
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Induced fac–mer rearrangements in {M(CO)3}+ complexes (M = Re, 99(m)Tc) by a PNP ligand†
Manuel Luca
Besmer
 *,
Flurina
Schwitter
,
Federica
Battistin‡
,
Henrik
Braband
,
Thomas
Fox
,
Bernhard
Spingler
and
Roger
Alberto
*,
Flurina
Schwitter
,
Federica
Battistin‡
,
Henrik
Braband
,
Thomas
Fox
,
Bernhard
Spingler
and
Roger
Alberto
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: manuel.besmer@chem.uzh.ch
First published on 2nd January 2024
Abstract
The fac–mer rearrangements in [MX3(CO)3]2− (M = Re, 99Tc) induced by a pincer-type ligand (PNP) and a “halide scavenger” are reported. The reactions of fac-[99mTc(CO)3(OH2)3]+ or [99mTcO4]− in saline both yield mer-[99mTc(PNP)(CO)3]+, the first example of a mer-{99mTc(CO)3}+ type complex. In contrast, reactions with terpyridine (terpy) only gave the facial κ2-terpy complexes with Re and 99Tc.
Transition metal carbonyl complexes form a large class of compounds with multiple applications.1 The CO ligand is electronically flexible and tolerates a range of metal oxidation states. Its π-accepting ability stabilises metals in low oxidation states in particular. In the manganese triad, Re and Tc, form highly stable fac-{M(CO)3}+-based complexes. First examples for the study of such tricarbonyl complexes were reported nearly sixty years ago.2 A convenient synthesis of (NEt4)2[ReBr3(CO)3] was published in 1994,3 the synthesis of its homologue 99Tc (half-life ∼ 2 × 105 years, β− decay Emax = 293 keV) was reported shortly thereafter starting from (NBu4)[TcO4].4 Commercial interest induced an elegant pathway to [99mTc(CO)3(OH2)3]+ in aqueous media, available as a kit (Isolink® Kit).5,6 Access to [99mTc(CO)3(OH2)3]+ triggered a plethora of compounds with mono-, bi- or tridentate ligands towards the goal of new radiopharmaceuticals.7,8
The remarkable stability of the fac-{99mTc(CO)3}+ motif and its capability to coordinate readily a wide range of ligands led to many compounds with Re and 99Tc. In turn, the meridional isomer (mer-{M(CO)3}+) has not been in the focus of 99mTc research since it is not accessible along a facile pathway. For Re(I), a few examples of mer-{Re(CO)3}+-type complexes have been reported,9,10 amongst which Langer et al. presented the light induced fac–mer rearrangement of a carbodiphosphorane complex.11 One of the first mer-{Tc(CO)3}+ complexes was obtained from [TcOCl4]− with PPh3 under a CO atmosphere.12 Intramolecular isomerisation has in fact been observed recently upon coordination of sterically highly demanding isonitrile ligands13 and a variety of mer-{Tc(CO)3}+-type complexes have been reported based on the mer-[Tc(CO)3(OH2)(PPh3)2]+ starting material.14,15
In an attempt to explore scarce fac–mer isomerisations for Re and Tc tricarbonyl complexes, we chose pincer-type ligands.16 The desired mer arrangement in the complexes refers solely to three carbonyl ligands, rather than three monodentate ligands of which some are carbonyl ligands or the meridional coordination of the pincer ligand. These tridentate PNP-based pincer systems are preorganised for meridional coordination with various d-elements, also with 99Tc, as recently reported by us.17,18 The distinct preference of pincer ligands such as 2,6-bis((di-tertbutylphosphino) methyl)pyridine (PyrPNPtBu) for coordinating the metal centre in a meridional fashion prompted us to explore their interactions with Re and 99(m)Tc tricarbonyl cores. Of special interest was the question if the mer-{99mTc(CO)3}+ motif was accessible for radiopharmaceuticals as well.
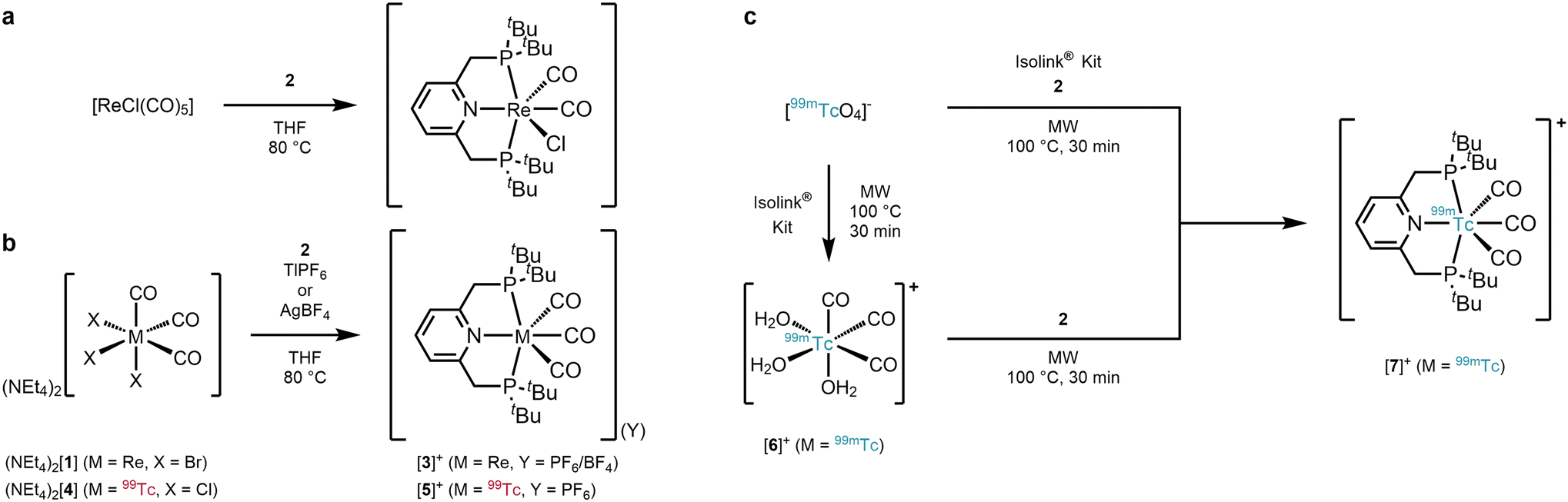
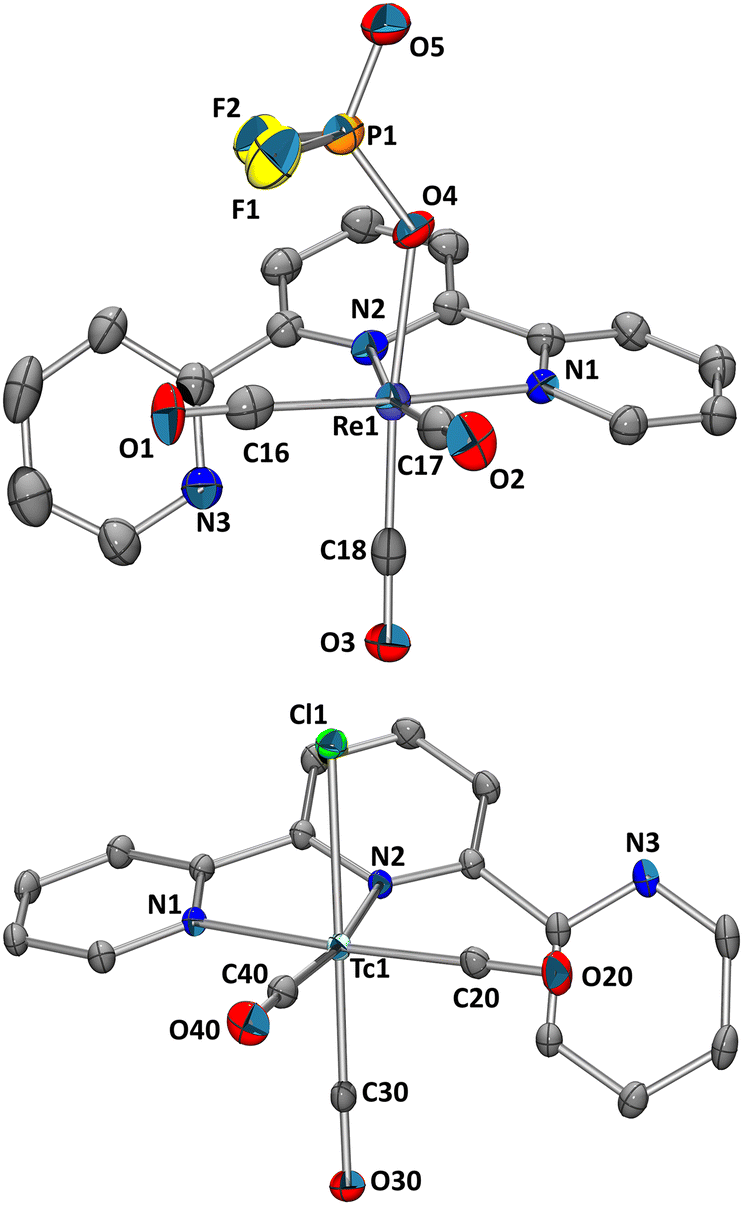
The reaction of PyrPNPtBu (2) with e.g. [ReCl(CO)5] in refluxing THF yields cis-[Re(PyrPNPtBu)(CO)2Cl] after loss of three CO's (Scheme 1a).19 When starting from fac-[ReBr3(CO)3]2− (fac-[1]2−) for inducing a fac–mer rearrangement, heating of fac-[1]2− with 2 in THF gave the expected yellowish product cis-[Re(PyrPNPtBu)(CO)2Br] after loss of two Br− and one CO ligand. Obviously, cleavage of a 3rd halide from fac-[1]2− and CO rearrangement is disfavoured over dissociation of one CO. Conceptually, we hypothesised that a “halide scavenger” would change this preference, facilitate halide removal and lead to the desired rearrangement. Indeed, performing the reaction of fac-[1]2− with 2 in THF (80 °C, 81 h) and with 3 equiv. of TlPF6 gave the rearrangement to mer-[Re(PyrPNPtBu)(CO)3](PF6) (mer-[3](PF6)) in 34% yield (Scheme 1b). The spectroscopic analysis of mer-[3](PF6) displayed the expected symmetric signals in the 1H-, 13C{1H}- and 31P{1H} NMR spectra (ESI Fig. S2–S4†). The carbon signals of the mer-{Re(CO)3}+ moiety appear as two triplets at δ 197.3 (2JP–C = 7.8 Hz) and 196.5 ppm (2JP–C = 3.6 Hz). For the PF6− anion a doublet at δ −72.9 ppm (1JF–P = 710.8 Hz) was found in the 19F NMR spectrum and a septet at δ −144.4 ppm (1JP–F = 710.8 Hz) in the 31P{1H} NMR spectrum. The IR spectrum (KBr pellet) of mer-[3](PF6) displayed a set of three bands υCO at 2047, 1946 and 1913 cm−1 in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2. The observed pattern coincides with a mer-{Re(CO)3}+ moiety and differs distinctly from the characteristic facial starting material ((NEt4)2[1]: 2000 and 1867 cm−1). A single crystal analysis confirmed mer-[Re(PyrPNPtBu)(CO)3](PF6) (Fig. 1, top). The analogous reaction with AgBF4 (instead of TlPF6) yielded mer-[3](BF4). Mechanistically, we hypothesise that the pincer ligand initially coordinates in a bidentate fashion forming the intermediate fac-[ReBr(PyrPNPtBu)(CO)3], which has been observed in UHPLC-ESI-MS. Upon abstraction/dissociation of the third halide ligand, the isomerisation takes place with concomitant tridentate pincer coordination. Following these results of an intramolecular fac–mer rearrangement, the homologous 99Tc reaction was performed. The technetium complex fac-[4]2− reacted with 2 in THF (80 °C, 5 h) in the presence of 3 equiv. of TlPF6 straight to mer-[Tc(PyrPNPtBu)(CO)3](PF6) (mer-[5](PF6)) in 27% yield as an off-white crystalline solid. The spectroscopic analysis revealed analogous features as compared to mer-[3](PF6) with a broad 99Tc NMR signal at δ −1574 ppm (Δ½ = 2.2 kHz). As in mer-[3]+, the IR spectrum evidences a mer-{Tc(CO)3}+-moiety with three bands υCO at 2053, 1960 and 1923 cm−1, only shifted slightly as compared to its rhenium congener. The solid-state structure of (mer-[5](PF6)) (Fig. 1, bottom) finally confirms structurally the rearrangement from fac-[Tc(CO)3]+ in fac-[4]2− to mer-[Tc(CO)3]+ in mer-[5]+. The products of these isomerisations with the PyrPNPtBu ligand add to the limited number of examples for meridional tricarbonyl complexes. We hypothesise that the strong preference for planar binding and the rather bulky phosphine substituents of the pincer, similar to the reported isonitrile study,13 induce an intramolecular rearrangement.
:2:2. The observed pattern coincides with a mer-{Re(CO)3}+ moiety and differs distinctly from the characteristic facial starting material ((NEt4)2[1]: 2000 and 1867 cm−1). A single crystal analysis confirmed mer-[Re(PyrPNPtBu)(CO)3](PF6) (Fig. 1, top). The analogous reaction with AgBF4 (instead of TlPF6) yielded mer-[3](BF4). Mechanistically, we hypothesise that the pincer ligand initially coordinates in a bidentate fashion forming the intermediate fac-[ReBr(PyrPNPtBu)(CO)3], which has been observed in UHPLC-ESI-MS. Upon abstraction/dissociation of the third halide ligand, the isomerisation takes place with concomitant tridentate pincer coordination. Following these results of an intramolecular fac–mer rearrangement, the homologous 99Tc reaction was performed. The technetium complex fac-[4]2− reacted with 2 in THF (80 °C, 5 h) in the presence of 3 equiv. of TlPF6 straight to mer-[Tc(PyrPNPtBu)(CO)3](PF6) (mer-[5](PF6)) in 27% yield as an off-white crystalline solid. The spectroscopic analysis revealed analogous features as compared to mer-[3](PF6) with a broad 99Tc NMR signal at δ −1574 ppm (Δ½ = 2.2 kHz). As in mer-[3]+, the IR spectrum evidences a mer-{Tc(CO)3}+-moiety with three bands υCO at 2053, 1960 and 1923 cm−1, only shifted slightly as compared to its rhenium congener. The solid-state structure of (mer-[5](PF6)) (Fig. 1, bottom) finally confirms structurally the rearrangement from fac-[Tc(CO)3]+ in fac-[4]2− to mer-[Tc(CO)3]+ in mer-[5]+. The products of these isomerisations with the PyrPNPtBu ligand add to the limited number of examples for meridional tricarbonyl complexes. We hypothesise that the strong preference for planar binding and the rather bulky phosphine substituents of the pincer, similar to the reported isonitrile study,13 induce an intramolecular rearrangement.
 | ||
| Scheme 1 a) Synthesis of fac-[Re(PyrPNPtBu)(CO)2Cl] from [ReCl(CO)5];19 (b) syntheses of mer-[M(PyrPNPtBu)(CO)3]+ (mer-[3](BF4 or PF6) and mer-[5](PF6) by reaction of fac-[1]2− or fac-[4]2− with PyrPNPtBu (2) in presence of TlPF6 (3 equiv.) and/or AgBF4 (3 equiv.); (c) synthetic pathways towards mer-[99mTc(PyrPNPtBu)(CO)3]+ (mer-[7]+) in one step (top) or two steps via fac-[6]+ (bottom). MW refers to “microwave reactor”. | ||
 | ||
| Fig. 1 Ellipsoid displacement plots20 of mer-[Re(PyrPNPtBu)(CO)3](PF6) ([3](PF6), top) and mer-[Tc(PyrPNPtBu)(CO)3](PF6) ([5](PF6), bottom). Ellipsoids represent a 35% probability. Hydrogen atoms and counterions have been omitted for clarity. | ||
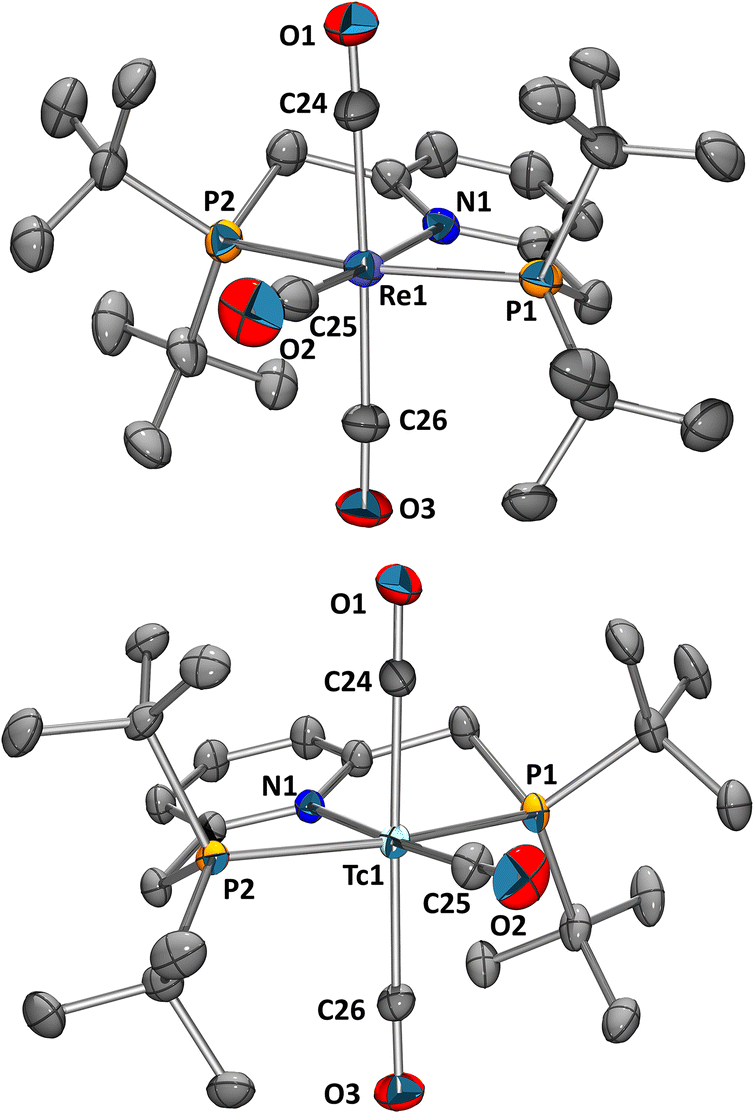
Having established the macroscopic reactions in organic solvents with Re and 99Tc, the question arose if a similar pathway was accessible for 99mTc in aqueous saline (0.9% NaCl) media. In an ideal case, the labelling of the pincer ligand requires a short reaction time and forms the product in high yield and radiochemical purity (RCP). In a first approach, fac-[99mTc(CO)3(OH2)3]+ (fac-[6]+) was prepared with the Isolink® Kit in the microwave reactor within 30 min at 100 °C. The aqueous solution containing fac-[6]+ was degassed (to remove CO), a solution of PyrPNPtBu in EtOH (degassed) was added and the mixture heated in a microwave reactor for 30 min at 100 °C (1:1 H2O/EtOH). The radio-HPLC analysis of the product solution showed one main product peak at 25.5 min (92% RCP). Coinjection of a purified sample with the previously obtained mer-[5](PF6) confirmed the identity of the only product as mer-[99mTc(PyrPNPtBu)(CO)3]+ (Fig. 2, the separation of about 1.5 min corresponds to the separation of the UV/vis- and γ-detector). To the best of our knowledge, this is the first example of a mer-{99mTc(CO)3}+-type complex. For practical purposes, a one-step preparation is preferable over the two-step synthesis described above. For this purpose, the saline [99mTcO4]− eluate was degassed and mixed with the boranocarbonate (Na2H3BCO2), tartrate (Na2C4H4O6) and tetraborate (Na2B4O7).21 A degassed EtOH solution with the pincer ligand was added. After heating in the microwave reactor for 30 min at 100 °C, the solution was analysed by HPLC, showing the equivalent product peak at 25.5 min. A true one-pot preparation is therefore possible. In contrast to the macroscopic scale with Re and 99Tc, where the challenge was the removal of three halide ligands from the starting material, the three aqua ligands are readily replaced by the tridentate pincer, which induced a fac–mer rearrangement. Mechanistically, we cannot exclude in the one-step procedure an initial PNP coordination in one of the oxidation states between 99mTc(VII) and 99mTc(I), followed by the coordination of the CO ligands. For the two-step approach however (vide supra), the reaction starts from a fac-{99mTc(CO)3}+ unit in the presence of a large excess of Cl−. The final meridional product must thus be the result of an intramolecular rearrangement induced by the PNP ligand.
 | ||
| Fig. 2 HPLC traces for the coinjection of mer-[5]+ (top, UV-trace) and mer-[7]+ (bottom, γ-trace). | ||
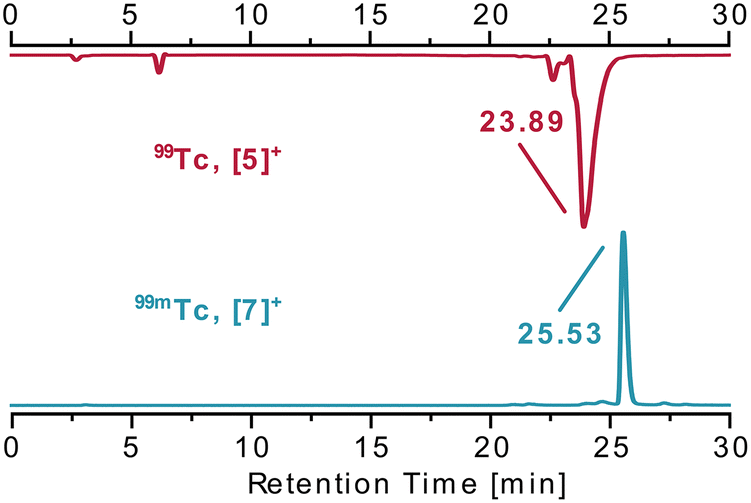
To assess if the mer-{M(CO)3}+ structure can be generalised to other ligands with meridional preference, terpyridine (terpy) as a more flexible tridentate ligand is an obvious choice. Terpy commonly binds in a meridional fashion as is known from many complexes.22 Following the pathway described above, fac-[1]2− was reacted with terpy in the presence of TlPF6. After work-up, the analysis of the pale yellowish, crystalline material by NMR and IR indicated a κ2-terpy complex (Scheme 2). Three bands in the CO region υCO at 2021, 1916 and 1895 cm−1 were observed, out of which the two lower energy vibrations are poorly resolved. In contrast to mer-[3]+ and mer-[5]+ these spectral signatures are indicative for fac-{M(CO)3}+ moieties. The 31P{1H} NMR spectrum revealed a triplet at δ −16.2 ppm (1JP–F = 961.7 Hz) and in the 19F NMR spectrum a doublet at δ −83.0 ppm (1JF–P = 964.6 Hz) was found. The latter signature is not compatible with the one expected for PF6−, indicating a phosphorous nuclei coupling only to two neighbouring fluorine nuclei. An X-ray diffraction analysis of single crystals allowed the identification of the product as fac-[Re(κ2-terpy)(CO)3(PO2F2)] (fac-[9]) (Fig. 3, top). The fac–mer isomerisation was not induced by terpy, but an uncommon difluorophosphate (PO2F2−) anion occupied the sixth coordination site. The comparison of the recorded IR spectrum with reported values for PO2F2− led to the identification of characteristic bands (1313, 1158, 842 and 498 cm−1).23 The PF6− anion hydrolysed, probably as a result of either residual traces of water in the glassware24 or a transition metal assisted pathway as observed similarly for other metals.25,26 The decomposition reaction was not further explored in detail and the analogous reaction with 99Tc indicated the formation of a similar product. The flexibility and reduced steric demand of the terpyridine ligand, compared to PNP, is evidenced in the lack of isomerisation and the isolation of facial tricarbonyl complexes. It cannot be excluded that a thermodynamic driving force for halide substitution, while forming a M–P bond in comparison to a M–N, contributes to the different reactivities. Similar complexes with terpy have been reported starting from [ReBr(CO)5] via reaction in toluene at 100 °C.27,28 The reaction of fac-[1]2− with terpyridine in the presence of AgBF4 represents thus an alternative pathway towards fac-[Re(κ2-terpy)Br(CO)3] (fac-[10], (Scheme 2)) (71%).27,28 Analogously for 99Tc, the reaction of fac-[4]2− with terpy in MeOH (no halide scavenger present) gave greenish fac-[Tc(κ2-terpy)Cl(CO)3] (fac-[11]) (Fig. 3, bottom) in quantitative yield. In contrast to fac-[9], the IR spectrum shows two bands for the CO ligands υCO at 2022 and 1926 cm−1 with the latter broader and not fully resolved. The fac-{M(CO)3}+ arrangement thus follows the rhenium model and is expected in the absence of halide scavengers. The 99Tc NMR spectrum of complex fac-[11] shows a comparable sharp signal at δ −1020 ppm (Δ½ = 420 Hz). The 1H NMR spectrum at 298 K revealed mostly broad signals except for one sharp triplet. Performing the measurements at 235 K allowed the observation of distinct signals and multiplicities. The broadening of the signals at ambient temperature stems from fluxional behaviour of the κ2-terpy ligand, as has been observed with the rhenium congener (Scheme S1, ESI†).27 Interestingly, the rhenium complex [9] with the uncommon (PO2F2−) anion does not show any fluxional behaviour at 298 K.
 | ||
| Fig. 3 Ellipsoid displacement plots20 of fac-[Re(κ2-terpy)(CO)3(PO2F2)] ([9], top) and fac-[Tc(κ2-terpy)(CO)3Cl] ([11], bottom). Ellipsoids represent a 35% probability. Hydrogen atoms are omitted for clarity. | ||
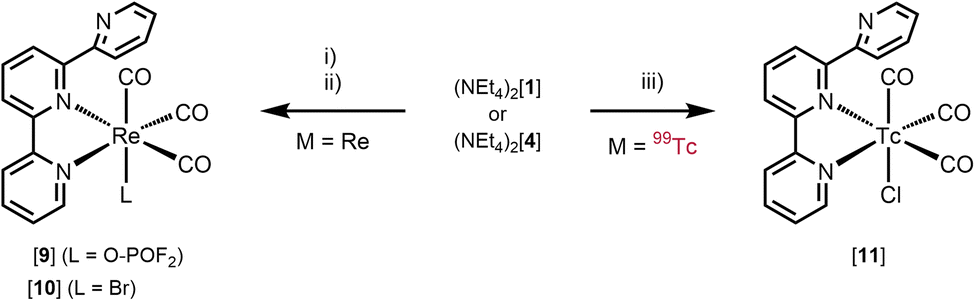 | ||
| Scheme 2 Syntheses of fac-[Re(κ2-terpy)(CO)3L] ([9](L = O-POF2), [10](L = Br)) and fac-[Tc(κ2-terpy)(CO)3Cl] ([11]). Conditions: (i) (NEt4)2[ReBr3(CO)3], 3 TlPF6, terpy, THF, 80 °C, 39 h; (ii) (NEt4)2[ReBr3(CO)3], 3 AgBF4, terpy, THF, 80 °C, 24 h; (iii) (NEt4)2[TcCl3(CO)3], terpy, MeOH, 50 °C, 4 h. | ||
We report herein one of the rare examples of an intramolecular fac-{M(CO)3}+ to mer-{M(CO)3}+ rearrangement with rhenium and technetium, induced by the bulky PyrPNPtBu pincer ligand. To enable these rearrangements with Re and 99Tc (fac-[1]2− and fac-[4]2−), concomitant halide precipitation with Ag(I) or Tl(I) is mandatory. The adaption of this chemistry from THF to saline and 99mTc chemistry gave a mer-{99mTc(CO)3}+ structure for the first time. The product mer-[99mTc(PyrPNPtBu)(CO)3]+ is water stable and can be made along a one- or a two-step synthesis. Large excesses of chloride do not interfere with the mer-product formation, implying the opportunity of making further mer-{99mTc(CO)3}+ complexes, in extension to the usual fac-{99mTc(CO)3}+ compounds. The observed reactivities with 99mTc thus enclose a potential to broaden the scope for tricarbonyl complexes. Potential decarbonylation in meridional tricarbonyl complexes could be exploited as a synthetic strategy for new dicarbonyl-based radiopharmaceuticals. We are currently investigating if reactivities of sterically less demanding ligands such as terpy and 99mTc lead to the same structures, contrasting macroscopic results described above.
Author contributions
MLB edited and wrote the manuscript, conceptualised the project and performed experiments, FS performed experiments (Re and 99Tc), FB performed 99mTc labelling experiments, HB advised 99mTc experiments, TF performed low temperature NMR studies, BS advised and supervised all crystallographic data collections and refinements, RA revised the manuscript, performed 99Tc experiments and initiated the project.Conflicts of interest
There are no conflicts to declare.References
-
V. Bhatt, in Essentials Coord. Chem, Ed. V. Bhatt, Elsevier Inc., London, 2016, pp. 191–236 Search PubMed
.
- E. W. Abel, I. S. Butler, M. C. Ganorkar, C. R. Jenkins and M. H. B. Stiddard, Inorg. Chem., 1966, 5, 25–27 CrossRef CAS
- R. Alberto, A. Egli, U. Abram, K. Hegetschweiler, V. Gramlich and A. P. Schubiger, J. Chem. Soc., Dalton Trans., 1994, 2815–2820 RSC
- R. Alberto, R. Schibli, A. Egli, W. A. Herrmann, G. Artus, U. Abram and T. A. Kaden, J. Organomet. Chem., 1995, 492, 217–224 CrossRef
- R. Alberto, R. Schibli, A. Egli, A. P. Schubiger, U. Abram and T. A. Kaden, J. Am. Chem. Soc., 1998, 120, 7987–7988 CrossRef CAS
- R. Alberto, ChemBioChem, 2020, 21, 2743–2749 CrossRef CAS
- R. Schibli, R. La Bella, R. Alberto, E. Garcia-Garayoa, K. Ortner, U. Abram and P. A. Schubiger, Bioconjugate Chem., 2000, 11, 345–351 CrossRef CAS PubMed
- R. Alberto, J. K. Pak, D. Van Staveren, S. Mundwiler and P. Benny, Biopolymers, 2004, 76, 324–333 CrossRef CAS PubMed
- D. Wei, O. Sadek, V. Dorcet, T. Roisnel, C. Darcel, E. Gras, E. Clot and J. B. Sortais, J. Catal., 2018, 366, 300–309 CrossRef CAS
- F. Salsi, M. Neville, M. Drance, A. Hagenbach, C. Chan, J. S. Figueroa and U. Abram, Chem. Commun., 2020, 56, 7009–7012 RSC
- L. Maser, M. Vogt and R. Langer, Z. Anorg. Allg. Chem., 2021, 647, 1518–1523 CrossRef CAS
- R. Alberto, W. A. Herrmann, P. Kiprof and F. Baumgärtner, Inorg. Chem., 1992, 1, 895–899 CrossRef
- G. Claude, F. Salsi, A. Hagenbach, M. Gembicky, M. Neville, C. Chan, J. S. Figueroa and U. Abram, Organometallics, 2020, 39, 2287–2294 CrossRef CAS
- M. R. Jungfer, L. Elsholz and U. Abram, Organometallics, 2021, 40, 3095–3112 CrossRef CAS
- M. R. Jungfer, L. Elsholz and U. Abram, Inorg. Chem., 2022, 61, 2980–2997 CrossRef PubMed
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC
- M. L. Besmer, H. Braband, S. Schneider, B. Spingler and R. Alberto, Inorg. Chem., 2021, 60, 6696–6701 CrossRef CAS PubMed
- M. L. Besmer, H. Braband, T. Fox, B. Spingler, A. P. Sattelberger and R. Alberto, Inorg. Chem., 2023, 62, 10727–10735 CrossRef CAS
- M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–565 CrossRef CAS
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 3, 3135–3136 CrossRef PubMed
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1–19 CrossRef CAS PubMed
- R. C. Thompson and W. Reed, Inorg. Nucl. Chem. Lett., 1969, 5, 581–585 CrossRef CAS
- S. Nayeri, S. Jamali, A. Jamjah, J. R. Shakirova, S. P. Tunik, V. Gurzhiy, H. Samouei and H. R. Shahsavari, Inorg. Chem., 2020, 59, 5702–5712 CrossRef CAS PubMed
- D. Dermitzaki, C. P. Raptopoulou, V. Psycharis, A. Escuer, S. P. Perlepes and T. C. Stamatatos, Dalton Trans., 2014, 43, 14520–14524 RSC
- S. J. Thompson, P. M. Bailey, C. White and P. M. Maitlis, Angew. Chem., Int. Ed. Engl., 1976, 15, 490–491 CrossRef
- E. W. Abel, V. S. Dimitrov, N. J. Long, K. G. Orrell, A. G. Osborne, H. M. Pain, V. Sik, M. B. Hursthouse and M. A. Mazidc, J. Chem. Soc., Dalton Trans., 1993, 597–603 RSC
- P. Bulsink, A. Al-Ghamdi, P. Joshi, I. Korobkov, T. Woo and D. Richeson, Dalton Trans., 2016, 45, 8885–8896 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2307669–2307674. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03992g |
| ‡ Current address: Univ Brest, UMR CNRS 6521 CEMCA, Brest 29200, France. |
| This journal is © The Royal Society of Chemistry 2024 |