21(19), B5′–C1′–Co3 122.67(18), C5–C6–B8′ 110.5(2), B11–B7–B8 109.2(2), C1–B4–B8 106.4(2), C1–B4–B9 105.7(2), B5–B4–B8 108.6(2), C1′–B4′–Co3 63.32(13), C1′–B4′–B8′ 106.5(2), C1′–B4′–B9′ 105.4(2), B5′–B4′–B8′ 109.3(2), B7–B8–B4 106.0(2), B4–B8–B12 106.2(2), C3–B8–Co3 105.42(18), C3–B8–B7 121.8(2), C3–B8–B4 119.8(2), C3–B8–B12 128.6(2), C3–B8–B9 127.3(2), B12–B8–Co3 115.19(19), B9–B8–Co3 115.32(17), B9–B8–B7 105.8(2), C6–B8′–Co3 105.38(18), C6–B8′–B4′ 120.6(2), C6–B8′–B7′ 121.1(2), C6–B8′–B12′ 128.3(2), B4′–B8′–B12′ 105.6(2), B7′–B8′–B9′ 105.4(2), B12′–B8′–Co3 115.45(18), B9′–B8′–Co3 114.74(18), B12′–B7′–Co3 118.1(2), B11′–B7′–B8′ 109.1(2), B10–B12–B8108.7(2), B11–B12–B8 109.1(2), C1′–B6′–B11′ 105.0(2), C1′–B6′–B10′ 105.8(2), B11′–B12′–B8′ 108.5(2), B10′–B12′–B8′ 109.1(2), C1–B6–B10 106.1(2), C1–B6–B11 105.4(2), C1–B6–B5 59.02(17), B10–B9–B8 109.3(2), B5–B9–B8 108.8(2), and C1′–B5′–B10′ 106.8(2). | ||
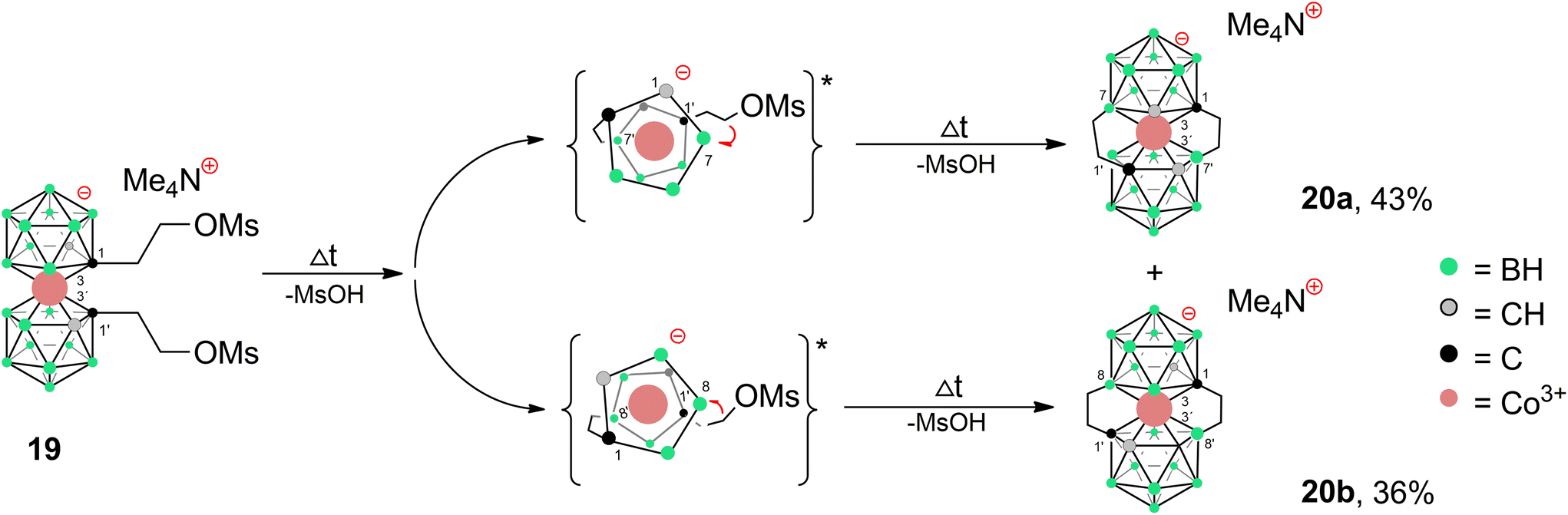 | ||
| Scheme 5 Thermally induced cyclization reaction of rac-dimesyl ester 19Me4N38 leading to doubly-bridged products 20a,b. Reaction conditions: 95 °C, 60 h; two step formation of isomers is assumed. Two transient singly-bridged isomers with a restrained geometry and a favourably short distance between C(1′)–B(7) and C(1′)–B(8) sites (around 2.9 Å in the XRD pattern of products 20a,b) are expected to form in the first step. This is considered as the main factor responsible for the regioselective formation of the second bridge, because of the significantly longer distance (over 3.5 Å in 20a,b) between C(1′) and other boron sites. *Only the ligand planes around the cobalt atoms are drawn for clarity in the reaction intermediates. | ||
Both isomers were separated by RP chromatography and crystallized (see the ESI†). The molecular structures determined by XRD are depicted in Fig. 5 and 6. Such a type of doubly bridged substitution has no precedent in the chemistry of the COSAN ion and produces an even more rigid and highly twisted arrangement of boron planes adjacent to the cobalt atom (Fig. 7). The bonds between carbon atoms have aliphatic character, the C–C distance in the ethylene bridges correspond to 1.53 and 1.54 Å in 20a− and 1.54 and 1.56 Å in 20b−, respectively. The substitution apparently forms uniformly via the doubly-repeated hydroboration reaction mechanism described above for the monosubstituted compound(s) and is shown in Fig. 2. Apparently, the mechanism is stepwise, which is supported by MS analysis of the reaction mixtures in early stages of thermal conversion, which shows clearly the molecular peak of a singly bridged {(μ-C2H4)(MsOC2H4)-(1,2-C2B9H10)2-3,3′-Co} intermediate product (not isolated). Also, the second cyclization step proceeds easily starting from two isomeric species bridged in positions identical to Me4N15a and Me4N15b (Fig. 2 and Scheme 5). This results in the clean formation of two isomers with the largest separations between both bridging groups. The two isomeric anions 20a,b− thus exhibit two ethylene “handles” on each side of the cluster anion that are mutually twisted forming a propeller shape. Such an arrangement conforms to axial chirality due to the twisting of the carbon positions and the zig-zag orientation of the ethylene units (see Fig. 7). The torsion angles between C1, C2 and C1′, C2′ atoms are 54.5° and 163.4° and the conformation is thus close to gauche- and transoid-arrangements, respectively.
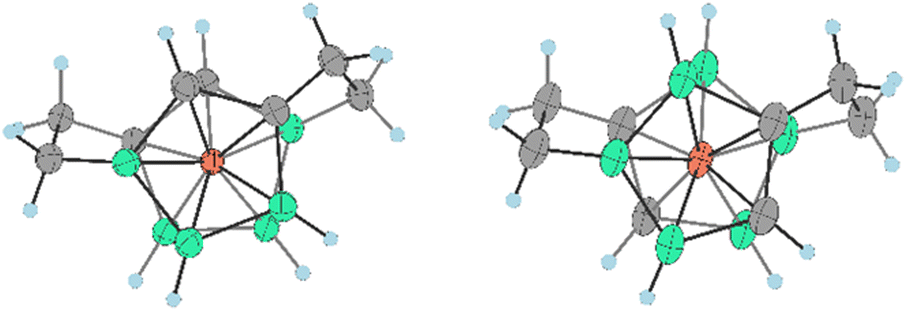 | ||
| Fig. 7 Orientation of skeletal carbon atoms in ligand planes adjacent to the cobalt atom and the ethylene bridging groups in the bis(bridged) anions 20a− (left) and 20b− (right). For selected angles see the Fig. 6 caption. | ||
Conclusions
We have successfully created new carbon-bound alkyl halides of the COSAN(1-) ion with excellent yields, avoiding the common issue of side skeletal halogenations on boron atoms that often occur with hydrohalogenic acids. The terminal halogen atoms in these products, especially for bromo derivatives, are poor leaving groups that can withstand the relatively high temperatures necessary for the subsequent nucleophilic substitution with low nucleophilic amino groups in anilines. These alkyl halides could be crucial for advancing the metallacarborane chemistry. The reactions with aromatic amines reliably yield good amounts of COSAN substituted with terminal propylaniline groups, a result hard to achieve through other methods. While propylene halides provided expected yields, congeners with a halogen atom adjacent to the anionic and bulky cage with a shorter pendant arm showed significantly different and unexpected reactivity. Here, we describe a previously unknown intramolecular cyclization reaction leading to substitution with an ethylene bridge between C(1) and B(7′) or B(8′) sites of the cobaltacarborane cage. The distinct reactivity of terminal groups, different from common organic or ferrocene chemistry, highlights the influence of the bulky anionic cluster anion. Beyond high steric strain and anionic charge, these pathways may involve reactions with electron-rich hydridic B–H bonds in the ligand planes attached to the cobalt atom. Under specific conditions, this can result in ansa-substitution with an ethylene bridge, producing compounds with a restrained geometry seen in isomeric products 15a,b−. Surprisingly, these pathways can also be utilized in the high-yield synthesis of bis(bridged) derivatives 20a,b. All ansa-substituted compounds exhibit a rigid arrangement of highly twisted ligand planes, potentially serving as an asymmetric platform for generating chiral COSAN derivatives with various functional groups. Preliminary observations suggest that aromatic amine derivatives of the COSAN(1-) ion effectively target several bacterial strains in ongoing studies.Data availability
The ESI† can be found at the RSC website including complete experimental methods, spectral data, 1H, 11B and 13C NMR spectra, and crystallographic data (pdf). Video (gif) and cif files of the modulated structure Me4N15 are attached as separate files.Author contributions
J. N. and B. G. designed experiments and supervised the experimental work, E. T. contributed to synthesis, separation of compounds and NMR characterization, and D. B. performed characterization using LC-MS and MS techniques. M. L. and M. D. performed X-ray structure measurements and refining of the structures. J. N. was responsible for preparing the final version of the MS, and B. G. and M. D. contributed to writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Czech Science Foundation, project number 21-14409S. The XRD analysis of Me4N15 involved the use of resources from the Czech Nanolab infrastructure (MEYS CR, project LM2023051).References
- M. F. Hawthorne and T. D. Andrews, Chem. Commun., 1965, 19, 443 RSC.
- R. N. Grimes, Carboranes, Academic Press, 2016 Search PubMed.
- B. P. Dash, R. Satapathy, B. R. Swain, C. S. Mahanta, B. B. Jena and N. S. Hosmane, J. Org. Chem., 2017, 849–850, 170–194 CrossRef CAS.
- R. N. Grimes, in Metallacarboranes of the transition and lanthanide elements, ed. A. Press, Elsevier, London, UK, 2016, pp. 711–903 Search PubMed.
- I. B. Sivaev and V. I. Bregadze, Collect. Czech. Chem. Commun., 1999, 64, 783–805 CrossRef CAS.
- L. Pazderová, E. Z. Tüzün, D. Bavol, M. Litecká, L. Fojt and B. Grűner, Molecules, 2023, 28, 6971 CrossRef PubMed.
- Z. J. Leśnikowski, E. Paradowska, A. B. Olejniczak, M. Studzińska, P. Seekamp, U. Schuessler, D. Gabel, R. F. Schinazi and J. Plešek, Bioorg. Med. Chem., 2005, 13, 4168–4175 CrossRef PubMed.
- I. Fuentes, T. García-Mendiola, S. Sato, M. Pita, H. Nakamura, E. Lorenzo, F. Teixidor, F. Marques and C. Viñas, Chem. – Eur. J., 2018, 24, 17239–17254 CrossRef CAS PubMed.
- P. Řezáčová, J. Pokorná, J. Brynda, M. Kožíšek, P. Cígler, M. Lepšík, J. Fanfrlík, J. Řezáč, K. Grantz Šašková, I. Sieglová, J. Plešek, V. Šícha, B. Grüner, H. Oberwinkler, J. Sedláček, H.-G. Kräusslich, P. Hobza, V. Král and J. Konvalinka, J. Med. Chem., 2009, 52, 7132–7141 CrossRef PubMed.
- P. Cígler, M. Kožíšek, P. Řezáčová, J. Brynda, Z. Otwinowski, J. Pokorná, J. Plesek, B. Grüner, L. Dolečková-Marešová, M. Mása, J. Sedláček, J. Bodem, H. G. Kräusslich, V. Král and J. Konvalinka, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15394–15399 CrossRef PubMed.
- B. Grüner, M. Kugler, S. El Anwar, J. Holub, J. Nekvinda, D. Bavol, Z. Růžičková, K. Pospíšilová, M. Fábry, V. Král, J. Brynda and P. Řezáčová, ChemPlusChem, 2021, 86, 352–363 CrossRef PubMed.
- B. Grüner, J. Brynda, V. Das, V. Šícha, J. Štěpánková, J. Nekvinda, J. Holub, K. Pospíšilová, M. Fábry, P. Pachl, V. Král, M. Kugler, V. Mašek, M. Medvedíková, S. Matějková, A. Nová, B. Lišková, S. Gurská, P. Džubák, M. Hajdúch and P. Řezáčová, J. Med. Chem., 2019, 62, 9560–9575 CrossRef PubMed.
- I. Bennour, M. N. Ramos, M. Nuez-Martinez, J. A. M. Xavier, A. B. Buades, R. Sillanpaa, F. Teixidor, D. Choquesillo-Lazarte, I. Romero, M. Martinez-Medina and C. Vinas, Dalton Trans., 2022, 51, 7188–7209 RSC.
- W. Swietnicki, W. Goldeman, M. Psurski, A. Nasulewicz-Goldeman, A. Boguszewska-Czubara, M. Drab, J. Sycz and T. M. Goszczynski, Int. J. Mol. Sci., 2021, 22, 6762 CrossRef CAS PubMed.
- I. Romero, M. Martinez-Medina, C. Camprubí-Font, I. Bennour, D. Moreno, L. Martínez-Martínez, F. Teixidor, M. A. Fox and C. Viñas, Organometallics, 2020, 39, 4253–4264 CrossRef CAS.
- Y. Zheng, W. Liu, Y. Chen, H. Jiang, H. Yan, I. Kosenko, L. Chekulaeva, I. Sivaev, V. Bregadze and X. Wang, Organometallics, 2017, 36, 3484–3490 CrossRef CAS.
- K. Fink and M. Uchman, Coord. Chem. Rev., 2021, 431, 213684–213694 CrossRef CAS.
- E. Vaňková, K. Lokočová, O. Maťátková, I. Křížová, J. Masák, B. Grüner, P. Kaule, J. Čermák and V. Šícha, J. Org. Chem., 2019, 899, 120891–120899 CrossRef.
- E. Kvasničková, J. Masák, J. Čejka, O. Maťátková and V. Šícha, J. Org. Chem., 2017, 827, 23–31 CrossRef.
- B. Grüner, J. Plešek, J. Báča, I. Císařová, J. F. Dozol, H. Rouquette, C. Viňas, P. Selucký and J. Rais, New J. Chem., 2002, 26, 1519–1527 RSC.
- B. Grüner, M. Kvíčalová, J. Plešek, V. Šícha, I. Císařová, M. Lučaníková and P. Selucký, J. Org. Chem., 2009, 694, 1678–1689 CrossRef.
- B. Grüner, J. Rais, P. Selucký and M. Lučaníková, in Boron Science, New Technologies and Applications, ed. N. S. Hosmane, CRC Press, Boca Raton, London, New York, 2012, ch. 19, pp. 463–490 Search PubMed.
- J. Rais and B. Grüner, in Solvent Extraction, Ion Exchange, ed. Y. Marcus and A. K. SenGupta, Marcel Dekker, New York, 2004, vol. 17, pp. 243–334 Search PubMed.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- I. Guerrero, Z. Kelemen, C. Viñas, I. Romero and F. Teixidor, Chem. – Eur. J., 2020, 26, 5027–5036 CrossRef CAS PubMed.
- J. Plešek, S. Heřmánek, A. Franken, I. Císařová and C. Nachtigal, Collect. Czech. Chem. Commun., 1997, 62, 47–56 CrossRef.
- J. Plešek, B. Štíbr and S. Heřmánek, Collect. Czech. Chem. Commun., 1984, 49, 1492–1496 CrossRef.
- V. S. Arderiu, C. Vinas and F. Teixidor, J. Organomet. Chem., 2015, 798, 160–164 CrossRef CAS.
- L. Matel, F. Macásek, P. Rajec, S. Heřmánek and J. Plešek, Polyhedron, 1982, 1, 511 CrossRef CAS.
- V. I. Bregadze, I. D. Kosenko, I. A. Lobanova, Z. A. Starikova, I. A. Godovikov and I. B. Sivaev, Organometallics, 2010, 29, 5366–5372 CrossRef CAS.
- I. D. Kosenko, I. A. Lobanova, I. V. Ananyev, I. A. Godovikov, L. A. Chekulaeva, Z. A. Starikova, S. Qi and V. I. Bregadze, J. Org. Chem., 2014, 769, 72–79 CrossRef CAS.
- P. Farras, A. D. Musteti, I. Rojo, C. Vinas, F. Teixidor and M. E. Light, Inorg. Chem., 2014, 53, 5803–5809 CrossRef CAS PubMed.
- J. Plešek, B. Grüner, V. Šícha, V. Böhmer and I. Císařová, Organometallics, 2012, 31, 1703–1715 CrossRef.
- J. Nekvinda, V. Šícha, D. Hnyk and B. Grüner, Dalton Trans., 2014, 43, 5106–5120 RSC.
- S. El Anwar, L. Pazderová, D. Bavol, M. Bakardjiev, Z. Růžičková, O. Horáček, L. Fojt, R. Kučera and B. Grüner, Chem. Commun., 2022, 58, 2572–2575 RSC.
- S. El Anwar, Z. Růžičková, D. Bavol, L. Fojt and B. Grüner, Inorg. Chem., 2020, 59, 17430–17442 CrossRef CAS PubMed.
- J. Nekvinda, J. Švehla, I. Císařová and B. Grüner, J. Org. Chem., 2015, 798, 112–120 CrossRef CAS.
- L.-W. Zhan, L. Han, P. Xing and B. Jiang, Org. Lett., 2015, 17, 5990–5993 CrossRef CAS PubMed.
- G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300–303 CrossRef CAS PubMed.
- J. Gour, S. Gatadi, S. Malasala, M. V. Yaddanpudi and S. Nanduri, J. Org. Chem., 2019, 84, 7488–7494 CrossRef CAS PubMed.
- P. Linciano, M. Pizzetti, A. Porcheddu and M. Taddei, Synlett, 2013, 2249–2254 CAS.
- Q.-Q. Li, Z.-F. Xiao, C.-Z. Yao, H.-X. Zheng and Y.-B. Kang, Org. Lett., 2015, 17, 5328–5331 CrossRef CAS PubMed.
- J. Nekvinda, M. Kugler, J. Holub, S. El Anwar, J. Brynda, K. Pospíšilová, Z. Růžičková, P. Řezáčová and B. Grüner, Chem. – Eur. J., 2020, 26, 16541–16553 CrossRef CAS PubMed.
- V. N. Kalinin, E. G. Rys, A. A. Tyutyunov, A. Z. Starikova, A. A. Korlyukov, V. A. Ol'shevskaya, D. D. Sung, A. B. Ponomaryov, P. V. Petrovskii and E. Hey-Hawkins, Dalton Trans., 2005, 903–908 RSC.
- S. Parihar, A. Kumar, A. K. Chaturvedi, N. K. Sachan, S. Luqman, B. Changkija, M. Manohar, O. Prakash, D. Chanda, F. Khan, C. S. Chanotiya, K. Shanker, A. Dwivedi, R. Konwar and A. S. Negi, J. Steroid Biochem. Mol. Biol., 2013, 137, 332–344 CrossRef CAS PubMed.
- Y. Chen, R. He, H. Song, G. Yu, C. Li, Y. Liu and Q. Wang, Eur. J. Org. Chem., 2021, 1179–1183 CrossRef CAS.
- S. Van Smaalen, Incommensurate crystallography, OUP, Oxford, 2007 Search PubMed.
- V. Petříček, L. Palatinus, J. Plášil and M. Dušek, Z. Kristallogr., 2023, 238, 271–282 Search PubMed.
- E. J. Juárez-Pérez, R. Núñez, C. Viñas, R. Sillanpää and F. Teixidor, Eur. J. Inorg. Chem., 2010, 2010, 2385–2392 CrossRef.
- I. Sivaev and I. Kosenko, Russ. Chem. Bull., 2021, 70, 753–756 CrossRef CAS.
- I. B. Sivaev, Molecules, 2017, 22, 2201 CrossRef PubMed.
- V. Petříček, V. Eigner, M. Dušek and A. Čejchan, Z. Kristallogr., 2016, 231, 301–312 Search PubMed.
- F. A. Gomez, S. E. Johnson, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1992, 31, 3558–3567 CrossRef CAS.
- I. Rojo, F. Teixidor, R. Kivekäs, R. Sillanpää and C. Viñas, J. Am. Chem. Soc., 2003, 125, 14720–14721 CrossRef CAS PubMed.
- A. P. Bento and F. M. Bickelhaupt, J. Org. Chem., 2008, 73, 7290–7299 CrossRef CAS PubMed.
- M. B. Smith and J. March, Advanced organic chemistry; March's advanced organic chemistry: reactions, mechanisms, and structure, Wiley & Sons, 2001 Search PubMed.
- H. R. Snyder, R. E. Carnahan and E. R. Lovejoy, J. Am. Chem. Soc., 1954, 76, 1301–1304 CrossRef CAS.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2246096, 2246097, 2246098, 2262251, 2313358 and 2313381. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00072b |
| This journal is © The Royal Society of Chemistry 2024 |