
Shake, shear, and grind! – the evolution of mechanoredox polymerization methodology
Sarah M.
Zeitler
 and
Matthew R.
Golder
*
and
Matthew R.
Golder
*
Department of Chemistry and Molecular Engineering & Science Institute, University of Washington, Seattle, WA 98195, USA. E-mail: goldermr@uw.edu
First published on 22nd November 2023
Abstract
In the last half decade, mechanoredox catalysis has enabled an entirely new genre of polymerization methodology. In this paradigm, mechanical force, such as ultrasonic cavitation bubble collapse or ball mill grinding, polarizes piezoelectric nanoparticles; the resultant piezopotential drives the redox processes necessary for free- and controlled-radical polymerizations. Since being introduced, evolution of these methods facilitates exploration of mechanistic underpinnings behind key electron-transfer events. Mechanical force has not only been identified as a “greener” alternative to more traditional reaction stimuli (e.g., heat, light) for the synthesis of commodity polymers, but also a potential technology to enable the production of novel thermoplastic and thermoset materials that are either challenging, or even impossible, to access using conventional solution-state approaches. In this Feature Article, significant contributions to such methods are highlighted within. Advances and ongoing challenges in both ultrasound and ball milling driven reactions for radical polymerization and crosslinking are identified and discussed.
Introduction
As polymer chemistry developed throughout the early twentieth century, the utility of synthetic polymers rapidly became apparent. However, methods for making such synthetic polymers were lacking, and polymer chemists sought to develop simple methods to access property-targeted polymeric materials. One such method that has grown exponentially in popularity is free radical polymerization (FRP) which is commonly used due to its versatility and simplicity.1 Utilizing this straightforward method which employs radical initiators and unsaturated monomers, polymers with numerous desirable properties and structures can be made for commercial purposes, from hard plastics to hydrophobic packaging to paints and coating products. While many high molecular weight species can be synthesized, FRP does not allow for molar mass or spatiotemporal control, both of which are critical considerations for advanced applications.2 Spatiotemporal control is particularly important as it allows for directed regulation of when and where polymer chains are growing; chain growth will only occur where stimulus is applied. Polymeric materials can then be programmed to have varying compositions at different locations and times. For example, Esser-Kahn demonstrated spatiotemporal distribution of engineered stress maps throughout soft materials using an electrodynamic shaker. This ability to pattern materials with force can be quantified using mechanical testing and corroborated with computational modelling, thereby demonstrating how stress is applied at different locations throughout the sample matrix.3To expand the utility of synthetic polymers, reversible deactivation radical polymerization (RDRP) techniques have been developed to allow for the synthesis of well-defined living polymers. The two most common RDRP methods are atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer polymerization (RAFT).4–6 Both of these RDRP methods utilize deactivation mechanisms to ensure uniform growth of all propagating polymer chains.
The mechanism for FRP is well established – stimuli-promoted (e.g., heat, light) homolytic cleavage of an initiator molecule commences the polymerization process. Chain growth continues via monomer propagation until termination occurs by H-atom abstraction or chain recombination.1,7 In FRP, all active chain ends propagate until such termination occurs.
Additionally, both ATRP and RAFT have well-studied mechanisms.5,8,9 Briefly, while the radical initiation steps are similar to those of FRP, ATRP and RAFT mechanisms introduce chain end “capping”, a deactivated dominant equilibrium state of the propagating polymer chain. In ATRP, this reversible deactivation process occurs through metal mediation, typically Cu(II)/Cu(I) or Fe(II)/Fe(I), to facilitate halide chain capping in the M(I) oxidation state and chain propagation in the M(II) oxidation state (Scheme 1(A)).10,11 As expanded upon below, the stimulus needs to be applied continuously for ATRP in order to regenerate the activator state of the transition metal catalyst. For example, as the stimulus is applied, Cu(I)X is consistently regenerated from oxidized Cu(II)X2, permitting subsequent electron transfer to reactivate dormant capped chain ends.12–15 In RAFT, this reversible deactivation process occurs via a chain transfer agent (CTA) (e.g., dithiocarbonate, trithiocarbonate). Growing chains quickly react with CTAs to establish the RAFT equilibrium; degenerate chain transfer facilitates piecemeal chain propagation of each growing polymer (Scheme 1(B)).8
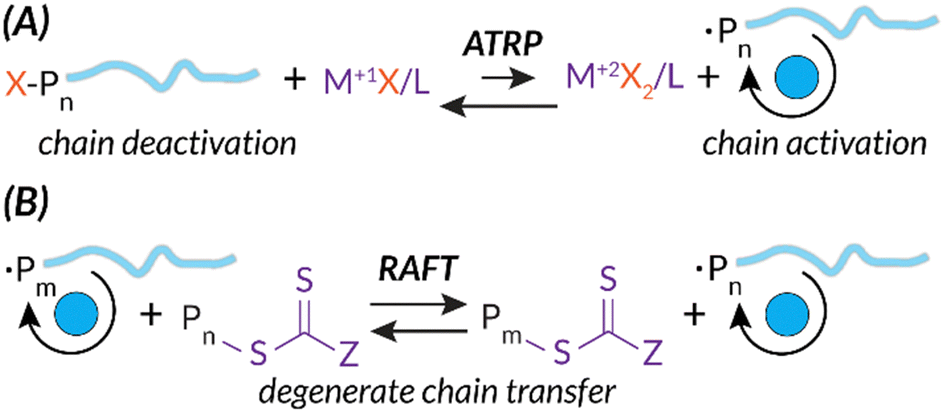 | ||
| Scheme 1 ATRP and RAFT mechanisms share similarities in deactivation via unique chain capping strategies leading to more control of chain length. | ||
Historically, traditional polymerizations in the field are subject to conventional reaction stimuli such as heat or light. Thermal initiations have been used readily both in academia and industry, and while unsaturated monomers can be thermally propagated, undesirable side reactions and degradation readily occur due to frequent thermal instability of polymer and reactant species.16 Thermal reactions also require a large amount of energy to apply heat uniformly in a reactor and consequently also suffer from poor spatiotemporal control.17
On the other hand, photoinduced radical polymerizations offer a unique alternative to thermal methods and due to an expansive history of photochemical methodology are currently of high interest to the polymer community.18–20 Photoinitiation offers increased spatiotemporal control compared to other stimuli (e.g., vat photopolymerizations21) and often requires less energy22 than thermal initiation. In recent years, photoredox methodologies pioneered by Hawker, Johnson, Fors, Boyer, Miyake, and Chen build on established energy transfer mechanisms established in small molecule synthesis to gain spatiotemporal control on the macromolecular scale.23–28 Still, a major limitation of photochemistry is that photon penetration is governed by the refractive index of a material or solution.29 Therefore, if colored or opaque media is employed, photochemical processes become more challenging.
Consistent with these ongoing challenges in the field to effectively apply stimuli to macromolecular systems, mechanochemistry is recognized as a reemerging area utilized by synthetic chemists, broadly defined.29 While relevant in the field of polymer chemistry since Staudinger in the 1930's,30 only recently have mechanically generated radicals become synthetically useful. Homolytic bond scission under mechanical stress generates so-called mechanoradicals that can then be used to initiate radical reactions, translating the mechanical energy into chemical energy.31–34 Grybowski exemplifies this paradigm by demonstrating that mechanoradicals can even be generated within commodity materials (e.g., tires, sneaker soles) upon mechanical flexing, akin to ordinary stress experienced by these commonplace polymers. In addition, due to mechanochemistry's unique ability to orthogonally transfer energy, it is rapidly being reevaluated as a useful method for polymer synthesis and upcycling processes traditionally performed in solution.35–44
Mechanochemical tools and early reports
Another way to utilize force as a stimulus for chemical reactions is to harness the power of piezoelectricity. Piezoelectricity is a phenomenon present in materials with non-centrosymmetric crystal lattice structures and uneven charge distribution (i.e., poling). This specific crystal structure translates mechanical force into a piezopotential.45 Alternatively, the inverse can occur within piezoelectric materials in which electric potential can cause a mechanical output (i.e., actuation).46 Piezoelectric materials (e.g., PVDF, BaTiO3, ZnO, etc.)47,48 are therefore able to translate mechanical force into electric potential to effect redox chemistry by disrupting the crystal structure equilibrium; material polarization facilitates single electron transfer from the surface (Fig. 1).29,48–50 At this time, piezoelectric nanoparticles or nanorods are typically utilized due to their high surface area to volume ratio which permits efficient charge transfer. Ongoing efforts suggest that incorporating piezoelectric components directly into engineered reaction vessels might be even more effective at capturing the maximum possible energy.51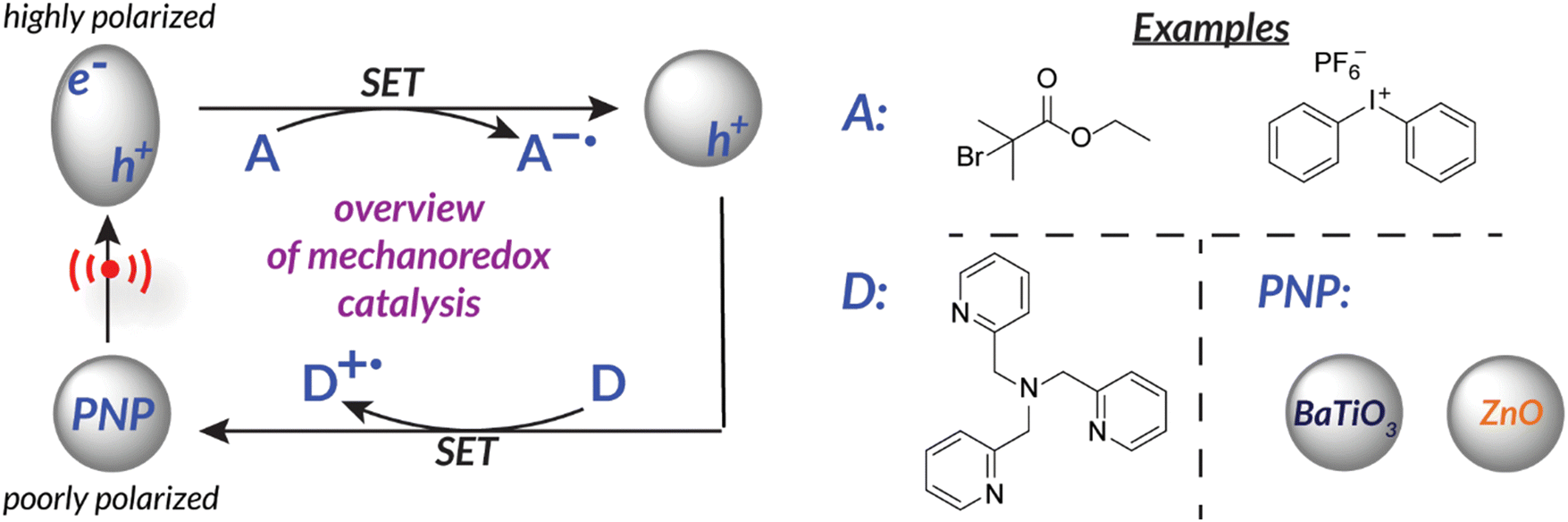 | ||
| Fig. 1 Piezoelectricity can be utilized for single electron transfer to reduce an “acceptor” initiator. Nanoparticles become activated upon application of force. | ||
The use of mechanical force to facilitate synthetic redox chemistry, specifically with piezoelectric materials (i.e., mechanoredox catalysis), draws inspiration from utilizing piezoelectric materials like ZnO and BaTiO3 for water splitting. In these works, piezoelectric nanorods are subjected to ultrasonic irradiation for the conversion of water into H2 and O2.52–54
The requisite force for mechanically induced processes can come from many different sources. Historically, a mortar and pestle or hammer may have been used for grinding reactions, and while sometimes utilized in recent times,55,56 more replicable sources include sonication horns, ultrasonic baths, ball mills, and even bench top vortexers.17,57
Sonication (i.e., ultrasonic irradiation), is a common “force source” for mechanochemistry.57 Ultrasound waves cause bubbles to form and expand where hot spots develop. Upon collapse, these cavitation bubbles exert a shear force within the reaction mixture to drive mechanochemical reactions.58 Ultrasonic horns apply ultrasound to a pinpointed region with higher power outputs than ultrasound baths. Ultrasonic baths are more readily available but apply force more broadly and can create hotspots.59 Both types of ultrasonic generators are readily employed in mechanochemistry.
Ball mills are alternative force sources consisting of a sealable reaction jar containing a specified number and size of ball bearings.60 The jars are then shaken, rolled, or spun at a particular frequency; ball bearings impact the sides of the reaction jar and transfer this energy to the reaction components. Ball milling reactions can uniquely occur in the solid state or near solid state due to the lack of dependence on solvent for energy transfer and mixing. Some reactions have been shown to be more efficient with miniscule amounts of solvent (0–2 μL mg−1) added to aid in mixing and solubility; this phenomenon is termed liquid assisted grinding (LAG) and has been utilized in mechanochemical reactions since the early 2000's.61–64
Previously, the intuitive use of force in polymer mechanochemistry was centered around bond scission, or destructive chemistry. Polymer degradation has been reported in an ultrasound bath, and applied stress has been repeatedly shown to activate bulk mechanophores, mechanically active molecules incorporated into polymers, via bond scission or molecular rearrangement. However, there have been some reports of constructive bond forming mechanochemistry to form polymers. Limited reports65,66 utilize high frequency ultrasound (>400 kHz) to induce homolytic solvent cleavage; the solvent radicals then induce radical polymerization. Other reports have shown that various types of post-polymerization functionalization and non-radical polymerizations can occur in the ball mill setting precedent for efficient constructive radical polymer chemistry to be developed in the ball mill without concern of structural degradation.35,67
While there have been limited reports of utilizing both sonication and ball milling to initiate radical polymerization reactions without inciting piezoelectricity,35 there is a growing demand for the accessible redox properties harnessed from piezoelectric materials in conjunction with force; therefore, the focus of this review will revolve primarily around these appropriately termed “mechanoredox polymerizations”.
Using this foundation for mechanoredox chemistry, in 2019 Kubota and Ito published work in which the “mechanically-activated” piezopotential from BaTiO3 was coupled to borylation reactions with aryl diazonium salts.55 This study demonstrates that force generated by ball bearings impacting the walls of ball milling jars induces the piezoelectric effect in the BaTiO3 nanoparticles. Then, single electron transfer from the BaTiO3 surface reduces the aryl diazonium salt; the resulting aryl radical (following N2 evolution) couples with bis(pinacolato)diboron for C–B bond formation; heteroarenes can also be utilized for C–C bond formation (Scheme 2). While this work was naturally motivated by earlier accounts of mechanoredox polymerizations (vide infra), the unique combination of mechanoredox catalysis under ball milling conditions served as a particularly important inspiration to our group when designing approaches to the macromolecular challenges described below.
 | ||
| Scheme 2 As demonstrated by Kubota and Ito, mechanoredox catalysis can be used to perform arylations and borylations. In these examples, aryl diazonium salts are reduced by piezoelectric nanoparticles in the ball mill. | ||
Ultrasonicated polymerizations
In 2017, the Esser-Kahn group published seminal work that utilized mechanoredox catalysis with ultrasonic irradiation to control ATRP. In this study (Fig. 2), Cu(II) is reduced by the mechanically-perturbed BaTiO3 piezoelectric nanoparticles. The Cu(II) system, Cu(OTf)2/Me6TREN/Bu4NBr, was specifically selected due to its reduction potential appropriately matching the generated reduction potential of the BaTiO3 (benchmarked to be at least −1.23 V via its water splitting ability),52 an important consideration for all mechanoredox polymerizations. An ultrasonic horn (20 kHz) was used to activate the BaTiO3 and subsequently reduce the Cu(II) catalyst. The produced Cu(I) can then activate the ATRP initiator to commence chain growth; continuous sonication is required for sustained monomer propagation. Overall, various polyacrylates were formed, although molar masses did not exceed 3 kDa. While limited in scope, this seminal work was crucial in demonstrating how mechanoredox catalysis can drive initiation and reversible activation/deactivation processes within a radical polymerization system.68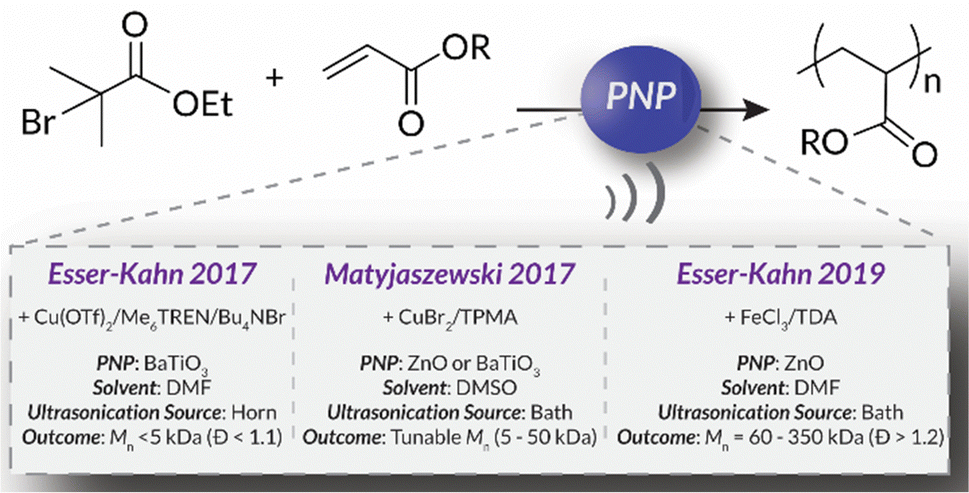 | ||
| Fig. 2 Comparison of the seminal Esser-Kahn/Matyjaszewski mechanoredox polymerization conditions highlighting important outcomes and progress. These works lay the foundation for the mechanoredox polymerization field. | ||
Following this initial ATRP report, the Matyjaszewski group optimized reaction conditions to utilize more readily accessible ultrasonic baths (rather than sonication horns), examine the specifics of mechanoredox catalyst nanoparticles (e.g., size, geometry, surface ligands), and expand the target molar mass range (Fig. 2).69 The monomer scope was also expanded to encompass myriad acrylate monomers with good temporal control demonstrated in on–off experiments (Fig. 3). Importantly, Matyjaszewski highlighted how it is possible to decrease catalyst loading and utilize ZnO instead of BaTiO3 as another lead-free piezoelectric material alternative.70,71
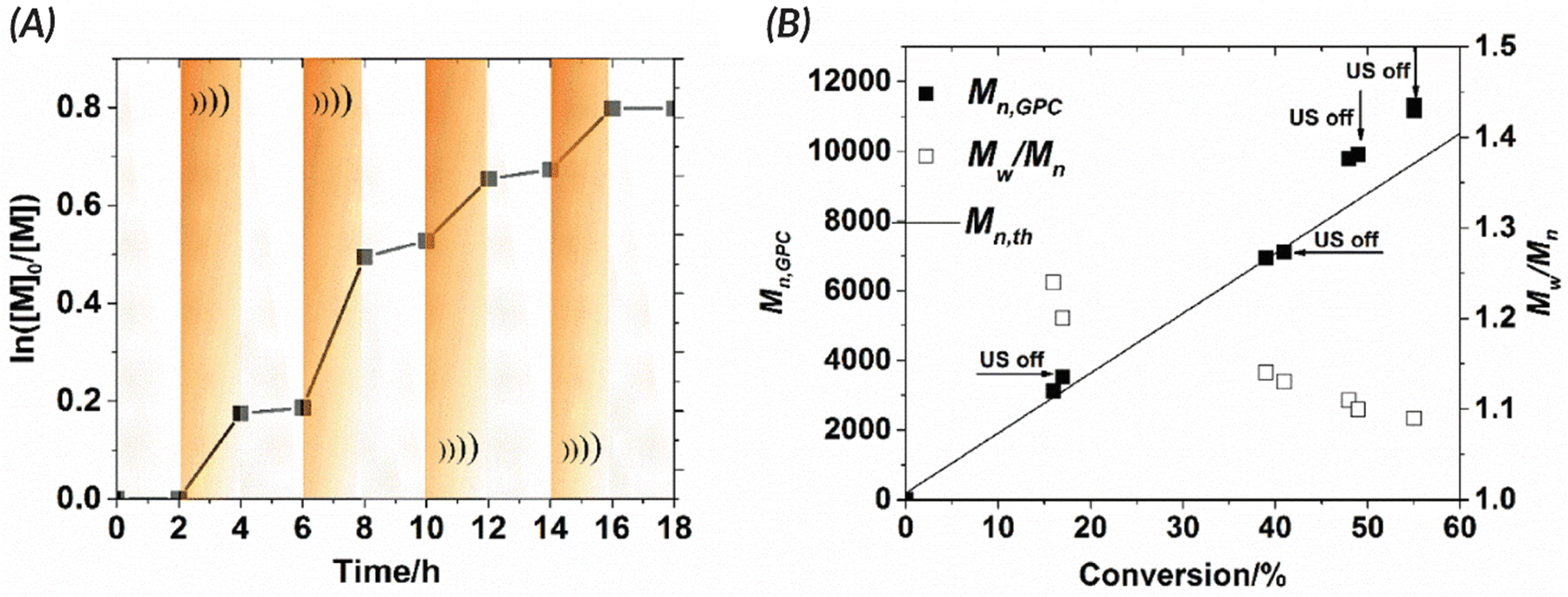 | ||
Fig. 3 Matyjaszewski refined the work of Esser-Kahn to create an optimized mechanoredox-catalyzed ATRP system which is demonstrated in the ability to “turn off” polymerization with the removal of stimuli. (A) Kinetic studies demonstrate lack of significant chain growth when ultrasound is turned off. (B) Molecular weight and dispersities match the expected ATRP values with low dispersities and consistent growth while sonication is applied. Reaction conditions: [monomer]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[initiator]0:[metal]0:[ligand]0 = 200:1:0.03:0.18 with 4.5 wt% BaTiO3 in DMSO. Reprinted with permission from ref. 69. Copyright 2017 American Chemical Society. :[initiator]0:[metal]0:[ligand]0 = 200:1:0.03:0.18 with 4.5 wt% BaTiO3 in DMSO. Reprinted with permission from ref. 69. Copyright 2017 American Chemical Society. | ||
In 2019, the Esser-Kahn group further expanded upon their initial ATRP work to demonstrate that mechanoredox FRP could also be conducted to access higher molar mass polymers. Utilizing the improvements outlined by the Matyjasweski reports, a low ZnO loading was used in conjunction with an ultrasound bath and a Fe catalyst to achieve polymerization (Fig. 2). While Fe-catalyzed ATRP has been reported,72 the system in this report better approximates FRP mechanistically. High molar masses (50–340 kDa) were achieved with a variety of monomers (Fig. 4); however, consistent with a free-radical mechanism there was only moderate control of dispersity (Đ = 1.2–1.3) (Table 1). The versatility of mechanoredox catalysis was also demonstrated in the synthesis of block copolymers emanating from macroinitiators.73
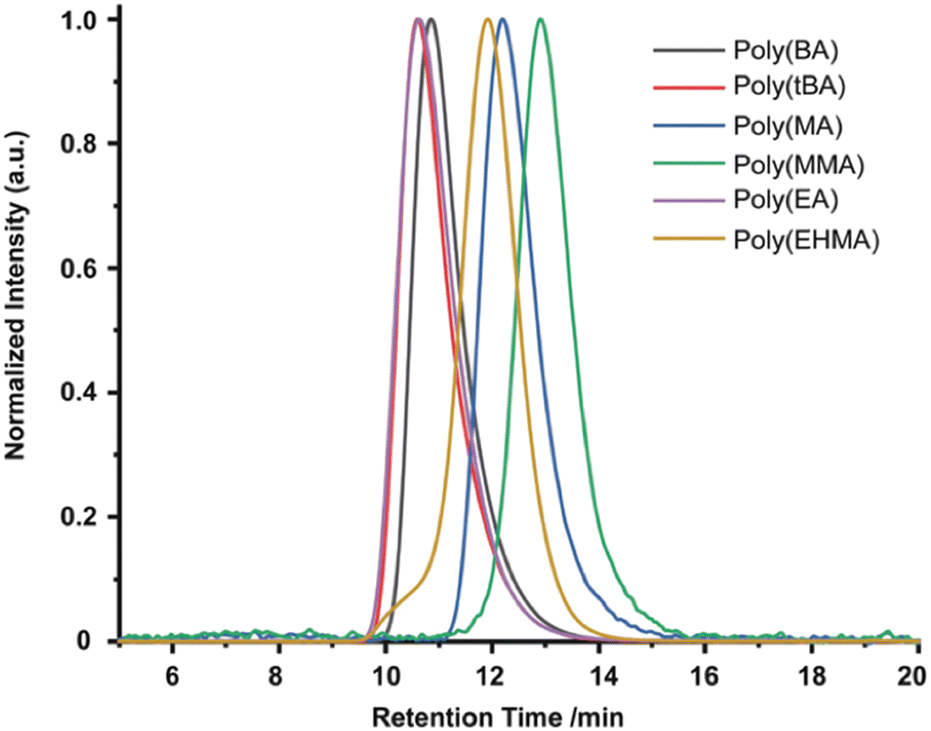 | ||
| Fig. 4 GPC-RI traces from Esser-Kahn's mechanoredox FRP methodology. Adapted with permission from ref. 73. Copyright 2019 Wiley Journals. | ||
:[initiator]0:[metal]0:[ligand]0 = 100:1:1:2 with 9 wt% ZnO in DMF. Adapted with permission from ref. 62. Copyright 2019 Wiley Journals
| Entry | Monomer | Conversiona (%) | M n b (kDa) | Đ b |
|---|---|---|---|---|
| 1 | tBA | 80 | 341 | 1.27 |
| 2 | BA | 74 | 246 | 1.22 |
| 3 | EA | 55 | 212 | 1.27 |
| 4 | MA | 71 | 62 | 1.18 |
| 5 | MMA | 52 | 50 | 1.30 |
| 6 | EHMA | 60 | 145 | 1.22 |
Most mechanoredox polymerizations require both the piezoelectric nanoparticles and an exogenous initiator, much like photoredox polymerizations that require a sensitizer and an initiator.74 Alternatively, Stenzel and coworkers were able to demonstrate initiation via water molecules adsorbed on the surface of the BaTiO3 mechanoredox catalyst (vide infra).56 In mechanoredox catalyzed polymerizations, the piezoelectric material is activated by force, allowing for single electron transfer to govern initiation. The mechanism following initiation is similar to conventional controlled radical chain growth mechanisms. For example, in the ATRP reactions demonstrated by the Esser-Kahn and Matyjaszewski groups, constant force is required to continue polymerization as the chain ends need to be continuously reactivated via a reduced metal species.68–70 These works demonstrate that temporal control observed with traditional stimuli can also be achieved with mechanical force for reversible-deactivation radical polymerizations.
As previously mentioned, the choice of initiator is important to consider. Although high frequency ultrasound can homolytically cleave solvent to produce radical initiators,75 the “effective force” generated in the aforementioned mechanoredox processes is generally not great enough to directly reduce solvent or other small molecules in solution. Instead, force must be transduced through the mechanoredox catalyst which subsequently generates an appropriate potential for downstream reactivity. Because the piezoelectric nanoparticles will only produce a measurable electric potential when activated, only certain radical initiators are eligible for use in initiation.
So far, mechanoredox FRP and ATRP require the presence of an activated alkyl halide initiator for direct or indirect mechanoredox activation. In other words, mechanoredox catalysis either directly reduces the initiator to an alkyl radical or facilitates reduction through a transition-metal (i.e., Cu) mediator. Alternatively, inspired by the work of both Esser-Kahn's FRP work and Kubota and Ito's aryl diazonium borylation reaction, our lab presented diaryliodonium salts as possible initiators for mechanoredox FRP with a BaTiO3 catalyst.76 These diaryliodonium salts, which are commercially available or easily synthesized, offer tunability as aryl substituents and counter ions can affect redox potentials, solubility, and monomer compatibility.77,78 Diaryliodoniums are less reactive aryl radical surrogates than aryl diazoniums due to their more negative reduction potentials.55 In our work, no metal additives were needed for mechanoredox initiation, thereby simplifying purification.
Also building upon the initial reports of mechanoredox ATRP,68–70,73,76,79 Pang recently expanded upon such previous work by studying the influence of ZnO nanoparticle shape and size on polymerization efficiency. Prior to this work, commercially available 20–200 nm nanoparticles with traditional morphologies (i.e. cubic, tetragonal, hexagonal) had primarily been utilized for mechanoredox polymerizations; however, it is known that nanoparticle morphology can impact the generated mechanoredox potential.80,81 Pang found that by substituting commercial ZnO for various sized nanorods (300–530 nm in diameter and 1.9–6.8 μm in length), less ZnO was needed to initiate polymerization with higher conversion and lower power ultrasonication than previously used (Fig. 5). The nanorods with the highest aspect ratio (L/D = 13) show an increase in piezeoelectric activity as they afforded polyacrylates with experimental Mn values closely matching theoretical Mn values in only 4 h; previous reports require at least 8 h for full monomer consumption. Uniquely when utilizing these nanorods, acrylonitrile monomers can also be polymerized to high molar mass and low dispersity, a shortcoming of prior mechanoredox ATRP processes.82 Pang also further expanded upon the study of ZnO nanoparticles by also identifying optimized BaTiO3 morphologies in a separate report.83
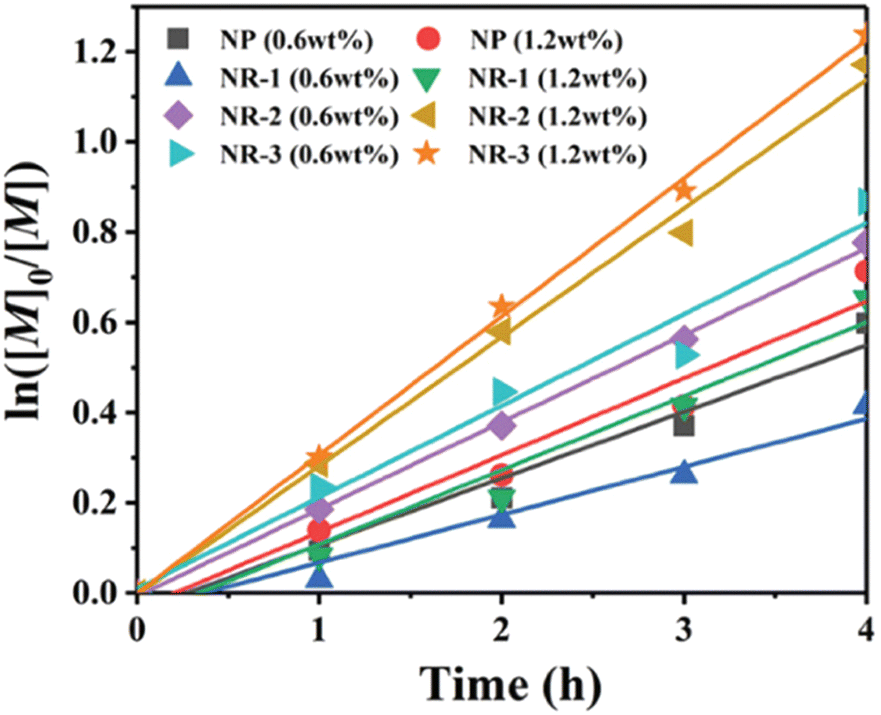 | ||
| Fig. 5 Pang compared ZnO nanoparticle sizes and shapes to their polymerization efficiencies. Reaction conditions: [monomer]0:[initiator]0:[metal]0:[ligand]0 = 100:1:0.03:0.18 with ZnO in DMSO. Adapted with permission from 82. Copyright 2022 American Chemical Society. | ||
Akin to solution-state mechanoredox ATRP processes, analogous RAFT polymerizations are also possible as recently demonstrated by Zhang. Using a bis(trithiocarbonate) bisulfide chain transfer agent in conjuction with previously described alkyl halide initiators,68–70,73 high monomer conversion was achieved when sonicating in the presence of ZnO. Controlled molar masses and dispersities were consistent with RAFT polymerizations (Fig. 6), and the high chain end fidelity was demonstrated by reactivating a macroinitiator synthesized via mechanoredox RAFT to form block copolymers. This report also introduces the use of a tris(2-pyridylmethyl) amine (TPMA) ligand that is hypothesized to fill holes following single electron transfer. TPMA is reported to be on the surface of the ZnO after sonication, as observed by X-ray photoelectron spectroscopy. Finally, the report uniquely shows the ability to cure opaque acrylic resins using force (Fig. 7). Recall that photochemistry is typically used to cure such resin formulations, but photons cannot penetrate deeply into opaque materials; mechanoredox catalysis increases the percentage of cured resin (ca. 95%) compared to that of photocuring (ca. 59%) and allows for filler and composite incorporation without decreasing stimulus penetration.84 This specific application-driven example is complemented by work from Esser-Kahn in which soft organogels were hardened via mechanoredox catalysis-induced disulfide bond formation (Fig. 8).85
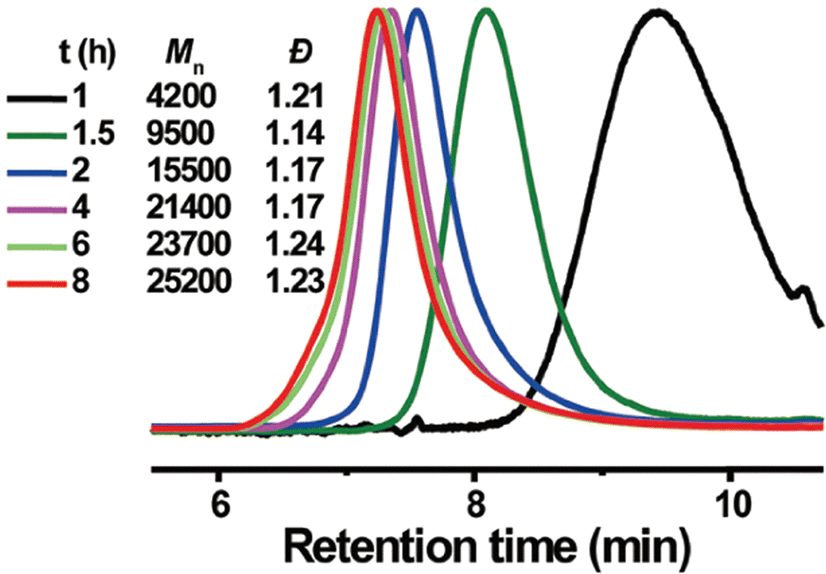 | ||
| Fig. 6 GPC-RI traces over time from Zhang's mechanoredox RAFT method demonstrating good control over Mn and dispersities. Reaction conditions: [monomer]0:[initiator]0:[chain transfer agent]0:[ligand]0 = 75:2:1:0.7 with 4.4 wt% ZnO in DMSO. Adapted with permission from ref. 84. Copyright 2022 American Chemical Society. | ||
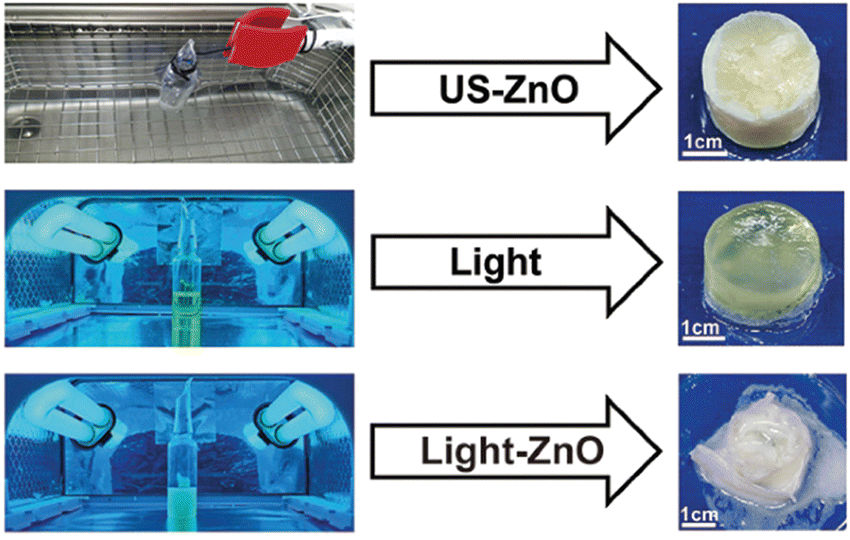 | ||
| Fig. 7 After showing that mechanoredox polymerizations can be translated to RAFT, Zhang further expanded the work to force cure ZnO opaque composites. Adapted with permission from ref. 84. Copyright 2022 American Chemical Society. | ||
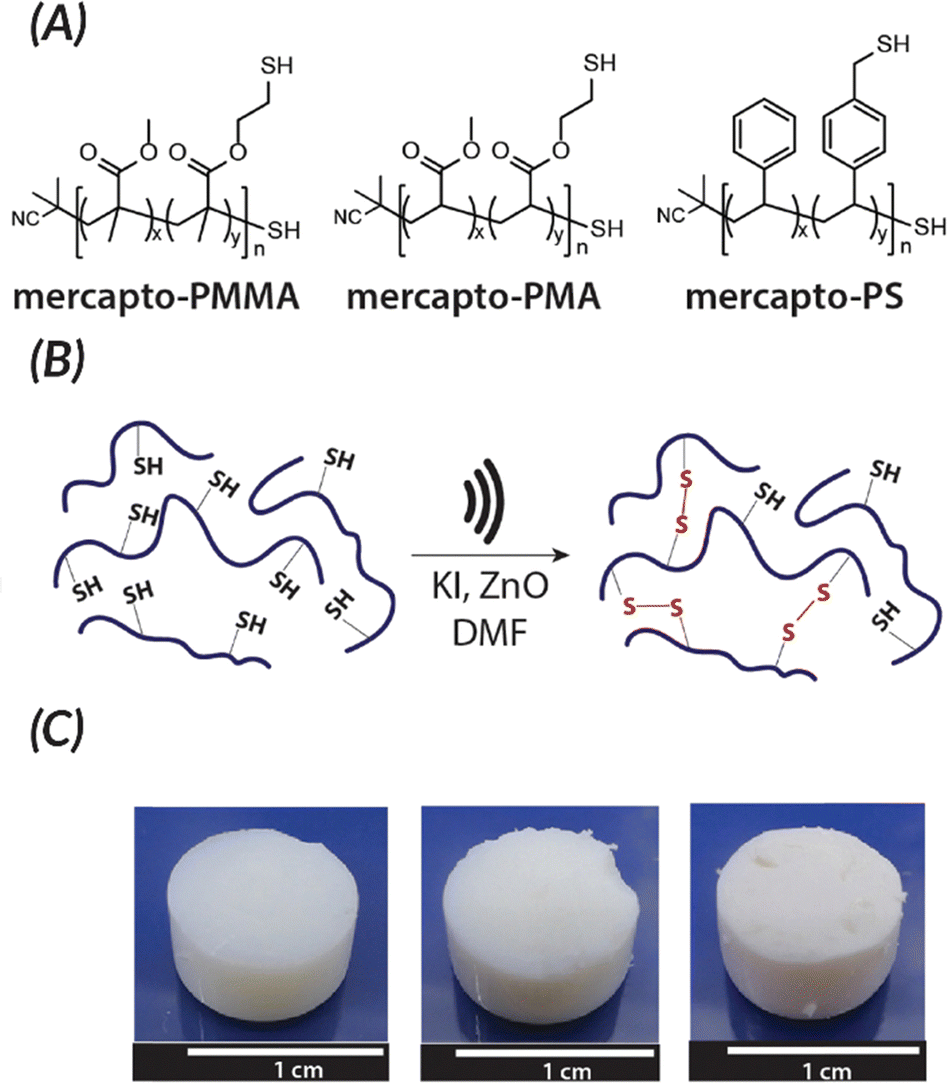 | ||
| Fig. 8 Esser-Kahn also demonstrated force can be used to induce crosslinking of organogels by causing the formation of disulfides. (A) Polymers used for force curing. (B) When force is applied in the presence of piezoelectric nanoparticles, disulfide bonds are formed. (C) Crosslinking for the different polymer systems after sonication occurs: PMMA, PMA, and PS (from left to right). Adapted with permission from ref. 85 Copyright 2019 American Chemical Society. | ||
A feature of the initial RAFT polymerizations by Zhang is the moderate initiator efficiencies ranging from ca. 6.1–44.8%. Following the initial report and inspired by the work of Pang examining the effects of the structure of ZnO on piezoelectric activity,82 the group improved the efficiency of the system by synthesizing “tuned” ZnO nanomaterials. ZnO hexagonal rods containing polar facets (i.e., crystal faces) improved piezoelectric catalytic activity and consequently increased initiator efficiency.86 In addition to improving initiator efficiency, this report also demonstrates that using an optimized trithiocarbonate chain transfer agent leads to better agreement of experimental Mn and theoretical Mn with lower dispersities.
Milled polymerizations
An especially unique aspect of our lab's initial work in mechanoredox polymerizations is introducing this paradigm to ball milling; our system can translate processes into the ball mill to produce high molar mass polymers with high monomer conversion in a fraction of the time as the sonicated reactions (3 h compared to 8–21 h). Notably, the ball milling reactions proceed without noticeable polymer degradation (Table 2) under these specific reaction conditions. We attribute this phenomenon to the LAG conditions being used; as demonstrated by Choi and Peterson, perturbing Tg through plasticization can drastically decrease the rate of mechanochemical scission in a ball mill.87 Also, our FRP reactions are set up in sealed jars prepared on the bench top opposed to an inert environment as all other mechanoredox polymerizations require. An induction period is observed when these reactions are sealed under air in which no monomer conversion is seen until 145 min, followed by full monomer consumption in the subsequent ca. 5 min. It is hypothesized that this incubation period is due to oxygen consumption (i.e., reduction to superoxide)88 under the reaction conditions, although the exact mechanism of action in our system is still under investigation.:[initiator]0:[BaTiO3]0 = 100:2:30 with 0.03 v/wt. DMF
|
|
||||
|---|---|---|---|---|
| Entry | Monomer | Conversiona (%) | M n b (kDa) | Đ b |
| 1 | tBA | >95 | 416 | 1.3 |
| 2 | BA | 90 | 556 | 1.6 |
| 3 | EA | >95 | 751 | 1.4 |
| 4 | MA | >95 | 937 | 1.2 |
| 5 | MMA | 86 | 56 | 1.7 |
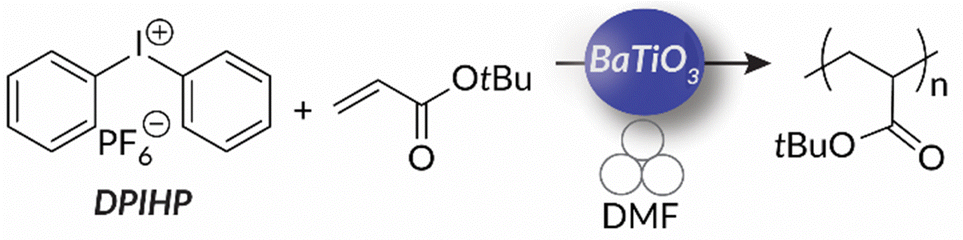
Building upon our group's previous work with FRP and the precedent of Zhang, in 2023 we reported the first example of mechanoredox-catalyzed RAFT conducted in a ball mill (Fig. 9). Utilizing a versatile trithiocarbonate chain transfer agent and our previously established diaryl iodonium initiators, we identified a straightforward synthetic route to well-controlled poly(meth)acrylates. Mechanistic studies demonstrate that mechanoredox reduction of a diaryliodonium (Fig. 9) affords phenyl radicals capable of initiating chains for subsequent degenerative chain transfer. In this RAFT example, once mechanoredox initiation occurs, continuous milling appears to be crucial for efficient mass transfer (i.e., mixing) rather than for subsequent redox processes. This chemistry offers unique advantages including short reaction times, low energy consumption, and the ability to copolymerize traditionally immiscible monomers by using LAG-assisted ball milling chemistry. We report the effective syntheses of poly(meth)acrylates with moderate conversion in the ultrasound bath, but target Mn values were larger than expected, ultimately leading to higher dispersities than are typically observed using thermal RAFT. This shortcoming was hypothesized to be due to previously reported chain scission75 and slow chain propagation. To combat these shortcomings, the system was transferred into the ball mall where the reaction reached high conversion in just 3 h with generally low dispersities (Đ = 1.0–1.1).
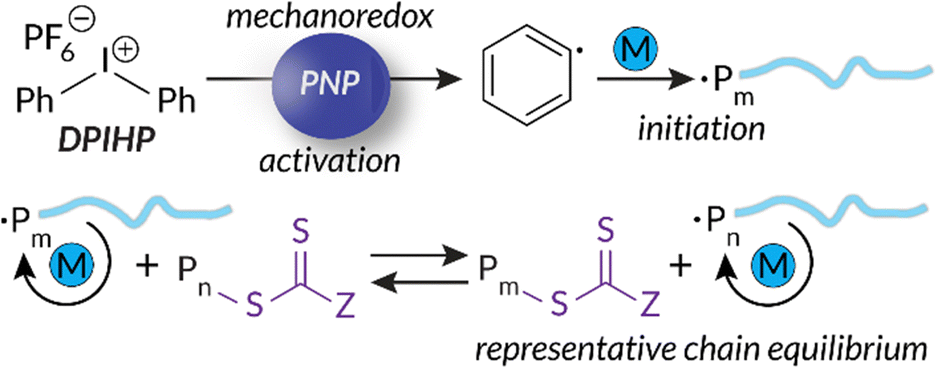 | ||
| Fig. 9 Based upon small molecule studies, Golder proposes a mechanism for mechanoredox-catalyzed RAFT polymerization. Adapted with permission from ref. 17. Copyright 2023 Wiley Journals. | ||
Additionally, the accessibility of this reaction was further enhanced by demonstrating that in 8 h, full conversion could be reached using a benchtop vortexer in place of the ball mill. Energy consumption studies also reported that ball milling and vortexing use at least a third of the energy a traditional thermal RAFT reaction would consume to reach full monomer conversion (Fig. 10). Finally, this work establishes a simple path in the ball mill towards block and random copolymers of chemically immiscible monomer feedstocks (e.g., fluorinated and hydrophilic acrylates). While solution-state and emulsion thermal RAFT methods have been used to access random semifluorinated acrylate copolymers, block copolymers from these feedstocks are considerably more challenging.89–91 Traditional synthetic pathways to blocky semifluorinated acrylate copolymers92,93 often require extended reaction times,94 unique catalysts,95 fluorinated initiators,96,97 fluorinated CTAs,96 and/or fluorinated solvent.98–100 Our proof-of-concept study will undoubtedly lead to the generation of high-χ polymers that are currently challenging, or even inaccessible, by traditional solution-state methods (Fig. 11).17
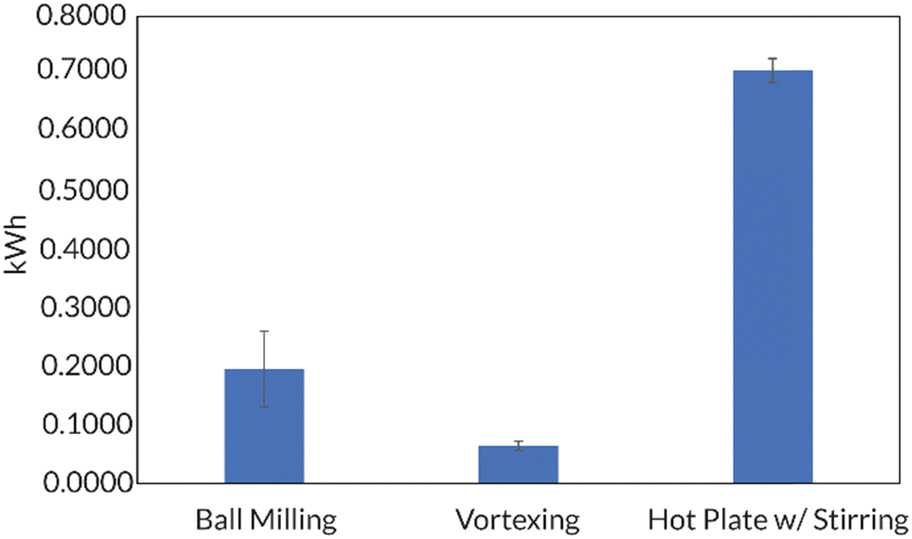 | ||
| Fig. 10 Golder shows that mechanochemical methods (e.g., ball milling and vortexing) consume less energy than a hot plate set to an appropriate temperature (ca. 70 °C) needed for a traditional thermal RAFT polymerization. Adapted with permission from ref. 17. Copyright 2023 Wiley Journals. | ||
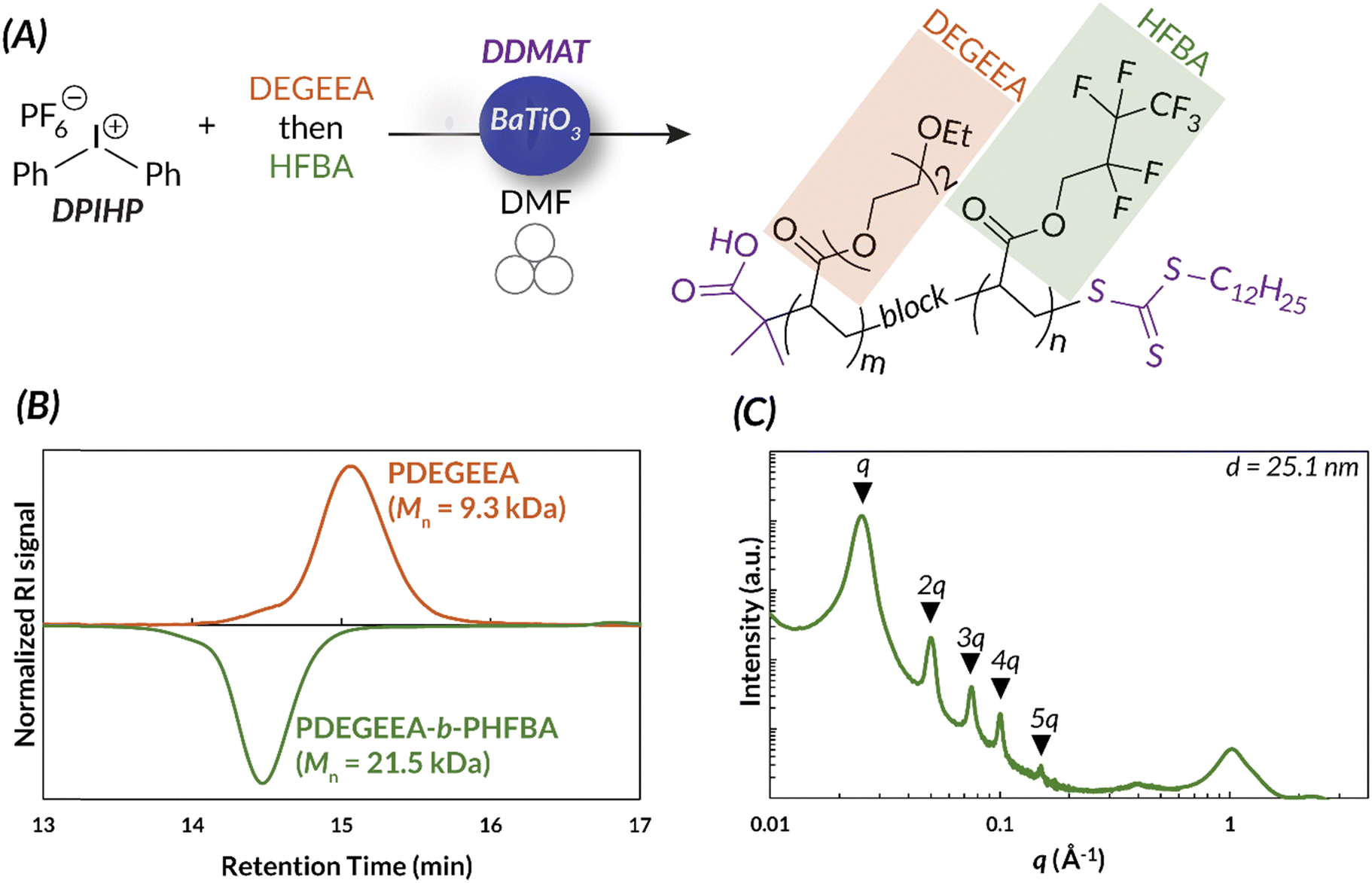 | ||
| Fig. 11 (A) The ability to synthesize immiscible monomers in the ball mill is a unique advantage of mechanoredox-mediated chemistry. (B) Immiscible monomer can be added in distinct blocks, as is shown by GPC. (C) The block copolymers self-assemble to form distinct blocks, shown by SAXS data. Adapted with permission from ref. 17. Copyright 2023 Wiley Journals. | ||
In 2023, Stenzel and coworkers further examine the utility of mechanoredox-mediated polymerizations by studying the nature of BaTiO3 nanoparticles in more depth. Looking into water adsorbed to the surface of the nanoparticles, they report the generation of hydroxy radicals when ball milled in the presence of a hydroxyl specific radical trap, terephthalic acid, with an increase in radical production in the presence of oxygen relative to an inert environment. The qualitative production of surface radicals was even possible by striking samples with a hammer! After demonstrating and quantifying radical formation, solid vinyl monomers were added to the reaction vessel and polymerized in high conversion to access high molar mass polymers (>250 kDa) without the need for any additional initiator or exogenous additives, although mechanochemical chain degradation is observed as milling time increases. While surface radical concentration increases in air, the polymerization system does need to be conducted in an inert environment.56 Interestingly, crosslinking is also possible in the ball mill (Fig. 12) to form an insoluble BaTiO3-poly(acrylamide) composite that is both swellable and moldable. It is hypothesized that interchain radical transfer causes the branching and physical self-crosslinking that would be required to form the insoluble material. Other solid-state polymerizations have now been developed as well.101,102
 | ||
| Fig. 12 Stenzel conducted polymerization of acrylamides in the ball mill which can then form physical crosslinks. Adapted with permission from ref. 56. Copyright 2023 Wiley Journals. | ||
Future outlook for mechanoredox-mediated polymerizations
The burgeoning field of mechanoredox polymerizations offers opportunities to optimize systems that provide more penetrable, energy efficient, and environmentally friendly opportunities for the synthesis of commodity and novel polymers alike. From FRP and RDRP reactions to crosslinking mechanisms using diverse mechanical and piezoelectric sources, we as a field have merely scratched the surface of using mechanoredox catalysis for constructive polymer chemistry.17,56,68–70,73,76,82,84–86,101 Yet, there are still many unanswered challenges that exist, including the quantification of “force” in a given system, recapitulating mechanoredox processes on large scale, exploring the scope of mechanoredox catalysts for specific applications (including the use of organic piezoelectric materials), and pushing the limits on accessing soft materials that are fully inaccessible using solution-state processes. Even more, the utility of mechanoredox chemistry will surely reach beyond polymer synthesis and into more applied areas of research such as the synthesis of anisotropic composites and layered materials. Finally, the life cycle and technoeconomic analyses of mechanoredox reactions need to be evaluated; a significant advantage of using mechanical force is the diminished need for toxic solvents and lower energy consumption. Building upon the work of small molecule chemists and physicists, we can continue to improve our systems and incorporate the concepts of mechanoredox into new methods and applications, developing more sustainable methods to make novel materials that are impossible, or at least challenging, and/or inefficient to make using the current state of the art.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by generous start-up funds from the University of Washington and University of Washington MEM-C, an NSF MRSEC funded under DMR-1719797.References
-
B. Yamada and P. B. Zetterlund, Handbook of Radical Polymerization, John Wiley and Sons Inc., Hoboken, NJ, 2002 Search PubMed
.
- D. Colombani, Prog. Polym. Sci., 1997, 22, 1649–1720 CrossRef CAS
- Z. Wang, J. Wang, J. Ayarza, T. Steeves, Z. Hu, S. Manna and A. P. Esser-Kahn, Nat. Mater., 2021, 20, 869–874 CrossRef CAS
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed
- H. S. Bisht and A. K. Chatterjee, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 2001, 41, 139–173 CrossRef
- G. Moad, J. Chiefari, R. T. A. Mayadunne, C. L. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromol. Symp., 2002, 182, 65–80 CrossRef CAS
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS
- K. Matyjaszewski, Isr. J. Chem., 2012, 52, 206–220 CrossRef CAS
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS
- P. Chmielarz, A. Sobkowiak, M. Fantin, S. Park, K. Matyjaszewski, A. A. Isse, A. Gennaro and A. J. D. Magenau, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC
- S. Dadashi-Silab, M. Atilla Tasdelen, A. Mohamed Asiri, S. Bahadar Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35, 454–459 CrossRef CAS
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC
- P. Chakma, S. M. Zeitler, F. Baum, J. Yu, W. Shindy, L. D. Pozzo and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202215733 CrossRef CAS
- T. Otsu, J. Polym. Sci., 1956, 221, 559–561 CrossRef
- M. A. Tasdelen, B. Kiskan and Y. Yagci, Macromol. Rapid Commun., 2006, 27, 1539–1544 CrossRef CAS
- M. A. Tasdelen, N. Moszner and Y. Yagci, Polym. Bull., 2009, 63, 173–183 CrossRef CAS
- F. Zhang, L. Zhu, Z. Li, S. Wang, J. Shi, W. Tang, N. Li and J. Yang, Addit. Manuf., 2021, 48 Search PubMed
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef CAS PubMed
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. J. Korley, A. C. Balazs and J. A. Johnson, ACS Cent. Sci., 2017, 3, 124–134 CrossRef CAS
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed
- K. Chen, Y. Zhou, S. Han, Y. Liu and M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202116135 CrossRef CAS PubMed
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 10665–10668 CrossRef CAS PubMed
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read De Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS
- J. A. Leitch and D. L. Browne, Chem. – Eur. J., 2021, 27, 9721–9726 CrossRef CAS PubMed
- W. H. Binder, Polymer, 2020, 202, 122639 CrossRef CAS
- H. T. Baytekin, B. Baytekin and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 3596–3600 CrossRef CAS
- J. Kwiczak-Yiǧitbaşı, M. Demir, R. E. Ahan, S. Canlı, U. Ö. Şafak Şeker and B. Baytekin, ACS Sustainable Chem. Eng., 2020, 8, 18879–18888 CrossRef
- Z. J. Wang, J. Jiang, Q. Mu, S. Maeda, T. Nakajima and J. P. Gong, J. Am. Chem. Soc., 2022, 144, 3154–3161 CrossRef CAS
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC
- H. Y. Cho and C. W. Bielawski, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS
- G. S. Lee, H. W. Lee, H. S. Lee, T. Do, J.-L. Do, J. Lim, G. I. Peterson, T. Friščić and J. G. Kim, Chem. Sci., 2022, 68, 42–61 Search PubMed
- N. Ohn, J. Shin, S. S. Kim and J. G. Kim, ChemSusChem, 2017, 10, 3529–3533 CrossRef CAS
- C. V. Oprea and F. Weiner, Acta Polym., 1987, 38, 429–432 CrossRef CAS
- G. S. Lee, B. R. Moon, H. Jeong, J. Shin and J. G. Kim, Polym. Chem., 2019, 10, 539–545 RSC
- M. B. Larsen and A. J. Boydston, Macromol. Chem. Phys., 2016, 217, 354–364 CrossRef CAS
- S. Grätz, M. Oltermann, E. Troschke, S. Paasch, S. Krause, E. Brunner and L. Borchardt, J. Mater. Chem. A, 2018, 6, 21901–21905 RSC
- H. Y. Cho and C. W. Bielawski, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS
- O. Maurin, P. Verdié, G. Subra, F. Lamaty, J. Martinez and T. X. Métro, Beilstein J. Org. Chem., 2017, 13, 2087–2093 CrossRef CAS PubMed
- M. B. Starr and X. Wang, Sci. Rep., 2013, 3, 2160 CrossRef PubMed
- A. Aabid, M. A. Raheman, Y. E. Ibrahim, A. Anjum, M. Hrairi, B. Parveez, N. Parveen and J. Mohammed Zayan, Sensors, 2021, 21, 4145 CrossRef
- T. D. Usher, K. R. Cousins and S. Ducharme, Polym. Int., 2018, 67, 790–798 CrossRef CAS
- A. Cafarelli, A. Marino, L. Vannozzi, J. Puigmartí-Luis, S. Pané, G. Ciofani and L. Ricotti, ACS Nano, 2021, 15, 11066–11086 CrossRef CAS
- H. Xia and Z. Wang, Science, 2019, 366, 1450–1451 CrossRef
- Z. Ren, Y. Peng, H. He, C. Ding, J. Wang, Z. Wang and Z. Zhang, Chin. J. Chem., 2023, 41, 111–128 CrossRef
- E. M. Marrero, C. J. Caprara, C. N. Gilbert, E. E. Blanco and R. G. Blair, Faraday Discuss., 2022, 241, 91–103 RSC
- K. S. Hong, H. Xu, H. Konishi and X. Li, J. Phys. Chem. Lett., 2010, 1, 997–1002 CrossRef CAS
- S. Ikeda, T. Takata, M. Komoda, M. Hara, J. N. Kondo, K. Domen, A. Tanaka, H. Hosono and H. Kawazoe, Phys. Chem. Chem. Phys., 1999, 1, 4485–4491 RSC
- J. M. Wu, Y.-G. Sun, W.-E. Chang and J.-T. Lee, Nano Energy, 2018, 46, 372–382 CrossRef CAS
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed
- M. D. Nothling, J. E. Daniels, Y. Vo, I. Johan and M. H. Stenzel, Angew. Chem., Int. Ed., 2023, 62, e202218955 CrossRef CAS
- K. S. Suslick, Science, 1990, 247, 1439–1445 CrossRef CAS PubMed
- G. Cravotto, E. C. Gaudinoa and P. Cintas, Chem. Soc. Rev., 2013, 42, 7521–7534 RSC
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2019, 59, 1018–1029 CrossRef
- T. Friščić, L. Fábián, J. C. Burley, W. Jones and W. D. S. Motherwell, Chem. Commun., 2006, 5009–5011 RSC
- T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546–7550 CrossRef
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS
- J. Collins, T. G. McKenzie, M. D. Nothling, M. Ashokkumar and G. G. Qiao, Polym. Chem., 2018, 9, 2562–2568 RSC
- G. S. Lee, H. W. Lee, H. S. Lee, T. Do, J. L. Do, J. Lim, G. I. Peterson, T. Friščić and J. G. Kim, Chem. Sci., 2022, 13, 11451–11698 RSC
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS
- Z. Wang, X. Pan, J. Yan, S. Dadashi-Silab, G. Xie, J. Zhang, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2017, 6, 546–549 CrossRef CAS
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS
- P. Zhu, Y. Chen and J. Shi, Adv. Mater., 2020, 32, e202001976 Search PubMed
- Z. Xue, D. He and X. Xie, Polym. Chem., 2015, 6, 1660–1687 RSC
- Z. Wang, J. Ayarza and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2019, 58, 12023–12026 CrossRef CAS
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS
- T. G. McKenzie, F. Karimi, M. Ashokkumar and G. G. Qiao, Chem. – Eur. J., 2019, 25, 5372–5388 CrossRef CAS
- S. M. Zeitler, P. Chakma and M. R. Golder, Chem. Sci., 2022, 13, 4131–4138 RSC
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed
- H. E. Bachofner, F. M. Beringer and L. Meites, J. Am. Chem. Soc., 1958, 80, 4274–4278 CrossRef CAS
- Z. Ren, C. Ding, R. Ding, J. Wang, Z. Li, R. Tan, X. Wang, Z. Wang and Z. Zhang, ACS Macro Lett., 2023, 12, 1159–1165 CrossRef CAS PubMed
- L. Kou and W. Guo, IEEE, 2008, 354–359 Search PubMed
- S. Goel and B. Kumar, J. Alloys Compd., 2020, 816 Search PubMed
- K. Liu, W. Zhang, L. Zong, Y. He, X. Zhang, M. Liu, G. Shi, X. Qiao and X. Pang, J. Phys. Chem. Lett., 2022, 13, 4884–4890 CrossRef CAS PubMed
- S. Xu, W. Zhang, C. Wang, W. Peng, G. Shi, Z. Cui, P. Fu, M. Liu, Y. He, X. Qiao and X. Pang, Polymer, 2022, 252, 124949 CrossRef CAS
- C. Ding, Y. Yan, Y. Peng, D. Wu, H. Shen, J. Zhang, Z. Wang and Z. Zhang, Macromolecules, 2022, 55, 4056–4063 CrossRef CAS
- J. Ayarza, Z. Wang, J. Wang and A. P. Esser-Kahn, ACS Macro Lett., 2021, 10, 799–804 CrossRef CAS PubMed
- C. Ding, Z. Ren, J. Wang, L. Zhang, Y. Yan, D. Wu, Z. Wang and Z. Zhang, Chin. J. Chem., 2023, 41, 2691–2696 CrossRef CAS
- G. I. Peterson, W. Ko, Y. J. Hwang and T. L. Choi, Macromolecules, 2020, 53, 7795–7802 CrossRef CAS
- G. Wang, J. Jia, Y. He, D. Wei, M. Song, L. Zhang, G. Li, H. Li and B. Yuan, RSC Adv., 2022, 12, 18407–18411 RSC
- W. Yao, Y. Li and X. Huang, Polymer, 2014, 55, 6197–6211 CrossRef CAS
- Y. Koda, T. Terashima, M. Sawamoto and H. D. Maynard, Polym. Chem., 2015, 6, 240–247 RSC
- L. Chen and F. Wu, J. Appl. Polym. Sci., 2012, 125, 376–381 CrossRef CAS
- H. Gong, Y. Gu and M. Chen, Synlett, 2018, 1543–1551 CAS
- K. Chen, X. Guo and M. Chen, Angew. Chem., Int. Ed., 2023, 62, e202310636 CrossRef CAS PubMed
- Y. Inoue, J. Watanabe, M. Takai, S. I. Yusa and K. Ishihara, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6073–6083 CrossRef CAS
- S. Perrier, S. G. Jackson, D. M. Haddleton, B. Améduri and B. Boutevin, Macromolecules, 2003, 36, 9042–9049 CrossRef CAS
- H. Gong, Y. Zhao, X. Shen, J. Lin and M. Chen, Angew. Chem., Int. Ed., 2018, 57, 333–337 CrossRef CAS PubMed
- J. Xia, T. Johnson, S. G. Gaynor, K. Matyjaszewski and J. DeSimone, Macromolecules, 1999, 32, 4802–4805 CrossRef CAS
- E. H. Discekici, A. Anastasaki, R. Kaminker, J. Willenbacher, N. P. Truong, C. Fleischmann, B. Oschmann, D. J. Lunn, J. Read De Alaniz, T. P. Davis, C. M. Bates and C. J. Hawker, J. Am. Chem. Soc., 2017, 139, 5939–5945 CrossRef CAS PubMed
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. Van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483–14487 CrossRef CAS PubMed
- Z. Ma and P. Lacroix-Desmazes, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2405–2415 CrossRef CAS
- M. Zhou, Y. Zhang, G. Shi, Y. He, Z. Cui, X. Zhang, P. Fu, M. Liu, X. Qiao and X. Pang, ACS Macro Lett., 2023, 12, 26–32 CrossRef CAS PubMed
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2022, 59, 1010–1013 RSC
| This journal is © The Royal Society of Chemistry 2024 |