
Singlet oxygen is an emissive ligand†
Paul Asselin ,
Adrien Schlachter and
Pierre D. Harvey*
,
Adrien Schlachter and
Pierre D. Harvey*
Département de Chimie, Université de Sherbrooke, 2500 Boul. de l’Université, Sherbrooke, QC J1K 2R1, Canada. E-mail: Pierre.Harvey@USherbrooke.ca
First published on 8th July 2024
Abstract
Unprecedented experimental evidence shows that gaseous singlet oxygen (1O2) acts as an emissive ligand following collisional photosensitization. This evidence was obtained by monitoring 1O2 phosphorescence intensity at ≈1275–1280 nm and the excited state lifetime of singlet oxygen generated by known tetraphenylporphyrin photosensitizers, while varying the atmospheric environment.
Singlet oxygen (specifically O2(1Δg), hereafter 1O2) is the first electronic excited state of the dioxygen molecule1–3 and has been known as a strong oxidizing agent for decades, with recent studies demonstrating a rich potential of applications in a wide variety of fields, including cancer therapy4 and antimicrobial activity.5 While 1O2 can be prepared in several ways, the most popular is photosensitization, a catalytic means of using ambient or supplied light, absorbed by an intermediate molecule, the photosensitizer (PS), whose triplet excited state (3PS*) is quenched by ground state dioxygen (O2(3Σ−g), hereafter 3O2).6–8 This Dexter-type energy transfer9,10 requires such proximity between 3PS* and 3O2 that a collision is all but implied. Photosensitization often readily occurs in solution from dissolved PS and 3O2 (homogeneous process) but also at the solid–liquid or solid–gas interfaces (heterogeneous process), using either neat, physiosorbed or otherwise attached PS on a substrate, and either dissolved or atmospheric 3O2.
It is known that 3O2 can act as a ligand in metallic complexes, notably in heme found in human blood cells. Similar synthetic complexes have been documented,11–13 usually taking advantage of significant steric hindering to keep the weak O2 ligand in place. Crystallographic elucidations and computational analyses often supplement these studies, and some entirely theoretical work has also been conducted on the subject,14 establishing the possibility of a transient complexed state as the energy transfer takes place (eqn (1) or (4) in ref. 14) when zinc(II) porphine (ZnP) is used as photosensitizer. A kinetic study of the homogeneous process also suggests the existence of transient exciplexes in freebase tetraphenylporphyrin and several other entirely organic photosensitizers.14,15
| 3ZnP + 3O2 → 1(3ZnP3O2) → 1(3ZnP1O2) → 1(3ZnP1O2) → 1ZnP + 1O2 | (1) |
Preliminary tests: first, 1O2 phosphorescence from dilute ZnTPP solutions in DCM and MeOH was recorded to demonstrate that both solvents allow 1O2 phosphorescence, and therefore must allow prior photosensitization (Fig. S1, ESI†). The emission band observed at ≈1275 nm is consistent with those typically reported in the literature.16 The 1O2 excitation spectra appear like the absorption spectrum of ZnTPP, meaning that more singlet oxygen is produced as ZnTPP absorbs more photons, and in turn produces more populated excited states able to transfer energy to 3O2. This proves that ZnTPP is acting as the photosensitizer. It is noteworthy that the 1O2 emission signal is roughly 15× weaker in MeOH than in DCM, suggesting a significant level of static quenching in the former solvent: MeOH does not quench 1O2 itself, but rather hinders its generation, likely by coordinating the Zn(II) atom and generating steric hindering.
Atmosphere cycling experiment: the detailed description can be found in the ESI.† A sealed 10 × 10 mm quartz cuvette containing drop-cast ZnTPP was analysed by measuring 1O2 phosphorescence intensity in a pure oxygen atmosphere, followed by argon, air, then methanol-saturated air,‡ and finally methanol-saturated oxygen‡ (Fig. 1). A fresh dry sample under pure O2 produced the highest intensity. After purging the cuvette with argon, the signal significantly decreased but did not disappear completely. The cuvette was then purged with air, and the signal intensity increased. Following this, the introduction of MeOH-saturated air resulted in a slow but complete disappearance of 1O2 phosphorescence. Finally, purging with MeOH-saturated O2 vapour, a very slight signal increase was noted, however considering noise levels, it cannot be confidently attributed to signal resurgence.
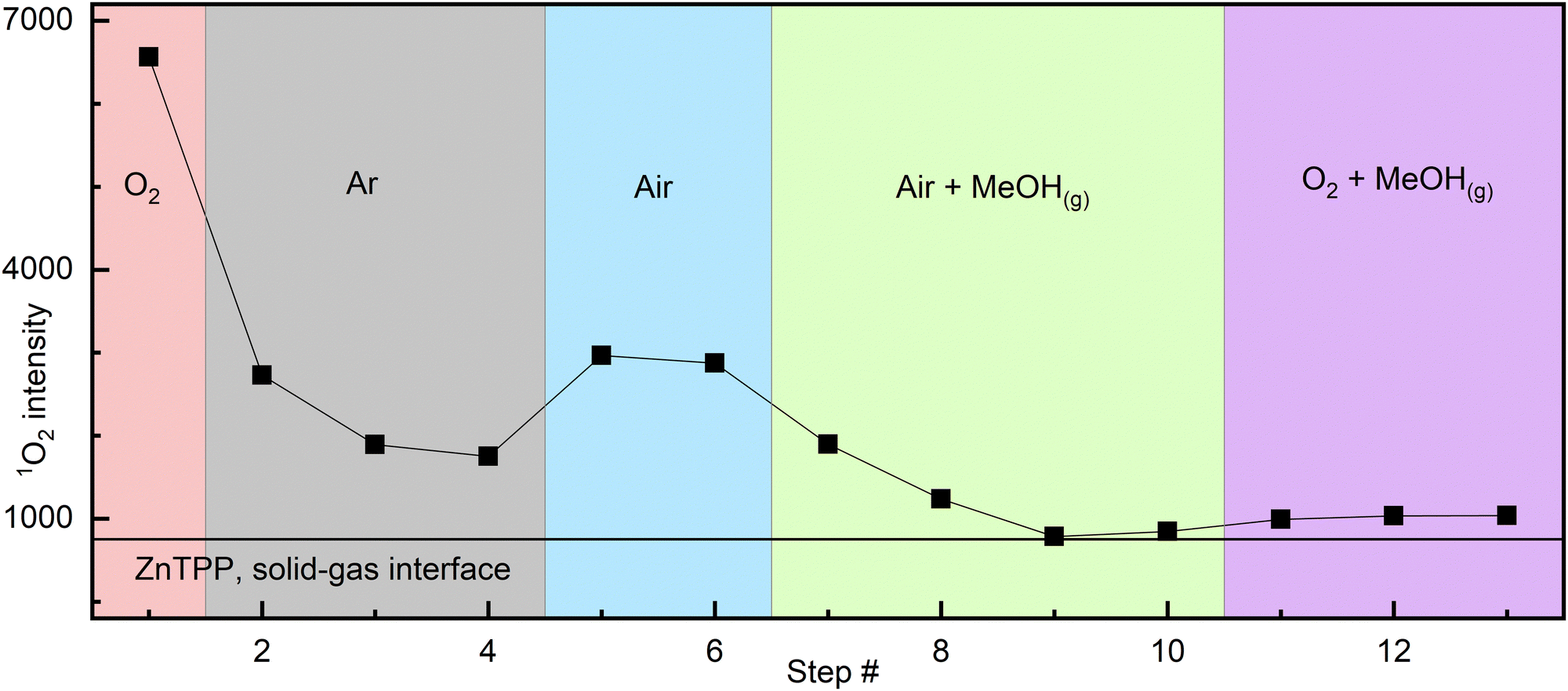 | ||
| Fig. 1 Interfacial 1O2 signal intensity varying with atmosphere composition (from drop cast ZnTPP). Each step (point) is separated by 4–8 min to account for the time to record each scan and gas purges. The solid line represents the average baseline value throughout the experiment. See Fig. S3 (ESI†) for raw spectra and additional details. | ||
Analysis: we know from X-ray structures of metalated porphyrins that MeOH and, in two reported cases, presumably triplet O2 can bind lone, heavily distorted and sterically hindered zinc porphyrins.12,13 PCN-224-Fe(II)–O2,11 with the framework itself generating steric hindering, has also been reported. Knowing this, there is a compelling case to be made that the behavior described by Fig. 1 is that of a coordination complex: the observation of a residual signal in the argon atmosphere after any gaseous O2 is carried away, followed by its disappearance when MeOH is present, indicates the presence of a relatively stable intermediate. As any gaseous O2 would be carried away upon successive argon purges, the remaining option for 1O2 persisting in the cuvette is the existence of this intermediate, the most likely form of which is coordinated [ZnTPP–O2]. Methanol, whilst known to be a weak ligand, is stronger than O2. Its introduction in the cell allows it to slowly replace any coordinated O2, thus preventing its subsequent photosensitization, explaining the slow and complete disappearance of 1O2 phosphorescence over 5–10 min (Fig. 2).
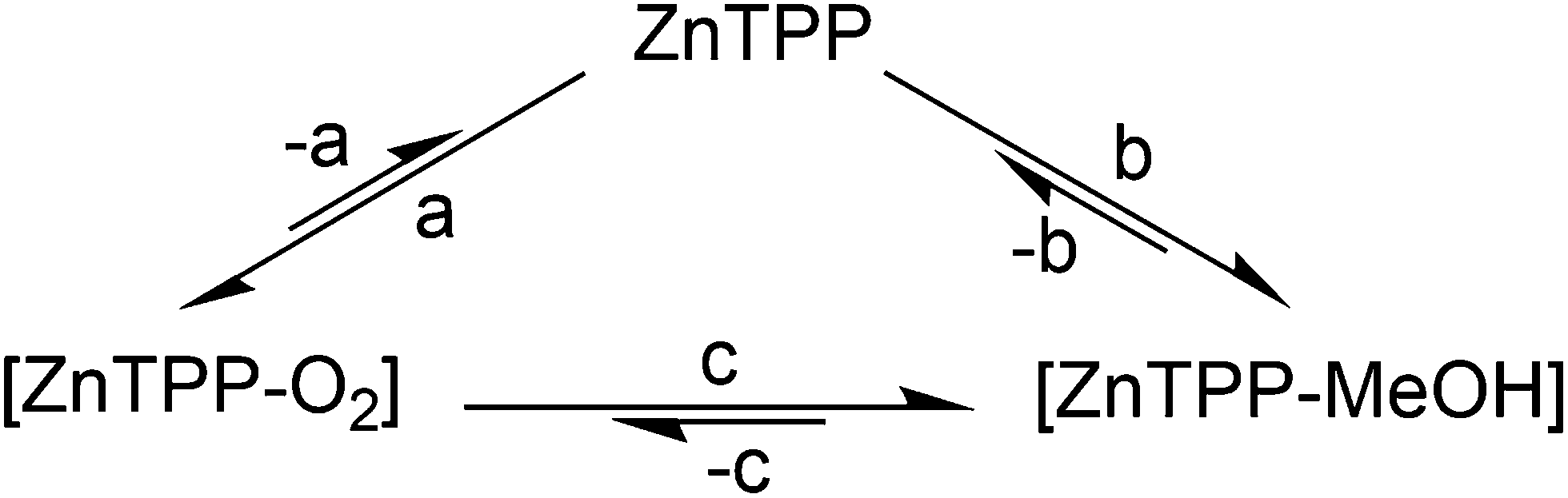 | ||
| Fig. 2 Proposed qualitative equilibrium between non-complexed ZnTPP, the [ZnTPP–O2] species and the [ZnTPP–MeOH] complex in the ground state consistent with the behavior shown in Fig. 1. (a) +O2; (b) +MeOH; (c) ligand exchange. | ||
Excited state lifetime measurements: two other pieces of evidence are obtained upon changing the metallic site (M = 2H+, Zn(II), Pd(II)): first, when comparing ZnTPP and H2TPP, a significant decrease in signal intensity is observed, and the position of the 1O2 emission band changes from 1280 to 1275 nm respectively (Fig. 3A). This shift suggests a change in the environment of 1O2, which can be explained by the coordinated vs. non-coordinated variants of 1O2.
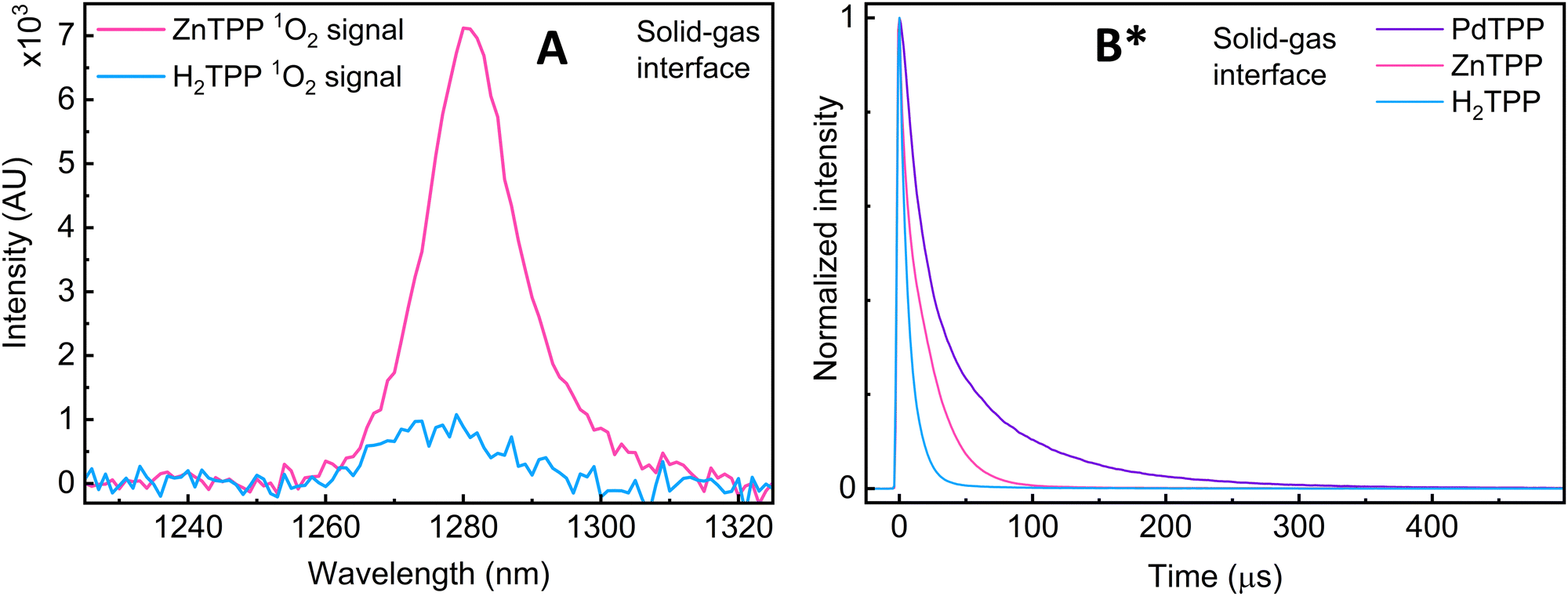 | ||
Fig. 3 (A) Comparison of the 1O2 phosphorescence signal intensity at the solid-state/gas interface of H2TPP and ZnTPP samples (pure O2(g)). (B) Comparison of the normalized 1O2 phosphorescence decays of H2TPP, ZnTPP and PdTPP samples, pure O2(g). *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The data presented in subfigure B has already been published in ref. 16. The data presented in subfigure B has already been published in ref. 16. | ||
Second, the presence of a metal results in a significant increase in one of the lifetime components (Fig. 3B and Table 1). All three porphyrins show a component in the roughly 10 μs range, and another at 5 μs, 218 μs, and 74 μs for H2-, Zn-, and PdTPP respectively. The microsecond values are perfectly coherent with known and documented values recorded from solution and solid-state samples,17 and are still well within the theoretically predicted and experimentally observed purely gas-phase maxima going to full second (direct from EPR and calculated from diffused distance).18,19 The changes in band position and lifetime duration can be explained by the change in 1O2's interfacial environment (metal coordination vs. H-bonding). This further supports the 1O2 ligand explanation.
| H2TPP | ZnTPP | PdTPP |
|---|---|---|
| The uncertainties stem from the fitting model: the lower limit of binning was 1 μs, so very few points were available for H2TPP, resulting in higher uncertainties. *The data presented in this table has already been published in ref. 16. |
||
| 5.3 ± 0.9 | 10.0 ± 0.5 | 9.8 ± 0.5 |
| 9.6 ± 1.5 | 218 ± 0.5 (minor) | 73.8 ± 0.5 |
While the nature of the [ZnTPP–1O2] emissive compound is now convincingly proven, two questions remain: first, our experiments cannot determine whether coordination occurs before photosensitization (ligand is 3O2, eqn (2)) which we call early coordination, or after photosensitization (ligand is 1O2, eqn (3)) which we designate late coordination. Note that these possibilities are not mutually exclusive. Second, emissive lifetimes do not follow the trend expected of the heavy-atom effect: the long ZnTPP lifetime, while minor, is roughly 3× that of PdTPP.
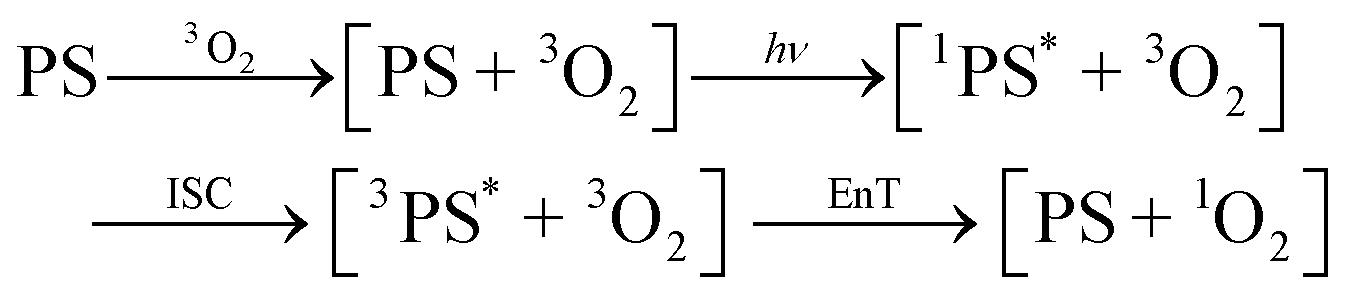 | (2) |
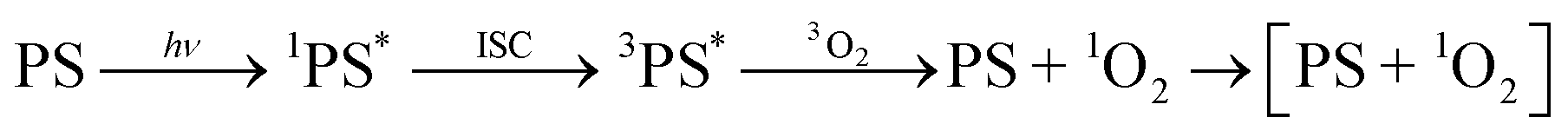 | (3) |
We have attempted to imitate Rusidy's computations using undistorted ZnTPP. Our calculated intermolecular interactions between optimized geometries of 1ZnTPP° + 3O2° (S0 + T0, both ground states), 3ZnTPP* + 3O2° (T1 + T0, before energy transfer), and 1ZnTPP° + 1O2* (S0 + S1, after energy transfer), hereafter identified [1ZnTPP° + 1O2*]. They resulted in Zn–O distances of 3.05 Å, 3.08 Å, and 2.36 Å respectively. The first two distances are in good agreement with Rusidy's 2.948 Å for triplet oxygen, which indicate very weak, if any interaction between the porphyrins and 3O2, and 2.36 Å also compares well with Rusidy's 2.411 Å between porphyrin and 1O2, indicating weak coordination. 1ZnTPP° + 1MeOH° (S0 + S0) was optimized as well (hereafter [1ZnTPP° + 1MeOH°]), resulting in a Zn–O distance of 2.30 Å (comparable to the 2.12 Å Zhou et al.12 and the 2.07 Å Zhang et al.13 obtained by X-ray), meaning that the ZnTPP–MeOH bond is slightly stronger than the ZnTPP–1O2 bond. This suggests that while both early and late coordination are plausible, late coordination seems more likely.
The calculated ΔE (Fig. 4) between 1ZnTPP° + 3O2° and 3ZnTPP* + 3O2° is 1.58 eV, corresponding to a 0–0 transition at 785 nm, which compares well to experimental results,20 at 77 K in toluene, which record the transition at 770 nm. After energy transfer, the forming of the [1ZnTPP° + 1O2*] complex's Zn–O bond seems indeed a downhill process by 0.17 eV, or 16.4 kJ mol−1, which is in the magnitude of H-bonding strength.
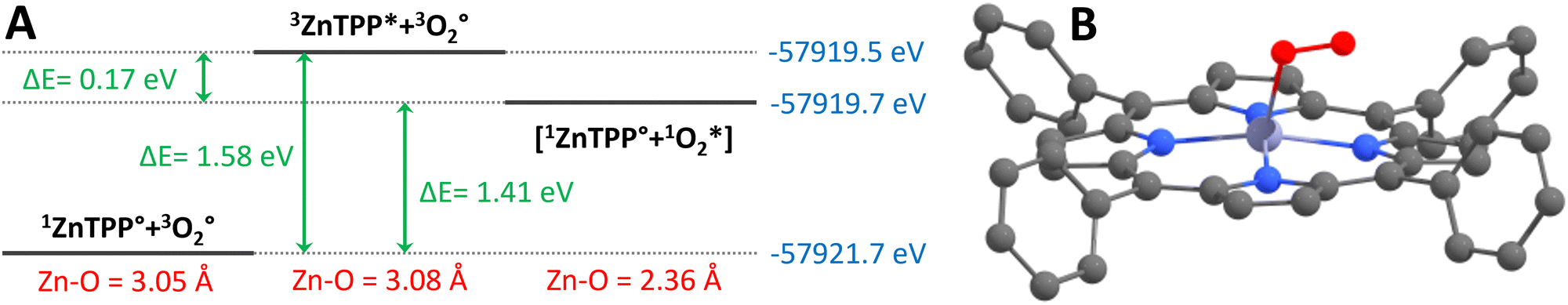 | ||
| Fig. 4 (A) Relative energies from the DFT computations on 1ZnTPP° + 3O2°, 3ZnTPP* + 3O2° and [1ZnTPP° + 1O2*]: blue = total energy of the optimized geometry, green = ΔE between the various species, red = Zn–O distance. (B) Optimized geometry of [1ZnTPP° + 1O2*]. | ||
As to the second question of why the 1O2 phosphorescence lifetime does not follow the trend expected of the heavy atom effect, hypotheses remain mostly speculative. The fact that the Zn–O distance is anticipated to be long may decrease the effect of spin–orbit coupling. It is also possible that we see both the coordination (or H-bonding for H2TPP) component of the lifetime (variable) and non-interaction component (≈10 μs) of 1O2. Finally, the limited number of points (fastest binning is 1 μs due to hardware limitations) undoubtably influences the quality of the fit, and by extension the exact values.
We have reported spectra showing the persistence of interfacial singlet oxygen (1O2(g)) phosphorescence in an argon atmosphere, followed by signal extinction when methanol vapors are introduced in an air atmosphere. This behavior proves the existence of an emissive coordinated complex, either occurring (late coordination) or persisting (early coordination) after the heterogeneous photosensitization process, which is then broken by competition from the comparatively stronger MeOH ligand. Rare crystal structures and excited-state lifetime measurements support the existence of this complex, and while DFT seem to point toward late coordination being more likely, the current level of experimental evidence cannot distinguish between the early and late coordination hypotheses. EPR measurements might help in this regard, as it could enable one to distinguish between non-coordinating and coordinating 3O2 and 1O2. If future experiments determine that the reported behavior is not unique to Zn, a better understanding of 1O2(g)'s interactions with metal-containing molecules could enable some of control over its diffusion distance, and by extension, over its action radius. The action of MeOH also demonstrates that it is possible to disable singlet oxygen production in an environment where the presence of oxygen and light are otherwise favorable to photosensitization. Finally, O2's loss of molecular symmetry when acting as a ligand is certain to break the degeneracy of it's π orbitals, leading to new electronic behaviours which may in time prove useful.
P. Asselin: conceptualization, formulating methodology performing experiments, data treatment, analysis of results, literature review, writing and proofreading manuscript; A. Schlachter: formulating methodology, performing experiments, analysis of results, proofreading manuscript; P. D. Harvey: conceptualization, analysis of results, literature review, writing and proofreading manuscript, funding acquisition, overall supervision of the work.
P. D. Harvey thanks Canada's Natural Sciences and Engineering Research Council (NSERC/CRSNG) for research grant RGPIN-2019-05289. P. Asselin thanks La Fondation de l’Université de Sherbrooke for the Bourse d’excellence Hydro-Québec doctoral scholarship grant and the Fonds de Recherche du Québec-Nature et Technologies for the Bourse de doctorat en recherche #329072 doctoral scholarship grant funding his main research project. Mr Kevin Tanner is posthumously thanked for performing computations.
Data availability
The data supporting this article has been included as part of the ESI.† The data presented in Fig. 4B and Table 1 is reused from Schlachter et al. https://doi.org/10.1002/adfm.202404111.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. U. Khan and M. Kasha, J. Am. Chem. Soc., 1970, 92, 3293–3300 CrossRef CAS.
- A. U. Khan, J. Phys. Chem., 1976, 80, 2219–2228 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS PubMed.
- G. Bauer, Anticancer Res., 2016, 36, 5649–5664 CrossRef CAS PubMed.
- A. Schlachter, P. Asselin and P. D. Harvey, ACS Appl. Mater. Interfaces, 2021, 13, 26651–26672 CrossRef CAS PubMed.
- M. DeRosa, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- J. Wahlen, D. E. De Vos, P. A. Jacobs and P. L. Alsters, Adv. Synth. Catal., 2004, 346, 152–164 CrossRef CAS.
- S. Faure, C. Stern, R. Guilard and P. D. Harvey, Inorg. Chem., 2005, 44, 9232–9241 CrossRef CAS PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- R. Schmidt, Photochem. Photobiol., 2007, 82, 1161–1177 CrossRef.
- J. S. Anderson, A. T. Gallagher, J. A. Mason and T. D. Harris, J. Am. Chem. Soc., 2014, 136, 16489–16492 CrossRef CAS PubMed.
- Z. Zhou, X. Zhou, Q. Liu, X. Zhang and H. Liu, Org. Lett., 2015, 17, 4078–4081 CrossRef CAS PubMed.
- J. Zhang, M. Tang, D. Chen, B. Lin, Z. Zhou and Q. Liu, Inorg. Chem., 2019, 58, 2627–2636 CrossRef CAS PubMed.
- F. Rusydi, M. Kemal Agusta, A. Gandaryus Saputro and H. Kasai, J. Phys. Soc. Jpn., 2012, 81, 124301 CrossRef.
- M. Bodesheim, M. Schütz and R. Schmidt, Chem. Phys. Lett., 1994, 221, 7–14 CrossRef CAS.
- A. Schlachter, P. Asselin, A. Chatelain and P. D. Harvey, Adv. Funct. Mater., 2024, 2404111 CrossRef.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663–677 CrossRef CAS.
- K.-K. Wang, S. Song, S.-J. Jung, J.-W. Hwang, M.-G. Kim, J.-H. Kim, J. Sung, J.-K. Lee and Y.-R. Kim, Phys. Chem. Chem. Phys., 2020, 22, 21664–21671 RSC.
- M. Ruzzi, E. Sartori, A. Moscatelli, I. V. Khudyakov and N. J. Turro, J. Phys. Chem. A, 2013, 117, 5232–5240 CrossRef CAS PubMed.
- V. A. Walters, J. C. de Paula, B. Jackson, C. Nutaitis, K. Hall, J. Lind, K. Cardozo, K. Chandran, D. Raible and C. M. Phillips, J. Phys. Chem., 1995, 99, 1166–1171 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The PDF file contains extra characterisation data, further experimental details and extra computation results. See DOI: https://doi.org/10.1039/d4cc02011a |
| ‡ Safety: see ESI† for safety details. |
| This journal is © The Royal Society of Chemistry 2024 |