
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt(III)-containing penta-dentate “helmet”-type phthalogens: synthesis, solid-state structures and their thermal and electrochemical characterization†
Rasha K.
Al-Shewiki
a,
Saddam
Weheabby
b,
Nell
Uhlig
a,
Marcus
Korb
 c,
Tom
Pester
d,
Stefan
Zahn
e,
S.
Grecchi
f,
P. R.
Mussini
f,
Tobias
Rüffer
*a and
Heinrich
Lang
g
c,
Tom
Pester
d,
Stefan
Zahn
e,
S.
Grecchi
f,
P. R.
Mussini
f,
Tobias
Rüffer
*a and
Heinrich
Lang
g
aChemnitz University of Technology, Department of Inorganic Chemistry, Strasse der Nationen 62, 09107, Chemnitz, Germany. E-mail: tobias.rueffer@chemie.tu-chemnitz.de
bChemnitz University of Technology, Measurement and Sensor Technology, 09126 Chemnitz, Germany
cUniversity of Western Australia, School of Molecular Sciences, M310, 6009 Perth, WA, Australia
dChemnitz University of Technology, Department of Organic Chemistry, Strasse der Nationen 62, 09111, Chemnitz, Germany
eLeibniz Institute of Surface Engineering (IOM), Permoserstraße 15, 04318 Leipzig, Germany
fUniversity of Milan, Department of Chemistry, Via Golgi 19, 20133 Milano, Italy
gResearch Center for Materials, Architectures and Integration of Nano-membranes (MAIN) Research Group Organometallic Chemistry, Technische Universität Chemnitz Rosenbergstraße 6, 09126 Chemnitz, Germany
First published on 29th January 2024
Abstract
Treatment of unsubstituted and substituted phthalonitrile (1a–d) with appropriate equivalents of sodium methoxide and ammonia afforded the corresponding 1,3-diiminoisoindolines (2a–d), which were converted to cobalt(III)-containing penta-dentate “helmet”-type phthalogens (3a–d) by the reaction with CoCl2·6H2O as templating agent in the inert solvent 1,2,4-trichlorobenzene. The identities of 2a–d and 3a–d were established by elemental analysis, infrared spectroscopy (IR), nuclear magnetic resonance (NMR), and electrospray ionization mass spectrometry (ESI-MS). A computational study was performed to determine the most stable tautomeric form of 2a–c in the gas phase. The solid-state structures of 2b and 2c were determined by single crystal X-ray diffraction (SC-XRD) studies to confirm their existence in the stereoisomeric anti-form, which is aligned with quantum chemical computations. SC-XRD studies of 3a and 3b revealed a slightly distorted octahedral geometry around the CoIII ions which are coordinated by five N-donor atoms and one extra co-ligand, resulting in a coordination environment of CoN5Cl (3a) and CoN5O (3b), respectively. The thermal stabilities of 2a–d and 3a–d were investigated by thermogravimetric analysis (TGA) in the temperature range of 40–500 °C and 40–800 °C, respectively, revealing that 3a–d were converted to the parent cobalt(II)-containing phthalocyanines (4a–d), which was verified independently by furnace heating experiments. Moreover, the electrochemical behavior of 3a was studied exemplarily for the phthalogens by cyclic voltammetry and square wave voltammetry. This study showed that 4a (CoPc) is formed irreversibly by reducing 3a electrochemically.
Introduction
Within the large family of porphyrinoids, (metallo)phthalocyanines (MPcs) enjoy a privileged position mainly due to their distinctive photophysical, structural, electronic, magnetic and catalytic properties combined with an exceptionally high thermal and chemical stability.1,2 They found numerous applications in different fields, including nonlinear optics,3 optical recording media,4 light harvesting systems,5 semiconductors,6 chemical sensors,7 photodynamic therapy8 and catalysis.9Phthalogens, as displayed in Fig. 1, are another member of the large porphyrinoid family and can be used, for example, to produce MPcs either by thermal or photochemical treatment, or by electrochemical reduction.10–12 Phthalogens possess different chemical and physical properties compared to MPcs which is due to skeletal modification resulting in the loss of the aromaticity of the latter.11 The already known phthalogens can be classified according to a recent review by some of us11 into four types: (i) “box”-type phthalogens (Type I),13–18 (ii) “helmet”-type phthalogens (Type II),19–32 (iii) “antenna”-type phthalogens (Type III)33–37 and (iv) “curled”-type phthalogens (Type IV),38cf. Fig. 1. The first example of a crystallographically characterized phthalogen was reported by Ercolani and coworkers in 1990,13 and later in 199814 for a Type I phthalogen (Fig. 1). The molecular structure determination revealed that Type 1 phthalogens have a sandwich-type structure with the metal ion in the centre of the molecule and the two ligands ‘stapled’ together by two interligand C–C σ-bonds, leading to the formation of sp3 hybridized and chiral carbon atoms indicated with (*) in Fig. 1. Due to the presence of the interligand σ-bonds both the planarity and resonance of the two formally fused MPcs got lost.13–18
 | ||
| Fig. 1 Chemical structures of crystallographically characterized phthalogens.11 The sign * indicate asymmetrically substituted carbon atoms but refer to racemic mixtures. | ||
The first example of a hexadentate Type II19 “helmet”-type phthalogen was reported in 1990 by Strähle and Gingl, in which two carbon atoms of the inner skeleton are bonded covalently to a 3,3′-iminobis(1-isoindolylideneamino) unit.19 As discussed before, this interrupts the resonance within the inner macrocycle ring and aromaticity got lost.19
Within Type II phthalogens the functionalized ligand act as a hexadentate one (by six isoindole-nitrogen donor atoms), while a chloride ion in the 7th position completes the coordination sphere of metal ions with higher charge, cf. Fig. 1.19 Further examples of Type II phthalogens with M = InIII,20 GdIII,21 TlIII,22 and CdII,25 on the one, and with BiIII,24 on the other hand (Fig. 1) were reported as well. Furthermore, a NbIV containing representative with the inner skeleton bridged at different positions was observed as well.14–16
The group of McGaff contributed with two further types of crystallographically characterized phthalogens, namely the one of Type II (pentadentate bicyclic “Helmet”-type phthalogen) and Type III (Fig. 1).19–37 In the case of the Type III representatives19–32 the modification of the ligand took place by incorporation of two alkoxy groups at the inner skeleton. As reported before for the other phthalogens, that resulted in re-hybridization of the respective carbon atoms from sp2 to sp3. Once again, the planarity of the formally phthalocyaninato core and its π-conjugation is lost, the latter is proven by the disappearance of the Q band in UV/Vis spectra.33 In case of the Type II pentadentate bicyclic “helmet”-type phthalogen,26 the ligand consist of five isoindoline units which coordinates to the trivalent metal ions FeIII and CoIII by five N-donor atoms, one from each isoindoline unit. As their synthesis made use of FeII or CoII salts as starting compounds a one-electron oxidation did obviously occur, and it seems as if this oxidation is a prerequisite for the formation of Type II compounds. So far, no Type II representative possessing a bivalent metal ion has been reported. The coordination sphere around the trivalent metal ions is fulfilled by an additional donor as a coordinating methanol, water or 4-hydroxypyridine molecule (Fig. 1).26 The iron(III) containing compounds attracted attention through the years due to their close relevance to biological activities involving heme proteins27–30 and have been used in oxidation catalysis to generate alcohols from cycloalkanes,27,30 epoxides from olefins28 and ketones and aldehydes from unactivated non-benzylic alcohols.29 All phthalogens reported so far have been synthesized from the reaction of a metal source as templating agent and a phthalonitrile precursor.13–38 However, another simpler synthetic route involve the direct use of 1,3-diiminoisoindoline have been reported.10,39 Additionally, 1,3-diiminoisoindoline is also useful for the synthesis of various phthalocyanine analogues, such as subphthalocyanine,40 hemiporphyrazines,41 metal chelates such as the bis(iminopyridyl)isoindoline42 and phthalazine ligands.43 In early 1950s, Linstead synthesized 1,3-diiminoisoindoline from the addition of liquid ammonia in a methanolic solution of phthalonitrile; the reaction mixture was heated in an autoclave for four hours to yield a beige/light green product.39a Later on 1,3-diiminoisoindoline was synthesized by using a catalytic amount of sodium in methanol,10 and since then several methods have been developed for the preparation of further 1,3-diiminoisoindolines.44
Here we report on an novel synthetic approach to prepare 1,3-diiminoisoindolines in very high yields and on an straightforward and easy-to-follow approach to synthesize cobalt(III) containing penta-dentate “helmet” type phthalogens (3a–d, Type II) under mild conditions. The compounds were characterized by a comprehensive set of techniques, including elemental analysis, IR, NMR, ESI-MS, UV/VIS, X-ray, and TGA. The process of converting 3a–d into the parent MPcs 4a–d through thermal decomposition and electrochemical reduction was examined.
Results and discussion
Synthesis
In order to have suitable precursors to synthesize cobalt(III) containing penta-dentate “helmet”-type phthalogens 3a–d, the corresponding phthalonitriles 1a–d were first converted to the 1,3-diiminoisoindoline's 2a–d (Scheme 1). | ||
| Scheme 1 Synthesis of 2a–d, 3a–d and 4a–d. (i) NH3, NaOMe, MeOH, 60–70 °C, 4 h, (ii) CoCl2·6H2O, 1,2,4-trichlorobenzene, Δ.† Comment: 2d was obtained as 2d·H2O, 3a as [3a(HCl)(dmf)]·2dmf, 3b as [3b(dmf)]·dmf·H2O and as [3b(dmf)]·2dmf in singly crystalline form, 3c as [3c(HCl)]·dmf and 3d as [3d(MeOH)]·dmf. | ||
Compounds 2a–d were synthesized by a one-pot method in excellent yields under mild solvothermal reaction conditions by reacting 1a–d with five equivalents of ammonia in a methanolic solution (see ESI†). When using less than five equivalents of ammonia in methanol as solvent the yields were always lower, the reaction times became longer, and the isolated materials were not analytically pure.
The synthesis of 3a–d included two steps: (i) the reaction of CoCl2·6H2O as templating agent with five equivalents of the corresponding 1,3-diiminoisoindoline's 2a–d in 1,2,4-trichlorobenzene as an inert and high boiling solvent and (ii) recrystallization of solid reaction products from dmf. With respect to (i): the reaction temperature was raised slowly up to 200–210 °C (cf. Experimental section and ESI†) to avoid boil over due to release of NH3. Stirring at that temperature was continued as long as NH3 developed. After the NH3 development ceased the reaction mixture was filtered off. In step (II) the filter cake was transferred into dmf and stirred for two hours. The resulting suspension was filtered again and allowed to separate purple or bluish-purple solids from an intense green coloured solution. The solid materials were identified as the cobalt(II) phthalocyanines 4a–d, whereby further purification39f was needed to obtain them analytically pure (see ESI†). The green filtrate was crystallized with et2O to obtain 3a–d as dark-red (3a) or orange crystals (3b–d) (see ESI†). In case of 3a no further purification was required and the crystallization afforded 3a in form of [3a(HCl)(dmf)]·2dmf, while for 3b–d complexes further purification efforts were required. Thereby, 3b complex was purified by two subsequently performed column chromatography using CH2Cl2 and EtOAc as eluent; respectively, affording 3b in form of [3b(dmf)]·dmf·H2O based upon elemental analysis studies. In case of 3c, separated orange microcrystals were noticed to be contaminated with an undetermined colourless material, therefore it was sonicated in MeOH for 15 min to afford 3c afforded in form of [3c(HCl)]·dmf. Crystals of 3d were purified by column chromatography by using a CH2Cl2–MeOH mixture (ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to afford 3d in form of [3d(MeOH)]·dmf (see ESI†).
:1, v/v) to afford 3d in form of [3d(MeOH)]·dmf (see ESI†).
In addition, another purification experiment of 3a was performed. The obtained crystals were dissolved in a minimum amount of dichloromethane and loaded onto a short silica gel flash column. An orange band was quickly eluted when a CH2Cl2–MeOH mixture (ratio 3:1, v/v) was passed through the column. The band was collected and upon slow evaporation of the solvent mixture dark red crystals of 3a·MeOH could be separated (see ESI†).
Characterization of 2a–d
Compounds 2a–d have three ionizable protons (H+) present on the imine and/or isoindoline nitrogen atoms45 and are therefore involved in a tautomeric equilibrium between the diimino and amino-imino form (Scheme 2). | ||
| Scheme 2 Principal tautomerism and stereoisomerism in 1,3-diiminoisoindolines on example of 2a.45 | ||
Both tautomeric forms might be involved in syn–anti equilibria (Scheme 2) with respect to the orientation of the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–H hydrogen towards the aromatic ring (syn) or away from it (anti). The room temperature 1H NMR spectra of 2a–d in DMSO-d6 confirmed the presence of mainly the C2 symmetric diimino tautomers, while broad resonances at 8.52, 8.71, 8.37 and 8.41 ppm for the N–H protons of 2a, 2b, 2c and 2d indicate a dynamic behavior in solution (see ESI†). ESI-TOF mass-spectrometric spectra of 2a–d showed in all cases the molecular ion peaks as basis peaks in the form of [M + H]+ (see ESI†), while for 2d, which was isolated as the adduct 2d·H2O, the peak of [2d + H2O + H]+ was observed in addition (see ESI†). In the FT-IR spectra of 2a–d taken from KBr pellets the intense absorption band around 2231 cm−1, corresponding to the –C
N–H hydrogen towards the aromatic ring (syn) or away from it (anti). The room temperature 1H NMR spectra of 2a–d in DMSO-d6 confirmed the presence of mainly the C2 symmetric diimino tautomers, while broad resonances at 8.52, 8.71, 8.37 and 8.41 ppm for the N–H protons of 2a, 2b, 2c and 2d indicate a dynamic behavior in solution (see ESI†). ESI-TOF mass-spectrometric spectra of 2a–d showed in all cases the molecular ion peaks as basis peaks in the form of [M + H]+ (see ESI†), while for 2d, which was isolated as the adduct 2d·H2O, the peak of [2d + H2O + H]+ was observed in addition (see ESI†). In the FT-IR spectra of 2a–d taken from KBr pellets the intense absorption band around 2231 cm−1, corresponding to the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group of phthalonitriles 1a–d, disappeared as a further prove of their conversion to analytically pure 2a–d (see ESI†).
N group of phthalonitriles 1a–d, disappeared as a further prove of their conversion to analytically pure 2a–d (see ESI†).
Molecular and crystal structure of 2b and 2c
The single crystal X-ray crystallographic studies (SC-XRD) of 2b and 2c confirmed both compounds as being isomorphic and isostructural. Therefore, the following discussion will include mostly 2b, which applies in particular to the discussion of the crystal structures. A comparison of related bond lengths of 2b and 2c reveals no further significant differences, while related bond angles show marginal differences only. Much likely, this is due to the formation of intermolecular interactions of 2b and 2c, especially by means of hydrogen bond formation, which possess a larger impact on these structural parameters than the different electronic nature of the chloro (2b) vs. the methyl (2c) substituents, respectively. The molecular structure of 2b is shown in Fig. 2 as a dimer in order to visualize the importance of hydrogen bond formation. Selected bond lengths and angles, in comparison to the ones of 2c, are summarized in Table 1, selected geometrical data of hydrogen bonds of 2b and 2c are given in Table 2 while Table S9† gives selected data collection and structural refinement data. | ||
| Fig. 2 ORTEP (50% probability ellipsoids) of a dimer formed by 2b in the solid state by means of hydrogen bond formation. Symmetry code: “A” = −x, −y, −z. | ||
| Bond length | Bond angles | ||||
|---|---|---|---|---|---|
| 2b | 2c | 2b | 2c | ||
| N1–C1 | 1.308(3) | 1.320(5) | N1–C1–N2 | 123.7(2) | 122.7(4) |
| C1–N2 | 1.327(3) | 1.330(5) | N1–C1–C2 | 124.8(2) | 125.2(4) |
| C1–C2 | 1.492(3) | 1.477(5) | C2–C1–N2 | 111.4(2) | 112.1(3) |
| N2–C8 | 1.382(3) | 1.396(5) | C1–N2–C8 | 108.2(2) | 107.2(3) |
| C8–N3 | 1.282(3) | 1.286(5) | N2–C8–N3 | 126.2(2) | 125.9(4) |
| C7–C8 | 1.494(3) | 1.493(5) | N2–C8–C7 | 109.2(2) | 109.0(3) |
| C2–C7 | 1.393(3) | 1.381(5) | N3–C8–C7 | 124.5(2) | 125.1(4) |
| C2–C3 | 1.384(3) | 1.389(5) | C6–C7–C8 | 133.5(2) | 133.0(3) |
| C3–C4 | 1.393(3) | 1.395(5) | C6–C7–C2 | 120.6(2) | 120.9(4) |
| C4–C5 | 1.398(4) | 1.403(5) | C8–C7–C2 | 105.9(2) | 106.1(3) |
| C5–C6 | 1.393(4) | 1.397(5) | C1–C2–C7 | 105.3(2) | 105.7(3) |
| C6–C7 | 1.380(3) | 1.383(5) | C1–C2–C3 | 132.9(2) | 133.4(4) |
| C3–C2–C7 | 121.9(2) | 121.0(3) | |||
| D–H⋯Aa | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| a Symmetry codes: “A” = −x, −y, −z. “B” = −x, y + ½, −z + ½. “C” = x, ½ − y, z + ½. b For a graphical representation cf. Fig. S1.† | ||||
| 2b: | ||||
| N1–H1⋯N2Ab | 0.91(3) | 1.98(3) | 2.891(3) | 173(3) |
| N1C–H2C⋯N3b | 0.87(3) | 2.00(3) | 2.848(3) | 165(3) |
| 2c: | ||||
| N1–H2⋯N2A | 0.96(5) | 1.94(5) | 2.896(5) | 173(4) |
| N1C–H1C⋯N3 | 0.99(4) | 1.89(4) | 2.858(4) | 167(4) |
The formal C–NH2/N–C single bonds (d(C1–N1)/(N2–C8) = 1.308(3)/1.382(3) Å) are just marginally longer compared to the formal CNH/CN double bonds (d(C8–N3)/(C1–N2) = 1.282(3)/1.327(3) Å). This observation indicates the H2N–CN–CNH unit of 2b as stabilized by mesomerism, but not in resonance with the substituted benzene ring. The latter is verified by bond lengths of 1.492(3) and 1.494(3) Å for C1–C2 and C7–C8, which both corresponds well to expected values for a C–C single bond and by which the two resonant groups are separated. Both 2b and 2c are planar as revealed from very small root-mean-square deviations from planarity of calculated mean planes of all atoms of 0.024 and 0.022 Å, respectively, with the highest deviation from planarity observed for N2 with 0.060(5) Å for 2b and for C9 with 0.056(8) Å for 2c.
 | ||
| Fig. 3 ORTEP (20% probability ellipsoids) of two different perspective views on a selected part of one of the layers formed by 2b in the solid state due to hydrogen bond formation (layer I). Dotted lines refer to intermolecular hydrogen bonds. Intermolecular π-interactions are not indicated. Symmetry codes: “A” = −x, −y, −z. “B” = −x, y + ½, −z + ½. “C” = x, ½ − y, z + ½. “D” = −x, 1 − y, −z. “E” = x, 1 + y, z. “F” = −x, 3/2 − y, z + ½. “G” = −x, 2 − y, −z. “H” = −x, 2 − y, −z + ½. “J” = −x, 3/2 + y, z + ½. “K” = −x, −½ + y, −z + ½. “L” = x, −y − ½, z + ½. | ||
Computational investigation of 2a–c
The five possible different stereo isomers of 2a–c (cf.Scheme 2) were calculated with ORCA (cf. Experimental section) in the gas phase and in dmf solution. It was found that the syn isomers of the diimino tautomeric forms, namely DIss (Scheme 2, and Fig. 4), possess the lowest relaxed interaction energy (ΔEgas) for the here investigated representatives in the gas phase (Table 3). In case of 2c the stability of the 2c-DIss compared to the other conformers is more pronounced, while in case of 2b the inverse behaviour is observed. This is attributed to the electron pushing properties of the methyl groups of 2c compared to the electron withdrawing properties of the chloro substituents of 2b. | ||
| Fig. 4 Ball-and-stick-model of investigated isomers of monomers and dimers. X is H in case of 2a, Cl in case of 2b or Me in case of 2c. | ||
| Isomer | ΔEgas | ΔGgas | ΔEdmf | ΔGdmf |
|---|---|---|---|---|
| 2a-DIaa | 5.4 | 11.3 | 2.7 | 3.7 |
| 2a-DIsa | 1.1 | 8.4 | 1.5 | 1.0 |
| 2a-Dlss | 0.0 | 0.0 | 0.0 | 0.0 |
| 2a-AIa | 7.4 | 10.8 | 2.5 | 0.7 |
| 2a-AIs | 23.1 | 26.1 | 5.3 | 6.2 |
| H-D1 | –68.8 | 9.7 | –34.4 | 29.1 |
| H-D2 | –58.9 | 12.3 | –32.2 | 30.5 |
| 2b-Dlaa | 3.8 | 4.1 | 1.0 | 0.9 |
| 2b-Dlsa | 0.4 | 0.0 | 0.3 | 1.8 |
| 2b-DIss | 0.0 | 0.4 | 0.0 | 2.8 |
| 2b-AIa | 5.4 | 2.3 | 0.2 | 0.0 |
| 2b-AIs | 22.0 | 16.6 | 3.8 | 3.7 |
| Cl-D1 | –64.9 | 1.5 | –37.0 | 23.0 |
| Cl-D1 | –61.9 | 1.3 | –34.8 | 29.4 |
| 2c-DIaa | 11.0 | 8.0 | 2.2 | 0.8 |
| 2c-Dlsa | 1.4 | 1.8 | 1.0 | 0.4 |
| 2c-DIss | 0.0 | 0.0 | 0.0 | 0.0 |
| 2c-AIa | 8.7 | 2.3 | 3.1 | 3.4 |
| 2c-AIs | 23.6 | 17.6 | 10.6 | 8.2 |
| Me-D1 | –59.6 | –3.5 | –33.7 | 26.5 |
| Me-D2 | –56.1 | 0.2 | –31.6 | 25.5 |
Considering solvation effects and thermodynamic corrections a significant decrease of the energy splitting between all conformers of 2a–c is noticed, cf. ΔEdmf in Table 3. Moreover, in case of 2b the 2b-AIa tautomer is more stable compared to the 2b-DIss tautomer, although the difference of ΔEdmf between both tautomers is small. For comparison, the reactions from the most stable monomer to two dimers, X-D1 and X-D2, were investigated as well. The methyl as well as the chlorine substituent increases the stability of the investigated dimers in the gas phase (ΔGgas) and in solution (ΔGdmf).
Molecular structures of 3a and 3b
Single crystals of [3a(HCl)(dmf)]·2dmf and of [3b(dmf)]·2dmf were studied by SC-XRD. Data will be compared with the ones of 3a in form of [3a(MeOH)]·2MeOH.26 In the following to the fragments [3a(HCl)(dmf)], [3a(MeOH)] and [3b(dmf)] is referred to as 3a′, 3aL26 and 3b′. The molecular structures of 3a′ and 3b′ are shown in Fig. 5. Selected bond lengths and angles are summarized in Table 4, while Table S9† gives selected data collection and structural refinement data. | ||
| Fig. 5 ORTEP (50% probability ellipsoids) of the molecular structure of 3a′ (above) and 3b′ (below) in two different perspective views. | ||
| Bond lengths | Bond angles | ||||||
|---|---|---|---|---|---|---|---|
| 3a | 3b | 3aL | 3a | 3b | 3aL | ||
| a D = Cl1 (3a′), O1D (3b′, κO-dmf) and O1 (3aL, κO-MeOH).26 | |||||||
| Co1–N1 | 1.906(3) | 1.899(4) | 1.896(2) | N1–Co1–N3 | 88.9(1) | 89.0(2) | 89.31(7) |
| Co1–N3 | 1.865(3) | 1.859(4) | 1.861(2) | N1–Co1–N5 | 177.1(1) | 174.9(2) | 174.93(7) |
| Co1–N5 | 1.896(3) | 1.878(4) | 1.898(2) | N1–Co1–N7 | 91.1(1) | 91.0(2) | 90.72(7) |
| Co1–N7 | 1.871(3) | 1.857(4) | 1.854(2) | N1–Co1–N10 | 91.0(2) | 92.6(2) | 92.39(7) |
| Co1–N10 | 1.889(3) | 1.850(4) | 1.858(2) | N1–Co1–D | 88.9(1) | 87.1(2) | 84.88(6) |
| Co1–Da | 2.2942(9) | 2.012(3) | 1.998(1) | N3–Co1–N5 | 91.2(1) | 91.4(2) | 90.87(7) |
| C16–N9 | 1.495(5) | 1.461(6) | 1.481(3) | N3–Co1–N7 | 173.6(1) | 173.8(2) | 174.36(7) |
| C16–N4 | 1.453(5) | 1.459(6) | 1.465(3) | N3–Co1–N10 | 86.8(1) | 87.0(2) | 87.08(7) |
| C16–N3 | 1.466(5) | 1.485(6) | 1.473(3) | N3–Co1–D | 93.0(1) | 95.9(2) | 93.67(6) |
| C16–C15 | 1.523(5) | 1.522(6) | 1.525(3) | N5–Co1–N7 | 89.1(1) | 89.1(2) | 89.59(7) |
| C32–N7 | 1.481(4) | 1.483(7) | 1.473(2) | N5–Co1–N10 | 91.8(2) | 92.4(2) | 92.39(7) |
| C32–N8 | 1.452(5) | 1.458(6) | 1.462(2) | N5–Co1–D | 88.2(1) | 87.7(2) | 84.88(6) |
| C32–N11 | 1.485(5) | 1.460(6) | 1.479(2) | N7–Co1–N10 | 86.9(1) | 86.8(2) | 87.28(7) |
| C32–C31 | 1.518(5) | 1.520(6) | 1.523(3) | N7–Co1–D | 93.3(1) | 90.2(1) | 91.97(7) |
| C1–C2 | 1.481(6) | 1.489(6) | 1.482(3) | N10–Co1–D | 179.8(1) | 177.0(2) | 177.17(6) |
| C7–C8 | 1.465(5) | 1.477(7) | 1.479(3) | C15–C16–N3 | 102.4(3) | 101.6(4) | 102.0(2) |
| C9–C10 | 1.478(5) | 1.472(7) | 1.474(3) | C15–C16–N4 | 112.5(3) | 111.5(4) | 111.9(2) |
| C17–C18 | 1.485(6) | 1.477(7) | 1.482(3) | C15–C16–N9 | 109.3(3) | 107.8(4) | 108.1(2) |
| C23–C24 | 1.475(5) | 1.477(7) | 1.475(3) | N3–C16–N4 | 116.3(3) | 113.0(4) | 114.0(2) |
| C25–C26 | 1.481(5) | 1.476(7) | 1.472(3) | N3–C16–N9 | 106.7(3) | 111.9(4) | 111.5(2) |
| C33–C34 | 1.482(5) | 1.491(6) | 1.483(3) | N4–C16–N9 | 109.2(3) | 110.7(4) | 109.0(2) |
| C39–C40 | 1.481(5) | 1.481(6) | 1.486(3) | C31–C32–N7 | 101.5(3) | 101.4(4) | 102.2(2) |
| C1–N8 | 1.289(5) | 1.276(6) | 1.284(3) | C31–C32–N8 | 112.1(3) | 111.1(4) | 112.8(2) |
| C1–N1 | 1.394(5) | 1.391(6) | 1.407(3) | C31–C32–N11 | 108.9(3) | 109.2(3) | 107.4(2) |
| N1–C8 | 1.353(5) | 1.350(5) | 1.353(3) | N7–C32–N8 | 115.8(3) | 114.7(4) | 113.8(2) |
| C8–N2 | 1.324(5) | 1.319(6) | 1.327(3) | N7–C32–N11 | 111.3(3) | 111.0(4) | 111.6(2) |
| N2–C9 | 1.360(5) | 1.354(6) | 1.364(3) | N8–C32–N11 | 107.1(3) | 109.1(4) | 108.7(2) |
| C9–N3 | 1.301(5) | 1.323(6) | 1.311(3) | ||||
| N3–C16 | 1.466(5) | 1.485(6) | 1.473(3) | ||||
| C16–N4 | 1.453(5) | 1.459(6) | 1.465(3) | ||||
| N4–C17 | 1.286(5) | 1.272(6) | 1.284(3) | ||||
| C17–N5 | 1.393(5) | 1.408(6) | 1.402(2) | ||||
| N5–C24 | 1.358(5) | 1.363(5) | 1.358(2) | ||||
| C24–N6 | 1.320(5) | 1.318(6) | 1.322(3) | ||||
| N6–C25 | 1.356(5) | 1.337(7) | 1.365(3) | ||||
| C25–N7 | 1.294(5) | 1.315(6) | 1.309(2) | ||||
The CoIII ions of 3a′, 3b′ and 3aL26 are coordinated by five N-donor atoms and by one additional co-ligand, which is in case of 3a′ an anionic chloro-ligand and in case of 3b′ and 3aL26 a neutral dmf and MeOH molecule, respectively (Fig. 5). The coordination setups of 3a′ (CoN5Cl) and of 3b′/3aL (CoN5O) possess all a slightly distorted octahedral geometry as referred, for example, from bond angles between trans- and cis-bonded donor atoms ranging from 173.6(1)° (3a′) to 179.8(1)° (3a′) and from 84.88(6)° (3aL) to 95.9(2)° (3b′). Comparing related bond lengths of the CoN5Cl and CoN5O coordination setups with each other reveals significant differences, which can be attributed to the different nature/charge of the co-ligands. Due to the larger charge input of the anionic chloro ligand of 3a′, which is compensated by an H+ ion bonded to N9 (Fig. 5), the trans-aligned Co–N bond is substantially elongated compared to the related bonds of 3b′ and 3aL (d(Co1–N10): 3a′ = 1.889(3) Å vs. 3b′/3aL = 1.850(4)/1.858(2) Å). Furthermore, some cis-aligned Co–N bonds of 3a′ are significantly elongated if compared to 3b′ and 3aL (Co1–N7 and Co1–N5, see Table 4), which is attributed to the charge input as well, although the bonds Co1–N1 and Co1–N3 of all three compounds are equal in length.
The helmet, thus the bridging fifth 1,3-diimino-isoindolene unit including the atoms N9–N11, C33–C40 (3a′) and additionally Cl9, Cl10 (3b′) is negatively charged and bonded via the above mentioned Co1–N10 bond to the CoIII ions. The helmet is bonded via the two covalent single bonds (see below) N9–C16/N11–C32 to the CoC32N8H16 (3a′, 3aL) or CoC32N8Cl8H8 (3b′) core. As a consequence, the aromaticity of these cores got lost, although 4a (CoPc) and 4b(CoPcCl10) possess cores with identical sum formulas. Additionally, the carbon atoms C16 and C32 obtain a fourth different bonding partner and thus chiral information. In the here reported cases of 3a′, 3b′ and 3aL the solid state comprises racemates composed of the S,S and R,R enantiomers. The imprint of a chiral information onto the previous non-chiral phthalocyanin core is a unique feature common to nearly all so far reported phthalogens.11
In case of 3a′ the nitrogen atom N9 is protonated and involved in a hydrogen bond with one dmf molecule (Fig. 5, (d(N1⋯O3D) = 2.771(4) A; ∡(N1–H1N⋯O3D) = 171(4)°)), although this feature does not induce any difference into related bond lengths of 3a′ compared to 3b′ and 3aL,26 (Table 4). Next, selected features of the molecular structure of 3a′ shall be compared with the ones of 4a and of 2b. Thereby, selected bond lengths and angles of 4a will be taken from a SC-XRD study performed along with the here reported work. Single crystals of 4a and of many others pristine metallophthalocyanines were so far exclusively fabricated by means of physical vapour transport methods (see Table S10†) and allows to obtain (needle like) crystals with dimensions up to 20 × 1 × 1 mm. When crystallizing the filtrated and concentrated solutions directly after the synthesis of all here reported helmet type phthalogens in all cases the formation of the related CoPc's was observed. The formation of crystalline CoPc's was noticed by their extraordinary large brightness and reflectivity in single crystalline state (see Fig. S70†). Only in case of 4a, however, the crystals were sufficiently large (up to 5 × 5 × 1 mm) for a SC-XRD study. The molecular structure of 4a is displayed in Fig. S71, and Table S11† gives selected bond lengths, bond and torsion angles while Table S9† summarizes selected crystal and structural refinement data of 4a. The most obvious difference between the molecular structure of 3a′ compared to the one of 4a, beside the bridging helmet, is the non-planarity of the CoN8C32 unit, which both compounds have in common. In case of 3a′ this unit has a root-mean-square deviation (rmsd) from planarity of 0.638 Å, with the highest deviation from planarity (hdp) observed for C21 with 1.197 Å which indicates severe distortion. For 4a the rmsd from planarity amounts to 0.050 Å with a hdp of 0.092 observed for C16. Thus, as expected, 4a is flat. For a graphical visualization of the distorted (3a′) vs. the flat geometry (4a) of these units Fig. 6 displays in two perspective views their overlay.
 | ||
| Fig. 6 Best fit of the CoC32N8 units of 4a (dashed lines) onto the one of 3a′ (full line, hydrogen atoms omitted) in two different perspective views. All non-hydrogen atoms were used for fitting with the program XP.50 | ||
The Co–N bond lengths of 4a (1.922(2) and 1.924(2) Å) are significantly longer compared to the ones of 3a′ (1.865(3)–1.906(3) Å), which might be attributable to the larger ionic radius of CoII in square-planar vs. CoIII in distorted octahedral coordination geometry. The differences in the bond lengths and angles between 4a and 3a′ are determined by the bridging helm, by which the atoms C16 and C32 are sp3 hybridized. Consequently, all bonds of these two atoms to its neighbouring atoms are single bonds with bond lengths in the range between d = 1.452(2) Å (C32–N8) to 1.523(5) Å (C15–C16). The bond angles around sp3 hybridized C16/C32 deviate substantially from the ideal 109.47° angle (range: ∡(C31–C32–N7) = 101.5(3) to ∡ (N3–C16–N4) = 116.3(3)°), although the sum of all angles around C16/C32 is with 656.4(7)/656.7(7)° very close to the value for an ideally sp3 hybridized atom of 656.82°. Because of the solely single bond character of the bonds C16–N4/C32–N8 the other bonds involving the N atoms, namely N4–C17/N8–C1 (1.286(5)/1.289(5) Å) are classic double bonds in character. Among all other bonds of 3a′ the latter two bonds are by far the shortest ones, see Table 4. These structural features are common to the other two helmet compounds discussed here, namely 3b and 3aL.26 Moreover, such short bonds as observed for N4–C17/N8–C1 cannot be found in 4a as all bonds are here aromatic in character. The more pronounced aromatic character of 4a compared to 3a′ and 2b can be checked by a further comparison of analogous bonds. The average bond lengths of C1–C2 and its analogous and symmetry generated equivalents amounts to 1.4525(11) Å. For 3a′ a significantly larger value of 1.4775(13) Å is observed, whereby the two pure single bonds C15–C16/C31–C32 were excluded from averaging, which displays less pronounced aromatic character. This tendency is even more pronounced in 2b, the precursor of both 4a and 3a′, as the averaged bond lengths is here 1.493(4) Å.
1H NMR spectroscopy of 3a and 3b
The compounds 3a–d are all diamagnetic; but due to solubility reason, well-resolved 1H NMR spectra could be recorded only for 3a and 3b in DMF-d7 and CD2Cl2, respectively (see ESI for details†). The 1H NMR spectra provide evidence for their C2 symmetry in solution. Noteworthy, for the NiII-containing alkoxy-substituted “antenna”-type phthalogens of Type III a related observation was made,33,34 whereas for all further literature-known phthalogens (Fig. 1) no well-resolved NMR spectra were reported. The electronic ESI display in Fig. S25† the aliphatic and aromatic regions of 1H NMR spectra of [3a(HCl)(dmf)]·2dmf in DMF-d7, while Fig. S26† shows the 1H and 13C{H} NMR spectra of [3a(MeOH)]·2MeOH in CD2Cl2.The 1H NMR of [3a(HCl)(dmf)]·2dmf shows in the aliphatic region two sets of signals related to DMF-d7 residual signals and of dmf as coordinating and packing solvent.51 In the aromatic region a complex series of resonances is observed, corresponding to the twenty aromatic protons of [3a(HCl)(dmf)]·2dmf (see Table S1†). A more detailed assignment is, due to their complexity, not possible. However, well-resolved 1H and 13C{1H} NMR spectra of [3a(MeOH)]·2MeOH were accessible from measurements carried out in CD2Cl2. The 1H NMR pattern in the aromatic region, corresponding to 20 aromatic protons, could be interpreted by assuming a C2 symmetry, which manifests additionally in the 13C{1H} NMR spectrum by 19 different resonances for the aromatic and one slightly upfield shifted resonance belonging to the sp3-hybridized carbon atoms (Table S1†).
The full assignments of the protons and the carbon atoms was achieved with the aid of 2D homo- and heteronuclear NMR techniques (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC and 1H–13C HSQC-TOCSY (Fig. S27–S30†)). As part of this assignment the 1H NMR resonances of H19 and H20 and the 13C NMR signals of C17 to C20 belonging to the bridging “helm” was straightforward. However, the assignment of the NMR resonances belonging to the atoms of the C32H16N8 core was more complicated, whereby the shifts of H11/C11 to H14/C14 could be assigned as belonging to the same chain via COSY techniques. However, a final assignment of the order of the atoms within the chain, even with the help of NOE-based 1D and 2D NMR techniques (NOESY, ROESY) based on an expected nuclear-overhauser-effect between H6 and H11, was not possible. On the contrary, C2 symmetric 3b displays in its 1H NMR spectra five singlets only, corresponding to its ten aromatic protons. Due to the poorer solubility of 3b compared to [3a(HCl)(dmf)]·2dmf and [3a·MeOH]·2MeOH, 13C NMR measurements did not lead to evaluable results.
ESI-MS spectrometry of 3a–d
The four phthalogens [3a(HCl)(dmf)]·2dmf, [3b(dmf)]·(dmf)·H2O, [3c(HCl)]·dmf and [3d(MeOH)]·dmf were characterized further by ESI-TOF-HRMS, whereby the insets displayed in Fig. 7 show the experimentally obtained isotope patterns of the fragments [3a–HCl–3dmf + H]+ (m/z = 714.1440, 12C401H2154Co14N11), [3b–2dmf–H2O + H]+ (m/z = 1053.7411, 12C401H1135Cl1054Co14N11), [3c–HCl–dmf + H]+ (m/z = 854.2917, 12C501H4154Co14N11) and [3d–MeOH–dmf + H]+ (m/z = 1014.2383, 12C501H4154Co14N1116O10), confirming their molecular structure identities. | ||
| Fig. 7 Enlarged part of the HRMS (ESI-TOF) spectra of 3a–d of the mole peak region. | ||
All peaks are isotopically resolved and are in agreement with their calculated isotopic patterns. The labile axial ligands (chloride in the case of 3a, 3c; dmf in the case of 3b and MeOH in case of 3d); as well as dmf packing solvents, are appears to dissociate from the complexes 3a–d under the conditions of the mass spectrometric measurements (Fig. S31, S35, S37 and S40†).
IR spectroscopy of 3a–d
The FT-IR spectra in the range between 500–4000 cm−1 of 3a–d are displayed in Fig. S32, S33, S36, S39 and S41, while the assignment of selected vibrational frequencies is given in Table S2.† Accordingly, sp2 C–H stretching vibrations (rings), were observed for all compounds between 3094 and 3008 cm−1, while the spectral range from 3000 to 2800 cm−1 is governed by C–H (sp3) symmetric and asymmetric stretching vibrations,(νas(C–H) and νs(C–H) absorptions) of either the dmf methyl groups in all 3a–d complexes or by the aliphatic substituents R of 3c (R = CH3) and 3d (R = OCH3). The presence of dmf is indicated further by the presence of carbonyl absorption band (ν(CO) vibrations) at 1664 cm−1, 1652 cm−1, 1660 cm−1 and 1661 cm−1 for 3a–d, respectively. The lower frequency of 3b compared to 3a, 3c and 3d is attributed to the different nature of dmf. Thereby, 3b possesses a datively bonded dmf molecule, while in case of 3a, 3c and 3d dmf acts as packing solvent. The ν(CO) vibrations overlap partially with the ν(CN) stretching vibrations and a large number of further aromatic ν(CC) or ν(CN) absorptions adds substantial complication for a more precise assignment.24 Moreover, the absorptions at 1607, 1615, 1598 and 1603 cm−1 for 3a–d, respectively, might be attributed to skeletal C–C vibrations (1580–1610 cm−1).24,52 A high intensity band at 1533, 1534, 1532 and 1533 cm−1 correlate to the vibrations of nitrogen bridging atoms (–N).52 In the range of 1430–1480 cm−1, a bands varies in intensity; from low to high, were observed and attributed to the ν(C–C) stretching vibrations of isoindole skeleton (vibrations of the pyrrole and benzene residues).52 The absorptions between 720 and 780 cm−1 for 3a–d, are attributed to out-of-plane C–H bending of the arylic groups. The number of arylic C–H is for 3b–d much smaller (with 10 aromatic C–H) as compared to 3a (20 aromatic C–H). This difference is nicely reflected in the intensities and shapes of C–H absorptions (see ESI† for further details).24,52
UV/Vis spectroscopy of 3a, 3b and 3d
The UV/Vis spectra of 3a, 3b and 3d (Fig. 8) display impressively the non-aromatic nature of the “helmet” type phthalogens compared to their CoII-containing phthalocyanine counterparts, as the typical intense Q and Soret band of the latter are not observed. The principal features of the spectra are then consistent with the one reported for 3a by McGaff and Sorokin.26,30 | ||
| Fig. 8 UV/Vis spectra (CH2Cl2, 220–800 nm) of 3a (c = 1.1333 × 10−5 M, black), 3b (c = 1.6576 × 10−5 M, red), and 3d (c = 2.1422 × 10−5 M, purple). | ||
Initially, all three phthalogens 3a, 3b and 3d exhibit similar features in their spectra. For 3a intense bands at 234 and 248 nm and another band at 318 nm followed by two shoulders at 388 nm and at approximately 472 nm were observed. For 3b we distinguish intense band at 262 nm, with a shoulder at 243 nm, a band at 318 nm followed by two shoulders at 398 and 475 nm respectively, while 3d displays an intense band at 272 nm with a shoulder at 251 nm, a band at 323 nm and a shoulder at 384 nm (cf. Fig. S47–49 and Tables S4–6† for more details).
The intense higher energy bands observed around 248, 262 and 272 nm for 3a, 3b and 3d respectively, might be due to π → π* transitions of phenolic group within the ligand.53 Based on Goutermann's and Lever's assignments,54,55 the shoulders between 380 and 480 nm; which contains two in the case of 3a and 3b and one in the case of 3d, were assigned to metal-to-ligand (MLCT) or ligand-to-metal (LMCT) charge-transfer transitions. Noticeable, absorptions solely based on d–d absorptions are not observed at all.56
Thermal behaviour of 2a–d
The thermal behaviour of 2a–d in the solid state was studied by thermogravimetry (TG) and by differential scanning calorimetry (DSC). The appropriate TG traces are depicted in Fig. 9. The thermal decomposition of 2a takes place in two steps (Fig. 9), but before decomposition melting at 140 °C is observed (Fig. S21†). | ||
| Fig. 9 TGA traces of 2a–d under argon (40–500 °C, heating rate: 10 K min−1, Ar gas flow: 20 ml min−1). | ||
The onset temperature of the first decomposition of 2a is 200 °C and this endothermic process is finished at 339 °C to leave a residue of 20.4%. This agrees well with a compound of the sum formula “CH3N” (theoretical residue: 20.0%). There are two chemical species possessing this formula, namely methylenimine (H2CNH),57 and the carbene aminomethylene (H–C|–NH2).58 While the latter specie could be isolated by matrix techniques at 12 K only,58 methylenimine is available by gas phase pyrolysis of methylamine (CH3NH2) at 1000 °C, but is only stable within the gas phase.57 Methylenimine is, however, unstable towards polymerization on surfaces57 and much likely the residue obtained out of 2a corresponds to (H2CNH)n. However, this residue decomposes to gaseous reaction products or sublimes with an onset temperature of 511 °C to leave no residue in the TGA crucible (Fig. S21†).
In case of 2b no melting is observed and the first endothermic decomposition occurred between 166 and 253 °C (Fig. 9 and S22†) to leave a mass residue of 93.2%. Most likely this decomposition liberates NH3, leading to the formation of H2PcCl8 in analogy to urea melt reactions using 4,5-dichlorophthalic acid or related starting compounds.39f The theoretical mass residue for the formation of the phthalocyanine H2PcCl8 is with 91.8% close to the experimentally observed one. However, an exclusive tetra-cyclization of 2b is unlikely and pristine-like phthalocyanines have in general a higher thermal stability39f compared to the second endothermic decomposition of 2b in the range of 268–298 °C leaving 74.2% mass residue. The first decomposition is regarded as a fusion of 2b to form higher oligomers (e.g. an hexamer, cf. Fig. S22†), while the nature of the second and third decomposition, starting at 345 °C (Fig. 9 and S22†), remains unknown.
Compound 2c decomposes in a first endothermic decomposition in the temperature range of 214–289 °C (onset: 281 °C; Fig. 9 and S23†) to leave a mass residue of 32.7%. This agrees well with a compound of sum formula “C3H7N”, which would correspond to, e.g., acetone imine (theoretical mass loss 32.9%). In analogy to 2a this hypothetical intermediate of 2c is highly reactive and prone to polymerization,59 and much likely “C3H7” compares therefore rather to a polymer of acetone imine. In a subsequent endothermic thermal decomposition at 340–460 °C a further mass loss to 23.0% is observed, which would correspond to a further decomposition of the “C3H7N” intermediate to a “C2N” species corresponding to a theoretical mass loss of 22.0%. While, for compound 2d in form of 2d·H2O adduct no melting is observed and the first mass loss between 57 and 96 °C (onset: 80 °C,, mass loss: theoretical: 8.1%, experimental: 9.7%; Fig. 9 and S24†), is due to the desorption of H2O.60 The nature of the second and third endothermic thermal decomposition, starting at 162 °C with a mass loss of 3.7%; (Fig. 9 and S24†), leaving 86.4% mass residue remains unknown. A further exothermic thermal decomposition in the range between 229–306 °C (onset: 279 °C, Fig. 9 and S24†) is observed; to leave a mass residue of 42.9%. This agrees well with a compound of the sum formula of “C5H5NO” (theoretical mass residue 42.6%).
Thermal behaviour of 3a–d
The thermal decomposition of 3a–d phthalogens occurs in multistep process with several endothermic processes, as shown in Fig. 10 and S42–S46.† The onset temperatures and all further temperature ranges are summarized in Table S3.† Initially, for the phthalogens 3a–d the mass decline at lower temperatures, precisely, for 3a between 65 and 205 °C, for 3b between 77 and 126 °C, for 3d between 75 and 148 °C, is attributed to the loss of packing or coordinating solvents, as well as HCl (Fig. 10, S42–46 and Table S3†), depending on the nature of the investigated phthalogens, while for 3c no mass declines were observed in these ranges. | ||
| Fig. 10 TGA traces of 3a–d under argon (40–800 °C, heating rate: 10 K min−1, Ar gas flow: 20 ml min−1). | ||
Rising the temperature for 3a–d a noticeable mass decline at 306–407 °C (3a), of 215–291 °C (3b), 256–301 °C (3c) and 245–263 °C for 3d is observed. This mass decline is due to the “cleave off” or the release of the bridged diiminoisoindolino units, thus of the “helm”.11 The weight loss of the phthalogens 3a–d fits to this assumption (Table S3†).
In order to confirm this both 3a and 3b were subjected to heating in a temperature-controlled furnace. The heating process consisted of two stages, whereby in step one the temperature was raised slowly (5 °C min−1) to 380 °C and in step two this temperature was left constant for 2 hours, giving rise to bright blue-coloured residues. Afterwards it was allowed to cool down to room temperature slowly (5 °C min−1). The comparative FT-IR analysis of the obtained residues of 3a and 3b (Fig. 11, S51 and S52†), accompanied with a subsequently further thermogravimetric characterization (Fig. S53, S54 and S55†) confirmed them to correspond to the cobalt(II)-containing phthalocyanines 4a (CoPc) and 4b (CoPcCl8). For example, the IR spectra of 4a and of 4b obtained by the two different chemical approaches compares well with each other as well as with already reported IR spectra.39f
 | ||
| Fig. 11 IR spectra of 4a/4b obtained out of thermal treatment of 3a/3b (left/right, above) and of 4a/4b obtained by work-up according to Scheme 1 (left/right, below) in the region of 550–1800 cm−1. | ||
After the “helms” of 3a–d are cleaved-off to give the parent cobalt(II)-phthalocyanines 4a–d the further mass decline is due to the thermal decomposition of the latter. This becomes obvious when comparing especially the TG traces of 3a and 3b with results reported of 4a and 4b.39f
Electrochemistry
The redox properties of phthalogens have been so far just sparingly described.11 In order to shed more light on this topic the electrochemical properties of 3a were determined and will be discussed compared to the ones of 4a.61 Therefore, 3a was studied electrochemically by cyclic voltammetry (CV) and square wave voltammetry (SWV) in anhydrous DMF solutions containing 0.1 mol L−1 of [N(nBu)4][B(C6F5)4] as supporting electrolyte.62 The data of the CV experiments of 3a at a scan rate of 100 mV s−1 are summarized in Table 5.| Couple |
E°′a [mV] |
ΔEpb [mV] |
|---|---|---|
| a E°′ = formal potential = ((Epa + Epc)/2). b ΔEp = (Epa − Epc). c E pc = cathodic peak potential of the irreversible process. d Values taken from the square-wave voltammogram. | ||
| Ia | −1319c | — |
| Ib | −1550c | — |
| II | −1754c | — |
| III | −1919d | — |
| IV | −1975d | — |
| V | −2075 d | — |
| VI | −2454 | 81 |
| VII | −953 | 70 |
The cyclic voltammogram of 3a (Fig. 12) clearly displays three irreversible reduction process at −1319 mV (E1pc) (Ia) and −1550 mV (E2pc) (Ib) and −1754 mV (E3pc) (II). They are followed by four quasi-reversible one-electron reductions at −1919 mV  (III), −1975 mV
(III), −1975 mV 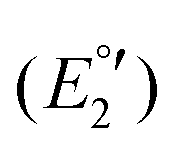 (IV), −2075
(IV), −2075  (V) and −2454 mV
(V) and −2454 mV  (VI) during the cathodic scan. The processes (III–V) take place in a very close potential range and they are not resolvable by CV; nonetheless, the SWV confirms the exiting of three processes (Fig. 12, inset). In the anodic scan an additional one-electron oxidation at −953 mV is observed
(VI) during the cathodic scan. The processes (III–V) take place in a very close potential range and they are not resolvable by CV; nonetheless, the SWV confirms the exiting of three processes (Fig. 12, inset). In the anodic scan an additional one-electron oxidation at −953 mV is observed  (VII).
(VII).
 | ||
| Fig. 12 Left: cyclic voltammograms of 3a; inset: the square-wave voltammogram of 3a (duration 5 s, amplitude 5 mV, pulse 25 mV); right: set of cyclic voltammograms of 3a. Scan rate: 100 mV s−1; in DMF solutions (1.0 mmol L−1) at 25 °C, supporting electrolyte 0.1 mol L−1 of [N(nBu)4][B(C6F5)4], working electrode: platinum electrode. Roman numbers in black correspond to processes in the initial and in red to the 1st cycle. | ||
There are noticeable differences in the electrochemical processes between the initial and the 1st cycle (Fig. 12, left). The reduction processes Ia/Ib of the initial cycle are not observed in the 1st cycle, while this cycle comprises a reduction process denoted as # in Fig. 12. Measuring up to the 5th full cycle reveals no further changes compared to the observations made for the 1st cycle (Fig. 12, right). The slight decline in current in due course of the 1st to the 5th indicates that the material generated electrochemically in the initial cycle remains stable throughout the applied potential range.
These observations are interpreted as follows: in the initial cycle the reduction processes Ia and Ib are due to the CoIII/II reduction of 3a, followed by the cleavage of the helm by two one electron bond breaking processes of the two N–Csp3 bonds. These two reductions, however, convert 3a to 4a (CoPc). This conversion is electrochemically irreversible, as it was already shown to be chemically irreversible.10 Subsequently a reduction at −1754 mV and a set of reductions (III–VI, Fig. 12, and Table 5) is observed. They are attributed to a CoII/I reduction and four reductions of the phthalocyanato core, as already reported.61 The same electrochemical behaviour of 3a has been observed using a glassy carbon working electrode (Fig. S66, and Table S8†).
Experimental section
Reagents and materials
1,2-Dicyanobenzene (phthalonitrile), 1,2-dimethylbenzene, 1,2-dimethoxybenzene and 4,5-dichlorophthalonotrile were purchased from TCI Chemical and used as received. Ammonia (NH3) solution (7 M in methanol (MeOH)) and sodium methoxide (NaOMe) solution (0.5 M in MeOH) were purchased from Sigma-Aldrich and used as received. Extra dry MeOH (99.8%, over Molecular Sieve, AcroSeal®) and extra dry N,N-dimethylformamide (DMF), (99.8%, AcroSeal®) were purchased from Acros Organics and used as received. 1,2,4-Trichlorobenzene was purchased from Acros Organics and used as received. Cobalt(II) chloride hexahydrate, 98% was purchased from Alfa Aesar (Johnson Matthey company) and used as received. The solvents were purified and dried using standard methods prior to use.63 Dichloromethane (CH2Cl2) was dried with an MBraun MB SPS-800 system (double column solvent filtration, working pressure 0.5 bar) and further purified by distillation from calcium hydride.63 Tetrahydrofuran (THF) was distilled prior to use. 1,2-Dibromo-4,5-dimethylbenzene,64 4,5-dimethylphthalonitrile,65 1,2-dibromo-4,5-dimethoxybenzene,66 4,5-dimethoxyphthalonitrile,65 and [N(nBu)4][B(C6F5)4],67 were synthesized in accordance with literature procedures. Column chromatography was performed on silica gel with a particle size of 200–300 mesh. Columns were packed as slurry of silica gel in n-hexane or CH2Cl2 and equilibrated solution using the appropriate solvent system.Apparatus and measurements
Proton nuclear magnetic resonance (1H NMR) and carbon NMR (13C{1H} NMR) were recorded in DMSO-d6; otherwise stated, at ambient temperature using a Bruker Avance III 500 Ultra Shield Spectrometer in the Fourier transform mode. The spectra were recorded at 500.300 MHz (1H), 125.813 MHz (13C{1H}), respectively. The chemical shifts δ are reported in ppm, using the residual solvent signal as an internal standard: (CDCl3: 1H NMR, δ = 7.26 ppm, 13C{1H} NMR, δ = 77.16 ppm); (DMSO-d6: 1H NMR, δ = 2.50 ppm; and 13C{1H} NMR, δ = 39.52 ppm); (DMF-d7: 1H NMR, δ = 2.75, 2.92 ppm, 13C{1H} NMR, δ = 29.76, 34.89 ppm); (CD2Cl2: 1H NMR, δ = 5.32 ppm, 13C{1H} NMR, δ = 53.84 ppm). Multiplicities were given as: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplets), dd (doublet of doublets), dt (doublet of triplets), and br (broad). A Bruker Avance 400 spectrometer equipped with double-tuned probes capable of MAS (magic angle spinning) was used to record the solid-state NMR spectra (1H-MAS NMR (400.1 MHz) and 13C{1H} CP-MAS NMR (100.6 MHz)). Spinning 3.2 mm zirconium oxide rotors are used for sample packing at 15 kHz. 1H-MAS NMR was obtained with single pulse excitation (90° pulse, pulse length 2.4 μs) and a recycle delay of 6 s. Cross-polarization (CP) technique with a contact time of 3 ms is used to enhance sensitivity for acquiring the spectra. The 13C{1H}-CP-MAS NMR is collected with 1H decoupling using a TPPM (two-pulse phase modulation) pulse sequence with a 6 s recycle delaying. The spectra are referenced to tetramethylsilane using TTSS (tetrakis(trimethylsilyl)silane) as an internal secondary standard (3.55 ppm for 13C, 0.27 ppm for 1H). Infrared spectroscopy (FT-IR) studies in solid state using KBr disc were performed in the range of 400–4000 cm−1 using a PerkinElmer 1000 FT-IR spectrometer. Bands are characterized as broad (br), strong (s), medium (m), and weak (w). C,H,N elemental analyses were performed using a Thermo FlashAE 1112 series analyzer. High resolution electrospray ionization mass spectrometry HRMS (ESI-TOF) were recorded in units of mass of charge ratio (m/z) with a Bruker micrOTOF QII equipped with an Apollo II ESI source. Mass samples were dissolved in DMF/MeCN or DMSO/MeCN or CH2Cl2/MeCN (HPLC grade). Ultraviolet/visible (UV/Vis) absorption spectra were recorded using a Spectronic GENESYS 6 UV/Vis spectrophotometer (Thermo Electron Corporation) between 200–800 nm. TG experiments were performed using a Mettler Toledo TGA/DSC 11600 system with a MX1 balance. The melting points of 2a–d were determined by a Gallenkamp MFB 595 010 M melting point apparatus.Electrochemical measurements were performed with 1.0 mmol L−1 solutions of the analytes in anhydrous DMF solutions containing 0.1 mol L−1 of [N(nBu)4][B(C6F5)4] as supporting electrolyte under a blanket of purified argon at 25 °C. The instrumentation consists of a Radiometer Voltalab PGZ 100 electrochemical workstation interfaced with a personal computer. The measurement cell contains three electrodes, a Pt auxiliary electrode, a glassy carbon working electrode (surface area 0.031 cm2), and an Ag/Ag+ (0.01 mol L−1 AgNO3) reference electrode. The working electrode was pretreated by polishing on a Buehler microcloth subsequently with 1 μm and 1/4 μm diamond paste. The reference electrode was constructed from a silver wire inserted in a 0.01 mmol L−1 AgNO3 and a 0.1 mol L−1 [N(nBu)4][B(C6F5)4] MeCN solution in a luggin capillary with a Vycor tip. This luggin capillary was inserted into a second luggin capillary with a Vycor tip filled with a 0.1 mol L−1 [N(nBu)4][B(C6F5)4] in DMF. Under these conditions all experiments showed that all oxidation and reduction processes were reproducible in the range of ±5 mV. All experimental potentials were internally referenced against a Ag/Ag+ reference electrode, whereas all presented results are referenced against FcH (as internal standard) as recommended by IUPAC.49,68 When Fc* [Fc* = Fe(η5-C5Me5)2] was used as an internal standard, the experimentally measured potential was converted into E vs. FcH/FcH+ (under our conditions the Fc*/Fc*+ couple was at −614 mV vs. FcH/FcH+, ΔEp = 60 mV).69 Data were then manipulated with a Microsoft Excel worksheet to set the formal redox potential of the FcH/FcH+ couple to 0 V.
Single crystal X-ray diffraction studies
Data of 2b, 3a, 3b and of 4a were collected with a Rigaku Oxford Gemini S diffractometer, while the one of 2c were collected with a Bruker Venture D8 diffractometer. All structures were solved by direct methods and refined by full-matrix least-squares procedures on (F2)70 as implemented in the WINGX v2013.3 suite.71 All non-hydrogen atoms were refined anisotropically and all carbon-bonded hydrogen atoms isotropically by using appropriate riding models. The atomic positions of nitrogen-bonded hydrogens were taken from Difference Fourier Maps and refined freely and isotropically. Crystallographic data have been deposited with Cambridge Crystallographic Data Centre: CCDC 2298395 (2b), 2298396 (2c), 2298397 (3a), 2298398 (3b) and 2298399 (4a).†Theoretical calculations
ORCA72 was employed for structure optimizations in the gas phase and in DMF solution in combination with the spin-component scaled second-order Møller–Plesset perturbation theory (SCS-MP2)73 and the aug-cc-pVTZ basis set.74,75 The resolution of identity approximation was employed to speed up the Hartree–Fock (RI-JK)76–79 as well as the SCS-MP2 part (RI-MP280,81) of the calculation. Solvation effects were considered by the conductor-like polarizable continuum model (C-PCM).82 TURBOMOLE83 was used to obtain thermodynamic corrections. This includes the following steps: structure optimization employing the BP86 functional84,85 and the TZVP basis set.86 Seminumerical frequency calculation employing NumForce. Subsequently, thermodynamic corrections were calculated by freeh, a tool provided by Turbomole. All calculations with TURBOMOLE employed the RI-approximation76,77,87 and solvation effects were considered by the conductor like screening model (COSMO).88General synthetic procedure for 3a–d and 4a–d
The 1,3-diiminoisoindoline's 2a–d were synthesized from the corresponding phthalonitriles 1a–d as described in detail in the ESI.† For the synthesis of 3a–d to a 500 mL one-neck (ground glass joint, NSH 29) round bottom flask containing a suspension of the corresponding 1,3-diiminoisoindolines 2a–d (5 equiv.) in 1,2,4-trichlorobenzene (150 mL), CoCl2·6H2O (1 equiv.) was added in a single portion at ambient temperature. Afterwards, a ca. 30 cm long reflux condenser (NSK 29) was mounted, and the reaction mixture temperature was raised slowly to 80 °C (within 1 h) under permanent stirring. Then, from 80 °C on, the heating rate was reduced (20 °C per 2 h), and the reaction mixture was heated gradually up to 210 °C. Noteworthy, by reaching ca. 100–120 °C NH3 was developed. The mixture was then heated at 210 °C as long as the NH3 development was noticed (heating may take up to 5 days). After the NH3 gas development had stopped, the reaction mixture was allowed to cool down to ambient temperature, filtrated and washed with CH2Cl2 (40 mL). The filter cake was next treated with DMF to separate between 3a–d and 4a–d. Out of crystallizations, dark red or orange-red crystals were grown by slow diffusion of et2O into DMF solution, which were identified as 3a–d. Crystals were collected, washed first with small amount of DMF then n-hexane, air dried in a fume hood and were used without further purification unless specified. While, the violet filter cake; which was identified later as the corresponding cobalt(II)phthalocyanines (4a–d), was washed first with DMF (5 × 30 mL) until no further green color, then washed with freshly distilled THF (4 × 50 mL) and air dried; (see ESI for full details†).Conclusions
In this paper we have demonstrated that the synthesis of the “helmet”-type phthalogens 3a–d with a rather broad variation of their substituents is possible via the use of 1,3-diiminoisoindolines (2a–d) as precursors. This synthetic route is easy-to-follow and affords the phthalogens in much higher yields compared to so far reported synthetic routes and makes this unique member of the porphyrinoid family broadly accessible beyond lab curiosities. With this larger set of pentadenate “helmet” phthalogens thermal decomposition studies were performed which verified that the “helms” are cleaved-off to give the parent cobalt(II)-containing phthalocyanines 4a–d. Furthermore, it was shown for 3a as example that a related cleavage occurs electrochemically. Besides potential applicability of 3a–d in homogenous catalytic oxidation reactions the easy accessibility of these precursors of metallophthalcyanines offers now a broader use of them for engineering optical media, coloured fibres or other material science application. Our future work is devoted to the synthesis of further series of “helmet”-type phthalogens with different metal centre.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft through project FOR 1154 “Towards Molecular Spintronics”. R. K. Al-Shewiki and S. W. thank FOR 1154 for a grant. Solid state NMR spectroscopic measurements were carried out by Dr Andreas Seifert at the Polymer chemistry department of the Technical University of Chemnitz. S. Z. is grateful to the Center for Information Services and High Performance Computing [Zentrum für Informationsdienste und Hochleistungsrechnen (ZIH)] for providing its facilities for computational studies.References
- For examples, see: (a) B. D. Berezin, Coordination Compounds of Porphyrins and Phthalocyanines, Wiley, Chichester, 1981 Search PubMed; (b) N. B. McKeown, Phthalocyanine materials: Synthesis, Structure and Function, Cambridge University Press, 1998 Search PubMed; (c) C. C. Leznoff and A. B. P. Lever, Phthalocyanines: Properties and Applications, Wiley-VCH, New York, 1989, 1993, 1993, 1996 Search PubMed; (d) The porphyrin handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic, San Diego, 2003 Search PubMed; (e) M. S. Rodriguez-Morgade, G. de la Torre and T. Torres, in The Porphyrin Handbook, Academic Press, New York, 2003, pp. 125–160 Search PubMed.
- For examples, see: (a) W. M. Sharman and J. E. van Lier, in The Porphyrin Handbook, Academic Press, New York, 2003; 15, 1–60 Search PubMed; (b) N. B. McKeown, in The Porphyrin Handbook, Academic Press, New York, 2003, pp. 61–124 Search PubMed; (c) K. Sakamoto and E. Ohno-Okumura, Materials, 2009, 2, 1127–1179 CrossRef CAS; (d) V. N. Nemykin and E. A. Lukyanets, ARKIVOC, 2010, 136–208 Search PubMed; (e) E. A. Lukyanets and V. N. Nemykin, J. Porphyrins Phthalocyanines, 2010, 14, 1–40 CrossRef CAS; (f) D. Arıcan, M. Arıcı, A. L. Ugur, A. Erdogmus and A. Koca, Electrochim. Acta, 2013, 106, 541–555 CrossRef.
- For examples, see: (a) Y. Chen, M. Hanack, W. J. Blau, D. Dini, Y. Liu, Y. Lin and J. Bai, J. Mater. Sci., 2006, 41, 2169–2185 CrossRef CAS; (b) D. Dini, M. Calvete and M. Hanack, Chem. Rev., 2016, 116, 13043–13233 CrossRef CAS PubMed.
- Y. C. Tsai, U.S. Pat. ApplUS 20060292494A1 20061228, 2006 Search PubMed.
- (a) C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed; (b) M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 760–792 CrossRef; (c) D. Wöhrle, G. Schnurpfeil, S. G. Makarov, A. Kazarin and O. N. Suvorova, Macroheterocycles, 2012, 5, 191–202 CrossRef; (d) Y. Matsuo, K. Ogumi, I. Leon, H. Wang and T. Nakagawa, RSC Adv., 2020, 10, 32578–32689 Search PubMed.
- For examples, see: (a) M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed; (b) M. L. Esqueda, M. E. S. Vergara, J. R. Á. Bada and R. Salcedo, Materials, 2019, 12, 434–451 CrossRef CAS PubMed.
- (a) Z. Z. Öztürk, N. Kılınça, D. Atilla, A. G. Gürek and V. Ahsen, J. Porphyrins Phthalocyanines, 2009, 13, 1179–1187 CrossRef; (b) M. Bouvet, P. Gaudillat and J.-M. Suisse, J. Porphyrins Phthalocyanines, 2013, 17, 913–919 CrossRef CAS; (c) T. Kerdcharoen and S. Kladsomboon, in Applications of Nanomaterials in Sensors and Diagnostics, 2013, pp. 237–255 Search PubMed.
- T. Nyokong, Pure Appl. Chem., 2011, 83, 1763–1779 CAS.
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS.
- F. Baumann, B. Bienert, G. Rösch, H. Vollmann and W. Wolf, Angew. Chem., 1956, 68, 133–150 CrossRef CAS , and patents cited therein.
- H. Lang and T. Rüffer, Applications of Porphyrinoids as functional materials, The royal Society of Chemistry, CPI group, UK, 2021 Search PubMed.
- F. Gund, J. Soc. Dyers Colour., 1953, 69, 671–682 CrossRef.
- C. Ercolan, A. M. Paoletti, G. Pennesi, G. Rossi, A. Chiesi-Villa and C. Rizzoli, J. Chem. Soc., Dalton Trans., 1990, 1971–1977 RSC.
- M. P. Donzello, C. Ercolani and P. J. Lukes, Inorg. Chim. Acta, 1997, 256, 171–172 CrossRef CAS.
- M. P. Donzello, C. Ercolani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1998, 37, 1347–1351 CrossRef CAS PubMed.
- E. M. Bauer, M. P. Donzello, C. Ercolani, E. Masetti, S. Panero, G. Ricciardi, A. Rosa, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 2003, 42, 283 CrossRef CAS PubMed.
- J. Janczak and R. Kubiak, Polyhedron, 2003, 22, 313 CrossRef CAS.
- J. Janczak and R. Kubiak, J. Porphyrins Phthalocyanines, 2017, 21, 1 CrossRef.
- F. Gingl and J. Strähle, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1841–1843 CrossRef.
- J. Janczak and R. Kubiak, J. Chem. Soc., Dalton Trans., 1994, 2539–2543 RSC.
- J. Janczak and R. Kubiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 2039–2042 CrossRef.
- J. Janczak and R. Kubiak, Acta Chem. Scand., 1995, 49, 871–877 CrossRef CAS.
- K. Benihya, M. Mossoyan-Deneux, F. Hahn, N. Boucharat and G. Terzian, Eur. J. Inorg. Chem., 2000, 1771 CrossRef CAS.
- K. Benihya, M. Mossoyan-Déneux and M. Giorgi, Eur. J. Inorg. Chem., 2001, 1343–1352 CrossRef CAS.
- T. Fukuda, N. Shigeyoshi, A. Fuyuhiro and N. Ishikawa, Dalton Trans., 2013, 42, 16486–16489 RSC.
- H. M. Kieler, M. J. Bierman, I. A. Guzei, P. J. Liska and R. W. McGaff, Chem. Commun., 2006, 3326–3328 RSC.
- E. S. Brown, J. R. Robinson, A. M. McCoy and R. W. McGaff, Dalton Trans., 2011, 40, 5921–5925 RSC.
- B. M. Peterson, M. E. Herried, R. L. Neve and R. W. McGaff, Dalton Trans., 2014, 43, 17899–17903 RSC.
- R. L. Neve, M. C. Eidenschink, I. A. Guzei, B. M. Peterson, G. M. Vang and R. W. McGaff, ChemistrySelect, 2016, 1, 5182–5186 CrossRef CAS.
- I. Y. Skobelev, E. V. Kudrik, O. V. Zalomaeva, F. Albrieux, P. Afanasiev, O. A. Kholdeeva and A. B. Sorokin, Chem. Commun., 2013, 49, 5577–5579 RSC.
- R. W. McGaff, U. S. Pat, US 10065980B2, 2018 Search PubMed.
- R. W. McGaff, WO 2020/041284A1, 2020.
- C. D. Molek, J. A. Halfen, J. C. Loe and R. W. McGaff, Chem. Commun., 2001, 2644–2645 RSC.
- J. R. Robinson, K. A. Bahr, M. J. Bierman, I. A. Guzei, H. M. Kieler-Ferguson, A. M. McCoy and R. W. McGaff, Dalton Trans., 2011, 40, 11809–11814 RSC.
- Y. Y. Karabach, M. N. Kopylovich, K. V. Luzyanin, M. F. C. Guedes da Silva, V. Yu. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2016, 455, 696–700 CrossRef.
- Y. Kikukawa, T. Fukuda, A. Fuyuhiro, N. Ishikawa and N. Kobayashi, Chem. Commun., 2011, 47, 8518–8520 RSC.
- T. Fukuda, Y. Kikukawa, R. Tsuruya, A. Fuyuhiro, N. Ishikawa and N. Kobayashi, Inorg. Chem., 2011, 50, 11832–11837 CrossRef CAS PubMed.
- T. Fukuda, Y. Kikukawa, A. Fuyuhiro, N. Kobayashi and N. Ishikawa, Chem. Lett., 2014, 43, 925–927 CrossRef CAS.
- For examples, see: (a) J. A. Elvidge and R. P. Linstead, J. Chem. Soc., 1952, 5000–5007 RSC; (b) J. A. Elvidge and R. P. Linstead, J. Chem. Soc., 1952, 5008–5012 RSC; (c) P. F. Clark, J. A. Elvidge and R. P. Linstead, J. Chem. Soc., 1953, 3593–3601 RSC; (d) J. A. Elvidge and R. P. Linstead, J. Chem. Soc., 1955, 3536 RSC; (e) P. J. Brach, S. J. Grammatica, O. A. Ossanna and L. Weinberger, J. Heterocycl. Chem., 1970, 7, 1403 CrossRef CAS; (f) T. Rüffer, D. Nurpeisova, Z. Jakupova, A. Tashenov, N. Uhlig, A. Khalladi, L. Mertens, A. Gonser, M. Mehring and H. Lang, Z. Naturforsch., B: J. Chem. Sci., 2017, 72, 589–601 CrossRef.
- (a) C. G. Claessens, D. Gonzalez-Rodriguez and T. Torres, Chem. Rev., 2002, 102, 835–853 CrossRef CAS PubMed; (b) T. Torres, Angew. Chem., Int. Ed., 2006, 45, 2834–2837 CrossRef CAS PubMed; (c) A. Medina and C. G. Claessens, J. Porphyrins Phthalocyanines, 2009, 13, 446–454 CrossRef CAS.
- (a) F. Fernandez-Lazaro, T. Torres, B. Hauschel and M. Hanack, Chem. Rev., 1998, 98, 563–575 CrossRef CAS PubMed; (b) W. S. Durfee and C. J. Ziegler, J. Porphyrins Phthalocyanines, 2009, 13, 304–311 CrossRef CAS.
- (a) W. O. Siegl, Inorg. Chim. Acta, 1977, 25, L65–L66 CrossRef CAS; (b) W. O. Siegl, J. Heterocycl. Chem., 1981, 18, 1613–1618 CrossRef CAS; (c) W. O. Siegl, J. Org. Chem., 1977, 42, 1872–1878 CrossRef CAS.
- (a) A. B. P. Lever, B. S. Ramaswamy and S. R. Pickens, Inorg. Chim. Acta, 1980, 46, L59–L61 CrossRef CAS; (b) L. K. Thompson, V. T. Chacko, J. R. Elvidge, A. B. P. Lever and R. V. Parish, Can. J. Chem., 1969, 47, 4141–4152 CrossRef CAS; (c) A. B. P. Lever, L. K. Thompson and W. M. Reiff, Inorg. Chem., 1972, 11, 104–109 CrossRef CAS.
- (a) H. Hopff and P. Gallegra, Helv. Chim. Acta, 1968, 51, 253–260 CrossRef CAS; (b) R. Sato, T. Senzaki, Y. Shikazaki, T. Goto and M. Saito, Chem. Lett., 1984, 1423–1426 CrossRef CAS.
- O. V. Shishkin, I. S. Konovalova, R. I. Zubatyuk, G. V. Palamarchuk, S. V. Shishkina, A. V. Biitseva, I. V. Rudenko, V. A. Tkachuk, M. Yu. Kornilov, O. V. Hordiyenko and J. Leszczynski, Struct. Chem., 2013, 24, 1089–1097 CrossRef CAS.
- A. Acker, H.-J. Hofmann and R. Cimiraglion, J. Mol. Struct., 1994, 315, 43–51 CrossRef.
- A. V. Lyubimtsev, A. Baranski, M. K. Isayaikin and R. P. Smirnov, Chem. Heterocycl. Compd., 1997, 33, 937–941 CrossRef CAS.
- Zh.-Q. Zang, J. M. Njus, D. J. Sandman, C. Guo, B. M. Foxman, P. Erk and R. Van Gelder, Chem. Commun., 2004, 5, 886–887 RSC.
- J. T. Engle, A. N. Allison, J. M. Standard, I.-S. Tamgho and C. J. Ziegler, J. Porphyrins Phthalocyanines, 2013, 17, 1–10 CrossRef.
- XP ‘A graphical program’. VS. SHELXTL-5.0, Bruker AXS GmbH Search PubMed.
- L. Wang, L. Lin, G. Zhang, K. Kodama, M. Yasutake and T. Hirose, Chem. Commun., 2014, 50, 14813–14816 RSC.
- A. V. Ziminov, S. M. Ramsh, E. I. Terukov, I. N. Trapeznikova, V. V. Shamanin and T. A. Yurre, Semiconductors, 2006, 40, 1131–1136 CrossRef CAS.
- D. K. Rastogi, S. K. Sahnis, V. B. Rana and S. K. Dua, J. Coord. Chem., 1918, 8, 97–104 CrossRef.
- P. Sayer, M. Gouterman and C. R. Connell, Acc. Chem. Res., 1982, 15, 73–79 CrossRef CAS.
- A. B. P. Lever, S. R. Pickens, P. C. Minor, S. Licoccia, B. S. Ramaswamy and K. Magnell, J. Am. Chem. Soc., 1981, 103, 6800–6806 CrossRef CAS.
- Inorganic Electronic Spectroscopy, A. B. P. Lever, American Elsevier publishing company, New York, 1968, pp. 299–314 Search PubMed.
- R. Pearson Jr. and F. J. Lovas, J. Chem. Phys., 1977, 66, 4149–4156 CrossRef.
- A. K. Eckhardt and P. R. Schreiner, Angew. Chem., Int. Ed., 2018, 57, 5248–5252 CrossRef CAS PubMed.
- J. M. Casas, J. Fornies, A. Martin and A. J. Rueda, Organometallics, 2002, 21, 4560–4563 CrossRef CAS.
- E. Pousaneh, M. Korb, V. Dzhagan, M. Weber, J. Noll, M. Mehring, D. R. T. Zahn, S. E. Schulze and H. Lang, Dalton Trans., 2018, 47, 10002–10016 RSC.
- (a) D. W. Clark, N. S. Hush and I. S. Woolsey, Inorg. Chim. Acta, 1967, 19, 129–132 Search PubMed; (b) A. Koca, H. A. Dincer, M. B. Kocak and A. Gül, Russ. J. Electrochem., 2006, 42, 31–37 CrossRef CAS.
- W. E. Geiger and F. Barrière, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Vol. 3, Pergamon, New York, 1988 Search PubMed.
- A. V. Ivanov, K. V. Kabanova, M. O. Breusova, I. V. Zhukov, L. G. Tomilova and N. S. Zefirov, Russ. Chem. Bull., 2008, 57, 1665–1670 CrossRef CAS.
- Z. Iqbal, A. Lyubimtsev and M. Hanack, Synlett, 2008, 2287–2290 CAS.
- L. Babel, T. N. Y. Hoang, L. Guenee, C. Besnard, T. A. Wesolowski, M. Humbert-Droz and C. Piguet, Chem. – Eur. J., 2016, 22, 8113–8123 CrossRef CAS PubMed.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 CrossRef.
- (a) A. Nafady and W. E. Geiger, Organometallics, 2008, 27, 5624–5631 CrossRef CAS; (b) I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem B, 1999, 103, 6713–6722 CrossRef CAS; (c) J. Ruiz and D. Astruc, C. R. Acad. Sci. Paris Ser. IIc., 1998, 21–27 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- S. J. Grimme, Phys. Chem., 2003, 118, 9095–9102 CAS.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- E. J. Baerends, D. E. Ellis and P. Ros, J. Chem. Phys., 1973, 2, 41–51 CAS.
- B. I. Dunlap, J. W. D. Connolly and J. R. Sabin, J. Chem. Phys., 1979, 71, 3396–3402 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. Feyereisen, G. Fitzgerald and A. Komornicki, Chem. Phys. Lett., 1993, 208, 359–363 CrossRef CAS.
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175–3183 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthesis of 2a–d, 3a–d and 4a–d, characterization data (1H,13C{1H} and 2D NMR, ESI-MS, IR spectra, and UV/Vis), crystal and structural refinement data. CCDC 2298395 (2b), 2298396 (2c), 2298397 (3a), 2298398 (3b) and 2298399 (4a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03950a |
| This journal is © The Royal Society of Chemistry 2024 |