
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vibrational spectroscopy of dispersed ReVIIOx sites supported on monoclinic zirconia†
Chrysanthi
Andriopoulou
a,
Theocharis
Kentri
a and
Soghomon
Boghosian
 *abc
*abc
aDepartment of Chemical Engineering, University of Patras, Patras, Greece. E-mail: bogosian@chemeng.upatras.gr
bInstitute of Chemical Engineering Sciences, FORTH/ICE-HT, Patras, Greece
cSchool of Science and Technology, Hellenic Open University, GR-26335 Patras, Greece
First published on 1st February 2024
Abstract
In situ Raman and FTIR spectra complemented by in situ Raman/18O isotope labelling are exploited for deciphering the structural properties and configurations of the (ReOx)n phase dispersed on monoclinic ZrO2 at temperatures of 120–400 °C under oxidative dehydration conditions and coverages in the range of 0.71–3.7 Re nm−2. The dispersed (ReOx)n phase is heterogeneous, consisting of three distinct structural units: (a) Species-I with mono-oxo termination O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Re(–O–Zr)m (ReO mode at 993–1005 cm−1); (b) Species-IIa with di-oxo termination (O)2Re(–O–Zr)m−1 (symmetric stretching mode at 987–998 cm−1); and (c) Species-IIb with di-oxo termination (O)2Re(–O–Zr)u (symmetric stretching mode at 982–991 cm−1); all terminal stretching modes undergo blue shifts with increasing coverage. With increasing temperature, a reversible temperature-dependent Species-IIa ↔ Species-I transformation is evidenced. At low coverages, below 1 Re nm−2, isolated species prevail; at 400 °C the mono-oxo ORe(–O–Zr)m Species-I is the majority species, the di-oxo Species-IIa occurs in significant proportion and di-oxo Species-IIb is in the minority. At coverage ≥1.3 Re nm−2, at 400 °C the di-oxo Species-IIa prevails clearly over mono-oxo Species-I. Below 80 °C and at a low coverage of 0.71 Re nm−2, the occurrence of a fourth structural unit, Species-III taking on a tri-oxo configuration (symmetric stretching mode at 974 cm−1) is evidenced. All temperature-dependent structural and configurational transformations are fully reversible and interpreted by mechanisms at the molecular level.
Re(–O–Zr)m (ReO mode at 993–1005 cm−1); (b) Species-IIa with di-oxo termination (O)2Re(–O–Zr)m−1 (symmetric stretching mode at 987–998 cm−1); and (c) Species-IIb with di-oxo termination (O)2Re(–O–Zr)u (symmetric stretching mode at 982–991 cm−1); all terminal stretching modes undergo blue shifts with increasing coverage. With increasing temperature, a reversible temperature-dependent Species-IIa ↔ Species-I transformation is evidenced. At low coverages, below 1 Re nm−2, isolated species prevail; at 400 °C the mono-oxo ORe(–O–Zr)m Species-I is the majority species, the di-oxo Species-IIa occurs in significant proportion and di-oxo Species-IIb is in the minority. At coverage ≥1.3 Re nm−2, at 400 °C the di-oxo Species-IIa prevails clearly over mono-oxo Species-I. Below 80 °C and at a low coverage of 0.71 Re nm−2, the occurrence of a fourth structural unit, Species-III taking on a tri-oxo configuration (symmetric stretching mode at 974 cm−1) is evidenced. All temperature-dependent structural and configurational transformations are fully reversible and interpreted by mechanisms at the molecular level.
1. Introduction
Several technologically important catalytic reaction processes benefit from supported rhenium oxide catalysts. Pertinent paradigms of such processes include olefin metathesis over ReOx/Al2O3,1–3 selective catalytic oxidation of light alcohols over ReOx supported on TiO2 and SnO2,4 methanol conversion to dimexothymethane over ReOx dispersed on TiO2(anatase), ZrO2 (monoclinic), SiO2 and Al2O3,5–8 as well as hydrodesulfurization and hydrodenitrogenation processes over ReOx/Al2O3 catalysts.9,10Diverging and often contradictory reports have been published in the literature of supported rhenia catalysts concerning the speciation and the molecular structure of the dispersed dehydrated ReVIIOx phase. Combined Raman and IR spectra recorded after cooling at 45 °C under dehydrated conditions were interpreted to suggest the existence of two distinct tri-oxo (O)3–Re–O species on Al2O3, TiO2 and ZrO2 with relative amounts varied by surface coverage,11 whereas the same group proposed the existence of one single tri-oxo (O)3–Re–O species based on Raman spectra obtained at room temperature after dehydration at 500 °C for 1 wt% Re2O7 on SiO2, Al2O3, TiO2 and ZrO2.12 Consistently with ref. 12, one single isolated species with tri-oxo termination configuration is proposed for ReOx dispersed at surface densities of 0.3–2.2 Re nm−2 on TiO2 and Al2O3 based on in situ Raman and FTIR spectroscopy under oxidative dehydrating conditions (at 400 °C and 300 °C, respectively).13 Later, the same laboratory, based on an elegant study employing in situ Raman spectroscopy coupled with 18O/16O isotope exchange at 450 °C on one single sample (3 wt% Re2O7/ZrO2) suggested the occurrence of a polymeric species with mono-oxo termination configuration.14 Additionally, a theoretical (DFT and X-ray spectroscopy simulations) study combined with experimental work (XAFS, STEM, TPR, XPS) performed at room temperature after cooling under controlled conditions on one single sample (0.7 wt% Re on Al2O3) suggested the existence of isolated tri-oxo ReOx.15 Separately, the occurrence of two distinct di-oxo (O)2–Re(–O–)2 species on Al2O3 has been convincingly shown following a sound experimental (in situ Raman coupled with 18O/16O exchange, in situ IR, UV-Vis, XANES) investigation combined with theoretical analysis (DFT).16Operando Raman studies have also provided evidence for heterogeneity of the dispersed ReOx phase on TiO2.7,8 An in situ Raman spectroscopic study coupled with 18O/16O isotope exchange at 450 °C showed that a tri-oxo (O)3–Re–O– configuration prevails on SiO2.17 A heterogeneity and an unprecedented reversible temperature-dependent evolution of prevailing structures and configurations (mono-oxo vs. di-oxo) has been established for the dispersed MoOx, WOx and ReOx phases on TiO2 by means of in situ molecular vibrational (Raman and FTIR) spectroscopy at temperatures of 175–430 °C.18 Unlimited cycling between the mono-oxo configuration (prevalent at high temperature) and the di-oxo configuration (predominant at intermediate temperature) is feasible by appropriate temperature control and adequate sample exposure.19 The extent of temperature dependence is connected to the support (TiO2) surface hydroxylation as demonstrated by complementary Raman spectra in sealed cells under forced dehydrated conditions.19 The dispersed ReOx phase on TiO2(P25) in the submomolayer 0.16–2.5 Re nm−2 range consists of a mono-oxo ORe(–O–Ti)n species and a di-oxo (O)2Re(–O–Ti)m species, in relative amounts that depend on temperature and coverage, as shown by means of in situ molecular vibrational (Raman and FTIR) spectroscopy complemented by 18O/16O isotope exchange and Raman spectra in sealed cells under forced dehydrated conditions.20 The occurrence of a mono-oxo and a di-oxo species has also been demonstrated by means of in situ molecular vibrational spectroscopy (Raman and FTIR) coupled with 18O/16O exchange for the ReOx phase dispersed on CeO2.21 An overview of selected reports on the molecular structure of dispersed ReOx sites is contained in a recent review on vibrational spectroscopy of supported transition metal oxides.22 Molecular vibrational spectroscopy is particularly suited for studying the transition metal oxide phases dispersed on oxide carriers since it can be applied in absence of long range order. Moreover, the combined use of Raman and FTIR spectroscopy, complemented also by isotope exchange provides a comprehensive approach that can differentiate amongst distinct molecular configurations exhibited by (MOx)n sites (M = e.g. V, Mo, W, Nb, Re etc.).22 A recently demonstrated neoteric concept of Raman spectroscopy of supported transition metal oxides in sealed cells under forced dehydrated conditions has also contributed in validating results obtained by in situ vibrational (Raman, IR) spectroscopies.19,20,23,24
In our view, the reasons for the controversy on the reported molecular structures of dispersed transition metal oxides in some pre-2015 reports are to be found in the deficiencies of the research strategies adopted in most of the pertinent investigations. Such deficiencies may include one or more of the following shortcomings: (i) studying only one type of vibrational spectra and/or not complementing the study with isotope exchange measurements; (ii) recording of vibrational spectra at room temperature after cooling; (iii) examining the structural properties of one single sample instead of examining samples in a wide range of loadings; (iv) recording of vibrational spectra at one single temperature etc. Hence, temperature and loading effects have often been overlooked. Indeed, temperature dependent speciations of distinct dispersed oxometallic species are established for MoOx, WOx, ReOx and VOx sites supported on TiO2,18–20,23,24 whereas the prevalence of particular configurations depending on coverage is also demonstrated.16,20,23 A holistic approach in the protocol followed for studying the molecular structure of dispersed metal oxide overlayers should include the use of both in situ Raman and in situ FTIR spectroscopies, ideally complemented by 18O/16O isotope exchange.
The present work is concerned with unraveling the molecular structure and termination configuration of the species constituting the (ReOx)n phase dispersed on monoclinic ZrO2. The ReO terminal stretching vibrational wavenumbers of dispersed ReOx sites with mono-oxo, di-oxo and tri-oxo termination configuration lie close to each other.16,20,25,26 Hence, locating the band wavenumbers with sufficient accuracy is not adequate for differentiating between mono-oxo, di-oxo and tri-oxo configurations. To this end, in situ Raman and in situ FTIR spectroscopies (the latter in the overtone region) are used in the temperature range of 400–80 °C for a wide range of surface coverage (0.71–3.7 Re nm−2) in order to shed light in the issue of heterogeneity (i.e. determine the number of species present and their relative presence depending on temperature and coverage). Moreover, by exploiting the vibrational selection rules of Raman and FTIR spectroscopy as well as the vibrational isotope effects, a differentiation between the various termination configurations is proposed.
2. Experimental section
2.1. Synthesis and textural characterization of ReOx/ZrO2 catalysts
The preparation of the ReOx/ZrO2 materials took place by incipient wet impregnation of monoclinic ZrO2 (Alfa Aeasar), which was calcined prior to its use at 600 °C (heating rate, 2 °C min−1) for 4 h. Pre-weighed amounts of precursor NH4ReO4 (Alfa Aeasar) corresponding to targeted coverages were dissolved in triply distilled water resulting to Re(VII) concentrations in the range of 1.6 × 10−3–8.2 × 10−3 M at pH = 7.5 that was adjusted by dropwise adding NH3 or HNO3 solutions. The solution speciation of oxo-Re(VII) species was examined by Raman spectroscopy (Fig. S1, ESI†) and it was confirmed that all precursor solutions contained Re exclusively in the form of ReO4− ions.For each synthesis, a weighed amount of the pre-calcined support was added in the precursor solution that was equilibrated at 45 °C for 1 h by continuous monitoring and periodic adjustment of the pH (pH = 7.5), and subsequently the solvent was removed by rotary evaporation. The obtained materials were dried for 16 h at 120 °C and were afterwards calcined in a muffle furnace at 450 °C for 3 h under static air. The resulting Re loading (wt% Re) for each sample was determined by icp-analysis and was found -as expected-20 lower than previewed from the weighed-in amounts of NH4ReO4 and ZrO2 due to partial loss of the volatile rhenium oxide by vaporisation.
The specific surface area of the catalysts was determined by the BET method based on N2 adsorption–desorption isotherms as described previously.20 Hence, the actual Re surface density for each sample, ns (expressed as Re nm−2), could be calculated as
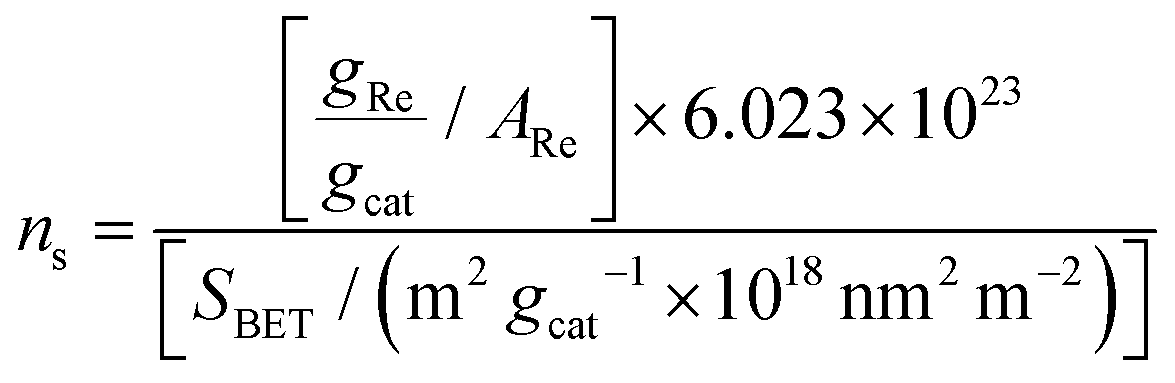 | (1) |
The only crystalline phase contained in the prepared samples was, as determined by powder XRD diffraction, monoclinic ZrO2. Table 1 compiles the principal characteristics for each catalyst sample, denoted as uReZrO2, where u is the surface density in Re nm−2.
| Catalysts | C Re(VII) (M) | Loading (wt% Re)b | S BET (m2 g−1) | n s (Re nm−2) |
|---|---|---|---|---|
| a Support ZrO2(m) pre-calcined at 600 °C for 4 h; SBET,ZrO2: 50 m2 g−1. b As per icp analysis. | ||||
| 0.71ReZrO2 | 1.6 × 10−3 | 1.09 | 50 | 0.71 |
| 1.3ReZrO2 | 3.3 × 10−3 | 2.08 | 52 | 1.3 |
| 1.5ReZrO2 | 4.1 × 10−3 | 2.43 | 52 | 1.5 |
| 1.9ReZrO2 | 5.0 × 10−3 | 3.08 | 53 | 1.9 |
| 2.4ReZrO2 | 5.8 × 10−3 | 3.64 | 49 | 2.4 |
| 3.7ReZrO2 | 8.2 × 10−3 | 5.33 | 46 | 3.7 |
2.2. Raman and FTIR spectroscopy. Protocols of measurements
To ascertain that the calcined uReZrO2 samples do not undergo further gradual ReOx loss due to ReOx vaporization, the following procedure was followed for each sample. First, each sample was heated to 400 °C for 1 h in the in situ Raman cell under flowing (30 cm3 min−1) 20% O2/He gas mixture (He 99.999% and O2 99.999% form L′ Air Liquide). Subsequently, temperature was lowered to 250 °C under flowing 20% O2/He gas and the in situ Raman spectrum was recorded after 1 h and 45 min on stream. Temperature was then raised to 400 °C and an in situ Raman spectrum was obtained under flowing 20% O2/He gas as reference at 400 °C after 1 h and 45 min on stream. Finally, temperature was lowered again to 250 °C and after 1 h and 45 min exposure to flowing 20% O2/He gas the in situ Raman spectrum at 250 °C was reproduced. Fig. S2 (ESI†) show the pertinent in situ Raman spectra obtained at 250 °C for three representative samples (0.71ReZrO2, 1.3ReZrO2 and 2.4ReZrO2), before and after the intermediate exposure of the sample at 400 °C under flowing feed gas for ∼3 h and 15 min. It is evident that the spectra are fully reproduced and hence following the initial partial loss of ReOx due to vaporization during the calcination stage, no further loss of ReOx had taken place.
Recording of in situ Raman spectra under continuous dehydrated feed conditions (30 cm3 min−1 20% O2/He) started at 400 °C after 1 h and 45 min on stream. Temperature was subsequently lowered to 250, 225, 200, 175, 145, 120, 100, 80, 60 and 35 °C using both a sequential and a random order of temperatures, and steady state in situ Raman spectra were collected after 1 h and 45 min sample treatment at each temperature to ensure steady state attainment. After recording the in situ Raman spectra at a certain temperature, the sample was first heated to 400 °C for 1 h and then cooled to the next temperature. After completing each sequence, the sample under study was heated to 400 °C and the full reinstatement of its structural state was checked and confirmed by reproducing the pertinent in situ Raman spectrum at 400 °C. The spectral resolution was set to 7 cm−1 by appropriate adjustments of the entrance and exit monochromator vertical slits’ apertures. A high signal-to-noise ratio could be achieved by performing slow scans with a 1.2 s photon counting per point at 0.25 cm−1 increments. A normalisation procedure of the obtained Raman spectra, previously described in detail,20,29 was implemented for normalising the spectra and compensate the “path length” effect caused by the coloured samples in the wide 0.71–3.7 Re nm−2 coverage range studied. Very briefly, first the background of each spectrum was zeroed by subtracting a rectangle, thereby zeroing the counts of the high wavenumber tail of the spectrum and subsequently the whole spectrum was divided by the area under the spectrum trace in the 270–1100 cm−1 wavenumber range that features the main Raman bands of structural relevance.
O stretching spectral range is studied due to strong absorption in the corresponding fundamental stretching region. Reference in situ FTIR spectra under flowing 20% O2/He were recorded at each temperature also for the pure support ZrO2 and were subtracted from the in situ FTIR spectra obtained for the samples studied. Hence, difference spectra pertaining to the dispersed (ReOx)n phase are reported below.
3. Results and discussion
The ReOx deposition on ZrO2(m) takes place inside the precursor suspension that contain Re exclusively in the form of ReO4− moieties (see Fig. S1†) and proceeds via Zr–O–Re(–O)3 and/or (Zr–O)2–Re(–O)3 anchoring (where in the latter case one of the Zr–O–Re anchors involves also a ZrO2 lattice O). Drying and calcination follows the well-established mechanism of gradual titration of surface hydroxyls by means of successive esterification-like steps and concomitant condensation of water molecules that remain retained by means of H-bonding to the surface layer.18–20,23,24,32–38 Hence, after the initial anchoring, bipedal and gradually multipedal ReOx units are formed at the expense of terminal Re–O sites. Therefore, as established for several supported MOx transition metal oxides (M = V, Mo, W, Re) for surface coverages below the respective monolayer limit, formation of distinct species takes place in the course of the calcination resulting in heterogeneity of the dispersed MOx phase.16,18–21,23,24,38 Additionally, when coverage exceeds a certain limit, which is characteristic for each system, M–O–M associations take place resulting in (MOx)n domain growth.223.1. In situ molecular vibrational (Raman, FTIR) spectra of ReOx/ZrO2(m) catalysts. Coverage and temperature effects
O and Re–O– modes. First observations of spectral effects in Fig. 1 and 2 include: (i) the convoluted band envelope due to terminal ReO stretching is asymmetric with evidence of several overlapping bands in the ReO stretching region that are known to occur at wavenumbers typically higher than 940 cm−1;16,25,26 (ii) the peak maximum of the ReO band envelope undergoes a pronounced blue shift with increasing coverage, e.g. from 991 cm−1 for 0.71 Re nm−2 to 1002 cm−1 for 3.7 Re nm−2 at 400 °C; (iii) with increasing coverage, a broad band at ∼885 cm−1 progressively loses intensity relative to the ca. 990–1000 cm−1 ReO stretching band envelope; and (iv) a broad, however obscured, band at ∼935 cm−1 emerges and progressively gains intensity with increasing coverage, becoming discernible for nS ≥ 1.3 Re nm−2. The wavenumber location and broad nature of the latter two bands (i.e. at ∼885 and ∼935 cm−1) are suggestive of Re–O– provenance (i.e. Re–O–Re, O–Re–O and Re–O–Zr).
 | ||
| Fig. 1 Effect of coverage on the in situ Raman spectra obtained for ReOx/ZrO2 catalysts under flowing 20% O2/He at temperatures of: (A) 400 °C; (B) 250 °C; and (C) 175 °C at coverages in the 0.71–3.7 Re nm−2 range as indicated by each spectrum. The full spectral range of 200–1100 cm−1 is shown. Recording parameters: laser wavelength, λ0 = 491.5 nm; laser power, w = 10 mW; time constant, τ = 0.3 s; spectral slit width, ssw = 7 cm−1. | ||
 | ||
| Fig. 2 Effect of coverage on the in situ Raman spectra obtained for ReOx/ZrO2 catalysts under flowing 20% O2/He at temperatures of: (A) 400 °C; (B) 250 °C; and (C) 175 °C at coverages in the 0.71–3.7 Re nm−2 range as indicated by each spectrum. The Re–O stretching region of 800–1050 cm−1 is shown. Recording parameters: laser wavelength, λ0 = 491.5 nm; laser power, w = 10 mW; time constant, τ = 1.2 s; spectral slit width, ssw = 7 cm−1. | ||
With increasing coverage, the ReOx domains are expected to grow by means of Re–O–Re associations at the expense of Re–O–Zr anchors. Hence, with increasing coverage the Re–O–Re/ReO ratio is expected to increase and the Re–O–Zr/ReO ratio is expected to decrease. In view of the above, the ∼885 cm−1 band that loses intensity relative to the ReO envelope with increasing coverage is assigned to Re–O–Zr modes and the ∼935 cm−1 band that gains intensity relative to the ReO envelope with increasing coverage is assigned to Re–O–Re modes. Additional evidence strengthening the assignment of the ∼885 cm−1 band to Re–O–Zr modes will be presented and discussed below, in the context of the temperature dependence of in situ Raman spectra.
A first discussion of the blue-shifting tendency observed with increasing coverage for the band envelope ascribed to terminal ReO stretching should include a consideration of factors affecting the wavenumber of the terminal ReO stretching. Such factors may include inter alia: (a) the termination configuration (i.e. mono-oxo, di-oxo etc.); (b) the Re coordination number; (c) vibrational coupling (particularly in case of occurrence of Re–O–Re associations); and (d) variations in basicity of support dative O atoms. In the initial steps of ReOx deposition, the most basic surface hydroxyls are titrated first, hence with increasing coverage hydroxyls with lower basicity come gradually into play and by that means – in a cascade effect – the terminal ReO stretching becomes gradually stronger thereby partly justifying the corresponding blue shift with increasing coverage (e.g., from 991 to 1002 cm−1 at 400 °C, Fig. 2(A)).
Fig. 3 shows the evolution of the in situ FTIR spectra obtained at constant temperature of 400 °C (Fig. 3A), 250 °C (Fig. 3B) and 175 °C (Fig. 3C) for ReOx/ZrO2 catalysts with coverages of 0.71, 1.3 and 2.4 Re nm−2 under dehydrating feed conditions of flowing (30 cm3 min−1) 20% O2/He gas. The spectra are obtained in the overtone region, hence the wavenumber span of the terminal stretching region is approximately doubled. Likewise, the mutual distance between the infrared bands corresponding to the counterpart Raman components comprising the terminal stretching band envelope observed in the Raman spectra (Fig. 2) is also approximately doubled resulting in clearly distinct bands in Fig. 3, thereby ascertaining the multiple character ascribed to the observed “broad” terminal stretching Raman band envelopes (Fig. 2) and excluding the possibility for certain bands as being simply of broad nature.
 | ||
| Fig. 3 Effect of coverage on the in situ FTIR spectra obtained for ReOx/ZrO2 catalysts under flowing 20% O2/He at temperatures of: (A) 400 °C; (B) 250 °C; and (C) 175 °C at coverages of 0.71, 1.3 and 2.4 Re nm−2 as indicated by each spectrum. The corresponding spectra obtained for ZrO2 have been subtracted. The dotted lines mark the band wavenumbers for the symmetric stretching modes of Species-I, Species-IIa and Species-IIb deduced by exploiting the respective temperature dependent features portrayed in Fig. 4(B), 5(B) and 6(B). Resolution, 4 cm−1. | ||
O and Re–O– stretching modes are portrayed. Panels 4(A), 5(A) and 6(A) pertain to in situ Raman spectra, whilst panels 4(B), 5(B) and 6(B) pertain to the counterpart in situ FTIR spectra. All spectra are recorded at a sequential order of decreasing temperatures. Notably, as mentioned also in the Experimental section, reheating the samples to 400 °C under flowing 20%O2/He results in full reinstatement of the corresponding spectrum in each case. Additionally, Fig. S3(A–F)† shows the temperature dependence of the in situ Raman spectra obtained for all six samples studied in the full temperature range of 400–35 °C. We shall, however, confine the discussion mainly to Fig. 4–6 that are adequate for inferring the number of species present and their respective termination configurations. The extent of the temperature-dependent changes observed vary with surface coverage, as discussed below.
 | ||
| Fig. 4 ReOx/ZrO2 with surface density of 0.71 Re nm−2: (A) sequential (430 → 250 → 175 → 145 → 120 °C) in situ Raman spectra obtained under flowing 20%O2/He at temperatures as indicated by each spectrum. Spectral recording parameters: see Fig. 2 caption. (B) Sequential (430 → 250 → 175 → 120 °C) in situ FTIR spectra obtained under flowing 20% O2/He at temperatures as indicated by each spectrum. The corresponding spectra obtained for ZrO2 have been subtracted. Resolution, 4 cm−1. | ||
 | ||
| Fig. 5 ReOx/ZrO2 with surface density of 1.3 Re nm−2: (A) sequential (430 → 250 → 175 → 145 → 120 °C) in situ Raman spectra obtained under flowing 20%O2/He at temperatures as indicated by each spectrum. Spectral recording parameters: see Fig. 2 caption. (B) Sequential (430 → 250 → 175 → 120 °C) in situ FTIR spectra obtained under flowing 20% O2/He at temperatures as indicated by each spectrum. The corresponding spectra obtained for ZrO2 have been subtracted. Resolution, 4 cm−1. | ||
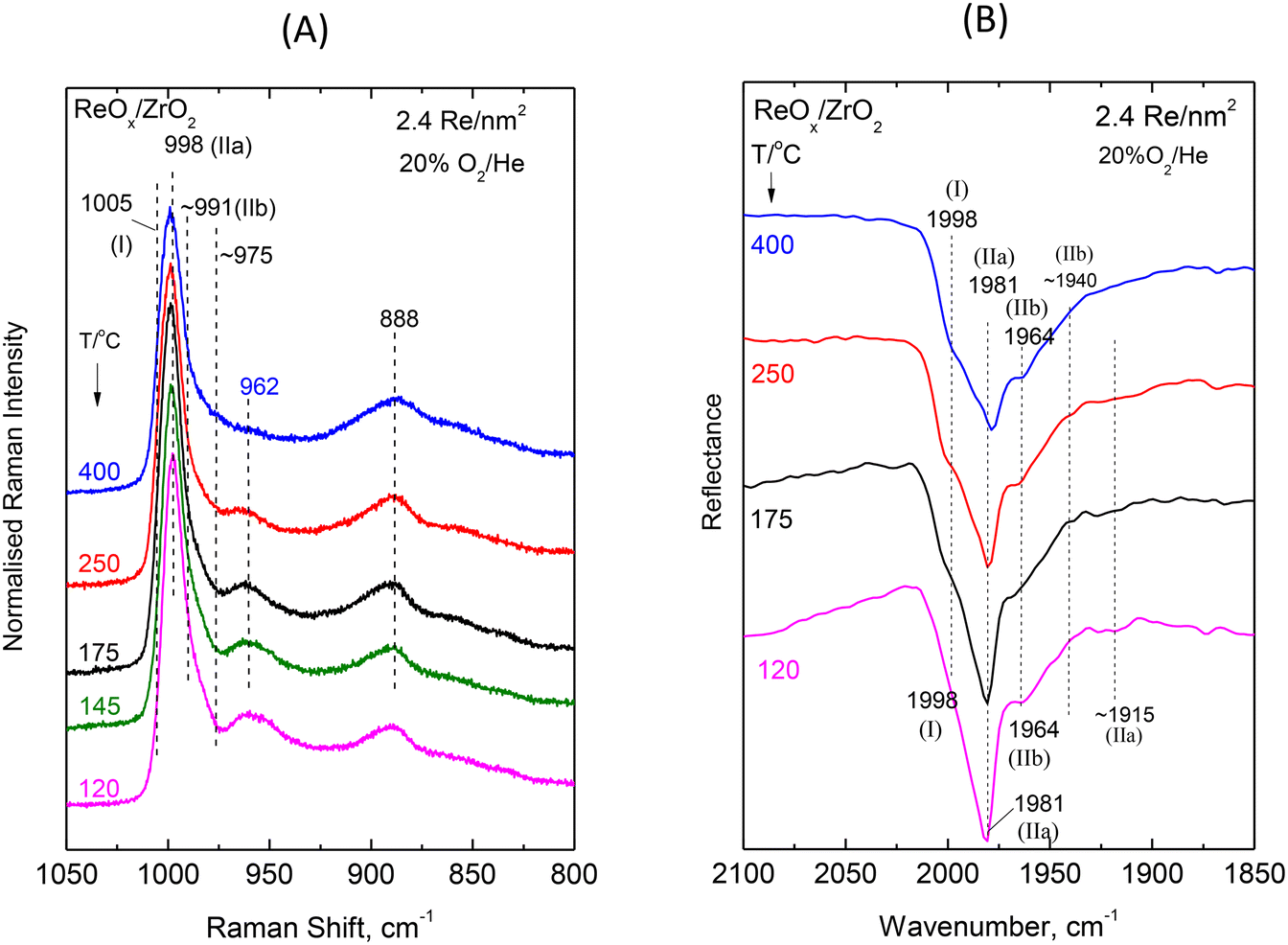 | ||
| Fig. 6 ReOx/ZrO2 with surface density of 2.4 Re nm−2: (A) Sequential (430 → 250 → 175 → 145 → 120 °C) in situ Raman spectra obtained under flowing 20%O2/He at temperatures as indicated by each spectrum. Spectral recording parameters: see Fig. 2 caption. (B) Sequential (430 → 250 → 175 → 120 °C) in situ FTIR spectra obtained under flowing 20% O2/He at temperatures as indicated by each spectrum. The corresponding spectra obtained for ZrO2 have been subtracted. Resolution, 4 cm−1. | ||
Low coverage ReOx/ZrO2 with 0.71 Re nm−2. The rhenium–oxygen terminal stretching modes are known to occur in the 940–1010 cm−1 region depending on the termination configuration and on the identity of the surrounding ligands.16,20,25,26Fig. 4(A) shows a pertinent multi-component band envelope exhibiting a strong feature at 995–975 cm−1 (presumably due to symmetric terminal stretching modes)20,25,26 and a 975–940 cm−1 tail, probably due to antisymmetric modes (of e.g. di-oxo sites) that are known to be weaker and to occur at 10–40 cm−1 lower than their respective symmetric counterpart modes.20,25,26,28 The temperature-dependent variations of the relative band intensities in Fig. 4(A) imply a corresponding temperature-dependent prevalence of distinct dispersed ReOx species, thereby corroborating the proposal for the heterogeneity of the dispersed rhenia phase. The counterpart in situ FTIR spectra displayed in Fig. 4(B) show a better separation amongst the respective component bands due to the approximate doubling of their mutual distances in the overtone region. Hence, the main FTIR band envelope due to Re–O terminal symmetric stretching has three clearly discerned components at 1971, 1963 and 1955 cm−1 plus two weaker broad features at ∼1920 and ∼1870 cm−1. The temperature dependent features of the relative intensities observed for the 1971, 1963 and 1955 cm−1 bands in Fig. 4(B) show that their prevalence is due to three distinct ReOx species. Hence, the asymmetric 975–995 cm−1 Raman band envelope in Fig. 4(A) is reasonably thought to consist of three component bands.
In order to corroborate the evidence supporting the above proposal we performed a peak analysis in the terminal stretching region of the in situ Raman spectra shown in Fig. 4(A) and the results are shown in Fig. 7(A). The criteria for the peak analysis of the Re(O)2 terminal stretching region invoke:
(1) The occurrence of five bands due to three Species, as evidenced from the combined observations of the in situ Raman and FTIR spectra as follows: (i) one band at the highest wavenumber due to the terminal ReO stretching mode of Species-I; (ii) two bands due to the νs/νas pair due to Species-IIa with νs − νas ≈ 10–40 cm−1; and (iii) two bands due to the νs/νas pair due to Species-IIb with νs − νas ≈ 10–40 cm−1
(2) Fixed band widths and positions (with allowance for the measurement precision, i.e. ±1 cm−1) for each studied sample with surface densities of 0.71, 1.3 and 2.4 Re nm−2
(3) Fixed relative Iνs/Iνas intensities for each dioxo species (IIa, IIb), and
(4) Allowance for broader νas bands compared to their respective νs counterparts.
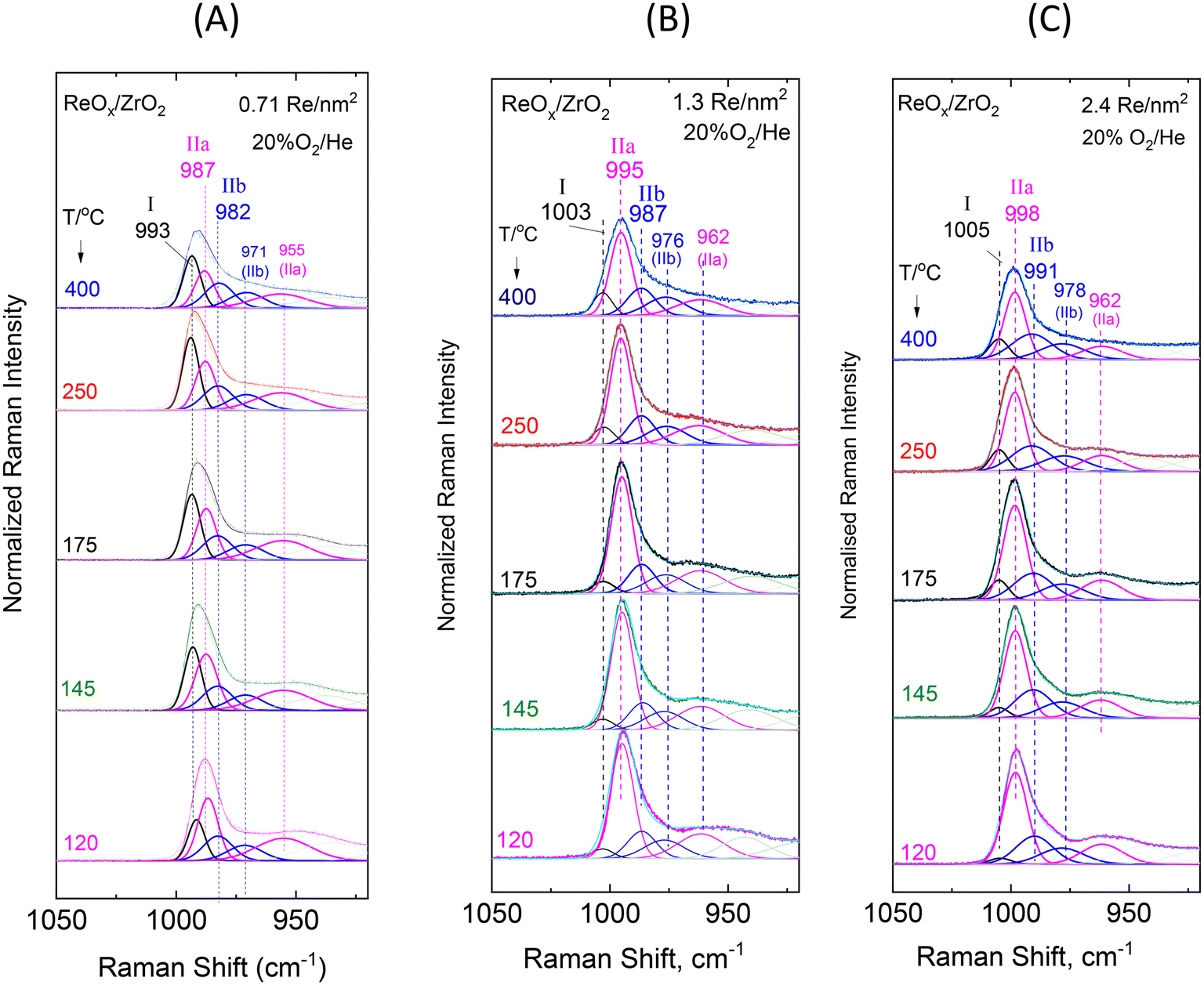 | ||
| Fig. 7 Sequential (430 → 250 → 175 → 145 → 120 °C) in situ Raman spectra obtained under flowing 20% O2/He at temperatures as indicated by each spectrum for ReOx/ZrO2 with surface densities of: (A) 0.71 Re nm−2; (B) 1.3 Re nm−2; and (C) 2.4 Re nm−2. Peak analysis is shown. The corresponding spectra obtained for ZrO2 have been subtracted. Spectral recording parameters: see Fig. 2 caption. | ||
The results of the peak analysis show that three peaks are found in the 975–1000 cm−1 symmetric terminal stretching region, at 993, 987 and 982 cm−1, whilst the low wavenumber tail is fitted with two further broader bands at 971 and 955 cm−1, presumably due to antisymmetric terminal stretching modes. Pairing of bands (i.e. assignment of two bands to the same species) can be justified when their mutual relative intensities are maintained in the full temperature range. In view of this principle, the 993 cm−1 band is solely assigned to Species-I, the 987/955 cm−1 pair is assigned to Species-IIa and the 982/971 cm−1 pair to Species-IIb.
The temperature-dependent behavior of the 993 (I), 987 (IIa) and 982 (IIb) cm−1 bands (Fig. 7) shows that with increasing temperature the relative intensity of the 993 cm−1 band due to Species-I is increased at the expense of the 987 cm−1 band (Species-IIa), whilst the 982 cm−1 component (Species-IIb) does not undergo discerned relative intensity changes in the 120–400 °C range. As shown previously for ReOx/TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and ReOx/CeO221 catalysts a OReOn species with mono-oxo termination configuration is favored at low coverage and high temperature, hence Species-I with a single ReO stretching mode at the highest wavenumber of 993 cm−1 among all bands has a mono-oxo OReOn configuration. Species IIa and IIb, both possess terminal stretching band pairs at 987/955 cm−1 and 982/971 cm−1 conforming to the νs/νas (i.e. symmetric/antisymmetric) expected pair for sites that take on a di-oxo termination configuration that is expected to give rise to a symmetric stretching mode, νs, with higher intensity compared to an antisymmetric stretching mode, νas, at wavenumbers conforming to the νs − νas = ∼10–40 cm−1 rule.25 The above observations in the temperature range of 120–400 °C are interpreted to indicate the following temperature-dependent reversible transformation
20 and ReOx/CeO221 catalysts a OReOn species with mono-oxo termination configuration is favored at low coverage and high temperature, hence Species-I with a single ReO stretching mode at the highest wavenumber of 993 cm−1 among all bands has a mono-oxo OReOn configuration. Species IIa and IIb, both possess terminal stretching band pairs at 987/955 cm−1 and 982/971 cm−1 conforming to the νs/νas (i.e. symmetric/antisymmetric) expected pair for sites that take on a di-oxo termination configuration that is expected to give rise to a symmetric stretching mode, νs, with higher intensity compared to an antisymmetric stretching mode, νas, at wavenumbers conforming to the νs − νas = ∼10–40 cm−1 rule.25 The above observations in the temperature range of 120–400 °C are interpreted to indicate the following temperature-dependent reversible transformation
 | (A) |
A molecular-level mechanism for the above reaction scheme (A) portraying plausible molecular models for Species–I and Species–IIa is shown in Fig. 8. Water molecules retained at the surface layer of oxide carriers by means of H-bonding have previously also been shown to mediate temperature-dependent transformations among distinct MOx sites in ReOx/TiO2, WOx/TiO2, MoOx/TiO2 and VOx/TiO2 catalysts.18–20,23,24 Such water molecules retained at the support oxide surface layers by H-bonding are formed during the titration of surface hydroxyls that takes place by the precursor oxometallic species during the deposition steps.32–37
 | ||
| Fig. 8 Molecular-level mechanism accounting for the reversible temperature-dependent Species-I ↔ Species-IIa transformation mediated by water molecules retained at the surface layer and surface hydroxyls. For simplicity, mononuclear representations are shown. The number of anchoring bonds and the resulting coordination number, CNRe = 5 are reasonable in view of the oxidation state VII for Re. Gold spheres, Re; red spheres, O; grey spheres, Zr. H atoms (white spheres) are included to account for hydroxylation (see text). | ||
Notably, the reaction scheme shown in Fig. 8 implies that with increasing temperature a gradual increase of Re–O–Zr anchors will take place thereby resulting in an increase of the Re–O–Zr/ReO ratio. Indeed, as seen in Fig. 4A, a relative strengthening of the ∼885 cm−1 band is observed with increasing temperature, thereby corroborating the initial assignment of the ∼885 cm−1 band (see section 3.1.1 pertaining to the effect of coverage) to Re–O–Zr anchors.
The temperature-dependent features of the in situ FTIR spectra obtained for the low coverage 0.71 Re nm−2 sample are in full conformity with the above scenario of temperature-dependent interplay involving the mono-oxo Species-I and di-oxo Species-IIa as well as the relative stability of di-oxo Species-IIb in the temperature range of 120–400 °C. On grounds of anharmonicity, which allows the observation of first overtones, the 993, 987 and 982 cm−1 stretching fundamentals due to Species-I (mono-oxo), Species-IIa (dioxo) and Species-IIb (dioxo) are expected to give overtones at wavenumbers slightly below the respective doubled fundamentals,25,40i.e. the ∼1971, ∼1963 and ∼1955 cm−1 observed wavenumbers conform to the above rule. With increasing temperature, the 1963 cm−1 band due to Species-IIa diminishes and the 1971 cm−1 Species-I band prevails, whilst the 1955 cm−1 Species-IIb band remains unaffected and is better discerned at 400 °C where the 1963 cm−1 Species-IIa band has diminished. The ∼1920 and ∼1870 cm−1 broad bands are assigned to the first overtones of the antisymmetric 971 and 955 cm−1, νas, modes of Species-IIb and Species-IIa, respectively.
Intermediate and high coverage ReOx/ZrO2 with 1.3 and 2.4 Re nm−2. Decoding of the temperature-dependent features of the in situ Raman and in situ FTIR spectra obtained for the ReOx/ZrO2 catalysts with 1.3 and 2.4 Re nm−2 shown in Fig. 5(A, B) and 6(A, B) becomes straight-forward in view of the findings discussed for the temperature dependent spectral features for the low coverage 0.71ReZrO2 sample (vide ante) and the general coverage effect trends presented in section 3.1.1.
The splitting pattern of the FTIR overtones in the symmetric stretching overtone 1960–2000 cm−1 region of the spectra shown in Fig. 5(B) and 6(B) reveals in each case the occurrence of three distinct bands due to Species-I, Species-IIa and Species-IIb. As discussed in section 3.1.1 (Fig. 3) all stretching band wavenumbers exhibit a gradual blue shift with increasing coverage in the range 0.71–3.7 Re nm−2 studied. Moreover, it is evident that whereas for a low coverage of 0.71 Re nm−2 the mono-oxo Species-I prevails at e.g. 400 °C, this is no longer the case for higher coverages of 1.3 and 2.4 Re nm−2 where, as best seen in the in situ FTIR spectra shown in Fig. 5B and 6B, Species-I is a minority species at 400 °C.
The interpretation and the pertinent discussions are facilitated by performing a peak analysis in the terminal stretching region of the in situ Raman spectra shown in Fig. 5A and 6A, obtained for 1.3ReZrO2 and 2.4ReZrO2, and the results of the peak analysis are shown in Fig. 7B and C. The band wavenumbers assigned to the different species for 1.3ReZrO2 are at 1003 cm−1 (Species-I, ReO, mono-oxo), 995/962 cm−1 (νs/νas, Species-IIa, di-oxo) and 987/976 cm−1 (νs/νas, Species-IIb, di-oxo). The respective wavenumbers for 2.4ReZrO2 are at 1005 cm−1 (Species-I, ReO, mono-oxo), 998/962 cm−1 (νs/νas, Species-IIa, di-oxo) and 991/978 cm−1 (νs/νas, Species-IIb, di-oxo). Table 2 compiles the band wavenumbers and assignments for the various species.
| Re nm−2 | Species-I, mono-oxo | Species-IIa, di-oxo | Species-IIb, di-oxo | O–Re–O | Re–O–Zr | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ORe(–O–Zr)n |
(O)2Re(–O–Zr)n |
(O)2Re(–O–Zr)n |
||||||||||
| ν 1←0,R | ν 2←0,IR | ν s,1←0,R | ν as,1←0,R | ν s,2←0,IR | ν as,2←0,IR | ν s,1←0,R | ν as,1←0,R | ν s,2←0,IR | ν as,2←0,IR | |||
| 0.71 | 993 | 1971 | 987 | 955 | 1963 | ∼1870 | 982 | 971 | 1955 | ∼1920 | 885 | |
| 1.3 | 1003 | 1994 | 995 | 962 | 1977 | ∼1910 | 987 | 976 | 1962 | ∼1940 | 935–940 | 890 |
| 2.4 | 1005 | 1998 | 998 | 962 | 1981 | ∼1915 | 991 | 978 | 1964 | ∼1940 | 935–940 | 890 |
As seen in Fig. 7B and C, with increasing temperature band (I) due to Species-I (mono-oxo) gains intensity at the expense of the Species-IIa νs mode for both 1.3ReZrO2 and 2.4ReZrO2. Contrary, however, to the case of the low coverage 0.71ReZrO2 (Fig. 8A), Species-IIa remains clearly predominant up to 400 °C. Reasonably, the reason lowering the progress of reaction scheme (A) to the left is the increased coverage with its consequent shortage of surface hydroxyls adjacent to Species-IIa caused by their extensive titration during the deposition process.
3.2. In situ Raman spectra and 18O/16O isotopic substitution. Vibrational isotope effects
Complementing in situ Raman spectroscopy with 18O/16O isotopic labelling sheds additional light into the issue of termination configuration in supported transition metal oxide catalysts.14,28,41 The vibrational isotope effects, i.e. the isotopic splitting pattern of terminal stretching bands and the isotopic shift allow for valid discussions on band assignments to (M18O)n modes (n = 1, 2, 3) and on the issue of discrimination between e.g. mono-oxo, di-oxo and tri-oxo configurations.14,41
The theory of vibrational isotope effects based on the harmonic oscillator approximation, i.e. based on a strictly quadratic potential function, results in justified theoretical predictions for the fundamental stretching ν(Re18O)n (n = 1, 2) modes’ wavenumbers of 18O/16O-substituted terminal Re18O and/or 18ORe18O sites.40,42 As a paradigm, the diatomic harmonic approximation for a MO (M: transition metal) terminal site results in the following formula for the isotopic ratio
Isotopic ratio:
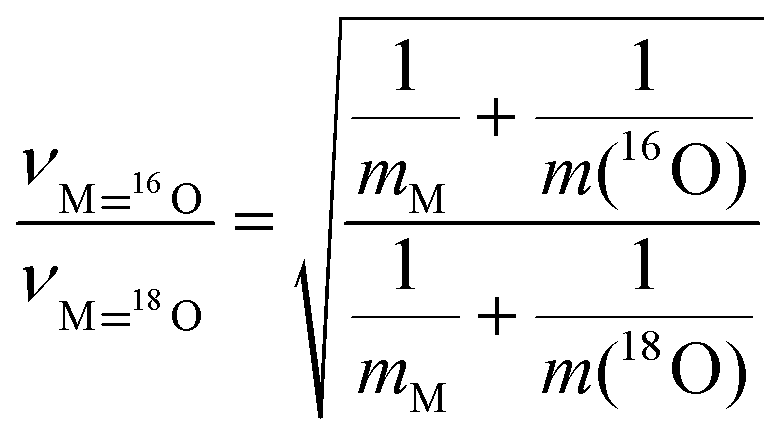 | (2) |
Indicative values for the isotopic ratio for several MO terminations have been reported.14 For the ReO diatomic harmonic oscillator, the isotopic ratio equals 1.0555. The theory and pertinent equations for triatomic M(O)2 “molecules” become more involved and elaborate,42 however leading to an approximately equal isotopic ratio for the symmetric stretching terminal mode. In accordance, DFT calculations for the effect of 18O/16O substitution on the symmetric stretching wavenumber for the di-oxo (O)2Mo(–O–Si)2 structure result in an isotopic ratio of 1.0507,43 effectively identical to the 1.0513 isotopic ratio calculated for a diatomic mono-oxo MoO configuration based on eq. (2). Hence, whereas calculations for theoretically predicted wavenumbers for ν(Re18O)n modes can be used for verifying the validity of experimentally observed isotopic shifts, such calculations are not adequate for discriminating between e.g. mono-oxo and di-oxo termination configurations. Additionally, experimental results pertaining to well-documented di-oxo Re(O)2 terminations on TiO2 and CeO2 have shown that the use of the isotopic ratio to calculate the wavenumber of the 18O/16O substituted site (e.g. Re18O or Re(18O)2) cannot be used for differentiating between mono-oxo and di-oxo termination configurations.20,21
Fig. 9(A–C) shows the evolution of the in situ Raman spectra obtained at 400 °C under flowing (10 cm3 min−1) 2% 18O2/He upon successive reduction/oxidation cycles using flowing 5% H2/He and 2% 18O2/He (see Experimental section) for samples with coverage of 0.71 Re nm−2 (Fig. 9A), 1.3 Re nm−2 (Fig. 9B) and 2.4 Re nm−2 (Fig. 9C). The number of redox 18O/16O substitution cycles is indicated by each spectrum. The gradual progress of the 18O/16O substitution and the consequent evolution of the in situ Raman spectra corroborate the aforementioned interpretations and partial conclusions for the occurrence of the three main structural units, namely mono-oxo Species-I, di-oxo Species-IIa and di-oxo Species IIb. Moreover, the observed effects are adequate for further discussions on the reducibility and the relative ease of 18O/16O substitution for each individual species.
 | ||
| Fig. 9 Sequential in situ Raman spectra obtained at 400 °C under flowing 2% 18O2/He after subsequent H2/18O2 reduction/oxidation cycles as indicated by each spectrum. (A) ReOx/ZrO2 with surface density of 0.71 Re nm−2; (B) ReOx/ZrO2 with surface density of 1.3 V nm−2; and (C) ReOx/ZrO2 with surface density of 2.4 Re nm−2; spectral recording parameters: see Fig. 2 caption. | ||
At 400 °C the ReOx phase dispersed on the low coverage 0.71ReZrO2 sample consists of Species-I (mono-oxo, prevalent species), Species-IIa (di-oxo, in significant amount) and Species-IIb (di-oxo, minority species). The following observations are made in Fig. 9A:
(i) After 21 18O/16O substitution redox cycles the band envelope of the convoluted (Re16O)n modes at ∼991 cm−1 exhibits a discerned shoulder at ∼982 cm−1. Such a band is assigned to partially 18O/16O substituted di-oxo sites (18ORe16O).16,20,21 Theoretical calculations for the wavenumber of a partially substituted di-oxo 18OMo16O site justify further such an assignment.43
(ii) During the initial steps of the 18O/16O substitution, i.e. after 1, 5 and 8 cycles, the band emerging due to the isotopic shift (eventually at ∼940 cm−1 after 21 cycles) exhibits a low wavenumber shoulder at ∼934 cm−1 indicating a relative ease for the 18O/16O substitution of the di-oxo terminal sites of the minority Species-IIb.
(iii) A facile Re–16O–Zr → Re–18O–Zr substitution is evidenced for the anchoring site, justified by the isotopic shift undergone by the ∼885 cm−1 Re–O–Zr band already from the 1st 18O/16O cycle.
For both 1.3ReZrO2 and 2.4ReZrO2 samples with coverages of 1.3 and 2.4 Re nm−2, the di-oxo Species-IIa prevails at 400 °C, whilst the mono-oxo Species-I and the di-oxo Species-IIb also exist in minority. The pertinent isotopic effects observed are similar for the two samples and differ from the ones observed for 0.71ReZrO2 only due to the different speciation of the dispersed ReOx phase. More specifically:
(i) A facile Re–16O–Zr → Re–18O–Zr substitution is evidenced already after the 1st 18O/16O cycle (and largely after the fifth cycle), justified by the isotopic shift undergone by the ∼888 cm−1 Re–O–Zr mode.
(ii) The bands emerging due to (Re18O)n at 942 cm−1 for 1.3ReZrO2 (Fig. 9B) and 946 cm−1 for 2.4ReZrO2 (Fig. 9C) exhibit in each case a shoulder on their low wavenumber wing in the initial stages of the isotopic exchange clearly seen after the 1st cycle at 936 cm−1 for 1.3ReZrO2 (Fig. 9B) and 940 cm−1 for 2.4ReZrO2 (Fig. 9C), thereby indicating a facile 16ORe16O → 18ORe18O substitution for the minority di-oxo Species–IIb. With further progress in the 18O/16O substitution the main bands’ masses due to the prevailing Species-IIa at 942 and 946 cm−1 obscure the shoulders.
(iii) After 25 18O/16O substitution cycles the band envelope due to the non-substituted (Re16O)n modes appears red-shifted (995 → 992 cm−1 for 1.3 ReZrO2, Fig. 9B and 999 → 995 cm−1 for 2.4 ReZrO2, Fig. 9C) due to the earlier described28,30 next-nearest-neighbor vibrational isotope effect caused by the facile Re–16O–Zr → Re–18O–Zr substitution on the anchoring sites. Moreover, the convolutions of the 992 and 995 cm−1 bands due to the non-substituted (Re16O)n modes after 25 cycles possess each a low wavenumber shoulder (at 985 and 988 cm−1, respectively for 1.3ReZrO2 and 2.4ReZrO2), assigned to partially substituted 18ORe16O sites.16,20,21,43
(iv) The Re16O modes due to the mono-oxo Species-I at 1003 cm−1 (1.3ReZrO2, Fig. 9B) and at 1005 cm−1 (2.4ReZrO2, Fig. 9C) are entirely diminished after 8 and 9 18O/16O cycles, confirming a facile Re16O → Re18O substitution for the mono-oxo Species-I.
3.3. Synopsis of structural interpretations
In the temperature range of 120–400 °C, the (ReOx)n phase dispersed on ZrO2 is built of three main structural units, namely mono-oxo Species-I, di-oxo Species-IIa and di-oxo Species-IIb. At low coverage of ns = 0.71 Re nm−2 most ReOx sites are isolated on ZrO2. At higher coverages of ns ≥ 1.3 Re nm−2, the occurrence of ReOx in vicinal sites may favor the formation of associated species by means of Re–O–Re linkages. Hence, with increasing loading, a lowering of the Raman band intensity due to Re–O–Zr at ∼885–890 cm−1 relative to Re(O)n (n = 1, 2 for mono-oxo and di-oxo species, respectively) at 991–1003 cm−1 is observed (Fig. 2A–C) and a concomitant increase of a partly obscured broad feature at ∼935–940 cm−1 (Fig. 2A–C) that has previously also been assigned as due to bridging modes (Re–O–Re or O–Re–O).44
A reversible temperature-dependent interconversion is evidenced between mono-oxo Species–I and di-oxo Species–IIa mediated by water molecules retained at the surface layer according to reaction scheme (A), portrayed also in Fig. 8. The Species-IIa → Species-I transformation taking place with increasing temperature requires the occurrence of two OH sites adjacent to a Species-IIa. Such a mechanism is limited at coverages of ns ≥ 1.3 Re nm−2 as seen in Fig. 7B and C due to larger extent of preceded titration of surface hydroxyls, thereby limiting the availability of surface hydroxyls and lowering of the possibility for occurrence of two surface hydroxyls adjacent to each Species-IIa.
As discussed, the Species–IIb di-oxo structural unit does not undergo transformations in the temperature range of 120–400 °C and its presence remains stable, as seen in Fig. 7(A–C). However, as seen e.g. in Fig. S3(A)† pertaining to the low coverage 0.71ReZrO2 sample, there is evidence for a temperature-dependent reversible Species-IIb ↔ Species-III interconversion. Species-III is represented by a main Re(O)n stretching band at ∼974 cm−1 (see spectrum (a) in Fig. S3(A)† pertaining to 35 °C), of which the appearance is observed with decreasing temperature together with a concomitant diminishing of the ∼885 cm−1 Re–O–Zr band. Hence, the ∼974 cm−1 band is assigned to an isolated tri-oxo site possessing less Re–O–Zr anchors, formed according to the scheme:
 | (B) |
Fig. 10 shows a molecular-level mechanism accounting for the above reaction scheme (B). The occurrence of tri-oxo Species-III units is not favored at coverages of ns ≥ 1.3 Re nm−2 where associated species possessing Re–O–Re linkages prevail.
 | ||
| Fig. 10 Molecular-level mechanism accounting for the reversible temperature-dependent Species-IIb ↔ Species-III transformation mediated by water molecules retained at the surface layer and surface hydroxyls. For simplicity, mononuclear representations are shown. The number of anchoring bonds and the resulting coordination number, CNRe = 4 are reasonable in view of the oxidation state VII for Re and the tri-oxo termination configuration of Species-III. Gold spheres, Re; red spheres, O; grey spheres, Zr. H atoms (white spheres) are included to account for hydroxylation (see text). | ||
Finally, apart from the temperature and coverage effects, it is noteworthy to also address in synopsis the most recently reported and well-documented effects of metal oxide carrier type on the speciation of the ReOx dispersed phase. Significantly, the heterogeneity of the dispersed phase is hitherto established also for ReOx deposited on Al2O3,16 TiO220 and CeO2.21 In particular, the existence of two di-oxo species has been proposed for ReOx/Al2O3 catalysts,16 whilst the occurrence of a mono-oxo and a di-oxo species has been evidenced for ReOx/TiO220 and ReOx/CeO221 catalysts. To the contrary, one single ReOx species taking on a tri-oxo configuration was found for ReOx/SiO2.17
4. Conclusions
In situ molecular spectroscopy is used for studying the vibrational properties and unravelling the structural and configurational characteristics of the (ReOx)n phase dispersed on monoclinic ZrO2. In situ Raman and in situ FTIR spectra (the former also coupled with 18O/16O exchange) are exploited under dehydrated feed conditions, covering a surface density span of 0.71–3.7 Re nm−2.(a) The (ReOx)n phase dispersed on ZrO2 is heterogeneous, consisting of distinct structural units. In the temperature range of 120–400 °C, three main structural units are identified: (i) Species–I with mono-oxo termination configuration, O(Re–O–Zr)m; (ii) Species–IIa with di-oxo termination configuration, (O)2(Re–O–Zr)m−1; and (iii) Species–IIb with di-oxo termination configuration, (O)2(Re–O–Zr)u.
(b) At low coverage of 0.71 Re nm−2, isolated (monomeric) ReOx species prevail. At 400 °C, the mono-oxo Species-I predominates, Species-IIa is significantly present, whilst Species-IIb occurs in minority.
(c) A reversible temperature-dependent Species-I ↔ Species-IIa transformation is evidenced, shifting to the right with decreasing temperature.
(d) At coverages of ns ≥ 1.3 Re nm−2, Species-IIa clearly prevails over Species-I, in the full temperature range of 120–400 °C.
(e) The terminal stretching wavenumbers of all Species undergo a blue shift with increasing coverage, presumably due to variations in the basicity of dative O support atoms and/or vibrational coupling in associated (ReOx)n moieties. As an example, at 400 °C, the terminal ReO stretching mode for Species-I is found at 993–1005 cm−1, the symmetric stretching mode for Species-IIa is found at 987–998 cm−1 and the counterpart symmetric stretching mode for Species-IIb at 982–991 cm−1.
(f) At temperature below 80 °C and for a low coverage of 0.71 Re nm−2, the occurrence of a fourth structural unit Species-III is evidenced, taking on a tri-oxo termination configuration with a symmetric stretching mode at 974 cm−1.
(g) The isotopic 18O/16O substitution experiments show a facile Re–16O–Zr → Re–18O–Zr substitution and a relative ease for (16O)2(Re–O–Zr)u → (18O)2(Re–O–Zr)u substitution for the minority Species-IIb.
(h) All temperature-dependent changes and structural transformations are fully reversible under dehydrated feed conditions.
(i) It is feasible to tune the speciation of the (ReOx)n phase dispersed on ZrO2 by suitably controlling the temperature and the coverage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Professor Dimitrios Tsiplakidis (Department of Chemistry, Aristotle University of Thessaloniki) is thanked for assisting the authors with the icp-analysis of the calcined samples. This study was supported by the project “Materials and Processes for Energy and Environment Applications” (MIS 5002556), which was implemented under the “Action for the Strategic Development on the Research and Technological Sector”, funded by the Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund).References
- J. A. Moulijn and J. C. Mol, Structure and Activity of Rhenium-Based Metathesis Catalysts, J. Mol. Catal., 1988, 46, 1–14 CrossRef.
- J. C. Mol, Olefin Metathesis over Supported Rhenium Oxide Catalysts, Catal. Today, 1999, 51, 289–299 CrossRef CAS.
- S. Lwin and I. E. Wachs, Olefin Metathesis by Supported Metal Oxide Catalysts, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- C.-B. Wang, Y. Cai and I. E. Wachs, Reaction-Induced Spreading of Metal Oxides onto Surfaces of Oxide Supports During Alcohol Oxidation: Phenomenon, Nature, and Mechanisms, Langmuir, 1999, 15, 223–1235 Search PubMed.
- Y. Yuan, T. Shido and Y. Iwasawa, The New Catalytic Property of Supported Rhenium Oxides for Selective Oxidation of Methanol to Methylal, Chem. Commun., 2000, 1421–1422 RSC.
- Y. Yuan, K. Tsai, H. Liu and Y. Iwasawa, Selective Methanol Conversion to Methylal on Re-Sb-O Crystalline Catalysts: Catalytic Properties and Structural Behavior, Top. Catal., 2003, 22, 9–15 CrossRef CAS.
- X. Secordel, E. Berier, M. Capron, S. Cristol, J.-F. Paul, M. Fournier and E. Payen, TiO2-Supported Rhenium Oxide Catalysts for Methanol Oxidation: Effect of Support Texture on The Structure and Reactivity Evidenced by an Operando Raman Study, Catal. Today, 2010, 155, 177–183 CrossRef CAS.
- A. Yoboue, A. Susset, A. Tougerti, D. Gallego, S. V. Ramani, M. Kalyanikar, D. S. Dolzhnikov, S. G. Wubshet, Y. Wang, S. Cristol, V. Briois, C. La Fontaine, R. M. Gauvin, J.-F. Paul and E. Berier, An Easily Accessible Re-Based Catalyst for the Selective Conversion of Methanol: Evidence for an Unprecedented Active Site Structure Through Combined Operando Techniques, Chem. Commun., 2011, 47, 4285–4287 RSC.
- R. Thomas, E. M. van Oers, V. H. J. de Beer, J. Medema and J. A. Moulijn, Characterization of γ-alumina-supported Molybdenum Oxide and Tungsten Oxide; Reducibility of the Oxidic State Versus Hydrodesulfurization Activity of the Sulfided State, J. Catal., 1982, 76, 241–253 CrossRef CAS.
- N. Escalona, J. Ojeda, R. Cid, G. Alves, A. L. Agudo, J. L. G. Fierro and F. J. G. Llambias, Characterization and Reactivity of Re(x)/γ-Al2O3 Catalysts in Hydrodesulfurization and Hydrodenitrogenation of Gas Oil: Effect of Re Loading, Appl. Catal., A, 2002, 234, 45–54 CrossRef CAS.
- M. Vuurman, D. J. Stufkens, A. Oskam and I. E. Wachs, Structural determination of surface rhenium oxide on various oxide supports (Al2O3, ZrO2, TiO2 and SiO2), J. Mol. Catal., 1992, 76, 263–285 CrossRef CAS.
- D. S. Kim and I. E. Wachs, Surface Rhenium Oxide-Support Interaction for Supported Re2O7 Catalysts, J. Catal., 1993, 141, 419–429 CrossRef CAS.
- B. Mitra, X. Gao, I. E. Wachs, A. M. Hirt and G. Deo, Characterization of Supported Rhenium Oxide Catalysts: Effect of Loading, Support and Additives, Phys. Chem. Chem. Phys., 2001, 3, 1144–1152 RSC.
- B. M. Weckhuysen, J.-M. Jehng and I. E. Wachs, In Situ Raman Spectroscopy of Supported Transition Metal Oxide Catalysts: 18O2-16O2 Isotopic Labeling Studies, J. Phys. Chem. B, 2000, 104, 7382–7387 CrossRef CAS.
- S. R. Bare, S. D. Kelly, F. D. Vila, E. Boldingh, E. Karapetrova, J. Kas, G. E. Mickelson, F. S. Modica, N. Yang and J. J. Rehr, Experimental (XAS, STEM, TPR, and XPS) and Theoretical (DFT) Characterization of Supported Rhenium Catalysts, J. Phys. Chem. C, 2007, 111, 5740–5745 CrossRef PubMed.
- S. Lwin, C. Keturakis, J. Handzlik, P. Sautet, Y. Li, A. I. Frenkel and I. E. Wachs, Surface ReOx Sites on Al2O3 and their Molecular Structure-Reactivity Relationships for Olefin Metathesis, ACS Catal., 2015, 5, 1432–1444 CrossRef CAS.
- E. L. Lee and I. E. Wachs, In Situ Raman Spectroscopy of SiO2-Supported Transition Metal Oxide Catalysts: An Isotopic 18O–16O Exchange Study, J. Phys. Chem. C, 2008, 112, 6487–6498 CrossRef CAS.
- C. Andriopoulou and S. Boghosian, Heterogeneity of Deposited Phases in Supported Transition Metal Oxide Catalysts: Reversible Temperature-Dependent Evolution of Molecular Structures and Configurations, Phys. Chem. Chem. Phys., 2018, 20, 1742–1751 RSC.
- C. Andriopoulou and S. Boghosian, Tuning the Configuration of Dispersed Oxometallic Sites in Supported Transition Metal Oxide Catalysts: A Temperature Dependent Raman Study, Catal. Today, 2019, 336, 74–83 CrossRef CAS.
- C. Andriopoulou and S. Boghosian, Molecular Structure and Termination Configuration of Oxo-Re(VII) Catalyst Sites Supported on Titania, Catal. Today, 2020, 355, 665–677 CrossRef CAS.
- B. MacQueen, B. Ruiz-Yi, M. Royko, A. Heyden, Y. J. Pagan-Torres, C. Williams and J. Lauterbach, In situ Oxygen Isotopic Exchange Vibrational Spectroscopy of Rhenium Oxide Surface Structures on Cerium Oxide, J. Phys. Chem. C, 2020, 124, 7174–7181 CrossRef CAS.
- J. Strunk, M. A. Banares and I. E. Wachs, Vibrational Spectroscopy of Oxide Overlayers, Top. Catal., 2017, 60, 1577–1617 CrossRef CAS.
- Th. Kentri, A. Trimpalis, A. Misa, E. Kordouli, Th. Ramantani and S. Boghosian, Rethinking Molecular Structures of WviOx Sites Dispersed on Titania. Distinct Mono-Oxo Configurations at 430 °C and Temperature-Dependent Transformations, Dalton Trans., 2022, 51, 7455–7475 RSC.
- Th. Kentri, A. Tsevis and S. Boghosian, Heterogeneity of the vanadia phase dispersed on titania. Co-existence of distinct mono-oxo VOx sites, Dalton Trans., 2023, 52, 7495–7511 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley – Interscience, New York, 6th edn, 2009 Search PubMed.
- C. Andriopoulou, I. Anastasiou and S. Boghosian, Di-Oxo and Tri-Oxo Re(VII)-Oxosulfato Complexes in the Re2O7-K2S2O7 Molten System. Molecular Structure, Vibrational Properties and Temperature-Dependent Interconversion, Vib. Spectrosc., 2019, 100, 14–21 CrossRef CAS.
- A. Christodoulakis and S. Boghosian, Molecular Structure and Activity of Molybdena Catalysts Supported on Zirconia for Ethane Oxidative Dehydrogenation Studied by Operando Raman Spectroscopy, J. Catal., 2008, 260, 178–187 CrossRef CAS.
- G. Tsilomelekis and S. Boghosian, On the Configuration, Molecular Structure and Vibrational Properties of MoOx Sites on Alumina, Zirconia, Titania and Silica, Catal. Sci. Technol., 2013, 3, 1869–1888 RSC.
- C. Andriopoulou, D. Harris, H. Stephenson, A. M. Eftsathiou and S. Boghosian, In situ Raman Spectroscopy as a Tool for Discerning Subtle Structural Differences Between Commercial (Ce,Zr)O2-Based OSC Materials of Identical Composition, Catalysts, 2020, 10, 462 CrossRef CAS.
- G. Tsilomelekis and S. Boghosian, In Situ Raman and FTIR Spectroscopy of Molybdenum(VI) Oxide Supported on Titania Combined with 18O/16O Exchange: Molecular Structure, Vibrational Properties and Vibrational Isotope Effects, J. Phys. Chem. C, 2011, 118, 2146–2154 CrossRef.
- G. Tsilomelekis and S. Boghosian, Structural and Vibrational Properties of Molybdena Catalysts Supported on Alumina and Zirconia Studied by in Situ Raman and FTIR Spectroscopies Combined with 18O/16O Isotopic Substitution, Catal. Today, 2010, 158, 146–155 CrossRef CAS.
- K. Bourikas, K. Ch. Kordulis and A. Lycourghiotis, Titanium Dioxide (Anatase and Rutile): Surface Chemistry, Liquid–Solid Interface Chemistry, and Scientific Synthesis of Supported Catalysts, Chem. Rev., 2014, 114, 9754–9823 CrossRef CAS PubMed.
- G. D. Panagiotou, Th. Petsi, K. Bourikas, Ch. Kordulis and A. Lycourghiotis, The Interfacial Impregnation Step Involved in the Preparation of Tungsten(VI) Supported Titania Catalysts, J. Catal., 2009, 262, 266–279 CrossRef CAS.
- G. D. Panagiotou, Th. Petsi, K. Bourikas, A. G. Kalampounias, S. Boghosian, Ch. Kordulis and A. Lycourghiotis, Interfacial Impregnation Chemistry in the Synthesis of Molybdenum Catalysts Supported on Titania, J. Phys. Chem. C, 2010, 114, 11868–11879 CrossRef CAS.
- G. Tsilomelekis, G. D. Panagiotou, P. Stathi, A. G. Kalampounias, K. Bourikas, Ch. Kordulis, Y. Deligiannakis, S. Boghosian and A. Lycourghiotis, Molybdena Deposited on Titania by Equilibrium Deposition Filtration: Structural Evolution of Oxo–Molybdenum(VI) Sites with Temperature, Phys. Chem. Chem. Phys., 2016, 18, 23980–23989 RSC.
- A. Tribalis, G. D. Panagiotou, G. Tsilomelekis, A. G. Kalampounias, K. Bourikas, Ch. Kordulis, S. Boghosian and A. Lycourghiotis, Temperature-Dependent Evolution of the Molecular Configuration of Oxo-Tungsten(VI) Species Deposited on the Surface of Titania, J. Phys. Chem. C, 2014, 118, 11319–11332 CrossRef CAS.
- E. Tella, A. Trimpalis, A. Tsevis, Ch. Kordulis, A. Lycourghiotis, S. Boghosian and K. Bourikas, Advanced Synthesis and Characterization of Vanadia/Titania Catalysts Through a Molecular Approach, Catalysts, 2021, 11, 322 CrossRef CAS.
- S. Loridant, C. Feche, N. Essayem and F. Figueras, WOx/ZrO2 Catalysts Prepared by Anionic Exchange; In situ Raman Investigation from the Precursor Solutions to the Calcined Catalysts, J. Phys. Chem. B, 2005, 109, 5631–5637 CrossRef CAS PubMed.
- B.-K. Kim and H. Hamaguchi, Mode Assignments of the Raman Spectrum of Monoclinic Zirconia by Isotopic Exchange Technique, Phys. Status Solidi B, 1997, 203, 557–563 CrossRef CAS.
- G. Herzberg, Molecular Spectra and Molecular Structure, I. Spectra of Diatomic Molecules, Van Nostrand Company Inc., Princeton, 2nd edn, 1950 Search PubMed.
- G. Busca, Differentiation of Mono-oxo and Polyoxo and of Monomeric and Polymeric Vanadate, Molybdate and Tungstate Species in Metal Oxide Catalysts by IR and Raman Spectroscopy, J. Raman Spectrosc., 2002, 33, 348–358 CrossRef CAS.
- G. Herzberg, Molecular Spectra and Molecular Structure, II. Infrared and Raman Spectra of Polyatomic Molecules, Van Nostrand Company Inc., Princeton, 2nd edn, 1950 Search PubMed.
- S. Chempath, Y. Zhang and A. T. Bell, DFT Studies of the Structure and Vibrational Specttra of Isolated Molybdena Species Supported on Silica, J. Phys. Chem. C, 2007, 111, 1291–1298 CrossRef CAS.
- M. A. Banares and I. E. Wachs, Raman Spectroscopy of Catalysts, in Encyclopedia of Analytical Chemistry, 2010, John Wiley & Sons, pp. 1–30 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt04270g |
| This journal is © The Royal Society of Chemistry 2024 |