
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The emerging chemistry of self-electrified water interfaces
Fernando
Galembeck
 *ab,
Leandra P.
Santos
b,
Thiago A. L.
Burgo
c and
Andre
Galembeck
d
*ab,
Leandra P.
Santos
b,
Thiago A. L.
Burgo
c and
Andre
Galembeck
d
aDepartment of Physical Chemistry, University of Campinas, Institute of Chemistry, 13083-872, Campinas, Brazil. E-mail: fernagal@unicamp.br
bGalembetech Consultores e Tecnologia, 13080-661, Campinas, Brazil
cDepartment of Chemistry and Environmental Sciences, São Paulo State University (Unesp), 15054-000, São José do Rio Preto, Brazil
dDepartment of Fundamental Chemistry, Federal University of Pernambuco, 50740-560, Recife, Brazil
First published on 2nd February 2024
Abstract
Water is known for dissipating electrostatic charges, but it is also a universal agent of matter electrification, creating charged domains in any material contacting or containing it. This new role of water was discovered during the current century. It is proven in a fast-growing number of publications reporting direct experimental measurements of excess charge and electric potential. It is indirectly verified by its success in explaining surprising phenomena in chemical synthesis, electric power generation, metastability, and phase transition kinetics. Additionally, electrification by water is opening the way for developing green technologies that are fully compatible with the environment and have great potential to contribute to sustainability. Electrification by water shows that polyphasic matter is a charge mosaic, converging with the Maxwell–Wagner–Sillars effect, which was discovered one century ago but is still often ignored. Electrified sites in a real system are niches showing various local electrochemical potentials for the charged species. Thus, the electrified mosaics display variable chemical reactivity and mass transfer patterns. Water contributes to interfacial electrification from its singular structural, electric, mixing, adsorption, and absorption properties. A long list of previously unexpected consequences of interfacial electrification includes: “on-water” reactions of chemicals dispersed in water that defy current chemical wisdom; reactions in electrified water microdroplets that do not occur in bulk water, transforming the droplets in microreactors; and lowered surface tension of water, modifying wetting, spreading, adhesion, cohesion, and other properties of matter. Asymmetric capacitors charged by moisture and water are now promising alternative equipment for simultaneously producing electric power and green hydrogen, requiring only ambient thermal energy. Changing surface tension by interfacial electrification also modifies phase-change kinetics, eliminating metastability that is the root of catastrophic electric discharges and destructive explosions. It also changes crystal habits, producing needles and dendrites that shorten battery life. These recent findings derive from a single factor, water's ability to electrify matter, touching on the most relevant aspects of chemistry. They create tremendous scientific opportunities to understand the matter better, and a new chemistry based on electrified interfaces is now emerging.
 Fernando Galembeck | Fernando Galembeck FRSC was educated at the University of São Paulo and was a post-doc at the University of Colorado Medical Center and the University of California Davis. He became an assistant professor at USP in 1970, moved to the University of Campinas in 1980, retired as a professor of chemistry in 2011, and became the director of the National Laboratory of Nanotechnology in Campinas till 2015. He then founded the Galembetech consulting and R&D company and continued to lecture as a volunteer at Unicamp. He is a member of the Brazilian Academy of Sciences and TWAS and has received many prizes for his scientific, educational, and technological contributions. His current research interests are matter electrification processes and consequences, and nanostructured materials. |
 Leandra P. Santos | Leandra Pereira dos Santos, a CNPq Level A Human Resources Fellow, holds a PhD degree in Chemistry from the University of Campinas. She specializes in physicochemical properties of surfaces, focusing on electrical properties, surface modification, and atomic force microscopy. With postdoctoral research at Unicamp and USP, she has contributed to projects on elastoelectricity, optoelectrochemical materials, and multifunctional coatings. Leandra's work extends to environmentally friendly materials, including fire retardants. Her impactful research is evident in publications on water reactivity, ice formation, and hygroelectric generators. Recognized for her contributions, she received the 21st Abrafati Science in Paints Award in 2020. |
 Thiago A. L. Burgo | Thiago Augusto de Lima Burgo obtained his PhD degree from the University of Campinas, specializing in the triboelectrification of dielectric polymers. Following this, he conducted postdoctoral research at the Argonne National Laboratory in Chicago, USA, where his focus was on studying the exchange of electric charge at metal–insulator interfaces during fluctuations in frictional forces. Dr Burgo has held positions as a Professor in the Department of Physics at the Federal University of Santa Maria. In 2022, he transitioned to the State University of São Paulo (UNESP), where he currently serves as an Associate Professor. In 2020, he was honored with the Rising Star Award by the Electrostatics Society of America. |
 Andre Galembeck | Andre Galembeck finished his PhD in Chemistry in 1998, at the University of Campinas, Brazil. He is a Full Professor at University Federal de Pernambuco, a former Director of the Center of Strategic Technologies of the Northeast (2011–2018), and a member of the National Advisory Council in Nanotechnology of the Ministry of Science and Technology. He is a CNPq fellow in Technological Development & Innovation. He carries out research activities on Colloid Chemistry and Materials Science focusing on antibacterial activity of silver nanoparticles and the development of hybrid compounds and nanostructures based on inorganic nanoparticles, conducting polymers, self-healable supramolecular structures and latex with applications in energy generation and storage and hygroelectricity. He filed more than 10 patents and founded a deeptech startup in 2020. |
Introduction
Electrostatic charging is often observed under low relative humidity and temperature conditions when the water vapor pressure is very low. High humidity produces water films at solid surfaces, conducting charge to the ground.However, water is also an active electrifying agent, and it can also acquire electricity in many environments, as in the case of ice in thunderclouds or water spray. Accepted explanations of water electrification were lacking until the present century, when the electric variants of AFM and practical Kelvin electrodes associated with inexpensive Faraday cups made observing electrified solids, liquids, and gases much easier, producing large amounts of reliable data.
This review shows that static electrification mediated by water pervades natural and anthropic environments. It addresses key past events, the types of charge carriers in water, evidence of water electrification, its effects on water properties, and water's ability to mediate the electrification of any material systems, shielding electromagnetic fields. The following sections present the effect of water on phase change and mass transfer, chemical reactivity, and energy harvesting. Mechanochemical electrification events, along with the interplay between electricity, friction, wear, and water, create positive feedback loops that further contribute to electrification. These phenomena exacerbate environmental, human, and property losses everywhere.
The new perspectives opened by recognizing water as an active electrification agent and creating a new chemistry and conclusions close the text.
History
The discovery of electricity is usually credited to Thales of Miletus but without support from historical evidence.1 Thales is also credited with a theorem in Geometry and other feats.During the first three centuries of experimental science following Francis Bacon, electricity attracted experimenters’ attention, producing provocative experiments.2 Many of these still lack acceptable explanations. Electrostatic and electrochemical tools were the relevant electric power sources until electromagnetism emerged during the 19th century. The importance of electrochemistry grew during the 20th and 21st centuries and is still increasing. Today, batteries and other electrochemical devices are found all over us. On the other hand, electrostatics lost relevance in the past century: the last important electrostatic generators were the van der Waals and Pelletron high-voltage sources (used in Nuclear Physics until the 1950s), while a few technologies like the Xerox copiers, electrostatic filters, and some problems encountered in space research justified continuing investigation, exemplified by the impressive NASA facilities.3
The electroneutrality of matter and electrons as the sole important carriers were the dominant paradigms during the 20th century. Adopting these ideas did not produce consensus on atmospheric electricity, spontaneous fires, and powder explosions that cause significant environmental, human, and property losses everywhere. It also led to unsustainable but widespread explanations offered to students and the public on practical issues like dust deposition and environmental electrification's health effects.
Electrostatics did not attract great attention from chemists, other than in Debye–Hückel and related theories, in studying the electric double-layer in interfaces or the Hamiltonian operator of quantum mechanics. Chemical thermodynamics books and courses virtually neglect matter's electrification, and the standard chemical potential definition assumes that the electric potential is zero.4 Calculation of mass balance and stoichiometry usually assumes that every solution or multiphase system is electroneutral, prompting McCarthy and Whitesides's challenging question: “…what is the chemistry of materials that bear a net electrostatic charge? ”5
The situation started to change in the 1990s with the emergence of new tools and ideas that are now flourishing. New ideas opened fascinating research opportunities whose results challenged established views on the structure and properties of matter. The authors described facts, ideas, and new opportunities in a book published in 2017.6 Many important new findings have created exciting scientific and technological opportunities since then.
The context
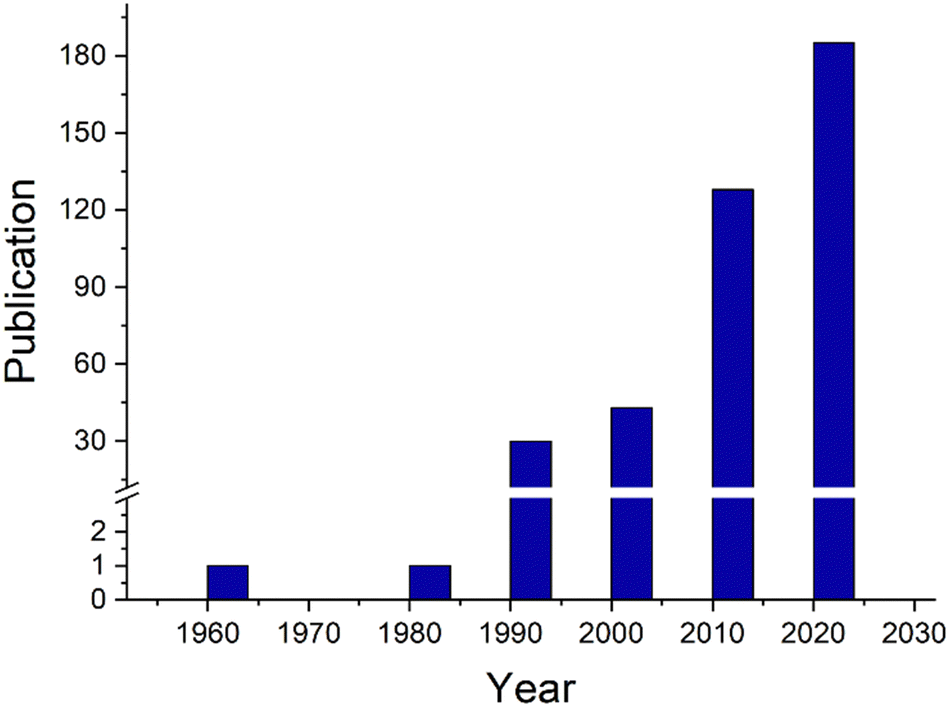 | ||
| Fig. 1 Number of publications about the Maxwell–Wagner effect in the indexed literature. Data extracted from the Web of Science using the search term “Maxwell–Wagner” also includes “Maxwell–Wagner–Sillars” (data extracted from the Web of Science). | ||
This effect appears in dielectric spectroscopy, explaining the large frequency effect on the dielectric response due to charge accumulation. This is often referred to as Maxwell–Wagner polarization, and the credit to Sillars recognizes that the equations earlier obtained by Wagner are a special case from Sillars' equations. The charge accumulation within a sample is expected at the sample interfaces due to the differences in electrical conductivity or dielectric constant. Then, when a polyphasic material is under an electric field, the charge carriers within each phase have different mobilities at the two sides of any interface, producing charge accumulation or depletion even at a significant distance from the interface. The contribution to polarization may exceed the contribution made by the molecular orientation within the field and its fluctuations.
Representing each phase by its parallel conductance and dielectric constant defines a relaxation time τ = ε/σ. Then, an electric current j across any interface produces interfacial polarization, caused by charge accumulation represented in Fig. 2.
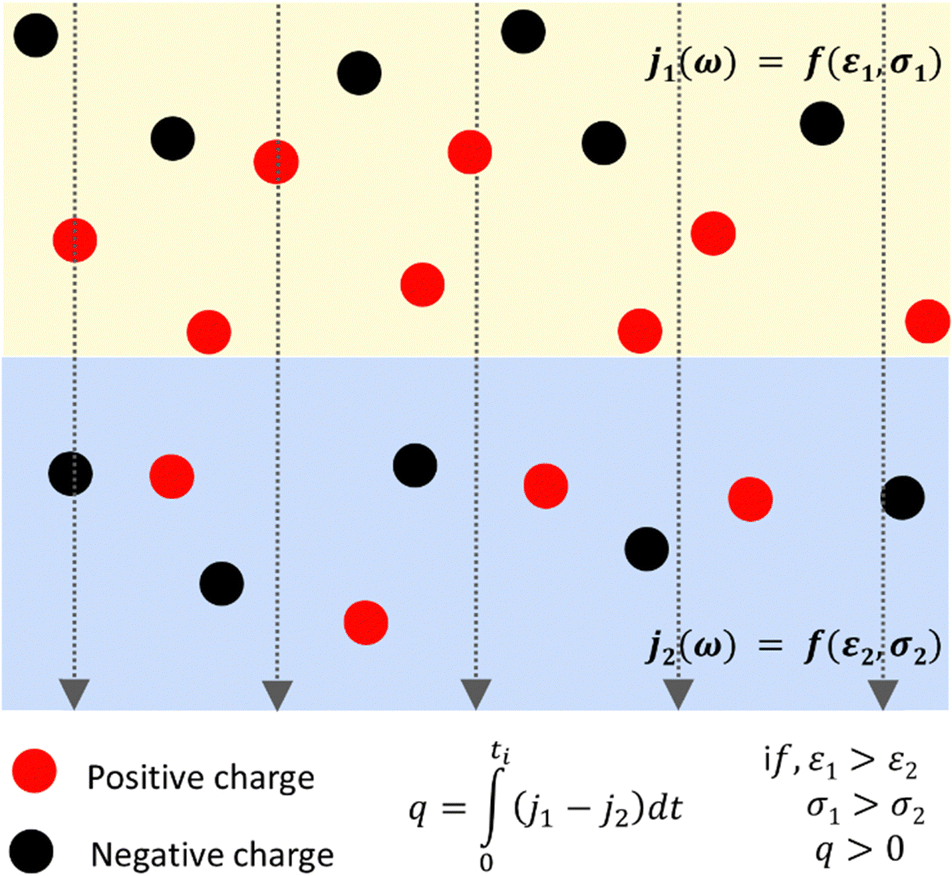 | ||
| Fig. 2 Schematic representation of the Maxwell–Wagner–Sillars effect. Excess charge builds up at an interface due to the difference in conductivity and dielectric constant between the adjacent phases, within an external electric field represented by dotted lines and arrows. | ||
Relaxation times still depend on geometric factors because the equivalent capacitances and resistance for each adjacent domain depend also on their interfacial area and thickness. Thus, every interface and its surroundings can show positive or negative charge excess, creating electric mosaics everywhere on the Earth's surface, where electric fields abound.
Experimental verifications of the Maxwell–Wagner effect10 show the model's validity but reveal quantitative deviations. For instance, the interfacial charge density in an FEP–PP interface was two orders of magnitude higher than the model prediction. The authors attributed the difference to an electret effect.11
Given the generality and the consequences of this effect, it is intriguing to understand why it has been ignored during the twentieth century when STEM disciplines dominated the scientific research. The best explanation the present authors can find is a combination of three factors: (i) the properties of any phase represented in textbooks and the literature are only the bulk properties of chemicals, leaving surface properties to specialized texts on colloids, surfaces, electrochemistry, and related subjects; (ii) electroneutrality is a prevailing belief; and (iii) the pervasive electric fields in any ambient on the Earth do not receive attention.
A consequence of the Maxwell–Wagner effect is the production of electric mosaics12 in any heterogeneous material, dividing its surface into domains of distinct electric charge. A related effect is the modulation of water surface charge under three-phase contact where the junction or contact potential depends on pH-dependent contact potential difference, contributing to water surface charge in various experimental environments.13
Recognizing that interfaces are always electrified invites researchers to stay alert for the ensuing effects while approaching any system. This attitude now contributes to discoveries and better explanations for challenging experimental facts observed in many research areas.
The inner organs of humans and other organisms are complex dynamic arrays of charged particles, membranes, and electric gates switching ion currents. These structures create and control an intense electric activity driving the living sensors and actuators responsible for human conscious or unconscious responses to environmental stimuli, actions, and thoughts. Excess charges in every section of the human electric arrays are always ions, not electrons, short-lived in condensed matter. Thus, all living hardware operates based on ion accumulation and transfer. The workings of human electrical structures are thus sensitive to electric fields that provoke ion mobility, membrane deformation, and changes in the conformation of biomolecules. Life protection is thus dependent on effectively shielding any living body (Fig. 3). This function is performed by water retained on the outermost dead skin cells. The shielding ability of water is familiar to electrical engineers as an impairment to electromagnetic wave communication in the water body subsurface: U-boats communicate using high-frequency acoustic waves, not the radio or microwaves used in the atmosphere. The forthcoming section entitled “Water shielding ability” describes a direct experimental proof of water shielding.
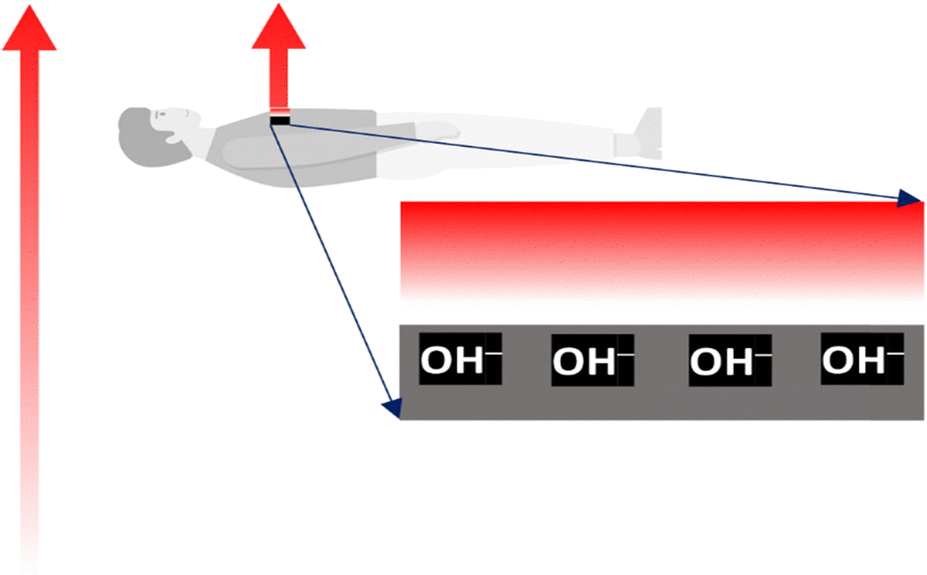 | ||
| Fig. 3 How a human body blocks the field in the Earth capacitor. An initially neutral body above the Earth's surface accumulates hydroxide ions from the skin moisture, protecting the electrified body interior from the outer fields. | ||
However, considering the ambient electric fields and the Maxwell–Wagner effect, every unshielded interface within the Earth capacitor is somewhat electrified.
Common events and specific situations exhibiting spontaneous electrification are:
– Contact between solid surfaces provokes their electrification that increases under friction.15,16 The triboelectric series17 represent the relative tendencies of different materials to acquire an excess positive or negative charge, when they are in static or shearing contact.
– Contact between water and solid surfaces18–20 also provokes mutual electrification, producing many fascinating phenomena that are collectively designated as “hydrovoltaic”,21 further discussed in the section “Water on solid surfaces”.
– Aerosol particles are always charged, depending on the particle size, composition, and formation mechanism. Charged aerosol is detected on the seaside, waterfalls,22 nebulizers,23 and sprayers of any type. Unexpected reactions occur in aerosol surfaces, e.g., hydrogen peroxide formation from water.24
– Any phase separation process produces electricity. Lord Armstrong and Faraday first reported this as “vapor electricity”,25 and it has been verified in evaporation, lyophilization, condensation, freezing,26 and melting; no exception has been detected. The reciprocal effect, phase separation speeding by electric fields, is explored as electrocrystallization27 in the food industry.
– Electrification during milling28 or rupturing solids is familiar to practitioners. In milling operations, the fines may strongly adhere to the walls, impeding the continuation of the operation.
– Dispersed particles of poorly soluble salts between polarized electrodes always migrate to one of the electrodes, depending on pH, and adsorbing ion concentrations. This is explored in chemical separation processes like electrodecantation.29
– Sedimentation and centrifugation of colloidal dispersions and polyelectrolyte solutions produce a potential difference30 between the top and bottom of the container because the settling mass transfer is coupled to charge transfer. Negative particles settle, imparting a negative potential to the bottom region relative to the top. This is why protein molecular weight (MW) determination by sedimentation yields low values unless the solution contains salt sufficient to shield the intermolecular electrostatic repulsion.31
– An asymmetric capacitor exposed to moisture spontaneously acquires charge,32 depending on the nature of the electrodes.33 This has the potential for energy harvesting that is currently being intensively investigated.
– Shoe soles, chair seats, clothes, and other objects made from insulators acquire charge during use, and their electrified surfaces attract dust.34 They may also cause electric discharges. In extreme cases, that may trigger the explosion35 of common powders, like wheat flour and sugar.
– The electrification of vehicles used in dry weather is the source of electric shock in the passengers when they touch the ground to enter or exit the car. This may be dangerous in a gas station or other places with fuel vapor in the atmosphere.
– Periodic stretching and contracting rubber produces alternate electric current with the same frequency of the mechanical action.36
Thus, natural and anthropic activities produce electricity, anywhere and at any time. In most cases, the electrification mechanism is poorly understood, but in some cases, the proposed mechanisms were used to predict results verified experimentally. The appearance of electrostatically driven programmed water droplet transfer procedures is a striking example of the progress achieved.37
Water with excess charge
Charge carriers
Electricity in the ambient matter is carried and stored in two kinds of particles: cations, anions, and electrons. Positrons and other charged entities are not usually considered since their lifetimes are very low, and their presence is insignificant under ambient conditions. Holes are always mentioned in the semiconductor area, but they are virtual entities or “quasi-particles,” representing a missing electron in the valence band of a crystal lattice.Ions are simple atomic or molecular entities carrying excess charge due to an excess or a deficiency of electrons. They are also found in macromolecular or supramolecular arrangements: polyelectrolytes, natural and synthetic membranes, micelles, coacervates, clays, silica, and mineral particles. Beyond, they are also present on the surfaces of macroscopic materials like glass, wood, paint, fibers, and whatever is part of everyday life.
Charge carrier mobility, cross-sections, and interactions
Electron lifetimes are in the picosecond to nanosecond range, in condensed matter and gases, except under high vacuum. It can be higher in semiconductors, reaching the microsecond range. Thus, free electrons cannot play a significant role in the electrification of any material lasting for microseconds or longer times.On the other hand, it has been suggested that contact electrification (CE) in water/hydrophobic interfaces occurs by electron transfer38 and can trigger redox chemical reactions that do not proceed in bulk water.39 CE of microdroplets at water–gas and water–solid leads also to the formation of reactive oxygen species (ROS) and the so-called hydrated electrons.40
In their turn, ions can last for very long times, within the geological range, and they constitute or are found in every mineral (with a few exceptions, like gold and other precious metals, sulphur, and a few other examples), including seawater that covers more than half of the Earth's surface. Free or surface-bound ions also account for the electrification of the proteins, cell membranes, sensors, motion actuators, and any other microstructure in living entities.
However, the formation, persistence, and effects of ions in the atmosphere are not consensual. It is generally acknowledged that solar radiation and cosmic rays can ionize atmospheric neutral molecules. We can often read phrases like “The high electric fields associated with lightning can cause the ionization of air molecules, generating ions”, without discussing ion participation in creating the atmospheric electric fields that cause lightning. It is still largely ignored that lightning takes place within the dust and gases emitted during volcano eruptions, during sandstorms41 in the desert and it is uninterrupted for long periods in a few places on the Earth, like the Catatumbo Lightning over the Maracaibo Lake, in Venezuela.
Properties of electrified water
Immobile charge carriers contribute excess charge to bulk matter, but electrostatic repulsion drives mobile water ions to the surfaces of liquids and solids. For this reason, electrification produces major changes at aqueous interfaces, with important consequences.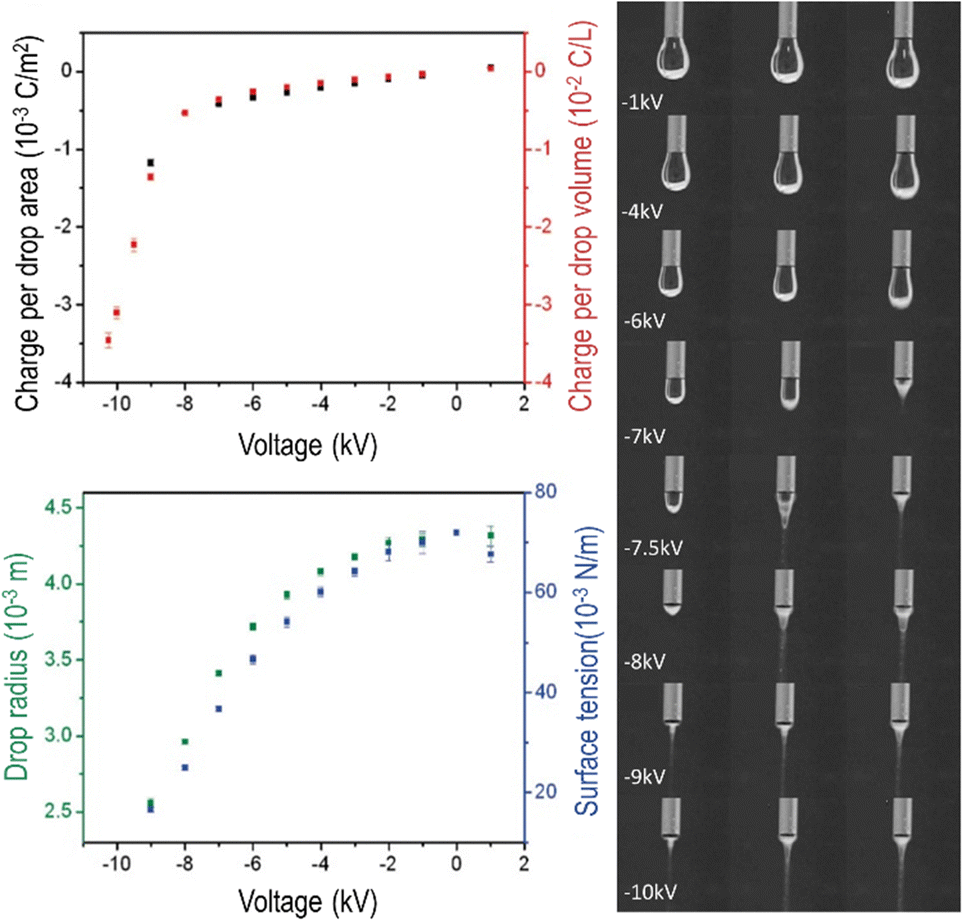 | ||
| Fig. 4 Water dropping from a biased needle acquires excess electric charge that changes the drop shapes. Top left: Area and volume charge density increase with the biasing potential. Bottom left: Drop radius and the calculated surface tension decrease as the needle electric potential departs from zero. Right: Frames extracted from a video showing the needle voltage effect on drop shapes. Electrification increases surface charge density, enhancing electrostatic repulsion among ions adsorbed at the surface. The consequence is a decrease in water surface tension that becomes negative at higher voltages, evidenced by Taylor cone formation and Coulombic explosion. Adapted with permission from ref. 42. Copyright 2023. American Chemical Society. | ||
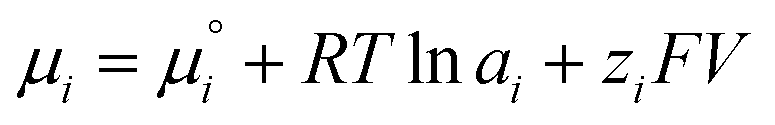 | (1) |
 is the chemical potential of species i under standard conditions (ai = 1 and V = 0), R is the gas constant, T is the temperature, and F is the Faraday constant. The ziFV term is usually neglected (except in electro- and colloid chemistry), assuming that V is always equal to zero, anywhere. Non-zero V results from external bias or redox reactions in electrodes or self-charging due to selective ion partition at interfaces. In the latter case, zi and V have the same signal, and ziFV is always positive, contributing positively to μi. Since μi measures the tendency of ions i to transform by any means, self-charging always increases the chemical reactivity of the i ions, and their tendency to transfer to other domains with lower V, including other phases. Thus, electrification may change the water's phase diagram, which is expected from extensive computation. However, this has not yet been experimentally demonstrated, and the experimental results presented in support of this idea do not present results on the thermodynamic equilibrium temperatures (Tv, Tm, Tc) but on nucleation temperatures determined by metastability during phase transition.45,46 Moreover, electric field penetration in water is limited to a few nanometres only. Thus, it may affect very thin films and particles but not bulk water.
is the chemical potential of species i under standard conditions (ai = 1 and V = 0), R is the gas constant, T is the temperature, and F is the Faraday constant. The ziFV term is usually neglected (except in electro- and colloid chemistry), assuming that V is always equal to zero, anywhere. Non-zero V results from external bias or redox reactions in electrodes or self-charging due to selective ion partition at interfaces. In the latter case, zi and V have the same signal, and ziFV is always positive, contributing positively to μi. Since μi measures the tendency of ions i to transform by any means, self-charging always increases the chemical reactivity of the i ions, and their tendency to transfer to other domains with lower V, including other phases. Thus, electrification may change the water's phase diagram, which is expected from extensive computation. However, this has not yet been experimentally demonstrated, and the experimental results presented in support of this idea do not present results on the thermodynamic equilibrium temperatures (Tv, Tm, Tc) but on nucleation temperatures determined by metastability during phase transition.45,46 Moreover, electric field penetration in water is limited to a few nanometres only. Thus, it may affect very thin films and particles but not bulk water.
Water as a universal electrifying agent
Water is abundant on the Earth's surface and in the atmosphere. It flows, condenses, evaporates, penetrates pores, and is adsorbed or absorbed even in hydrophobic substances like plastics and crystalline ionic materials like clays and cement, increasing the system's entropy by occupying additional microstates.Water has a finite equilibrium solubility47 in hydrophobic liquids but these are often quoted as “immiscible” with water. Other effects appear under non-equilibrium conditions. For instance, volatile liquids pick moisture from the air during unprotected storage or transfer, due to the cooling effect of evaporation.
Water is absorbed within polymers containing polar groups like polycarbonates, polyesters, and polyamides. It is also adsorbed in the polyolefins’ surfaces that usually contain polar groups due to oxidation under air or processing.48 For this reason, polymer processing plants usually dry their raw plastic pellets prior to processing.49
Two widespread but controversial assumptions on the electric properties of water are its electroneutrality and role in electrical phenomena, as a passive poor conductor. However, reports from Armstrong,25 Faraday,50 Wilson, Kelvin,51,52 Vonnegut,53 and their contemporaries showed that water is an active electrifying agent. These results were treated as scientific curiosity or largely ignored and forgotten, for over a century. However, several findings54–57 have been reported since the 1990s, showing the active role of water contributing to electrification in natural or anthropic environments.
Water evaporation involves mass transfer across the water–air interface, comprising water molecules and their ions. Beyond this, transferring the positive water ions to the atmosphere involves less bond breaking than transferring the negative ions, as shown in Fig. 5.
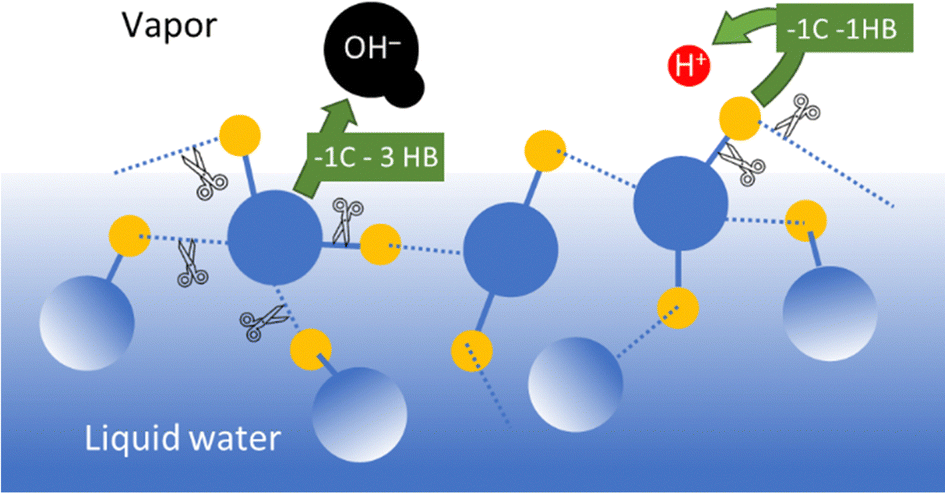 | ||
| Fig. 5 Schematic representation of covalent and hydrogen bonds needed to release a hydronium ion or a hydroxide ion from a neutral water surface to the atmosphere, independent of the solvation of the ions. Releasing the positive ions requires less bond-breaking and thus lower energy than releasing hydroxide ions. C refers to a covalent bond and HB refers to a hydrogen bond. | ||
The result is excess OH− concentration at water surfaces. Thus, the self-electrifying properties of water and its ability to electrify other chemicals and materials derive from water self-ionization, predicting that water–air interfaces are negative. Indeed, careful measurements show that water samples collected from the environment are negative.58 However, positive water ions emerge at droplet surfaces at pH (of the bulk liquid) lower than 4,59 and the system is buffered at pH 3.60
A complicating factor in water electrification is its position at the top of the triboelectric series. Thus, water is at the negative side of the electric double layer in water–air interfaces, but at the positive side of most water–solid interfaces.
Electric field intensifies water sorption in hydrophobic polymers producing surprising results, like the simple technique for charging thin foils of poly(fluoroethylene-propylene) (FEP) at 23 °C in a laboratory atmosphere, with decay time constants exceeding 20 years.61 The author interpreted the results by a charging mechanism due to aqueous ions stemming from field-induced water adsorption. This is an accepted electrowetting mechanism that is effective even in PTFE.62
An investigation by scanning probe microscopy63 revealed polymer electrification by water ion injection into amorphous polymers and the formation of charge mosaics during water drying on a polymer surface.12
Charge in water within other media produces interesting electro-hydrodynamic effects with relevant applications, like programmed water transport on charge-printed surfaces.37
Summing up, many electrification mechanisms have been identified in different systems. Relevant cases are schematically described in Fig. 6, but this is not exhaustive. Other cases not included in this figure are the S/L phase change, L/L liquid partition equilibrium, elastoelectricity, and the various moisture-enabled power generation effects.
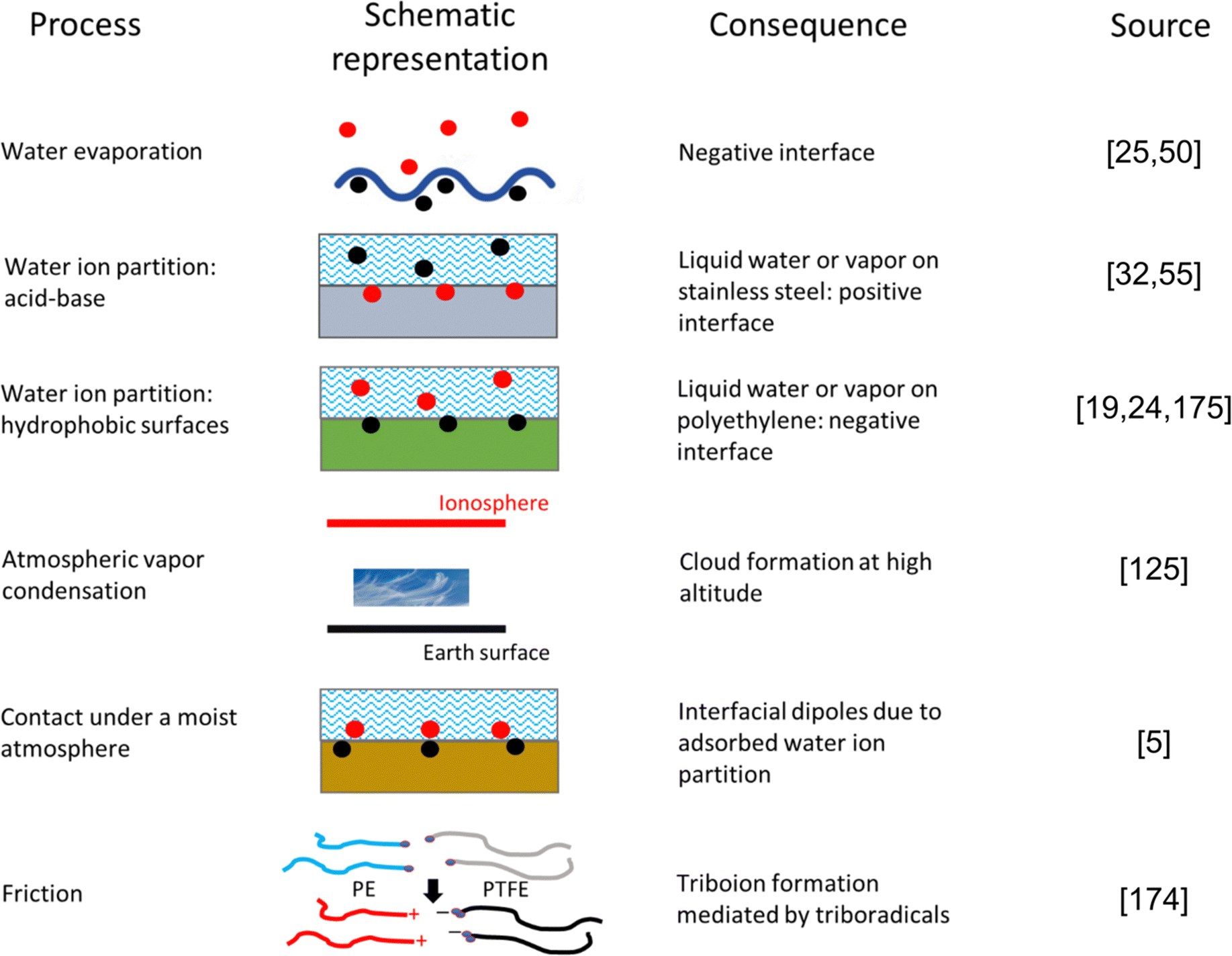 | ||
| Fig. 6 Schematic representation of some mechanisms or electrostatic charging. Many practical situations allow the occurrence of two or more mechanisms, preventing the use of broad explanations for seemingly similar situations with different outcomes. | ||
In the case of common metals, the sign of the charge acquired under exposure to water vapor depends on the acid–base features of the oxide layer that always coats the metal surface, that is schematically represented in Fig. 7.
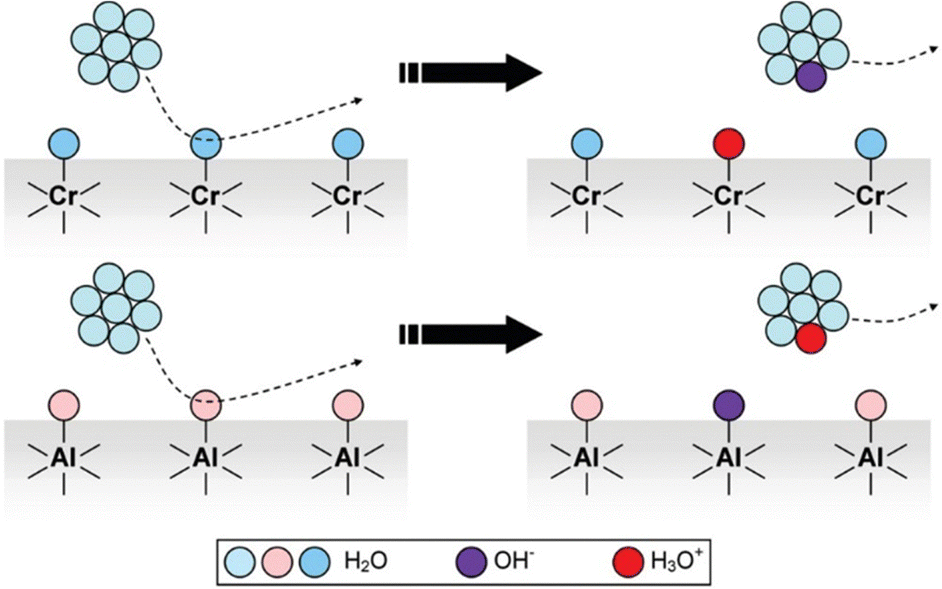 | ||
| Fig. 7 Schematic description of metal charging under a moist atmosphere. Aluminum metal is always coated by an acidic oxide layer that picks hydroxide ions from impinging water clusters, acquiring negative charge. Stainless steel and other alloys contain basic chromium oxide and bind hydronium ions from atmospheric water, acquiring positive charge. Reproduced with permission from ref. 32. Copyright 2023. American Chemical Society. | ||
Water shielding ability
Following Debye's theory, the large water molar polarization under static conditions and low frequency is due to its large molecular dipole. Under an electric field, the dipoles align collectively, opposing the field. This explanation is now complemented by the formation of water ion concentration gradients, with positive ions migrating to the negative electrode and vice versa, reaching the equilibrium state described by eqn (1).Measuring the rates of electric potential change in a cellulose film provides an experimental demonstration of charge accumulation within a field. The plots in Fig. 8 show that the film acquires charge fast under high RH, slowing down under lower RH.64
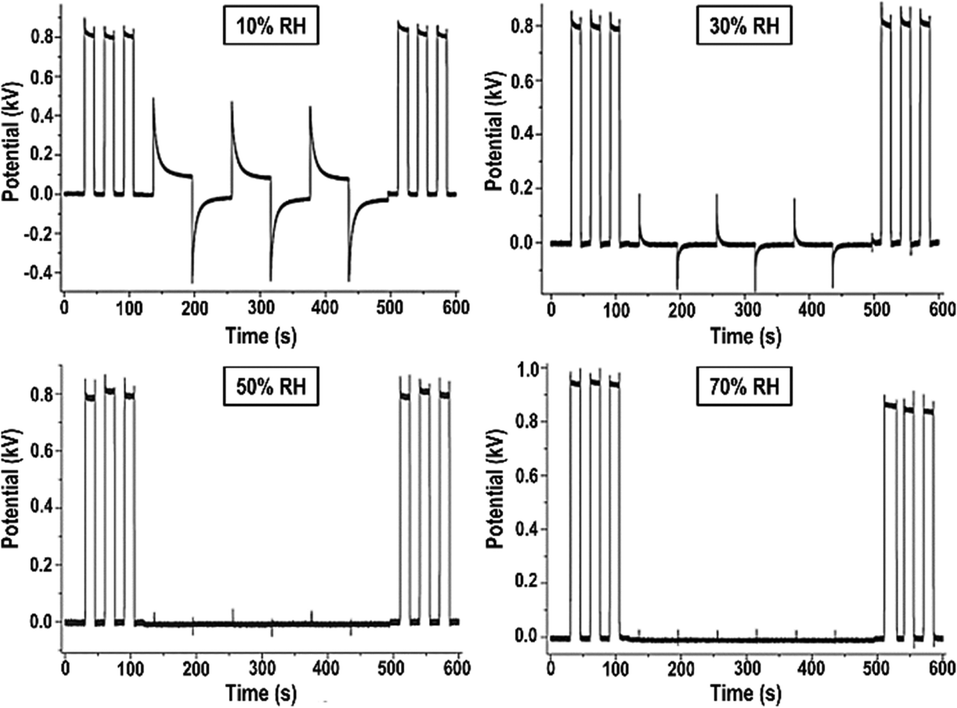 | ||
| Fig. 8 Non-contact potential as a function of time recorded for a paper sheet under different relative humidities. The experiment consists in recording the electrostatic potential of a piece of an electrified acrylic plate, an inductor, covered or uncovered by a sheet of cellulose paper and exposed to different atmospheric conditions. In the first 100 seconds the inductor was periodically introduced beneath the kelvin electrode, showing quasi-squared waves. From 100 seconds to 500 seconds the inductor continued to be periodically introduced beneath the electrode, but a sheet of paper sample always remained between them. After 500 seconds just the inductor was introduced beneath the electrode restoring the initial behavior. This shows a shielding effect based on the electrification of water adsorbed on cellulose due to the ion partition under a non-zero electric potential. Adapted from ref. 64. | ||
This time-dependence is incompatible with polarization deriving from the collective alignment of dipoles, whose relaxation times are in the microsecond time range. Moreover, the time dependence of charge building up and dissipation is the same, except under low RH when charge accumulation on a paper is faster than charge dissipation. Considering the rates of collision of atmospheric water molecules explains the symmetry in the polarization times of the films and charge build-up in the film follows eqn (1).
Consequences of water electrification
There is now ample evidence of the effects of electrification on phase change, chemical reactivity, and energy harvesting through hydrovoltaic effects and friction.Phase change and mass transfer
The first demonstration of electrostatic charging during a phase change process was the “vapor electricity” discovered by a railway engineer and studied by Armstrong and Faraday.25,50 Workman and Reynolds65 later found a potential difference across ice–water interfaces, and Costa Ribeiro66 found solid–liquid interfaces in carnauba wax and naphthalene.The reciprocal effect is the change in the melting or condensation rates, under an applied field. The investigation of ice formation in the atmosphere67 revealed that fields in the 1 kV cm−1 range quickly change supersaturated water vapor into 36 millimeters long ice needles68 (Fig. 9).
 | ||
| Fig. 9 Fast needle growth under a 15 kV potential difference between the electrodes. (a) and (b) Needles’ size increases with time. Pictures in (c)–(e) are magnified sections of (a) and (b). Copyright 2023 IEEE. Adapted, with permission, from ref. 68. | ||
The field has two effects: increasing the rate of vapor crystallization and producing an unusual ice crystal habit. Moreover, the ice needles carry excess electric charge, evidenced by their electrophoretic motion in the air and shape change when the field is withdrawn. These observations are explained considering (i) the effect of the electric potential on ice surface tension that decreases or eliminates the energy barrier to ice nucleation, and (ii) the elongated ice habit with a large area for charge accumulation, reaching the same range of the fields measured in thunderstorm clouds.
Moreover, these experiments also revealed the formation of ice from vapour by a non-classical crystallisation mechanism akin to the spinodal phase separation (Fig. 10).69 In this case, the electric field effect on water molecule clusters produces elongated tenuous structures that grow by densification rather than germ growth.
 | ||
| Fig. 10 Video frames from the same area, showing an ice needle formation by a non-classical crystallization69 mechanism. There is no sign of a needle in frame (a) but it appears faintly in the following frame. Needle contrast and thickening increases from (b)–(d). The time difference between consecutive frames is 0.04 s. | ||
The positive feedback between ice formation and increased atmospheric electric field explains the production of atmospheric electricity, showing how this is produced and stored in humid air under pressure and temperature gradients and how the electrified ice needles produce the electric fields that trigger lightning (Fig. 11).
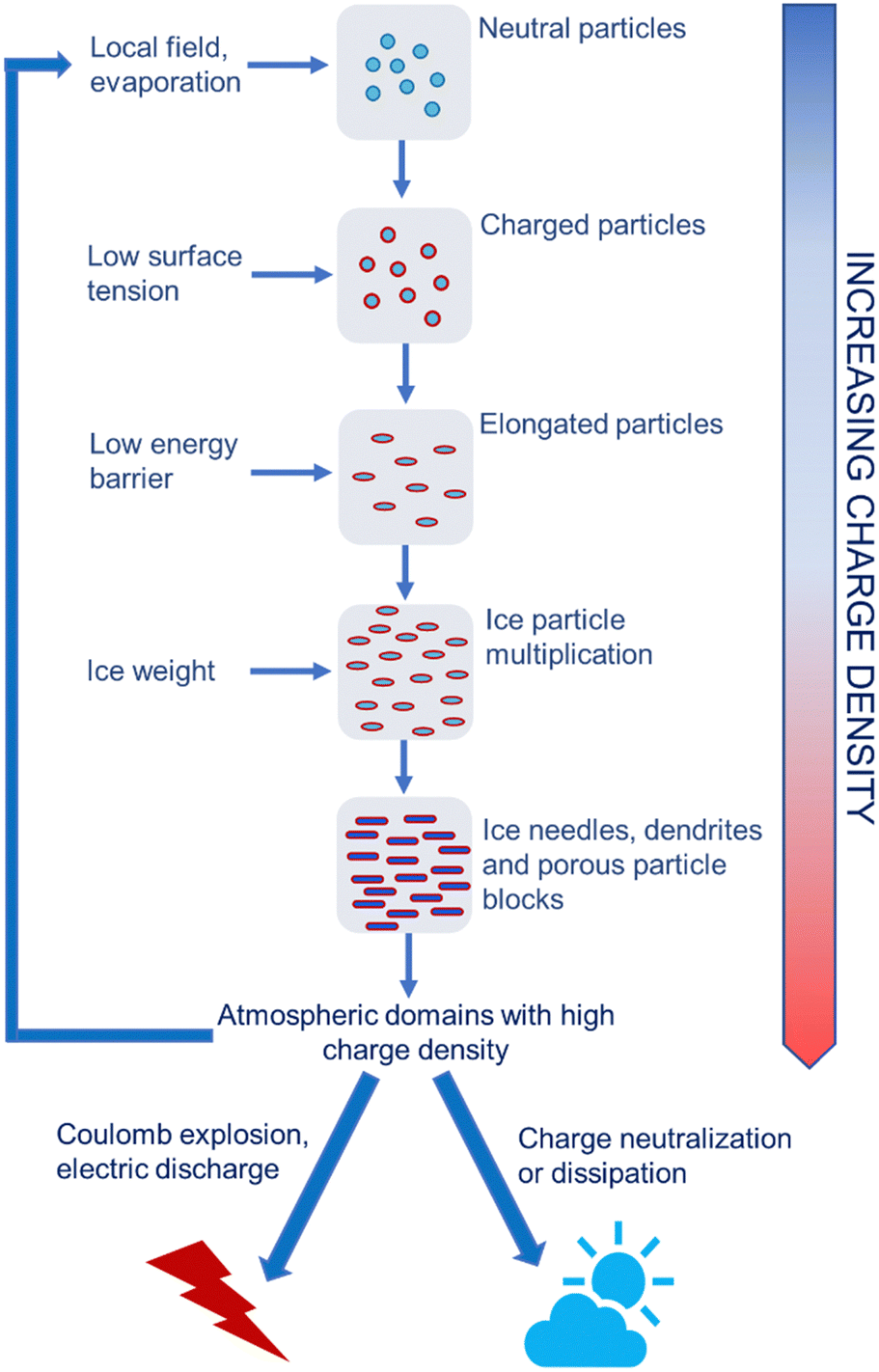 | ||
| Fig. 11 Schematic representation of high voltage production in the atmosphere. Electrified water droplets, ice crystals or aerosol particles create local electric fields. This changes the electrochemical potential of ions on the surface of or in atmospheric vapor surrounding cooled water droplets. The surface tension of electrified water droplets decreases also lowering the energy barrier to ice nucleation. Electrified ice crystals show extended shapes with high specific surface area that allows needles and dendrites to store more charge. Many electrified dendrites form electrified blocks with high charge density that can further contribute to rapidly multiplying the electrified particles in the atmosphere. These create positive feedback loops67 that terminate as electric discharges or explosions during thunderstorms. | ||
Applying this information to other crystallization phenomena explains puzzling observations ranging from internal short-circuits in batteries70 to the difficulty in handling dry powders and damaging electric discharges, in industrial plants and products.
Phase change has paramount importance in chemical separation processes, but there are not yet published studies on its application in distillation, sublimation, and the purification of solids by crystallization.
Charge separation during a phase change process is a feasible procedure for harvesting environmental energy.71 This success suggests that the tropical forest, oceans, and any warm wet areas on the Earth's surface contribute to atmospheric electrification and electrofluidodynamic effects that did not yet receive scientific attention.
The gel-based new devices for water purification and desalination72 offer new opportunities to harvest ambient energy from water evaporation and condensation as in the water evaporation-driven73–76 electric generators.
Reactivity in electrified interfaces
The detection of unexpected chemical reactions was made in aerosols, hydrophobic particle dispersions, emulsions and in surfaces showing Lewis acid–base properties.Aqueous aerosols77 are widely found in the Earth's atmosphere and in any anthropic environment. Aerosols produced by a nebulizer or resulting from water splashing contain both positive and negative droplets,78,79 this means, they are bipolar.23 Measuring charge in a Faraday cup crossed by flowing aerosol produces fractal time series plots, as expected considering the previously observed fractal structure in charged solid surfaces.23 Current explanations for aerosol bipolarity are based on charge partition at interfaces combined with the interfacial area/volume ratio variability in aqueous droplets. Negative charge predominates at water–air interfaces imparting an excess negative charge to the finer aerosol particles, leaving excess positive charge in bulk water or larger particles. The excess charge also appears in the water droplets and ice particles resulting from water vapor condensation, previously discussed in the “Phase Change” section.
Atmospheric processes at aerosol surfaces are observed to follow mechanisms that are quite different from those in the gas phase.80
When the water or solution droplets land on solid surfaces and dry, they produce charge mosaics that were detected but not understood, two decades ago.
Water on hydrophobic surfaces
The water position in the tribochemical series is at the positive end, meaning that most materials in contact with pure water adsorb OH− ions18 thus acquiring a negative charge. This agrees with previous observation of the migration of air bubbles and oil droplets to the positive electrode, during careful electrophoretic experiments.19,20 However, other factors previously described in Fig. 6 intervene and the outcome may be a combination of the various effects according to the superposition principle of Electrostatics.The still widespread electroneutrality paradigm ignores the electrification of macroscopic matter and, consequently, the effects of electrification on chemical reactivity. The influential book of G.N. Lewis on Thermodynamics shows that the change in the Gibbs energy for a chemical reaction under standard conditions is not affected by changing the overall electric potential of the system. However, any natural system is out of equilibrium, formed by many phases and a multitude of interfaces whose electrification produces domains with non-zero electric potential and complex potential gradients. Thus, chemical reactivity at interfaces may show large departures from the reactivity of the same chemicals in the bulk of any phase.
Under these conditions, many substances exhibit unexpected chemical behavior at interfaces that has not been acknowledged since the past decade.
“On-water” reactions
A precursor of the “on water”81 rate acceleration findings is the Diels–Alder reaction between cyclopentadiene and butanone studied by Rideout and Breslow,82 which is 58-fold and more than 700-fold faster than in methanol and hydrophobic solvents, respectively. The authors postulated that this is due to the hydrophobic effect, and comparing the rates of other reactions in water, polar and non-polar solvents led to the exclusion of a polarity effect.Other early evidence for the effect of matter electrification on chemical reactivity comes from mechanochemistry.83 However, this topic was neglected during the 20th century and is treated in a separate section of this article.
In 2005, Sharpless et al.84 showed extensive results on the acceleration of cycloaddition reactions proceeding faster when the reagents were dispersed in water than when they were dissolved in polar or non-polar solvents, shown in Fig. 12. Then, he introduced the term “on water” referring to this peculiar behavior.
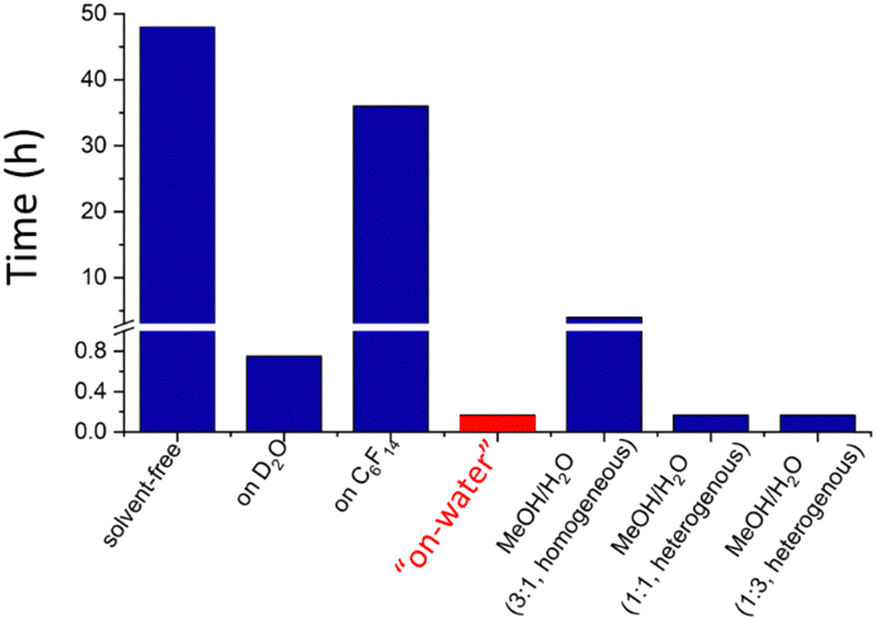 | ||
| Fig. 12 Time to complete the reaction between quadricyclane and dimethyl azodicarboxylate in different solvents. Data from Sharpless 2005.84 | ||
For instance, the “on-water” reaction of quadricyclane with dimethyl azodicarboxylate took 10 min only, compared to 48 h and more than 18 h required in the absence of a solvent and in organic solvents. Adding solvents to water did not have a significant effect as long as the reaction heterogeneity was conserved. Even using a critical concentration of methanol producing a homogeneous environment, the reaction time was 24 times the “on-water” reaction time. Since then, there has been growing recognition of the importance of interfacial effects in atmospheric, environmental, biological, prebiotic, or synthetic organic chemistry,85,86 and participation in reactions driven by other factors.87
Environmental, health, and sustainability concerns call for substituting water for organic solvents in organic synthesis. This is decisive in the chemical industry, which has faced tougher regulations designed to protect the public and the environment. Moreover, using large amounts of solvents increases production costs for their acquisition and recycling or proper disposal. On the other hand, nature makes complex chemicals precisely in water, showing that water is a suitable environment for organic synthesis when it is distributed within compartments or interfaces in complex multiphase systems. This stimulated the emergence of biotechnology as a source of chemicals and a fast change in the approaches and perspectives of synthetic chemistry in laboratories and plants.
In this context, interest in “on water” chemical reactivity grew fast. However, achieving fast reaction rates without dissolving the reagents is counter-intuitive because direct contact between the reagent molecules in a non-solvent is much lower than in a mutual solvent. However, the positive experimental results from many authors on various systems are impressive.
These results attracted the attention of many researchers showing the advantages of performing organic reactions in water.
A consequence was the introduction of surfactants as reaction aids by creating micelles that multiply the interfacial area within the system,88,89 producing exciting results. However, it created the need to discard surfactants, a new problem. The authors acknowledge that physicochemical concepts from the bulk are not always applicable at interfaces, as the latter are disordered systems of nanometric thickness displaying sharp configurational fluctuations. The reasons underlying rate acceleration at aqueous interfaces remain unclear, stimulating theoretical work90,91 and the proposal of new effects to explain the catalytic role of the interface beyond the hydrophobic effect: the formation of hydrogen bonds with dangling protons at the interface,89,92 partial solvation,93,94 preferential orientations,95 curvature in nanodroplets,96,97 water surface pH,98 the “reservoir effect”,88 lipophilic ligand design,99,100 and the “nano-to-nano” effect,101 showing that “on water” reactions can follow different rules from those guiding synthetic chemistry thinking, until recently.
The acceleration of chemical reactions within water microdroplets has been reviewed by Wei et al.,93 assuming that partial solvation of the reagents at the interface reduce the reaction critical energy.
Many reactions usually considered non-spontaneous proceed spontaneously in microdroplets,102–110 including biotic or prebiotic reactions.111 Phosphorylation reactions generate fundamental cell components, including building blocks for RNA and DNA, phospholipids for cell walls, and adenosine triphosphate (ATP) are thermodynamically unfavorable in solution. Zare112 found that the yield for D-ribose-1-phosphate was 6% at room temperature. The Gibbs function of this reaction changes by −1.1 kcal mol−1, lower than ΔG for the same reaction but in solution (+5.4 kcal mol−1). The phosphorylation in microdroplets is exothermic (−0.9 kcal mol−1) and ΔS is negligible (0.0007 kcal (mol K)−1). Thus, the spontaneous phosphorylation reaction is enthalpy-driven. Aqueous microdroplets containing D-ribose, phosphoric acid, and uracil produce uridine, attracting the attention of researchers interested in prebiotic synthesis.
Electric effects at the droplet water surfaces were also considered. The first authors calling attention to the participation of the ever-present surface charges were Beattie et al.113 They proposed a mechanism for reactions of emulsified reagents based on the electrification of hydrophobic oil in water by OH− ion adsorption, leaving an aqueous acidic environment where the substrate is protonated. Other effects explored in the literature are surface potential fluctuations,114 spatial distribution of ions,115 and electric fields at microdroplet surfaces.116–118
Richard Zare and collaborators studied many reactions in aerosols.119–124 A striking example is the formation of hydrogen peroxide24,125 from pure water whose formation from water is not expected, considering the standard Gibbs energies of water and H2O2. These results were challenged,126 but their confirmation came from additional experimental data and computational work.40,127,128 The authors proposed a mechanism based on excess OH− ions at the droplet interface losing electrons and forming hydroxyl radicals that dimerize, producing H2O2. However, water oxidation forming the peroxide should also yield a product of water reduction. Nguyen et al.129 proposed that hydrogen is the reduced coproduct of hydrogen peroxide formation and asked for its experimental verification that was recently published,130 analogous to hygroelectricity.131 A different mechanism proposed by Colussi identifies the mechanical energy required for droplet formation as the energy source for hydrogen peroxide formation.132
A general perspective was proposed by Chamberlayne and Zare, considering the electrical double layer (EDL) in water microdroplets in such a way they can be treated as electrochemical cells.133 This is an interesting approach, but it is necessary to keep in mind that EDLs are sensitive to seemingly minor changes in the system composition.
H2O2 also results from contact electrification in the interfaces of poly(tetrafluoroethylene) particles and deionized water/O2 interfaces, particularly under a mechanical force. The authors134 proposed that the process initiates by electron transfer generating reactive free radicals (˙OH and ˙O2−). Following their proposed mechanism, any contact-charged water interfaces are catalysts for H2O2 production from water. This is a new simple possibility to generate H2O2in situ for sanitation and other purposes as in the Fenton aqueous membrane reactor fabricated based on the porous aqueous thin membrane supported by a Fe2O3 modified non-woven.135
Other experimental work confirmed the formation of H2O2 at solid–water interfaces,39 within microfluid channels of a polydimethylsiloxane (PDMS) slab on a flat glass substrate and nine other types of substrates: calcium oxide, zirconium oxide, silicon oxide, aluminum oxide, manganese dioxide, titanium oxide, copper oxide, zinc oxide, and graphene. They measured the difference in zeta potential before and after the contact between water and the solids and observed a positive difference in zeta potential. Moreover, the increasing density of hydroxyl groups on SiO2 substrates enhances the production of H2O2.
Other interesting examples reported recently include the synthesis of gold nanoparticles136 and the spontaneous reduction of Cu2+ and Fe3+, respectively, to Cu+ and Fe2+.105 In both cases, the reducing agent is the electron itself.
The diversity of “On-water” reactions prevents proposing any general mechanism. In every case, the structure of the charged double-layer or interface and the participating ions should be kept in mind. Moreover, there is extensive documentation for free-radical formation on electrified interfaces,137 but it is not always clear if radicals are formed from ions, or vice versa.
Hydrovoltaics and energy harvesting
Electric potential gradients found all over challenge researchers to further devise and explore new techniques to produce electricity from moist air, water, plants,138 and ground, introducing hydrovoltaic (HV) technologies73–76,139–144 that are recent, but already show great potential for energy production, including its storage.145 That may provoke surprise among researchers that were educated within the electroneutrality paradigm, but the recent overlapping but converging findings contribute to the growing acceptance of non-electroneutrality and pervasive electrification as normal features of matter.146Although the literature is full of different names and acronyms for water-driven energy conversion devices, HV harvesters can be categorized into three primary types: hygroelectric generators, which generate electricity through the sorption/desorption of water vapor or liquid;131,147 evaporation-driven generators, which convert the latent heat of vaporization into electricity without the need for additional mechanical work;148,149 and generators that harvest energy due to streaming potentials at liquid water–solid interfaces.150,151 On the other hand, given the similarities and tenuous boundaries between each phenomenon, more than one mechanism can happen concurrently for a given device.
Beyond the water itself, HV technologies also share the initial steps on the solid surface: the adsorption of ions and, for most surfaces, this adsorption is asymmetric. Originally, the hygroelectricity was described for metal surfaces with different Brønsted acid–base character where hydronium ions adsorb on basic sites while hydroxide ions adsorb on acidic sites.32 Later, a steady current output was observed in a hygroelectric device,33 which would necessarily require redox reactions at the solid–liquid interface. Thus, hygroelectric cells are also chemical reactors.131
The first experimental demonstration of simultaneous hydrogen and H2O2 production in a water–solid interfacial reaction was carried out using hygroelectric cells.131 In this case, the authors presented a thermodynamic analysis showing how this reaction, which is non-spontaneous under standard conditions, takes place in an open system showing electric potential gradients.
Thermodynamic analysis
The cell electrodes are separated, but connecting them with a wire allows electron flow, thus delivering an electric current. Water dehydrogenation is a non-spontaneous reaction under standard conditions but proceeds spontaneously in hygroelectric cells that are open, non-electroneutral systems. This is analogous to the hydrogen peroxide formation in charged aqueous aerosol droplets and solid surfaces.Extracting hydrogen from water is now a major research objective that is pursued resorting to electrolysis, photocatalysis, and other approaches to split the water molecule into the elements, or by reducing water with aluminum metal and other reducing substances.
Water-splitting always requires a significant additional energy input because the Gibbs function of this endothermic reaction is positive.
The energy requirements are much lower for another reaction (eqn 2) that produces hydrogen from water:
| 2H2O → H2 + H2O2 | (2) |
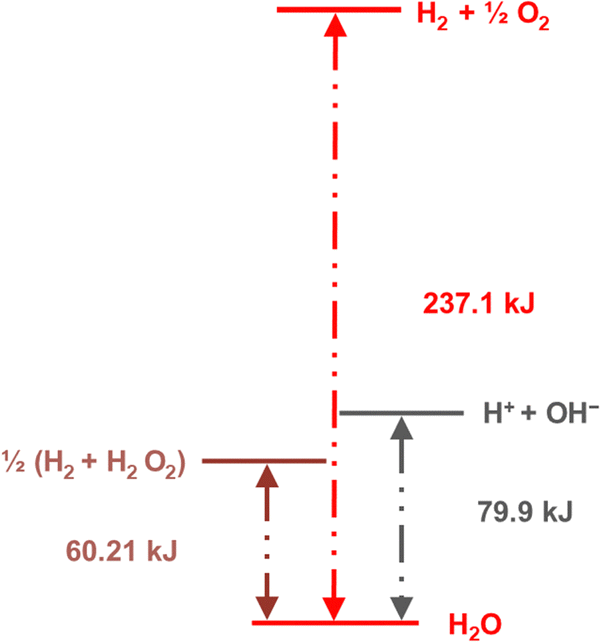 | ||
| Fig. 13 Diagram showing the standard Gibbs energy levels for water, hydrogen peroxide, the ions formed by water self-ionization, and H2 + ½O2. Reprinted with permission from ref. 131. Copyright 2023. American Chemical Society. | ||
Adsorption and partition of water ions on two surfaces showing different Lewis acid–base behavior enhances water ion separation, producing an electric potential difference between the surfaces. The local electrochemical potentials ![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e0.gif) i change according to eqn (1). Thus, the chemical stability of any ion decreases when ziFV > 0. This term is positive in eqn (1) as written for H+, and OH−, increasing the Gibbs function and thus increasing the tendency of H+ and OH− to undergo further reactions.
i change according to eqn (1). Thus, the chemical stability of any ion decreases when ziFV > 0. This term is positive in eqn (1) as written for H+, and OH−, increasing the Gibbs function and thus increasing the tendency of H+ and OH− to undergo further reactions.
Assuming that the potential difference between the solid surfaces is 1 volt, the Gibbs function for water ionization increases, exceeding water dehydrogenation (H2 + H2O2), by 96.5 kJ per mole of water facilitating hydrogen formation (3):
| 4H+ + 4OH− → H2 + H2O2 | (3) |
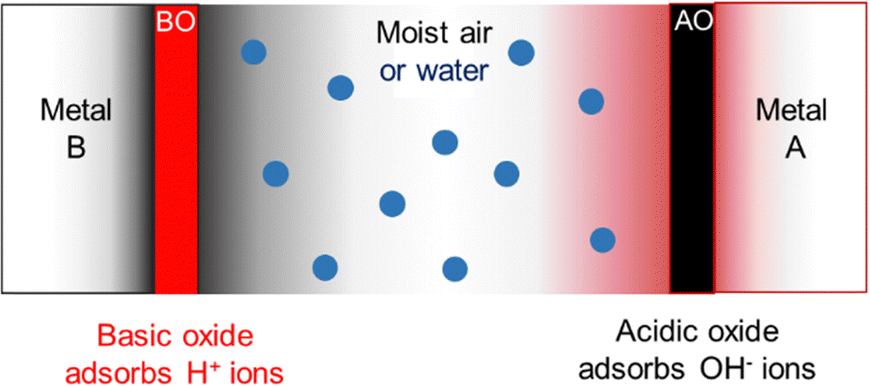 | ||
| Fig. 14 Electrified compartments in a hygroelectric cell made from parallel plates of conductive materials, metals A and B. The oxide AO adsorbs OH− ions becoming negative and repelling negative charge in the supporting metal, while BO becomes positive by adsorbing H+ ions. Excess charge in the oxides produces a potential difference between metals A and B. Adapted with permission from ref. 131. Copyright 2023. American Chemical Society. | ||
Thus, ion partition produces a positive compartment filled with H+ ions and a negative compartment filled with OH− ions represented by eqn (4) and (5). Then, the electrochemical potential of both ions increases, compared to an electroneutral system:
| BS(s) + H+ → BS(H+) | (4) |
| AS(s) + OH− → AS(OH−) | (5) |
| 2AS(OH−) → 2AS + H2O2 + 2e− | (6) |
| 2BS(H+) + 2e− → 2BS + H2 | (7) |
| 2H2O + 2AS + 2BS → 2AS(OH−) + 2BS(H+) → 2AS + 2BS + H2O2 + H2 | (8) |
| 2H2O → H2O2 + H2 | (9) |
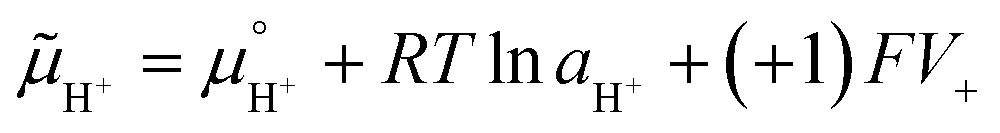 | (10) |
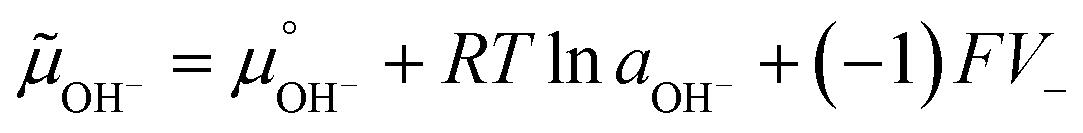 | (11) |
| ΔGelect. = (+1)FV+ + (−1)FV− = FΔV | (12) |
| ΔV/V | F × ΔV/J | ΔrG/J |
|---|---|---|
| 0 | 0.0 | 1.20 × 105 |
| 1 | 9.65 × 104 | 2.35 × 104 |
| 1.2 | 1.16 × 105 | 4.22 × 103 |
| 1.3 | 1.25 × 105 | −5.43 × 103 |
| 1.4 | 1.35 × 105 | −1.51 × 104 |
Shortly, H+ and OH− ion partition in separate solid surfaces or other compartments changes a non-spontaneous reaction into one that easily takes place, in an open system. Feeding H2O to the electrodes while removing the reaction products, H2O2 + H2, produces a steady current output between the electrodes.
A stepwise description of the formation of hydrogen peroxide in the negative electrode of the hygroelectric cell is shown in Fig. 15.
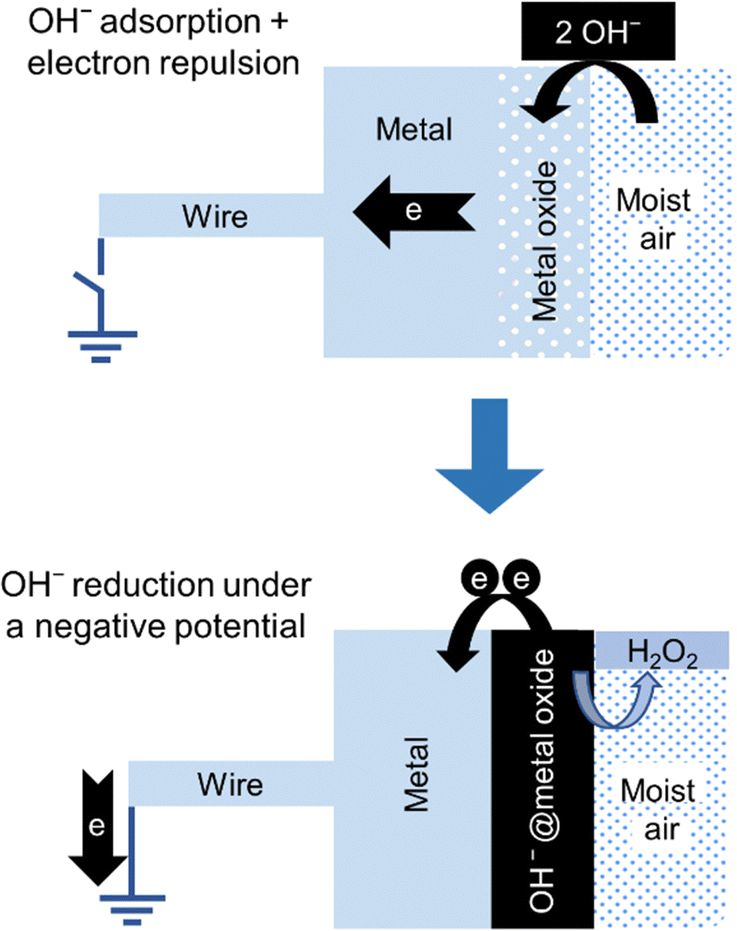 | ||
| Fig. 15 Stepwise description of the two steps in the negative electrode of a hygroelectric cell: (top) adsorption: an isolated electroneutral acidic oxide surface in contact with moisture adsorbs OH− ions acquiring negative charge. (bottom) Redox reaction: electron transfer from OH− ions to the electrode produces hydrogen peroxide. Adapted with permission from ref. 131. Copyright 2023. American Chemical Society. | ||
Hydrogen and hydrogen peroxide formation from water under electroneutral conditions is not predicted using standard thermodynamic data and assuming electroneutrality. But the potential differences within the cell allow us to understand this unexpected change in water chemical behavior. Thus, a HG cell is an example of the effect of intrinsic non-electroneutrality on the chemical behavior of a multiphase system producing a surprising chemical behavior.
The present analysis shows the pertinence of the question raised by McCarthy and Whitesides5 calling for an effort to examine the effects of non-zero potential on chemical thermodynamics. It also suggests an approach to understand “on-water” reactions and strategies “to utilize the pervasively present electrostatic charges in nature in an eco-friendly way, in on any multiphase system”.
For instance, the reductive ability of OH− ions producing gold nanoparticles demonstrated by Zare122 suggests that positive droplets carrying excess H+ ions are suitable sites for oxidation reactions. Positive water droplets appear under different conditions. Moreover, the surface charge density of positive or negative water is easily controlled and we can now envision droplets and other interfacial systems as fine-tuned oxidant or reducing agents, depending on the electric potential applied during their formation.
A question arises: can other anions also became reductive in droplet and other hydrophobic-water interfaces, analogous to hydroxide ions? Bicarbonate adsorption in hydrophobic surfaces competing with hydroxide ions has already been shown, in the 5–10 pH region where bicarbonate153 ions predominate, but water ions displace it outside this range, and its reduction has not yet been demonstrated. This reaction could find great application in decarbonization. The Zare group recently showed a related reaction that is the formation of urea from droplets in the presence of N2 and CO2.154
Mechanochemical reactions and catalysis: the interplay between electricity, friction, wear, and water
The simplest case of mechanical action between two solids is their mutual contact which often produces detectable electrification. Many authors attempted to explain it, considering electron transfer determined by the difference in their respective work functions.155 These explanations apply to metals but did not resist experimental verification when applied to adhesion.5,156,157 However, electrostatic adhesion mediated by ions is now well established and successfully used in self-assembled materials, polymer composites, and blends derived from latex5,158 and in layer-by-layer fabrication. Williams15 discussed three mechanisms for contact electrification: electron transfer, ion transfer, and mass transfer, showing the assumptions and evidence for each mechanism and its implications. However, electron wavefunction overlap between states of coupled potential wells is considered in the case of triboelectric nanogenerators.159Chemical reactions triggered by friction and milling have been known since the late 1800s,160,161 but they did not receive great attention during the past century. This is intriguing because it was then known that mechanochemical reactions often yield different products from the same reactions but under the action of heat.162
Moreover, in the last few decades of the past century, the emerging fields of materials chemistry, molecular biology, and nanotechnology often used milling, renewing the interest in mechanochemistry.163–169
Mechanical stress and sonication techniques, applying high energy densities on molecules and particles, are enough to achieve extensive molecular breakdown. A well-known case is the reduction in viscosity observed when passing viscous native DNA solutions through narrow bores using syringes with needles, due to breaking of DNA into smaller pieces.
Electrification during thermoplastic processing by extrusion, injection, and other large-scale processing techniques is familiar to practitioners. Powder electrification during milling, sieving, pneumatic transportation, and other standard methods is easily observed, provoking deep impressions in plant or shop visitors and the risk of electric discharges. A puzzling question is why a scientific topic posing challenging and deeply related to widely used industrial processes was neglected for many decades. A tentative answer is the difficulty of an experimental and theoretical study of mechanochemical reactions, compared to reactions in the same systems but constrained only by pressure and temperature.170,171
Without mechanical agents, Boltzmann energy distribution among molecular entities applies. Little is known about the distribution of mechanical energy applied to a solid, but we must consider many questions: what is the spatial distribution of any mechanical agent? How do the applied forces affect the microstructures in the solid? Which are the local excess energies, and how are they spatially distributed? The energy distribution introduced by a mechanical agent in a condensed phase depends on geometric, microstructural, and energy transfer details that produce enormous complexity. Under these conditions, quantum mechanics is not helpful. Computational methods can consider only a few participating molecular-size entities, and are insufficient to handle many problems. Additional complications are raised by ambient water and air humidity. Knowledge of their role in surface electrification evolved parallel to the growth of “on-water” reactions, and the only attempt to connect these two topics was a work conducted by Beattie et al., concluding that on-water catalysis is facilitated by the strong adsorption of the hydroxide ion by-product at the oil–water interface.113
Heinicke introduced the word “triboplasma” to designate the short-lived molecular arrays undergoing chemical transformation triggered by mechanical action.83 This name is appropriate and reminds us of the systems that are shortly exposed to high energy densities, like flames and any materials exposed to gamma or X-rays. Flame reaction products include several high-energy species not observed in simpler thermal reactions. Free radicals abound, and the fire electrification is evidenced by flame distortion under an electric field. Specific products’ appearance and relative amounts depend on seemingly minor details. This complexity creates unusual demands for experimental research, but many new experimental tools with high spatial, temporal, and chemical speciation resolution now bring exciting results.
Electricity and friction. Rubbing two materials imparts electric charges to both. The dominating idea is that one object acquires positive charges while the other becomes negative. Contrary to this idea, the coexistence of opposite charges on the same surface of an insulator was revealed by Scanning Kelvin Probe Microscopy (SKPM),172 (Fig. 16).
 | ||
| Fig. 16 KPM images of some polymers. Used with permission of Institute of Physics (Great Britain), from ref. 173, permission conveyed through Copyright Clearance Center, Inc. | ||
The results were not understood initially, but they were taken seriously following many calibration and control experiments. The accuracy of the results was verified by comparing the Kelvin micrographs of latex particles with elemental maps acquired in an analytical transmission electron microscope fitted with an energy filter (EF-TEM). However, it was then unclear why the surfaces of any insulators examined appeared as electric charge mosaics.
These have a non-integer fractal dimension, meaning that the adjacent charge domains should show scale symmetry. Examining macroscopic polymer surfaces of lab and office materials confirmed this hypothesis: their surfaces are usually charged but in non-uniform patterns. The surfaces are thus a maze of electric potential gradients that are removed or largely decreased by rinsing with ethanol. This allowed the preparation of charged surfaces by rubbing two polymer sheets and mapping their surfaces with a macroscopic scanning Kelvin electrode system that produced images shown in Fig. 17a. Tribocharged polytetrafluoroethylene (PTFE) rubbed with a polyethylene (PE) foam disc displays macroscopically patterned positive and negative domains,174 indicating fractal geometry. This result contradicts the idea assumed from the triboelectric series, whereupon one material charges uniformly positively and the other negatively. The positive and negative species in PE and PTFE were identified as hydrocarbocations and fluorocarbanions (Fig. 17b), formed through mechanochemical chain rupture and subsequent electron transfer (Fig. 17c). Self-assembly of polymer ions creates the observed patterns. These findings highlighted the importance of considering complex chemical events and well-established physicochemical concepts in understanding tribocharging. Moreover, due to the fractal nature of electrostatic potentials on insulators,175 such mosaics of surface charge have scale symmetry and were also identified at the nanometric scale.176
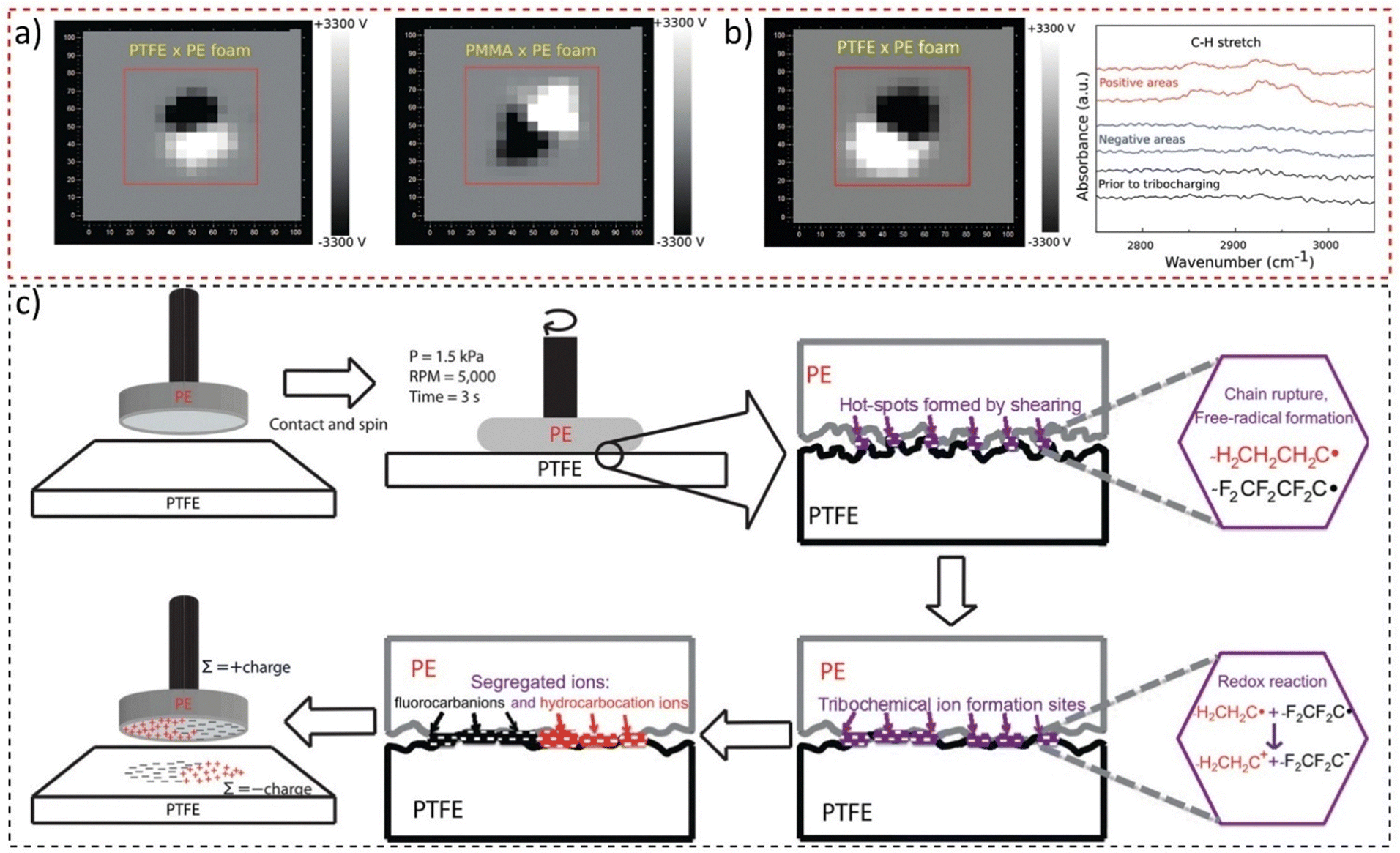 | ||
| Fig. 17 (a) Macroscopic potential patterns on PTFE and PMMA rubbed with a PE foam disk. (b) Potential maps of PTFE charged by shearing with a PE foam disk, showing the IR reflectance spectra of positive and negative areas on the PTFE sheet. (c) The mechanism proposed for contact triboelectrification of insulating polymers highlights that redox reactions following material transfer and ion segregation are responsible for the large charged domains displayed on PTFE. Adapted with permission from ref. 174. Copyright 2023 American Chemical Society. | ||
For most cases, the range where the van der Waals forces act is typically on the order of a few nanometers. On the other hand, the range of Coulombic forces is much larger (virtually acting over any distance) and capable of moving even macroscopic objects, as seen when children move small fragments of a paper with a plastic ruler. Friction between dielectric surfaces produces stable electric charges that contribute to surface interactions. Friction coefficients were measured on tribocharged PTFE, and the results showed that it significantly increases friction coefficients at both macro- and nanoscales.177 Thus, tribocharging may dominate friction coefficients in PTFE and other insulating polymers.
Again, the effect of the fractal nature of surface charges on the friction coefficient was verified using specific setups for friction tests such as the ball-on-disc geometry (Fig. 18). Friction and triboelectrification of materials were strongly correlated during sliding contacts.178 Tribocurrent generated at the interface varies intermittently between positive and negative values (Fig. 18a), meaning that the flow of charges is constantly altered, but only during friction force fluctuations.
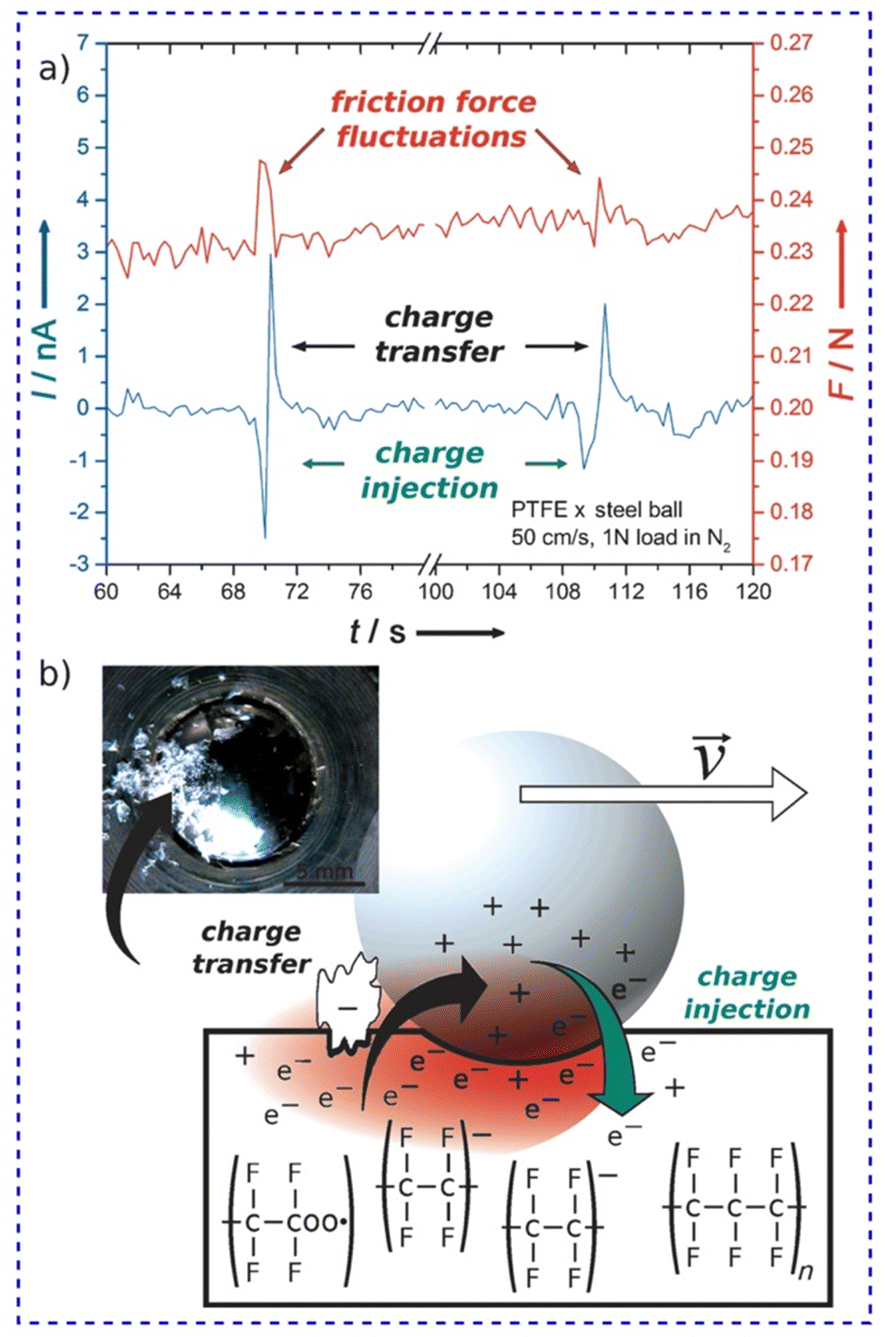 | ||
| Fig. 18 (a) Friction force fluctuations and tribocurrent at the metal–PTFE interface and (b) the suggested mechanism. Electron injection in PTFE, simultaneous to the mechanochemical homolytic rupture of PTFE bonds produces excess negative charge on PTFE. The attractive force between two surfaces with opposite charges increases the friction force. Reprinted from ref. 178, Copyright (2023), with permission from John Wiley and Sons. | ||
AFM adhesion maps reveal that contact between metal and PTFE interfaces increases the pull-off force from 10 to 150 nN, indicating resilient electrostatic adhesion. These findings suggested a common origin for friction and triboelectrification, likely associated with strong electrostatic interactions at the interface and thus also verifying the fractal nature of electrostatic charges.
Although its complex nature is often debated, mutual friction resulting in triboelectrification is a very familiar topic, even for non-scientific people. However, investigating surface charges on insulators requires special setups like the Faraday cup or Kelvin probes at various size scales.
Beyond bond-braking and the formation of high-energy species, mechanical action also provokes catalytic action mediated by excess charges. For instance, hydrogen is produced from water containing ZnO microfibers and BaTiO3 micro-dendrites. Sonicating a piezoelectric immersed in water produces excess charges that provoke redox reactions.179 Following this work, piezocatalysis was utilized for a number of other applications, including oxygen activation and water decontamination.180–182 These results are cited by Fan et al.,183 proposing the following tribocatalysis experiment: ball-milling a mixture of two powders could impart a positive charge to a group of particles and negative charge to the other. Excess charge allows the former to act as reducing agents while the latter may perform as oxidizers. Piezo- and tribocatalyses are two types of mechano-electrocatalysis, holding promise for relevant applications. Although their complex nature is often a matter of debate, mutual friction resulting in triboelectrification is a very familiar topic, even for non-scientific people.
Investigating surface charges on insulators requires special setups like the Faraday cup or Kelvin probes. During a routine laboratory experiment with a Kelvin probe turned on, we noticed very high potentials when playing with a rubber band. This intriguing accidental result motivated further experiments to properly understand this phenomenon.36
The experiment shown in Fig. 19a using a setup schematically shown in Fig. 19b demonstrated the conversion of mechanical energy to electricity by stretching rubber tubing periodically.36 The rubber surface shows reversible electrostatic potential variations, but amplitude of potential oscillations decreases with humidity (Fig. 19c).
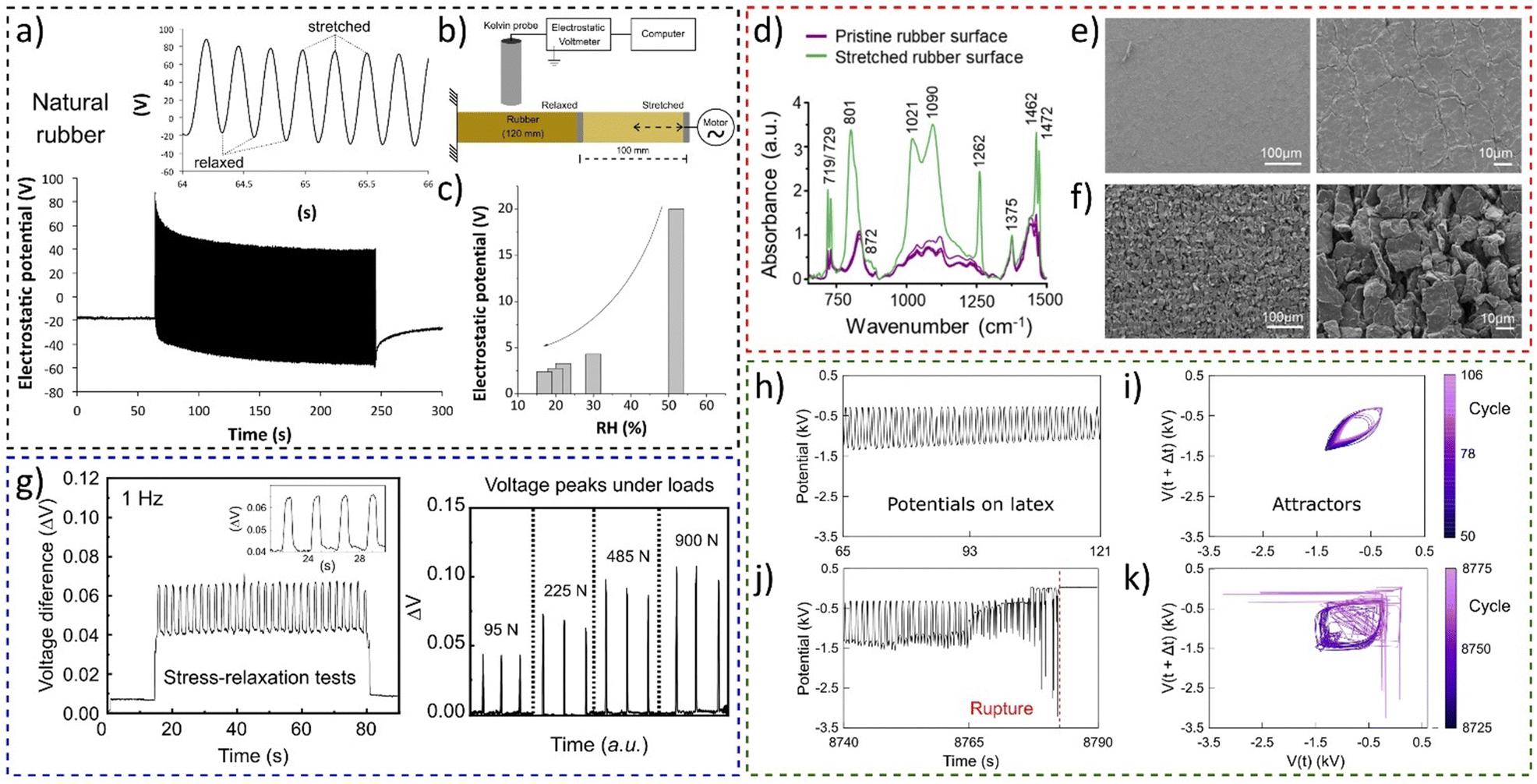 | ||
| Fig. 19 Electromechanical coupling in rubbers. (a) Electrostatic potential of natural under stretching−relaxation cycles using (b) the Kelvin electrode. (c) Experiments showed that the amplitude of electrostatic potential oscillations is dependent on the RH. (d) Normalized IR reflectance spectra of rubber periodically stretched, until rupture. SEM micrographs under two different magnifications for (e) the pristine rubber surface and (f) the surface of a rubber periodically stretched between 100 and 200% strain. (g) Periodic stress-relaxation tests on a rubber-based device showing the (a) open-circuit voltage difference and its electromechanical coupling dependence under different loads. (h) and (j) Sections of the recorded time series, of the electrostatic potential of the periodically stretched natural latex and (i) and (k) the respective attractors. (a)–(c) Adapted with permission from refence 36, Copyright 2023 American Chemical Society, (d)–(f) adapted from ref. 184, Copyright (2023), with permission from Elsevier, (g) adapted from ref. 187, with the permission of AIP Publishing, (h)–(k) adapted from ref. 188 with permission from the Royal Society of Chemistry. | ||
Rubber charging patterns are peculiar and cannot be understood only considering the framework of surface piezoelectricity and flexoelectricity. Thus, rubber potential arises from two main factors: hygroelectricity and mechanochemical reactions observed through spectroscopy and microscopy experiments (Fig. 19d–f).184
The electromechanical coupling of elastomers was found to be a universal phenomenon of rubber and was used to construct a low-cost transductor device using vulcanized rubber and a exfoliated and Kraft paper coated with reassembled graphite (ERG).185,186 The force transducer comprises a flexible graphite-based electrode between two vulcanized rubber parts, showing a linear relationship between the strain gradient and the electric response (Fig. 19g),187 a promising result for the use of elastomers in soft electronics, wearable healthcare devices, and various flexoelectric applications. Electromechanical coupling in elastomers revealed periodic electrification in sync with rubber stretching and regular patterns until fatigue failure, where waveforms become complex (Fig. 19h–k).
Attractors from the electromechanical transduction in rubber predict failure one minute ahead of breakdown. Thus, electromechanical coupling in rubber offers real-time prediction of fatigue failure and energy harvesting potential.188
Harvesting energy from triboelectricity has been achieved from different ways, but recently triboelectric nanogenerators (TENGs) have been emerged showing potential as an auxiliary electric power generation system.189 Despite their many uses and different designs, conventional TENGs described in the literature often use expensive materials and are fabricated following costly multistep processes. Solving these problems may allow TENGs to contribute to distributed power production.
Thus, we have used protocols for polyethylene oxidation190 and charge monitoring on PE surfaces191 developed in our laboratory to construct an efficient and low-cost triboelectric nanogenerator, converting mechanical energy into electrical energy using aseptic carton packages.
Material oxidation and assembly are shown in Fig. 20a, while the electrical performance during contact is displayed in Fig. 20b. The device achieves potentials up to 200 V under mechanical loads and can charge a 10 μF capacitor to 3.5 V in less than 10 minutes.192 Under pressure-relaxation cycles, it develops a continuous open-circuit voltage of roughly 80 V with an output peak current of ∼400 nA at 1.8 Hz. This is comparable to those of nanoengineered devices but at a fraction of the cost presenting a reliable and environmentally friendly solution for low-cost energy harvesting and the destination of food packaging.
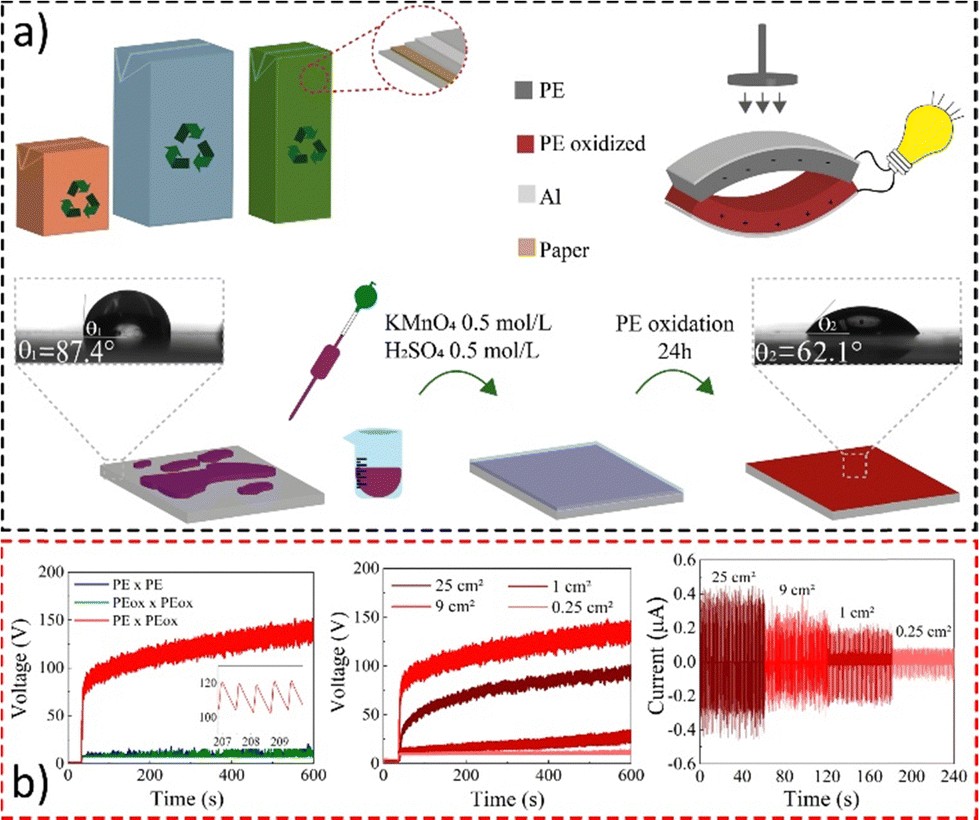 | ||
| Fig. 20 (a) Fabrication of a simple triboelectric energy harvesting device using a laminated food packaging film. LDPE surface oxidation with KMnO4 solution lowered the water contact angle. (b) Open-circuit voltage using different dielectric-to-dielectric pairs and the (d) open-circuit voltage and (e) short-circuit current on samples with different sizes. Adapted from ref. 192, Copyright (2023), with permission from Elsevier. | ||
Hygroelectric devices can also produce electricity through ionization, such as in the directional movement of spontaneously released H+ ions of graphene oxide in the presence of water vapor193 or due to the self-ionization of water molecules inside protein films of gelatine molecules. This ionization can be transferred through a conducting path formed by the hydrogen-bonded water molecules on the gelatine macromolecules.194
Electricity generation in evaporation-driven devices is attributed to continuous capillary water flow that induces convective ion flux and polarization during evaporation, driving charge carrier flows in contacting electrodes (in some cases referred to as the “ionovoltaic effect”), and enabling the generation of a continuous electrical current.195 Most evaporation-driven devices demand humidities below 30%. However, employing a thin layer chromatography system coated with α-Al2O3 allowed for consistent capillary-driven water flow, resulting in a continuous voltage of ∼0.33 V and a short-circuit current of ∼0.85 μA over a wide range of humidity (10–90%).196 Building on these concepts, we also propose a notion that a portion of atmospheric electricity may originate from evapotranspiration, ultimately associated with the Maxwell–Wagner–Sillars effect.12
While flow electrification in insulating liquids has been reported over the years, generators that harvest energy from streaming potentials at solid–liquid interfaces are a more recent development.197 In this case, the production of electricity comes from the movement of fluid across a solid surface, which causes ions to migrate, creating a charge difference and an electric field.198 This electricity generation, driven by electrokinetic effects, can harness energy wherever a water flow comes into contact with a charged solid, owing to the pressure-driven transport of counterions in the electrical double layer199 and has been used for the development of different devices that harvest energy from water in the form of raindrops.200–203
Electrified liquid water–solid interfaces204–206 including biosurfaces207 deliver electric current, in many ways.208,209 Moving a 0.6 mL aqueous (NaCl 0.6 M) single droplet on a graphene/polytetrafluorethylene surface produces an output power of 1.9 μW210 while raindrops211 on fluorinated super–hydrophobic greenhouse-modified films give a maximum output power density of 38 mW m−2 through a 16 MΩ load resistance.212
Many energy devices that use atmospheric electricity and water to deliver electric power have received other designations, beyond the HV technologies described before. The most frequent is “moisture enabled generator (MEG)”.213–216 However, the literature also presents “moist-electric generator”,217 “water-enabled electricity generation”,218 “wood-derived moist-induced electricity generator (WMEG)”,219,220 PMEG,221 and MIEG.215 Water interfaces stimulate photovoltaic effects in the vicinity of silicon surfaces,222 and in some reports, moisture associated with light is called LMEG.223,224 Other cases include adsorption–desorption (MADG),225 as in an agricultural waste-derived moisture absorber (AWH).226 The perspective of application in wearable electronics led to the development of a yarn-shaped moisture-induced electric generator (YMEG).227
A frequent explanation for its operation is water ion adsorption and diffusion accompanied by electric induction, but not considering the participation of redox reactions essential for converting ionic currents to electronic currents and vice versa. In fact, the potentials at the solid–liquid interfaces can have contributions of both hydrovoltaic effects228 and redox reactions229 and using both phenomena can substantially boost the effective power of hydrovoltaic devices.
Combining the electricity collected from water and moisture to materials showing shape memory or deformation triggered by water is now used for producing sensor-actuators that may find many applications.230
Positive feedback in electrification and chemical events
Ambient electrification often goes unnoticed by humans but produces catastrophic consequences, like lightning, powder explosions, and other accidents caused by electrostatic discharges. Catastrophic events are sudden and often extreme changes in a system leading to disruptions, collapses, or rapid transformations, out of control.They often result from the accumulation of positive feedback loops that push a system beyond a critical threshold, triggering an abrupt and often irreversible change, as in autocatalysis.231,232
The concepts of catastrophic behavior, positive feedback, and autocatalysis are often intertwined in various fields of science beyond chemistry, such as biology, ecology, and systems theory.
The electrostatic phenomena discussed in the previous sections provide interesting examples of positive feedback explained by the mutual interactions between electric fields and ions in a system. For instance, the enhancement of ice formation by electric fields is a process in which the disturbance caused by a field on water vapor leads to an amplified or intensified response, that is, the electrified needle-shaped ice that reinforces the initial field, driving the system further away from its equilibrium state. This can lead to exponential growth or destabilization of the system by electrostatic discharge or Coulomb explosion that can produce serious environmental, personal, and property losses.
Thus, the mutual interaction between electricity and matter may trigger processes akin to autocatalysis, where a substance catalyses its own production or transformation. Autocatalytic processes can lead to exponential growth, as the rate of production increases over time driven by positive feedback loops that cause fast temperature growth, often leading to explosion and fire.
The chemical mechanisms of matter electrification offer plausible explanations for catastrophic events, based on positive feedback analogous to autocatalysis. Lack of attention to these mechanisms impaired progress in understanding electrostatic phenomena but acknowledging the effect of water on the dynamics of matter electrification is now producing solid knowledge rooted in experimental results and consistent with all accepted concepts of matter structure and properties, at the supramolecular scale and upwards.
Dissemination of these ideas should stimulate the investigation of spontaneous fire, fire propagation, and powder explosions of other challenging phenomena, improving our ability to avoid major losses in natural or human environments.
Disaster prevention: electrification and explosions
The multitude of electrification mechanisms should be known to any person concerned with the protection of humans, properties, and the environment. The occurrence of challenging powder explosions arises from the accumulation of charges in high surface area materials such as wheat flour, sugar, polyethylene powder, and dust. This phenomenon is observed in silos, logistic operations, and due to unattended accumulation in air-conditioning duct traps. Why do materials handled in huge amounts in many places suddenly become powerful explosives? How can powder explosions be avoided?Accidental electrostatic charge accumulation due to friction should be expected considering that friction is a powerful charging operation that also produces heat. Friction was used as the standard technology for producing fire, for millennia. Industries that handle combustible powders, such as those in the food, chemical, or manufacturing sectors, often implement safety measures to control and mitigate the risks of dust explosions. This may include measures to prevent the accumulation of dust, proper ventilation systems, and the use of anti-static equipment to minimize electrostatic charging.
Notwithstanding all the preventive measures, there were an average of 31.8 dust explosions per year, 29 injuries and 2.6 fatalities over the 2016–2020 period, in the US. The global figure for the number of explosions is 127, in the same period.233 Lightning causes significant numbers of deaths234 and ca. 1 US Billion (2022) property losses annually, in the US only.235 Thunderstorms, hail and other meteorological events are always associated with atmospheric electricity, that is not often monitored.
This review showed the formation of oxidant hydrogen peroxide in aerosols and water-hydrophobic surfaces and hydrogen formation from electrified surfaces was revealed in two papers reported this year. These are highly reactive chemicals that can provoke fire and other undesirable events.
Perspectives and new hypotheses
The pervasive but often ignored electrification of matter at the macroscopic scale changes many important properties of chemical substances. There is now ample evidence of the effects of electrification on chemical reactivity, crystal habits, metastability in phase transition, surface tension, and friction. Many important discoveries made during the past two decades will probably be extended, and new effects will be discovered, attracting the attention of students, researchers, engineers, and the public to their ample horizons.Unfortunately, this subject does not yet appear in STEM curricula except for eventual mentions. Students do not receive recent but proven information showing the decisive role of chemical events in matter electrification. Instead, a phrase representative of the information transmitted during elementary and college classes or by textbooks and the internet is: “The concept of work function or electron affinity explains the specific cause of contact charging”. The role of water ions that Langmuir already considered236 many decades ago is still largely neglected, omitting abundant experimental evidence that has been appearing regularly in the literature.71,237–239
Thus, it is not surprising that researchers and professionals in many areas do not pay attention to the effects of macroscopic electrification on the systems and phenomena they are concerned with. Notable cases are fracture mechanics, animal and vegetal physiology, and health, where we expect interesting examples of important effects of electrification.
However, the lacunes of fundamental knowledge did not prevent the appearance of technologies like electrostatic separation, electrostatic coating, wetting, spraying, electrospinning, electrocrystallization, and others used in industrial processes. In some cases, these were developed empirically or based on incorrect mechanisms and theories but producing significant practical results.
Recent recognition of electric potential gradients in any real matter, the chemical events participating in matter electrification, and the recognition of water's reactivity under these conditions may bring new views and thinking to many scientific problems and research areas. New technologies are emerging, like the decontamination of the atmosphere by water aerosols containing H2O2 and power generation by hygroelectricity and the myriad moisture-enabled technologies.
The exciting new chemistry that is being discovered in “on-water” reactions and their applications appear at a critical time for chemical synthesis, when the pressure to abandon classical protocols increases all the time. New “on-water” technologies promise chemical production without producing rejects whose proper discard is too expensive. This may increase the competitiveness of chemical methods in face of biotech processes, eliminating rejection by the public and politicians.
The current “on-water” reactions are based on the spontaneous accumulation of hydroxide ions at water interfaces with the atmosphere and other hydrophobic media. The properties of the intervening chemicals determine the interfacial excess charge and potential and are hardly predicted, in a complex multicomponent system. For this reason, current results provide only a narrow window of possibilities. However, the electric potentials at the interfaces may be controlled over a broad range of positive and negative values, using blocked electrodes and bicarbonate or other anions, widening the scope and perspectives of “on-water” reactions.
Many “on-water” reactions now derive from the compartmented formation of oxidants like OH radicals and H2O2. However, aerosols carrying positive charge deliver the reducing H and H2, and they may thus trigger many other reactions. While the interface delivers oxidizing and reducing agents, the droplet interior becomes acidic or basic, acquiring the corresponding catalytic properties. These new possibilities open a wide frontier for chemical synthesis and for accelerating the degradation of undesirable chemicals.
For instance, the current issues of environmental degradation by improperly discarded plastics and microplastics raise a question. Many natural resins and monomers like isoprene240 undergo photochemical polymerization in the atmosphere. Other natural polymers, like the keratin of human hair, animal fur, and feathers, are extraordinarily resistant to degradation. These polymers have been produced on the Earth's surface for millions of years, in large amounts, but leaving no signs of accumulation, except in fossils like amber, coal, or petrol that only exist in special environments. Thus, there are powerful degradation mechanisms acting on natural hydrophobic polymers. Their disappearance is usually assigned to oxidation by atmospheric oxygen, but we can now consider also the hydrogen peroxide formed “on water.” Nearly one century ago, Langmuir reported on “Surface Electrification Due to the Recession of Aqueous Solutions from Hydrophobic Surfaces”. Today, we can speculate on “Surface electrification and the oxidizing power due to the recession of aqueous solutions from hydrophobic surfaces”. We find water receding every few minutes on beach shores worldwide. And beach shores are notably exempt of organic matter, in most places.
The authors hope that learning about the emerging new Chemistry of electrified interfaces will allow the development of effective tools for disaster prevention and climate engineering, contributing to face challenging current global problems.
Conclusions
Surprising experimental results published in the past twenty years in different areas of chemical research are explained by acknowledging two facts: water electrifies matter, and any real material system is a charge mosaic.Water electrifies by spontaneously self-charging and transferring charge to other materials, a unique feature derived from its amphoteric character. Water releases H+ and OH− ions on demand thus showing high electric polarization. Ion transfer from water to neighboring surfaces depends on their acid–base character: Lewis acid surfaces acquire negative charge from water and basic surfaces acquire positive charge.
Water abundance on the Earth allows its contact with most real materials. Water mixing, adsorption and adsorption spread its ability to produce self-electrification events, transforming matter into a mosaic of electrified interfaces. Charge mosaics change form and local charge density following the Maxwell–Wagner–Sillars effect.
Self-electrification creates niches where the electrochemical potentials of the charging ions are higher than those usually expected for electroneutral conditions. The net results are higher ion reactivity and driving force for mass transfer. This translates into chemical reactions that are never observed in neutral bulk materials. A striking example is a hygroelectric cell that delivers electricity, hydrogen, and hydrogen peroxide, with the sole inputs of water and ambient thermal energy.
Another powerful result of water electrification is lowering its surface tension down to negative values evidenced by Coulombic explosions. This lowers the energy barrier to nucleation, eliminating metastability and accelerating phase change processes. Electrification also modifies the habits of crystals, producing unusual needles and dendrites in ice and other crystals.
The recent learning on matter electrification has already penetrated many research areas, but the electroneutrality paradigm still prevails in many others that could largely benefit from abandoning it. Broader dissemination among faculties, students, professionals, and the public will probably contribute to solving long-standing problems and to the emergence of new applications.
New technologies created in this context fit within the desire for sustainability, in every respect. The important process input is water as found in the atmospheric humidity, oceans and other water bodies, soil, residues from food production, grey waters, and other widespread, low-cost sources. All the processes take place in the environment, under ambient temperature and pressure conditions, fully compatible with human life. The products of the various processes already devised are valuable chemicals like hydrogen peroxide and hydrogen and urea, and electricity, with no undesirable by-products. Challenging or energy-demanding reactions are easily carried out under accessible conditions, including impressive abiotic syntheses of biochemicals.
Eliminating metastability is key for avoiding destructive explosions and fires, and may contribute safe technologies for climate problem mitigation.
The recent knowledge of spontaneous electrification, its theoretical support, and its consequences, together with the absence of foreseeable risks, opens promising paths for research in many areas of science and technology, with potential for positive economic and social impacts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the INCT program of the Brazilian agencies MCTIC/CNPq (465452/2014-0), CAPES and FAPESP (2014/50906-9), and by FAPESP/FINEP/PIPE/PAPPE project (2022/01668-4).Notes and references
- P. Iversen and D. J. Lacks, J. Electrostat., 2012, 70, 309–311 CrossRef.
- A. K. T. Assis, The Experimental and Historical Foundations of Electricity, C. Roy Keys Inc., Montreal, 2010 Search PubMed.
- NASA, https://www.nasa.gov/exploration-research-and-technology/electrostatics-and-surface-physics-laboratory/, (accessed November 2023).
- Standard chemical potential' in IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry, 2006, Online version 3.0.1, 3rd edn, 2019 DOI:10.1351/goldbook.S05908.
- L. S. McCarty and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 2188–2207 CrossRef CAS PubMed.
- F. Galembeck and T. A. L. Burgo, Chemical Electrostatics, Springer International Publishing AG, Cham, 2017 Search PubMed.
- J. B. Dellinger, Nature, 1938, 142, 965 CrossRef.
- C. Haldoupis, M. Rycroft, E. Williams and C. Price, J. Atmos. Sol.-Terr. Phys., 2017, 164, 127–131 CrossRef.
- M. J. Rycroft, S. Israelsson and C. Price, J. Atmos. Sol.-Terr. Phys., 2000, 62, 1563–1576 CrossRef CAS.
- C. Li, G. Chen, X. Qiu, Q. Lou and X. Gao, AIP Adv., 2021, 11, 065227 CrossRef CAS.
- R. Gerhard-Mulhaupt, Electrets, Springer Verlag, New York, 3rd edn, 1999, vol. II Search PubMed.
- K. S. Moreira, D. Lermen, Y. A. S. da Campo, L. O. Ferreira and T. A. L. Burgo, Adv. Mater. Interfaces, 2020, 7, 2000884 CrossRef.
- V. Artemov, L. Frank, R. Doronin, S. P. Staerk, A. Schlaich, A. Andreev, T. Leisner, A. Radenovic and A. Kiselev, J. Phys. Chem. Lett., 2023, 14, 4796–4802 CrossRef CAS PubMed.
- E. Young, Super Senses: The Science of Your 32 Senses and How to Use Them, J. Murray, London, 2021 Search PubMed.
- M. W. Williams, Am. Sci., 2012, 100, 316–323 CrossRef.
- H. T. A. Awan and M. Khalid, Mater. Today: Proc., 2023, 73, 361–365 Search PubMed.
- A. F. Diaz and R. M. Felix-Navarro, J. Electrostat., 2004, 62, 277–290 CrossRef CAS.
- T. A. L. Burgo, F. Galembeck and G. H. Pollack, J. Electrostat., 2016, 80, 30–33 CrossRef.
- T. W. Healy and D. W. Fuerstenau, J. Colloid Interface Sci., 2007, 309, 183–188 CrossRef CAS PubMed.
- A. Gray-Weale and J. K. Beattie, Phys. Chem. Chem. Phys., 2009, 11, 10994–11005 RSC.
- Z. Zhang, X. Li, J. Yin, Y. Xu, W. Fei, M. Xue, Q. Wang, J. Zhou and W. Guo, Nat. Nanotechnol., 2018, 13, 1109–1119 CrossRef CAS PubMed.
- L. Laakso, A. Hirsikko, T. Grönholm, M. Kulmala, A. Luts and T.-E. Parts, Atmos. Chem. Phys., 2007, 7, 2271–2275 CrossRef CAS.
- T. A. L. Burgo and F. Galembeck, Colloids Interf. Sci. Commun., 2015, 7, 7–11 CrossRef CAS.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed.
- H. G. Armstrong, London Edinburgh Philos. Mag. J. Sci., 1840, 17, 370–374 CrossRef.
- R. Mukherjee, S. F. Ahmadi, H. Zhang, R. Qiap and J. B. Boreyko, ACS Nano, 2021, 15, 4669–4677 CrossRef CAS PubMed.
- P. K. Jha, M. Sadot, S. A. Vino, V. Jury, S. Curet-Ploquin, O. Rouaud, M. Havet and A. Le-Bail, Innovative Food Sci. Emerging Technol., 2017, 42, 204–219 CrossRef CAS.
- W. Kaialy, Int. J. Pharm., 2016, 503, 262–276 CrossRef CAS PubMed.
- B. M. Belongia, P. D. Haworth, J. C. Baygents and S. Raghavan, J. Electrochem. Soc., 1999, 146, 4124 CrossRef CAS.
- B. J. Marlow and R. L. Rowell, Langmuir, 1985, 1, 83–90 CrossRef CAS.
- K. E. Van Holde, in Sedimentation analysis of proteins. The Proteins, ed. H. Neurath and R. L. Hill, Academic Press, New York, 3rd edn, 1975, pp. 225–291 Search PubMed.
- T. D. Ducati, L. H. Simões and F. Galembeck, Langmuir, 2010, 26, 13763–13766 CrossRef CAS PubMed.
- K. S. Moreira, D. Lermen, L. P. Santos, F. Galembeck and T. A. L. Burgo, Energy Environ. Sci., 2021, 14, 353–358 RSC.
- C.-S. Jiang, H. R. Moutinho, B. To, C. Xiao, L. J. Simpson and M. M. Al-Jassim, Sol. Energy Mater. Sol. Cells, 2020, 204, 110206 CrossRef CAS.
- M. Glor, Powder Technol., 2003, 135–136, 223–233 CrossRef CAS.
- T. A. L. Burgo, B. C. Batista and F. Galembeck, ACS Omega, 2017, 2, 8940–8947 CrossRef CAS PubMed.
- Q. Sun, D. Wang, Y. Li, J. Zhang, S. Ye, J. Cui, L. Chen, Z. Wang, H.-J. Butt, D. Vollmer and X. Deng, Nat. Mater., 2019, 18, 936–941 CrossRef CAS PubMed.
- S. Lin, L. Xu, A. C. Wang and Z. L. Wang, Nat. Commun., 2020, 11, 399 CrossRef CAS PubMed.
- B. Chen, Y. Xia, R. He, H. Sang, W. Zhang, J. Li, L. Chen, P. Wang, S. Guo, Y. Yin, L. Hu, M. Song, Y. Liang, Y. Wang, G. Jiang and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209056119 CrossRef CAS PubMed.
- M. A. Mehrgardi, M. Mofidfar and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 7606–7609 CrossRef CAS PubMed.
- M. Sow, E. Crase, J. L. Rajot, R. M. Sankaran and D. J. Lacks, Atmos. Res., 2011, 102, 343–350 CrossRef.
- L. P. Santos, T. R. D. Ducati, L. B. S. Balestrin and F. Galembeck, J. Phys. Chem. C, 2011, 115, 11226–11232 CrossRef CAS.
- T. A. L. Burgo and F. Galembeck, J. Braz. Chem. Soc., 2016, 27, 229–238 Search PubMed.
- V. T. C. Paiva, L. P. Santos, D. S. da Silva, T. A. L. Burgo and F. Galembeck, Langmuir, 2019, 35, 7703–7712 CrossRef CAS PubMed.
- S. Hejazi, H. Pahlavanzadeh and J. A. W. Elliott, J. Phys. Chem. B, 2021, 125, 1271–1281 CrossRef CAS PubMed.
- C. Q. Sun, Adv. Colloid Interface Sci., 2020, 282, 102188 CrossRef CAS PubMed.
- IUPAC-NIST Solubility Database, series 37, https://srdata.nist.gov/solubility/IUPAC/SDS-37/SDS-37-pages_221.pdf, (accessed July 2023).
- T. A. L. Burgo and F. Galembeck, J. Braz. Chem. Soc., 2016, 27, 229–238 Search PubMed.
- Why Drying Polyolefins Might Be Right for Your Process, https://www.ptonline.com/articles/why-drying-polyolefins-might-be-right-for-your-process, (accessed July 2023).
- M. Faraday, Philos. Trans. R. Soc. London, 1940, 133, 17–32 Search PubMed.
- W. Thomson, Proc. R. Soc. London, 1867, 16, 67–72 Search PubMed.
- Á. G. Marín, W. van Hoeve, P. García-Sánchez, L. Shui, Y. Xie, M. A. Fontelos, J. C. T. Eijkel, A. van den Berg and D. Lohse, Lab Chip, 2013, 13, 4503–4506 RSC.
- B. Vonnegut, C. B. Moore, R. G. Semonin, J. W. Bullock, D. W. Staggs and W. E. Bradley, J. Geophys. Res., 1962, 67, 3909–3922 CrossRef.
- R. F. Gouveia and F. Galembeck, J. Am. Chem. Soc., 2009, 131, 11381–11386 CrossRef CAS PubMed.
- R. F. Gouveia, J. S. Bernardes, T. R. D. Ducati and F. Galembeck, Anal. Chem., 2012, 84, 10191–10198 CrossRef CAS PubMed.
- L. O. Ferreira and T. A. L. Burgo, J. Electrost., 2020, 106, 103453 CrossRef CAS.
- S. Lin and T. Shao, Phys. Chem. Chem. Phys., 2017, 19, 29418–29423 RSC.
- M. S. Amin, T. F. Peterson and M. Zahn, J. Electrostat., 2006, 64, 424–430 CrossRef.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2010, 1, 1599–1604 CrossRef CAS.
- A. Barišić, J. Lützenkirchen, N. Bebić, Q. Li, K. Hanna, A. Shchukarev and T. Begović, Colloids Interfaces, 2021, 5, 6 CrossRef.
- P. W. Chudleigh, Appl. Phys. Lett., 1972, 21, 547–548 CrossRef CAS.
- M. Vallet, B. Berge and L. Vovelle, Polymer, 1996, 37, 2465–2470 CrossRef CAS.
- N. Knorr, S. Rosselli, T. Miteva and G. Nelles, J. Appl. Phys., 2009, 105, 114111 CrossRef.
- L. C. Soares, S. Bertazzo, T. A. L. Burgo, V. Baldim and F. Galembeck, J. Braz. Chem. Soc., 2008, 19, 277–286 CAS.
- E. J. Workman and S. E. Reynolds, Phys. Rev., 1950, 78, 254–259 CrossRef CAS.
- J. C. Ribeiro, An. Acad. Bras. Cienc., 1950, 22, 325–347 Search PubMed.
- L. P. Santos, A. Galembeck and F. Galembeck, Electrified Ice Needles Fast Formation under the Influence of Electric Fields, International Conference on Lightning and Static Electricity 2022, Madrid, 2022 DOI:10.5281/zenodo.7090059.
- L. P. Santos, D. S. da Silva, A. Galembeck and F. Galembeck, IEEE Trans. Ind. Appl., 2022, 58, 2430–2435 CAS.
- L. P. Santos, D. S. da Silva, A. Galembeck and F. Galembeck, Colloids Interfaces, 2022, 6, 13 CrossRef.
- M. Woody, M. Arbabzadeh, G. M. Lewis, G. A. Keoleian and A. Stefanopoulou, J. Energy Storage, 2020, 28, 101231 CrossRef.
- W. Hong, Y. Zheng, C. Hao, T. Liu, Y. Hu, H. Jiang and C. Li, ACS Appl. Eng. Mater., 2023, 1, 1719–1729 CrossRef CAS.
- S. Yang, Y. He, J. Bai and J. Zhang, Small, 2023, 19, 2302526 CrossRef CAS PubMed.
- X. Chen, D. Goodnight, Z. Gao, A. H. Cavusoglu, N. Sabharwal, M. DeLay, A. Driks and O. Sahin, Nat. Commun., 2015, 6, 7346 CrossRef CAS PubMed.
- T. Ding, K. Liu, J. Li, G. Xue, Q. Chen, L. Huang, B. Hu and J. Zhou, Adv. Funct. Mater., 2017, 27, 1700551 CrossRef.
- J. Li, K. Liu, T. Ding, P. Yang, J. Duan and J. Zhou, Nano Energy, 2019, 58, 797–802 CrossRef CAS.
- Z. Sun, C. Han, S. Gao, Z. Li, M. Jing, H. Yu and Z. Wang, Nat. Commun., 2022, 13, 5077 CrossRef CAS PubMed.
- P. Lenard, Ann. Phys., 1982, 282, 584–586 CrossRef.
- E. W. B. Gill and G. F. Alfrey, Nature, 1952, 169, 203–204 CrossRef.
- P. Kolarž, M. Gaisberger, P. Madl, W. Hofmann, M. Ritter and A. Hartl, Atmos. Chem. Phys., 2012, 12, 3687–3697 CrossRef.
- J. Zhong, M. Kumar, J. S. Francisco and X. C. Zeng, Acc. Chem. Res., 2018, 51, 1229–1237 CrossRef CAS PubMed.
- M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- G. Heinicke, Tribochemistry, Carl Hanser Verlag, München-Wien, 1984 Search PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- M. F. Ruiz-Lopez, J. S. Francisco, M. T. C. Martins-Costa and J. M. Anglada, Nat. Rev. Chem., 2020, 4, 459–475 CrossRef CAS PubMed.
- S. Shaik, D. Danovich, J. Joy, Z. Wang and T. Stuyver, J. Am. Chem. Soc., 2020, 142, 12551–12562 CrossRef CAS PubMed.
- S. J. P. Marlton, B. I. McKinnon, N. S. Hill, M. L. Coote and A. J. Trevitt, J. Am. Chem. Soc., 2021, 143, 2331–2339 CrossRef CAS PubMed.
- S. Handa, M. P. Andersson, F. Gallou, J. Reilly and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 4914–4918 CrossRef CAS PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J.-Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 2169 CrossRef PubMed.
- Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS PubMed.
- O. Acevedo and K. Armacost, J. Am. Chem. Soc., 2010, 132, 1966–1975 CrossRef CAS PubMed.
- T. Kitanosono and S. Kobayashi, Chem. – Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed.
- Z. Wei, Y. Li, R. G. Cooks and X. Yan, Ann. Rev. Phys. Chem., 2020, 71, 31–51 CrossRef CAS PubMed.
- L. R. Pestana, H. Hao and T. Head-Gordon, Nano Lett., 2020, 20, 606–611 CrossRef CAS PubMed.
- D. Munoz-Santiburcio and D. Marx, Chem. Rev., 2021, 121, 6293–6320 CrossRef CAS PubMed.
- Z. Li, A. Kiyama, H. Zeng and X. Zhang, J. Phys. Chem. C, 2021, 125, 15324–15334 CrossRef CAS.
- C. M. Gabriel, N. R. Lee, F. Bigorne, P. Klumphu, M. Parmentier, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 194–197 CrossRef CAS PubMed.
- D. Munoz-Santiburcio and D. Marx, Chem. Rev., 2021, 121, 6293–6320 CrossRef CAS PubMed.
- N. Akporji, R. R. Thakore, M. Cortes-Clerget, J. Andersen, E. Landstrom, D. H. Aue, F. Gallou and B. H. Lipshutz, Chem. Sci., 2020, 11, 5205–5212 RSC.
- E. B. Landstrom, S. Handa, D. H. Aue, F. Gallou and B. H. Lipshutz, Green Chem., 2018, 20, 3436–3443 RSC.
- B. H. Lipshutz, Johnson Matthey Technol. Rev., 2017, 61, 196–202 CrossRef CAS.
- C. Gong, D. Li, X. Li, D. Zhang, D. Xing, L. Zhao, X. Yuan and X. Zhang, J. Am. Chem. Soc., 2022, 144, 3510–3516 CrossRef CAS PubMed.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2022, 61, e202210765 CrossRef CAS PubMed.
- H. Chen, R. Wang, J. Xu, X. Yuan, D. Zhang, Z. Zhu, M. Marshall, K. Bowen and X. Zhang, J. Am. Chem. Soc., 2023, 145, 2647–2652 CrossRef CAS PubMed.
- X. Yuan, D. Zhang, C. Liang and X. Zhang, J. Am. Chem. Soc., 2023, 145, 2800–2805 CrossRef CAS PubMed.
- D. Xing, X. Yuan, C. Liang, T. Jin, S. Zhang and X. Zhang, Chem. Commun., 2022, 58, 12447–12450 RSC.
- L. Qiu, M. D. Psimos and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2022, 33, 1362–1367 CrossRef CAS PubMed.
- L. Qiu, N. M. Morato, K.-H. Huang and R. G. Cooks, Front. Chem., 2022, 10, 903774 CrossRef CAS PubMed.
- Y. B. Vogel, C. W. Evans, M. Belotti, L. Xu, I. C. Russell, L.-J. Yu, A. K. K. Fung, N. S. Hill, N. Darwish, V. R. Gonçales, M. L. Coote, K. S. Iyer and S. Ciampi, Nat. Commun., 2020, 11, 6323 CrossRef CAS PubMed.
- S. Jin, R. Wang, H. Chen, X. Yuan and X. Zhang, J. Phys. Chem. A, 2023, 127(12), 2805–2809 CrossRef CAS PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- I. Nam, J. K. Lee, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12396–12400 CrossRef CAS PubMed.
- J. K. Beattie, C. S. P. McErlean and C. B. W. Phippen, Chem. – Eur. J., 2010, 16, 8972–8974 CrossRef CAS PubMed.
- M. T. C. Martins-Costa and M. F. Ruiz-López, J. Am. Chem. Soc., 2023, 145, 1400–1406 CrossRef CAS PubMed.
- C. F. Chamberlayne and R. N. Zare, J. Chem. Phys., 2020, 152, 184702 CrossRef CAS PubMed.
- H. Hao, I. Leven and T. Head-Gordon, Nat. Commun., 2022, 13, 280 CrossRef CAS PubMed.
- D. Zhang, X. Yuan, C. Gong and X. Zhang, J. Am. Chem. Soc., 2022, 144, 16184–16190 CrossRef CAS PubMed.
- M. T. C. Martins-Costa and M. F. Ruiz-López, Theor. Chem. Acc., 2023, 142, 29 Search PubMed.
- J. K. Lee, S. Banerjee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2015, 48, 437–444 CrossRef CAS PubMed.
- L. Zhao, X. Song, C. Gong, D. Zhang, R. Wang, R. N. Zare and X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200991119 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Nat. Commun., 2018, 9, 1562 CrossRef PubMed.
- X. Song, Y. Meng and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 16744–16748 CrossRef CAS PubMed.
- D. Gao, F. Jin, J. K. Lee and R. N. Zare, Chem. Sci., 2019, 10, 10974–10978 RSC.
- J. K. Lee, H. S. Han, S. Chaikasetsin, D. P. Marron, R. M. Waymouth, F. B. Prinz and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30934–30941 CrossRef CAS PubMed.
- A. Gallo Jr., N. H. Musskopf, X. Liu, Z. Yang, J. Petry, P. Zhang, S. Thoroddsen, H. Im and H. Mishra, Chem. Sci., 2022, 13, 2574–2583 RSC.
- D. Xing, Y. Meng, X. Yuan, S. Jin, X. Song, R. N. Zare and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207587 CrossRef CAS PubMed.
- J. P. Heindel, H. Hao, R. A. LaCour and T. Head-Gordon, J. Phys. Chem. Lett., 2022, 13, 10035–10041 CrossRef CAS PubMed.
- D. Nguyen, P. Lyu and S. C. Nguyen, J. Phys. Chem. B, 2023, 127, 2323–2330 CrossRef CAS PubMed.
- X. Chen, Y. Xia, Z. Zhang, L. Hua, X. Jia, F. Wang and R. N. Zare, J. Am. Chem. Soc., 2023, 145, 21538–21545 CrossRef CAS PubMed.
- L. P. Santos, D. Lermen, R. G. Yoshimura, B. L. da Silva, A. Galembeck, T. A. L. Burgo and F. Galembeck, Langmuir, 2023, 39, 5840–5850 CrossRef CAS PubMed.
- A. J. Colussi, J. Am. Chem. Soc., 2023, 145, 16315–16317 CrossRef CAS PubMed.
- C. F. Chamberlayne and R. N. Zare, J. Chem. Phys., 2022, 156, 054705 CrossRef CAS PubMed.
- J. Zhao, X. Zhang, J. Xu, W. Tang, Z. L. Wang and F. R. Fan, Angew. Chem., Int. Ed., 2023, 62, e202300604 CrossRef CAS PubMed.
- J. Li, Y. Xia, J. Li, K. Long, F. Chen, M. Feng, S. Guo and B. Chen, J. Water Process. Eng., 2023, 52, 103521 CrossRef.
- M. A. Saucedo-Espinosa, M. Breitfeld and P. S. Dittrich, Angew. Chem., Int. Ed., 2023, 62, e202212459 CrossRef CAS PubMed.
- B. Baytekin, H. T. Baytekin and B. A. Grzybowski, J. Am. Chem. Soc., 2012, 134, 7223–7226 CrossRef CAS PubMed.
- H. Wu, Z. Chen, G. Xu, J. Xu, Z. Wang and Y. Zi, ACS Appl. Mater. Interfaces, 2020, 12, 56060–56067 CrossRef CAS PubMed.
- M. M. Hasan, T. Zhang, H. Wu and Y. Yang, Adv. Energy Mater., 2022, 12, 2201383 CrossRef CAS.
- R. Kumar, T. Tabrizizadeh, S. Chaurasia, G. J. Liu and K. Stamplecoskie, Sustainable Energy Fuels, 2022, 6, 1141–1147 RSC.
- K. Rogdakis, N. Karakostas and E. Kymakis, Energy Environ. Sci., 2021, 14, 3352–3392 RSC.
- X. Wang, F. Lin, X. Wang, S. Fang, J. Tan, W. Chu, R. Rong, J. Yin, Z. Zhang, Y. Liu and W. Guo, Chem. Soc. Rev., 2022, 51, 4902–4927 RSC.
- H. U. Lee, C. Y. Chung, S. W. Han, C. Choi and S. B. Cho, J. Mater. Chem. A, 2023, 11, 1148–1158 RSC.
- X. Liu, H. Gao, L. Sun and J. Yao, Adv. Mater., 2023, 2300748 CrossRef PubMed.
- V. D. Trung, P.-A. Le, J. Natsuki and T. Natsuki, Adv. Mater. Technol., 2023, 8, 2300638 CrossRef CAS.
- Innovation Origins: Accidental discovery unveils revolutionary power generation option, https://innovationorigins.com/en/accidental-discovery-unveils-revolutionary-power-generation-option/, (accessed July 2023).
- Y. Huang, H. Cheng, C. Yang, P. Zhang, Q. Liao, H. Yao, G. Shi and L. Qu, Nat. Commun., 2018, 9, 4166 CrossRef PubMed.
- G. Xue, Y. Xu, T. Ding, J. Li, J. Yin, W. Fei, Y. Cao, J. Yu, L. Yuan, L. Gong, J. Chen, S. Deng, J. Zhou and W. Guo, Nat. Nano, 2017, 12, 317–321 CrossRef CAS PubMed.
- J. Li, K. Liu, T. Ding, P. Yang, J. Duan and J. Zhou, Nano Energy, 2019, 58, 797–802 CrossRef CAS.
- Z.-H. Lin, G. Cheng, S. Lee, K. C. Pradel and Z. L. Wang, Adv. Mater., 2014, 26, 4690–4696 CrossRef CAS PubMed.
- J. Yin, X. Li, J. Yu, Z. Zhang, J. Zhou and W. Guo, Nature Nanotechnol., 2014, 9, 378 CrossRef CAS PubMed.
- P. Linstrom, NIST Chemistry WebBook – SRD 69, (accessed September 2021) DOI:10.18434/T4D303.
- X. Yan, M. Delgado, J. Aubry, O. Gribelin, A. Stocco, F. B.-D. Cruz, J. Bernard and F. Ganachaud, J. Phys. Chem. Lett., 2018, 9, 96–103 CrossRef CAS PubMed.
- X. Song, C. Basheer, Y. Xia, J. Li, I. Abdulazeez, A. A. Al-Saadi, M. Mofidfar, M. A. Suliman and R. N. Zare, J. Am. Chem. Soc., 2023, 145, 25910–25916 CrossRef CAS PubMed.
- J. Lowell, J. Phys. D: Appl. Phys., 1975, 8, 53 CrossRef CAS.
- B. V. Deryagin, N. A. Krotova and V. P. Smilga, Adhesion of Solids, Consultant Bureau, New York, 1978 Search PubMed.
- D. A. Hays, in Fundamentals of Adhesion, ed. L. H. Lee, Springer, New York, 1991, vol. 8, pp. 249–278 Search PubMed.
- L. F. Valadares, C. A. P. Leite and F. Galembeck, Polymer, 2006, 47, 672–678 CrossRef CAS.
- D. Tan, M. Willatzen and Z. L. Wang, Nano Energy, 2021, 79, 105386 CrossRef CAS.
- M. C. Lea, Am. J. Sci., 1892, 43, 527 CrossRef.
- W. Spring, Bull. Soc. Chim. Fr., 1883, 39, 195 Search PubMed.
- L. Takacs, Acta Phys. Pol., A, 2014, 126, 1040–1043 CrossRef CAS.
- S. L. James and T. Friščić, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC.
- T. Friščić, S. L. James, E. V. Boldyreva, C. Bolm, W. Jones, J. Mack, J. W. Steed and K. S. Suslick, Chem. Commun., 2015, 51, 6248–6256 RSC.
- G. Kaupp, Cryst. Eng. Comm., 2009, 11, 388 RSC.
- P. Baláz, Mechanochemistry in Nanoscience and Minerals Engineering, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed.
- B. Bhushan and C. Kajdas, in Fundamentals of Tribology and Bridging the Macro- and Micro/Nanoscales, ed. B. Bhushan, Kluwer Academic Publishers, The Netherlands, 2001, p. 735 Search PubMed.
- S. N. Zhurkov, Int. J. Fract. Mech., 1965, 1, 311 CrossRef CAS.
- L. Takacs, Acta Phys. Pol., A, 2012, 121, 711 CrossRef CAS.
- G. S. P. Castle, J. Electrostat., 1997, 40–41, 13–20 CrossRef.
- D. J. Lacks and T. Shinbrot, Nat. Rev. Chem., 2019, 3, 465–476 CrossRef CAS.
- A. Galembeck, C. A. R. Costa, M. C. V. M. da Silva, E. F. Souza and F. Galembeck, Polymer, 2001, 42, 4845–4851 CrossRef CAS.
- C. A. Rezende, R. F. Gouveia, M. A. da Silva and F. Galembeck, J. Phys.: Condens. Matter, 2009, 21, 263002 CrossRef CAS PubMed.
- T. A. L. Burgo, T. R. D. Ducati, K. R. Francisco, K. J. Clinckspoor, F. Galembeck and S. E. Galembeck, Langmuir, 2012, 28, 7407–7416 CrossRef CAS PubMed.
- T. A. L. Burgo, C. A. Rezende, S. Bertazzo, A. Galembeck and F. Galembeck, J. Electrostat., 2011, 69, 401–409 CrossRef CAS.
- H. T. Baytekin, A. Z. Patashinski, M. Branicki, B. Baytekin, S. Soh and B. A. Grzybowski, Science, 2011, 333, 308–312 CrossRef CAS PubMed.
- T. A. L. Burgo, C. A. Silva, L. B. S. Balestrin and F. Galembeck, Sci. Rep., 2013, 3, 2384 CrossRef PubMed.
- T. A. L. Burgo and A. Erdemir, Angew. Chem., Int. Ed., 2014, 53, 12101–12105 CrossRef CAS PubMed.
- K. S. Hong, H. Xu, H. Konishi and X. Li, J. Phys. Chem. Lett., 2010, 1, 997–1002 CrossRef CAS.
- S. Li, Z. Zhao, J. Zhao, Z. Zhang, X. Li and J. Zhang, ACS Appl. Nano Mater., 2020, 3, 1063–1079 CrossRef CAS.
- M. B. Starr and X. Wang, Sci. Rep., 2013, 3, 2160 CrossRef PubMed.
- S. Tu, Y. Guo, Y. Zhang, C. Hu, T. Zhang, T. Ma and H. Huang, Adv. Funct. Mater., 2020, 30, 2005158 CrossRef CAS.
- F. R. Fan, S. Xie, G. W. Wang and Z. Q. Tian, Sci. China: Chem., 2021, 64, 1609–1613 CrossRef CAS.
- L. P. Santos, D. S. da Silva, B. C. Batista, K. S. Moreira, T. A. L. Burgo and F. Galembeck, Polymer, 2019, 171, 173–179 CrossRef CAS.
- E. S. Ferreira, D. S. da Silva, T. A. L. Burgo, B. C. Batista and F. Galembeck, Nanoscale, 2017, 9, 10219–10226 RSC.
- L. P. Santos, D. Soares Da Silva, J. P. Ferreira Bertacchi, K. S. Moreira, T. A. Lima Burgo, B. C. Batista, J. Dos Santos, P. A. De Paula and F. Galembeck, Faraday Discuss., 2021, 227, 105–124 RSC.
- K. S. Moreira, E. Lorenzett, A. L. Devens, Y. A. Santos da Campo, D. Mehler and T. A. L. Burgo, J. Appl. Phys., 2021, 129, 234502 CrossRef CAS.
- Y. A. Santos Da Campo, D. Mehler, E. Lorenzett, K. S. Moreira, A. L. Devens, L. P. dos Santos, F. Galembeck and T. A. L. Burgo, Phys. Chem. Chem. Phys., 2021, 23, 26653–26660 RSC.
- F.-R. Fan, Z.-Q. Tian and Z. L. Wang, Nano Energy, 2012, 1, 328–334 CrossRef CAS.
- A. F. Rubira, A. C. da Costa, F. Galembeck, N. F. Leite Escobar, E. C. da Silva and H. Vargas, Colloids Surf., 1985, 15, 63–73 CrossRef CAS.
- T. A. L. Burgo, L. B. S. Balestrin and F. Galembeck, Polym. Degrad. Stab., 2014, 104, 11–17 CrossRef CAS.
- K. S. Moreira, Y. A. Santos, E. Lorenzett and T. A. L. Burgo, Results Eng., 2023, 17, 100965 CrossRef CAS.
- T. Xu, X. Ding, C. Shao, L. Song, T. Lin, X. Gao, J. Xue, Z. Zhang and L. Qu, Small, 2018, 14, 1704473 CrossRef PubMed.
- S. Mandal, S. Roy, A. Mandal, T. Ghoshal, G. Das, A. Singh and D. K. Goswami, ACS Appl. Electron. Mater., 2020, 2, 780–789 CrossRef CAS.
- S. G. Yoon, Y. Yang, J. Yoo, H. Jin, W. H. Lee, J. Park and Y. S. Kim, ACS Appl. Electron. Mater., 2019, 1, 1746–1751 CrossRef CAS.
- S. Chaurasia, R. Kumar, T. Tabrizizadeh, G. Liu and K. Stamplecoskie, ACS Omega, 2022, 7, 2618–2623 CrossRef CAS PubMed.
- Z. H. Lin, G. Cheng, L. Lin, S. Lee and Z. L. Wang, Angew. Chem., Int. Ed., 2013, 52, 12545–12549 CrossRef CAS PubMed.
- Z. Zhang, X. Li, J. Yin, Y. Xu, W. Fei, M. Xue, Q. Wang, J. Zhou and W. Guo, Nat. Nanotechnol., 2018, 13, 1109–1119 CrossRef CAS PubMed.
- F. H. J. van der Heyden, D. J. Bonthuis, D. Stein, C. Meyer and C. Dekker, Nano Lett., 2007, 7, 1022–1025 CrossRef CAS PubMed.
- W. Xu, H. Zheng, Y. Liu, X. Zhou, C. Zhang, Y. Song, X. Deng, M. Leung, Z. Yang, R. X. Xu, Z. L. Wang, X. C. Zeng and Z. Wang, Nature, 2020, 578, 392–396 CrossRef CAS PubMed.
- C. Xu, X. Fu, C. Li, G. Liu, Y. Gao, Y. Qi, T. Bu, Y. Chen, Z. L. Wang and C. Zhang, Microsystems Nanoeng., 2022, 8, 30 CrossRef PubMed.
- Y. Zeng, Y. Luo, Y. Lu and X. Cao, Nano Energy, 2022, 98, 107316 CrossRef CAS.
- L. Li, X. Wang, W. Deng, J. Yin, X. Li and W. Guo, Droplet, 2023, 1–14 Search PubMed.
- J. Nauruzbayeva, Z. Sun, A. Gallo Jr., M. Ibrahim, J. C. Santamarina and H. Mishra, Nat. Commun., 2020, 11, 5285 CrossRef CAS PubMed.
- S. Lin, M. Zheng and Z. L. Wang, J. Phys. Chem. C, 2021, 125, 14098–14104 CrossRef CAS.
- S. Lin, X. Chen and Z. L. Wang, Chem. Rev., 2022, 122, 5209–5232 CrossRef CAS PubMed.
- A. Chatterjee, S. Lal, T. G. Manivasagam and S. K. Batabyal, Bioresour. Technol. Rep., 2023, 22, 101379 CrossRef CAS.
- S. Chatterjee, S. R. Burman, I. Khan, S. Saha, D. Choi, S. Lee and Z.-H. Lin, Nanoscale, 2020, 12, 17663–17697 RSC.
- K. Wang and J. Li, J. Mater. Chem. A, 2021, 9, 8870–8895 RSC.
- S. S. Kwak, S. Lin, J. H. Lee, H. Ryu, T. Y. Kim, H. Zhong, H. Chen and S.-W. Kim, ACS Nano, 2016, 10, 7297–7302 CrossRef CAS PubMed.
- J. Dong, F. R. Fan and Z. Q. Tian, Nanoscale, 2021, 13, 17290–17309 RSC.
- Q. Zhang, C. Jiang, X. Li, S. Dai, Y. Ying and J. Ping, ACS Nano, 2021, 15, 12314–12323 CrossRef CAS PubMed.
- Y. Long, P. He, Z. Shao, Z. Li, H. Kim, A. M. Yao, Y. Peng, R. Xu, C. H. Ahn, S. W. Lee, J. Zhong and L. Lin, Nat. Commun., 2021, 12, 5287 CrossRef CAS PubMed.
- J. Tan, S. Fang, Z. Zhang, J. Yin, L. Li, X. Wang and W. Guo, Nat. Commun., 2022, 13, 3643 CrossRef CAS PubMed.
- P. Wang, J. Xu, R. Wang and T. Li, Cell Rep. Phys. Sci., 2023, 4, 101517 CrossRef CAS.
- X. Yang, C. Xiang and R. Wang, Device, 2023, 1, 100016 CrossRef.
- T. Xu, X. Ding, Y. Huang, C. Shao, L. Song, X. Gao, Z. Zhang and L. Qu, Energy Environ. Sci., 2019, 12, 972–978 RSC.
- Q. Tang and P. Yang, J. Mater. Chem. A, 2016, 4, 9730–9738 RSC.
- J. Zhang, Y. Hou, Y. Li and S. Hu, J. Mater. Res. Technol., 2022, 17, 1822–1830 CrossRef.
- T. Cai, L. Lan, B. Peng, C. Zhang, S. Dai, C. Zhang, J. Ping and Y. Ying, Nano Lett., 2022, 22, 6476–6483 CrossRef CAS PubMed.
- T. He, H. Wang, B. Lu, T. Guang, C. Yang, Y. Huang, H. Cheng and L. Qu, Joule, 2023, 7, 935–951 CrossRef CAS.
- J. Li, Y. Long, Z. Hu, J. Niu, T. Xu, M. Yu, B. Li, X. Li, J. Zhou, Y. Liu, C. Wang, L. Shen, W. Guo and J. Yi, Nat. Commun., 2021, 12, 4998 CrossRef CAS PubMed.
- J. Bai, Y. Huang, H. Wang, T. Guang, Q. Liao, H. Cheng, S. Deng, Q. Li, Z. Shuai and L. Qu, Adv. Mater., 2022, 34, 2103897 CrossRef CAS PubMed.
- J. Bai, Q. Liao, H. Yao, T. Guang, T. He, H. Cheng and L. Qu, Energy Environ. Sci., 2023, 16, 3088–3097 RSC.
- H. Wang, T. He, X. Hao, Y. Huang, H. Yao, F. Liu, H. Cheng and L. Qu, Nat. Commun., 2022, 13, 2524 CrossRef CAS PubMed.
- F. Gong, H. Li, Q. Zhou, M. Wang, W. Wang, Y. Lv, R. Xiao and D. V. Papavassiliou, Nano Energy, 2020, 74, 104922 CrossRef CAS.
- Z. Sun, X. Wen, S. Guo, M. Zhou, L. Wang, X. Qin and S. C. Tan, Nano Energy, 2023, 116, 108748 CrossRef CAS.
- P. Dhiman, F. Yavari, X. Mi, H. Gullapalli, Y. Shi, P. M. Ajayan and N. Koratkar, Nano Lett., 2011, 11, 3123–3127 CrossRef CAS PubMed.
- J. Yin, Z. Zhang, X. Li, J. Zhou and W. Guo, Nano Lett., 2012, 12, 1736–1741 CrossRef CAS PubMed.
- T. Xu, Q. Han, Z. Cheng, J. Zhang and L. Qu, Small Methods, 2018, 2, 1800108 CrossRef.
- P. Schuster, Monatsh. Chem., 2019, 150, 763–775 CrossRef CAS.
- C. Wäckerlin and K.-H. Ernst, J. Phys. Chem. C, 2021, 125, 13343–13349 CrossRef.
- Canadian Forest Industries, https://www.woodbusiness.ca/combustible-dust-incidents-from-around-the-world-in-2020/, (Accessed November 2023).
- R. L. Holle, Annual Rates of Lightning Fatalities by Country, 20 th International Lightnning Detection Conference 2008, Arizona 2008, https://www.vaisala.com/sites/default/files/documents/Annual_rates_of_lightning_fatalities_by_country.pdf.
- Insurance Information Institute, https://www.iii.org/fact-statistic/facts-statistics-lightning. (Accessed November 2023).
- I. Langmuir, J. Am. Chem. Soc., 1938, 60, 1190–1194 CrossRef CAS.
- W. S. Y. Wong, P. Bista, X. Li, L. Veith, A. Sharifi-Aghili, S. A. L. Weber and H.-J. Butt, Langmuir, 2022, 38, 6224–6230 CrossRef CAS PubMed.
- A. F. Diaz and D. Fenzel-Alexander, Langmuir, 1993, 9, 1009–1015 CrossRef CAS.
- G. W. Gross, J. Geophys. Res., 1965, 70, 2291–2300 CrossRef CAS.
- S. Enami, H. Mishra, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2012, 116, 6027–6032 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |