
Fine-tuning of thermally induced SCO behaviors of trinuclear cyanido-bridged complexes by regulating the electron donating ability of CCN-terminal fragments†
Ying-Ying
Huang
ab,
Yong
He
ab,
Yang
Liu
ab,
Jin-Hui
Fu
ab,
Xiao-lin
Liu
ab,
Xin-Tao
Wu
 a and
Tian-Lu
Sheng
*a
a and
Tian-Lu
Sheng
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: tsheng@fjirsm.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing, 100049, China
First published on 24th January 2024
Abstract
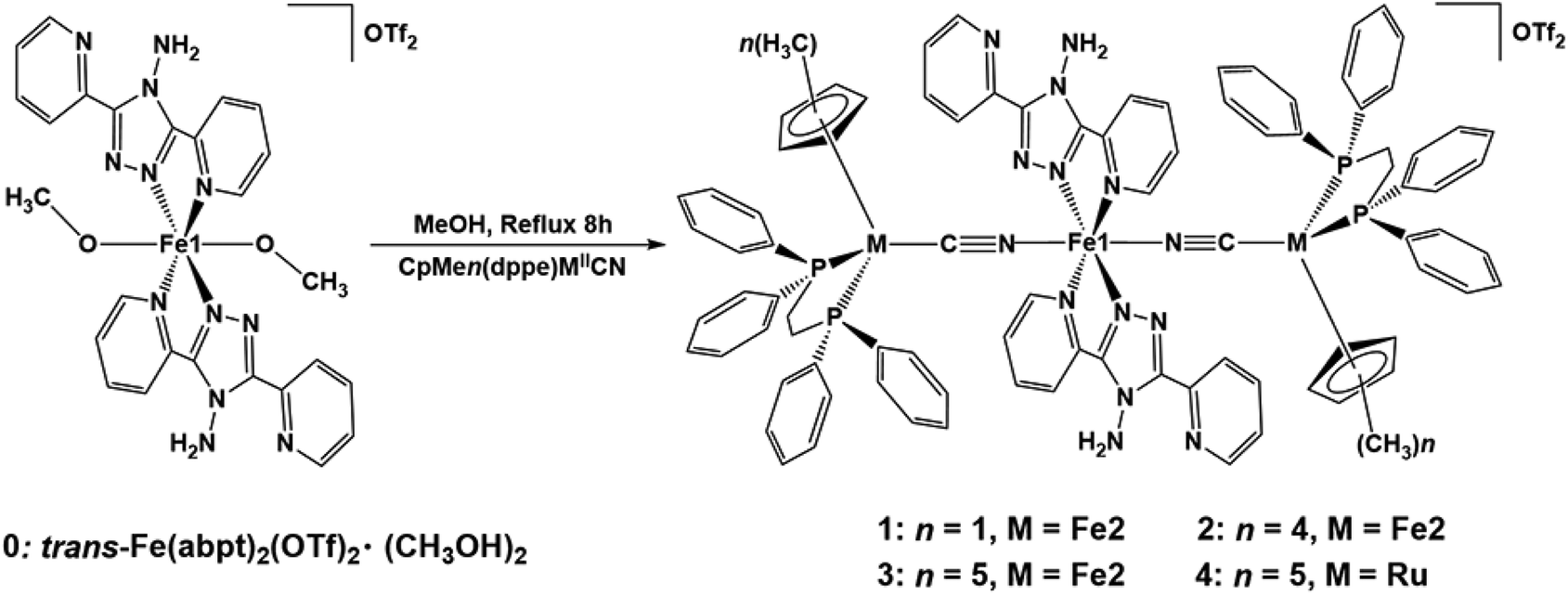
To achieve fine regulation of FeII SCO behavior, a series of trinuclear cyanido-bridged complexes trans-[CpMen(dppe)MII(CN)]2[Fe1II(abpt)2](OTf)2 (1–4) (1, M = Fe2 and n = 1; 2, M = Fe2 and n = 4; 3, M = Fe2 and n = 5; 4, M = Ru and n = 5; CpMen = alkyl cyclopentadienyl with n = 1, 4, 5; dppe = 1,2-bis-(diphenylphosphino)ethane; abpt = 4-amino-3,5-bis-(pyridin-2-yl)-1,2,4-triazole and OTf = CF3SO3−) were synthesized and fully characterized by using elemental analysis, X-ray crystallography, magnetic measurements, variable-temperature IR spectroscopy and Mössbauer spectroscopy. It is worth mentioning that different from many mononuclear Fe(abpt)2X2 (X = NCS, NCSe, N(CN)2, C(CN)3, (NC)2CC(OCH3)C(CN)2, (NC)2CC(OC2H5)C(CN)2, C16SO3 and Cl) complexes with more than one polymorph, only one polycrystalline form was found in complexes 1–4. Moreover, the thermally induced SCO behaviors of these four complexes are independent of intermolecular π–π interactions. The electron-donating ability of the CCN-terminal fragment of CpMen(dppe)MIICN can be flexibly regulated by changing the methyl number (n) of the cyclopentadiene ligand or metal ion type (MII). These investigations indicate that the electron-donating ability of the CCN-terminal fragment has an influence on the SCO behavior of Fe1II. The spin transition temperature (T1/2) of the complexes decreases with the increase of the electron-donating ability of the fragment CpMen(dppe)MII. This study provides a new strategy to predict and precisely regulate the behaviors of SCO complexes.
Introduction
Spin crossover (SCO) complexes can easily undergo transition between high spin (HS) and low spin (LS) states through external stimuli such as temperature, pressure, light, magnetic fields, and guest molecules,1–10 and this interesting spin state transition is usually accompanied by changes in the physical properties of the materials, including their color, structural phase transition, dielectric constant and magnetism. Therefore, as controllable functional molecular materials, these novel properties of SCO complexes have been widely studied and applied in high-performance devices such as molecular switches, information storage devices, optical devices, and sensors in the past few decades.8,11–18 Among them, predicting the SCO behavior of complexes to precisely control their spin transition temperature (T1/2) is important for practical applications. Due to the direct influence of the ligand field on the SCO properties of complexes, modifying organic ligands directly bound to spin transition metal centers to regulate SCO behaviors is a common research method that has been extensively studied.19–28 However, the spatial and/or substituent effects caused by the direct modification of ligands often have complicated and unpredictable effects on the properties of SCO.26,29–34 Thus, although some relevant reports have been published, regulating the SCO behavior of complexes remains a significant challenge.SCO-active materials based on octahedral complexes with [FeIIN6] coordination centers have been most frequently reported, and ligands containing 1,2,4-triazoles can usually provide suitable ligand field strength for iron(II) to induce SCO.35,36 The complex [Fe(abpt)2(NCS)2] (abpt = 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole) is one particularly interesting example, which has four reported polymorphs (A–D) that show slightly different spin crossover behaviors.37,38 Polymorph A has been initially reported to have an independent Fe(II) center (Z′ = 0.5) in the asymmetric unit which undergoes a thermal spin transition without hysteresis, with T1/2 = 180 K. The complex [Fe(abpt)2(NCSe)2] has also been reported along with polymorph A of [Fe(abpt)2(NCS)2].37 The two derivatives are isostructural, they crystallize in the monoclinic P21/n space group, and [Fe(abpt)2(NCSe)2] exhibits thermally induced SCO behavior without hysteresis, with T1/2 = 224 K. Afterwards, many mononuclear SCO complexes with the chemical formula Fe(abpt)2X2 (X = NCS, NCSe, N(CN)2, C(CN)3, (NC)2CC(OCH3)C(CN)2, (NC)2CC(OC2H5)C(CN)2, C16SO3 and Cl) have been reported.35,39–41 The different magnetic behaviors of these complexes provide us with research ideas for this work. As cyanide (CN−) possesses a unique coordination mode, the structure of cyanide-bridged complexes with spin crossover can be more conveniently modified, and this modification is far away from the spin transition metal center, which can partly reduce the complicated effects of steric and substituent effects on SCO behavior. In addition, the stretching frequency of the cyanido group (CN−) is characterized by infrared spectroscopy and can be easily observed, particularly because it can sensitively reflect the spin state of iron(II) complexes.
Herein, we report the synthesis and characterization of four trinuclear cyanido-bridged complexes trans-[CpMen(dppe)MII(CN)]2[Fe1II(abpt)2](OTf)2 (1, M = Fe2 and n = 1; 2, M = Fe2 and n = 4; 3, M = Fe2 and n = 5; 4, M = Ru and n = 5; CpMen = alkyl cyclopentadienyl with n = 1, 4, 5; dppe = 1, 2-bis-(diphenylphosphino)ethane; abpt = 4-amino-3,5-bis- (pyridin-2-yl)-1,2,4-triazole and OTf = CF3SO3−) (Scheme 1). The N-terminal of the CN bridge is the spin transition center (Fe1II), and ligand field strength is regulated by modifying the CpMenMII(dppe) fragment at the C-terminal of the bridge, namely changing the methyl number (n) of the cyclopentadiene ligand or metal ion type. The magnetic behaviors of these four complexes have been investigated by X-ray crystallography, magnetic measurements, variable-temperature IR, and Mössbauer spectroscopy. The experimental results demonstrate that the electron-donating ability of the CpMen(dppe)MII fragment has a significant influence on the SCO behavior of Fe1II. The T1/2 of the complex can be fine-tuned by regulating the electron-donating ability of the metal organic fragment at the C-terminal of CN.
 | ||
| Scheme 1 Synthesis of complexes 1–4. | ||
Results and discussion
Synthesis and X-ray structure determination
Complexes 1–4 were synthesized by reacting CpMen(dppe)MIICN with trans-Fe(abpt)2(OTf)2·(CH3OH)2 in methanol as shown in Scheme 1 and obtained by slow diffusion of diethyl ether into a mixed solvent of methanol and dichloromethane. Only one polycrystalline form was found for complexes 1–4. Single-crystal X-ray data of 1, 2 and 4 were collected at 293 K. In order to describe the thermal magnetic properties in detail and relate these structures to the crystal structure of 3, three data sets were collected at 100, 293, and 400 K (along the thermal spin transition). Unfortunately, however, the useful crystal data of 3 at 400 K could not be obtained due to the poor crystal quality at high temperatures. 1 and 2 crystallize in the monoclinic P21/n space group, whereas 3 and 4 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Because central Fe1II is located at an inversion centre, half of the molecules in the four complexes are in the asymmetric unit (Z′ = 0.5). The crystallographic data and structural details are listed in Tables S1 and S2 (see the ESI†), and the structures of the four complexes are shown in Fig. 1 and Fig. S3–S6.† The main structural features of these four complexes are very similar. Each asymmetrical unit of the molecular structures of 1–4 consists of a discrete [MIILCNFe1II(abpt)2NCMIIL] molecule where two equivalent chelating abpt ligands stand in the equatorial plane and the two equivalent metal–organic fragments (MIILCN) complete the coordination sphere at trans positions (Fig. 1). Central Fe1II is in a distorted [FeN6] octahedral environment and coordinated by six nitrogen atoms: two from the two CN groups and four from the two abpt ligands (two pyridyl nitrogen and two triazole nitrogen).
. Because central Fe1II is located at an inversion centre, half of the molecules in the four complexes are in the asymmetric unit (Z′ = 0.5). The crystallographic data and structural details are listed in Tables S1 and S2 (see the ESI†), and the structures of the four complexes are shown in Fig. 1 and Fig. S3–S6.† The main structural features of these four complexes are very similar. Each asymmetrical unit of the molecular structures of 1–4 consists of a discrete [MIILCNFe1II(abpt)2NCMIIL] molecule where two equivalent chelating abpt ligands stand in the equatorial plane and the two equivalent metal–organic fragments (MIILCN) complete the coordination sphere at trans positions (Fig. 1). Central Fe1II is in a distorted [FeN6] octahedral environment and coordinated by six nitrogen atoms: two from the two CN groups and four from the two abpt ligands (two pyridyl nitrogen and two triazole nitrogen).
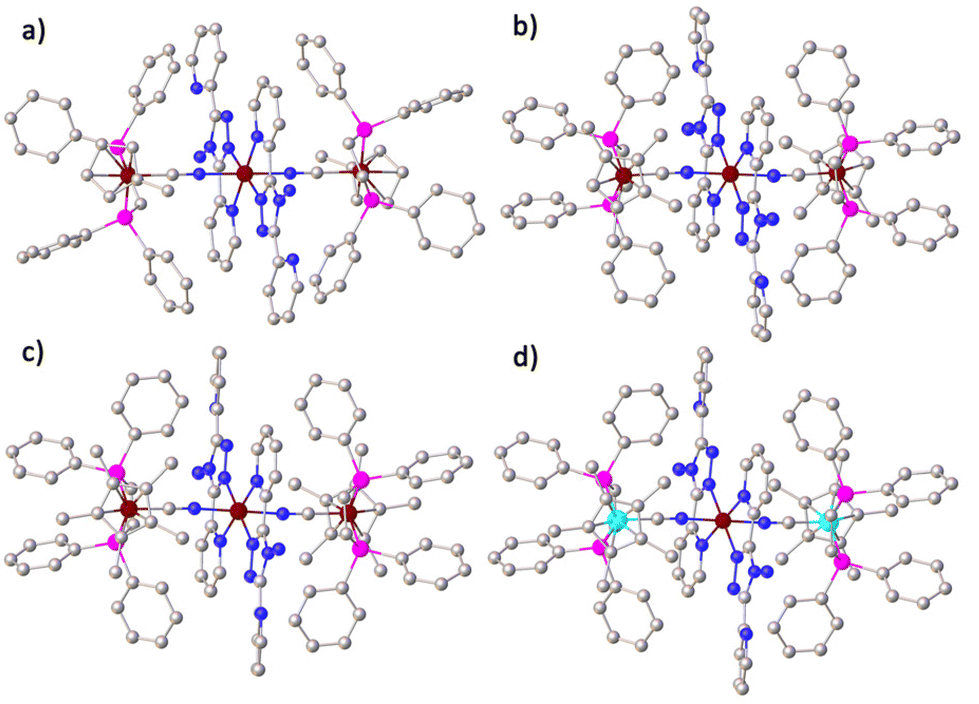 | ||
| Fig. 1 Molecular structure of 1–4 (a–d). Hydrogen atoms and CF3SO3− anions have been removed for clarity. Reddish brown, Fe; Tiffany blue, Ru; pink, P; gray, C; dark blue, N. | ||
The Fe1–N bond lengths of these four complexes are summarized in Table 1. At room temperature, the average Fe1–N bond lengths of 1 and 4 are about 2.071 Å and 2.103 Å, and the Fe–Npy (coordinated with pyridyl nitrogen) bond lengths are 2.103 Å and 2.155 Å, respectively. The average Fe1–N bond lengths are between the typical Fe–N bond lengths of LS and HS Fe(abpt)2X2 complexes,35,37–39 indicating that the Fe1II centers of 1 and 4 undergo SCO at room temperature. The average length of the Fe1–N bond of 3 is about 1.979 Å (Fe1–Npy = 2.007 Å) at 100 K, which is characteristic of LS FeII. When the temperature reached at 293 K, the average Fe1–N and Fe1–Npy bond lengths of 3 are 2.152 Å and 2.201 Å, that are very close to the characteristic bond lengths of HS Fe(abpt)2X2. This result may imply that complex 3 undergoes a thermally induced spin state transition from 100 K to 293 K. Similar to 3, the average length of the Fe1–N bond of 2 is about 2.150 Å (Fe1–Npy = 2.221 Å) at 293 K, very close to the typical Fe–N bond lengths of HS Fe(abpt)2X2.
| Complex | T (K) | Fe1–N (dicyan) | Fe1–N (py) | Fe1–N (trz) | Fe1–Na | Space group | Z′ | Spin state (Fe1II) | T 1/2 (K) |
|---|---|---|---|---|---|---|---|---|---|
| a The average length of the Fe1–N bond. | |||||||||
| 1 | 293 | 2.037(5) | 2.102(4) | 2.071(4) | 2.070 | P21/n | 0.5 | Mix LS/HS | 300 |
| 2 | 293 | 2.073(2) | 2.220(2) | 2.155(2) | 2.150 | P21/n | 0.5 | Mix LS/HS | 232 |
| 3 | 293 | 2.076(2) | 2.201(3) | 2.179(3) | 2.152 |
P |
0.5 | Mix LS/HS | 222 |
| 3 | 100 | 1.937(15) | 2.007(14) | 1.995(14) | 1.980 |
P |
0.5 | LS | — |
| 4 | 293 | 2.026(3) | 2.155(3) | 2.131(3) | 2.104 |
P |
0.5 | Mix LS/HS | 275 |
The molecular packing of 1 and 2, and 3 and 4 is same (Fig. 2), but that of 1 and 3 is quite different. Both 3 and 4 have a similar 1D molecular chain (Fig. S9 and S10†) often observed in abpt complexes, which are formed by π–π interactions between neighboring parallel abpt planes (Fig. 2c and d). The pyridyl rings on abpt of 3 and 4 are involved in π–π interactions between the intermolecular pyridyl rings with the distances of 3.813 Å for 3 and 3.833 Å for 4. However, in complexes 1 and 2, the abpt planes between the molecules are nearly perpendicular to each other, and no π–π interaction is found (Fig. 2a and b). According to published reports, the SCO behaviors of the mononuclear Fe(abpt)2X2 complexes are generally related to the intermolecular π–π interactions. All of the mononuclear Fe(abpt)2X2 complexes that undergo at least a partial thermal SCO displayed π–π interactions between pyridyl rings on abpt ligands. Besides, all of the mononuclear Fe(abpt)2X2 complexes without intermolecular π–π interactions did not undergo a thermal spin crossover.42
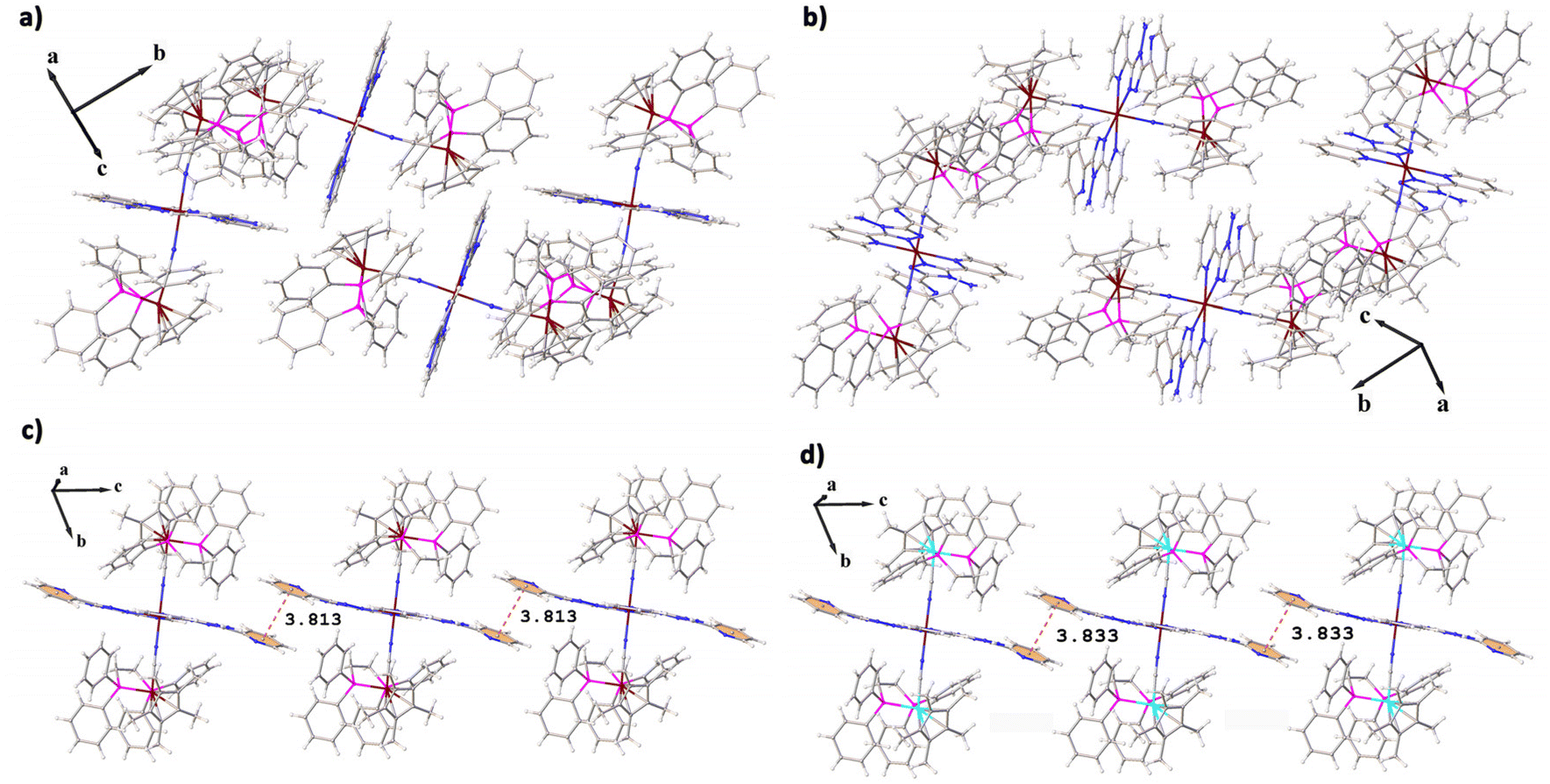 | ||
| Fig. 2 Diagrams showing the molecular packing of 1–4 (a–d); CF3SO3− anions have been removed for clarity. The red dashed lines represent the π⋯π interactions. | ||
Obviously, compared with reported mononuclear Fe(abpt)2X2, the influence of intermolecular π–π interactions on the SCO behavior is very weak for the trinuclear complexes 1–4. All 1–4 exhibit thermally induced SCO behaviors (see below), although there is no intermolecular π–π interaction in complexes 1 and 2. In addition, the π–π interactions in 3 and 4 (3.813 Å and 3.833 Å) are significantly weaker than those of previously reported mononuclear Fe(abpt)2X2 (3.256–3.684 Å),35,37–39,41 which may be related to the modification away from the spin transition metal center.
Solid-state magnetic properties
The temperature dependence of magnetic susceptibilities of 1–4 were investigated on the crystals under an applied field of 1000 Oe in the temperature range of 2–400 K during both heating and cooling processes, and the χMT vs. T plots of 1–4 in the range of 2–400 K are shown in Fig. 3. Complexes 2 and 3 exhibit a complete thermal spin crossover in the temperature range of 2–400 K. The χMT value is about 3.23 cm3 K mol−1 for 2 and 3 at 400 K, which is in the range of the expected value for one HS FeII, indicating one HS Fe1II in the central fragment and two LS Fe2II in the terminal CpMen(dppe)FeII fragments in 2 and 3 at 400 K. As the temperature decreases, the χMT values first smoothly decrease down to 3.0 cm3 K mol−1 at 290 K for 2 and 2.94 cm3 K mol−1 at 280 K for 3. Then, the χMT values drop more rapidly to reach 0.058 cm3 K mol−1 at 170 K (for 2) and 0.049 cm3 K mol−1 at 150 K (for 3). These are the characteristic SCO behaviors of the transition from a HS state to a LS state. Compared with 2 and 3, complexes 1 and 4 undergo a partial thermal spin crossover in the range of 2–400 K. At 400 K, the χMT values of 1 and 4 are about 2.97 and 3.13 cm3 K mol−1, suggesting that neither has reached a complete HS state compared to 2 and 3. During the cooling process, the χMT values decrease gradually to 0.03 cm3 K mol−1 for 1 at 200 K and 0.045 cm3 K mol−1 for 4 at 188 K, indicating that 1 and 4 have completely transformed into a LS state. Furthermore, the χMT vs. T plots of 1–4 in the cooling mode and the heating mode are basically consistent without thermal hysteresis loops.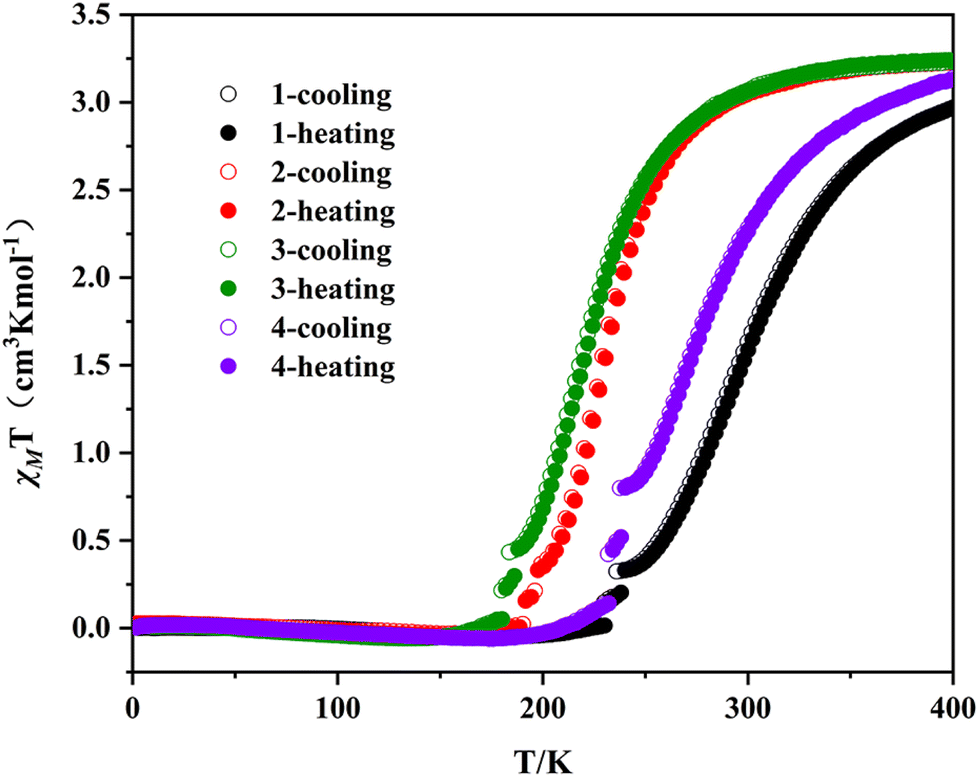 | ||
| Fig. 3 Temperature-dependent susceptibilities under 1000 Oe for the polycrystalline samples of 1–4. | ||
The spin transition temperature T1/2 of 1–4 are calculated to be 300, 232, 222 and 275 K (see Table 1). Obviously, from 1 to 3, the spin transition temperature decreases with the increase of the number of methyl substituents on the cyclopentadiene ligand, and as for 3 and 4, after replacing CCN-terminal FeII with RuII in the fragment (CpMe5(dppe)MII), the spin transition temperature increases from 222 K to 274 K. This can be attributed to the stronger electron donating ability of FeII than that of RuII, which also further explain the similarity in magnetic behaviors between complexes 2 and 3, and complexes 1 and 4. The similarity in magnetic behaviors between 2 and 3 is due to their very similar number (n) of methyl on the CpMenMII(dppe) fragment (4 and 5, respectively), resulting in similar electron donating abilities of the CCN-terminal fragment. However, for complexes 1 and 4, their CCN-terminal fragments are CpMeFeII(dppe) and CpMe5RuII(dppe), respectively. Obviously, the electron donating ability of five methyl groups (for complex 4) is stronger than that of one methyl group (for complex 1), but the electron donating ability of RuII (for complex 4) is weaker than that of FeII (for complex 1), which results in the similar electron donating abilities of the two fragments, resulting in the similarity in magnetic behaviors of 1 and 4. Therefore, it is evident that the T1/2 of complexes 1–4 can be regulated by the electron donating ability of the fragment CpMen(dppe)MII. When the electron donating ability of CpMen(dppe)MII weakens, that is, upon reducing the number of methyl substituents on the cyclopentadiene ligand or upon replacing FeII with RuII in the metal organic fragment, the spin transition temperature of the complex obviously increases.
Infrared spectra
The spin states of the cyanido-bridged complexes can be sensitively reflected by the cyanide stretching frequency (νCN). Therefore, variable temperature infrared spectra have been performed for 1–4 and the results are shown in Fig. 4. For complex 1–4, clear changes of the cyanide stretching frequency occurred during the spin transition. Only one strong νCN peak is observed in 1 (2096 cm−1) and 4 (2115 cm−1) at 170 K, and in 2 (2090 cm−1) and 3 (2084 cm−1) at 150 K, which is attributed to the stretching modes of CN in the LS states. As the temperature increases, the CN stretching peaks for the LS state of 1–4 begin to weaken and a new CN stretching peak at a lower energy appears, attributed to the appearance of the HS state component. Upon further increasing the temperature, the CN stretching peaks of the LS state weaken obviously, while the CN stretching peaks of HS state are significantly enhanced. Furthermore, the integrated peaks of LS and HS states exhibit a regular red shift as the temperature decreases, which may be due to the increase of the high spin state components. Only one strong νCN peak remains in 1 (2065 cm−1) and 4 (2073 cm−1) at 450 K, and in 2 (2051 cm−1) and 3 (2044 cm−1) at 400 K, suggesting that complexes 1–4 have completely transformed from the LS to HS state. Evidently, the measurement results of the variable temperature IR of 1–4 are consistent with their magnetic measurements.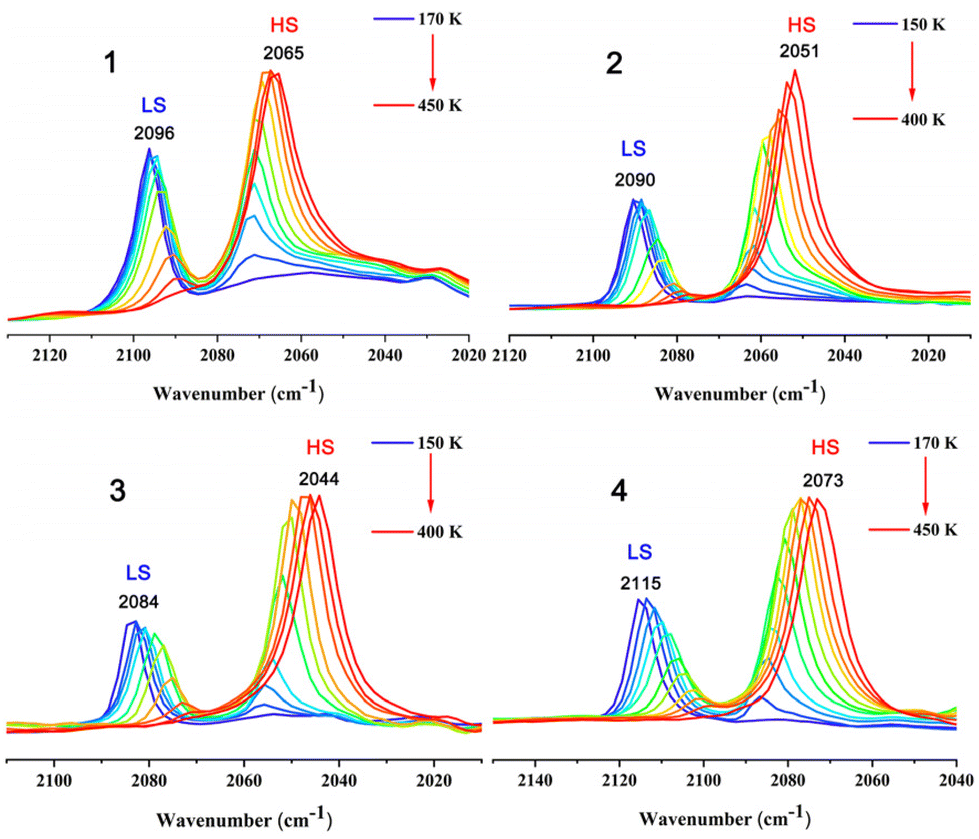 | ||
| Fig. 4 Variable-temperature infrared spectra of 1–4. | ||
Mössbauer spectra
The 57Fe Mössbauer spectra can effectively determine the spin states and proportion of FeII in the complexes, further explaining its magnetic behaviors. Spectra of complexes 1–4 have been obtained at room temperature, and the results are shown in Fig. 5. The isomer shift (δ), quadrupole splitting (ΔEQ), peak width (Γ) and the HS/LS area fractions are summarized in Table 2. The spectra of 1 can be deconvoluted into three sets, one with δ = 0.17 mm s−1 and ΔEQ = 1.84 mm s−1, the second one with δ = 0.36 mm s−1 and ΔEQ = 0.48 mm s−1 and the last one with δ = 1.02 mm s−1 and ΔEQ = 2.31 mm s−1. The former is typical of LS Fe2II of the CpMen(dppe)FeCN fragment,43,44 the second one is attributed to LS Fe1II of the FeIIN6 motif, and the last one is assigned to HS Fe1II. The three peak area ratio is 66.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19.3:14.0, where the peak area ratio of Fe1II to Fe2II is about 1:2, which is consistent with the magnetic measurement result and the structural analyses on three FeII centers (one Fe1II and two Fe2II) of 1. Similar to complex 1, three quadrupole doublets are observed in 2 and 3. The three peak area ratios of the LS Fe2II/LS Fe1II/HS Fe1II species in 2 and 3 are 66.7/3.5/29.8 and 66.7/3.3/30, respectively, which are also in good agreement with their magnetic measurement results. Due to the presence of only one FeII center in the structure of 4, only two doublets are observed for LS Fe1II [δ = 0.39 mm s−1, ΔEQ = 0.45 mm s−1] and HS Fe1II [δ = 1.04 mm s−1, ΔEQ = 2.22 mm s−1], respectively. The two peak area ratio (LS Fe1II/HS Fe1II) is 0.35/0.65, which further supports the magnetic susceptibility data of 4.
:19.3:14.0, where the peak area ratio of Fe1II to Fe2II is about 1:2, which is consistent with the magnetic measurement result and the structural analyses on three FeII centers (one Fe1II and two Fe2II) of 1. Similar to complex 1, three quadrupole doublets are observed in 2 and 3. The three peak area ratios of the LS Fe2II/LS Fe1II/HS Fe1II species in 2 and 3 are 66.7/3.5/29.8 and 66.7/3.3/30, respectively, which are also in good agreement with their magnetic measurement results. Due to the presence of only one FeII center in the structure of 4, only two doublets are observed for LS Fe1II [δ = 0.39 mm s−1, ΔEQ = 0.45 mm s−1] and HS Fe1II [δ = 1.04 mm s−1, ΔEQ = 2.22 mm s−1], respectively. The two peak area ratio (LS Fe1II/HS Fe1II) is 0.35/0.65, which further supports the magnetic susceptibility data of 4.
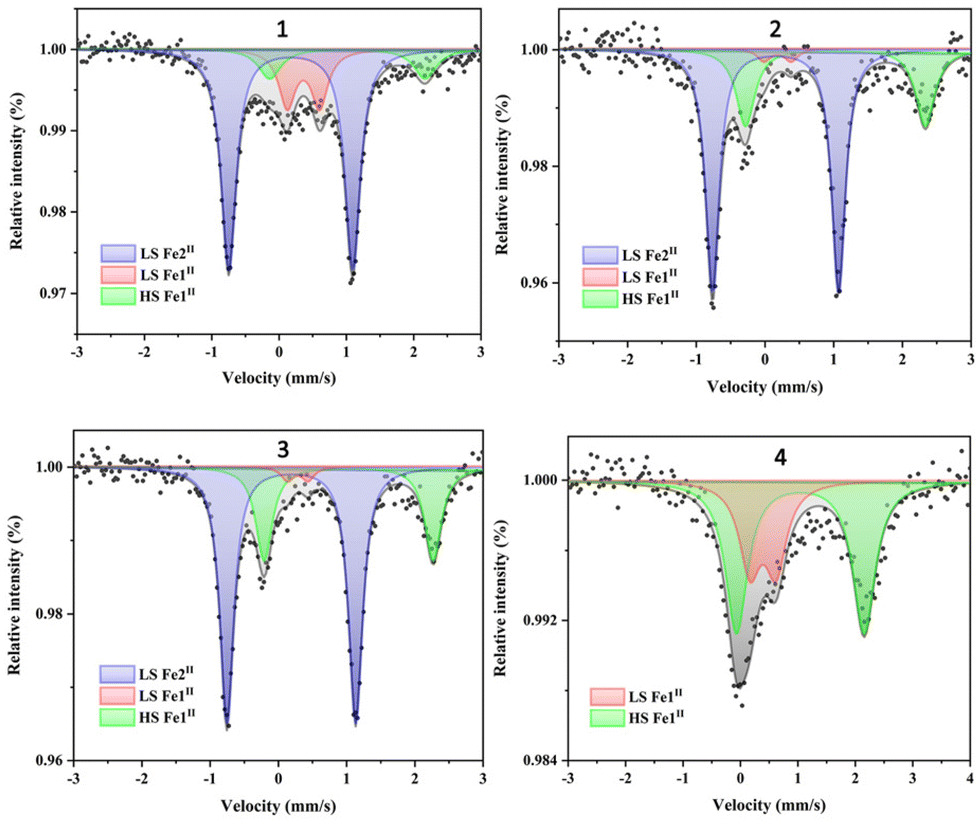 | ||
| Fig. 5 57Fe Mössbauer spectra of 1–4; the black line represents the sum of contributions of all types of Fe signals in the sample. | ||
| Complex | Fe type | δ (mm s−1) | ΔEQ (mm s−1) | Area (%) | Γ (mm s−1) |
|---|---|---|---|---|---|
| 1 | LS Fe2II | 0.17 | 1.84 | 66.7 | 0.271 |
| LS Fe1II | 0.36 | 0.48 | 19.3 | 0.302 | |
| HS Fe1II | 1.02 | 2.31 | 14 | 0.411 | |
| 2 | LS Fe2II | 0.15 | 1.84 | 66.7 | 0.239 |
| LS Fe1II | 0.18 | 0.40 | 3.5 | 0.240 | |
| HS Fe1II | 1.03 | 2.62 | 29.8 | 0.334 | |
| 3 | LS Fe2II | 0.19 | 1.89 | 66.7 | 0.244 |
| LS Fe1II | 0.28 | 0.30 | 3.3 | 0.221 | |
| HS Fe1II | 1.03 | 2.48 | 30 | 0.297 | |
| 4 | LS Fe1II | 0.39 | 0.45 | 35 | 0.451 |
| HS Fe1II | 1.04 | 2.22 | 65 | 0.468 |
Conclusions
In summary, we have synthesized and characterized four trinuclear cyanido-bridged complexes trans-[CpMen(dppe)MII(CN)]2[Fe1II(abpt)2](OTf)2 (n = 1, 4, 5; M = Fe and Ru). Therein, only one polycrystalline form was found in complexes 1–4 and it was different from the reported mononuclear Fe(abpt)2X2 (X = NCS, NCSe, N(CN)2, C(CN)3, (NC)2CC(OCH3)C(CN)2, (NC)2CC(OC2H5)C(CN)2, C16SO3 and Cl), in which the intermolecular π–π interactions play a key role in the presence of spin crossover. Magnetic measurement results indicate that all four trinuclear cyanido-bridged complexes undergo thermally induced SCO, which is influenced by the electron donating ability of the CpMen(dppe)MII fragment but not by the intermolecular π–π interactions. The spin transition temperature (T1/2) of the complexes decreases with the increase of the electron-donating ability of the fragment CpMen(dppe)MII, and the variable temperature IR and 57Fe Mössbauer spectra have further confirmed this conclusion. This study indicates that this modification away from the spin transition metal center successfully weakens the decisive influence of intermolecular π–π interactions on SCO behaviours, providing a new strategy to predict and precisely regulate the behaviors of SCO complexes.Experimental
Materials and synthesis
All manipulations were performed under an argon atmosphere with the use of standard Schlenk techniques unless otherwise stated. The ligand abpt45 and metal organic fragment CpMen(dppe)MIICN46–48 were prepared according to published papers. All other chemical solvents were available commercially at reagent grade and used without further purification.Synthesis of trans-Fe(abpt)2(OTf)2·(CH3OH)2 (0)
FeII(OTf)2 (0.0354 g, 1 mmol) and abpt (0.0476 g, 2 mmol) were added in 10 mL of methanol. The reaction mixture was refluxed with stirring at 80 °C under a nitrogen atmosphere for 6 h. After that, the mixture solution was concentrated under reduced pressure, and then the product was precipitated by the addition of diethyl ether. The obtained deep red powder was redissolved in methanol (6 mL). The faint yellow crystals of trans-Fe(abpt)2(OTf)2·(CH3OH)2 were obtained by slow diffusion of diethyl ether into the methanol solution. Yield: 0.052 g, 58%. Anal. Calcd (%) for C28H28F6FeN12O8S2: C, 37.59; H, 3.15; N, 18.79. Found (%): C, 37.16; H, 2.93; N, 18.97.Synthesis of trans-[CpMe(dppe)FeII(CN)]2[Fe1II(abpt)2](OTf)2 (1)
CpMe(dppe)FeCN (0.1118 g, 2 mmol) and trans-Fe(abpt)2(OTf)2·(CH3OH)2 (0.0895 g, 1 mmol) were added in 15 mL of methanol. The reaction mixture was refluxed with stirring at 80 °C under a nitrogen atmosphere for 6 h. After that, the mixture solution was concentrated under reduced pressure, and then the product was precipitated by the addition of diethyl ether. The obtained product was redissolved in a mixed solvent of methanol (6 mL) and dichloromethane (2 ml). The black-red block shaped crystals of 1 were obtained by slow diffusion of diethyl ether into the mixed solvent of methanol and dichloromethane. Yield: 0.150 g, 77%. Anal. Calcd (%) for C92H82F6Fe3N14O6P4S2: C, 56.69; H, 4.24; N, 10.06. Found (%): C, 56.22; H, 4.03; N, 10.17.Synthesis of trans-[CpMe4(dppe)FeII(CN)]2[Fe1II(abpt)2](OTf)2 (2)
CpMe4(dppe)FeCN (0.1202 g, 2 mmol) and trans-Fe(abpt)2(OTf)2·(CH3OH)2 (0.0895 g, 1 mmol) were added in 15 mL of methanol. The reaction mixture was refluxed with stirring at 80 °C under a nitrogen atmosphere for 6 h. After that, the mixture solution was concentrated under reduced pressure, and then the product was precipitated by the addition of diethyl ether. The obtained product was redissolved in a mixed solvent of methanol (6 mL) and dichloromethane (2 ml). The deep-red block shaped crystals of 2 were obtained by slow diffusion of diethyl ether into the mixed solvent of methanol and dichloromethane. Yield: 0.142 g, 70%. Anal. Calcd (%) for C98H94F6Fe3N14O6P4S2: C, 57.88; H, 4.66; N, 9.64. Found (%): C, 57.62; H, 4.64; N, 9.82.Synthesis of trans-[CpMe5(dppe)FeII(CN)]2[Fe1II(abpt)2](OTf)2 (3)
CpMe5(dppe)FeCN (0.1230 g, 2 mmol) and trans-Fe(abpt)2(OTf)2·(CH3OH)2 (0.0895 g, 1 mmol) were added in 15 mL of methanol. The reaction mixture was refluxed with stirring at 80 °C under a nitrogen atmosphere for 6 h. After that, the mixture solution was concentrated under reduced pressure, and then the product was precipitated by the addition of diethyl ether. The obtained product was redissolved in a mixed solvent of methanol (6 mL) and dichloromethane (2 ml). The red block shaped crystals of 3 were obtained by slow diffusion of diethyl ether into the mixed solvent of methanol and dichloromethane. Yield: 0.136 g, 66%. Anal. Calcd (%) for C100H98F6Fe3N14O6P4S2: C, 58.26; H, 4.79; N, 9.51. Found (%): C, 57.96; H, 4.88; N, 9.72.Synthesis of trans-[CpMe5(dppe)RuII(CN)]2[Fe1II(abpt)2](OTf)2 (4)
CpMe5(dppe)RuCN (0.1320 g, 2 mmol) and trans-Fe(abpt)2(OTf)2·(CH3OH)2 (0.0895 g, 1 mmol) were added in 15 mL of methanol. The reaction mixture was refluxed with stirring at 80 °C under a nitrogen atmosphere for 6 h. After that, the mixture solution was concentrated under reduced pressure, and then the product was precipitated by the addition of diethyl ether. The obtained product was redissolved in a mixed solvent of methanol (6 mL) and dichloromethane (2 ml). The deep-red block shaped crystals of 4 were obtained by slow diffusion of diethyl ether into the mixed solvent of methanol and dichloromethane. Yield: 0.155 g, 72%. Anal. Calcd (%) for C100H98F6FeN14O6P4Ru2S2: C, 55.81; H, 4.59; N, 9.11. Found (%): C, 55.50; H, 4.68; N, 9.30.As reported, many Fe(abpt)2X2 complexes have more than one polymorph, and usually, the different polymorphs can be easily distinguished with the color and morphology of the crystals. However, it appears that only one polymorph could be found during the preparation of complexes 1–4.
Physical measurements
Author contributions
T. L. S. conceived and supervised the project. Y-Y. H. synthesized and characterized the complexes, and performed the data analysis. Y. H., Y. L., J-H. F. and X-L. L. assisted in characterizing the complexes. The paper was written by Y-Y. H. and T-L. S., and all authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21973095 and 92261116), and we acknowledge Lanzhou University for providing the Mössbauer measurements.References
- P. Gütlich, Y. Garcia and T. Woike, Coord. Chem. Rev., 2001, 219–221, 839–879 CrossRef.
- A. Bousseksou, G. Molnár, J. A. Real and K. Tanaka, Coord. Chem. Rev., 2007, 251, 1822–1833 CrossRef CAS.
- J. A. Real, A. B. Gaspar and M. C. Munoz, Dalton Trans., 2005, 2005, 2062–2079 RSC.
- A. B. Gaspar, M. C. Muñoz and J. A. Real, J. Mater. Chem., 2006, 16, 2522–2533 RSC.
- N. Hoshino, F. Iijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Nat. Chem., 2012, 4, 921–926 CrossRef CAS PubMed.
- S. Chorazy, T. Charytanowicz, D. Pinkowicz, J. Wang, K. Nakabayashi, S. Klimke, F. Renz, S. I. Ohkoshi and B. Sieklucka, Angew. Chem., Int. Ed., 2020, 59, 15741–15749 CrossRef CAS PubMed.
- N. T. Yao, L. Zhao, H. Y. Sun, C. Yi, Y. H. Guan, Y. M. Li, H. Oshio, Y. S. Meng and T. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208208 CrossRef CAS PubMed.
- L. Zhao, Y. S. Meng, Q. Liu, O. Sato, Q. Shi, H. Oshio and T. Liu, Nat. Chem., 2021, 13, 698–704 CrossRef CAS PubMed.
- S. Bonnet, G. Molnár, J. S. Costa, M. A. Siegler, A. L. Spek, A. Bousseksou, W.-T. Fu, P. Gamez and J. Reedijk, Chem. Mater., 2009, 21, 1123–1136 CrossRef CAS.
- S. Ohkoshi, K. Imoto, Y. Tsunobuchi, S. Takano and H. Tokoro, Nat. Chem., 2011, 3, 564–569 CrossRef CAS PubMed.
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, P. Lazic, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blugel, M. Munzenberg and J. S. Moodera, Nature, 2013, 493, 509–513 CrossRef CAS PubMed.
- Y. S. Meng and T. Liu, Acc. Chem. Res., 2019, 52, 1369–1379 CrossRef CAS PubMed.
- E. Coronado, Nat. Rev. Mater., 2019, 5, 87–104 CrossRef.
- K. S. Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef CAS PubMed.
- M. Estrader, J. S. Uber, L. A. Barrios, J. Garcia, P. Lloyd-Williams, O. Roubeau, S. J. Teat and G. Aromi, Angew. Chem., Int. Ed., 2017, 56, 15622–15627 CrossRef CAS PubMed.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- G. Molnar, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 17003862 CrossRef PubMed.
- Y.-Y. Zhu, H.-Q. Li, Z.-Y. Ding, X.-J. Lü, L. Zhao, Y.-S. Meng, T. Liu and S. Gao, Inorg. Chem. Front., 2016, 3, 1624–1636 RSC.
- C. Bartual-Murgui, S. Vela, M. Darawsheh, R. Diego, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem. Front., 2017, 4, 1374–1383 RSC.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- C.-J. Zhang, K.-T. Lian, S.-G. Wu, Y. Liu, G.-Z. Huang, Z.-P. Ni and M.-L. Tong, Inorg. Chem. Front., 2020, 7, 911–917 RSC.
- J. Yuan, M.-J. Liu, S.-Q. Wu, X. Zhu, N. Zhang, O. Sato and H.-Z. Kou, Inorg. Chem. Front., 2019, 6, 1170–1176 RSC.
- C.-F. Wang, Z.-S. Yao, G.-Y. Yang and J. Tao, Inorg. Chem., 2019, 58, 1309–1316 CrossRef CAS PubMed.
- P. Hamon, J.-Y. Thépot, M. Le Floch, M.-E. Boulon, O. Cador, S. Golhen, L. Ouahab, L. Fadel, J.-Y. Saillard and J.-R. Hamon, Angew. Chem., 2008, 120, 8815–8819 CrossRef.
- A. Kimura and T. Ishida, ACS Omega, 2018, 3, 6737–6747 CrossRef CAS PubMed.
- B. Fei, X. Q. Chen, Y. D. Cai, J.-K. Fang, M. L. Tong, J. Tucek and X. Bao, Inorg. Chem. Front., 2018, 5, 1671–1676 RSC.
- S. Rodríguez-Jiménez, M. Yang, I. Stewart, A. L. Garden and S. Brooker, J. Am. Chem. Soc., 2017, 139, 18392–18396 CrossRef PubMed.
- L. J. K. Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55, 4327–4331 CrossRef PubMed.
- A. Kimura and T. Ishida, Inorganics, 2017, 5, 52 CrossRef.
- H.-J. Lin, D. Siretanu, D. A. Dickie, D. Subedi, J. J. Scepaniak, D. Mitcov, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2014, 136, 13326–13332 CrossRef CAS PubMed.
- K. Nakano, N. Suemura, K. Yoneda, S. Kawata and S. Kaizaki, Dalton Trans., 2005, 2005, 740–743 RSC.
- J. G. Park, I.-R. Jeon and T. D. Harris, Inorg. Chem., 2014, 54, 359–369 CrossRef PubMed.
- W. Phonsri, D. S. Macedo, K. R. Vignesh, G. Rajaraman, C. G. Davies, G. N. L. Jameson, B. Moubaraki, J. S. Ward, P. E. Kruger, G. Chastanet and K. S. Murray, Chem. – Eur. J., 2017, 23, 7052–7065 CrossRef CAS PubMed.
- J. A. Kitchen, A. Noble, C. D. Brandt, B. Moubaraki, K. S. Murray and S. Brooker, Inorg. Chem., 2008, 47, 9450–9458 CrossRef CAS PubMed.
- H. L. C. Feltham, A. S. Barltrop and S. Brooker, Coord. Chem. Rev., 2017, 344, 26–53 CrossRef CAS.
- N. Moliner, M. C. Muñoz, S. Létard, J.-F. Létard, X. Solans, R. Burriel, M. Castro, O. Kahn and J. A. Real, Inorg. Chim. Acta, 1999, 291, 279–288 CrossRef CAS.
- C.-F. Sheu, S.-M. Chen, S.-C. Wang, G.-H. Lee, Y.-H. Liu and Y. Wang, Chem. Commun., 2009, 2009, 7512–7514 RSC.
- G. Dupouy, M. Marchivie, S. Triki, J. Sala-Pala, J.-Y. Salaün, C. J. Gómez-García and P. Guionneau, Inorg. Chem., 2008, 47, 8921–8931 CrossRef CAS PubMed.
- N. Moliner, A. B. Gaspar, M. C. Muñoz, V. Niel, J. Cano and J. A. Real, Inorg. Chem., 2001, 40, 3986–3991 CrossRef CAS PubMed.
- N. G. White, H. L. C. Feltham, C. Gandolfi, M. Albrecht and S. Brooker, Dalton Trans., 2010, 39, 3751–3758 RSC.
- H. E. Mason, J. R. C. Musselle-Sexton, J. A. K. Howard, M. R. Probert and H. A. Sparkes, New J. Chem., 2021, 45, 14014–14023 RSC.
- N. Le Narvor, L. Toupet and C. Lapinte, J. Am. Chem. Soc., 2002, 117, 7129–7138 CrossRef.
- Y.-Y. Yang, X.-Q. Zhu, Y. Wang, X.-T. Wu and T.-L. Sheng, Inorg. Chem. Front., 2022, 9, 4732–4740 RSC.
- W. J. Wang, C. H. Lin, J. S. Wang and S. W. Tang, Mol. Cryst. Liq. Cryst., 2006, 456, 209–219 CrossRef CAS.
- C. Nataro, J. Chen and R. J. Angelici, Inorg. Chem., 1998, 37, 1868–1875 CrossRef CAS.
- S.-H. Li, Y. Liu, Y.-Y. Yang, Y.-X. Zhang, Q.-D. Xu, S.-M. Hu, X.-T. Wu and T.-L. Sheng, Polyhedron, 2019, 173, 114109 CrossRef CAS.
- Y. Y. Yang, X. Q. Zhu, J. P. Launay, C. B. Hong, S. D. Su, Y. H. Wen, X. T. Wu and T. L. Sheng, Angew. Chem., Int. Ed., 2021, 60, 4804–4814 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- J. Hesse and A. Rubartsch, J. Phys. E: Sci. Instrum., 1974, 7, 526–532 CrossRef.
- Z. Klencsár, E. Kuzmann and A. Vértes, J. Radioanal. Nucl. Chem., 1996, 210, 105–118 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2313059–2313064. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04226j |
| This journal is © The Royal Society of Chemistry 2024 |