
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dual encapsulation strategy to generate anion-responsive luminescent lanthanide hydrogels†
Samantha E.
Bodman
 ab,
Colum
Breen
ab,
Annaliese Rebecca
Hambleton
a,
Stephen J.
Butler
*b and
Helen
Willcock
*a
ab,
Colum
Breen
ab,
Annaliese Rebecca
Hambleton
a,
Stephen J.
Butler
*b and
Helen
Willcock
*a
aDepartment of Materials, Loughborough University, Epinal Way, Loughborough, LE11 3TU, UK. E-mail: h.willcock@lboro.ac.uk
bDepartment of Chemistry, Loughborough University, Epinal Way, Loughborough, LE11 3TU, UK. E-mail: s.j.butler@lboro.ac.uk
First published on 21st November 2023
Abstract
We report a new method to generate ion-responsive luminescent hydrogels, involving encapsulation of a luminescent lanthanide probe within crosslinked amphiphilic polymer particles and subsequent entrapment within a hydrogel. The resulting hydrogels are capable of reversible bicarbonate sensing, exhibit no leaching, and can be tuned for a range of sensing applications.
Chemical sensing is a huge growing market in biomedical and healthcare sectors based on the increased demand for quick, precise, and portable diagnostic equipment for monitoring disease progression and in-home healthcare. The global market is predicted to reach over USD 29 billion by 2030.1 Chemical sensors (or chemosensors) report on the concentration of a specific analyte or total composition of a sample providing a measurable signal,2 most commonly an electrochemical or luminescent signal due to their high sensitivity and rapid response.3,4 Chemosensors that bind reversibly and selectivity to their target analyte offer the potential for continuous analysis of health status and disease progression.5,6 However, most solution-based chemosensors are restricted to single use measurements which limits their utility in biological and biomedical applications. This issue may be addressed by the incorporation of chemosensors into materials that can be embedded within existing diagnostic and analytic devices.7
Different methods to incorporate chemosensors within materials have been devised including Langmuir Blodgett layers,8,9 silica nanoparticles,10 metal–organic frameworks11 and polymeric materials, including hydrogels.12–14 Hydrogel materials offer several advantages including their ability to absorb large amounts of water, relative ease of synthesis, and tuneable functionality and mechanical properties based on the composition of monomers used. The ability of hydrogels to swell and absorb water is advantageous for biological sensing applications provided that the system is designed to enable rapid diffusion of the analyte to the incorporated chemosensor. However, the challenge remains to integrate a chemosensor into a hydrogel material without perturbing its selectivity, sensitivity and emission response.
A chemosensor may be incorporated into a hydrogel through encapsulation15 or covalent attachment. Direct encapsulation of the chemosensor during hydrogel synthesis offers the simplest method in principle; however, this is likely to cause significant leaching of the sensor from the hydrogel over time. Indeed, this approach has been used for slow and controlled release of drug molecules.16,17 Notably, Gunnlaugsson non-covalently entrapped a pH-responsive europium probe into HEMA-based hydrogels demonstrating minimal leaching at room temperature, which increased at 37 °C.18 Alternatively, the covalent attachment of a chemosensor to the hydrogel can overcome leaching issues, but this approach requires additional synthesis and sensor modification, which can negatively impact its analyte affinity, selectivity, and sensitivity.19
Herein we present a new hydrogel sensing platform20 based on a dual encapsulation strategy, wherein the luminescent chemosensor is trapped within an amphiphilic polymer particle that is subsequently embedded within the hydrogel matrix. The performance of the hydrogel sensor is exemplified using a bicarbonate responsive europium complex.21 We demonstrate that this chemosensor does not leach out of the hydrogel, is capable of reversible sensing and thus multiple re-use cycles. In addition, the material is flexible, robust and can be stored for extended time periods. The design strategy presented here can be used to encapsulate a range of chemosensors without the need for chemical modification, thus may be applied to a range of sensing applications.
The particles were carefully chosen to promote the encapsulation of the bulky aromatic complex [Eu.1]+ within the more hydrophobic polymethyl methacrylate (PMMA) core. A polyoligoethylene glycol methyl ether methacrylate (POEGMEM) hydrophilic shell was chosen with a relatively high degree of polymerisation (DP 75) to induce stable spherical particle formation.22 We have previously demonstrated that incorporation of >1 wt% of the crosslinker ethylene glycol dimethacrylate (EGDMA) ensures concentration independent stability of the particles.23 The hydrogel was designed to allow rapid uptake of the analyte solution, whilst preserving the encapsulated particles and thus the probe itself. The particle and hydrogel chemical and physical properties are easily tuned to control uptake and sensing of biological anions. We selected the stable monocationic europium complex [Eu.1]+ for encapsulation, which we have previously shown to reversibly bind bicarbonate producing a large enhancement in europium-centred luminescence.21 Emissive lanthanide complexes have attractive optical properties for use in bioassays and diagnostics, including long emission lifetimes that permit time-resolved measurements to eliminate background fluorescence and light scattering.24,25 Complex [Eu.1]+ serves as an appropriate model for encapsulation and represents a family of lanthanide-based sensors that respond to biological anions.26–29
The synthesis of the particles begins with formation of hydrophilic polymer p(OEGMEM)75 (Scheme S1, ESI†) and subsequent chain extension of the hydrophilic shell (Scheme S2, ESI†) according to our previously disclosed procedures.20,22 Full details of the polymer synthesis and characterisation are provided in the ESI.† The molecular weight (DP = 75) of the polymer was confirmed by 1H NMR spectroscopy, and the low dispersity and controlled RAFT-polymerisation confirmed by GPC (Fig. S1b, ESI†). The particles were analysed by DLS, with particle sizes of 65–270 nm produced (Fig. S2b and S2c, ESI†).
The proof of concept of probe encapsulation was exploited with the emissive europium complex [Eu.1]+ previously reported by the Butler group,30 as well as commercially available luminescent complex tris(2,2′-bipyridyl)dichlororuthenium(II)hexahydrate, [Ru(bpy)3]2+ (Fig. S3, ESI†). The anion responsive europium(III) probe contains a quinoline-functionalised macrocycle which has been shown to induce an emission increase upon binding certain anions, in particular bicarbonate21 and ADP.31 [Eu.1]+ was encapsulated within the core of the particles during the extended-chain polymer step (Fig. 1 and Scheme S2, ESI†). After dialysis, the synthesised particles displayed the expected europium(III) emission of the encapsulated probe in the absence of anions (Fig. S4a, ESI†).
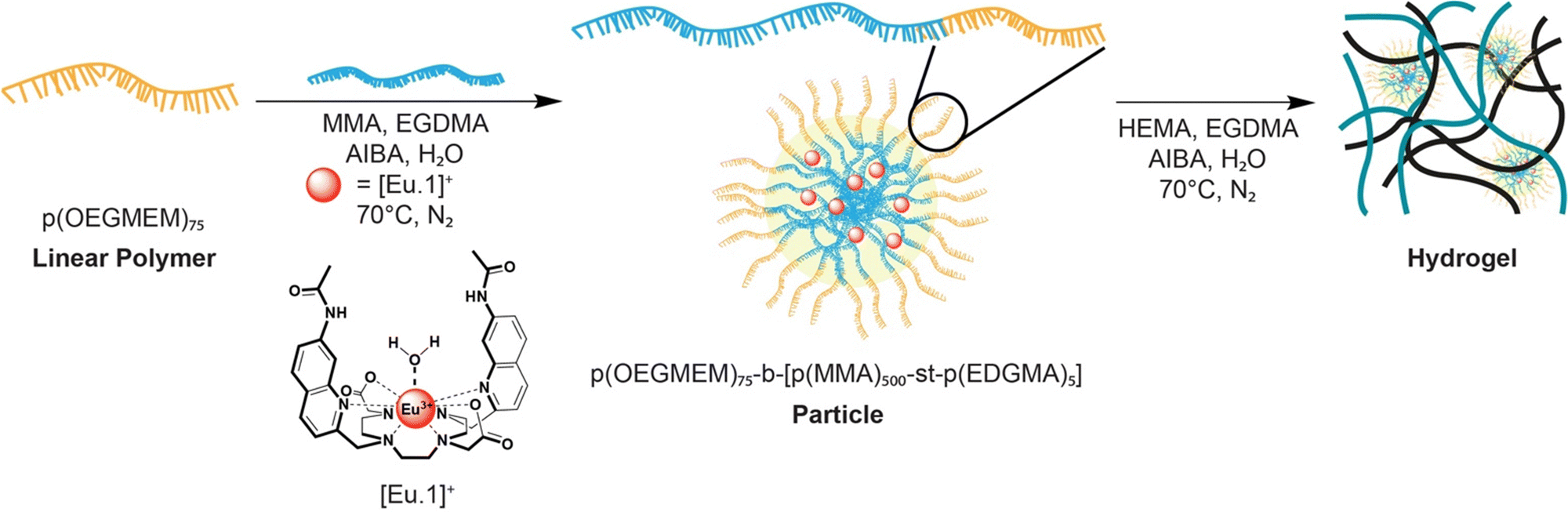 | ||
| Fig. 1 Schematic representation of the synthesis of the particles with europium(III) probe encapsulation and subsequent hydrogel synthesis using our dual encapsulation method. | ||
Next, we examined the emission response of the particles containing the encapsulated [Eu.1]+ probe upon addition of increasing amounts of bicarbonate (Fig. S4, ESI†). Pleasingly, the probe-loaded particles showed a sensitive emission enhancement and distinctive changes in spectral shape in the presence of bicarbonate, very similar to the emission response observed for the molecular probe in aqueous solution. An apparent binding constant was determined for the encapsulated [Eu.1]+ with bicarbonate of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka = 3.4, which is comparable with that previously reported for the probe alone under the same conditions,30 indicating that the particles did not prevent the anion from diffusing and interacting with the complex and the affinity of the complex for bicarbonate is unchanged. A large increase in emission lifetime of the Eu-loaded particles (from 0.01 ms to 0.42 ms following addition of 30 mM bicarbonate) indicated displacement of the coordinated water as expected (Fig. S5, ESI†).
Ka = 3.4, which is comparable with that previously reported for the probe alone under the same conditions,30 indicating that the particles did not prevent the anion from diffusing and interacting with the complex and the affinity of the complex for bicarbonate is unchanged. A large increase in emission lifetime of the Eu-loaded particles (from 0.01 ms to 0.42 ms following addition of 30 mM bicarbonate) indicated displacement of the coordinated water as expected (Fig. S5, ESI†).
Stable particles with diameters between 65–270 nm were achieved by controlling the amount of MMA and monitoring the extent of polymerisation,22 and subsequently employed in the formation of responsive gels (Fig. S6, ESI†). This range of particle sizes gave the best response towards bicarbonate. We verified that the [Eu.1]+ probe does not leach out from the optimised particles over extended time periods, and we demonstrated that a structurally different complex [Ru(bpy)3]2+ can be effectively encapsulated using the same methodology. The particles were analysed by SEM displaying the desired spherical shapes as previously reported.22
Next, we incorporated the emissive particles containing [Eu.1]+ into hydrogels (Scheme S3, ESI†). The hydrogels were synthesised using silicon moulds sandwiched between two microscope slides (Fig. S7a, ESI†). Details of the synthesis are provided in the ESI.† Briefly, the particle solution, 2-(hydroxyethyl)methacrylate (HEMA) and crosslinker were stirred at room temperature, the initiator was added, and solution purged with nitrogen. After five minutes the reaction mixture was transferred via syringe to a silicon mould, cured in the oven at 80 degrees and removed from the mould while still warm to prevent fracture during removal. Following successful formation of the hydrogel and dialysis, the hydrogels were analysed by fluorimetry. The hydrogel was cut and then placed diagonally in a 1 cm × 1 cm quartz cuvette, as shown in Fig. S8 (ESI†). The characteristic Eu(III) emission of the probe can be seen clearly in Fig. S9 (ESI†), confirming successful incorporation of the particles into the hydrogel. The steady-state emission spectra of the [Eu.1]+-loaded particles and the hydrogel showed significant light scattering that obscured the Eu(III)-centred emission (Fig. S9a, ESI†). However, this issue was overcome readily by recording time-resolved emission spectra resulting in the complete removal of light scattering (Fig. S9b, ESI†). Furthermore, the hydrogel displayed clear red emission under a standard laboratory UV-lamp (Fig. S7b, ESI†), indicating the presence of the hydrated Eu(III) complex. When the hydrogel was dehydrated on exposure to air, stronger luminescence was observed under the UV-lamp due to a reduction in non-radiative energy transfer to surrounding water molecules.
SEM images of the material reveal that the particles appear to be clustered within the pores of the hydrogel (Fig. 2a). A hydrogel control sample was also synthesised (Scheme S3, ESI†) containing no particles; in this case the SEM images show unoccupied pores throughout the hydrogel network (Fig. 2b).
 | ||
| Fig. 2 Scanning electron microscopy (SEM) images of (a) hydrogels containing [Eu.1]+ loaded particles embedded in the pores of the hydrogel, and (b) hydrogels lacking particles showing unoccupied pores. | ||
Hydrogels with various initial crosslinker concentrations (1–20% v/v) were synthesised to analyse the effect on the rheological characteristics, mechanical properties, swelling behaviour and potential particle/probe leaching from the material. Rheological studies of the hydrogels show the gel point decreases with increasing crosslinker, when hydrogels containing 1%, 5% and 10% crosslinker were compared (Fig. S10, ESI†). Furthermore, three different 10% crosslinker hydrogels were synthesised and analysed containing different weight-percentage of particles (2 wt% and 10 wt%). The gel point of the hydrogels further decreases upon increasing the particle loading from 2 wt% to 10 wt% (Fig. S10, ESI†). Comparison of the tensile strength of the differing crosslinked hydrogels display an increase with increasing crosslinker (1% to 10%) and the addition of particles appears to increase this further (Fig. S12a, ESI†).32 The same trend was observed for the toughness of the hydrogels (Fig. S12b, ESI†). We further investigated the effect of the crosslinker within the hydrogels by studying the uptake of water. It was found that increasing the crosslinker amount decreases the initial rate of swelling (Fig. S14, ESI†),33 whereas the particle loading level did not appear to influence the uptake of water of hydrogels composed of 10% crosslinker.34–36
The physical properties of the hydrogels confirm that as expected as more crosslinker is added, the less flexible and more brittle the material becomes (Fig. S15, ESI†).37 Moreover the amount of crosslinker affected the anion response of the [Eu.1]+ probe. When 20% crosslinker was added, it appeared that the anion could not permeate the hydrogel within a 30 minute time period, resulting in very little bicarbonate response. In contrast, when 1% crosslinker was used the material became very flexible (Fig. S15a, ESI†) but displayed leaching with a decrease in emission response over time. The optimum amount of 5–10% crosslinker showed a sensitive and stable response to bicarbonate with no leaching observed over 72 hours (Fig. S16a, ESI†). The stability of emission of the hydrogel containing [Eu.1]+ loaded particles with 2 wt% crosslinker was tested over a two-month period, confirming that the particles did not leach from the hydrogel over extended time periods (Fig. S16, ESI†). These hydrogels can be made within different sized moulds, are flexible and can be cut into different shapes.38
Finally, we demonstrated the ability of the hydrogel to respond to changes in bicarbonate in a reversible manner. We emersed the hydrogel containing [Eu.1]+ loaded particles into a buffered solution containing 30 mM bicarbonate. Upon binding of bicarbonate to the [Eu.1]+ within the hydrogel the Eu(III) emission visibly increases (Fig. 3b), reaching maximum intensity after 15 minutes (Fig. 3c, cycle 1). The emission lifetime of the Eu-loaded hydrogel increased to a similar value to that of the particles in the presence of bicarbonate (Fig. S17, ESI†). The hydrogel can then be washed with buffered aqueous solution to remove the anion and the hydrogel is then ready to perform another sensing measurement. We showed that the sensing of bicarbonate can be performed over three cycles, with consistent maximum emission values obtained for each cycle (Fig. 3c). These data demonstrate the reversibility and thus re-usability of the hydrogel-based sensors.
 | ||
| Fig. 3 (a) Emission titration of [Eu.1]+ loaded particles with bicarbonate. (b) Photos of hydrogels containing [Eu.1]+ loaded particles under UV-light, showing the visual increase in red emission intensity upon 15 minute incubation with bicarbonate. (c) Reversible increase in emission of the hydrogel upon three repeat sensing cycles, involving incubation with 30 mM bicarbonate, followed by washing in buffered aqueous solution. | ||
In summary, we have developed a new dual encapsulation method to generate stable anion-responsive hydrogels, involving encapsulation of a discrete luminescent lanthanide complex within amphiphilic polymer particles and subsequent entrapment within a crosslinked hydrogel. Our dual encapsulation ensures that no leaching of the emissive probe occurs over extended time periods. The particles are synthesised through RAFT emulsion polymerisation and the resulting hydrogels are easy to prepare, flexible and easily manipulated into different shapes and sizes. We demonstrate that the hydrogels are capable of reversible sensing in three repeated cycles of bicarbonate exposure followed by a washing step. The hydrogel composition can be tuned to encapsulate different emissive probes and sensors without the need for chemical modification. These combined features open the door for a range of sensing applications, with both dip-and-read and microfluidic devices envisaged.
This work was supported by the Loughborough University EPSRC Impact Accelerator Account (EP/X525595/1) and a Royce Undergraduate Research Internship Scheme. The authors acknowledge use of the facilities and the assistance of Sam Davis in the Loughborough Materials Characterisation Centre, and thank Roshna Mistry from the Research and Innovation office for support obtaining funding. We gratefully acknowledge support from High Force Research regarding scale-up synthesis of [Eu.1]+.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Market Research Future (MRFR), Chemical Sensors Market Projected to Hit USD 29.27 Billion at a CAGR of 5.52% by 2030, New York, 2022.
- A. Hulanicki, S. Glab and F. Ingman, Pure Appl. Chem., 1991, 63, 1247–1250 CrossRef.
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- B. Daly, J. Ling and A. P. De Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS PubMed.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- M. Clemente-León, E. Coronado, A. Soriano-Portillo, C. Mingotaud and J. M. Dominguez-Vera, Adv. Colloid Interface Sci., 2005, 116, 193–203 CrossRef.
- J. A. Kitchen, D. E. Barry, L. Mercs, M. Albrecht, R. D. Peacock and T. Gunnlaugsson, Angew. Chem., 2012, 124, 728–732 CrossRef.
- M. Comes, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, L. A. Villaescusa, A. Graefe and G. J. Mohr, J. Mater. Chem., 2008, 18, 5815–5823 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- I. N. Hegarty, S. J. Bradberry, J. I. Lovitt, J. M. Delente, N. Willis-Fox, R. Daly and T. Gunnlaugsson, Mater. Chem. Front., 2023, 7, 906–916 RSC.
- X. Ji, W. Chen, L. Long, F. Huang and J. L. Sessler, Chem. Sci., 2018, 9, 7746–7752 RSC.
- P. Anzenbacher, Y. L. Liu and M. E. Kozelkova, Curr. Opin. Chem. Biol., 2010, 14, 693–704 CrossRef CAS.
- S. J. Bradberry, J. P. Byrne, C. P. McCoy and T. Gunnlaugsson, Chem. Commun., 2015, 51, 16565–16568 RSC.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- M. A. Elsawy, J. K. Wychowaniec, L. A. Castillo Díaz, A. M. Smith, A. F. Miller and A. Saiani, Biomacromolecules, 2022, 23, 2624–2634 CrossRef CAS PubMed.
- E. M. Surender, S. J. Bradberry, S. A. Bright, C. P. McCoy, D. Clive Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2017, 139, 381–388 CrossRef CAS PubMed.
- S. Xu, A. C. Sedgwick, S. A. Elfeky, W. Chen, A. S. Jones, G. T. Williams, A. T. A. Jenkins, S. D. Bull, J. S. Fossey and T. D. James, Front. Chem. Sci. Eng., 2020, 14, 112–116 CrossRef CAS.
- H. Willcock, S. J. Butler and M. Rolph, Hydrogel, WO 2020/044022, 2023 Search PubMed.
- L. Martínez-Crespo, S. H. Hewitt, N. A. De Simone, V. Šindelář, A. P. Davis, S. Butler and H. Valkenier, Chem. – Eur. J., 2021, 27, 7367–7375 CrossRef PubMed.
- C. J. Marsden, C. Breen, J. D. Tinkler, T. R. Berki, D. W. Lester, J. Martinelli, L. Tei, S. J. Butler and H. Willcock, Polym. Chem., 2022, 13, 4124–4135 RSC.
- A. B. Mabire, Q. Brouard, A. Pitto-Barry, R. J. Williams, H. Willcock, N. Kirby, E. Chapman and R. K. O’Reilly, Polym. Chem., 2016, 7, 5943–5948 RSC.
- J. C. G. Bünzli, J. Lumin., 2016, 170, 866–878 CrossRef.
- D. Parker, J. D. Fradgley and K. L. Wong, Chem. Soc. Rev., 2021, 50, 8193–8213 RSC.
- R. Mailhot, T. Traviss-Pollard, R. Pal and S. J. Butler, Chem. – Eur. J., 2018, 24, 10745–10755 CrossRef CAS PubMed.
- S. H. Hewitt, G. Macey, R. Mailhot, M. R. J. Elsegood, F. Duarte, A. M. Kenwright and S. J. Butler, Chem. Sci., 2020, 11, 3619–3628 RSC.
- S. E. Bodman, C. Breen, S. Kirkland, S. Wheeler, E. Robertson, F. Plasser and S. J. Butler, Chem. Sci., 2022, 13, 3386–3394 RSC.
- M. L. Shipton, F. A. Jamion, S. Wheeler, A. M. Riley, F. Plasser, B. V. L. Potter and S. J. Butler, Chem. Sci., 2023, 4979–4985 RSC.
- S. J. Butler, Chem. Commun., 2015, 51, 10879–10882 RSC.
- S. H. Hewitt, J. Parris, R. Mailhot and S. J. Butler, Chem. Commun., 2017, 53, 12626–12629 RSC.
- T. A. Asoh, T. Yamamoto and H. Uyama, Sci. Rep., 2020, 10, 17173 CrossRef CAS PubMed.
- A. Dodero, L. Pianella, S. Vicini, M. Alloisio, M. Ottonelli and M. Castellano, Eur. Polym. J., 2019, 118, 586–594 CrossRef CAS.
- H. Chavda and C. Patel, Int. J. Pharm. Invest., 2011, 1, 17 CrossRef CAS.
- R. S. H. Wong, M. Ashton and K. Dodou, Pharmaceutics, 2015, 7, 305–319 CrossRef CAS.
- T. Cui, Y. Sun, Y. Wu, J. Wang, Y. Ding, J. Cheng and M. Guo, LWT, 2022, 161, 113374 CrossRef CAS.
- B. D. Johnson, D. J. Beebe and W. C. Crone, Mater. Sci. Eng., C, 2004, 24, 575–581 CrossRef.
- Loughborough University, Ion Sensing Technology – https://www.youtube.com/watch?v=2AaGlsiAd3U.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of polymer particles and hydrogels, photophysical properties, swelling data and mechanical properties of hydrogels. See DOI: https://doi.org/10.1039/d3cc04877b |
| This journal is © The Royal Society of Chemistry 2024 |