
Self-assembled nanodrug delivery systems for anti-cancer drugs from traditional Chinese medicine
Qiao
Li
a,
Yuan
Lianghao
a,
Gao
Shijie
a,
Wang
Zhiyi
a,
Tang
Yuanting
a,
Chen
Cong
*b,
Zhao
Chun-Qin
 *c and
Fu
Xianjun
*d
*c and
Fu
Xianjun
*d
aExperimental Centre, Shandong University of Traditional Chinese Medicine, Jinan 250355, P. R. China
bInnovative Institute of Chinese Medicine and Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan 250355, P. R. China. E-mail: keaidedacong@163.com
cAcademy of Chinese Medicine Literature and Culture, Key Laboratory of Classical Theory of Traditional Chinese Medicine, Ministry of Education, Shandong University of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, 250355, PR China. E-mail: CQZhao21@126.com
dMarine Traditional Chinese Medicine Research Centre, Qingdao Academy of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Qingdao 266114, P. R. China. E-mail: xianxiu@hotmail.com; qcwei@htu.edu.cn
First published on 12th February 2024
Abstract
Traditional Chinese medicine (TCM) is a combination of raw herbs and herbal extracts with a plethora of documented beneficial bioactivities, which has unique advantages in anti-tumor therapy, and many of its major bioactive molecules have been identified in recent years due to advances in chemical separation and structural analysis. However, the major chemical classes of plant-derived bioactive compounds frequently possess chemical properties, including poor water solubility, stability, and bioavailability, that limit their therapeutic application. Alternatively, natural small molecules (NSMs) containing these components possess modifiable groups, multiple action sites, hydrophobic side chains, and a rigid skeleton with self-assembly properties that can be exploited to construct self-assembled nanoparticles with therapeutic effects superior to their individual constituents. For instance, the construction of a self-assembled nanodrug delivery system can effectively overcome the strong hydrophobicity and poor in vivo stability of NSMs, thereby greatly improving their bioavailability and enhancing their anti-tumor efficacy. This review summarizes the self-assembly methods, mechanisms, and applications of a variety of NSMs, including terpenoids, flavonoids, alkaloids, polyphenols, and saponins, providing a theoretical basis for the subsequent research on NSMs and the development of SANDDS.
1. Introduction
Malignant tumors are a major cause of premature death, and mortality rates are rising in many regions around the world despite the advances in treatment.1,2 According to the latest data from the International Agency for Research on Cancer (IARC), there were 19.29 million new cancer cases and 9.96 million cancer-related deaths globally in 2020, including 4.57 million new cases and 3 million deaths in China. In fact, China accounts for the greatest proportion of new cases (23.7%) and deaths (30%) worldwide.3 The pathogenesis of malignancy is complex, and both prevention and control are difficult due to the multitude of risk factors. Further, many tumor types still lack effective treatment strategies, especially at advanced stages. At present, the main treatment strategy is surgical resection of the tumor supplemented by chemotherapy and (or) radiotherapy because surgical resection alone frequently does not completely eradicate all tumor tissue. However, traditional chemotherapy drugs are also variably toxic against non-cancerous tissues, resulting in intolerable side effects.4In this case, traditional Chinese medicine (TCM) is a promising alternative treatment for many cancers given that these plant-based formulations demonstrate multiple synergistic or additive modes of action, fewer and less severe side effects, and reduced propensity for inducing drug resistance than synthetic anti-cancer agents.5 Moreover, recent advances in chemical separation and structural analysis have led to the identification of many major anti-tumor agents in TCM.6 Another potential significant advantage of the natural small molecules (NSMs) present in Chinese medicinal herbs is their higher biological safety when used as either primary drugs or carriers for other anti-cancer drugs due to their lower inherent cytotoxicity and tissue accumulation. However, many potent anti-tumor agents from TCM have physical and chemical properties that limit their medicinal use, such as poor water solubility, stability, and bioavailability. Conversely, some of these agents, including various terpenoids, flavonoids, alkaloids, polyphenols, and saponins, have chemical moieties that facilitate self-assembly into multicomponent nanoparticles (NPs) with enhanced bioavailability.7
Nanotechnology has numerous applications in medicine, especially for the targeted delivery of anti-cancer drugs. Nanodrug delivery systems (NDDSs) can effectively improve the physical and chemical properties of parent drugs, thereby overcoming physiological barriers to tumor accumulation, reducing off-target toxicity, prolonging the bioavailability, and ultimately enhancing the treatment efficacy.8–10 Nanodrug delivery systems can be constructed without an inactive dedicated carrier, which are termed carrier-free drug-delivery systems (CFDDSs).11 Nonetheless, most nanodrugs still require ionic materials, surfactants, or amphiphilic structures to maintain their stability and function. In recent years, CFDDSs have been developed with high drug-loading capacity, low off-target toxicity, and minimal immunogenicity for cancer treatment.12–14 Further, self-assembly technology holds enormous potential for the development of new drugs.15,16 In self-assembled nanodrug delivery systems (SANDDSs), NSMs form nanocarrier complexes spontaneously through non-covalent bonds (including van der Waals forces, hydrogen bonds, π–π stacking, halogen bonds, hydrophobic interactions, cation–π bonds, ionic bonds, CH–π bonds and solvation) under equilibrium conditions and can be combined with photosensitizers or other drug molecules to form nano-sized multifunctional drug complexes targeting tumor tissues.17
The chemical structures of TCM components are often complex, including side chains with self-assembly characteristics, and many previous studies have used these features to improve the clinical applicability of the core bioactive NSMs for the development of SANDDSs.18,19 Additional structural components can also be added, although these modifications must be simple to preserve the inherent biological safety and pharmacological activities of the parent compound.20,21 The construction of carrier-free SANDDSs based on the self-assembly characteristics of NSMs in particular can greatly improve the drug curative effects by conferring greater tissue targeting and water solubility than traditional synthetic anti-cancer drugs.22 Recently, Xie and colleagues reviewed the self-assembly mechanisms of TCM-derived NSMs and the clinical potential of these NSMs for the treatment of cancers and neurological diseases. They concluded that further research on the self-assembly of NSMs will accelerate the development of these compounds for clinical treatment.23
The construction of SANDDSs can effectively mitigate the clinical limitations caused by the strong hydrophobicity and poor in vivo stability of many TCM-derived NSMs, thereby enhancing their bioavailability and anti-tumor efficacy.17,24 The self-assembly processes may involve the active components of TCM preparations, other clinical anti-cancer compounds, metal chelators, prodrugs, tracers, inhibitors, and (or) photosensitizers, but in all cases the aim is to overcome the suboptimal physical and chemical properties of NSMs. In addition to improved clinical anti-cancer activity, the self-assembly of TCM compounds has great research value, given that it may reveal specific interactions between components and provide clues to even more effective drug combinations. Moreover, Hou and coworkers proposed that the spontaneous formation of multicomponent complexes through self-assembly holds the greatest promise for improved therapeutic efficacy because free drugs seldom possess the bioactivity of the complete TCM preparation.25 Specifically, the interactions among ingredients within the same complex has a great impact on the therapeutic efficacy. For instance, heat reflux and water decoction inadvertently create appropriate conditions for the formation of natural assemblies, and these assemblies both eliminate the toxicity of component drugs and improve the water solubility of the bioactive components.26 Herein, we describe the SANDDSs of TCM-derived terpenoids, flavonoids, alkaloids, polyphenols, and saponins (as shown in Fig. 1). By summarizing the self-assembly applications of existing NSMs and further exploring the possible mechanisms of self-assembly (as shown in Table 1), this review aims to provide a theoretical basis for the future research and development of SANDDSs.
| Category | Natural product | Plant source | Self-assembly mechanism | Self-assembled systems | Advantages | Ref. |
|---|---|---|---|---|---|---|
| Terpenoids | Oleanolic acid | Ligustrum lucidum | Electrostatic, π–π stacking, hydrophobic interactions | OA/GA NPs | Good stability and sustained release | 36 |
| π–π stacking and hydrophobic interaction | OC | Improved solubility, efficacy and safety of the OA | 40 | |||
| Betulinic acid | π–π stacking, hydrophobic interactions | BA-S-S/Ce6 NPs | Improved multiple combination antitumor therapies | 29 | ||
| Betulinic acid | White birch | Hydrogen bonding, hydrophobic interactions | BA/PTX NPs | Improved cancer therapeutics efficiency | 44 | |
| π–π stacking | BA-Ce6 NPs | Enhanced synergistic anticancer efficacy | 45 | |||
| Dihydroartemisinin | Artemisia annua | Hydrogen bonding, hydrophobic interactions | DHA NPs | Good stability, high drug loading efficiency, and responsive release. | 59 | |
| Dihydroartemisinin | Artemisia annua | Hydrophobic interactions | CD NPs | Prolonged blood circulation time | 61 | |
| Flavonoids | Quercetin | Panax notoginseng | Hydrophilic-hydrophobic interactions | HA-Que micelles | Improved the drug targeting | 69 |
| hydrophobic interactions | TQ-Que NPs | Accumulate better in mitochondria | 72 | |||
| Myricetin | Myrica rubra | Hydrophobic interactions | Casein nanocapsules | Good biodegradability | 76 | |
| Coordination interactions | MZG nanoparticles | Good bioavailability and biocompatibility, | 78 | |||
| Baicalin | Scutellaria baicalensis Georgi | Electrostatic interactions, hydrogen bonding | DOX/SA-SS-BAI NPs | Improving the tumor targeting | 83 | |
| Dynamic covalent bonds | BAI hydrogels | Improved antibacterial activities | 85 | |||
| Alkaloids | Berberine | Coptis chinensis | Hydrogen bonds and π–π stacking | BBR and CA co-assembly | Good therapeutic efficacy against | 92 |
| Electrostatic and hydrophobic interactions | BBR-BAI NPs | Good biocompatibility | 93 | |||
| Electrostatic and π–π stacking interactions | The self-assembly of BBR and AA | Blocked the toxicity of AA | 100 | |||
| Camptothecin | Camptotheca acuminata | Hydrophilic-hydrophobic interactions | Camptothecin-Dipeptide Nanotubes | Improved the loading and the stability | 107 | |
| Hydrophilic-hydrophobic and π–π stacking interactions | Self-assembly of CPT and carbamoyl mannose conjugates | Reduced acute renal injury | 108 | |||
| Hydrophobic interactions | SN38/Ce6 NPs | Enhanced cell absorption and tumor accumulation | 114 | |||
| Hydrophobic interaction and π–π stacking | Taxol-CPP nanospheres | Exhibit efficient cytotoxicity | 121 | |||
| Paclitaxel | Taxus brevifolia | Hydrophobic interactions | The self-assembly of PTX-S-S-VE prodrug | High drug loading efficiency | 123 | |
| π–π stacking | SePTX NPs | Selectivity towards cancer cells | 124 | |||
| Hydrophobic force and halogen bonds | F8-SS-PTX NPs | Exhibited good antitumor effect | 125 | |||
| Polyphenols | Curcumin | Curcuma longa | Coordination interactions | Cur-Ca@DMSNs-FA | Enhanced anti-tumor effect | 138 |
| Coordination interactions | ICG@cur-GdNPs | Enhanced the MR and fluorescence imaging and anticancer treatments | 140 | |||
| Tannic acids | Chinese gallnut | Coordination interactions | DOX-Al3+-TA capsule | Reduced cytotoxicity and strengthened targeting | 151 | |
| Coordination interactions | siRNA/EGCG/protamine nanocomposites | Overcome the drug resistance | 159 | |||
| Other Polyphenols | Sm3+-EGCG nanocomposite | Induced apoptosis in cancer cell through mitochondrial dysfunction | 163 | |||
| Saponins | Ginsenosides | Ginseng-derived compounds | Hydrogen bonding and hydrophobic interactions | GSN | Enhanced anti-tumor efficacy and drug targeting, and reduces systemic toxicity | 189 |
| Ginsenosides | Ginseng-derived compounds | Hydrogen bonding and both hydrophobic and electrostatic interactions | INS@GS NPs | Improved the stability and good hypoglycemic efficiency | 191 | |
| Ginsenoside Rg3 | Ginseng-derived compounds | Hydrogen bond interaction | Rg3-PTX-LPs | Enhanced glioma targeting and intratumoral diffusion capability | 204 | |
| Glycyrrhetinic acid | Licorice | Hydrophilic-hydrophobic interactions | CpG-EXO/TGM | Increased the solubility of drug | 212 |
 | ||
| Fig. 1 Schematic diagram of natural small molecules (NSMs) (terpenoids, flavonoids, alkaloids, polyphenols, and saponins) commonly used to construct self-assembled nanodrug delivery systems. | ||
2. NSMs-based SANDDS
2.1 Terpenoids
All derivatives of isoprene (terpene) with the general molecular formula C5H8(n) are called terpenoids. According to the number of isoprene units, these compounds are classified as monoterpenes, sesquiterpenes, diterpenes, and triterpenes. Terpenoids are widely distributed in gymnosperms, and most are aromatic liquids. Many natural terpenoid small molecules (NTSMs) have anti-cancer activity and important applications in drug research and development, such as betulinic acid (BA) extracted from white birch and oleanolic acid (OA) extracted from Ligustrum lucidum. In addition to anti-tumor activity, terpenoids possess strong self-assembly capacity,27 which is attributed to their multi-hydroxyl and multi-carboxyl structures. Moreover, terpenoids can be used to assemble SANDDSs containing photosensitizers and other anti-cancer drugs (including other NSMs from TCMs) through hydrogen bonding, hydrophobic interaction, and π–π stacking. Terpenoids can complete self-assembly in aqueous solution through hydrophobic and hydrogen bond interactions due to their unique hydrophobic skeleton and the hydrogen bond donors and acceptors of NTSMs.28 The UV-vis spectra of some assembly systems also show that π–π stacking is a critical driving force in the molecular self-assembly of terpenoids.Further, NTSMs can also self-assemble with other NSMs to form natural drug carriers with better biosafety and compatibility as well as broader functionality than conventional anti-cancer agents due to their unique biological activities such as hepatoprotection and suppression of drug resistance.29 Through self-assembly technology, NTSMs can also improve the bioavailability of commonly used clinical anti-tumor drugs such as paclitaxel (PTX) and cisplatin. In addition, NTSMs can be efficiently self-assembled with photosensitizers, possibly due to the π–π stacking of the isoprene structure, to produce nanostructures that can combine chemotherapy and photodynamic therapy (PDT) for an enhanced anti-tumor effect.30
Bag35et al. confirmed the potential of self-assembly for drug development with statistics, especially using OA and the triterpene BA. Liu et al.34 showed that the chemical structure of terpenoids gives them strong self-assembly capacity, completing self-assembly with photosensitizers or other anticancer drugs through the hydrogen bonding interaction of hydroxyl and π–π stacking and hydrophobic interactions of the skeleton, between which π–π stacking is a key driving force for the self-assembly of terpenoid molecules. Also, the differences in the spatial orientation of the basic backbone and the carboxyl, such as nano- or micro-sized fibers, tubes, spheres, and flower- and grass-like fibers facilitate their self-assembly. Furthermore, OA can be co-assembled with glycyrrhetinic acid (GA) to construct drug carriers. Wang and colleagues developed a nanocarrier with multiple biological activities via self-assembly of OA and GA, and reported a drug loading efficiency of 15% for the traditional anti-cancer drug PTX. Moreover, OA, GA, and PTX showed synergistic anti-tumor activities. In addition, the drugs loaded on OA/GA NPs also demonstrated good stability and sustained release characteristics, while the NSMs conferred hepatoprotective and anti-inflammatory activities.36
Tumors are a heterogeneous disease with uncontrolled evolution over time, and in many cases, a single treatment strategy cannot completely inhibit tumor growth.37 Furthermore, tumors are prone to relapse, drug resistance and spreading, and drug therapy of tumors has problems such as difficult real-time monitoring of the drug processes in vivo, multi-drug resistance of tumors, and insufficient carrier targeting. However, traditional cancer treatment methods have limitations such as many adverse reactions, poor specificity, and easy to cause drug resistance. Accordingly, the combination of traditional therapy with photothermal therapy (PTT), chemodynamic therapy (CDT), sonodynamic therapy (SDT) and immunotherapy has been proven to be an effective way to treat cancer.38 For instance, sono-photodynamic therapy (SPDT) is a combination of PDT and sonodynamic therapy (SDT) with better anti-cancer efficacy than its individual modalities.39 Similarly, the combination of chemotherapeutic drugs and PDT may possess superior efficacy than each alone. Chloroe6 (Ce6) is a chlorophyll derivative with photodynamic and sonodynamic effects, which is often used as an initiator in cancer treatment. Zheng and colleagues reported that a carrier-free nanosensitizer drug formed using OA and Ce6 through self-assembly produced effective combined chemotherapy and SPDT (Fig. 2). After intravenous injection of OC, OC reaches the tumor site through the EPR effect, and then enters cancer cells. Under stimulation from light and ultrasound, OC can exert synergistic chemotherapy and SPDT by producing ROS, decreasing MMP, and inducing cell apoptosis. The self-assembly of OA with Ce6, possibly via electrostatic, π–π stacking, or hydrophobic interactions, was not only relatively simple, but the product demonstrated good monodispersity and photostability in aqueous solution. Compared to free drugs, drugs combined with OC via self-assembly have demonstrated greatly improved solubility, efficacy, potency, and safety. More importantly, OC shows potent toxicity against tumor cells, and thus OC-mediated synergistic therapy has great potential to improve cancer therapeutics.40
 | ||
| Fig. 2 Schematic diagram of the self-assembly of oleanolic acid and chlorin e6 nanoparticles (OC) and their application for anti-tumor therapy. Reprinted with permission from ref. 40. Copyright (2021), Elsevier. | ||
The bioavailability, side-effect profile, and therapeutic efficacy of the first-line anti-cancer agent PTX can be improved by self-assembly with BA. Wang and coworkers formed supramolecular co-assembled nanoparticles from BA and PTX through hydrogen and hydrophobic interactions and found that these BA/PTX NPs retained both the characteristics of the nanoparticles and the activities of the constituent NSMs. Further, BA/PTX NPs showed good anti-tumor efficacy in vivo and vitro due to the improved biocompatibility and safety conferred by using NSMs as carriers, as well as the synergy between BA and PTX due to their distinct modes of action. Infrared and ultraviolet spectra also indicated that hydrogen bonding and hydrophobic interactions are the main driving force for the co-assembly of BA and PTX. This study by Wang and colleagues provides a template for future research on NSM carrier drugs and a rationale for the choice of pentacyclic triterpenoids as NSM carriers.44
Cheng and colleagues screened eleven small molecules that can self-assemble with Ce6 for combined chemotherapy/PDT and found that BA-based nano-co-assemblies (BA-Ce6 NPs) combined with PDT achieved 93.6% tumor cell eradication. In addition, NTSMs can be constructed through π–π stacking without any other external force for self-assembly. Compared to the free drug and Ce6, BA-Ce6 NPs also demonstrated better stability and dispersion, longer blood circulation time, and greater tumor accumulation, highlighting the advantages of self-assembled NSMs as a new drug-delivery platform.45
The efficacy of PDT is dependent on the generation of reactive oxygen species (ROS); however, the ROS produced by PDT in the tumor microenvironment (TME) may be consumed by glutathione (GSH), which is overproduced in tumor cells.46 Thus, Cheng and coworkers designed and synthesized a natural pentacyclic triterpene BA modified with a disulfide bond (BA–S–S), and then constructed a new carrier-free delivery system with BA–S–S and Ce6 for synergistic anti-cancer enhancement and improved PDT biosafety. Also, given that the disulfide bond enables the drug to respond to GSH, Ce6 produces sufficient ROS for efficient PDT, and both in vitro and in vivo treatment studies demonstrated that this modified NP preparation significantly enhanced the synergistic anti-tumor effect, while still maintaining good biodegradability and biocompatibility.29
Cheng and colleagues designed a multifunctional SANDDS for combined chemotherapy and PTD by assembling Ce6 with disulfide-modified UA, yielding UASS-Ce6 NPs, and reported good self-assembly using simple preparation methods and a better therapeutic effect on tumor cells than individual Ce6 and UA treatment. Moreover, the introduction of disulfide bonds ensured robust ROS-induced cytotoxicity even in the presence of GSH. In addition, depending on the characteristics of the NSM used, these dual-function nanodrugs can show better biocompatibility, safety, and biodegradability than raw NSMs given that the whole assembly structure acts as a therapeutic drug.51
UA possesses unique drug characteristics compared to other terpenoids, which can prevent drug resistance by tumor cells, and thus improve the therapeutic effect. For instance, the combination of UA and cisplatin can effectively overcome multiple cisplatin inactivation pathways in ovarian cancer cells. Wang and colleagues constructed a reductive reactive platinum(IV) (Pt(IV))/UA/polyethylene glycol (PEG) dual prodrug (Pt(IV)-UA PEG) complex to treat cisplatin-resistant ovarian cancer and reported that the released UA and cisplatin had distinct anti-cancer mechanisms and that the synergistic effects overcame the detoxification and anti-apoptosis mechanisms of cancer cells. In addition, the Pt(IV)-UA NPs in A2780/DDP showed a prolonged blood circulation time, enhanced tumor accumulation, and significantly improved anti-tumor efficacy without detectable side effects in tumor-bearing mice.52
UA itself could be formulated into nanosize or microsize carriers, and many SANDDSs based on UA have been constructed for drug delivery against various cancers.53 However, the poor tumor targeting and low solubility of UA still affect its curative effect.54 Some studies have shown that structural modification of drugs can alter their physical and chemical properties. The carboxyl and hydroxyl groups on the UA structure can be used for polymerization to form poly UA.55 Then, poly UA can self-assembly into NPs via the nano-precipitation method. Ou et al. synthesized poly(ursolic acid) (PUA) utilizing the hydroxyl and carboxyl groups of UA via polycondensation, where PUA could self-assemble into nanoparticles (PUA-NPs).50 PTX was selected as a model drug to generate PTX-loaded nanoparticles (PUA-NPs@PTX) against colorectal cancer. In vitro studies showed that PUA-NPs@PTX has strong cytotoxicity against colorectal cancer CT26 cells, while the in vivo results indicated that PUA-NPs@PTX can prolong the blood circulation time, enhance the tumor accumulation, and significantly improve antitumor efficacy in CT26 tumor-bearing mice (Fig. 3).
 | ||
| Fig. 3 Illustration of the major steps involved in PTX delivery by PUA-NPs@PTX to treat colorectal cancer. Reprinted with permission from ref. 50. Copyright (2020), Wiley. | ||
In recent research, a carrier-free DHA nanodrug (DHA NPs) was prepared by the self-assembly of DHA molecules via hydrogen bonding and hydrophobic interactions (Fig. 4).59 The DHA NPs possessed a monodispersed spherical morphology with a mean diameter of 93 nm. The DHA NPs also exhibited good stability, high drug loading efficiency, and responsive release. The in vitro and in vivo experiments showed that the DHA NPs exhibited significantly higher therapeutic efficacy than the free DHA. Furthermore, they revealed the potential molecular mechanism of the DHA NPs by RNA-seq and Western blotting analysis. Some hydrophobic prodrugs could self-assemble into prodrug nanoparticles, which have shown great potential in drug delivery systems with high drug-loading efficiency and favorable therapeutic effect.60 Luo and colleagues designed albumin-binding and light-triggered core–shell dimeric prodrug nanoparticles (Ce6&DHA-S-DHA@CMN NPs, CDC NPs).61 Firstly, the ROS-sensitive single thioether bridge DHA dimer (DHA-S-DHA) was prepared. Then, the dimeric prodrug DHA-S-DHA self-assembled into nanoparticles (CD NPs) via a simple nanoprecipitation method. Finally, CD NPs were encapsulated with Chlorin e6 (Ce6) and stabilized by albumin-capturing maleimide- and hypoxia-sensitive 2-nitroimidazole-modified carboxymethyl chitosan (CMCTS-MAL&NI, CMN), and CDC NPs were obtained. The results showed that the novel CDC NPs with long blood circulation and light-triggered drug release were successfully established for chemo-photodynamic combination therapy against hypoxia-induced metastasis of lung cancer (Fig. 5).
 | ||
| Fig. 4 Schematic diagram of the preparation of self-assembled DHA NPs and their application in cancer treatment. Reprinted with permission from ref. 59. Copyright (2023), The Royal Society of Chemistry. | ||
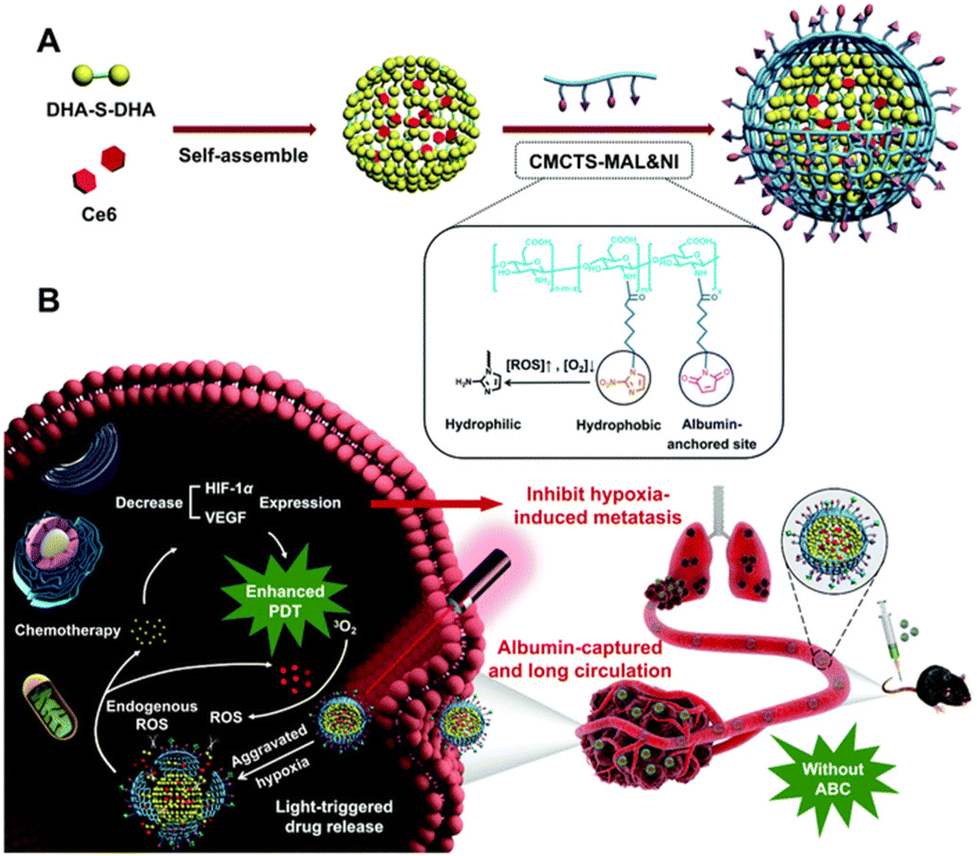 | ||
| Fig. 5 Schematic illustration of DHA-S-DHA self-assembling into CDC nanoparticles (A) and its applications for chemo-photodynamic combination therapy (B). Reproduced from ref. 61 with permission from (Elsevier), Copyright (2021). | ||
2.2 Flavonoids
Flavonoids are abundant and diverse secondary metabolites of plants.62 Originally, ‘flavonoid’ was the general name given to the class of compounds derived from 2-phenylchromone, but now refers to a series of compounds formed by connecting two benzene rings through three carbon atoms (general structure C6–C3–C6). Similar to most NSMs, flavonoid small molecules have numerous biological activities, including antibacterial, anti-viral, hypoglycemic, anti-tumor, anti-inflammatory, and antioxidant effects, but their use in the pharmaceutical industry and clinical applications is limited due to their low water solubility.63–65 In drug research and development, self-assembly is mainly used to improve the physical and chemical properties of flavonoids. Amphiphilic compounds are obtained by connecting hydrophilic groups, and self-assembly is completed by hydrophobic interactions to obtain nanoparticles with improved water solubility and bioavailability. Through self-assembly, flavonoids can be connected to phospholipids, lecithins, hydrophilic macromolecular proteins, or borates to form water-soluble vesicles or micellar drug molecules in which the hydrophobic components act as a container for central hydrophobic drugs, and the hydrophilic components contact water in biological fluids to form a shell.To further enhance the anti-tumor efficacy of Que, Pang and colleagues constructed a coupling polymer to improve its water solubility.69 The hydroxyl group of Que was connected to HA premodified by adipic acid diamine (ADH) through succinate ester, yielding an amphiphilic polymer (adipic acid-modified hyaluronic acid). Subsequently, hyaluronic acid–Que micelles were synthesized through self-assembly. In addition to the hydrophobic effect of the amphiphilic polymer, the driving force for self-assembly of the coupling compound, i.e., the skeleton in HA, can form a hydrophilic shell by hydrogen bonding with the surrounding water molecules. The cytotoxicity of HA–Que micelles was 4-fold greater against MCF-7 cells (a CD44-overexpressing cell line) compared to free Que. In addition, HA–Que micelles demonstrated a 20-fold greater in vivo half-life and good inhibitory effects on tumor growth in H22 tumor-bearing mice. Hemolytic toxicity and intravenous stimulation tests further indicated that the HA–Que micelles were safe and effective NDDSs for targeted anti-tumor therapy. Although HA can increase cell uptake and targeting and has good biocompatibility and biodegradability, the modification with ADH can further improve drug targeting by forming ester bonds with Que to achieve pH-controlled release.
Chen and coworkers developed a Que-loaded micellar anti-cancer drug-delivery system based on lecithin, which demonstrated good therapeutic effects against breast cancer cells70 due to the improved physical and chemical properties of Que and concomitantly higher bioavailability. The addition of lecithin to the micellar system also increased the volume of the hydrophobic core, thereby providing more space for the dissolution of hydrophobic drugs and a higher drug loading capacity.
Mitochondrial signaling and metabolic output are critical for the evolution and progression of cancer, and thus targeted mitochondrial NDDSs may be particularly effective treatments.71 Xing and colleagues constructed a mitochondrial-targeted SANDDS based on amphiphilic TPP–Que conjugates.72 Triphenylphosphine (TPP) is the most commonly used mitochondrial ligand and has shown high efficacy for delivering drugs to mitochondria. To improve the stability of the coupling compound, PEG was introduced in the catechol structure of phenylboronic acid (PBA) and Que in the form of coordination bonds. The special catechol structure of Que makes it possible to polyglycolate with PBA–PEG through coordination bonds. A TPP–Que coupling compound was successfully synthesized through a nucleophilic substitution reaction. After PEGylation, the obtained TQ–PEG NPs exhibited better stability, thus prolonging the circulation time and enhancing the tumor-targeting effect. In addition, the coordination bond made the polymer pH responsive, thereby enhancing PEG removal for increased activity on tumor mitochondria. Compared to free Que, TQ–PEG NPs accumulated in mitochondria in vivo and in vitro and showed significant anti-cancer effects. More importantly, the TPP–Que conjugates self-assembled into nanoparticles, indicating that self-assembly is mainly driven by hydrophobic interactions rather than hydrogen bonds or ion interactions.
To improve the bioavailability of Que, Liu and colleagues constructed Que-Quinoa protein (QP)-Lotus root amylopectin (LRA) nanoparticles (Que-QP-LRA).73 It was shown that Que can self-assemble with QP to greatly improve its solubility. However, the bioavailability of Que was not greatly improved due to the influence of active enzymes and gastric acid in the stomach. Therefore, Liu and colleagues coated Que-QP with LRA to obtain nanoparticles with a more regular spherical shape. In vitro simulated digestion experiments showed that Que-QP-LRA NPs improved the stability of Que in the stomach and absorption across the gastrointestinal tract. Molecular interaction analysis also showed that hydrogen bonding and hydrophobic interactions are the main driving forces for the formation of Que-QP-LRA nanoparticles.
Macromolecular proteins have better biocompatibility, biodegradability, and biosafety, and thus Guo and colleagues combined Myr with macromolecular proteins to improve its physical and chemical properties, while retaining its efficacy.76 The dietary protein casein also has good biodegradability and biocompatibility when taken orally, and in addition is an amphiphilic protein with good self-assembly characteristics. The hydrophobic sites of these proteins can be self-assembled with Myr through hydrophobic interactions, and the preparation process is simple. Guo and coworkers used the self-assembly method to prepare casein Myr nanocapsules and observed that the absorption efficiency was four-times that of standard Myr samples, thus greatly improving its utilization efficiency.
Oxidative stress is caused by an imbalance between the cellular generation of prooxidants (from oxidative phosphorylation in mitochondria and various other metabolic reactions) and cellular antioxidant capacity, which can cause irreversible damage to cell components and even lead to cancer and other diseases.77 Moreover, oxidative stress is a critical pathomechanism in many chronic diseases. Therefore, the development of antioxidant agents is an important strategy to slow the development of chronic diseases and to maintain redox balance for optimal cell function and general health. Myricetin has strong antioxidant capacity given that it not only can chelate intracellular transition metal ions to prevent Fenton-like reactions, but can also upregulate antioxidant enzymes and related signaling pathways to increase total cellular antioxidant capacity. Glutathione is the primary free antioxidant peptide in cells and is used as an electron donor by several antioxidant enzymes. To obtain an effective and practical antioxidant, Ma and colleagues78 co-assembled Myr, Zn2+, and GSH into Myr–Zn2+–GSH (MZG) nanoparticles, which demonstrated high water solubility, good bioavailability and biocompatibility, and a stable antioxidant effect. The main driving force for the formation of MZG was coordination interactions, which are the strongest non-covalent interactions, thus ensuring better stability under physiological conditions and stable antioxidant performance without affecting the clearance of ROS by Myr and GSH.
Although BAI has good curative effects on tumors, its poor water solubility and permeability also limit clinical applications. Therefore, it is usually combined with macromolecules to form amphiphilic compounds as drug carriers for improved therapeutic effects. Zhang and coworkers83 used cystamine as the connecting arm to graft BAI with sodium alginate (SA), forming a SA-BAI NDDS through hydrophobic or electrostatic action, which was used as a carrier for the anti-cancer agent doxorubicin (DOX) to obtain DOX/SA-SS-BAI NPs. SA greatly enhanced the water solubility of this drug, while cysteamine, as a connecting arm, could be specifically induced by GSH at the tumor site to release DOX and BAI, thereby improving the tumor targeting and reducing exposure of healthy tissue. Indeed, the inhibitory effect of DOX/SA-SS-BAI NPs on cancer cells was higher than that of free BAI or DOX, while the drug loading and encapsulation efficiency of the carrier were satisfactory.
O-carboxymethyl chitosan (OCMC) has a large number of active amino groups that are amenable to chemical modification, while lactic acid (LA) can be specifically recognized by ASGP-R receptors on the membrane of liver parenchyma cells and quickly absorbed by endocytosis. Therefore, Shao and colleagues connected BAI and LA to OCMC through an amidation reaction and loaded DOX to produce a new anti-cancer treatment84 that releases BAI and DOX in the TME through active targeting and enhanced permeability and retention (EPR) for a synergistic effect on the tumor but reduced toxic side effects. To improve the physical and chemical properties of BAI for greater bioavailability, Wang and colleagues prepared a dynamic covalent hydrogel by combining BAI directly with borate through the boric acid α-hydroxycarboxylic acid group and catechol structure.85 The dynamic boric acid bond is reversible and covalent, which endowed the hydrogel with excellent stability, plasticity, and self-healing. Further, BAI could also contribute to therapeutic effects after intravenous injection due to its endogenous antibacterial activity, including good therapeutic efficacy against Staphylococcus aureus infection.
The self-assembly strategy can improve the bioavailability and the clinical efficacy of the NSMs. Coptidis Rhizoma (CR) and Scutellariae Radix (SR) are classical TCM combinations in clinic, which are used as the herb pair CR-SR. Huang et al. selected the CR-SR main component berberine-baicalin (BBR-BAI) as examples to reveal the differences and mechanisms of self-assemblies originating from the co-decoction and physical mixture, respectively.86 This study showed that the physical mixture morphology of both the herb pair (CR-SR) and the phytochemicals ((BBR-BAI)) was nanofibers (NFs), while their co-decoction morphology was nanospheres (NPs). This result indicates that temperature can affect the arrangement of the assembly (Fig. 6).
 | ||
| Fig. 6 Illustration displaying the differences between berberine–baicalin self-assemblies formed by physical mixing and decocting. Reprinted with permission from ref. 86. Copyright (2022), BMC. | ||
2.3. Alkaloids
Alkaloids are natural alkaline organic compounds, most of which have complex ring structures, containing nitrogen atoms. They are important active ingredients in many TCM with numerous bioactivities but are poorly soluble in water.87 However, alkaloids such as berberine, PTX, and camptothecin (CPT) are widely studied for potential clinical use and SANDDS development.28 Berberine (BBR) from Coptis chinensis is often self-assembled with NSMs of other TCMs to improve its inherently disadvantageous physical and chemical properties, while enhancing its anti-tumor and antibacterial efficacy. Camptothecin derived from Camptotheca acuminata is often conjugated to polypeptide molecules by self-assembly through π–π stacking and hydrophobic interactions to improve bioavailability. Moreover, the ester bond and succinyl bond used for conjugation can be hydrolyzed at the tumor site to improve the drug targeting and release of the CTP precursor for enhanced tumor accumulation. In addition, due to their structural properties and metal chelation capacity, some alkaloids can be conjugated with photosensitizers to produce molecules for combined chemotherapy plus PDT. Alkaloids can also be self-assembled with NSMs and other substances to yield multifunctional drugs that retain their original bioactivity but with improved water solubility and bioavailability.Huang and colleagues92 reported a two-component self-assembly model for combining plant chemicals such as BBR and cinnamic acid (CA) into nanoparticles through intermolecular hydrogen bonding and π–π stacking. The success of this self-assembly was successfully verified through infrared spectroscopy, ultraviolet spectroscopy, field-emission scanning electron microscopy, and transmission electron microscopy (TEM). It is speculated that the initial self-assembly involves two molecules of BBR and two molecules of CA, which form a butterfly-like structure with CA as the body and BBR as the wings. The conjugation of two molecules depends on the formation of hydrogen bonds between the CA carbonyl group and BBR nitrogen atom, while their respective aromatic rings π–π stack to form spherical nanoparticles with stronger biofilm removal capacity than each compound individually. The authors further demonstrated that these NPs can spontaneously adhere to the bacterial surface, penetrate cells, and induce a polymerization attack on methicillin-resistant Staphylococcus aureus (MRSA). Through transcriptome analysis and quantitative polymerase chain reaction analysis, the multipathway bactericidal mechanism of NPs against MRSA was described. In addition, these NPs were found to be non-hemolytic in vivo and vitro, with low toxicity for the effective and safe treatment of MRSA infection.
Due to the clinical misuse of antibiotics, bacterial resistance is rising, creating an urgent need for novel antibiotic drugs with distinct mechanisms of action. The design of new drugs through self-assembly technology of natural molecules may be one solution, given that these compounds not only have high drug loading capacities and multiple bioactivities, but also good safety profiles. Li and colleagues developed new antibiotics by self-assembling BBR and BAI into nanoparticles.93 BBR is a natural hydrophobic cation, which first combines via the electrostatic interaction between the –COO– of the flavonoid glycoside and the BBR N+ to form a one-dimensional complex unit. Then the hydrophobic flavonoid nuclei of BAI and BBR approach each other to form a basic unit that can further form NPs. Hydrophobic interactions further drive their self-assembly into three-dimensional nanostructures with hydrophilic glucuronic acid directed outward and the hydrophobic parent nucleus inward. With the extension of the hydrophilic glucuronic acid, the NPs showed stronger affinity for bacteria and greater efficacy for biofilm reduction. In vitro hemolysis tests, cytotoxicity tests, and in vivo toxicity evaluation of zebrafish demonstrated that the self-assembled products are highly biocompatible, which provides a foundation and direction for the production of other nano-antibacterial drugs.
BBR and BAI are the active components of the Huanglian Jiedu Decoction (consisting of rhubarb and Scutellaria baicalensis). A comprehensive analysis of the supernatant and natural sediment of the Huanglian Jiedu Decoction by Chen and coworkers94 revealed a significantly higher BAI and BBR content in the sediment than in the aseptic precipitation of the supernatant, suggesting their self-assembled complexation in this TCM prescription. The formation of a BAI-BBR complex was studied by electrospray ionization (ESI)-mass spectrometry, nuclear magnetic resonance, ultraviolet visible (UV vis), Fourier transform infrared (FTIR), and fluorescence spectroscopy. Subsequently, the morphology and size distribution of the BAI-BBR self-assembled nanoparticles were characterized by SEM, TEM, and dynamic light scattering. This study provides strong evidence for self-assembled phytochemical compounds in natural sediments and provides a better understanding of Huanglian Jiedu Decoction activities.
The development of mitochondrial-targeting probes for the induction of metabolic dysfunction in tumor cells combined with tumor imaging is a promising strategy for cancer diagnosis and treatment,95 where BBR has several advantageous characteristics for this application, including natural mitochondrial targeting, a cationic structure, and fluorescence properties from its unique aromatic rings suitable for imaging.96 However, due to its π-conjugated structure, BBR shows low anti-cancer efficacy and low fluorescence quantum yield in practical applications. Thus, to improve both its anti-cancer efficacy and quantum yield, An and colleagues synthesized dehydroberberine (DH-BBR), a planar molecule with a complete π-conjugated four-ring structure97 for longer fluorescence emission wavelengths and higher fluorescence quantum yield. Moreover, the planar structure could allow for better intermolecular interactions to promote self-assembly. In fact, DH-BBR was found to self-assemble into monodispersed organic nanoparticles (DTNPs) after integration with the lipophilic anion tetraphenylborate (TPB). After intravenous injection, DTNPs entered the tumor tissue more effectively, selectively accumulated in mitochondria, emitted stronger fluorescence, and caused greater mitochondrial dysfunction than BBR, thus inducing more effective tumor shrinkage.
Berberine can also be modified with TMC-derived NSMs to reduce the drug toxicity. Aristolochic acid (AA) is a nitrophenanthrene organic acid compound isolated from Aristolochia plants such as Aristolochia and Asarum.98 It is reported to cause a series of health problems, such as acute kidney injury and liver cancer. Therefore, the use of medicinal plants containing AA is restricted or prohibited.99 However, if the toxicity of AA can be neutralized by molecular modification, the application scope of AA-containing medicinal plants can be expanded. Wang and colleagues speculated that the self-assembly of BBR and AA could neutralize the toxicity of AA.100 This assembly was initially driven by the electrostatic attraction between the carboxyl group of AA and the quaternary ammonium ion of BBR, and then by π–π stacking and hydrophobic interaction, yielding a linear, large-scale, and strong self-assembly. Due to the large assembly binding constant, a series of assembly driving forces and various π–π stacking sites are closely assembled. Notably, the assembly structure protects the carboxyl sites of AA from being metabolized into the toxic aristolochic lactam. Thus, this self-assembly strategy provides a chemical basis for detoxification. Indeed, systemic toxicological studies in zebrafish and mice showed that the supramolecular self-assembly of BBR and AA significantly reduced acute renal injury induced by AA.
Camptothecin is difficult to dissolve but easy to form aggregates because of the strong π–π accumulation of its quinoline ring. Guo and colleagues105 enhanced the water solubility of CPT by conjugation to hydrophilic arginine via ester bonds, which allowed CPT to be selectively released by hydrolysis. It was also found that this modified-CPT could self-assemble into helical nanofibers through π–π stacking and hydrophilic–hydrophobic interactions. The nanofiber could be effectively taken up by cells and were easily absorbed by the nuclei of tumor cells. Moreover, in vivo, nanofibers showed the best tumor inhibition effect, and this phenomenon was attributed to the assembled nanofibers better accumulating in the tumor and entering cells.
Sun and colleagues synthesized a CPT lysine A coupling compound through succinic acid conjugation106 and found that its self-assembled into water-soluble uniform nanotubes suitable as drug carriers due to its high loading capacity (60.5%). The nanotube structure also provides a good hydrophobic environment for CPT, and indeed CPT in this formation showed good cell uptake and toxicity in vitro against several tumor cell lines.
Kim reported a self-assembly system of two CPT-coupled dipeptides, in which CPT molecules were linked to amino acids by succinyl bonds.107 Fourier transform-IR showed that the main driving force for self-assembly was amphiphilic phase separation in aqueous solution. Subsequently, these CPT dipeptides were assembled into nanotubes with diameters in the range of 80–120 nm in PBS. The nanotube structure isolates the CPT segment in the hydrophobic nanotube wall, thereby protecting the drug from the opening of hydrolyzed lactones. Further, the succinyl bond permits the easy release of the active CPT via hydrolysis, and the rate of release is strongly dependent on its concentration. This dipeptide nanostructure can improve drug loading and CPT stability without excipients or macromolecular carriers.
Wang and colleagues developed a new method to conjugate CPT with carbamoyl mannose108 and reported good self-assembly. In this design, CPT is meant to act as an anti-tumor agent and promote self-assembly, while carbamoyl mannose was chosen to enhance the selectivity for tumor cells. The carbamoyl mannose group points to water as a hydrophilic group, while the CPT group turns in the opposite direction as a hydrophobic tail to form nanotubes. Self-assembly is mainly mediated by hydrophobic–hydrophilic interactions and π–π interactions between the CPT skeletons. Through this assembly, the anti-cancer activity and water solubility of CPT greatly increased, and cancer cell apoptosis could be efficiently induced.
Anti-cancer regimens that include drugs with imaging functions are valuable given that they permit the monitoring of the biological distribution and circulation of therapeutic drugs as well as therapeutic responses in real time. Recent studies have shown that camptothecin can be self-assembled into a drug with imaging function. DOTA is a widely used metal chelating agent that can effectively chelate lanthanide metals, and thus used in a variety of imaging and radiotherapy applications. Su and colleagues directly conjugated CPT to DOTA, and then self-assembled the complex under physiological conditions to obtain the prodrug SPAD with glutathione responsiveness for improved tumor targeting.109 The two hydrophobic CPT molecules provide strong π–π interaction to promote the directional growth of monomers, while DOTA increases the water solubility, promotes the self-assembly of the prodrugs into nanostructures, and can bind metal ions at the outer surface. SAPD was reported to have significant in vitro toxicity against a variety of cancer cell lines and effectively inhibit the growth of tumor globules.
The CPT analog 10,11-methylenedioxy-camptothecin (FL188) has even better anti-cancer effects and selective activity, but due to its low solubility and adverse side effects, it needs to be further optimized.110 ES-285 is an aliphatic amino alcohol with long saturated hydrophobic hydrocarbon side chains. It has good anti-proliferation activity against cancer cells, but also has low water solubility.111 Zhang and colleagues112 used FL188 and ES-285 to prepare two hydrophobic prodrugs (DHPs, FL-1 and FL-2), which were self-assembled into nanoparticles to improve their water solubility. The obtained DHPs and FL118 promoted the formation of nanoparticles (FL-2NPs) through π–π stacking of the conjugated aromatic rings. The polar high charge densities of DHPs and FL118 form stable hydrogen bonds with surrounding water molecules and stabilize the particle-solution interface. When FL-2NPs are triggered by endogenous reductive stimulation in the TME, they rapidly release the bioactive FL118 for anti-cancer therapy.
Guo and coworkers self-assembled the CPT derivative irinotecan with the photosensitizer CUR to develop a new dual-action anti-cancer drug.113 Although a variety of anti-cancer drugs inhibit tumors via distinct mechanisms, appropriate combinations can achieve synergistic effects for improved anti-tumor efficacy. An ion pair complex (ICN) composed of CUR and irinotecan hydrochloride in polar organic solvent displayed a dispersion stability time profile indicative of hydrogen bonds between CUR and its derivatives. This unique nanoparticle not only overcomes the hydrophobicity of CUR, but also enhances the anti-tumor effect of irinotecan by integrating multiple therapeutic modes. Similarly, Zhao and colleagues assembled the CPT analog 7-ethyl-10-hydroxycamptothecin (SN38) with the PDT compound Ce6, yielding the carrier-free nanoparticle SN38/Ce6 for combined chemical photodynamic therapy (CDT) against cancer.114 Both visual images of molecular dynamics simulations (MDS) and experimental data revealed that the main driving forces for self-assembly were hydrophobic interactions and van der Waals forces, which constitute a novel strategy for the development of NDDSs. The stability and dispersion of the SN38/Ce6 NPs were significantly improved compared to free Ce6 NPs. In addition, the compound demonstrated higher ROS production efficiency under irradiation and enhanced cell absorption and tumor accumulation in vivo and in vitro.
An and colleagues122 designed a SANDDS that exploits the hydrophobicity of PTX to assemble gallic acid (GA)Fe(III)-coated bovine serum albumin (BSA) nanoparticles (GA-Fe@BSA-PTX) with a consistent size (∼115 nm), high water dispersibility and stability, and low non-target cytotoxicity. Moreover, GA-Fe@BSA-PTX demonstrated a good magnetic resonance imaging performance and tumor accumulation equivalent to GA-e@BSA-PTX. The self-assembled nanoparticles showed good therapeutic efficacy due to the combined effects of chemotherapy and photothermal therapy (PTT).
Amphiphilic molecules can easily self-assemble into nanoparticles in water, while hydrophobic molecules easily form precipitates in water and are difficult to self-assemble into nanoparticles. However, inserting disulfide bonds between two hydrophobic molecules can promote their self-assembly to form stable nanodrugs. Wang and colleagues conjugated the hydrophobic PTX and the hydrophobic vitamin E (VE) through an S–S bond to form the PTX-S-S-VE prodrug molecule and further showed its self-assembly into uniform and stable nanoparticles with a drug loading efficiency for Taxol of up to 60%.123
Zhou and coworkers constructed a self-destructive dimer PTX prodrug connected by a hypoxia-sensitive azobenzene bond and used the dimer prodrug with Ce6 for combined chemotherapy and PDT, while exploiting the hypoxic characteristics of the TME for targeting.124 When the drug reaches the hypoxic tumor site, the prodrug will undergo biological reduction due to the overexpression of nitroreductase and azoreductase, releasing active PTX. Simultaneously, the photosensitizer will produce 1O2 in response to light stimulation for further tumor cell destruction. This process requires oxygen consumption, which further aggravates hypoxia in the TME, and thus promotes the release of more dimer prodrug. Through this cycle, synergy is achieved for a superior anti-tumor effect.
Wang and colleagues reported that fluorination can further improve the self-assembly properties of PTX prodrug nano-assemblies.125 A fluorine-modified prodrug was developed by combining PTX with perfluorooctanol (F8-SS-PTX) through disulfide bonds and compared to PTX octanol prodrugs (C8-SS-PTX). The results showed that fluoroalkylation can improve the self-assembly stability, in vivo fate, and anti-tumor efficacy. Fluorination may be an especially effective strategy for the rational design of advanced nanodrugs. This assembly process was further demonstrated by MDS, which revealed that alkyl–π interaction is the main force for C8-SS-PTX assembly given that there are more hydrogen bonds, while halogen bond formation (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯F) is the main force for F8-SS-PTX assembly. The only difference between these two prodrug nanomodules is the substitution of fluorine atoms. It has been reported that fluoroalkylation can promote the self-assembly of amphiphilic polymers and greatly improve the self-assembly and colloidal stability of prodrug nanomodules due to the fluorine-mediated hydrophobic force and the halogen bond of fluoroamphiphilic polymers.
O⋯F) is the main force for F8-SS-PTX assembly. The only difference between these two prodrug nanomodules is the substitution of fluorine atoms. It has been reported that fluoroalkylation can promote the self-assembly of amphiphilic polymers and greatly improve the self-assembly and colloidal stability of prodrug nanomodules due to the fluorine-mediated hydrophobic force and the halogen bond of fluoroamphiphilic polymers.
2.4 Polyphenols
Polyphenols are organic molecules containing o-dihydroxy or o-trihydroxy groups directly connected to aromatic hydrocarbon groups by one or more hydroxyl groups. These structures are complex and diverse, and widely distributed in vegetables, fruits, spices, herbs, tea, and grapes among other sources.126 The demonstrated benefits of these compounds include anti-inflammatory, anti-tumor, anti-vascular, antioxidant, and antibacterial bioactivities.127 The distinct properties of these molecules, including antioxidant activity, anti-aging effects, substrate adsorption, adhesion, enzyme inhibition, and biocompatibility, appear to depend on the positions of various functional groups such as carbon–carbon double bonds, phenolic hydroxyl moieties, and carbonyl groups. In recent years, polyphenols have attracted extensive attention due to their excellent properties for various biomedical applications such as imaging and drug delivery.128 Polyphenols can form metal polyphenolic networks (MPNs) by complexing with metal ions through coordination bonds. These MPN structures can be used to construct core–shell structures to transport hydrophobic drugs and can also improve the efficacies of protein drugs and inhibitors.129 The polyphenols commonly used for self-assembly modification include tannic acid (TA), catechin (CA), epigallocatechin gallate (EGCG), oligomeric epigallocatechin gallate (OEGCG), and gallic acid (GA).130The catechol structure of natural polyphenol compounds can coordinate with a variety of metal ions, and the MPNs formed are important resources for new drug research.131 MPNs have attracted much attention in the drug research and development fields due to their simple fabrication, excellent physical and chemical properties, high biocompatibility, and numerous potential therapeutic applications.132 Drug delivery by MPNs has many advantages, including the potential for drug-controlled release, tumor metastasis inhibition, bioimaging, and tumor targeting. Through the coordination network, many types of drugs can be encapsulated for targeted release, and different types of capsules and films can be obtained by selecting different metals to construct MPNs, thereby expanding their functional diversity.133 The selection of metal ions is also critical for effective tumor visualization. In addition, MPNs have good pH responsiveness. Acidic conditions can weaken the coordination between polyphenols and metals to achieve specific drug release in the TME, reduce exposure and toxicity to normal cells, and improve bioavailability. At present, MPNs composed of metal ions and polyphenols have aroused the interest of researchers due to their pH responsiveness, flexible structures for the incorporation of various small and macromolecules and good thermal stability. Currently, these systems are being used for the development of novel antibacterial drugs, anti-tumor therapies, PDTs, CDTs, and metal ion imaging.134
Wang and colleagues synthesized multifunctional dendritic mesoporous nanoparticles (DMSNs) for carrying CUR through Ca2+ chelation (Cur-Ca@DMSNs-FA).138 This carrier system demonstrated pH-sensitive drug release as well as good tumor cell targeting and biocompatibility. Cur-Ca@DMSNs-FA also induced apoptosis of breast cancer cells by inhibiting the PI3K/AKT/mTOR and Wnt/B-catenin signaling pathways and by activating the mitochondrial apoptosis pathway. Further, the carrier significantly improved the bioavailability of CUM in vivo. Thus, these initial findings provide a theoretical basis for the development of new drugs to treat breast cancer.
Chelation with metal ions can also be used to combine CUM with contrast agents for simultaneous tumor destruction and imaging application. Cancer cells have high energy requirements, and thus inhibiting mitochondrial respiration is a powerful means of cancer treatment. Recent studies have reported that CUM can inhibit mitochondrial function and induce endoplasmic reticulum stress in cancer cells, causing the release of Ca, which leads to the loss of mitochondrial membrane integrity, triggering of apoptosis.139 Wen and colleagues140 constructed Moss PNs with CUR and Gd3+ and loaded indocyanine green (ICG) through electrostatic attraction, yielding ICG@cur-GdNPs. The degradation properties of these NPs led to the specific release of CUR, Gd3+ and ICG in cancer cells, greatly improving the efficacy of chemotherapy/PDT, while minimizing damage to normal tissues. Curcumin has also been found to induce mitochondrial damage in cancer cells, while Gd3+ integration in ICG ICG@CUR-GdNPs allowed for magnetic resonance (Mr)/fluorescence imaging guided chemotherapy/PDT. Bimodal MR/fluorescence imaging using ICG@CUR-GdNPs may provide accurate tumor visualization to guide combined chemotherapy/PDT. Indeed, both in vivo and in vitro experiments have shown that this system has superior efficacy for the treatment of triple negative breast cancer.
Wong and colleagues found that CUR can complete self-assembly with sugar as the catalyst.141 When fructose and CUR are added to water, the hydrogen bonds of fructose can guide the self-assembly of the highly insoluble (hydrophobic) CUR to form a well-defined capsule through non-covalent forces. This simple method has the potential for template polymerization and construction of nanocarriers in water.
Many protein drugs have demonstrated potent pharmacological effects, but have limited clinical utility due to their potential immunogenicity, high degradability, and rapid removal from the circulation.147 In this case, PEG is widely used to improve the clinical utility of protein drugs, but some proteins cannot bind to PEG because they do not contain the necessary functional groups, such as amino, carboxyl, and mercaptan groups. In addition, PEGylation may reduce the activity of the original protein due to spatial interference at the active site. Alternatively, polyphenols are composed of hydrophobic and hydrophilic groups, which can form complexes with biological macromolecules such as proteins through non-covalent interactions. Honda and colleagues148 modified a green fluorescent protein with TA to improve the stability of the protein delivery system. However, there was a non-specific interaction between the two, and thus they used a copolymer modified with tannic acid and PBA to conduct sequential self-assembly, mixed the protein and TA in aqueous solution to cover the protein with TA, and then added the copolymer PEG (PEG poly(amino acid)) to form borate esters between TA and the copolymer, yielding a core–shell ternary complex. In a subcutaneous tumor model, this ternary complex covered by PEG prevented adverse interactions with serum components, thereby significantly prolonging its blood circulation and enhancing its accumulation at the tumor site. This supramolecular self-assembly technique can be used as a new method to design protein delivery systems.
In recent years, hollow capsules assembled with MPNs have attracted interest due to their ideal performance, selective permeability, high mechanical and thermal stability, and pH-responsive decomposition. Guo and colleagues developed a library of MNP capsules with distinct properties by combining TA with different metal ions.149 For example, the film thickness, disassembly characteristics, and fluorescence behavior were all dependent on the metal ions. In addition, customized functions such as drug delivery, suitability for positron emission tomography and magnetic resonance imaging, and catalysis can be introduced through the selection of the appropriate TA/metal combination. For instance, the thickness and stability of the MPN film was controlled by metal selection and the metal feed concentration. The intermediate disassembly kinetics of the Al3+-TA capsules compared with Cu2+-TA capsules and Zr4+-A capsules was consistent with the ideal drug-delivery profile because the capsules were relatively stable at the blood pH (7.4) and gradually disassembled in lower pH regions such as lysosomes (pH of 5.0–6.0). Moreover, the Al3+-TA capsules showed extremely low cytotoxicity and better-controlled release potential. The addition of 2-monoacyltrifluoroacetone (TTA) and acetylacetone (AA) to the EuIII-TA and TbIII-TA networks enhanced the imaging characteristics of MPN capsules for use in tracking the biological distribution of the loaded drug and the carrier itself. The authors also assembled (CuII/UuIIITTA)-TA capsules and studied the function of mixed bimetallic capsules as biomedical imaging agents. Analysis using X-ray photoelectron spectroscopy confirmed that electrons were transferred from TA to metal, and also confirmed the coordination between TA and metal. Thus, the addition of a variety of metals to MPN capsules can produce a variety of functional materials and clinically useful properties.
Ping and coworkers studied the pH response of MPN capsules designed as self-assembled film of TA and Al3+ to support the anti-cancer drug DOX using PSS-doped CaCO3 as the template and demonstrated that the obtained DOX-AlIII-TA showed a good drug loading ability and pH response as well as reduced non-target cytotoxicity and improved tumor targeting.150 The formation of coordination bonds between polyphenols and Al3+ is pH dependent, and these bonds also exhibit pH-dependent decomposition. Simulation of the pH environment inside and outside cells and in lysosomes revealed that the DOX-AlIII-TA capsule stability was reduced at acid pH values. Also, the permeability of the DOX-AlIII-TA capsule allowed free DOX to diffuse through its permeation shell due to the reduced hydrogen bond interaction between DOX and poly(styrene) sulfonate (PSS), and the increased hydrophilicity of DOX at low pH. This characteristic reduces the cytotoxicity in the circulation and strengthens the targeting.
Ejima and coworkers reported a simple, rapid, and robust conformal coating method involving the one-step assembly of coordination complexes to prepare various films and particles.151 Natural polyphenol tannic acid (TA) and FeIII were used as the organic ligand and inorganic crosslinking agent, respectively. This force of X depended on the adsorption capacity of polyphenols and was guided by pH-dependent multivalent coordination bonds. The three tolylpropyl groups in TA can react with each FeIII ion to form a stable octahedral complex, enabling each TA molecule to react with several FeIII centers to form a cross-linked film. TA has a general surface binding affinity, and thus this method is applicable to a variety of substrates. This film can cover substrates with different compositions, sizes, shapes, and structures in a conformal manner, and can even be used to produce hollow capsules. This system has extensive applications in drug and gene delivery as well as catalysis and biosensors.
Rahim and colleagues used metal polyphenol interactions to form a surface-restricted film, especially between tannic acid and Fe3+.152 They demonstrated a simple, rapid, and universal strategy for the formation of surface-confined films. These molecular hybrid films may provide a wide range of multifunctional platforms, including catalysis, energy, and optoelectronics, and the experimental results demonstrated that the presence of at least one adjacent diol group (i.e., double dentate) in the aromatic ring is a prerequisite for membrane formation (phenolic ligands).
EGCG is the main component of green tea polyphenols and catechin monomer isolated from tea leaves and has documented antibacterial, anti-viral, antioxidant, anti-atherosclerosis, anti-thrombosis, anti-angiogenesis, anti-inflammatory, and anti-tumor effects.156–158 EGCG molecules, with galloyl and catechol groups, can interact non-covalently with various biological molecules such as DNA and RNA to form complexes for synergistic therapy. However, EGCG is hydrophilic, while many anti-cancer drugs are hydrophobic, and thus it is difficult to directly load anti-cancer drugs onto EGCG. Ding and colleagues assembled EGCG, (si)RNA, and protamine sulfate into a nanogel carrier for siRNA.159 The siRNA/protamine complex was first formed into a dense nanocomposite via the electrostatic interaction of the positively charged protamine sulfate, and then the anti-cancer drug EGCG was absorbed through non-covalent interaction with siRNA. To prevent drug leakage, simultaneously HA was assembled into siRNA/EGCG/protamine nanocomposites. The carrier showed good drug loading and used an HA shell and tumor homing cell-penetrating peptide as target ligands, which has minor side effects. This design effectively overcomes the drug resistance problem caused by the overexpression of connective tissue growth factor in breast cancer and provides a template for further drug development for drug-resistant triple negative breast cancer.
Polyphenols can also inhibit the epithelial to mesenchymal transition, downregulate the expression of vascular endothelial growth factor and matrix metalloproteinases (MMP) levels, and regulate other molecular pathways. Based on these properties, polyphenols can be used as anti-tumor metastasis adjuvants to compensate for the limitations of chemotherapy.160,161 In this case, EGCG is the most effective polyphenol against tumor metastasis. Chen and colleagues developed a multifunctional drug-delivery system, EINP@DOX, composed of tea-derived polyphenol EGCG, iron ion, and DOX.162 The driving force for the formation of the EGCG iron nanoparticles (EINP) is the coordination bond rather than ionic, hydrogen bond, or hydrophobic interaction. Experimental tests revealed that EINP@DOX has good stability under physiological conditions and can be rapidly decomposed in acidic environments such as nuclear endosomes. This system integrates the functions of treatment, imaging, and anti-metastasis for systematic tumor treatment. In vitro and in vivo studies demonstrated its good tumor diagnostic performance and curative effect, including effective inhibition of metastasis.
Skin melanoma is one of the most lethal and fastest growing cancers in humanins. Li and colleagues designed a self-assembled Sm3+-EGCG nanocomposite with synergistic tumor inhibition properties by chelating EGCG with Sm3+.163 In a murine model, Sm3+-pigallocatechin-3-O-gallate effectively induced the apoptosis of melanoma cells through mitochondrial dysfunction, and also effectively inhibited metastasis to other major organs, which is the ultimate cause of mortality from advanced melanoma.
Bortezomib (BTZ) is a boric acid protease inhibitor used for cancer treatment, but its efficacy may be reduced by combining it with polyphenols in food. Thus, to solve this problem, Wang and colleagues constructed a supramolecular nanosystem to achieve drug-controlled release.164 They combined BTZ with natural catechol-containing polyphenols through borate ester bonds, formed dynamic drug amphiphiles through catechol–boric acid dynamic covalent bonds, introduced iron ions into supramolecular system for biological imaging and improved the molecular stability, yielding a new drug termed BTI. This agent demonstrated pH-dependent cytotoxicity by releasing BTZ, while significantly reducing the cytotoxicity of free BTZ. Studies have shown that supramolecular nanodrugs composed of natural polyphenols, BTZ, and iron ions can induce cancer cell apoptosis and inhibit tumor growth with fewer adverse reactions. This unique supramolecular design provides many advantages for the treatment of tumors, including highly efficient and accurately controlled drug loading, excellent biodegradability and biocompatibility, easy preparation, and simple chemical synthesis. The resulting BTI supramolecular nanodrugs have high biocompatibility with blood and normal tissues and can enhance the anti-tumor therapeutic effect of BTZ, which is especially useful for the treatment of bone tumors.
Zhang and colleagues designed another targeting drug through the self-assembly of bovine serum albumin (BSA) and tea polyphenol (TP), yielding nanospheres, which were then modified with folic acid (FA) to support doxorubicin, ultimately yielding DOX@BSA-TP-FA.165 The cysteine and sulfhydryl structure of NSs and BSA allow for glutathione targeting in tumor regions, while the DOX@BSA-TP-FA folic acid on the surface of NSs can bind to the folate receptor on the surface of tumor cells, which improves the drug targeting. Overall, this system demonstrated efficient drug loading, further supporting its possible clinical application.
Chung and coworkers composed self-assembled micellar nanocomposites of green tea catechin derivatives and protein drugs and demonstrated that both drugs have therapeutic effects equivalent to the corresponding carrier-free drug.166 Oligomeric EGCG derivatives were self-assembled with the anti-cancer protein Herceptin to form stable micelles and complexed with PEG-EGCG to form a shell. The assembled complex showed better tumor selectivity and growth suppression, a longer blood half-life, and better biocompatibility than the original protein. The prevention of Herceptin clearance not only improved the drug efficacy but also effectively reduced the carrier toxicity.
Anti-cancer drugs based on cisplatin often induce resistance and strong side effects, while anti-cancer drugs delivered through MPNs are generally less prone to resistance due to their synergistic actions and produce fewer side effects due to their superior tumor targeting. Dai and colleagues assembled polyphenols and cross-linked iron ions to form MPNs and coated (PEG)-modified Pt prodrug to deliver cisplatin drugs.167 In a mouse experiment, platinum nanoparticles showed better tumor inhibition than the equivalent dose of free platinum prodrug with no obvious side effects.
Compared to nanowires, nanoparticles, and nanosheets, flower-shaped nanomaterials (nanoflowers) have demonstrated several advantages as drug-delivery vehicles, including a larger surface area for drug loading and release as well as excellent catalytic ability. Xu and Liu et al. oxidized tea polyphenols in air to obtain tea polyphenol palmitate (TPP), and then self-assembled TPP with copper sulfate to form nanoflowers.168 Thorough characterization of the product revealed that the TPP oligomer was strongly coordinated with Cu2+. Tea polyphenol hybrid nanoflowers also possessed a flower-like porous structure and hydrophobic bags provided by lipophilic side chains, which conferred high CUR loading capacity and a demonstrated a robust pH-dependent drug release response. The nanoflower also protected CUR from ultraviolet radiation and heat treatment. Through derivatization and oxidative coupling of tea polyphenol components, the assembly behavior could be easily adjusted to produce particles with substantial structural versatility.
However, controlling the self-assembly process in a multicomponent environment remains a challenge. Lin and coworkers produced a metal phenolic network by adding Fe3+ into the solution of eucalyptus leaf extracts containing multiple phenolic compounds169 and found that the number of chelating sites on the phenolic compounds may determine the selective assembly observed in complex multicomponent systems. As the reaction proceeds, the target ligand with two chelating sites can grow and form a large scalable network. Surface adsorption and simultaneous crosslinking are more competitive if there are more sites in this network compared to those with fewer chelating sites. The selective metal–phenol assembly can recognize the small change in the number of possible chelating sites between different phenolic compounds, which makes it a simple method to separate required phenolic components from complex systems. In addition, MPNs and free phenolic compounds show a better free radical scavenging performance, making them potential candidates for antioxidant applications.
2.5. Saponins
Saponins are abundant in terrestrial plants such as Ginseng, Polygala tenuifolia, Platycodon grandiflorum, Glycyrrhiza, Anemarrhena asphodeloides and Bupleurum chinense.170,171 Common saponins with demonstrated pharmaceutical activities include ginsenoside from ginseng, GA from licorice, and platycodon from Platycodon grandiflorum. These compounds are used as antipyretics, sedatives, anti-cancer agents, and for the treatment of diabetes, depression, and other disorders.172–174Saponins are composed of aglycones and glycosides, and the specific structures and functional groups of these two components confer unique bioactivities.175 More than 100 ginsenosides have been described and investigated for pharmaceutical activities. These studies have found that most saponins with anti-cancer activity have fewer sugar groups, and thus low water solubility and bioavailability, which abrogates the unique advantages of these natural drugs. Saponins are natural surfactants due to their unique amphiphilic structure. The sugar chains of saponins have strong hydrophilicity, while the core sapogenin has strong lipophilicity. Therefore, similar to other synthetic surfactants, saponins have high surface activity and self-assembly performance, and can be used as natural drug carriers.176 It is undoubtedly a good strategy to encapsulate anti-cancer NSMs through simple synthesis to improve their poor physical and chemical properties. Compared to synthetic carriers, NDDSs based on saponin compounds have unique targeting and improved biological drug resistance, features that enhance their tumor cell toxicity. Compared to synthetic carriers, saponins also demonstrate higher safety and stability, are less toxic due to reduced carrier accumulation, and show better metabolic clearance. Therefore, saponins are a good choice for the construction of liposome drug-delivery systems.177,178
GSs have unique advantages for cancer treatment, but their poor water solubility severely limits their application.186 In this case, the development of SANDDS allows for a variety of GS applications. Amphiphilic GS can be self-assembled in aqueous solution to form unique core–shell structure nanoparticles, with hydrophobic chambers formed in the middle to transport hydrophobic drugs with improved solubility and stability.187,188 The unique characteristics of GS can improve the sensitivity of tumor cells to taxanes, and the glucose analogs in this structure can combine with glucose transporter-1 (GLUT1) on the surface of cancer cells to enhance targeted drug delivery.189 GSs can assemble into spherical nanoparticles (GSN) with an average diameter of 18.24 nm through hydrogen bonding and hydrophobic interactions between GS and anti-cancer drugs in aqueous solution. A hemolytic test indicated that GSN has good biocompatibility and biosafety (Fig. 7). In vitro, free GS exhibited higher cell toxicity to normal cells than GSN, which also indicated the relative biosafety of GSN. In vivo, GSN showed significant inhibitory effects on cancer metastasis with an H22 murine artificial pulmonary metastatic model.190 Similar to many other TCM ingredients, GSs have strong pharmacological effects but low water solubility, poor stability and low bioavailability. However, it is often possible to improve their physical and chemical properties and enhance their solubility by coupling these molecules to hydrophilic compounds, such as bovine serum albumin (BSA), PEG, and chitosan derivatives.
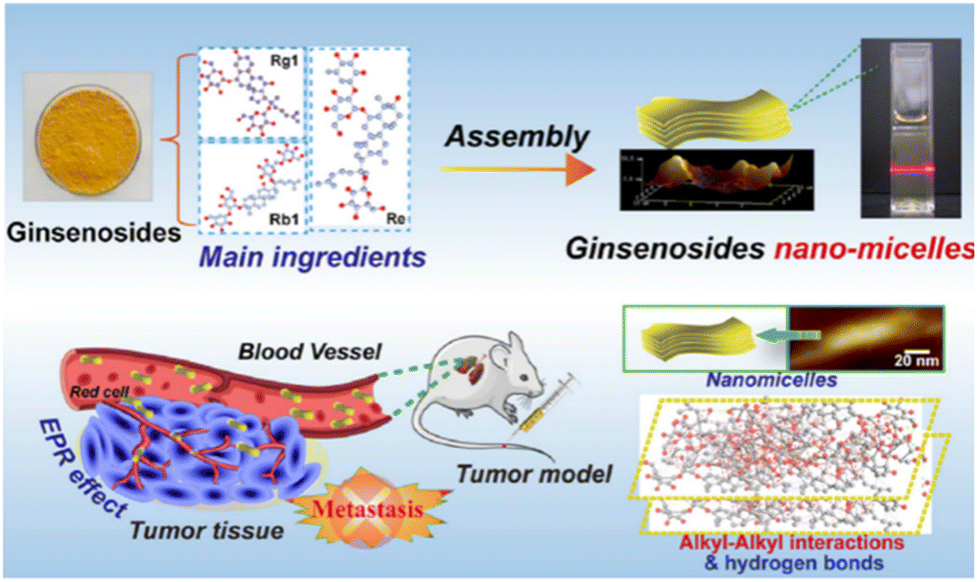 | ||
| Fig. 7 Schematic illustration of the process for the preparation of GSN and its anti-metastasis activity. Reprinted with permission from ref. 190. Copyright (2022), Elsevier. | ||
Alternatively, wrapping hydrophobic anti-cancer drugs in GS as a carrier can improve the solubility of both components and facilitate synergistic anti-tumor efficacy. Dai and colleagues proposed a universal hydrophobic drug-delivery system with GS as the carrier and encapsulated anti-cancer drugs to form nanoparticles through the unique self-assembly characteristics of GS.191 In this simple manufacturing process, hydrophobic drugs were encapsulated by the hydrophobic triterpene group of GS Rb1 NPs. The core can be used as a nanocontainer for hydrophobic drugs, and the shell can be formed by the sugar chains of GS to improve the water solubility and stability. Further, GS Rb1 itself has a variety of biological activities, including anti-cancer, antioxidant, and anti-inflammatory effects, inhibition of neointima hyperplasia, induction of angiogenesis, and vasodilation. Thus, the carrier itself can contribute to the therapeutic effect. Most importantly, it has good biosafety. In addition, the GS core–shell structure obtained by this method has the advantages of easy preparation, appropriate size, better tumor selectivity, efficient drug loading capacity, and good stability.
Ginsenoside-based drug-delivery systems also possess unique penetration characteristics. For instance, insulin (INS) is the first-line treatment for type 1 diabetes. However, traditional insulin administration requires frequent subcutaneous injection and can induce periodic hypoglycemia as well as fat atrophy, local tissue necrosis, nerve damage, and other diseases over the longer term. Transdermal drug delivery is an alternative that may mitigate these effects by supplying a more constant dose. Further, compared to subcutaneous injection, transdermal patches can improve patient compliance and avoid the gastrointestinal “first pass” metabolism following oral administration. Zou and colleagues developed a new nanopreparation consisting of insulin and GS for transdermal administration, namely, INS@GSNPs,192 which can pass through the cutin barrier in the skin. The system is self-assembled into a spherical shell structure through hydrogen bonding and both hydrophobic and electrostatic interactions. Ginsenoside is assembled into a shell to protect insulin from hydrolysis and improve its stability. The natural steroid structure of GS enhances the skin penetration given that the unique interactions between ginsenosides and membrane components such as cholesterol and phospholipid produce temporary pores. Further, experimental testing revealed sustained release characteristics and good hypoglycemic efficiency.
Dai and colleagues also193 studied the self-aggregation behavior of ginsenoside Ro (Ro) and the interaction between Ro and saikosaponin a (SSa) through mesoscopic simulations and dissipative particle dynamics, and further explained the mechanism of saponin aggregation and dissolution from different views as well as the principle of SSa solubilization by Ro. Ginsenoside Ro is a bimolecular saponin with two sugar chains connected to a triterpene aglycone. This special triblock copolymer-like structure enables the self-assembly of Ro in aqueous solution. MDS results showed that Ro can form spherical vesicles in aqueous solution with two hydrophilic regions, i.e., one in the core and the other on the surface, to obtain good carrier capacity for hydrophobic drugs. In contrast, the glucuronic acid component of Ro was located on the surface of the vesicle, where it was stabilized by strong hydrophilicity and electrostatic repulsion. At low concentrations, SSa molecules were found to dissolve in the palisade layer of Ro vesicles and the hydrophobic aglycone of the SSa molecule was inserted into the transition layer formed by the aglycone of Ro, while the SSa sugar was located on the surface and interacted with the Ro sugar through hydrogen bonds or dipole interactions. At higher SSa concentrations, Ro interacted with SSa to form mixed vesicles. Due to its stronger hydrophobicity, SSa molecules were preferentially absorbed by vesicles closer to the core. This study demonstrated for the first time that Ro can form vesicles through the closure of a flat layer membrane, a finding that broadens our understanding of the GS self-assembly behavior and may also expand the potential applications of GS in the construction of NDDSs.
Coupling saponins with hydrophilic molecules is also an effective method to improve their water solubility. Moreover, the polymers obtained through coupling often have strong self-assembly ability, which is conducive to drug delivery. Ginsenoside compound K (CK) is one of the main metabolites of PPD-type GS after intestinal enzyme metabolism. It has a wide range of pharmacological properties, such as anti-cancer, neuroprotective, anti-inflammatory, anti-angiogenesis, anti-allergen, anti-diabetes, anti-aging, and hepatoprotective activities. However, its low water solubility and poor permeability reduce its clinical efficacy.194,195 Mathiyalagan coupled CK to the backbone of the hydrophilic glycolchitosan (GC) through self-assembly to form spherical GC-CK nanoparticles in aqueous solution.196 Glycolchitosan has low toxicity and good biocompatibility, and thus can be used as a multifunctional carrier to deliver therapeutic drugs efficiently. The combination of anti-cancer drugs with hydrophilic polymers can increase the half-life of drugs in vivo and their anti-tumor effect. In this study, GC-CK was constructed to improve the drug solubility and anti-tumor efficacy, prolong blood circulation time, and protect against proteolysis. Zhang and colleagues197 synthesized deoxycholic acid-O carboxymethyl chitosan (DA-OCMC) for the encapsulation of CK into NPs by combined ultrasonic dialysis and self-assembly technology. The negatively charged OCMC was uniformly distributed on the spherical surface of the CK NPs as a hydrophilic shell layer. This method greatly improved the solubility of CK, and the anti-cancer activity was also enhanced by the chitosan structure, which endowed greatly enhanced drug release efficiency under slightly acidic conditions (such as in the TES) and reduced the systemic toxicity. In vivo and vitro experiments showed that the tumor cytotoxicity and apoptosis rate were greatly improved compared to free CK.
Ginsenoside Rg5 (Rg5) is a rare ginsenoside with unique biological characteristics, which has demonstrated curative effects against esophageal, gastric, breast, and cervical cancers.198 However, its clinical application is limited by its low water solubility, poor bioavailability, and short biological half-life. Dong and colleagues combined BSA with Rg5 by dispersing BSA in water, adjusting the pH to alkaline, and dehydrating Rg5 with ethanol to obtain a Rg5–BSA mixture, and then modified the surface BSA with folic acid to obtain FA-Rg5-BSA, a complex that improved the disadvantageous physical and chemical properties of Rg5.199 In addition, the complex demonstrated good stability over a wide range of temperatures, specific release in the weakly acidic TME, and potent induction of MCF-7 cell apoptosis. In summary, this drug-delivery system has potential to markedly improve the clinical efficacy of Rg5.
The use of water-soluble polymers is another method for improving the poor water solubility and bioavailability of GS. Mathiyalagan and coworkers conjugated hydrophilic PEG to the surface of protopanaxadiols (PPD) through a pH-sensitive ester bond to obtain PEG–PPD.200 Due to the internal hydrophobicity and external hydrophilicity of the polymer, these conjugates spontaneously formed SANDDS in an aqueous environment. In this formulation, PEG improved the water solubility, targeting, and bioavailability of GS, while reducing the cytotoxicity to non-targeted tissues. In addition, PEG protected PPD from proteolysis and greatly prolonged the drug circulation time compared to free PPD. Moreover, the PEG–PPD self-assembled nanoparticles demonstrated robust pH responsiveness, enabling their effective accumulation at pathophysiological sites.
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide. The most common cause of mortality is not the tumor itself, but complications of chronic cirrhosis, such as ascites, hepatic encephalopathy, variceal bleeding, and hepatorenal syndrome. Thus, by inhibiting these complications, the survival time of patients can be greatly prolonged. Ren and colleagues synthesized Fe@Fe3O4 nanoparticles conjugated with GS Rg3 (Rg3) (NpRg3) through self-assembly and found that they effectively inhibited HCC development and metastasis. Metal and metal oxide nanoparticles have good therapeutic functions, and iron-based nanoparticles have excellent biocompatibility and liver-targeting properties.201 Studies have shown that NpRg3 has good therapeutic effects on HCC complications. The application of NpRg3 also helps correct imbalances in the intestinal microbiota and metabolism during HCC treatment, can effectively inhibit HCC metastasis, and solve the problems of rapid gastrointestinal passage of Rg3 and its low liver absorption. Thus, the application of NpRg3 significantly prolonged the survival time of HCC model mice, inhibited the development of HCC, and eliminated metastasis of HCC to the lungs.
Liposomes are special nanocarriers that can improve the therapeutic efficacies of many different drugs.202,203 Liposomes demonstrate EPR effects and can preferentially deliver therapeutic drugs to the tumor site, thereby enhancing the therapeutic efficacy and reducing systemic toxicity. However, to date, no liposome delivery system has been fully approved by the United States Food and Drug Administration (FDA). Replacing cholesterol synthesized in liposomes with ginsenoside compounds can obviate some of the inherent dangers of liposome delivery systems, such as hyperlipidemia, pulmonary hypertension, and other diseases caused by excessive cholesterol absorption.204,205 Therefore, liposome delivery systems based on GS provide expanded possibilities for the delivery of liposome drugs for chronic diseases. Wang and colleagues conducted a detailed study of a ginsenoside delivery system, including a comparison of different GS types for encapsulation efficacy and the feasibility of preparation technologies.184 They concluded that GSs are good pharmaceutical excipients to develop new drugs. In addition to excellent biological activity, GSs can stabilize the amphiphilic structure of phospholipid bilayers due to their structural similarity with cholesterol. Ginsenosides Rg3, Rg5, and Rh2 can replace cholesterol in liposomes carriers, and these new liposome carriers have demonstrated improved drug solubility, reduced drug toxicity and side effects, improved drug stability, increased free drug concentration in the vascular system, and prolonged circulation time. In addition, this new liposome delivery system had high tumor-targeting efficiency due to the biological activity of ginsenoside itself, as well as a synergistic effect with chemotherapy drugs. Thus, this new liposome drug-delivery system may effectively overcome the side effects of traditional cholesterol-containing liposome drugs, while reducing the systemic toxicity caused by non-targeted aggregation. Also, further improvements in liposome drug-delivery systems are likely in the near future.
Wang and colleagues designed a new liposome delivery system for tumor targeted therapy that solved a series of problems by replacing cholesterol with ginsenoside Rh2 (Rh2) to obtain PTX-Rh2-lipo, in which Rh2 is not only used as a membrane stabilizer, but also as a chemotherapy adjuvant.178 Aglycones have a similar steroid structure to cholesterol, and thus can effectively replace cholesterol in the lipid delivery system. They have similar capacities to improve the physical and chemical properties of the phospholipid bilayer, which can effectively prevent allergic reactions to cholesterol and increase acceptance by vegetarians. In addition, Rh2(20 (S) – Rh2) can trigger a variety of signal transduction pathways to facilitate multiple synergistic anti-tumor mechanisms, including when loaded with the anti-cancer drug paclitaxel. However, because Rh2 is easily degraded in the gastrointestinal tract and blood, its bioavailability can be greatly enhanced by constructing a liposome delivery system. In vitro and animal model studies demonstrated that PTX-Rh2-lipo possesses good encapsulation and drug protection efficacy compared to traditional liposomes, with greatly prolonged drug circulation time and targeted release in the TME for improved treatment efficacy.
Triple-negative breast cancer continues to result in high mortality, with the proliferation of circulating tumor cells (CTCs) the main cause of death. Thus, Xia and colleagues proposed an improved liposome to assemble phospholipid, Rg3, and docetaxel (DTX),206 a first-line anti-metastasis drug approved by the FDA as an anti-cancer drug in 2000. This system effectively increased the tumor cell sensitivity to taxanes, thereby improving the anti-cancer efficacy. Through the structural analysis of Rg3, the authors found that its steroid ring structure, the side chain of C17, and the hydroxyl group of C3 site meet all the conditions required for the regulation of the liposome membrane proposed by researchers, which supports the potential of ginsenoside to replace cholesterol as a liposome membrane material. Ginsenoside Rg3 can be inserted into the liposome membrane and interact with phospholipids. The end pyran ring of the Rg3 hydrophilic group extends out of the phospholipid membrane plane and interacts with water in the solvent to reduce the mutual aggregation of water molecules. The hydroxyl group in the pyran ring of Rg3 can form a hydrogen bond with the amino group on the head of choline phosphate (PC), which increases the stability of the membrane and enables it to target and capture circulating tumor cells (CTCs). Compared to traditional liposomes and DTX micelles, the therapeutic effect of the drug was also significantly improved.
Zhu and colleagues used GS liposomes loaded with PTX for the treatment of glioma. Due to the multiple functions of G-Rg3, these liposomes not only effectively solved cholesterol-related obstacles, but also enhanced the BBB penetrability, GLUT-mediated active tumor targeting through immune enhancement, tumor cell killing activity, and TME remodeling.207 As shown in Fig. 8(A), ginsenoside Rg3 substitutes cholesterol as the membrane material in the preparation of Rg3-PTX-LPs. The glucose chains of ginsenoside Rg3 specifically bind to the GLUT of BBB and enhance the penetration of liposomes into the brain across the BBB. As shown in Fig. 8(B), Rg3-PTX-LPs target TAMs, promoting the normalization of M2 to M1 and eliciting anti-tumor immunity. Fig. 8(C) shows the release of the loaded drugs of Rg3-PTX-LPs and the induction of apoptosis of tumor cells. In vitro, the uptake of Rg3-PTX-LPs in C6 cells, BCEC and HUVEC cells was significantly higher than that of the common liposomes. Rg3-PTX-LPs could increase the penetration ability of BBB, promote tumor cell apoptosis, and extend the median survival time of glioma-bearing mice and rats from 27 days in the free PTX group to 54 days and 66.5 days, respectively. Rg3-PTX-LPs significantly improved the therapeutic effect on glioma. In addition, Rg3-based liposomes synergistically enhanced the chemotherapy efficacy of PTX, regulated the TEM, and activated the glioma immune microenvironment, significantly improving the anti-tumor effect.
 | ||
| Fig. 8 Schematic illustration of ginsenoside Rg3-based liposomes for glioma-targeting therapy. (A) Ginsenoside Rg3 substitutes cholesterol as the membrane material in preparing Rg3-PTX-LPs. (B) Rg3-PTX-LPs target to TAMs, promote the normalization of M2 to M1. (C) The apoptosis of tumor cells. (D) Rg3-PTX-LPs reduce the T-reg cells and MDSCs. Reprinted with permission from ref. 207. Copyright (2021), Elsevier. | ||
The clinical treatment of gastric cancer (GC) is hindered by the development of anti-cancer drug resistance, poor pharmacokinetics, off-target toxicity, and insufficient accumulation of current chemotherapy drugs in the tumor. Hong and colleagues synthesized liposomes with GS replacing cholesterol to carry paclitaxel. Three formulations were obtained, PTX-Rh2-lipo, PTX-Rg3-lipo, and PTX-Rg5-lipo, by replacing cholesterol with Rh2, Rg3, and Rg5, respectively,177 and their functions were compared, including blood circulation time, active recognition of GLUT1, and tumor cytotoxicity, to identify the most feasible for clinical treatment. The GS-containing liposomes have multiple functions in this new drug-delivery system, acting to stabilize the phospholipid bilayer and to promote the anti-tumor effects of PTX through synergy. Indeed, GS improved the therapeutic effect and enhanced the accumulation of liposomes at the tumor site, possibly by interacting with GLUT1. The main difference between liposomes with cholesterol or GS is the extra glycoside chain, which significantly changes the physical and chemical properties of the membrane. Compared to cholesterol, the addition of the hydrophilic glycoside chain significantly reduces the lipophilicity of ginsenoside, resulting in a significant difference in size and surface morphology (but not shape). In vivo and in vitro studies revealed that the blood circulation times of Rh2-lipo and Rg3-lipo were significantly prolonged compared to PTX-Rg5-lipo and cholesterol-based liposomes. The longer blood circulation time not only improved the stability and biological half-life of various components, but also exploited the passive targeting characteristics conferred by tumor blood vessels. Thus, compared to free Rh2 and Rg3, the ginsenoside liposomes also significantly enhanced tumor cell apoptosis, and the PTX leakage from PTX-Rh2-lipo and PTX-Rg3-lipo was only 25% after 48 h, indicating the good drug protection. Therefore, this liposome delivery system constructed with Rh2 and Rg3 can be an effective chemotherapy treatment.
Glycyrrhetinic acid and GS can both self-assemble into spherical nanoparticles to carry other drugs and in fact have many similar self-assembly applications. You and colleagues used GA nanoparticles to improve the oral bioavailability of the polyphenol BAI,212 the main bioactive components of Radix Scutellariae with documented neuroprotective, antioxidant, antibacterial, anti-viral, anti-allergic, anti-inflammatory, and anti-tumor activities. The three adjacent phenolic hydroxyl groups in BAI form intramolecular hydrogen bonds, and thus the water solubility of BAI is low, resulting in its low oral bioavailability. The main purpose of wrapping BAI with GA is to improve its water solubility. Furthermore, to improve the bioavailability of BAI, nanomicelles with hydrophobic inner cavities were prepared via the self-assembly of amphiphilic molecular GA in aqueous solution. Hydrophobic cavities of nanomicelles can form host complexes by self-assembly to encapsulate other hydrophobic compounds including BAI, thereby increasing its solubility and inhibiting its precipitation. GA can also change the characteristics of the cell membrane to enhance drug absorption. The formation of this composite micelle is likely to be driven by hydrophobic and hydrogen bonding interactions. Thus, the formulation of GA-based micelles is a promising strategy to improve the bioavailability of NSMs.
In professor Di Liuqing's team from Nanjing University of Traditional Chinese Medicine, immune exosomes loading self-assembled nanomicelles (CpG-EXO/TGM) were constructed for chemo-immunotherapy against glioblastoma (Fig. 9).213 Firstly, tanshinone II A (Tan II A), a lipid-soluble active component in Salvia miltiorrhiza, and glycyrrhizic acid (GL), an amphiphilic active component in TCM licorice, were used to construct Tan II A-GL micelles (TGM) through the self-assembly strategy. Then, TGM was loaded into the serum-derived EXO. Finally, CpG ODN 1826 was coupled to phospholipid DSPE and modified on the EXO membrane, and CpG-EXO/TGM was obtained for achieving the co-delivery of multiple components of traditional Chinese medicine and immune adjuvants. In cell experiments, this study demonstrated through confocal microscopy and flow cytometry that CpG-EXO/TGM is very easy for bEnd.3 cells and GL261 cells to uptake. After intravenous injection of CpG-EXO/TGM, the transferrin receptor on its surface can bind to free transferrin in the blood, and then cross the BBB/BBTB and enter cells through TfR-mediated endocytosis. In vivo, CpG-EXO/TGM could significantly prolong the median survival of orthotopic glioma mice and inhibit the postoperative recurrence of GBM.
 | ||
| Fig. 9 Schematic Illustration of the preparation and therapeutic mechanism of self-assembled nanomicelles (CpG-EXO/TGMa). (A) The preparationof CpG-EXO/TGM. (B) The mechanism of CpG-EXO/TGM traversing BBB. (C) The therapeutic mechanism of CpG-EXO/TGM. Reprinted with permission from ref. 213. Copyright (2023), the American Chemical Society. | ||
Paeoniflorin (Pae) is a water-soluble monoterpene glucoside with potential clinical value for autoimmune and inflammatory disease treatment. However, similar to many other saponins, its bioavailability is low due to glycoprotein efflux and its ease of hydrolysis in the gastrointestinal tract.214Glycyrrhiza uralensis Fisch is a commonly used material in TCM given that it can enhance the efficacy and reduce the toxicity of other components. Among the most important properties of GA for pharmaceutical applications are its self-assembly characteristics conferred by its amphiphilic structure. Shen and colleagues designed a spherical particle carrier by self-assembly with GA micelles, and successfully encapsulated paeoniflorin.215 This nanoparticle significantly improved the absorption of paeoniflorin in the intestinal tract, enhanced the tissue permeability, conferred its controlled release, and improved its oral bioavailability.
3.1. The mechanism of self-assembly
Herein, we describe the self-assembly of TCM-derived terpenoids, flavonoids, alkaloids, polyphenols, and saponins. These NSMs usually possess functional groups, multiple action sites, hydrophobic side chins, and rigid skeleton, which make it easier for them to form non-covalent bonds and self-assemble. The noncovalent bond interactions between these NSMs mainly include hydrogen bonds, π–π stacking interactions, van der Waals forces, hydrophobic interactions, electrostatic interactions, and coordination interactions. NSMs can directly form SANDDSs through single or multiple noncovalent interactions.2163.2. Hydrogen bonding interactions
Hydrogen bonding is a relatively weak force, which mainly exist between H atoms and electronegative atoms (e.g., O, F, and N).217 Hydrogen bonds play an important role in the self-assembly process of natural small molecules. Hydrogen bonding is the main driving force of self-assembly in natural small molecules containing hydroxyl groups, carboxyl groups, amino groups, and amide groups. Terpenoids (such as oleanolic acid, ursolic acid, and betulonic acid) possess structures containing carboxyl groups or hydroxyl groups, and thus hydrogen bond interactions often play the most important role in their assembly process.2183.3. Hydrophobic interactions
Hydrophobic interactions occur when hydrophobic groups (such as polycyclic alkyl skeleton structure containing multiple methyl molecules) aggregate close to each other to avoid water. Some terpenoids contain alkyl side chains and rigid skeletons, and thus hydrophobic interactions are the main driving force for the self-assembly of NSMs in aqueous solution.2193.4. π–π stacking interactions
The π–π stacking interaction is a weak noncovalent interaction, particularly in cases involving aromatic groups with bonded structures.220 Many natural small molecules contain alkene groups with carbon–carbon double bonds, carbon–oxygen double bonds, or aromatic ring structures, which can easily lead to π–π stacking effects. In this case, π–π stacking interactions are the driving force for the construction of SANDDSs.Rhein (Rhe) is an anthraquinone compound extracted from the dried roots and rhizomes of rhubarb. Zheng et al. reported a self-assembly of a rhein hydrogel via π–π stacking interactions.221 Bai et al. constructed a carrier-free doxorubicin-Rhe supramolecular, which was prepared by π–π stacking of Rhe and doxorubicin.222 Lei et al. reported the formation of BBR–Rhe nanoparticles via the direct self-assembly of BBR and Rhe, and the characterization of the Ber–Rhe NPs by UV–Vis spectroscopy, FT-IR spectroscopy, fluorescence spectroscopy indicated that their self-assembly is mediated by the hydrogen bonding or π–π stacking between BBR and Rhe.223 Many photosensitizers have aromatic-conjugated structures with C–C double bonds and electron-rich heterocycles, which can co-assemble with NSMs via π–π stacking interactions. Photosensitizer Ce6 consists of a hydrophobic porphyrin ring, and 10-hydroxycamptothecin (HCPT) is an indole alkaloid isolated from the Chinese tree Camptotheca acuminate, which can co-assemble into stable, discrete nanorods through the hydrophobic and π–π interaction between the porphyrin ring of Ce6 with the aromatic ring of HCPT.224
3.5. Electrostatic interactions
Electrostatic interaction is a non-covalent bond interaction based on the interaction between charges or dipoles, which is influenced by factors such as ion strength, pH level, charge ratio, and concentration. NSMs with charged groups, such as NH3+ and COO− can self-assemble to form nano-drug delivery systems via electrostatic interactions. Thus, BBR as a quaternary ammonium alkaloid can self-assemble into nanoparticles with the carboxyl groups in BAI via electrostatic interactions.93 Aristolochic acid is a component of traditional herbs such as Asarum and Aristolochia, which can be assembled with BBR to form linear supramolecules through electrostatic interactions and π–π stacking interactions.1003.6. Coordination interactions
Coordination interactions are medium-strength intermolecular forces that are weaker than covalent interactions and hydrogen bond interactions in strength.225 Catechol flavonoids or polyphenols with dihydroxyphenyl (catechol) or trihydroxyphenyl (galloyl) structures can coordinate with metal ions to self-assemble and form metal–phenolic networks.A single type of non-covalent bonding force is often weak and unstable, and thus the self-assembly between small molecules in TCMs is often caused by the combined action of multiple non-covalent bonding forces, thereby forming a stable supramolecular system. Zhang et al. constructed self-assembled Ce6-GA-FA NPs for chemotherapy-photodynamic therapy through the co-assembly of gambogic acid (GA), chlorin e6 (Ce6) and folic acid (FA). Ce6-GA-FA NPs were formed due to the π–π stacking, hydrophobic interactions and electrostatic interactions among the three substances.226 Yang et al. constructed composite nanoparticles using self-assembling abietic acid (AA) as a carrier to load curcumin. The FITR, UV, and molecular dynamics simulation revealed that hydrogen bonds and hydrophobic interactions are the assembly mechanism of AA loaded curcumin.227
3.7. The morphology of the self-assembly
NSMs can form a micro-structure feature with various morphologies ranging from nanometers to micrometers through self-assembly in different solvents. Natural small molecules can form fibrous, vesicular, spherical, nanosheets through self-assembly. They may also exhibit phase change behavior by forming nanoparticles, emulsion gels, low-molecular-weight gels, etc., which are influenced by pH, temperature, concentration, and the type of solvent used. Some NSMs can self-assemble into spherical nanoparticles through nanoprecipitation, such as oleanolic acid and ursolic acid, while betulinic acid, glycyrrhetinic acid, and betulic acid self-assembled into nanofibers through the same method.228 Glycoside baicalin (BA) and wogonoside (WOG) are the main active components in Scutellariae radix, which have a similar molecular skeleton. The two flavonoid glycosides could form different self-assemblies with BBR in aqueous solution, and BA and BBR formed NPs with diameters of 100 nm, whereas WOG and BBR assembled into nanofibers with diameters in the range of 50–100 nm. BA and WOG had similar structures, but their spatial conformation varied, causing them to generate two different self-assembling modes.88 In addition to the influence of spatial conception on self-assembly behavior, solvents also have a significant impact on the assembly of supramolecules, such as β-sitosterol can self-assemble into sheet-like, strip-shaped, and fibrous forms in methanol, DMSO, and cyclohexane, respectively.2294. Summary and future prospects
The active ingredients extracted from Chinese medicinal herbs for TCM are an important resource for new drug research and development. Due to advances in separation technology, numerous NSMs have been isolated and characterized in detail. However, although many of them have broad spectrum and potent pharmacological activities, they exhibit limited in vivo efficacy due to their poor water solubility, instability, and poor pharmacokinetics, and are often eliminated in new drug screens. The bioactive NSMs of TCM often have multiple hydroxyl or carboxyl structures amenable to hydrogen bond formation and self-assembly with various other molecules. Moreover, NSMs have a unique hydrophobic skeleton that also contributes to their self-assembly. In addition, small molecules involved in assembly often contain carbon-based double bonds (CO) and olefin double bonds (CC), even if no aromatic ring produces π–π stacking, which are conducive to π–π interaction and play an important role in molecular assembly. Given the structural heterogeneity of these bioactive molecules, the pathways of self-assembly are diverse. Terpenoids have strong π–π stacking ability and can be assembled with photosensitizers. Flavonoids usually have poor physical and chemical properties for free administration but can be connected to hydrophilic compounds and assembled into a core–shell structure via non-covalent bonds. In these core–shell structures, the inner hydrophobic core can store hydrophobic drugs, such as anti-cancer agents, while the outer hydrophilic shell can protect clinical drugs from destruction. Alkaloids are also common active ingredients of TCM preparations used to construct self-assembled drugs with other natural active ingredients or amino acids to achieve combined treatment or improve bioavailability. The metal phenolic network constructed by polyphenol compounds and metals is another promising system for new drug research and clinical development. By adjusting the types of polyphenols or metal ions, the thickness of MPNs and other physical properties can be controlled and these vehicles can be constructed to include multiple drugs with distinct functions, including anti-cancer agents, photosensitizers, and biological imaging agents. Some saponins have hydrophobic steroidal skeletons and hydrophilic glycosyl side chains that facilitate self-assembly into nanomicelles. In addition, GSs have a steroid structure and can replace cholesterol in the synthesis of liposomes for safer drugs to treat chronic diseases. The construction of appropriate nanodrug delivery systems based on the self-assembly of multiple components from TCM can improve the anti-tumor efficacy, while reducing side effects, thereby expanding the clinical applications of TMC components in modern medicine.
Although NSMs and SANDDSs hold great promise for the treatment of cancer, many theoretical and technical obstacles remain to be overcome. The first is the nature of natural small molecules themselves, such as the low content of natural chemical components in nature, their difficult extraction and separation, poor water solubility leading to poor pharmacokinetics, low bioavailability, and poor stability. For example, many natural components show potential anti-cancer activity in in vitro models, but have poor effect in in vivo models and low success rate in clinical transformation. Accordingly, SANDDSs open a new path for the application of traditional Chinese medicine small molecules, improving their stability, enhancing oral bioavailability, controlled release, and targeted delivery and reducing their toxic side effects. However, there are still many theories to be further studied. The self-assembly of natural small molecules is still in the stage of random and accidental discovery. However, previous self-assembly systems often require a complex preparation process and high cost. In addition, there is a small number of these natural small molecules and their structure type is single, which cannot provide effective clues for the summary of their trends. In addition, a more important point is that the self-assembly between natural small molecules is mostly driven by non-covalent forces. However, due to the weak and dynamic nature of non-covalent forces, it is difficult to determine the relationship between the introduced functional groups and the molecular assembly mode. Therefore, more in-depth studies are needed to clarify the assembly mechanism of molecules in different phase states. Making full use of their self-assembly capability makes it possible to develop new active ingredients of traditional Chinese medicine, promote the clinical transformation of nano-preparations, and improve the clinical application of NSMs.
In addition, we have found that the traditional ‘single chemical’ perspective has limitations for understanding the efficacy of TCM. Rather, the forms and phase states of all the active ingredients as well as the myriad interactions among these molecules influence the efficacy of TCM. Research on self-assembly applications for the active ingredients of TCM may connect TCM theory with modern science and technology and create important avenues for new drug research and development.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Shandong Natural Science Foundation (ZR2022QB075), the Project funded by China Postdoctoral Science Foundation (2022M711983).References
- R. Zheng, S. Zhang, H. Zeng, S. Wang, K. Sun, R. Chen, L. Li, W. Wei and J. He, Cancer incidence and mortality in China, J. Natl. Cancer Cent., 2022, 2, 1–9 CrossRef.
- J. Ferlay, M. Colombet, I. Soerjomataram, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Cancer statistics for the year 2020: An overview, Int. J. Cancer, 2021, 149, 778–789 CrossRef CAS PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- B. Li, H. Shao, L. Gao, H. Li, H. Sheng and L. Zhu, Nano-drug co-delivery system of natural active ingredients and chemotherapy drugs for cancer treatment: a review, Drug Delivery, 2022, 29, 2130–2161 CrossRef CAS PubMed.
- A. Mandal, R. Bisht, I. D. Rupenthal and A. K. Mitra, Polymeric micelles for ocular drug delivery: From structural frameworks to recent preclinical studies, J. Controlled Release, 2017, 248, 96–116 CrossRef CAS PubMed.
- J. Zhang, K. Hu, L. Di, P. Wang, Z. Liu, J. Zhang, P. Yue, W. Song, J. Zhang and T. Chen, Traditional herbal medicine and nanomedicine: Converging disciplines to improve therapeutic efficacy and human health, Adv. Drug Delivery Rev., 2021, 178, 113964–113995 CrossRef CAS PubMed.
- Y. Xie, C. Ma, X. Yang, J. Wang, G. Long and J. Zhou, Phytonanomaterials as therapeutic agents and drug delivery carriers, Adv. Drug Delivery Rev., 2021, 176, 113868–113879 CrossRef CAS PubMed.
- M. Souri, M. Soltani, F. M. Kashkooli and M. K. Shahvandi, Engineered strategies to enhance tumor penetration of drug-loaded nanoparticles, J. Controlled Release, 2022, 341, 227–246 CrossRef CAS PubMed.
- M. Souri, M. Soltani, F. M. Kashkooli, M. K. Shahvandi, M. Chiani, F. S. Shariati, M. R. Mehrabi and L. L. Munn, Towards principled design of cancer nanomedicine to accelerate clinical translation, Mater. Today Bio, 2022, 13, 100208–100237 CrossRef CAS PubMed.
- X. Qian, X. Xu, Y. Wu, J. Wang, J. Li, S. Chen, J. Wen, Y. Li and Z. Zhang, Strategies of engineering nanomedicines for tumor retention, J. Controlled Release, 2022, 346, 193–211 CrossRef CAS PubMed.
- H. Mei, S. Cai, D. Huang, H. Gao, J. Cao and B. He, Carrier-free nanodrugs with efficient drug delivery and release for cancer therapy:From intrinsic physicochemical properties to external modification, Bioact. Mater., 2021, 8, 220–240 Search PubMed.
- K. S. Man, S. Yang, I. C. Sun and K. Kim, Tumor-activated carrier-free prodrug nanoparticles for targeted cancer Immunotherapy: Preclinical evidence for safe and effective drug delivery, Adv. Drug Delivery Rev., 2022, 183, 114177–114198 CrossRef PubMed.
- Y. T. Zhong, Y. Cen, L. Xu, S. Y. Li and H. Cheng, Recent Progress in Carrier–Free Nanomedicine for Tumor Phototherapy, Adv. Healthcare Mater., 2023, 12, 2202307–2202338 CrossRef CAS PubMed.
- Y. Zhou, Y. Zhang, C. Jiang, Y. Chen, F. Tong, X. Yang, Y. Wang, X. Xia and H. Gao, Rosmarinic acid-crosslinked supramolecular nanoassembly with self-regulated photodynamic and anti-metastasis properties for synergistic photoimmunotherapy, Small, 2023, 19, 2300594–2300608 CrossRef CAS PubMed.
- L. Qiao, H. Yang, S. Gao, L. Li, X. Fu and Q. Wei, Research progress on self-assembled nanodrug delivery systems, J. Mater. Chem. B, 2022, 10, 1908–1922 RSC.
- W. Jia, R. Liu, Y. Wang, C. Hu, W. Yu, Y. Zhou, L. Wang, M. Zhang, H. Gao and X. Gao, Acta Pharm. Sin. B, 2022, 12, 3354–3366 CrossRef CAS PubMed.
- Z. Wang, W. Li, J. Lu, Z. Yuan, W. Pi, Y. Zhang, H. Lei, W. Jing and P. Wang, Revealing the active ingredients of the traditional Chinese medicine decoction by the supramolecular strategies and multitechnologies, J. Ethnopharmacol., 2023, 300, 115704–115717 CrossRef CAS PubMed.
- X. Feng, Q. Xie, C. Yang, L. Kong and Z. Zhang, Carrier-free nanoparticles based on self-assembly of active ingredients from Chinese medicin, Acta Pharm. Sin. B, 2021, 56, 3203–3211 Search PubMed.
- X. Lin, X. Huang, X. Tian, Z. Yuan, J. Lu, X. Nie, P. Wang, H. Lei and P. Wang, Natural Small-Molecule-Based Carrier-Free Self-Assembly Library Originated from Traditional Chinese Herbal Medicine, ACS Omega, 2022, 7, 43510–43521 CrossRef CAS PubMed.
- L. H. Liu and X. Z. Zhang, Carrier-free nanomedicines for cancer treatment, Prog. Mater. Sci., 2022, 125, 100919–110975 CrossRef CAS.
- S. Karaosmanoglu, M. Zhou, B. Shi, X. Zhang and X. Chen, Carrier-free nanodrugs for safe and effective cancer treatment, J. Controlled Release, 2021, 329, 805–832 CrossRef CAS PubMed.
- S. Jiang, Y. Fu, X. Zhang and J. Du, Research Progress of Carrier-Free Antitumor Nanoparticles Based on Phytochemicals, Front. Bioeng. Biotechnol., 2021, 9, 799806–199813 CrossRef PubMed.
- X. Ying, M. Chao, Y. Xin, D. Jwc, A. Gl and D. Jza, Phytonanomaterials as therapeutic agents and drug delivery carriers, Adv. Drug Delivery Rev., 2021, 176, 113868–113895 CrossRef PubMed.
- J. Wang, W. Qiao, X. Li, H. Zhao, H. Zhang, A. Dong and X. Yang, A directed co-assembly of herbal small molecules into carrier-free nanodrugs for enhanced synergistic antitumor efficacy, J. Mater. Chem. B, 2021, 9, 1040–1048 RSC.
- Y. Hou, L. Zou, Q. Li, M. Chen, H. Ruan, Z. Sun, X. Xu, J. Yang and G. Ma, Supramolecular assemblies based on natural small molecules: Union would be effective, Mater. Today Bio, 2022, 15, 100327–100344 CrossRef CAS PubMed.
- H. Qiao, L. Di, Q. Ping and L. Hu, Structural Chinese medicine: new research field on pharmacodynamic substance basis of traditional Chinese medicine, Zhongguo Zhongyao Zazhi, 2021, 46, 2443–2448 Search PubMed.
- J. Cheng and X. Yang, Self-assembly performance of triterpene natural small molecules and their application in synergistic antitumor chemotherapy, Acta Pharm. Sin., 2021, 56, 2102–2111 Search PubMed.
- J. Zhu, Z. Zhang, R. Wang, K. Zhong, K. Zhang, N. Zhang, W. Liu, F. Feng and W. Qu, Review of Natural Phytochemical-Based Self-Assembled Nanostructures for Applications in Medicine, ACS Appl. Nano Mater., 2022, 5, 3146–3169 CrossRef CAS.
- J. Cheng, X. Li, S. Wang, Y. Han, H. Zhao and X. Yang, Carrier-free triterpene prodrugs with glutathione response and biosafety for synergistically enhanced photochemotherapy, ACS Appl. Mater. Interfaces, 2020, 13, 245–256 CrossRef PubMed.
- A. Kaps, P. Gwiazdoń and E. Chodurek, Nanoformulations for delivery of pentacyclic triterpenoids in anticancer therapies, Molecules, 2021, 26, 1764 CrossRef CAS PubMed.
- L. Žiberna, D. Šamec, A. Mocan, S. F. Nabavi, A. Bishayee, A. A. Farooqi, A. Sureda and S. M. Nabavi, Oleanolic acid alters multiple cells signaling pathways: implication in cancer prevention and therapy, Int. J. Mol. Sci., 2017, 18, 643–658 CrossRef PubMed.
- Y. Bao, S. Zhang, Z. Chen, A. T. Chen, J. Ma, G. Deng, W. Xu, J. Zhou, Z. Yu, G. Yao and J. Chen, Synergistic Chemotherapy for Breast Cancer and Breast Cancer Brain Metastases via Paclitaxel-Loaded Oleanolic Acid Nanoparticles, Mol. Pharm., 2020, 17, 1343–1351 CrossRef CAS PubMed.
- B. G. Bag and S. S. Dash, First self-assembly study of betulinic acid, a renewable nano-sized, 6-6-6-6-5 pentacyclic monohydroxy triterpenic acid, Nanoscale, 2011, 3, 4564–4566 RSC.
- S. Liu, H. Liu, L. Zhang, C. Ma and A. M. Abd El-Aty, Edible pentacyclic triterpenes: A review of their sources, bioactivities, bioavailability, self-assembly behavior, and emerging applications as functional delivery vehicles, Crit. Rev. Food Sci., 2022, 1–17 Search PubMed.
- B. G. Bag and R. Majumdar, Self–assembly of renewable nano–sized triterpenoids, Chem. Rec., 2017, 17, 841–873 CrossRef CAS PubMed.
- J. Wang, H. Zhao, W. Qiao, J. Cheng and X. Yang, Nanomedicine-Cum-Carrier by Co-Assembly of Natural Small Products for Synergistic Enhanced Antitumor with Tissues Protective Actions, ACS Appl. Mater. Interfaces, 2020, 12, 42537–42550 CrossRef CAS PubMed.
- D. Pe'er, S. Ogawa, O. Elhanani, L. Keren, T. G. Oliver and D. Wedge, Tumor heterogeneity, Cancer Cell, 2021, 39, 1015–1017 CrossRef PubMed.
- Y. Duan, Y. Yu, P. Liu, Y. Gao, X. Dai, L. Zhang, L. Chen and Y. Chen, Reticular Chemistry–Enabled Sonodynamic Activity of Covalent Organic Frameworks for Nanodynamic Cancer Therapy, Angew. Chem., Int. Ed., 2023, 135, 202302146–202302157 CrossRef.
- A. Yz, B. Jy, A. Zl, B. Hc and G. Yu, Recent progress in sono-photodynamic cancer therapy: From developed new sensitizers to nanotechnology-based efficacy enhancing strategies, Acta Pharm. Sin. B, 2020, 11, 2197–2219 Search PubMed.
- Y. Zheng, Z. Li, Y. Yang, H. Shi, H. Chen and Y. Gao, A nanosensitizer self-assembled from oleanolic acid and chlorin e6 for synergistic chemo/sono-photodynamic cancer therapy, Phytomedicine, 2021, 93, 153788–153801 CrossRef CAS PubMed.
- S. Amiri, S. Dastghaib, M. Ahmadi, P. Mehrbod and S. Ghavami, Betulin and its derivatives as novel compounds with different pharmacological effects, Biotechnol. Adv., 2020, 38, 107409–107447 CrossRef CAS PubMed.
- U. Bildziukevich, Z. Zdemir and Z. Wimmer, Recent Achievements in Medicinal and Supramolecular Chemistry of Betulinic Acid and Its Derivatives, Molecules, 2019, 24, 3546–3565 CrossRef CAS PubMed.
- Y. Li, Y. Wang, L. Gao, Y. Tan, J. Cai, Z. Ye, A. T. Chen, Y. Xu, L. Zhao and S. Tong, Betulinic acid self-assembled nanoparticles for effective treatment of glioblastoma, J. Nanobiotechnol., 2022, 20, 39–51 CrossRef CAS PubMed.
- J. Wang, W. Qiao, H. Zhao and X. Yang, Paclitaxel and betulonic acid synergistically enhance antitumor efficacy by forming co-assembled nanoparticles, Biochem. Pharmacol., 2020, 182, 114232–114241 CrossRef CAS PubMed.
- J. Cheng, H. Zhao, J. Wang, Y. Han and X. Yang, Bioactive natural small molecule-tuned coassembly of photosensitive drugs for highly efficient synergistic and enhanced type I photochemotherapy, ACS Appl. Mater. Interfaces, 2020, 12, 43488–43500 CrossRef CAS PubMed.
- J. Lan, L. Liu, R. Zeng, Y. Qin, J. Hou, S. Xie, S. Yue, J. Yang, R. J. Ho and Y. Ding, Tumor-specific carrier-free nanodrugs with GSH depletion and enhanced ROS generation for endogenous synergistic anti-tumor by a chemotherapy-photodynamic therapy, Chem. Eng. J., 2021, 407, 127212–127225 CrossRef CAS.
- L. Wang, Q. Yin, C. Liu, Y. Tang and J. Zhuang, Nanoformulations of Ursolic Acid: A Modern Natural Anticancer Molecule, Front. Pharmacol., 2021, 12, 706121–706144 CrossRef CAS PubMed.
- A. Schito, D. Caviglia, G. Piatti, A. Zorzoli, D. Marimpietri, G. Zuccari, G. Schito and S. Alfei, Efficacy of Ursolic Acid-Enriched Water-Soluble and Not Cytotoxic Nanoparticles against Enterococci, Pharmaceutics, 2021, 13, 1976–1996 CrossRef CAS PubMed.
- P. Nahak, G. Karmakar, P. Chettri, B. Roy, P. Guha, S. E. Besra, A. Soren, A. G. Bykov, A. V. Akentiev and B. A. Noskov, Influence of lipid core material on physicochemical characteristics of an ursolic acid-loaded nanostructured lipid carrier: An attempt to enhance anticancer activity, Langmuir, 2016, 32, 9816–9825 CrossRef CAS PubMed.
- K. Ou, X. Xu, S. Guan, R. Zhang, X. Zhang, Y. Kang and J. Wu, Nanodrug carrier based on poly (ursolic acid) with self–anticancer activity against colorectal cancer, Adv. Funct. Mater., 2020, 30, 1907857–1907867 CrossRef CAS.
- J. Cheng, S. Wang, H. Zhao, Y. Liu and X. Yang, Exploring the self-assembly mechanism and effective synergistic antitumor chemophototherapy of a biodegradable and glutathione responsive ursolic acid prodrug mediated photosensitive nanodrug, Biomater. Sci., 2021, 9, 3762–3775 RSC.
- Y. Wang, Z. Luo, D. Zhou, X. Wang, J. Chen, S. Gong and Z. Yu, Nano-assembly of ursolic acid with platinum prodrug overcomes multiple deactivation pathways in platinum-resistant ovarian cancer, Biomater. Sci., 2021, 9, 4110–4119 RSC.
- P. Namdeo, B. Gidwani, S. Tiwari, V. Jain, V. Joshi, S. S. Shukla, R. K. Pandey and A. Vyas, Therapeutic potential and novel formulations of ursolic acid and its derivatives: an updated review, J. Sci. Food Agric., 2023, 103, 4275–4292 CrossRef CAS PubMed.
- V. Khwaza, O. O. Oyedeji and B. A. Aderibigbe, Ursolic acid-based derivatives as potential anti-cancer agents: An update, Int. J. Mol. Sci., 2020, 21, 5920–5946 CrossRef CAS PubMed.
- P. Hassanzadeh and E. Arbabi, Presenting a bioactive nanotherapeutic agent for colon cancer treatment, Eur. J. Pharmacol., 2022, 927, 175084–175093 CrossRef CAS PubMed.
- Z.-w. Zeng, D. Chen, L. Chen, B. He and Y. Li, A comprehensive overview of Artemisinin and its derivatives as anticancer agents, Eur. J. Med. Chem., 2022, 247, 115000–115019 CrossRef PubMed.
- J. Fan, X. Liu, Q. Wang, H. Wang, H. Liu, D. Han and J. Ren, A Metal-Organic Framework-Based Redox Homeostasis Disruptor Selectively Potentiate the Cytotoxicity of Dihydroartemisinin for Cancer Therapy, Nano Res., 2023, 16, 7489–7495 CrossRef CAS.
- K. H. Wong, D. Yang, S. Chen, C. He and M. Chen, Development of nanoscale drug delivery systems of dihydroartemisinin for cancer therapy: A review, Asian J. Pharm. Sci., 2022, 17, 475–490 CrossRef PubMed.
- Y. Li, W. Zhang, N. Shi, W. Li, J. Bi, X. Feng, N. Shi, W. Zhu and Z. Xie, Self-assembly and self-delivery of the pure nanodrug dihydroartemisinin for tumor therapy and mechanism analysis, Biomater. Sci., 2023, 11, 2478–2485 RSC.
- G. Li, B. Sun, Y. Li, C. Luo, Z. He and J. Sun, Small–molecule prodrug nanoassemblies: an emerging nanoplatform for anticancer drug delivery, Small, 2021, 17, 2101460–2101488 CrossRef CAS PubMed.
- R. Luo, Z. Zhang, L. Han, Z. Xue, K. Zhang, F. Liu, F. Feng, J. Xue, W. Liu and W. Qu, An albumin-binding dimeric prodrug nanoparticle with long blood circulation and light-triggered drug release for chemo-photodynamic combination therapy against hypoxia-induced metastasis of lung cancer, Biomater. Sci., 2021, 9, 3718–3736 RSC.
- K. Wen, X. Yang, J. Yang, Y. Yao, K. S. Nandakumar, M. L. Salem and K. Cheng, Recent Research on Flavonoids and their Biomedical Applications, Curr. Med. Chem., 2021, 28, 1042–1066 CAS.
- Y. Wei, J. Guo, X. Zheng, J. Wu, Y. Zhou, Y. Yu, L. Zhang and L. Zhao, Preparation, pharmacokinetics and biodistribution of baicalin-loaded liposomes, Int. J. Nanomed., 2014, 9, 3623–3630 CAS.
- K. Hao, X. Liu, G. Wang and X. Zhao, Pharmacokinetics, tissue distribution and excretion of gambogic acid in rats, Eur. J. Drug Metab. Pharmacokinet., 2007, 32, 63–68 CrossRef CAS PubMed.
- Q. Wu, S. Deng, L. Li, L. Sun, X. Yang, X. Liu, L. Liu, Z. Qian, Y. Wei and C. Gong, Biodegradable polymeric micelle-encapsulated quercetin suppresses tumor growth and metastasis in both transgenic zebrafish and mouse models, Nanoscale, 2013, 5, 12480–12493 RSC.
- A. Rauf, M. Imran, S. A. Gilani, Z. Mehmood and M. Mubarak, Anticancer Potential of Quercetin: A Comprehensive Review, Phytother. Res., 2018, 32, 2109–2130 CrossRef CAS PubMed.
- G. E.-S. Batiha, A. M. Beshbishy, M. Ikram, Z. S. Mulla, M. E. A. El-Hack, A. E. Taha, A. M. Algammal and Y. H. A. Elewa, The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: quercetin, Foods, 2020, 9, 374–389 CrossRef CAS PubMed.
- K. Hu, L. Miao, T. J. Goodwin, J. Li and Q. Liu, Quercetin Remodels the Tumor Microenvironment To Improve the Permeation, Retention, and Antitumor Effects of Nanoparticles, ACS Nano, 2018, 11, 4916–4925 CrossRef PubMed.
- X. Pang, Z. Lu, H. Du, X. Yang and G. Zhai, Hyaluronic acid-quercetin conjugate micelles: Synthesis, characterization, in vitro and in vivo evaluation, Colloids Surf., B, 2014, 123, 778–786 CrossRef CAS PubMed.
- L. Chen, Y. Chen, C. Su, C. Hong, H. Ho and M. Sheu, Development and characterization of self-assembling lecithin-based mixed polymeric micelles containing quercetin in cancer treatment and an in vivo pharmacokinetic study, Int. J. Nanomed., 2016, 11, 1557–1566 CAS.
- I. Noh, D. Lee, H. Kim, C. U. Jeong, Y. Lee, J. O. Ahn, H. Hyun, J. H. Park and Y. C. Kim, Enhanced photodynamic cancer treatment by mitochondria–targeting and brominated near–infrared fluorophores, Adv. Sci., 2017, 5, 1700481–1700491 CrossRef PubMed.
- L. Xing, J. Lyu, Y. Yang, P. Cui, L. Gu, J. Qiao, Y. He, T. Zhang, M. Sun and J. Lu, pH-Responsive de-PEGylated nanoparticles based on triphenylphosphine–quercetin self-assemblies for mitochondria-targeted cancer therapy, Chem. Commun., 2017, 53, 8790–8793 RSC.
- K. Liu, H.-L. Zhang, L.-H. Pan, Q.-M. Li, J.-P. Luo and X.-Q. Zha, The nanomicelles consisting of lotus root amylopectin and quinoa protein: Construction and encapsulation for quercetin, Food Chem., 2022, 387, 132924 CrossRef CAS PubMed.
- X. Song, L. Tan, M. Wang, C. Ren, C. Guo, B. Yang, Y. Ren, Z. Cao, Y. Li and J. Pei, Myricetin: A review of the most recent research, Biomed. Pharmacother., 2021, 134, 111017–111029 CrossRef CAS PubMed.
- G. Gupta, M. A. Siddiqui, M. M. Khan, M. Ajmal, R. Ahsan, M. A. Rahaman, M. A. Ahmad, M. Arshad and M. Khushtar, Current pharmacological trends on myricetin, Drug Res., 2020, 70, 448–454 CrossRef CAS PubMed.
- H. Guo, Y. Chen, Y. Tang and J. Qian, Method for enhancing bioavailability of myricetin based on self-assembly of casein-myricetin nanomicelles, IET Nanobiotechnol., 2020, 14, 239–244 CrossRef PubMed.
- T. Wang, Q. Fan, J. Hong, Z. Chen, X. Zhou, J. Zhang, Y. Dai, H. Jiang, Z. Gu and Y. Cheng, Therapeutic nanoparticles from grape seed for modulating oxidative stress, Small, 2021, 17, 2102485 CrossRef CAS PubMed.
- X. Ma, H. Gong, K. Ogino, X. Yan and R. Xing, Coordination-assembled myricetin nanoarchitectonics for sustainably scavenging free radicals, Beilstein J. Nanotechnol., 2022, 13, 284–291 CrossRef CAS PubMed.
- J.-y. Yang, M. Li, C.-l. Zhang and D. Liu, Pharmacological properties of baicalin on liver diseases: A narrative review, Pharmacol. Rep., 2021, 73, 1230–1239 CrossRef PubMed.
- L. Li, H. Cui, T. Li, J. Qi, H. Chen, F. Gao, X. Tian, Y. Mu, R. He and S. Lv, Synergistic effect of berberine-based Chinese medicine assembled nanostructures on diarrhea-predominant irritable bowel syndrome in vivo, Front. Pharmacol., 2020, 11, 1210–1220 CrossRef PubMed.
- X. Jia, Z. Yuan, Y. Yang, X. Huang, N. Han, X. Liu, X. Lin, T. Ma, B. Xu and P. Wang, Multi-functional self-assembly nanoparticles originating from small molecule natural product for oral insulin delivery through modulating tight junctions, J. Nanobiotechnol., 2022, 20, 116–132 CrossRef CAS PubMed.
- Z. Wang, J. Lu, Z. Yuan, W. Pi, X. Huang, X. Lin, Y. Zhang, H. Lei and P. Wang, Natural Carrier–Free Binary Small Molecule Self–Assembled Hydrogel Synergize Antibacterial Effects and Promote Wound Healing by Inhibiting Virulence Factors and Alleviating the Inflammatory Response, Small, 2023, 19, 2205528–2205544 CrossRef CAS PubMed.
- S. Zhang, Y. Shao, Q. Guo, F. Gong, C. Guo and Z. Liu, Preparation and in vitro evaluation of baicalin nanoparticles loaded with doxorubicin, Drugs Clinic., 2022, 37, 1482–1486 Search PubMed.
- Y. Shao, Q. Guo, S. Zhang, Y. Yang, F. Gong and Z. Liu, Preparation and in vitro evaluation of self-assembled micellar drug delivery system of baicalin modified with lactobionic acid, Chin. Tradit. Herb. Drugs, 2021, 20, 6216–6225 Search PubMed.
- Z. Z. Wang, Y. Jia, G. Wang, H. He, L. Cao, Y. Shi, M. Miao and X. M. Li, Dynamic covalent hydrogel of natural product baicalin with antibacterial activities, RSC Adv., 2022, 12, 8737–8742 RSC.
- X. Huang, X. Liu, X. Lin, Z. Yuan, Y. Zhang, Z. Wang, W. Pi, H. Zhao, H. Lei and P. Wang, Thermodynamics driving phytochemical self-assembly morphological change and efficacy enhancement originated from single and co-decoction of traditional chinese medicine, J. Nanobiotechnol., 2022, 20, 527–541 CrossRef CAS PubMed.
- S. Bhambhani, K. R. Kondhare and A. P. Giri, Diversity in Chemical Structures and Biological Properties of Plant Alkaloids, Molecules, 2021, 26, 3374–3402 CrossRef CAS PubMed.
- N. Puthdee, W. Seubwai, K. Vaeteewoottacharn, T. Boonmars and S. Wongkham, Berberine Induces Cell Cycle Arrest in Cholangiocarcinoma Cell Lines via Inhibition of NF-κB and STAT3 Pathways, Biol. Pharm. Bull., 2017, 40, 751–757 CrossRef CAS PubMed.
- H. Guo, C. Feng, W. Zhang, Z. Luo, H. Zhang, T. Zhang, C. Ma, Y. Zhan, R. Li and S. Wu, Liver-target nanotechnology facilitates berberine to ameliorate cardio-metabolic diseases, Nat. Commun., 2019, 10, 1981–1996 CrossRef PubMed.
- P. Wang, W. Guo, G. Huang, J. Zhen and A. Xu, Berberine-Based Heterogeneous Linear Supramolecules Neutralized the Acute Nephrotoxicity of Aristolochic Acid by the Self-Assembly Strategy, ACS Appl. Mater. Interfaces, 2021, 13, 32729–32742 CrossRef CAS PubMed.
- H. Majidzadeh, M. Araj-Khodaei, M. Ghaffari, M. Torbati and M. R. Hamblin, Nano-based delivery systems for berberine: A modern anti-cancer herbal medicine, Colloids Surf., B, 2020, 194, 111188 CrossRef CAS PubMed.
- X. Huang, P. Wang, T. Li, X. Tian, W. Guo, B. Xu, G. Huang, D. Cai, F. Zhou and H. Zhang, Self-assemblies based on traditional medicine berberine and cinnamic acid for adhesion-induced inhibition multidrug-resistant Staphylococcus aureus, ACS Appl. Mater. Interfaces, 2019, 12, 227–237 CrossRef PubMed.
- T. Li, P. Wang, W. Guo, X. Huang and H. Lei, Natural Berberine-Based Chinese Herb Medicine Assembled Nanostructures with Modified Antibacterial Application, ACS Nano, 2019, 13, 6770–6781 CrossRef CAS PubMed.
- M. Chen, P. Wang, T. Li, L. Li, J. Li, H. Bai, H. Lei and Q. Ma, Comprehensive analysis of Huanglian Jiedu decoction: Revealing the presence of a self-assembled phytochemical complex in its naturally-occurring precipitate, J. Pharm. Biomed. Anal., 2021, 195, 113820–113830 CrossRef CAS PubMed.
- C. J. Zhang, J. Wang, J. Zhang, M. Lee and B. Liu, Mechanism–Guided Design and Synthesis of a Mitochondria–Targeting Artemisinin Analogue with Enhanced Anticancer Activity, Angew. Chem., Int. Ed. Engl., 2016, 128, 13974–13978 CrossRef.
- N. L. Andreazza, C. Vevert-Bizet, G. Bourg-Heckly, F. Sureau, M. J. Salvador and S. Bonneau, Berberine as a photosensitizing agent for antitumoral photodynamic therapy: Insights into its association to low density lipoproteins, Int. J. Pharm., 2016, 510, 240–249 CrossRef PubMed.
- R. An, Z. Gu, H. Sun, Y. Hu, R. Yan, D. Ye and H. Liu, Self-assembly of Fluorescent Dehydroberberine Enhances Mitochondria-Dependent Antitumor Efficacy, Chem. – Eur. J., 2018, 24, 9812–9819 CrossRef CAS PubMed.
- B. Schaneberg and I. Khan, Analysis of products suspected of containing Aristolochia or Asarum species, J. Ethnopharmacol., 2004, 94, 245–249 CrossRef CAS PubMed.
- F. D. Debelle, J.-L. Vanherweghem and J. L. Nortier, Aristolochic acid nephropathy: a worldwide problem, Kidney Int., 2008, 74, 158–169 CrossRef CAS PubMed.
- P. Wang, W. Guo, G. Huang, J. Zhen, Y. Li, T. Li, L. Zhao, K. Yuan, X. Tian and X. Huang, Berberine-based heterogeneous linear supramolecules neutralized the acute nephrotoxicity of aristolochic acid by the self-assembly strategy, ACS Appl. Mater. Interfaces, 2021, 13, 32729–32742 CrossRef CAS PubMed.
- M. Wall, M. Wani, C. Cook, K. Palmer and G. Sim, Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata1,2, J. Am. Chem. Soc., 1986, 88, 2325 Search PubMed.
- Y. Pommier, Topoisomerase I inhibitors: camptothecins and beyond, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed.
- H. Wu, M. Hu, L. Luo, H. Zhou, J. Hu, J. Xia, Z. Ke, Y. Wang and J. Song, Research progress on camptothecin and its derivative nano-prodrugs based on tumor microenvironment response, Zhongguo Zhongyao Zazhi, 2022, 47, 2852–2865 Search PubMed.
- W. He, X. Hu, W. Jiang, R. Liu, D. Zhang, J. Zhang, Z. Li and Y. Luan, Rational design of a new self–codelivery system from redox–sensitive camptothecin–cytarabine conjugate assembly for effectively synergistic anticancer therapy, Adv. Healthcare Mater., 2017, 6, 1700829–1700841 CrossRef PubMed.
- Z. Guo, L. Lin, K. Hao, D. Wangdand and X. Chen, Helix Self-Assembly Behavior of Amino Acid-Modified Camptothecin Prodrugs and Its Antitumor Effect, ACS Appl. Mater. Interfaces, 2020, 12, 7466–7476 CrossRef CAS PubMed.
- Y. Sun, A. Shieh, S. H. Kim, S. King, A. Kim, H.-L. Sun, C. M. Croce and J. R. Parquette, The self-assembly of a camptothecin-lysine nanotube, Bioorg. Med. Chem. Lett., 2016, 26, 2834–2838 CrossRef CAS PubMed.
- Se H. Kim, J. A. Kaplan, Y. Sun, A. Shieh, C. M. Croce, M. W. Grinstaff and J. R. Parquette, The Self-Assembly of Anticancer Camptothecin-Dipeptide Nanotubes: A Minimalistic and High Drug Loading Approach to Increased Efficacy, Chemistry, 2015, 21, 101–105 CrossRef CAS PubMed.
- J. Wang, K. Wang, T. Han, J. Li, X. He, R. Rong, Z. Cao, X. Li and K. Wang, Antitumour properties based on the self–assembly of camptothecin and carbamoylmannose conjugates, Chem. Biol. Drug Des., 2020, 96, 870–877 CrossRef CAS PubMed.
- M. Li, Z. Xu, L. Zhang, M. Cui, M. Zhu, Y. Guo, R. Sun, J. Han, E. Song, Y. He and Y. Su, Targeted Noninvasive Treatment of Choroidal Neovascularization by Hybrid Cell-Membrane-Cloaked Biomimetic Nanoparticles, ACS Nano, 2021, 15, 9808–9819 CrossRef CAS PubMed.
- Qi Weng, L. Zhou, L. Xia, Y. Zheng, X. Zhang, F. Li and Q. Li, In vitro evaluation of FL118 and 9-Q20 cytotoxicity and cellular uptake in 2D and 3D different cell models, Cancer Chemother. Pharmacol., 2019, 84, 527–537 CrossRef CAS PubMed.
- R. Cuadros, E. Garcini, F. Wandosell, G. T. Faircloth and J. Avila, The marine compound spisulosine, an inhibitor of cell proliferation, promotes the disassembly of actin stress fibers, Cancer Lett., 2000, 152, 23–29 CrossRef CAS PubMed.
- Y. Zhang, R. Yin, G. Wu, M. Yu, J. Liu, X. Wang, X. Liu, H. Guan, R. Yu and T. Jiang, Self-assembling nanoparticles of dually hydrophobic prodrugs constructed from camptothecin analogue for cancer therapy, Eur. J. Med. Chem., 2020, 200, 112365–112375 CrossRef CAS PubMed.
- Y. Guo, H. Liu, H. Xiao, M. Yuan and C. Li, Self-assembled Camptothecin derivatives – Curcuminoids conjugate for combinatorial chemo-photodynamic therapy to enhance anti-tumor efficacy, J. Photochem. Photobiol., B, 2021, 215, 112124–112131 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhao, Q. Ma, H. Zhang and J. Han, Novel carrier-free nanoparticles composed of 7-ethyl-10-hydroxycamptothecin and chlorin e6: Self-assembly mechanism investigation and in vitro/in vivo evaluation, Colloids Surf., B, 2019, 188, 110722–110729 CrossRef PubMed.
- Q. Chen, S. Xu, S. Liu, Y. Wang and G. Liu, p Emerging nanomedicines of paclitaxel for cancer treatment, Journal of Controlled Release, J. Controlled Release, 2022, 342, 280–294 CrossRef CAS PubMed.
- A. M. Sofias, M. Dunne, G. Storm and C. Allen, The battle of “nano” paclitaxel, Adv. Drug Delivery Rev., 2017, 122, 20–30 CrossRef CAS PubMed.
- J. Liu, P. Wang, B. Huang, Q. Cheng, Y. Duan, L. Chen, T. Ma, C. Zhu, D. Li and W. Fan, Effective suppression of triple negative breast cancer by paclitaxel nanoparticles conjugated with transmembrane TNF-α monoclonal antibody, Int. J. Pharm., 2022, 624, 121969–121981 CrossRef CAS PubMed.
- J. Yu, Y. Wang, S. Zhou, J. Li, J. Wang, D. Chi, X. Wang, G. Lin, Z. He and Y. Wang, Remote loading paclitaxel-doxorubicin prodrug into liposomes for cancer combination therapy, Acta Pharm. Sin. B, 2020, 10, 1730–1740 CrossRef CAS PubMed.
- C. Luo, J. Sun, B. Sun and Z. He, Prodrug-based nanoparticulate drug delivery strategies for cancer therapy, Trends Pharmacol. Sci., 2014, 35, 556–566 CrossRef CAS PubMed.
- R. Lin, A. G. Cheetham, P. Zhang, Y. A. Lin and H. Cui, Supramolecular filaments containing a fixed 41% paclitaxel loading, Chem. Commun., 2013, 49, 4968–4970 RSC.
- R. Tian, H. Wang, R. Niu and D. Ding, Drug delivery with nanospherical supramolecular cell penetrating peptide–taxol conjugates containing a high drug loading, J. Colloid Interface Sci., 2015, 45, 15–20 CrossRef PubMed.
- L. An, C. Yan, X. Mu, T. Cheng, Q. Tian, J. Lin and S. Yang, Paclitaxel-Induced Ultrasmall Gallic Acid-Fe@BSA Self-Assembly with Enhanced MRI Performance and Tumor Accumulation for Cancer Theranostics, ACS Appl. Mater. Interfaces, 2018, 10, 28483–28493 CrossRef CAS PubMed.
- Y. Wang, D. Liu, Q. Zheng, Q. Zhao, H. Zhang, Y. Ma, J. K. Fallon, Q. Fu, M. T. Haynes, G. Lin, R. Zhang, D. Wang, X. Yang, L. Zhao, Z. He and F. Liu, Disulfide Bond Bridge Insertion Turns Hydrophobic Anticancer Prodrugs into Self-Assembled Nanomedicines, Nano Lett., 2014, 14, 5577–5583 CrossRef CAS PubMed.
- M. Zhou, Y. Xie, S. Xu, J. Xin, J. Wang, T. Han, R. Ting, J. Zhang and F. An, Hypoxia-activated nanomedicines for effective cancer therapy, Eur. J. Med. Chem., 2020, 195, 112274 CrossRef CAS PubMed.
- X. Wang, B. Yang, L. Li, T. Liu, S. Zuo, D. Chi, Z. He, B. Sun and J. Sun, Probing the fluorination effect on the self-assembly characteristics, in vivo fate and antitumor efficacy of paclitaxel prodrug nanoassemblies, Theranostics, 2021, 11, 7896–7910 CrossRef CAS PubMed.
- H. Wang, D. Wang, J. Yu, Y. Zhang and Y. Zhou, Applications of metal–phenolic networks in nanomedicine: a review, Biomater. Sci., 2022, 10, 5786–5808 RSC.
- W. Xie, Z. Guo, L. Zhao and Y. Wei, Metal-phenolic networks: facile assembled complexes for cancer theranostics, Theranostics, 2021, 11, 6407–6426 CrossRef CAS PubMed.
- S. Liu, X. Xu, J. Ye, J. Wang, Q. Wang, Z. Liu, J. Xu and Y. Fu, Metal-coordinated nanodrugs based on natural products for cancer theranostics, Chem. Eng. J., 2023, 456, 140892–140920 CrossRef CAS.
- H. Geng, Q.-Z. Zhong, J. Li, Z. Lin, J. Cui, F. Caruso and J. Hao, Metal ion-directed functional metal–phenolic materials, Chem. Rev., 2022, 122, 11432–11473 CrossRef CAS PubMed.
- Q. Li, Z. Dong, M. Chen and L. Feng, Phenolic molecules constructed nanomedicine for innovative cancer treatment, Coord. Chem. Rev., 2021, 439, 213912–213928 CrossRef CAS.
- Y. Feng, P. Li and J. Wei, Engineering functional mesoporous materials from plant polyphenol based coordination polymers, Coord. Chem. Rev., 2022, 468, 214649–214673 CrossRef CAS.
- Q. Dai, H. Geng, Q. Yu, J. Hao and J. Cui, Polyphenol-Based Particles for Theranostics, Theranostics, 2019, 9, 3170–3190 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, Y. Zhao, M. Pu, X. Song, L. Yu, X. Yan, J. Wu and Z. He, Innovations and challenges of polyphenol-based smart drug delivery systems, Nano Res., 2022, 15, 8156–8184 CrossRef CAS.
- G. Fan, J. Cottet, M. R. Rodriguez-Otero, P. Wasuwanich and A. L. Furst, Metal–phenolic networks as versatile coating materials for biomedical applications, ACS Appl. Bio Mater., 2022, 5, 4687–4695 CrossRef CAS PubMed.
- Z. Ma, N. Wang, H. He and X. Tang, Pharmaceutical strategies of improving oral systemic bioavailability of curcumin for clinical application, J. Controlled Release, 2019, 316, 359–380 CrossRef CAS PubMed.
- Z. Li, M. Shi, N. Li and R. Xu, Application of Functional Biocompatible Nanomaterials to Improve Curcumin Bioavailability, Front. Chem., 2020, 8, 589957–589966 CrossRef CAS PubMed.
- K. Khezri, M. Saeedi and H. Mohammadamini, Abbas and Khezri, A comprehensive review of the therapeutic potential of curcumin nanoformulations, Phytother. Res., 2021, 35, 5527–5563 CrossRef CAS PubMed.
- J. Wang, Y. Wang, Q. Liu, L. Yang, R. Zhu, C. Yu and S. Wang, Rational Design of Multifunctional Dendritic Mesoporous Silica Nanoparticles to Load Curcumin and Enhance Efficacy for Breast Cancer Therapy, ACS Appl. Mater. Interfaces, 2016, 8, 26511–26523 CrossRef CAS PubMed.
- A. Moustapha, P. Pérétout, N. Rainey, F. Sureau, M. Geze, J. Petit, E. Dewailly, C. Slomianny and P. Petit, Curcumin induces crosstalk between autophagy and apoptosis mediated by calcium release from the endoplasmic reticulum, lysosomal destabilization and mitochondrial events, Cell Death Discovery, 2015, 1, 15017–15031 CrossRef CAS PubMed.
- Y. Wen, J. Hu, J. Liu and M. Li, Degradable Carrier-Free Metal–Phenolic Network Theranostic Agent with Targeted Mitochondrial Damage for Efficient Cancer Theranostics, Chem. Mater., 2021, 33, 7089–7099 CrossRef CAS.
- S. Wong, J. Zhao, C. Cao, C. K. Wong, R. P. Kuchel, S. De Luca, J. M. Hook, C. J. Garvey, S. Smith and J. Ho, Just add sugar for carbohydrate induced self-assembly of curcumin, Nat. Commun., 2019, 10, 582–590 CrossRef CAS PubMed.
- R. Xu, W. Deng, Y. Dai and J. Hu, pH-responsive citral microcapsules with tannic acid-FeIII coordination complexes, Food Chem., 2022, 397, 133715–133724 CrossRef CAS PubMed.
- S. A. Abouelmagd, N. H. A. Ellah, O. Amen, A. Abdelmoez and N. G. Mohamed, Self-assembled tannic acid complexes for pH-responsive delivery of antibiotics: Role of drug-carrier interactions, Int. J. Pharm., 2019, 562, 76–85 CrossRef CAS PubMed.
- T. Liu, J. Feng and X. Zhang, Metal-Polyphenol Network Based Multifunctional Materials for Theranostic Applications, J. Funct. Polym., 2019, 32, 421–433 CAS.
- J. Li, Y. Zhou, J. Liu, X. Yang, K. Yang, L. Lei, H. Hu, H. Zhang, L. Ouyang and H. Gao, Metal-phenolic networks with ferroptosis to deliver NIR-responsive CO for synergistic therapy, J. Controlled Release, 2022, 352, 313–327 CrossRef CAS PubMed.
- P. Liu, X. Shi, S. Zhong, Y. Peng, Y. Qi, J. Ding and W. Zhou, Metal-phenolic networks for cancer theranostics, Biomater. Sci., 2021, 9, 2825–2849 RSC.
- S. Mitragotri, P. A. Burke and R. Langer, Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed.
- Y. Honda, T. Nomoto, M. Matsui, H. Takemoto and N. Nishiyama, Sequential Self-Assembly Using Tannic Acid and Phenylboronic Acid-Modified Copolymers for Potential Protein Delivery, Biomacromolecules, 2020, 21, 3826–3835 CrossRef CAS PubMed.
- J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Engineering Multifunctional Capsules through the Assembly of Metal–Phenolic Networks, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- Y. Ping, J. Guo, H. Ejima, X. Chen, J. J. Richardson, H. Sun and F. Caruso, pH–responsive capsules engineered from metal–phenolic networks for anticancer drug delivery, Small, 2015, 11, 2032–2036 CrossRef CAS PubMed.
- H. Ejima, J. J. Richardson, L. Kang, J. P. Best, M. Koeverden, G. K. Such, J. Cui and F. Caruso, One-Step Assembly of Coordination Complexes for Versatile Film and Particle Engineering, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- M. A. Rahim, K. Kempe, M. Müllner, H. Ejima, Y. Ju, M. V. Koeverden, T. Suma, J. A. Braunger, M. G. Leeming and B. F. Abrahams, Surface-Confined Amorphous Films from Metal-Coordinated Simple Phenolic Ligands, Chem. Mater., 2015, 27, 5825–5832 CrossRef CAS.
- I. Valle, H. G. Roweth, M. W. Malloy, S. Moco, D. Barron, E. Battinelli, J. Loscalzo and A. Barabási, Network medicine framework shows that proximity of polyphenol targets and disease proteins predicts therapeutic effects of polyphenols, Nat. Food, 2021, 2, 143–155 CrossRef PubMed.
- Y. Wang, J. Zhang, Y. Zhao, M. Pu, X. Song, L. Yu, X. Yan, J. Wu and Z. He, Innovations and challenges of polyphenol-based smart drug delivery systems, Nano Res., 2022, 15, 8156–8184 CrossRef CAS.
- C. Qi, G. Liu, Y. Ping, K. Yang, Q. Tan, Y. Zhang, G. Chen, X. Huang and D. Xu, A comprehensive review of nano-delivery system for tea polyphenols: construction, applications, and challenges, Food Chem., 2013, 17, 100571–100583 Search PubMed.
- K. Liang, J. E. Chung, S. Gao, N. Yongvongsoontorn and M. Kurisawa, Highly Augmented Drug Loading and Stability of Micellar Nanocomplexes Composed of Doxorubicin and Poly (ethylene glycol)–Green Tea Catechin Conjugate for Cancer Therapy, Adv. Mater., 2018, 30, 1706963–1706970 CrossRef PubMed.
- M. Alam, S. Ali, G. M. Ashraf, A. L. Bilgrami, D. K. Yadav and M. I. Hassan, Epigallocatechin 3-gallate: From green tea to cancer therapeutics, Food Chem., 2022, 379, 132135–132150 CrossRef CAS PubMed.
- B. Lsa, A. Gg, A. Ww, T. B. Wei, B. Zw, Y. B. Zhen, B. Wf, B. Gz, A. Kz and B. Oj, Self-assembled green tea polyphenol-based coordination nanomaterials to improve chemotherapy efficacy by inhibition of carbonyl reductase 1, Biomaterials, 2019, 210, 62–69 CrossRef PubMed.
- J. Ding, T. Liang, Q. Min, L. Jiang and J. Zhu, “Stealth and Fully-Laden” Drug Carriers: Self-Assembled Nanogels Encapsulated with Epigallocatechin Gallate and siRNA for Drug-Resistant Breast Cancer Therapy, ACS Appl. Mater. Interfaces, 2018, 10, 9938–9948 CrossRef CAS PubMed.
- L. Crascì, M. R. Lauro, G. Puglisi and A. Panico, Natural antioxidant polyphenols on inflammation management: Anti-glycation activity vs metalloproteinases inhibition, Crit. Rev. Food Sci. Nutr., 2018, 58, 893–904 CrossRef PubMed.
- B. Das, S. Nivedita, B. Anupam and S. Dona, Dietary phytochemicals in the regulation of epithelial to mesenchymal transition and associated enzymes: A promising anticancer therapeutic approach, Semin. Cancer Biol., 2019, 56, 196–218 CrossRef CAS PubMed.
- S. Chen, J. Fan, D. Zheng, F. Liu, X. Zeng, G. Yan and X. Zhang, A multi-functional drug delivery system based on polyphenols for efficient tumor inhibition and metastasis prevention, Biomater. Sci., 2020, 8, 702–711 RSC.
- K. Li, G. Xiao, J. J. Richardson, B. L. Tardy, H. Ejima, W. Huang, J. Guo, X. Liao and B. Shi, Targeted Therapy against Metastatic Melanoma Based on Self–Assembled Metal–Phenolic Nanocomplexes Comprised of Green Tea Catechin, Adv. Sci., 2019, 6, 1801688–1801694 CrossRef PubMed.
- Y. P. Wang, Z. J. Luo, D. Zhou, X. Wang and Z. Yu, Nano-assembly of Ursolic Acid with Platinum Prodrug Overcomes Multiple Deactivation Pathways in Platinum-Resistant Ovarian Cancer, Biomater. Sci., 2021, 9, 4110–4119 RSC.
- J. Zhang, X. Ren, X. Tian, P. Zhang, Z. Chen, X. Hu and X. Mei, GSH and enzyme responsive nanospheres based on self-assembly of green tea polyphenols and BSA used for target cancer chemotherapy, Colloids Surf., B, 2018, 173, 654–661 CrossRef PubMed.
- J. E. Chung, S. Tan, S. J. Gao, N. Yongvongsoontorn, S. H. Kim, J. H. Lee, H. S. Choi, H. Yano, L. Zhuo and M. Kurisawa, Self-assembled micellar nanocomplexes comprising green tea catechin derivatives and protein drugs for cancer therapy, Nat. Nanotechnol., 2014, 9, 907–912 CrossRef CAS PubMed.
- Y. Dai, J. Guo, T. Y. Wang, Y. Ju, A. J. Mitchell, T. Bonnard, J. Cui, J. J. Richardson, C. E. Hagemeyer and K. Alt, Self–Assembled Nanoparticles from Phenolic Derivatives for Cancer Therapy, Adv. Healthcare Mater., 2017, 6, 1700467–1700473 CrossRef PubMed.
- L. Xu and S. Liu, Rapid Cu(II)-Directed Self Assembly of Esterified Tea Polyphenol Oligomers to Controlled Release Nanoflower Carrier, J. Agric. Food Chem., 2021, 69, 7725–7732 CrossRef CAS PubMed.
- G. Lin, M. A. Rahim, M. G. Leeming, C. Cortez-Jugo, Q. A. Besford, Y. Ju, Q. Zhong, S. T. Johnston, J. Zhou and F. Caruso, Selective metal–phenolic assembly from complex multicomponent mixtures, ACS Appl. Mater. Interfaces, 2019, 11, 17714–17721 CrossRef CAS PubMed.
- I. Góral and K. Wojciechowski, Surface activity and foaming properties of saponin-rich plants extracts, Adv. Colloid Interface Sci., 2020, 279, 102145–110268 CrossRef PubMed.
- M. Sochacki and O. Vogt, Triterpenoid Saponins from Washnut (Sapindus mukorossi Gaertn.)—A Source of Natural Surfactants and Other Active Components, Plants, 2022, 11, 2355–2378 CrossRef CAS PubMed.
- Z. A. Ratan, M. F. Haidere, Y. H. Hong, H. P. Sang and J. Y. Cho, Pharmacological potential of ginseng and its major component ginsenosides, J. Ginseng Res., 2020, 45, 199–210 CrossRef PubMed.
- D. Singh and P. K. Chaudhuri, Structural characteristics, bioavailability and cardioprotective potential of saponins, Integr. Med. Res., 2018, 7, 33–43 CrossRef PubMed.
- Q. Li, D. Yu, L. Guo, L. Guo and X. Dou, Research progress on mechanism of transformation of saponins by microbial fermentation, Chin. Tradit. Herb. Drugs, 2022, 53, 7264–7278 Search PubMed.
- A. Wong, C. Che and K. Leung, Recent advances in ginseng as cancer therapeutics: a functional and mechanistic overview, Nat. Prod. Rep., 2015, 32, 256–272 RSC.
- M. Wang, Y. Xu, J. Xie, Z. Jiang and L. Peng, Ginsenoside as a new stabilizer enhances the transfection efficiency and biocompatibility of cationic liposome, Biomater. Sci., 2021, 9, 8373–8385 RSC.
- C. Hong, D. Wang, J. Liang, Y. Guo, Y. Zhu, J. Xia, J. Qin, H. Zhan and J. Wang, Novel ginsenoside-based multifunctional liposomal delivery system for combination therapy of gastric cancer, Theranostics, 2019, 9, 4437–4449 CrossRef CAS PubMed.
- C. Hong, J. Liang, J. Xia, Y. Zhu and J. Wang, One Stone Four Birds: A Novel Liposomal Delivery System Multi-functionalized with Ginsenoside Rh2 for Tumor Targeting Therapy, Nanomicro Lett., 2020, 12, 129–146 CAS.
- M. Murugesan, R. Mathiyalagan, V. Boopathi, B. M. Kong, S. K. Choi, C. S. Lee, D. C. Yang, S. C. Kang and T. Thambi, Production of minor ginsenoside CK from major ginsenosides by biotransformation and its advances in targeted delivery to tumor tissues using nanoformulations, Nanomaterials, 2022, 12, 3427–3450 CrossRef CAS PubMed.
- Z. A. Ratan, M. F. Haidere, Y. H. Hong, S. H. Park, J. O. Lee, J. Lee and J. Y. Cho, Pharmacological potential of ginseng and its major component ginsenosides, J. Ginseng Res., 2021, 45, 199–210 CrossRef PubMed.
- X. Piao, H. Zhang, J. P. Kang, D. U. Yang and Y. Wang, Advances in Saponin Diversity of Panax ginseng, Molecules, 2021, 45, 199–210 Search PubMed.
- J. H. Jeon, J. Lee, M. K. Choi and I. S. Song, Pharmacokinetics of ginsenosides following repeated oral administration of red ginseng extract significantly differ between species of experimental animals, Arch. Pharmacal Res., 2020, 43, 1335–1346 CrossRef CAS PubMed.
- S. A. Nag, J. J. Qin, W. Wang, M. Wang and R. Zhang, Ginsenosides as Anticancer Agents: In vitro and in vivo Activities, Structure–Activity Relationships, and Molecular Mechanisms of Action, Front. Pharmacol., 2021, 3, 25–42 Search PubMed.
- H. Wang, H. Wang, Y. Zheng, Y. Zheng, Q. Sun, Q. Sun, Z. Zhang, Z. Zhang, M. Zhao and M. Zhao, Ginsenosides emerging as both bifunctional drugs and nanocarriers for enhanced antitumor therapies, J. Nanobiotechnol., 2021, 19, 322–361 CrossRef CAS PubMed.
- J. F. Xu, Y. Wan, F. Tang and C. Peng, Emerging Significance of Ginsenosides as Potentially Reversal Agents of Chemoresistance in Cancer Therapy, Front. Pharmacol., 2021, 12, 720474 CrossRef CAS PubMed.
- Y. Ke, L. Huang, Y. Song, Z. Liu, L. Liang, L. Wang and T. Wang, Preparation and pharmacological effects of minor ginsenoside nanoparticles: a review, Front. Pharmacol., 2022, 13, 974274–974288 CrossRef CAS PubMed.
- J. Zhao, Z. Duan, X. Ma, Y. Liu and D. Fan, Recent advances in systemic and local delivery of ginsenosides using nanoparticles and nanofibers, Chin. J. Chem. Eng., 2022, 30, 291–300 CrossRef.
- M. Li, J. Lan, X. Li, M. Xin, H. Wang, F. Zhang, X. Lu, Z. Zhuang and X. Wu, Novel ultra-small micelles based on ginsenoside Rb1: a potential nanoplatform for ocular drug delivery, Drug Delivery, 2019, 26, 481–489 CrossRef CAS PubMed.
- Y. Yi, H. J. Kim, M. Zheng, P. Mi, M. Naito, B. S. Kim, H. S. Min, K. Hayashi, F. Perche and K. Toh, Glucose-linked sub-50-nm unimer polyion complex-assembled gold nanoparticles for targeted siRNA delivery to glucose transporter 1-overexpressing breast cancer stem-like cells, J. Controlled Release, 2019, 295, 268–277 CrossRef CAS PubMed.
- X. Tan, L. Chao, K. Feng, J. Le, J. Shen and J. Shao, Self-assembled micelles of the natural medicine ginsenosides for cancer metastasis therapy, J. Ind. Eng. Chem., 2022, 116, 303–309 CrossRef CAS.
- L. Dai, K. Liu, C. Si, L. Wang, J. Liu, J. He and J. Lei, Ginsenoside nanoparticle: a new green drug delivery system, J. Mater. Chem. B, 2016, 4, 529–538 RSC.
- J. Zou, J. Le, B. Zhang, M. Yang, J. Jiang, J. Lin, P. Wu, C. Li, L. Chen and J. Shao, Accelerating Transdermal Delivery of Insulin by Ginsenoside Nanoparticles with Unique Permeability, Int. J. Pharm., 2021, 605, 120784–120795 CrossRef CAS PubMed.
- X. Dai, X. Shi, Y. Wang and Y. Qiao, Solubilization of saikosaponin a by ginsenoside Ro biosurfactant in aqueous solution: Mesoscopic simulation, J. Colloid Interface Sci., 2012, 384, 73–80 CrossRef CAS PubMed.
- Z. Zhang, G. Du, C. Wang, X. Wen, T. Calway, Z. Li, T. He, W. Du, M. Bissonnette and M. W. Musch, Compound K, a ginsenoside metabolite, inhibits colon cancer growth via multiple pathways including p53-p21 interactions, Int. J. Mol. Sci., 2013, 14, 2980–2995 CrossRef CAS PubMed.
- T. Chen, B. Li, Y. Qiu, Z. Qiu and P. Qu, Functional mechanism of Ginsenosides on tumor growth and metastasis, Saudi J. Biol. Sci., 2018, 25, 917–922 CrossRef CAS PubMed.
- R. Mathiyalagan, S. Subramaniyam, Y. J. Kim, Y. C. Kim and D. C. Yang, Ginsenoside compound K-bearing glycol chitosan conjugates: Synthesis, physicochemical characterization, and in vitro biological studies, Carbohydr. Polym., 2014, 112, 359–366 CrossRef CAS PubMed.
- J. Zhang, Y. Wang, Y. Jiang, T. Liu, Y. Luo, E. Diao, Y. Cao, L. Chen, L. Zhang, Q. Gu, J. Zhou, F. Sun, W. Zheng, J. Liu, X. Li and W. Hu, Enhanced cytotoxic and apoptotic potential in hepatic carcinoma cells of chitosan nanoparticles loaded with ginsenoside compound K, Carbohydr. Polym., 2018, 198, 537–545 CrossRef PubMed.
- X. Gao, J. Zhang, X. Gong, K. Li, L. Zhang and W. Li, Ginsenoside Rg5: A Review of Anticancer and Neuroprotection with Network Pharmacology Approach, Am. J. Chin. Med., 2022, 50, 2033–2056 CrossRef CAS PubMed.
- Y. Dong, R. Fu, J. Yang, P. Ma and D. Fan, Folic acid-modified ginsenoside Rg5-loaded bovine serum albumin nanoparticles for targeted cancer therapy in vitro and in vivo, Int. J. Nanomed., 2019, 14, 6971–6988 CrossRef CAS PubMed.
- R. Mathiyalagan, Y. J. Kim, C. Wang, Y. Jin, S. Subramaniyam, P. Singh, D. Wang and D. Yang, Protopanaxadiol aglycone ginsenoside-polyethylene glycol conjugates: synthesis, physicochemical characterizations, and in vitro studies, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1803–1809 CrossRef CAS PubMed.
- Z. Ren, X. Chen, L. Hong, X. Zhao, G. Cui, A. Li, Y. Liu, L. Zhou, R. Sun, S. Shen, J. Li, J. Lou, H. Zhou, J. Wang, G. Xu, Z. Yu, Y. Song and X. Chen, Nanoparticle Conjugation of Ginsenoside Rg3 Inhibits Hepatocellular Carcinoma Development and Metastasis, Small, 2020, 16, 1905233–1905246 CrossRef CAS PubMed.
- A. Guimaraes, Design of liposomes as drug delivery system for therapeutic applications, Int. J. Pharm., 2021, 601, 120571–120585 CrossRef PubMed.
- P. Liu, G. Chen and J. Zhang, A review of liposomes as a drug delivery system: current status of approved products, regulatory environments, and future perspectives, Molecules, 2022, 27, 1372–1394 CrossRef CAS PubMed.
- X. Zhang, S. Li, Y. Dong, H. Rong, J. Zhao and H. Hu, A multifunctional cholesterol-free liposomal platform based on protopanaxadiol for alopecia therapy, Nano Res., 2022, 15, 9498–9510 CrossRef CAS.
- C. Chen, J. Xia, H. Ren, A. Wang, Y. Zhu, R. Zhang, Z. Gan and J. Wang, Effect of the structure of ginsenosides on the in vivo fate of their liposomes, Asian J. Pharm. Sci., 2022, 17, 219–229 CrossRef PubMed.
- J. Xia, S. Ma, X. Zhu, C. Chen, R. Zhang, Z. Cao, X. Chen, L. Zhang, Y. Zhu and S. Zhang, Versatile ginsenoside Rg3 liposomes inhibit tumor metastasis by capturing circulating tumor cells and destroying metastatic niches, Sci. Adv., 2022, 8, 1262–1280 CrossRef PubMed.
- Z. Ying, B. Jla, B. Cg, B. Aw, B. Jx, H. B. Chao, D. Zz, Z. E. Zhong, B. Jk and B. Hr, Multifunctional ginsenoside Rg3-based liposomes for glioma targeting therapy, J. Controlled Release, 2021, 330, 641–657 CrossRef PubMed.
- K. Chen, R. Yang, F. Shen and H. Zhu, Advances in Pharmacological Activities and Mechanisms of Glycyrrhizic Acid, Curr. Med. Chem., 2020, 27, 6219–6243 CrossRef CAS PubMed.
- O. Y. Selyutina and N. Polyakov, Glycyrrhizic acid as a multifunctional drug carrier–From physicochemical properties to biomedical applications: A modern insight on the ancient drug, Int. J. Pharm., 2019, 559, 271–279 CrossRef CAS PubMed.
- X. Su, L. Wu, M. Hu, W. Dong, M. Xu and P. Zhang, Glycyrrhizic acid: A promising carrier material for anticancer therapy, Biomed. Pharmacother., 2017, 95, 670–678 CrossRef CAS PubMed.
- I. M. Tucker, A. Ab, A. Rep, A. Slh, C. Jpb, B. Rkt, C. Pxl, C. Jrpw, C. Rw and C. Jd, Adsorption and self-assembly properties of the plant based biosurfactant, Glycyrrhizic acid, J. Colloid Interface Sci., 2021, 598, 444–454 CrossRef CAS PubMed.
- G. You, T. Teng, G. Zhang, M. Chen, F. Liu, L. Sun, M. Wang and X. Ren, Preparation, optimization, characterization and in vitro release of baicalein-solubilizing glycyrrhizic acid nano-micelles, Int. J. Pharm., 2021, 601, 120546–120554 CrossRef CAS PubMed.
- J. Cui, X. Wang, J. Li, A. Zhu, Y. Du, W. Zeng, Y. Guo, L. Di and R. Wang, Immune Exosomes Loading Self-Assembled Nanomicelles Traverse the Blood–Brain Barrier for Chemo-immunotherapy against Glioblastoma, ACS Nano, 2023, 17, 1464–1484 CrossRef CAS PubMed.
- Y. X. Zhou, X. H. Gong, H. Zhang and C. Peng, A review on the pharmacokinetics of paeoniflorin and its anti-inflammatory and immunomodulatory effects, Biomed. Pharmacother., 2020, 130, 110505–110516 CrossRef CAS PubMed.
- C. Shen, B. Shen, J. Zhu, J. Wang and X. Li, Glycyrrhizic acid-based self-assembled micelles for improving oral bioavailability of paeoniflorin, Drug Dev. Ind. Pharm., 2021, 47, 207–214 CrossRef CAS PubMed.
- S. Fu and X. Yang, Recent advances in natural small molecules as drug delivery systems, J. Mater. Chem. B, 2023, 11, 4584–4599 RSC.
- X. Zhang, S. Hu, L. Huang, X. Chen, X. Wang, Y. Fu, H. Sun, G. Li and X. Wang, Advance Progress in Assembly Mechanisms of Carrier-Free Nanodrugs for Cancer Treatment, Molecules, 2023, 28, 7065–7095 CrossRef CAS PubMed.
- H. Zhang, J. Guo, J. Hu and M. Zhou, Terpenoid-based supramolecular materials: fabrications, performances, applications, Supramol. Chem., 2023, 34, 105–131 CrossRef CAS.
- M. Chen, X. He, Z. Sun, X. Huo, Y. Hou, X. Xu, H. Wu, L. Shi and G. Ma, Natural carrier-free self-assembled diterpene nanoparticles with its efficient anti-inflammation through the inhibition of NF-κB pathway for accelerated wound healing, Biomed. Pharmacother., 2023, 165, 115041–115052 CrossRef CAS PubMed.
- J. Huang, Y. Zhu, H. Xiao, J. Liu, S. Li, Q. Zheng, J. Tang and X. Meng, Formation of a traditional Chinese medicine self–assembly nanostrategy and its application in cancer: a promising treatment, China's Med., 2023, 18, 66–88 CrossRef CAS PubMed.
- J. Zheng, R. Fan, H. Wu, H. Yao, Y. Yan, J. Liu, L. Ran, Z. Sun, L. Yi, L. Dang, P. Gan, P. Zheng, T. Yang, Y. Zhang, T. Tang and Y. Wang, Directed self-assembly of herbal small molecules into sustained release hydrogels for treating neural inflammation, Nat. Commun., 2019, 10, 1604–1615 CrossRef CAS PubMed.
- P. Bai, D. Li, M. Shi, L. Yang, M. Tang, N. Qiu and J. Wen, Carrier-free doxorubicin/rhein supramolecular co-assembly for cancer therapy, J. Drug Delivery Sci. Technol., 2023, 89, 105030 CrossRef CAS.
- X. Tian, P. Wang, T. Li, X. Huang, W. Guo, Y. Yang, M. Yan, H. Zhang, D. Cai, X. Jia, F. Li, B. Xu, T. Ma, C. Yan and H. Lei, Acta Pharm. Sin. B, 2020, 10, 1784–1795 CrossRef CAS PubMed.
- Y. Wen, W. Zhang, N. Gong, Y. Wang, H. Guo, W. Guo, P. Wang and X. Liang, Carrier-free, self-assembled pure drug nanorods composed of 10-hydroxycamptothecin and chlorin e6 for combinatorial chemo-photodynamic antitumor therapy in vivo, Nanoscale, 2017, 9, 14347–14356 RSC.
- L. Qiao, M. Han, S. Gao, X. Shao, X. Wang, L. Sun, X. Fu and Q. Wei, Research progress on nanotechnology for delivery of active ingredients from traditional Chinese medicines, J. Mater. Chem. B, 2020, 8, 6333–6651 RSC.
- J. Lan, L. Liu, R. Zeng, Y. Qin, J. Hou, S. Xie, S. Yue, J. Yang, R. J. Y. Ho, Y. Ding and T. Zhang, Chem. Eng. J., 2021, 407, 127212 CrossRef CAS.
- Y. Han, H. Zhang, H. Zhao, S. Fu, R. Li, Z. Wang, Y. Wang, W. Lu and X. Yang, Nanoparticle encapsulation using self-assembly abietic acid to improve oral bioavailability of curcumin, Food Chem., 2024, 436, 137676 CrossRef CAS PubMed.
- S. Fu, M. Wang, B. Li, X. Li, J. Cheng, H. Zhao, H. Zhang, A. Dong, W. Lu and X. Yang, Bionic natural small molecule co-assemblies towards targeted and synergistic Chemo/PDT/CDT, Biomater. Res., 2023, 27, 43–62 CrossRef CAS PubMed.
- N. He, K. Zhi, X. Yang, H. Zhao, H. Zhang, J. Wang and Z. Wang, Self-assembled fibrillar networks induced by two methods: a new unmodified natural product gel, New J. Chem., 2018, 42, 14170–14178 RSC.
| This journal is © The Royal Society of Chemistry 2024 |