
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduced chirality at the surface: fixation of a dynamic M/P invertible helical Co3 complex on SiO2†
Satoshi
Muratsugu
 *ab,
Kana
Sawaguchi
ac,
Takafumi
Shiraogawa
d,
Shunsuke
Chiba
e,
Yoko
Sakata
ef,
Sora
Shirai
a,
Hiroshi
Baba
a,
Masahiro
Ehara
*d,
Shigehisa
Akine
*ef and
Mizuki
Tada
*abc
*ab,
Kana
Sawaguchi
ac,
Takafumi
Shiraogawa
d,
Shunsuke
Chiba
e,
Yoko
Sakata
ef,
Sora
Shirai
a,
Hiroshi
Baba
a,
Masahiro
Ehara
*d,
Shigehisa
Akine
*ef and
Mizuki
Tada
*abc
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8602 Aichi, Japan. E-mail: smuratsugu@chem.nagoya-u.ac.jp; tada.mizuki.u6@f.mail.nagoya-u.ac.jp
bIntegrated Research Consortium on Chemical Science (IRCCS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8602 Aichi, Japan
cResearch Center for Materials Science (RCMS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8602 Aichi, Japan
dInstitute for Molecular Science/School of Physical Sciences, Graduate University for Advanced Studies, Myodaiji, Okazaki, 444-8585 Aichi, Japan. E-mail: ehara@ims.ac.jp
eGraduate School of Natural Science and Technology, Kanazawa University, Kakuma-machi, Kanazawa, 920-1192 Ishikawa, Japan. E-mail: akine@se.kanazawa-u.ac.jp
fNano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kakuma-machi, Kanazawa, 920-1192 Ishikawa, Japan
First published on 5th January 2024
Abstract
Dynamic M/P invertible helicity was successfully induced at a SiO2 surface immobilized with a dynamic helical trinuclear cobalt complex, [LCo3(NHMe2)6](OTf)3, using chiral ((R) or (S))-1-phenylethylamine. Solid-state CD spectra and theoretical calculations suggested that the fixation of the M/P helical complex on the surface via coordination interactions was the key factor of the induced chirality at the surface.
The helix is one of the chiral structures derived from spiral or propeller-like structures, and left- and right-handed orientations of spiral structures produce helicity.1,2 Helical molecules have been applied to molecular machines,3,4 molecular recognition,5 optical probes,6 and so on. In particular, helical molecules with a cage structure have attracted much attention, because these molecules could be used as a chiral host for enantioselective guest binding, a chiral nanoreactor, a chiral molecular switch triggered by guest binding, etc. The tris(saloph) organic cage (denoted as H6L)7–11 is one of the useful helical molecular cages, because metal complexes LM3 were found to show unique dynamic structural conversions in response to guest binding.9–11
Among them, trinuclear cobalt(III) helical cage complexes, denoted as [LCo3] unit, have great advantages for post-synthetic functionalization thanks to the additional coordination to the axial sites of the octahedral Co centres. When the complex has an achiral ligand such as NHMe2, the left-handed (M)-[LCo3(NHMe2)6]3+ and right-handed (P)-[LCo3(NHMe2)6]3+ are enantiomeric and their ratio is 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 at equilibrium (Scheme 1). This achiral ligand can be replaced with a chiral ligand X, which makes the M and P isomers diastereomeric and biased M/P equilibrium. Indeed, this ligand exchange reaction smoothly proceeds upon the addition of chiral ligands such as ((R) or (S))-1-phenylethylamine (denoted as A1) and ((R) or (S))-3-amino-1,2-propanediol (denoted as A2), to give the M/P diastereomeric pair of the complexes, which exhibits intense circular-dichroism (CD) signals due to the shifted M/P equilibrium in a solution.9 We expected that this additional coordination on the Co centre could be utilized both for the fixation of the helical structures of the [LCo3X6] complexes and for the equilibrium shift caused by the ligand exchange.9
:50 at equilibrium (Scheme 1). This achiral ligand can be replaced with a chiral ligand X, which makes the M and P isomers diastereomeric and biased M/P equilibrium. Indeed, this ligand exchange reaction smoothly proceeds upon the addition of chiral ligands such as ((R) or (S))-1-phenylethylamine (denoted as A1) and ((R) or (S))-3-amino-1,2-propanediol (denoted as A2), to give the M/P diastereomeric pair of the complexes, which exhibits intense circular-dichroism (CD) signals due to the shifted M/P equilibrium in a solution.9 We expected that this additional coordination on the Co centre could be utilized both for the fixation of the helical structures of the [LCo3X6] complexes and for the equilibrium shift caused by the ligand exchange.9
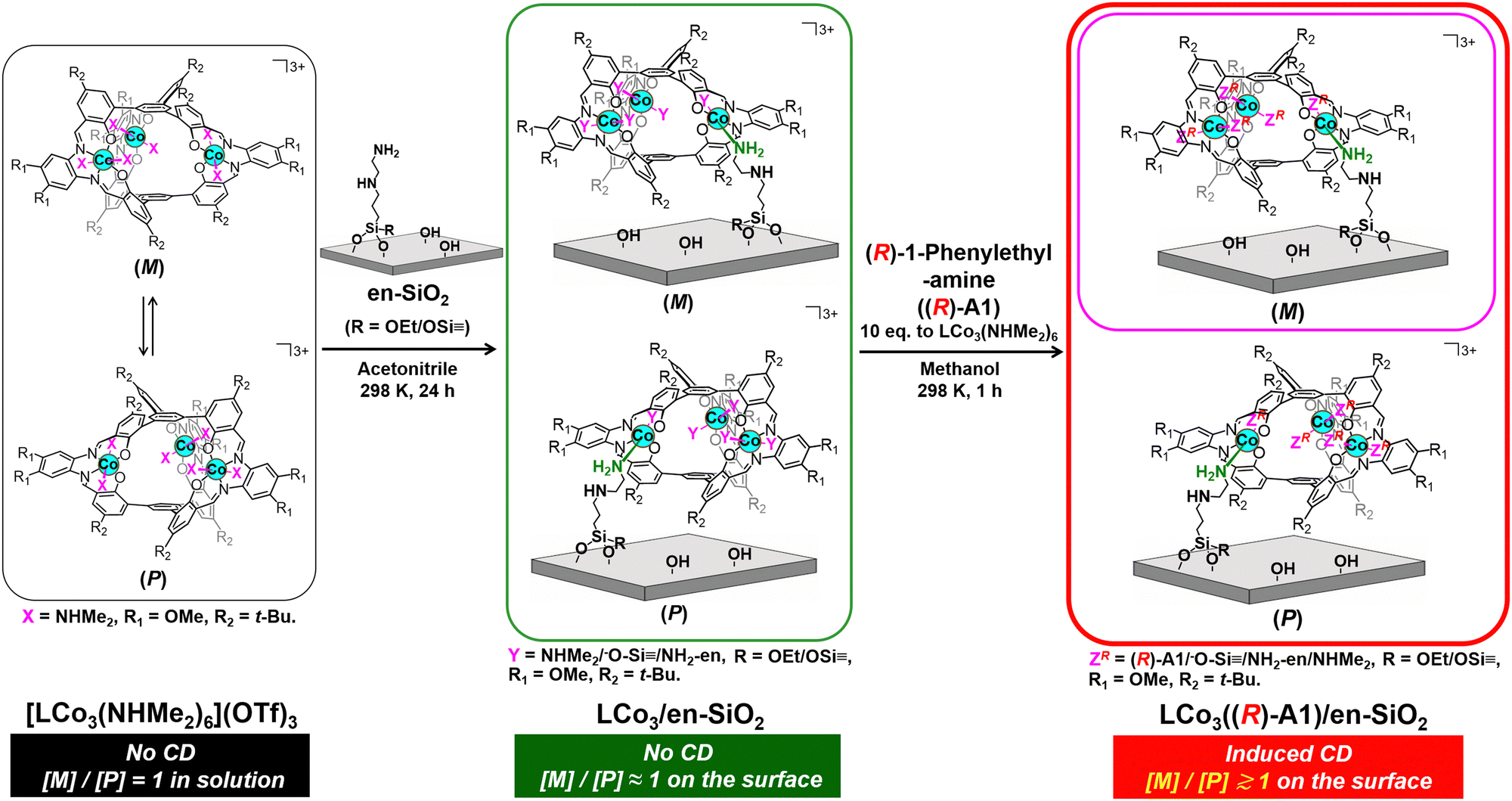 | ||
| Scheme 1 Preparation of a supported helical LCo3 complex, LCo3((R)-A1)/en-SiO2, by grafting of [LCo3(NHMe2)6](OTf)3 on an ethylenediamine-functionalized SiO2 surface (en-SiO2), followed by the addition of chiral (R)-1-phenylethylamine. Possible paired surface structures are presented. The whole scheme for LCo3((R) or (S)-A1)/en-SiO2 is presented in the ESI† (Scheme S1). | ||
A solid surface has been used for the support of functional compounds, and the attachment of metal complexes on a solid surface has been widely applied for functional materials such as heterogeneous catalysts and sensors.12–15 The chemical attachment of a metal complex to a solid surface is designed by the chemical reaction of a metal complex precursor and functional groups that originally existed on the surface (e.g. silanol (Si–OH)) or functional groups that are grafted on the surface.16,17 The attachment of metal complexes on solid surfaces often produces unique metal coordination structures, which are different from precursor complexes. This process is also expected to have a great influence on controlling the conformation of the helical structures of metal complexes.
In this paper, we investigated the fixation of the dynamic M/P invertible helicity of [LCo3(NHMe2)6](OTf)3 on a SiO2 surface on which ethylenediamine was functionalized (en-SiO2). The addition of the chiral amine ligands ((R) or (S))-A1 successfully induced the deviation of the dynamic helicity of the Co3 complex at the surface. We demonstrated that the M/P equilibration was almost suppressed on the solid surface even in the presence of the chiral ligand, because this showed a unique CD spectrum, which was significantly different from that in solution. The theoretical calculation of the surface-attached helical structures suggested the origin of these induced CD spectra.
[LCo3(NHMe2)6](OTf)3 was prepared according to the literature.9 The attachment of [LCo3(NHMe2)6](OTf)3 was investigated by using en-SiO2, whose surface ethylenediamine site was used as the coordination site of [LCo3(NHMe2)6](OTf)3 (Scheme 1). H2N(CH2)2NH(CH2)3Si(OCH3)3 was reacted with Si–OH groups on a SiO2 surface, and the surface density of the attached ethylenediamine moiety was regulated as low as 2.3 nm−2 to avoid the aggregation of the ethylenediamine branches at the SiO2 surface.
[LCo3(NHMe2)6](OTf)3 was impregnated with the prepared en-SiO2 in acetonitrile (Scheme 1; denoted as LCo3/en-SiO2). The averaged Co loading on LCo3/en-SiO2 was estimated to be 0.6 wt% by XRF, which corresponds to be 0.1 [LCo3] unit per nm2 at the SiO2 surface. Similar impregnation on SiO2 without the functionalization of the ethylenediamine moiety was conducted (LCo3/SiO2), but a substantial amount of the precursor was leached out to the solution phase by washing with acetonitrile, suggesting that [LCo3(NHMe2)6](OTf)3 cannot be stably immobilized on the SiO2 surface without the ethylenediamine moiety. Therefore, the surface ethylenediamine sites functionalized on SiO2 were essential to the attachment of the [LCo3(NHMe2)6](OTf)3 on SiO2.
In the diffuse-reflectance (DR) UV-vis spectrum of LCo3/en-SiO2, peaks at 401, 424, and 494 nm were observed, which were similar to those in the UV-vis spectrum of [LCo3(NHMe2)6](OTf)3 in methanol (397, 420, and 502 nm), indicating that the [LCo3] unit was kept after the attachment on en-SiO2 (Fig. 1(A)(a and b)). Similar Co K-edge energy in the X-ray absorption near edge structure (XANES) spectra of the [LCo3(NHMe2)6](OTf)3 precursor and LCo3/en-SiO2 suggest that the oxidation states of Co were similar to each other before and after the surface attachment on SiO2 (Fig. S1, ESI†).
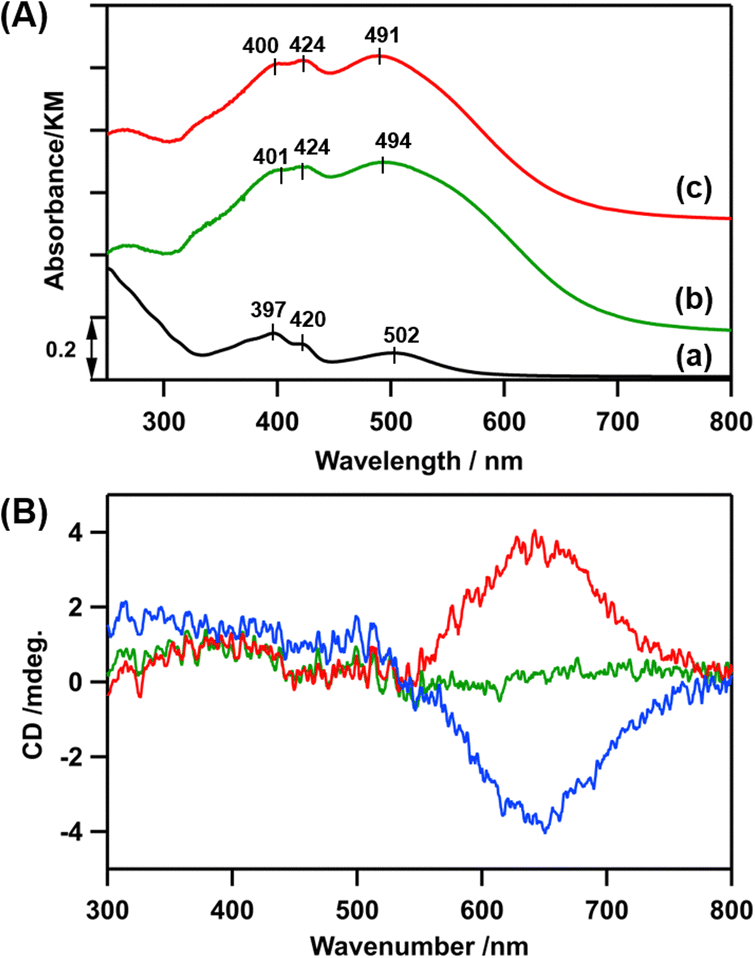 | ||
| Fig. 1 (A) (a) A transmission UV-vis spectrum of [LCo3(NHMe2)6](OTf)3 in methanol (c = 0.2 mol L−1). (b) and (c) DR UV-vis spectra of (b) LCo3/en-SiO2, and (c) LCo3((R)-A1)/en-SiO2. (B) DR CD spectra of LCo3/en-SiO2 (green), LCo3((R)-A1)/en-SiO2 (red) and LCo3((S)-A1)/en-SiO2 (blue). | ||
Then, the addition of chiral (R)- or (S)-A1 was conducted on LCo3/en-SiO2. 10 eq. (to [LCo3] unit) of (R)- or (S)-A1 was impregnated with LCo3/en-SiO2 in methanol, and the DR UV-vis, Co K-edge XANES, and DR CD spectra of the obtained samples (denoted as LCo3((R) or (S)-A1)/en-SiO2) were measured. The DR UV-vis and Co K-edge XANES spectra of LCo3((R) or (S)-A1)/en-SiO2 were almost similar to those of LCo3/en-SiO2 as shown in Fig. 1(A)(c) and Fig. S1 (ESI†), indicating that the [LCo3] unit was maintained after the coordination of the chiral A1.
On LCo3/en-SiO2 before the addition of the chiral A1, its DR CD spectrum was silent, suggesting that the amounts of the P and M conformations were comparable on the SiO2 surface (Scheme 1). In contrast, significant CD peaks at 645 nm, which were the positive/negative mirror peaks, were observed after the addition of (R)-/(S)-A1, as shown in Fig. 1(B). The CD peak top position of LCo3((R)-A1)/en-SiO2 was different from the peak top position observed in its UV-vis spectrum (491 nm) (Fig. 1(A and B)). The biased M/P equilibrium of [LCo3] unit was suggested but the CD spectra of LCo3((R)-A1)/en-SiO2 were remarkably different from those of the [LCo3((R)-A1)6](OTf)3 in methanol (Fig. S4(E), ESI†).9
We conducted DFT calculations to look for the possible origin of the unique CD spectra of LCo3((R) or (S)-A1)/en-SiO2. We modelled the plausible coordination structures of the SiO2-supported [LCo3] unit and simulated their UV-vis and CD spectra (see ESI†). First, the two helical structures of (M) and (P)-[LCo3(NHMe2)6]3+ were optimized, and the optimized structures agreed well with those obtained from single crystal X-ray diffraction (Fig. S3, ESI†). The TD-DFT calculations of the excited states of the complexes provided the simulated UV-vis and CD spectra of [LCo3(NHMe2)6]3+ (Fig. 2(A) and Fig. S3, ESI†), which suitably reproduced the experimentally observed UV-vis spectra of (M) and (P)-[LCo3(NHMe2)6]3+. [LCo3] unit at which all NHMe2 ligands were substituted to (R)-A1, (M)-[LCo3((R)-A1)6](OTf)3 and (P)-[LCo3((R)-A1)6](OTf)3 (Fig. S4, ESI†), were also optimized, and the calculated UV-vis and CD spectra of (M)-[LCo3((R)-A1)6](OTf)3,9 showed similar trends to those experimentally obtained (Fig. 2(B)).
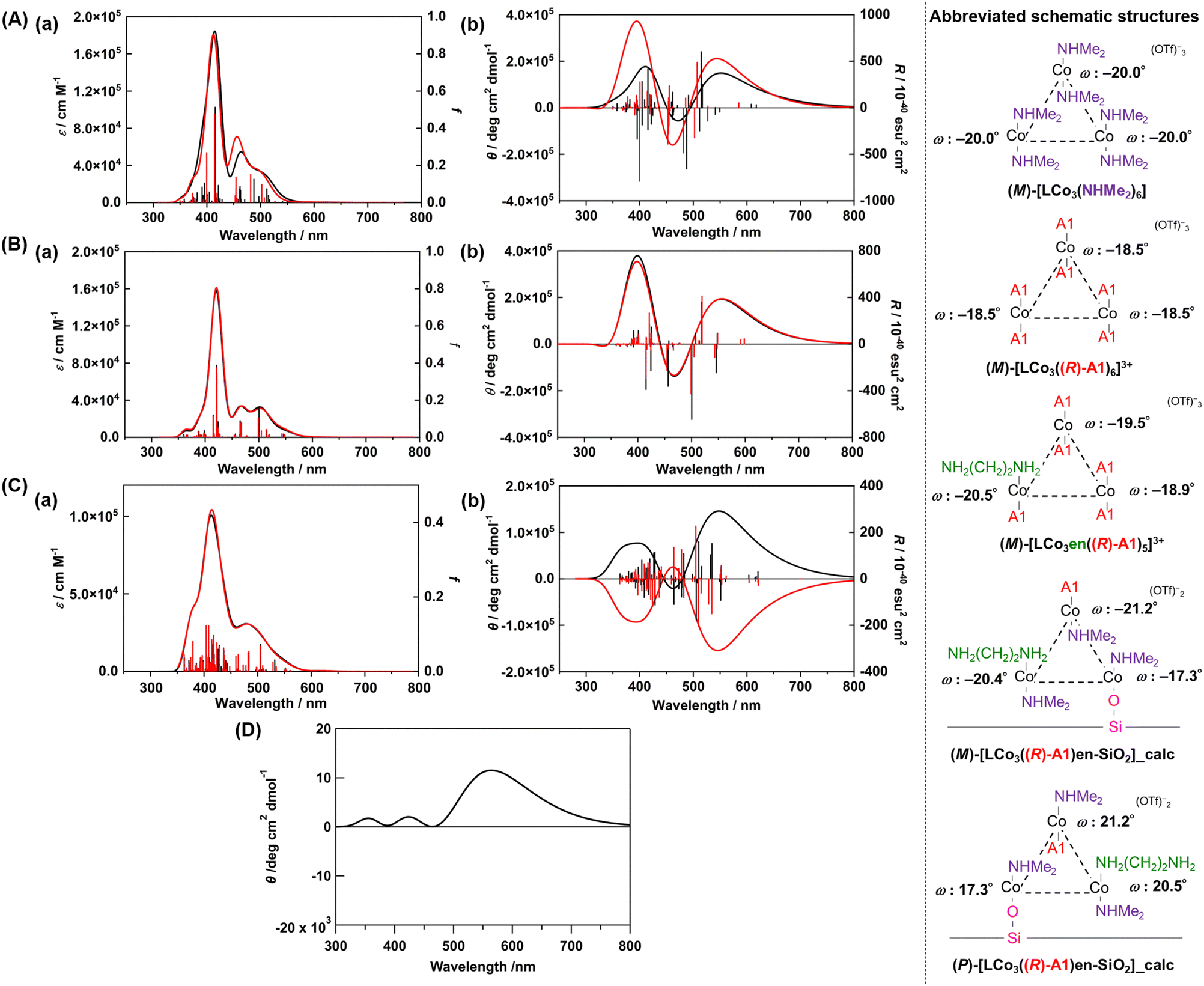 | ||
| Fig. 2 (A) Calculated (a) UV-vis and (b) CD spectra of (M)-[LCo3(NHMe2)6]3+ (black) and (M)-[LCo3((R)-A1)6]3+ (red) (in acetonitrile). (B) Calculated (a) UV-vis and (b) CD spectra of (M)-[LCo3((R)-A1)6]3+ (black) and (M)-[LCo3en((R)-A1)5]3+ (red) (in vacuum). (C) Calculated (a) UV-vis and (b) CD spectra of the modelled structure of LCo3((R)-A1)/en-SiO2_calc ((M) configuration) (black) and LCo3((R)-A1)/en-SiO2_calc ((P) configuration) (red) (in vacuum). (D) The summed simulated CD spectrum of the modelled structure of LCo3((R)-A1)/en-SiO2_calc ((M) configuration) and LCo3((R)-A1)/en-SiO2_calc ((P) configuration) with the ratio of 55/45. Abbreviated schematic structures (see ESI† and Fig. S2 for details) of the optimized trinuclear cobalt complexes are also presented. The torsion angles of each saloph moiety of the optimized structures under vacuum are also shown as ω. | ||
Then, ligand substitution from A1 to ethylenediamine at [LCo3] unit was investigated. (M)-[LCo3en((R)-A1)5]3+ was designed by using H2N(CH2)2NH2 as the model of the ethylenediamine site on SiO2 and coordinating it to one of the Co sites in [LCo3] unit (Fig. 2). The theoretical UV-vis and CD spectra of (M)-[LCo3((R)-A1)6]3+, (M)-[LCo3en((R)-A1)5]3+, and (M)-[LCo3(NHMe2)6]3+ were found to be almost similar to each other (Fig. 2(B)(a and b)). The ligand exchange to (R)-A1 and the ethylenediamine unit on SiO2 was suggested not to give significant changes in their UV-vis and CD spectra.
Finally, we investigated the interaction from the surface Si–OH group on LCo3((R)-A1)/en-SiO2. The structural models of LCo3((R)-A1)/en-SiO2 with the coordination of ethylenediamine and Si-O− (H+ was removed from Si–OH.) were calculated (denoted as LCo3((R)-A1)/en-SiO2_calc). By comparison between the calculated UV-vis spectra of (M)-[LCo3((R)-A1)6]3+, (M)-[LCo3en((R)-A1)5]3+, and (M)-LCo3((R)-A1)/en-SiO2_calc (Fig. 2(C)(a)), the peak intensity at around 480 nm of (M)-[LCo3((R)-A1)6]3+ and (M)-[LCo3en((R)-A1)5]3+ became small on that of (M)-LCo3((R)-A1)/en-SiO2_calc. In the CD spectra of (M)- and (P)-LCo3((R)-A1)/en-SiO2_calc (Fig. 2(C)(b)), it should be noted that the CD spectral intensities at 500–600 nm became large compared with those at 350–450 nm.
The summed CD spectra of (M)- and (P)-LCo3((R)-A1)/en-SiO2_calc were simulated by changing the molar ratio of (M) and (P) diastereomers (see ESI† for detailed calculation methods). It should be noted that the simulated CD spectra show a dominating peak at 500–600 nm and negligible peak at 350–450 nm and the observed CD intensities are well reproduced at the diastereomeric ratio of M/P = 55/45 (Fig. 2(D) and Fig. S5, ESI†). The estimated M/P ratio was less biased than the equilibrated ratio of M/P = 88:12 in a methanol solution (c = 0.2 mmol L−1), which was experimentally observed for [LCo3((R)-A1)6]3+.9 Thus, the emergence of this characteristic CD spectra suggested that the M/P equilibrium shift of [LCo3((R)-A1)6]3+ would be suppressed by the attachment to the en-SiO2 surface in the current system. Due to this equilibrium suppression, the differences in the peak top positions of the UV-vis and CD spectra of LCo3((R)-A1)/en-SiO2 emerged, which were supported by the current calculations.
When an excess amount (100 eq. to [LCo3] unit) of (S) or (R)-A2 was added to LCo3((R) or (S)-A1)/en-SiO2, respectively, the DR CD spectra completely inverted as shown in Fig. S6 (ESI†), and the peak tops that appeared at 564 nm and 467 nm were close to the CD spectrum of [LCo3((S) or (R)-A2)6](OTf)3 (538 nm and 467 nm) in methanol (c = 0.2 mmol L−1).9 These results suggested that all axial A1 ligands of the [LCo3] unit were exchanged to (S) or (R)-A2, and the helicity of the [LCo3] unit was totally inverted by the ligand exchange. These results also supported that the origin of the unique induced CD spectra would be derived from the dual coordination of the ethylenediamine unit and the surface Si–O− group on the SiO2 surface.
In summary, unique helicity induction was observed on a SiO2 surface by the attachment of the [LCo3(NHMe2)6](OTf)3 complex and the addition of chiral (R) or (S)-phenylethylamine A1. Solid-state CD spectra and theoretical calculations suggested that the coordination of both the ethylenediamine site and Si–OH would be key for the immobilized helical structures, and the M/P ratio of [LCo3] unit on en-SiO2 was found to be different from the corresponding ratio observed in a solution. The current results demonstrate that a solid surface acts as the field of fixation of dynamic helicity, and that oxide surfaces are promising to work as a tuner of the M/P ratio of the dynamic helical complexes.
This work was supported in part by MEXT/JSPS KAKENHI (Grant No. JP16H06510 (S. A.), JP16H06511 (M. E.), JP16H06512 (M. T.) (coordination asymmetry), JP18K05144 (S. M.), JP18H01940 (M. T.), JP20H02718 (M. E.), JP22H05133 (M. E.), JP21J00210 (T. S. JSPS Fellows)), and the Integrated Research Consortium on Chemical Sciences. XAFS measurements were performed with the approval of PF-PAC (no. 2016G078). The computations were partially performed in the Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C185).
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Mori, Chem. Rev., 2021, 121, 2373 CrossRef CAS PubMed.
- A. Tsurusaki and K. Kamikawa, Chem. Lett., 2021, 50, 1913 CrossRef CAS.
- N. Ousaka and E. Yashima, Chem. Lett., 2021, 50, 320 CrossRef CAS.
- E. Yashima and K. Maeda, Bull. Chem. Soc. Jpn., 2021, 94, 2637 CrossRef CAS.
- H. Gholami, D. Chakraborty, J. Zhang and B. Borhan, Acc. Chem. Res., 2021, 54, 654 CrossRef CAS PubMed.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673 RSC.
- S. Akine, M. Miyashita and T. Nabeshima, J. Am. Chem. Soc., 2017, 139, 4631 CrossRef CAS PubMed.
- S. Akine, M. Miyashita and T. Nabeshima, Chem. – Eur. J., 2019, 25, 1432 CrossRef CAS PubMed.
- Y. Sakata, S. Chiba, M. Miyashita, T. Nabeshima and S. Akine, Chem. – Eur. J., 2019, 25, 2962 CrossRef CAS PubMed.
- S. Akine, M. Miyashita and T. Nabeshima, Inorg. Chem., 2021, 60, 12961 CrossRef CAS PubMed.
- Y. Sakata, S. Chiba and S. Akine, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2113237119 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456 CrossRef CAS PubMed.
- J. M. Notestein and A. Katz, Chem. – Eur. J., 2006, 12, 3954 CrossRef CAS PubMed.
- M. Tada and Y. Iwasawa, Coord. Chem. Rev., 2007, 251, 2702 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323 CrossRef PubMed.
- S. Muratsugu, Z. Weng, H. Nakai, K. Isobe, Y. Kushida, T. Sasaki and M. Tada, Phys. Chem. Chem. Phys., 2012, 14, 16023 RSC.
- S. Muratsugu, M. H. Lim, T. Itoh, W. Thumrongpatanaraks, M. Kondo, S. Masaoka, T. S. A. Hor and M. Tada, Dalton Trans., 2013, 42, 12611 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Whole image of Scheme 1, experimental details, XANES and CD spectra, computational details, and calculated UV-vis and CD spectra. See DOI: https://doi.org/10.1039/d3cc05534e |
| This journal is © The Royal Society of Chemistry 2024 |