
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient procedure for biodiesel synthesis from waste oil and t-butylation of resorcinol using a porous microtube polymer-based solid acid
Zhijin
Guo
and
Xuezheng
Liang
 *
*
School of Chemistry and Chemical Engineering, Shaoxing University, Shaoxing, 312000, China. E-mail: liangxuezheng@126.com
First published on 18th October 2023
Abstract
A novel porous microtube polymer-based solid acid was synthesized via the polymerisation of naphthalene and subsequent sulfonation. The polymer structure could be easily adjusted by altering the amount of formaldehyde dimethyl acetal (FDA), and could be changed to thin slices, microtubes or microspheres. The high surface area of 128.92 m2 g−1 and special microtube structure indicated good mass transfer efficiency and a hydrophobic surface. The abundant naphthalene rings provided sufficient sulfonation sites, which resulted in a high acid density of over 3 mmol g−1. An efficient procedure for biodiesel synthesis and t-butylation of resorcinol were developed using the novel solid acid. The high Brunauer–Emmett–Teller (BET) surface area, special microtube structure and organic framework were the key factors for the high activities, providing great potential in green chemistry.
1. Introduction
Biodiesel is a well-known renewable resource, and has been regarded as an appropriate substitute for traditional mineral fuels.1 However, the high price of the vegetable oil raw material has restricted its wide commercial application.2 On the other hand, the consumption of large amounts of edible oil in China, with its large population, has resulted in abundant waste oil, which would be a better raw material with a much lower price.3 However, the waste oil has high contents of free fatty acids (FFAs) and water, which make base-catalyzed processes unsuitable for waste oils.4 Pre-esterification must be carried out to convert the FFAs. Several acid catalysts, such as sulfuric acid and acidic ionic liquids (ILs),5,6 have been used to catalyze the pre-esterification process. This results in a tedious two-step synthetic process that requires additional acid-catalyst removal. Acids could catalyze both esterification and transesterification reactions, and are quite suitable for waste oil raw materials.7 Solid acids, such as SO42−/SnO2,8 carbon-based solid acids,9 Zr-doped SBA-1510 and clay-based acids,11 have been used for one-step synthesis of biodiesel from waste oils. However, harsh reaction conditions, such as high temperature (220 °C) and high pressure, must be applied to get satisfactory yields using these solid acid catalysts. A low yield of only 46% was obtained using a zeolite-based catalyst at 70 °C.12 Bio-hydroxyapatite-supported mesoporous Cr heterogeneous catalysts showed high activities for waste oil raw materials.13,14 Efficient one-pot biodiesel synthesis from waste oils was developed in our previous work using a catalyst based on a hydrophobic acidic ionic liquid immobilized on polydivinyl benzene (PDVB).15–17 However, the high price of the ionic liquid moieties limits their practical application. Sulfonated PDVB has been applied to biodiesel synthesis, and showed high activities towards both fatty acids and triglycerides.18–20 The catalytic activity towards waste oil has been little investigated. Sulfonated porous polymers have also shown high activities for the reversible adsorption of ammonia.21,22 However, PDVB from vinyl polymerization has an alkyl chain framework, which is not as stable as a rigid aromatic ring framework.Alkylation of aromatics is very important in the chemical industry.23 Friedel–Crafts butylation of resorcinol to produce butylated products is one of the most noteworthy examples, because the products are used as antioxidants, polymer stabilizers and therapeutics.24
Porous materials have wide applications in gas storage, pollution treatment, drug delivery, catalysis and so on.25,26 Traditional porous materials, such as zeolites, activated carbon, and carbon nanotubes with inorganic frameworks, have less affinity for organic compounds.27–30 Porous organic polymers (POPs) have attracted much attention for the variable and multiple chemical functionalities of the organic building blocks. Hypercrosslinked polymers are one such POP and have a simple synthesis process. Tan and co-workers have made great achievements in polymerisation of rigid aromatic building blocks with external crosslinkers.31–33 The polymers have high surface areas, making them quite suitable for gas adsorption and storage. Also, this high surface area means that these polymers have great potential in catalysis. Acid-catalyzed reactions are among the most important in the chemical industry. Liquid acids, such as sulfuric acid and phosphoric acid, are still widely used, which causes various problems such as equipment corrosion, separation difficulty and heavy environmental pollution. The motivations of “green chemistry” have led to the development of highly active and recyclable catalysts. Solid acids, being difficult to dissolve in water and organic solvents, could avoid the drawbacks of liquid acids. Current solid acids are mainly zeolites, heteropolyacids, mineral clays, sulfonated metal oxides, and ion-exchange resins.34–36
Herein, porous polymers with an aromatic ring framework were presented, using naphthalene as the monomer and formaldehyde dimethyl acetal (FDA) as the external crosslinker. Then, –SO3H groups were introduced via sulfonation of the aromatic rings using 98% H2SO4. The novel porous polymer-based solid acid with an aromatic ring framework and special microtube structure was applied to biodiesel synthesis from waste oil and t-butylation of resorcinol, and showed very high activities and reusability for these reactions.
2. Experimental
2.1 Chemicals and reagents
All reagents were of analytical grade and used as purchased without further purification. Naphthalene, methanol and acetone were purchased from Sinopharm Chemical Reagent Co., Ltd. Dimethoxymethane (formaldehyde dimethyl acetal, FDA) and dichloroethane (DCE) were obtained from Energy Chemical. Ferric trichloride (FeCl3) was supplied by Aladdin.2.2 Catalyst preparation
2.3 The procedure for the biodiesel synthesis
Waste oil with 25% FFAs was used as the raw material. The waste oil was pretreated via filtration, dehydration and decolorization. The free fatty acid content in the waste oil is important for the biodiesel product quality.37,38 The fatty acid contents in the FFAs and triglycerides were analyzed using the ISO 5509:2000 method and the results are shown in Tables 1 and 2. The waste oil, methanol and catalyst were mixed and stirred at 70 °C for a certain time. The reaction progress was monitored via gas chromatography (GC) analysis at half-hour intervals. After completion, the solid acid was first recycled via filtration. Then, the reaction mixture was left to settle to form two layers, after distillation of excess methanol. The upper biodiesel layer was analyzed according to the method of Alcantara et al.39 on a Shimadzu 14B gas chromatograph using an SE-52-packed column, coupled with an FID.| Fatty acid | Palmitic acid (C16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) :0) |
Stearic acid (C18:0) |
Oleic acid (C18:1) |
Linolenic acid (C18:3) |
Linoleic acid (C18:2) |
|---|---|---|---|---|---|
| Content (wt%) | 25 | 32 | 30 | 7 | 8 |
| Fatty acid | Palmitic acid (C16:0) |
Stearic acid (C18:0) |
Oleic acid (C18:1) |
Linolenic acid (C18:3) |
Linoleic acid (C18:2) |
|---|---|---|---|---|---|
| Content (wt%) | 23 | 29 | 33 | 9 | 10 |
2.4 The tert-butylation of resorcinol
The tert-butylation was carried out in a 20 mL autoclave. A typical reaction mixture of 2.2 g resorcinol, 4.8 g t-butanol (TBA) and 0.05 g of catalyst was heated and stirred in an oil bath. After a certain time, the reaction mixture was cooled to room temperature and diluted with 5 mL ethanol. It was then filtered and the filtrate was analyzed using a Shimadzu 14B gas chromatograph using an SE-52-packed column, coupled with FID. The conversion was defined as the percentage of resorcinol converted into product.2.5 Catalyst characterization
Nitrogen isotherms were measured on a Micromeritics ASAP 2020M system. The samples were degassed for 10 h at 150 °C before measurement. The pore-size distribution of the mesopores was calculated using the Barrett–Joyner–Halenda (BJH) model. CHNS elemental analysis was performed on a PerkinElmer 2400 series CHNS analyzer. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker 66 V FT-IR spectrometer in the wave-number range of 4000–500 cm−1. The total acid densities of POP-x–SO3H-m were determined by acid–base titration using a KOH standard solution. Thermogravimetric analysis (TGA) was performed on an HTG-2, with the temperature increasing from room temperature to 700 °C at a rate of 10 °C min−1 under an air atmosphere. Morphological analysis was carried out using a JEOL JSM-6360LV.3. Results and discussion
3.1 Characterization of the polymers and solid acids
The polymer was synthesized by polymerisation of the naphthalene rings using an external crosslinking agent. The main reaction was the Friedel–Crafts alkylation. The elemental analysis gave the following results for POP-0.01: C 95.03%, H 4.97% (found), C 94.52% and H 5.48% (calculated). The element analysis results did not agree well with the calculated values (structure in Scheme 1a), which indicated that the Scholl reaction between the naphthalene rings also occurred. POP-0.01–SO3H-20 showed the following results: C 50.936%, H 3.412% and S 7.998%. The S content agreed well with the acid density, which indicated that the sulfur element existed in the form of –SO3H (Scheme 1b). | ||
| Scheme 1 The structures of the polymer and solid acid. (a) POP-0.01, (b) POP-0.01–SO3H-30. | ||
According to the polymer structure (Scheme 1a), the molar ratio of naphthalene and the external crosslinking agent FDA was quite important, as it determined the chemical structure and crosslinking extent. Fig. 1 shows that the amount of FDA had a great influence on the POP structure. POP showed a thin-slice structure with 0.005 mol FDA (Fig. 1a). There was not enough FDA for the alkylation crosslinking reaction and the Scholl reaction between the naphthalene rings also occurred, which reduced the crosslinking extent. The low crosslinking extent was unbeneficial to the stability. The slice structure changed to a microtube structure when the FDA amount increased to 0.01 mol (Fig. 1b). The crosslinking extent increased with the FDA amount, which induced the self-assembly of the polymer. The microtube structure would benefit the mass transfer efficiency. Thermally stable microsphere structures formed when the FDA amount further increased to 0.015 mol (Fig. 1c), which indicated that a high crosslinking extent benefited the thermal stability. The sphere size was almost even with 0.015 mol FDA. The microsphere sizes increased with the amount of FDA and the microsphere structures became uneven when the FDA amount reached 0.020 mol (Fig. 1d). Further crosslinking reactions caused the connection of the small spheres, which resulted in a sphere size increase (Fig. 1e). Since the microtube structure could benefit the mass transfer efficiency in the reaction, POP-0.01 with the microtube structure was chosen for further study.
 | ||
| Fig. 1 SEM images of POPs with different amounts of FDA. (a) POP-0.005, (b) POP-0.01, (c) POP-0.015, (d) POP-0.02, (e) POP-0.025. | ||
Fig. 2 shows the IR spectra of (a) naphthalene, (b) POP-0.01, (c) POP-0.01–SO3H-10, and (d) POP-0.01–SO3H-30. Comparing naphthalene (Fig. 2a) and POP-0.01 (Fig. 2b), there are some differences that confirm the polymerization. The peaks at 1638 cm−1, 1456 cm−1 and 1375 cm−1 are associated with naphthalene ring vibration, with no obvious differences between naphthalene and the polymer, which confirmed the naphthalene ring skeleton in the polymers. The 2933 cm−1 peak corresponded to alkane C–H stretching vibration, but the peak was very weak. The new peak at 1030 cm−1 for POP-0.01–SO3H-10 (Fig. 2c) and POP-0.01–SO3H-30 (Fig. 2d) corresponded to S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, confirming the successful introduction of sulfonic acid groups. Comparing the four IR spectra, the peak at 3420 cm−1 strengthened, and furthermore a blue shift occurred for POP-0.01–SO3H-30 (Fig. 2d). For POP-0.01–SO3H-10 (Fig. 2c) and POP-0.01–SO3H-30 (Fig. 2d), both polymers were grafted with sulfonic acid groups; however, the acid density of POP-0.01–SO3H-30 was as high as 3.71 mmol g−1, while that of POP-0.01–SO3H-10 was only 1.83 mmol g−1.
O stretching vibration, confirming the successful introduction of sulfonic acid groups. Comparing the four IR spectra, the peak at 3420 cm−1 strengthened, and furthermore a blue shift occurred for POP-0.01–SO3H-30 (Fig. 2d). For POP-0.01–SO3H-10 (Fig. 2c) and POP-0.01–SO3H-30 (Fig. 2d), both polymers were grafted with sulfonic acid groups; however, the acid density of POP-0.01–SO3H-30 was as high as 3.71 mmol g−1, while that of POP-0.01–SO3H-10 was only 1.83 mmol g−1.
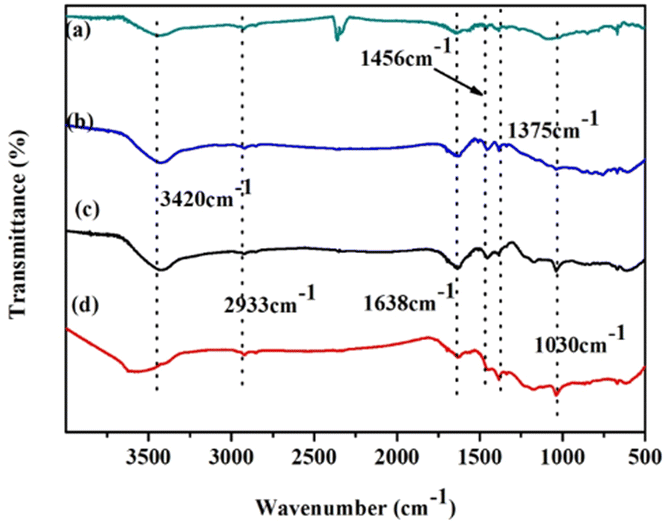 | ||
| Fig. 2 FT-IR spectra of monomer, polymer and catalysts. (a) Naphthalene, (b) POP-0.01, (c) POP-0.01–SO3H-10 and (d) POP-0.01–SO3H-30. | ||
The SEM images confirmed the differences between the solid acids (Fig. 3). POP-0.01–SO3H-10 (Fig. 3a) showed very long microtube structures. The microtube structure was well-retained when using a small amount of sulfuric acid and the long tube even became curved. For POP-0.01–SO3H-30 (Fig. 3b), using more sulfuric acid gave short microtube structures, which indicated that the microtube structure was corroded in a large amount of sulfuric acid. However, the short-microtube structure could make full use of the solid acid’s internal surface and allow reactants to access the active sites more easily. POP-0.015–SO3H-30 (Fig. 3c) retained the stable microsphere structure well and the microspheres gathered together during the sulfonation process.
 | ||
| Fig. 3 SEM images of the solid acids. (a) POP-0.01–SO3H-10, (b) POP-0.01–SO3H-30, (c) POP-0.015–SO3H-30. | ||
The thermal stability of the polymer and solid acids was studied through thermogravimetric (TG) analysis (Fig. 4). Fig. 4 shows the thermogravimetry of POP-0.01 (a), POP-0.01–SO3H-10 (b), POP-0.01–SO3H-30 (c) and POP-0.015–SO3H-30 (d). There were obvious weight losses at about 100 °C, which was due to the desorption of physically adsorbed water. The amount of physically adsorbed water in POP-0.01 was quite low compared to those in the sulfonated polymers. For POP-0.01, the weight even increased a little between 200–350 °C, which might be caused by the oxidation of the polymer. The weight loss above 350 °C was due to the destruction of the polymer framework. For POP-0.015–SO3H-30, the weight loss below 100 °C was almost 20%, which indicated that the solid acid had high polarity and adsorbed more water. POP-0.015–SO3H-30 showed the microsphere structure, which caused the sulfonic acid groups to attach to the external surface. As a result, the solid acid showed a certain hydrophilicity and easily adsorbed water. POP-0.01–SO3H-30 showed the least weight loss before 100 °C, which indicated that the solid acid with the microtube structure was more hydrophobic. For POP-0.01–SO3H-10, the solid acid with the long microtube structure also showed a relatively high weight loss before 100 °C, which indicated that the sulfonic acid groups were attached onto the external surface of the long microtubes. As a result, the solid acid showed relatively high weight loss since the polar surface adsorbed more water.
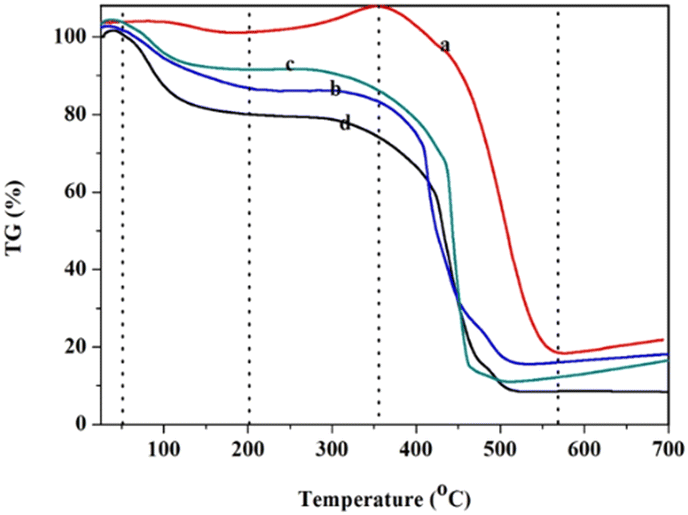 | ||
| Fig. 4 The thermogravimetry of the polymer and solid acids. (a) POP-0.01, (b) POP-0.01–SO3H-10, (c) POP-0.01–SO3H-30, (d) POP-0.015–SO3H-30. | ||
The X-ray photoelectron spectroscopy (XPS) analysis of POP-0.01–SO3H-30 showed a single S 2p peak at 169 eV, which indicated that the S element existed in the polymer in the single form of sulfonic acid groups (Fig. 5).21 The results agreed well with the S elemental analysis and the acid density.
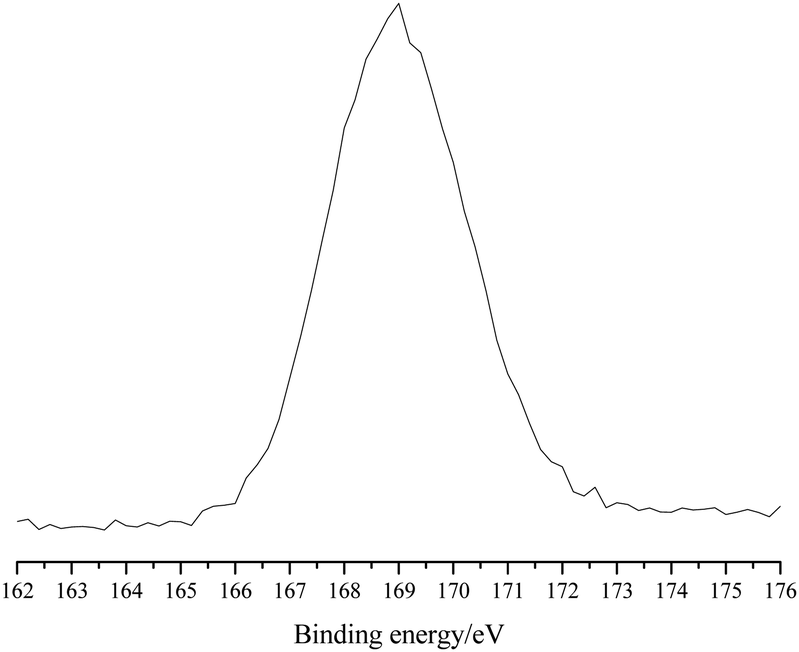 | ||
| Fig. 5 The S 2p XPS of POP-0.01–SO3H-30. | ||
The temperature programmed desorption of chemisorbed ammonia (NH3-TPD) was carried out for POP-0.01–SO3H-30 (Fig. 6). The result showed that the solid acid had single strong acid sites with the ammonia desorption occurring between 500–700 °C, which was assigned to sulfonic acid groups.
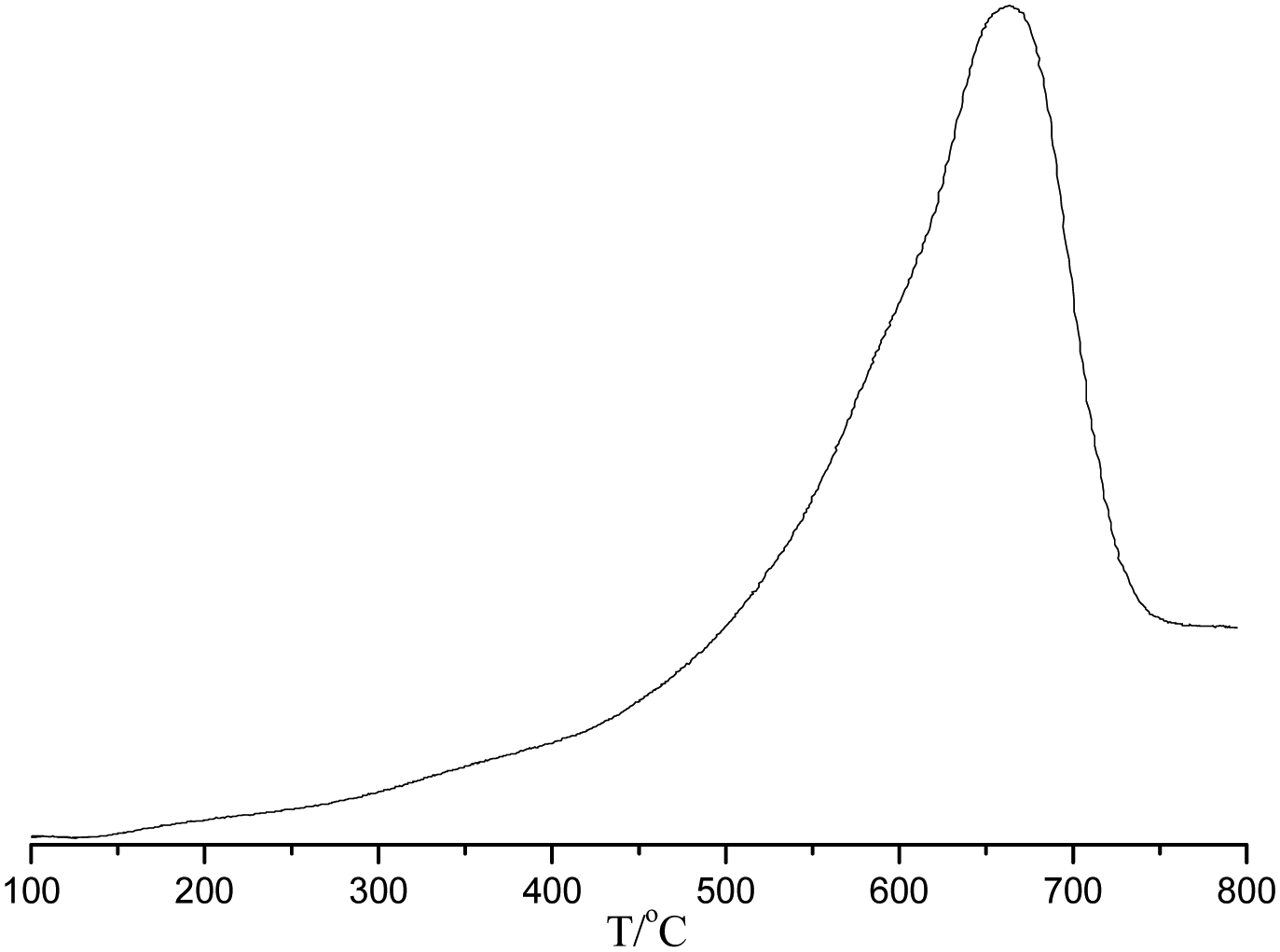 | ||
| Fig. 6 The NH3-TPD of POP-0.01–SO3H-30. | ||
The surface areas of the different polymers were investigated (Fig. 7). POP-0.01 showed a typical type-IV isotherm, which indicated that the polymer had a mesoporous structure. However, the curve was not closed, which indicated that the polymer had various pore sizes, from micropores to macropores. The pore-size distribution showed pore sizes ranging from 5 to 100 nm with an average size of 16 nm. POP-0.01 showed a high surface area of 562.74 m2 g−1. After sulfonation, the surface area of the solid acids decreased greatly. POP-0.01–SO3H-30 showed a surface area of 128.96 m2 g−1, which indicated that the sulfonic acid groups blocked some of the pores and reduced the surface area. POP-0.01–SO3H-10, with the long microtube structure, showed a smaller surface area of 84.79 m2 g−1, which indicated that the microtube structure was too long, rendering some of the inner surface useless.
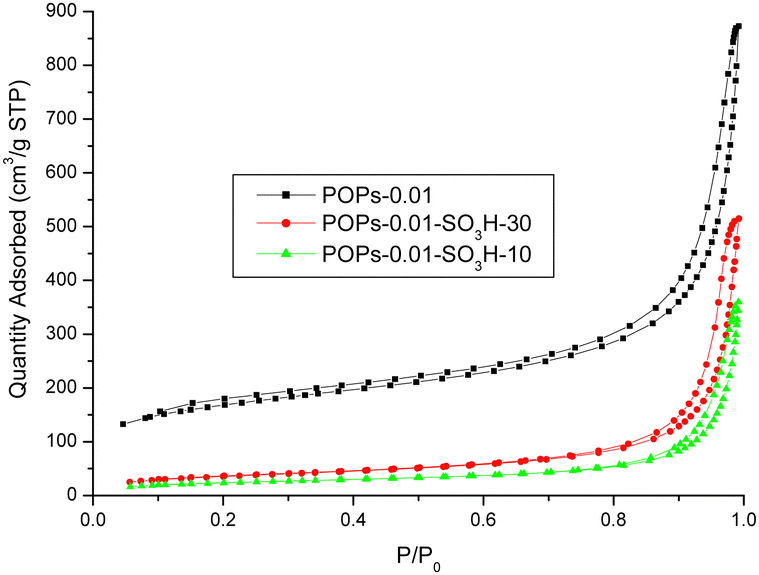 | ||
| Fig. 7 Nitrogen adsorption–desorption isotherms. | ||
The surface areas, pore sizes and pore volumes are summarized in Table 3. The surface area and pore volume decreased after sulfonation, which indicated that the sulfonic acid groups blocked the pores and reduced the pore volume. However, the sulfonation process would also destroy the polymer structure and cause the pore size to increase. The large pore size benefits the mass transfer efficiency of bulky molecules.
| Sample | S BET (m2 g−1) | d p (nm) | V p (cm3 g−1) |
|---|---|---|---|
| POP-0.01 | 562.74 | 12 | 1.24 |
| POP-0.01–SO3H-10-30 | 128.96 | 27 | 0.75 |
| POP-0.01–SO3H-10-10 | 84.79 | 24 | 0.56 |
3.2 The catalytic activities for tert-butylation of resorcinol
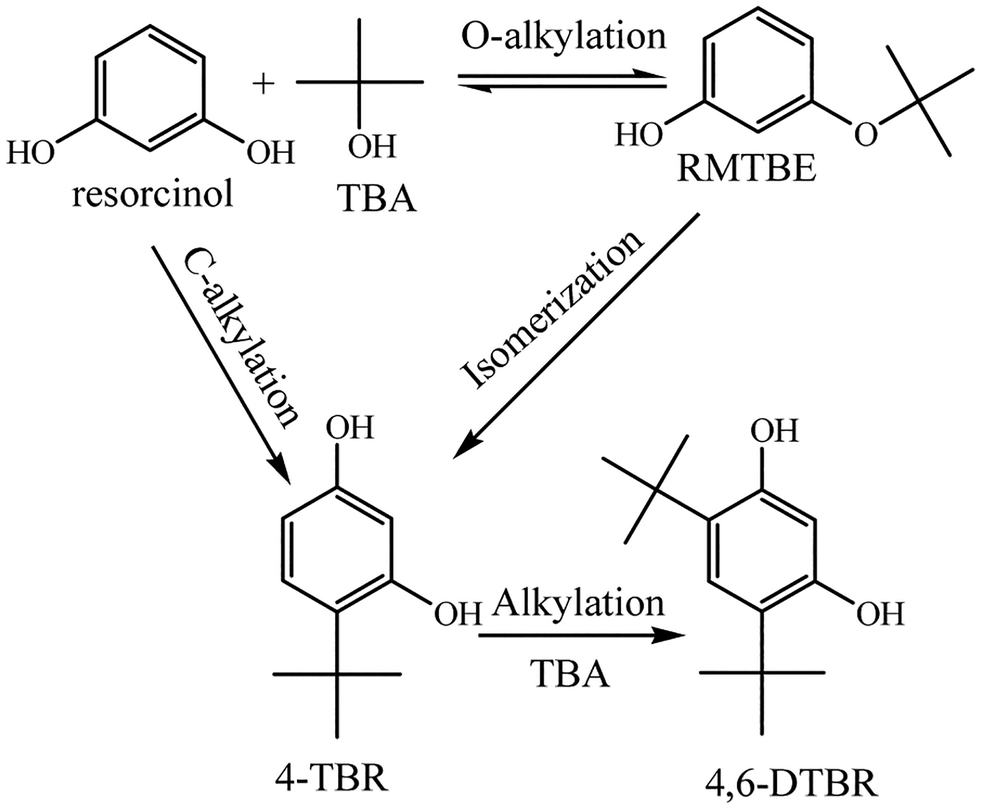
The solid acids were applied to catalyze the tert-butylation of resorcinol (Table 4). The results were analyzed by GC. The products obtained were 4-t-butylresorcinol (4-TBR) and 4,6-di-t-butylresorcinol (4,6-DTBR) (Scheme 2). 4,6-DTBR was the thermodynamically favoured product while 4-TBR was the target product with performance as an antioxidant.23| Catalysts | Acid density (mmol g−1) | Conversion (%) | Selectivity (%) |
|---|---|---|---|
| a Reaction conditions: 2.2 g resorcinol, 4.8 g t-butanol, 0.05 g catalyst, 120 °C, 4 h. | |||
| POP-0.01–SO3H-10 | 1.83 | 93.4 | 72.0 |
| POP-0.01–SO3H-30 | 3.71 | 97.8 | 85.4 |
| POP-0.015–SO3H-30 | 3.52 | 93.1 | 67.5 |
 | ||
| Scheme 2 The reaction process of t-butylation of resorcinol. | ||
The novel polymer-based solid acids showed high activities for the t-butylation of resorcinol with high conversions of over 90% (Table 4). Polymers with the microtube structure showed higher activity for the reaction. POP-0.01–SO3H-10 showed comparable conversion to POP-0.015–SO3H-30. Although POP-0.015–SO3H-30 had a high acid density, the solid acid showed high affinity to the byproduct water, which was harmful for the reversible t-butylation reaction. The solid acids with the microtube structure showed higher hydrophobicity, which favours the butylresorcinol products.23 As a result, the conversion was higher using POP-0.01–SO3H-10 and POP-0.01–SO3H-30. POP-0.01–SO3H-10 had long microtube structures, which rendered some of the inner surface useless during the catalytic process. Furthermore, the lower acidity offered fewer active sites for the reaction. POP-0.01–SO3H-30 had high acid density, which provided enough active sites for the reaction. The short-microtube structure could allow full use of the inner surface, which benefited the mass transfer efficiency. Furthermore, the microtube structure could provide shape selectivity for the product, with the bulky di-substituted products being harder to form due to steric hindrance. POP-0.01–SO3H-30, with high conversion and selectivity, was chosen for further investigation.
The effect of the reaction temperature was examined (Table 5). The results showed that the temperature was very important for the reaction. The reaction occurred at low temperature (80 °C) with low conversion, which indicated that the reaction could not be efficiently activated. The conversion increased with the temperature and reached the peak value at 120 °C. On further increasing the temperature, the selectivity greatly decreased, which was caused by further t-butylation. 4,6-DTBR is the thermodynamically favoured product and the high temperature benefited the thermally stable product.24 Therefore, 120 °C was chosen as the optimal reaction temperature.
| Temperature/°C | 80 | 100 | 120 | 140 |
|---|---|---|---|---|
| a Reaction conditions: 2.2 g resorcinol, 4.8 g t-butanol, 0.05 g catalyst, 4 h. | ||||
| Conversion/% | 84.3 | 94.3 | 97.8 | 98.9 |
| Selectivity/% | 87.7 | 86.3 | 85.5 | 82.2 |
The effect of catalyst amount on the reaction was also investigated (Table 6). The catalyst showed very high activity for the reaction and the reaction conversion reached 89.5% with only 0.03 g catalyst. The conversion increased while the selectivity decreased with the catalyst amount. The number of active sites increased with the catalyst amount, which meant that more reactions could be carried out simultaneously. The selectivity for the di-butylated product was low with a small amount of catalyst, which indicated that the butylation reaction proceeded step-by-step.23 For the reaction with a small amount of catalyst, the 4-t-butylresorcinol (4-TBR) formed as the main product. Then, the double-substituted product increased when the presence of more active sites accelerated the reaction. 4-TBR was further butylated to form the di-butylated product. The conversion changed little after the catalyst amount reached 0.05 g. The conversion and selectivity even decreased with 0.09 g catalyst, which was caused by the further butylation of 4-TBR. The reaction was optimal with 0.05 g catalyst.
| Catalyst amount/g | 0.01 | 0.03 | 0.05 | 0.07 | 0.09 |
|---|---|---|---|---|---|
| a Reaction conditions: 2.2 g resorcinol, 4.8 g t-butanol, 120 °C. | |||||
| Conversion/% | 71.2 | 89.5 | 97.8 | 97.9 | 97.4 |
| Selectivity/% | 86.3 | 85.8 | 85.5 | 85.3 | 84.1 |
The reaction process was investigated by varying the reaction time (Table 7). The results showed the high activity of the catalyst with a conversion of over 60% after only 1 h. The conversion increased quickly with time. The mono- and di-substitution reactions occurred simultaneously, with both products present at the beginning of the reaction. The selectivity was 89.3% at 1 h, while the conversion was 61.2%. There were still many raw materials in the reaction mixture and the double substituted product (4,6-DTBR) was also formed, which indicated that the double-substitution reaction occurred as a competitive reaction. 4,6-DTBR was the further t-butylation product of 4-TBR. Compared to resorcinol, 4-TBR with its electron-donating group (tert-butyl group) was more reactive.24 As a result, the di-substitution reaction occurred simultaneously with mono-butylation. The t-butylation reached equilibrium after 4 h with the highest conversion and selectivity.
| Reaction time/h | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a Reaction conditions: 2.2 g resorcinol, 4.8 g t-butanol, 0.05 g catalyst, 120 °C. | |||||
| Conversion/% | 61.2 | 80.4 | 94.8 | 97.8 | 97.4 |
| Selectivity/% | 89.3 | 87.5 | 86.5 | 85.5 | 84.1 |
The catalytic activity of the solid acid after recycling was also investigated (Table 8). The results showed that the novel solid acid had high stability with high activity after being recycled 5 times. The catalyst had high thermal stability, which ensured high activity after recycling when used at high temperature (120 °C). The activity after recycling decreased a little, which might be caused by the active sites being covered by adsorbed bulky products and some pores being blocked. Inductively coupled plasma mass spectrometry (ICP-MS) analysis of the filtrate showed no S element, which confirmed that little acid-site leaching occurred.40 Also, the activity after recycling could be refreshed by treating the recycled solid acid in refluxing ethanol for 4 h.
| Times recycled | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a Reaction conditions: 2.2 g resorcinol, 4.8 g t-butanol, 0.05 g catalyst, 120 °C, 4 h. | |||||
| Conversion/% | 97.5 | 97.2 | 96.5 | 96.2 | 95.3 |
| Selectivity/% | 84.4 | 85.1 | 84.3 | 84.1 | 84.2 |
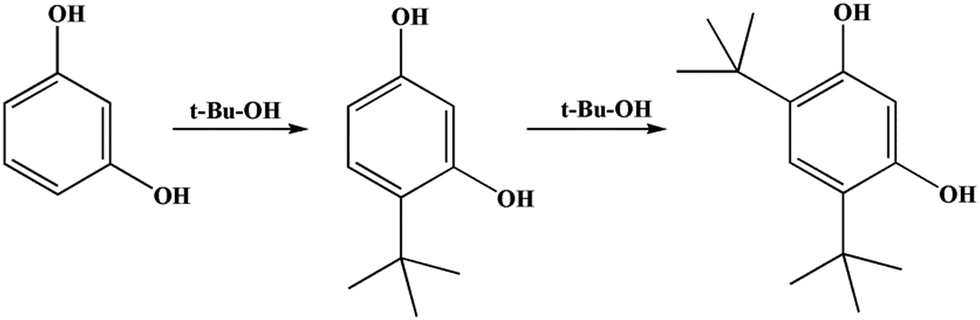
A possible reaction mechanism for t-butylation of resorcinol is shown in Scheme 3.41 The reaction inside the microtube might proceed through a direct C-alkylation pathway. TBA within the microtube underwent dehydration over the acid sites (to form an active carbocation), then the active carbocation attacked resorcinol to form 4-TBR. The reaction on the external surface may proceed through isomerization of resorcinol-mono-tert-butanol ether (RMTBE). RMTBE was formed over the external surface of the catalyst through O-alkylation of resorcinol, then isomerization of RMTBE occurred to form 4-TBR.
 | ||
| Scheme 3 The possible reaction mechanism for t-butylation. | ||
3.3 The catalytic activities for biodiesel synthesis from waste oil
The novel porous polymer-based solid acids were applied to biodiesel synthesis from waste oil (Table 9). The results showed that POP-0.01–SO3H-30 with the short-microtube structure had the highest activity for the reaction. The solid acids with the microtube structure had a hydrophobic surface, which could efficiently separate the byproduct water from the active sites.16 As a result, POP-0.01–SO3H-10 showed a higher activity than POP-0.015–SO3H-30, even with a lower acid density. POP-0.015–SO3H-30 had the microsphere structure and easily adsorbed the byproduct water, which benefited the reverse hydrolysis reactions. Therefore, the yield was much lower, even with a higher acidic density. For POP-0.01–SO3H-30, the short-microtube structure could make full use of the surface and the higher acidity could provide more active sites for the reaction. Both the FFA conversion and total yield were high when using POP-0.01–SO3H-30; therefore, POP-0.01–SO3H-30 was chosen for further investigation.| Catalysts | FFA conversion (%) | Yield (%) |
|---|---|---|
| a Reaction conditions: waste oil 5 g, methanol 2.94 g, catalyst amount 0.05 g, 70 °C, 7 h. b The yield was based on FFAs and triglycerides and calculated via GC using an internal standard. | ||
| POP-0.01–SO3H-10 | 96.4 | 97.4 |
| POP-0.01–SO3H-30 | 98.8 | 99.2 |
| POP-0.015–SO3H-30 | 93.4 | 94.3 |
The reaction progress was examined over time (Fig. 8). POP-0.01–SO3H-30 showed very high activity for the reaction with a yield of 99.2% after only 7 h. Both the FFAs and triglycerides were transformed into biodiesel products. POP-0.01–SO3H-30 had high acid density and a short-microtube structure, which benefited the interaction between the active sites and reactants. Furthermore, the acidic sites on the short-microtube surface were quite suitable for the bulky triglyceride molecules. Both esterification and transesterification are reversible reactions. The biodiesel concentration increases with the reaction, which is unbeneficial for the reaction. Therefore, the reactions using waste oils are often carried out at high temperature (220 °C) to promote the endothermic esterification and transesterification.8–11 POP-0.01–SO3H-30 with the short-microtube structure showed high compatibility with the hydrophobic products, which were distributed in the upper biodiesel layer after the products were left to settle for a certain time. The yield was only 30.0% after 1 h, while the FFA conversion reached 45%. The intermediate products, diglycerides and monoglycerides, were formed.16 These intermediates have both polar hydroxyl groups and hydrophobic fatty chains, which act as a compatibilizer for oil and methanol. The reaction rate then increased.17 POP-0.01–SO3H-30 had a high hydrophobic surface area and the special microtube structure, which ensured high activities. Polar byproducts such as glycerol and water tended to be released from the hydrophobic surface, which led to the reverse reaction being avoided. The peak yield was reached after 7 h.
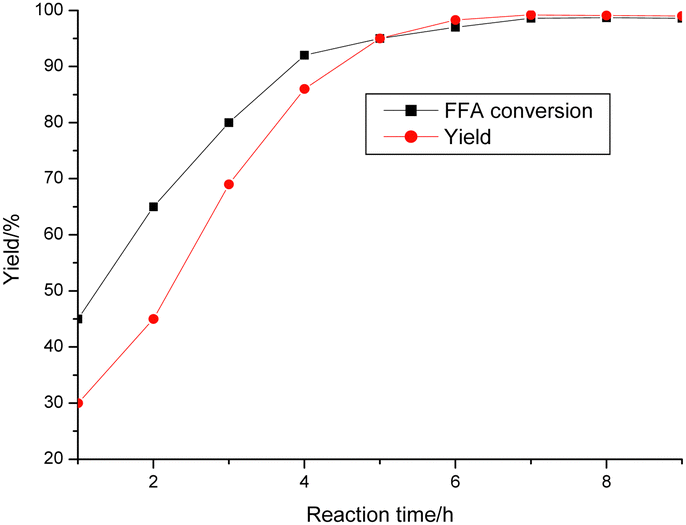 | ||
| Fig. 8 The catalytic activity of the porous microtube polymer-based solid acid for biodiesel synthesis. Reaction conditions: waste oil 5 g, methanol 2.94 g, catalyst amount 0.05 g, 70 °C. b. The yield was based on FFAs and triglycerides and calculated via GC using an internal standard. | ||
The effect of catalyst amount was investigated (Table 10). POP-0.01–SO3H-30 provided catalytic sites for the reaction, which was very important for both FFA and triglyceride conversion. Compared to the large amount of catalyst used for traditional solid acids (10%),9,10 the amount of the novel catalyst was quite low (1%). A 95.1% yield was obtained using only 0.01 g catalyst. Both FFAs and triglycerides were transformed to biodiesel products with high yields. The yield increased with the catalyst amount. The highest yield of 99.2% was reached with 0.05 g catalyst. The yield even decreased slightly with more catalyst. The acid catalyst could catalyze both the forward and reverse reactions.11 Too much catalyst could also accelerate the reverse reaction, which would shift the reaction equilibrium backward.15 Therefore, 0.05 g catalyst was chosen as the optimal amount.
The methanol amount was also investigated here (Table 11). The yield increased with the methanol amount as the interactions between the reactants increased. The conversion of FFAs was high with even a low methanol amount, because FFAs have low steric hindrance and are present in an equimolar ratio to methanol.6 The yield reached the highest value of 99.2% with 2.94 g methanol. The methanol amount was high compared to reactions using pure triglycerides or FFAs as raw materials.11 Here, the biodiesel concentration was higher, which was unbeneficial for the reversible reaction. As a result, more methanol was needed to promote the reaction in the forward direction. Therefore, the optimal methanol amount was 2.94 g.
The effect of reaction temperature was also investigated (Table 12). Using waste oils, a high temperature (>220 °C) was needed to promote the reversible reaction in the forward direction.9 The temperature was also very important for POP-0.01–SO3H-30. The yield was quite low at 25 °C, which indicated that the reaction could not be activated. The reaction also hardly occurred at 50 °C for the endothermic reaction.10 The reaction was activated quickly at 70 °C and the yield reached 99.2%. Restricted by the low boiling point of methanol, the reaction at 70 °C would ensure mild conditions (1 atm). Here, reactions at high temperature (up to 220 °C) were also carried out in an autoclave. A high yield of 99.3% was obtained within 3 h, which indicated that POP-0.01–SO3H-30 showed much higher activity than traditional solid acids (98%, 8 h).9,11 Also, POP-0.01–SO3H-30 gave a higher activity at a much lower reaction temperature (140 °C). POP-0.01–SO3H-30 had high thermal stability, which also makes it quite suitable for reactions at higher temperatures.
| T/°C | 25 | 50 | 70 | 140c | 220d |
|---|---|---|---|---|---|
| a The reaction conditions: waste oil 5 g, methanol 2.94 g, catalyst 0.05 g, 7 h. b FFA conversion was based on acidity and the yield was based on FFAs and triglycerides. c The reaction was carried out in an autoclave for 4 h. d The reaction was carried out in an autoclave for 3 h. | |||||
| FFA conversion/% | 12.4 | 35.7 | 98.6 | 99.0 | 99.1 |
| Total yield/% | 21.3 | 46.1 | 99.2 | 99.2 | 99.3 |
The activities of POP-0.01–SO3H-30 after recycling were examined carefully (Fig. 9). The catalyst was recycled via filtration and washed with ethyl acetate. The catalyst was reused after drying. POP-0.01–SO3H-30 had high stability and the activity after recycling changed little after reusing it eight times. The reaction filtrate was analyzed by ICP and no sulfur element was detected via element analysis, which confirmed little leaching of acid sites.40 The acid–base titration of the recycled POP-0.01–SO3H-30 gave an acid density of 3.56 mmol g−1, which was almost the same as that of the fresh catalyst. These results further confirmed the high stability of POP-0.01–SO3H-30. The novel solid acid had the special short-microtube structure and a high BET surface area, which benefited the mass transfer efficiency. Moreover, the hydrophobic surface separated the polar byproducts water and glycerol from the active sites, which led to the reverse reaction being avoided. The reaction was carried out under mild conditions, which also avoided the side reactions and ensured the high stability of POP-0.01–SO3H-30.
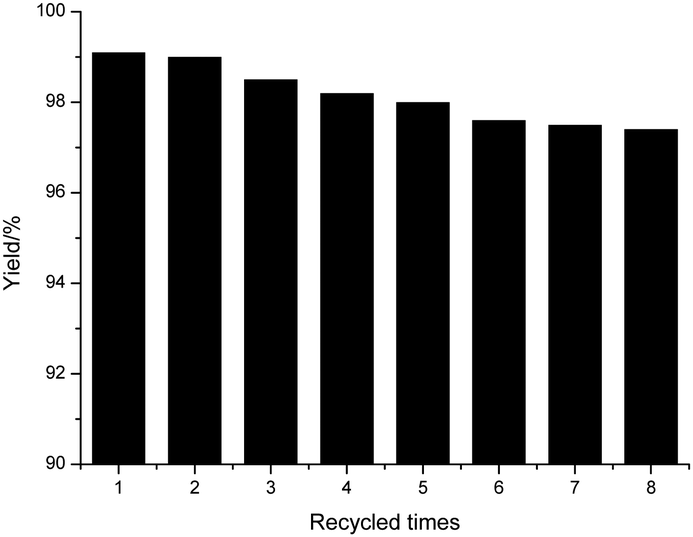 | ||
| Fig. 9 The reuse of POP-0.01–SO3H-30 for biodiesel sythesis. | ||
The activities of different catalysts were compared (Table 13). The catalyst amount and reaction time were optimized for each catalyst. For a Lewis-acidic catalyst ([Et3NH]Cl-AlCl3),42 the yield was quite low. The byproduct water from the esterification reaction dissociated the Lewis-acidic IL catalyst. The activity of H2SO4 was not very high, either. H2SO4 dissociated H+ ions in the reaction mixture, which caused the hydrolysis of the biodiesel and reduced the yield. A traditional carbon-based solid acid43 with a low surface area (2 m2 g−1) showed low activity (79.6%) at 70 °C. For a porous carbon-based solid acid with a higher surface area (563 m2 g−1), the yield increased to 91.2%. POP-0.01–SO3H-30, with the special short-microtube structure, had the highest activity (99.2%). Furthermore, the short-microtube surface offered hydrophobicity. The byproducts water and glycerol were effectively separated from the active sites, which led to the hydrolysis reaction being avoided. Therefore, the novel porous microtube polymer-based solid acid had higher activity with a smaller catalyst amount and shorter reaction time.
| Catalyst | Catalyst amount/g | Reaction time/h | FFA conversion/% | Yield/% |
|---|---|---|---|---|
| a The reaction conditions: waste oil 5 g, methanol 2.94 g, 70 °C. b FFA conversion was based on acidity and the yield was based on FFAs and triglycerides. | ||||
| POP-0.01–SO3H-30 | 0.05 | 7 | 98.6 | 99.2 |
| Carbon-based solid acid42 | 0.10 | 12 | 84.1 | 79.6 |
| Porous carbon-based solid acid43 | 0.07 | 12 | 86.3 | 91.2 |
| [Et3NH]Cl-AlCl3 | 0.06 | 23 | 85.7 | 79.3 |
| H2SO4 | 0.07 | 18 | 88.6 | 89.8 |
3.4 The quality of the refined biodiesel
After the reaction, the upper layer of biodiesel was refined by distillation under vacuum (130 °C, 0.01 torr). The quality of the refined biodiesel was examined carefully (Table 14). The refined biodiesel was high quality, similar to the value of biodiesel from fresh rapeseed oil.44–47 The biodiesel showed similar a fatty-acid composition to the raw waste oil, which indicated that almost all the raw oils were converted to biodiesel using POP-0.01–SO3H-30.| Entry | Value |
|---|---|
| Density (15 °C) (g cm−3) | 0.88 |
| Viscosity (40 °C) (mm2 s−1) | 4.2 |
| Sulfur content (%) | 1.3 mg kg−1 |
| Water content (mg kg−1) | 128 |
| Methanol content (%) | 0.02 |
| Biodiesel content (%) | >99.3 |
| Glycerol content (%) | 0.01 |
4. Conclusions
A novel porous microtube polymer-based solid acid was synthesized via the polymerisation of naphthalene and sulfonation. The novel solid acid had a high surface area and special short-microtube structure, which benefited the interaction of active sites and large hydrophobic reactants. The solid acid showed very high activities for tert-butylation of resorcinol using 2.2 g resorcinol, 4.8 g t-butanol and 0.05 g catalyst at 120 °C for 4 h, with a conversion of 97.8% and a selectivity of 85.5%. For biodiesel synthesis from waste oil, the novel solid acid gave 99.2% yield using 5 g waste oil, 2.94 g methanol and 0.05 g catalyst at 70 °C for 7 h. Besides the high activities, the solid acid had high stability and could be recycled five times for the tert-butylation of resorcinol and eight times for biodiesel synthesis from waste oils.Author contributions
Both authors contributed to the study. Xuezheng Liang contributed to the conception and design of the experiment. Material preparation, data collection and analysis was performed by Zhijin Guo. The first draft of the manuscript was written by both authors, and both authors commented on the different versions of the manuscript. Both authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper did not receive any funding.Notes and references
- R. Mohammadinejad, S. Karimi, S. Iravani and R. S. Varma, Green Chem., 2016, 18, 20 RSC.
- Y. Shi, M.-W. Kevin and X. Liang, J. Environ. Chem. Eng., 2018, 6, 6633 CrossRef CAS.
- L. Ranganatha Swamy, N. R. Banapurmath, T. K. Chandrashekar, M. E. M. Soudagar, M. Gul, N. N. Nik-Ghazali, M. A. Mujtaba, K. Shahapurkar, Ü. Ağbulut, H. M. Alshehri, A. M. Sajjan and M. Goodarzi, Chem. Eng. Commun., 2023, 210, 1060 CrossRef CAS.
- P. Mukhopadhyay and R. Chakraborty, Sustain. Energy Technnol., 2021, 44, 100994 Search PubMed.
- H. Khan, M. E. M. Soudagar, R. H. Kumar, S. M. Rafaei, M. Farooq, A. Khidmatgar, N. R. Banapurmath, R. A. Farade, M. M. Abbas, A. Afzal, W. Ahmed, M. Goodarzi and S. N. Taqui, Symmetry, 2020, 12, 961 CrossRef CAS.
- R. S. Gavhane, A. M. Kate, A. Pawar, M. R. Safaei, M. E. M. Soudagar, M. Mujtaba Abbas, H. Muhammad Ali, N. R. Banapurmath, M. Goodarzi, I. A. Badruddin, W. Ahmed and K. Shahapurkar, Symmetry, 2020, 12, 1042 CrossRef CAS.
- M. J. Bouchet, A. Rendon, C. G. Wermuth, M. Goeldner and C. Hirth, J. Med. Chem., 1987, 30, 2222 CrossRef CAS PubMed.
- M. K. Lam, K. T. Lee and A. R. Mohamed, Appl. Catal., B, 2009, 93, 134 CrossRef CAS.
- Q. Shu, J. Gao, Z. Nawaz, Y. Liao, D. Wang and J. Wang, Appl. Energy, 2010, 87, 2589 CrossRef CAS.
- J. A. Melero, L. F. Bautista, J. Iglesias, G. Morales and R. Sánchez-Vázquez, Catal. Today, 2012, 195, 44–53 CrossRef CAS.
- M. A. Olutoye and B. H. Hameed, Appl. Catal., A, 2013, 450, 57 CrossRef CAS.
- P. Pradhan and R. Chakraborty, Energy, 2018, 164, 35 CrossRef CAS.
- P. Pradhan, P. Karan and R. Chakraborty, Environ. Sci. Pollut. R, 2022, 29, 20064 CrossRef CAS PubMed.
- M. Hassani, G. D. Najafpour, M. Mohammadi and M. Rabiee, J. Sci. Ind. Res., 2014, 73, 129 CAS.
- X. Liang, Energy, 2013, 63, 103 CrossRef CAS.
- X. Liang, H. Xiao and C. Qi, Fuel Process. Technol., 2013, 110, 109 CrossRef CAS.
- X. Liang, Ind. Eng. Chem. Res., 2013, 52, 6894 CrossRef CAS.
- W. Chen, Z. Wu, Z. Wang, C. Chen and Z. Zhang, RSC Adv., 2022, 12, 12363 RSC.
- M. Arindam, G. Anindya, R. M. Akshay, P. Ashish, S. Manickam, K. P. Kamal, C. Biswajit and B. Asim, ACS Sustainable Chem. Eng., 2021, 9, 12431 CrossRef.
- M. Arindam, R. M. Akshay, R. R. Sonde and K. P. Kamal, Renewable Energy, 2023, 212, 97 CrossRef.
- X. Kan, Z. Liu, F. Liu, F. Li, W. Chen, X. Yi, A. Zheng, L. Jiang and F. S. Xiao, Chem. Eng. J., 2023, 451, 139085 CrossRef CAS.
- J. Mi, W. Peng, Y. Luo, W. Chen, L. Lin, C. Chen, Q. Zhu, F. Liu, A. Zheng and L. Jiang, Macromolecules, 2021, 54, 7010 CrossRef CAS.
- V. S. Marakatti and E. M. Gaigneaux, Catal. Commun., 2021, 152, 106291 CrossRef CAS.
- C. Pezzotta, G. Fleury, M. Soetens, S. Van der Perre, J. F. M. Denayer, O. Riant and E. M. Gaigneaux, J. Catal., 2018, 359, 198 CrossRef CAS.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703 CrossRef CAS PubMed.
- Y. Zhang, S. Wei, H. Zhang, S. Liu, F. Nawaz and F. S. Xiao, J. Colloid Interface Sci., 2009, 339, 434 CrossRef CAS PubMed.
- M. Kitano, K. Arai, A. Kodama, T. Kousaka, K. Nakajima, S. Hayashi and M. Hara, Catal. Lett., 2009, 131, 242 CrossRef CAS.
- C. D. Koolen, W. Luo and A. Züttel, ACS Catal., 2023, 13, 948 CrossRef CAS.
- J. Pang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935 RSC.
- L. Tan and B. Tan, Chem. Eng. J., 2020, 390, 124485 CrossRef CAS.
- H. Ouyang, K. Song, J. Du, Z. Zhan and B. Tan, Chem. Eng. J., 2022, 431, 134326 CrossRef CAS.
- Y. Qiao, Z. Zhan, Y. Yang, M. Liu, Q. Huang, B. Tan, X. Ke and C. Wu, Mater. Today Commun., 2021, 27, 102338 CrossRef CAS.
- L. Tan and B. Tan, Polym. Chem., 2021, 12, 2689 RSC.
- R. Ma, L. Wang, H. Wang, Z. Liu, M. Xing, L. Zhu, X. Meng and F.-S. Xiao, Appl. Catal., B, 2019, 244, 594 CrossRef CAS.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790 CrossRef CAS PubMed.
- Q. Sun, K. Hu, K. Leng, X. Yi, B. Aguila, Y. Sun, A. Zheng, X. Meng, S. Ma and F.-S. Xiao, J. Mater. Chem. A, 2018, 6, 18712 RSC.
- R. Chakraborty and S. K. Das, Ind. Eng. Chem. Res., 2012, 51, 8404 CrossRef CAS.
- R. Chakraborty, S. Das, P. Pradhan and P. Mukhopadhyay, Ind. Eng. Chem. Res., 2014, 53, 19681 CrossRef CAS.
- R. Alcantara, J. Amores, L. Canoira, E. Fidalgo, M. J. Franco and A. Navarro, Biomass Bioenergy, 2000, 18, 515 CrossRef CAS.
- P. Mukhopadhyay and R. Chakraborty, Catal. Commun., 2017, 94, 73 CrossRef CAS.
- V. S. Marakatti and E. M. Gaigneaux, Catal. Commun., 2021, 152, 106291 CrossRef CAS.
- X. Liang, G. Gong, H. Wu and J. Yang, Fuel, 2009, 88, 613 CrossRef CAS.
- M. Okamura, A. Takagaki, M. Toda, J. N. Kondo, K. Domen, M. Hara and S. Hayashi, Chem. Mater., 2006, 18, 3039 CrossRef CAS.
- K. Nakajima, M. Okamura, J. N. Kondo, K. Domen, T. Tatsumi, S. Hayashi and M. Hara, Chem. Mater., 2009, 21, 186 CrossRef CAS.
- M. Maitra, S. Chatterjee, P. Mukhopadhyay and R. Chakraborty, Indian Chem. Eng., 2021, 63, 152 CrossRef CAS.
- R. Chakraborty, S. Das and S. K. Bhattacharjee, Clean. Technol. Environ. Policy, 2015, 17, 455 CrossRef CAS.
- R. Chakraborty and A. Banerjee, Waste Biomass Valor, 2010, 1, 201 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |