
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the lithiation mechanism of Li2O-doped spinel high-entropy oxides as anode materials for Li-ion batteries
Guozhe
Ma
,
Yu
Zheng
,
Fanbo
Meng
 and
Renzong
Hu
*
and
Renzong
Hu
*
School of Materials Science and Engineering, Guangdong Provincial Key Laboratory of Advanced Energy Storage Mater., South China University of Technology, Guangzhou, 510640, China. E-mail: msrenzonghu@scut.edu.cn
First published on 22nd August 2023
Abstract
High-entropy oxides (HEOs) have started to attract widespread interest as anode materials for lithium-ion batteries (LIBs), because of their high theoretical discharge capacities, fast ionic conductivity, and stable structure caused by the unique entropy stabilization effect and the “cocktail” effect. However, the lithiation mechanism of HEO anode is still conversional, which prevents its further development. Herein, we propose an Li2O-doping strategy for improving the lithium storage ability of HEO anodes. Li2O is successfully introduced into the spinel high-entropy oxide to obtain a (FeMgNiCrMnLi)3O4 anode (Li-SHEO) via a solution combustion synthesis and ball milling method. Experimental results show Li doping would induce the growth of oxygen vacancies and regulate the conversion reactions during the discharge process, leading to improved electrochemical performance. As a result, the lithiation process of an Li-SHEO anode includes an enhanced Li+ ion intercalation process and a typical conversion reaction. Compared with the (FeMgNiCrMnLi)3O4 (SHEO) anode, the Li-SHEO anode shows a high reversible discharge capacity of 850.7 mA h g−1 after 200 cycles under a large current density of 2.0 A g−1.
Introduction
Among all energy storage technologies, LIBs have emerged as the preferred energy storage technology for mobile electronic devices and power systems, due to their high energy density, excellent cycling stability, and no memory effects.1–3 However, the rise of electric vehicles has heightened the demand for LIBs with even greater energy density. To this end, the development of novel battery materials has become a critical research endeavor.4,5 Although commercial LIBs currently utilize graphite as the anode material, the low theoretical capacity of 372 mA h g−1 of a graphite anode cannot satisfy increasing energy and power density demands. Safety issues such as lithium plating and dendrite formation on the graphite surface further constrain the development of LIBs.6,7 The exploration of high-capacity anode materials has thus become a research hotspot.In 2015, inspired by the study of high-entropy alloys (HMAs) and the basic principles of thermodynamics, Rost et al. introduced the concept of high entropy to a multi-component oxide. A synthesized novel metal oxide containing five metallic elements demonstrates the structural stabilization effect driven by entropy, which sparked research into and development of HEOs.8 Similar to the classification of HMAs, materials with a configurational entropy (Sconfig) ≥ 1.5R can be classified as “high-entropy”, and in the case of an oxide system with a near-equimolar composition of five cations, Sconfig can reach 1.61R.9–11 Thus, HEOs can be defined as single-phase oxide solid solutions consisting of five or more different cations occupying the same Wyckoff position in the crystal with similar molar proportions to obtain a high Sconfig for the system.8,12,13 The high Sconfig endows HEOs with excellent physicochemical properties, such as the most typical entropy-induced phase stabilization effect, a sluggish diffusion effect, a lattice distortion effect, and a “cocktail” effect.14–17 These unique properties mean HEOs exhibit great research value in the fields of energy storage, catalysts, and magnetic materials.
Then, Sarkar et al. first introduced the concept of an HEO anode and developed a transition-metal-based HEO (TM-HEO). It is found that the stabilization effect of entropy brought significant benefits to the capacity retention of the HEO, resulting in greatly improved cycling stability and rate capability.18 The five-cation HEO anode material has been shown to have superior cycling stability compared to four-cation “medium-entropy oxides”, as the exclusion of one element from the system reduces the Sconfig from ∼1.61R to ∼1.39R depriving it of the unique properties of HEOs.8,18 Thus, it is the unique high-entropy effect that accounts for the exceptional performance of high-entropy oxide anodes. Inspired by this groundbreaking work, a variety of HEO anode materials, including spinel-type HEO (S-HEO), perovskite-type HEO (PE-HEO), and rock-salt-type HEO (RS-HEO), were developed.18–21 Compared with single transition metal oxide anodes, HEOs have been found to exhibit higher dielectric properties and ionic conductivity, while the entropy-stabilized single-phase crystal structure can be maintained during the cycling process, ensuring good reversible energy storage ability.22–25 For HEO anode materials, choosing appropriate elemental components is critical to achieving better performance. For example, using Co, Ni, Zn, Mn, etc. as active species can provide cycling capacity, while using non-active species such as Mg and Al as buffer materials can stabilize the lattice structure, suppress volume expansion, and ensure good cycling stability.13,26 The performance of an HEO is closely related to the metal cations; therefore, by adjusting the types of cations, it is possible to regulate the crystal structure and properties of the HEO anode, providing a modular approach for the design and performance regulation of anode materials in the field of LIBs.16,18,25
The aggregation of multiple cations in the crystal lattice and the oxygen defects caused by lattice distortion are among the reasons for the improved electrochemical performance of an HEO.27 It is reported that oxygen vacancies can improve electronic and ionic conductivity for anode materials with more additional active sites for the intercalation/deintercalation of Li+ ions. In addition, the charge distribution imbalance resulting from oxygen vacancies can provide additional Coulombic forces, leading to a rapid Li+ ion transfer rate. An aliovalent ion-doped HEO can exhibit interesting properties due to internal charge compensation and defect formation (e.g., oxygen vacancies).26,28–30 Berardan et al. investigated a series of Li+/Na+-substituted (Mg0.2Ni0.2Co0.2Cu0.2Zn0.2)O HEOs and it was found that the Li-doped samples exhibited clear increased conductivities at room temperature.24 Previously, Ersu Lökçü et al. had introduced Li2O into a type of RS-HEO via a solid-state sintering method. As a result, the Li2O-doped HEO anode can deliver a higher capacity and rapid reaction kinetics. The enhanced performance can be attributed to the additional oxygen vacancies and charge distribution imbalance brought about by Li2O addition.26
Herein, a Li-doped (FeMgNiCrMn)3O4 (Li-SHEO) is prepared by an environmentally friendly and efficient solution combustion synthesis method (SCS) and applied as the anode material for LIBs. The Li-SHEO anode exhibits improved rate capability and cyclic properties. In particular, the Li-SHEO anode delivers a high reversible capacity of 850.7 mA h g−1 under a large current density of 2.0 A g−1 after 200 cycles. Characterization techniques show that the introduction of Li2O improves the valence state of Mn in SHEO and induces the formation of oxygen vacancies. During cycling, more Li+ ions can be embedded into the crystal structure of the SHEO anode, and the conversion reaction can occur over a broader voltage range. Finally, a two-step lithiation mechanism of the Li-SHEO anode is studied in detail and put forward.
Materials and methods
Preparation of the Li-SHEO powder
The Li-SHEO powder was synthesized via a solution combustion synthesis31 and a ball milling method. Initially, 0.01 mol metal nitrate LiNO3 (99%, Aladdin Industrial Inc., Shanghai, China), Fe(NO3)2·5H2O (99%, Aladdin Industrial Inc., Shanghai, China), Mg(NO3)2 (99%, Sinopharm Chemical Reagent Co., Ltd, Shanghai, China), Ni(NO3)2·6H2O (98%, Aladdin), 50 wt% Mn(NO3)2 solution (AR, Aladdin), and Cr(NO3)3·9H2O (99%, Aladdin) were dissolved in 50 mL of water, resulting in a yellow solution after stirring for 30 min. Afterward, a certain amount of glycine (99%, Macklin Biochem. Co., Ltd, Shanghai, China) fuel was added to the former yellow solution, which was then dried to a dark brown gel. After that, the gel was heated in a muffle furnace at 780 °C for 0.5 h to prepare the Li-SHEO precursor. In this process, the combustion of the precursor releases a considerable amount of heat, and the instantly vaporized water contributes to the formation of a porous material. Subsequently, planetary ball milling was employed to refine the powder for 10 hours and the powder was dispersed in an isopropanol environment. Finally, Li-SHEO anode material was obtained after drying in a vacuum drying oven at 70 °C for 6 h. For comparison, SHEO anode material was obtained through the same method without the addition of Li2O.Structure and morphology characterizations
An X-ray diffractometer (XRD, Empyrean Alpha 1, PANalytical B.V., Almelo, The Netherlands) using Cu Kα radiation operating at 45 kV and 40 mA was employed to determine the phase of the samples. The microstructure of the samples was observed through transmission electron microscopy (HRTEM, EOL JEM-2100, JEOL Ltd, Tokyo, Japan), where the electrons emitted from the cathode were accelerated by a voltage of 200 kV. The characterization of the element distribution in the sample was achieved through energy-dispersive X-ray spectroscopy (EDS, JEOL Ltd, Tokyo, Japan), which was assembled in the transmission electron microscope. The element species and their valence states on the surface of the samples were analysed using X-ray photoelectron spectroscopy (XPS, Escalab 250, Thermo Fisher Scientific Ltd). An Al Kα X-ray source (1486.6 eV) with 400 μm spot size was operated at a pass energy of 30 eV and an energy step of 0.05 eV to record the scan spectra of each element. The binding energy scale was calibrated on carbon set at 284.8 eV.Electrochemistry property measurements
To prepare the working electrode, the active material, acetylene black (Super C-45, Kejing Materials Technology Co., Hefeng, China), and sodium carboxymethyl cellulose (CMC, Kejing) were uniformly mixed with a mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 to form an electrode slurry in the water. The slurry was then coated onto copper foil and dried in a vacuum drying oven at 60 °C for 8 h. The dried electrode sheet was cut into working electrodes of approximately 12 mm in diameter using a punching machine. The working electrodes were then used to assemble a CR2016 coin cell. Each active material loading for the prepared working electrode was about 1.05–1.20 mg cm−2. The coin cells were assembled in a glove box (MIKROUNA Super 750, Shanghai, China) filled with Ar gas and with H2O and O2 contents of less than 0.01 ppm. Lithium metal flakes were used as the counter electrode during the assembly process. 1 M LiPF6 dissolved in ethylene carbonate (EC)/diethyl carbonate (DEC) was used as the electrolyte (Tianci Materials Technology Co., Guangzhou, China), and a polyolefin porous membrane (Celgard 2400) was selected as the separator. All cells were tested under galvanostatic conditions at room temperature using a cell testing system (LAND-CT2001A, Wuhan LAND Electronic Co., Ltd, Wuhan, China) at various current densities between 0.01 and 3 V (vs. Li/Li+). CV tests were performed using an electrochemical workstation (Gamry-Interface 1000, America Gamry Instruments, Inc.).
:2:1 to form an electrode slurry in the water. The slurry was then coated onto copper foil and dried in a vacuum drying oven at 60 °C for 8 h. The dried electrode sheet was cut into working electrodes of approximately 12 mm in diameter using a punching machine. The working electrodes were then used to assemble a CR2016 coin cell. Each active material loading for the prepared working electrode was about 1.05–1.20 mg cm−2. The coin cells were assembled in a glove box (MIKROUNA Super 750, Shanghai, China) filled with Ar gas and with H2O and O2 contents of less than 0.01 ppm. Lithium metal flakes were used as the counter electrode during the assembly process. 1 M LiPF6 dissolved in ethylene carbonate (EC)/diethyl carbonate (DEC) was used as the electrolyte (Tianci Materials Technology Co., Guangzhou, China), and a polyolefin porous membrane (Celgard 2400) was selected as the separator. All cells were tested under galvanostatic conditions at room temperature using a cell testing system (LAND-CT2001A, Wuhan LAND Electronic Co., Ltd, Wuhan, China) at various current densities between 0.01 and 3 V (vs. Li/Li+). CV tests were performed using an electrochemical workstation (Gamry-Interface 1000, America Gamry Instruments, Inc.).
Results and discussion
Structures and morphologies of the Li-SHEO materials
Fig. 1(a) shows the XRD pattern of the prepared Li-SHEO material. Clearly, all the diffraction peaks are related to the distinct F![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m spinel phase (ICDD 01-089-7827). To investigate the effect of the introduction of Li2O into the spinel structure, XRD patterns of Li-SHEO and SHEO mixed with standard Si were compared as shown in Fig. 1(b). As can be seen, the introduction of Li2O exhibits a shift in diffraction peaks towards high angles, due to the smaller Li atom replacing the larger metal atoms in the SHEO material. According to the Bragg diffraction equation, a larger diffraction angle indicates a decrease in the crystal plane spacing, which in turn implies that the introduction of Li2O leads to a decrease in the lattice constant of the SHEO crystal structure. Moreover, it has also been reported that this phenomenon may be associated with valence compensation and the formation of oxygen vacancies caused by the incorporation of monovalent metal elements.24
m spinel phase (ICDD 01-089-7827). To investigate the effect of the introduction of Li2O into the spinel structure, XRD patterns of Li-SHEO and SHEO mixed with standard Si were compared as shown in Fig. 1(b). As can be seen, the introduction of Li2O exhibits a shift in diffraction peaks towards high angles, due to the smaller Li atom replacing the larger metal atoms in the SHEO material. According to the Bragg diffraction equation, a larger diffraction angle indicates a decrease in the crystal plane spacing, which in turn implies that the introduction of Li2O leads to a decrease in the lattice constant of the SHEO crystal structure. Moreover, it has also been reported that this phenomenon may be associated with valence compensation and the formation of oxygen vacancies caused by the incorporation of monovalent metal elements.24
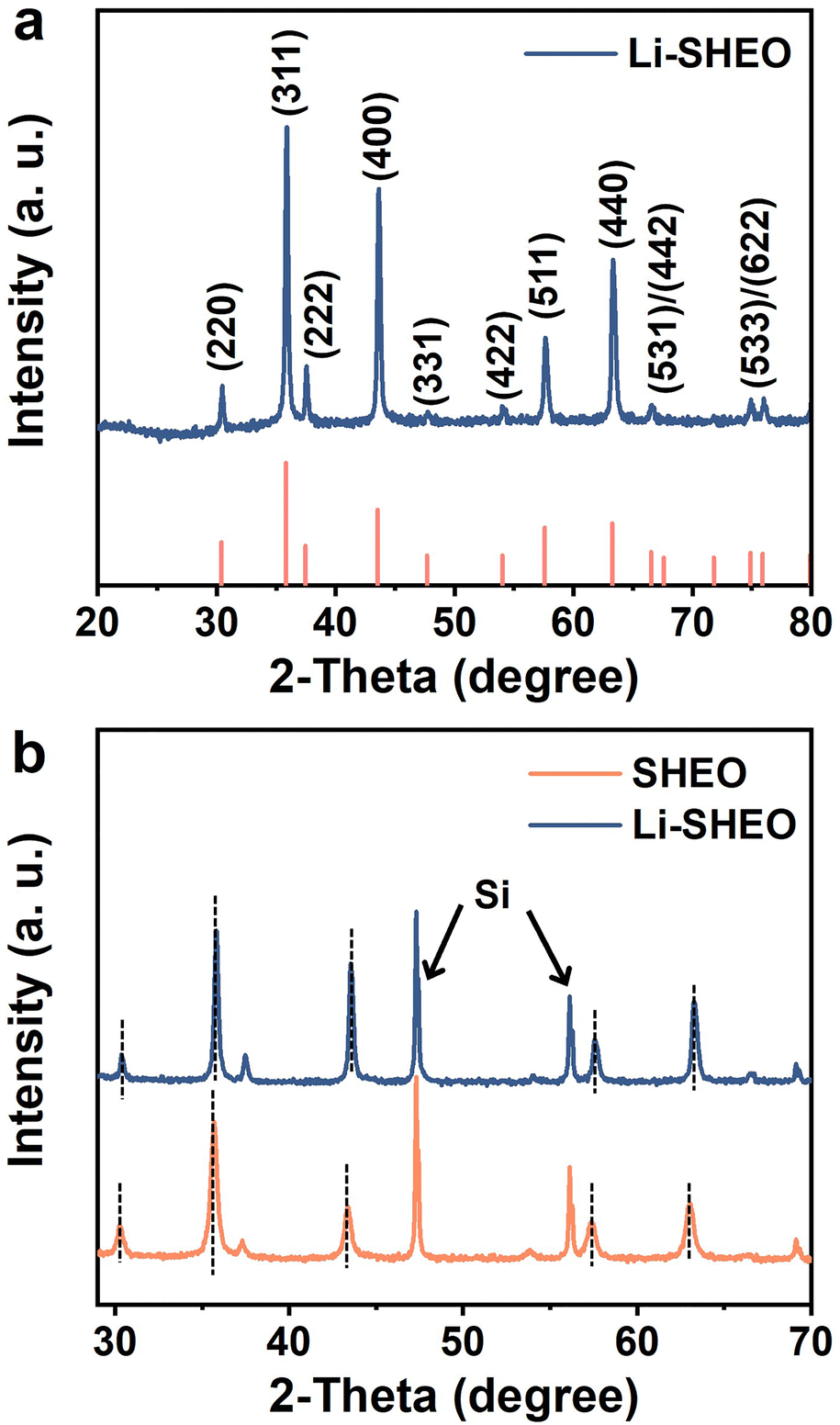 | ||
| Fig. 1 XRD patterns of (a) Li-SHEO material, and (b) SHEO and Li-SHEO materials for revealing the peak shift behaviour. | ||
TEM and TEM-EDS tests were performed to further confirm the phase structure, particle size distribution, and elemental distribution of Li-SHEO material. Fig. 2(a) shows the TEM image of Li-SHEO material, which reveals that the powder is composed of particles with sizes ranging from tens of nanometres, and the particle size distribution is relatively uniform. The HRTEM image of Li-SHEO powder in Fig. 2(b) exhibits a well-defined lattice fringe with d = 0.46 nm corresponding to the (111) plane of the spinel phase. Fig. 2(c) illustrates the EDS elemental scan analysis images of the Li-SHEO powder, in which Mn, Mg, Fe, Cr, O, and Ni elements are highly uniformly distributed, which is consistent with the characteristic random distribution of metal cations in lattice sites in an HEO.
 | ||
| Fig. 2 (a) TEM and (b) HR-TEM image, as well as (c) HADDF image with the related EDS mapping results of Li-SHEO material. | ||
To examine the chemical valence for all the metals and the oxygen vacancies of the Li-SHEO anode, an XPS test was performed on the Li-SHEO and SHEO materials and the results are exhibited in Fig. 3.
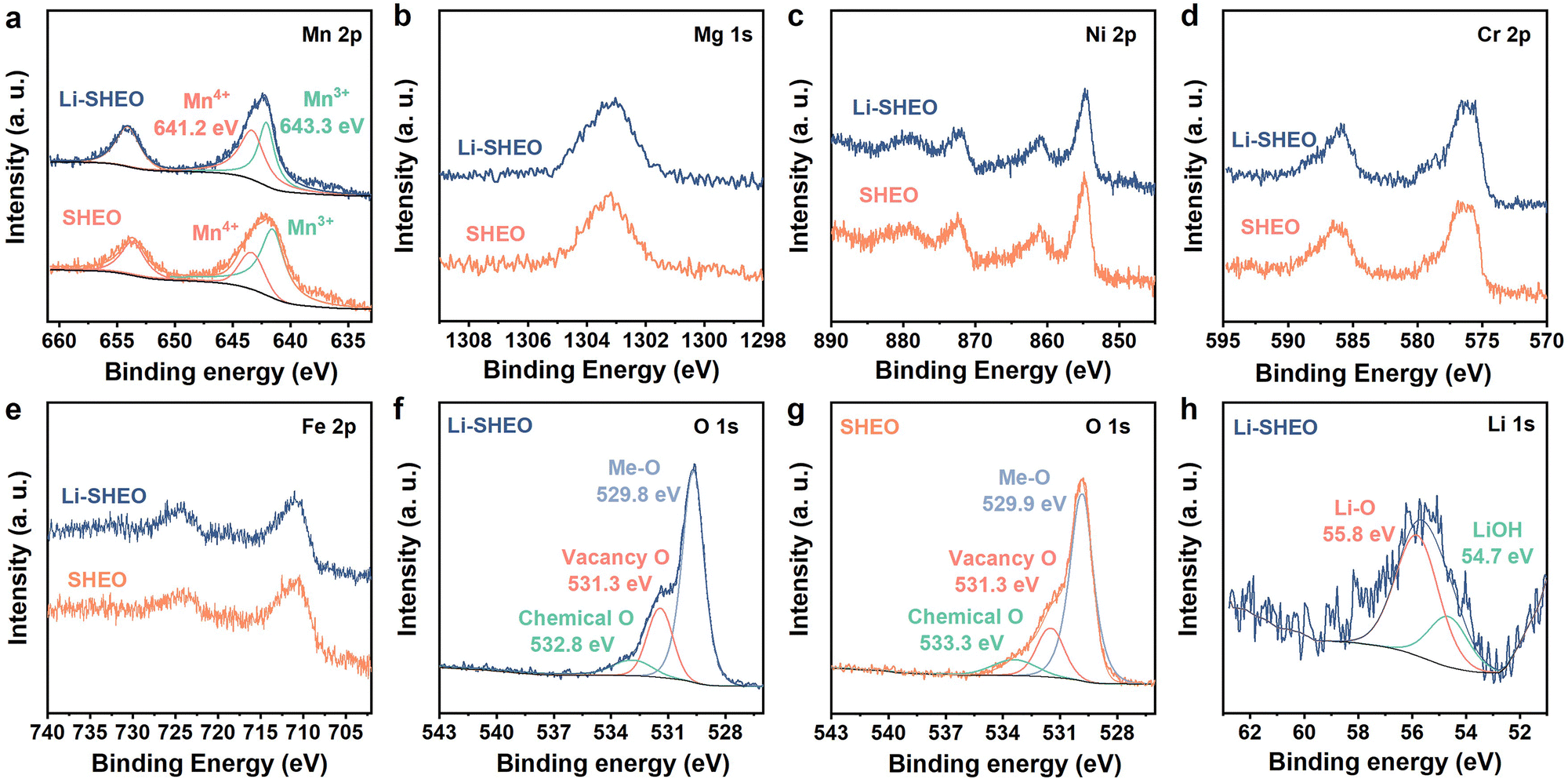 | ||
| Fig. 3 High-resolution XPS spectra of Li-SHEO and SHEO: (a) Mn 2p, (b) Mg 1s, (c) Ni 2p, (d) Cr 2p, (e) Fe 2p, (f), (g) O 1s, and (h) Li 1s. | ||
As exhibited in Fig. 3(a), the two main peaks at binding energies of 652.7 eV (Mn 2p1/2) and 641.2 eV (Mn 2p3/2) are considered to be signals of Mn3+, while the peak at a binding energy of 643.3 eV is considered to be highly correlated with Mn4+.20,32 Compared with the SHEO material, the peak at 643.3 eV of the Li-SHEO material shows a higher intensity with a larger area, indicating a higher valence state of Mn in the Li-SHEO material, which can be attributed to charge compensation caused by the successful introduction of Li2O. Fig. 3(b)–(e) display that other transition metals, such as Mg 1s, Ni 2p, Cr 2p, and Fe 2p, show similar spectral features in both Li-SHEO and SHEO, which suggests that the introduction of Li2O does not cause valence changes in the transition metal elements except for Mn. It is evident that only the valence state of Mn changes due to charge compensation upon the introduction of Li2O, which is related to the multivariable valence nature of Mn.
Fig. 3(f) and (h) present the spectra of O 1s of Li-SHEO and SHEO, respectively, which can be deconvoluted into three peaks located at 529.8, 531.3, and 532.8 eV, which correspond to Me–O bonds, oxygen vacancies, and chemical oxygen, respectively.18,33,34 There is a significant difference in oxygen vacancy content between the two samples. Specifically, the XPS peak representing oxygen vacancies is significantly enhanced in Li-SHEO after the introduction of Li2O. The increase in oxygen vacancies can be explained from multiple perspectives. From the perspective of crystal structure integrity, the introduction of Li2O increases defects in the SHEO crystal structure, thereby promoting the generation of more oxygen vacancies. Furthermore, from the perspective of crystal charge neutrality principles, the introduction of low-valence Li also leads to the production of oxygen vacancies to maintain charge neutrality. It is reported that the introduction of oxygen vacancies can simultaneously enhance the ionic and electronic conductivity of the anode material,29 whilst introducing extrinsic Coulombic forces that facilitate the migration of Li+ ions.30 As shown in Fig. 3(h), the XPS peak located at 55.8 eV is related to the Li–O bond, indicating that the Li+ ion was successfully introduced as a Li2O component into the Li-SHEO material. Another 54.7 eV peak corresponds to LiOH, which is due to the reaction between Li2O and water in the air.
Electrochemical performance of SHEO and Li-SHEO anodes
To explore the influence of the introduction of Li2O on the electrochemical performance of SHEO, both Li-SHEO and SHEO anodes were assembled into half-cells for electrochemical characterization. Fig. 4(a) illustrates the initial charge–discharge curves of the Li-SHEO and SHEO anodes at 0.1 A g−1. It can be observed that both Li-SHEO and SHEO anodes exhibit two discharge plateaus in the range 0.5–1.0 V and below 0.5 V. The first discharge plateau corresponds to the intercalation reaction of Li+ ions, while the latter discharge plateau represents the conversion reaction of transition metal components. The Li-SHEO anode exhibits a higher discharge capacity of 1242.7 mA h g−1, which is higher than the 1109 mA h g−1 of the SHEO anode. The increase in the discharge capacities of the Li-SHEO anode may be due to the introduction of more oxygen vacancies as previously mentioned, resulting in the insertion of a larger amount of Li+ ions during the discharge process. Obviously, the capacity contribution of the plateau below 0.5 V is significantly higher for both Li-SHEO and SHEO anodes, indicating that the reduction reaction is still the main lithium storage process. As for the first plateau, the Li-SHEO anode exhibits an increased potential plateau above 1.0 V, while the SHEO anode only shows a low plateau potential nearly 0.5 V. This higher lithium insertion plateau in the Li-SHEO anode can contribute to its own higher Li content. Regarding the second discharge plateau, the Li-SHEO anode shows a higher average reaction potential compared to the SHEO anode, which may be related to the decreased conversion reaction barrier due to the introduction of Li2O.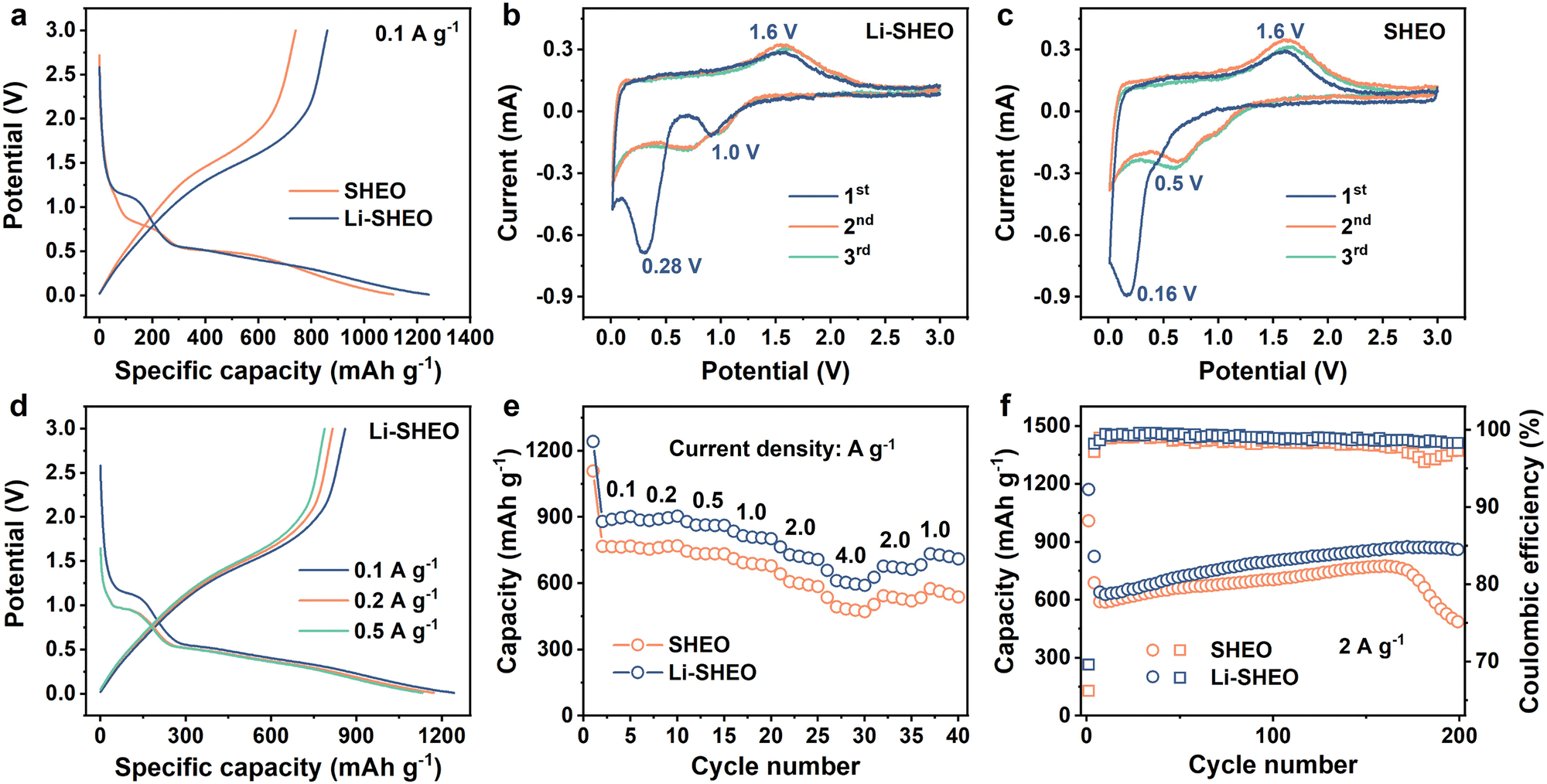 | ||
| Fig. 4 Electrochemical performance of the Li2O-doped SHEO anode: (a) initial discharge–charge profile of the Li-SHEO and SHEO anodes under 0.1 A g−1, CV curves of (b) Li-SHEO and (c) SHEO anodes, (d) initial discharge–charge curves of the Li-SHEO anode under 0.1, 0.2, and 0.5 A g−1, (e) rate capability and (f) cyclic property with related Coulombic efficiency of the Li-SHEO and SHEO anodes. | ||
A CV test was further carried out to investigate the impact of Li2O-introduction on the lithiation process of the SHEO anode at a scan rate of 0.2 mV s−1 in the scan voltage range of 0–3 V. As can be seen in Fig. 4(b) and (c), the Li-SHEO anode shows high reduction peaks at 1.0 and 0.28 V, which are much higher than the 0.5 and 0.16 V of the SHEO anode, demonstrating a smaller electrochemical overpotential and fast electrochemical reaction kinetics in both the Li+ ion intercalation process and the metallic reduction reaction after the addition of Li2O. Both Li-SHEO and SHEO anodes exhibit a similar anodic peak observed at 1.6 V, representing the oxidation reaction of the metallic components in SHEO during the charge process. In the subsequent cycles, the CV curves of the Li-SHEO anode show better overlap and repeatability, illustrating better electrochemical reversibility of the Li-SHEO anode.35
To investigate more accurate electrochemical reaction plateaus of the Li-SHEO anode, the charge–discharge curves at 0.1, 0.2, and 0.5 A g−1 were studied, and the results are shown in Fig. 4(d). With the increase in current density, the two discharge plateaus exhibited a downward shift. Under 0.1 A g−1, the position of the first discharge plateau related to the lithium insertion reaction is higher and broader, which is attributed to a more complete electrochemical reaction process at a lower current density. Fig. 4(e) displays the rate capability of Li-SHEO and SHEO anodes. The Li-SHEO anode delivers high discharge capacities of 1242.7, 886.9, 880, 836.1, 764.8, and 658.7 mA h g−1 under 0.1, 0.2, 0.5, 1.0, 2.0, and 4.0 A g−1, which are higher than those of the SHEO anode, showing good lithiation ability under a large density current.
For further estimating the cyclic properties of the Li-SHEO anode, galvanostatic discharge–charge measurements were carried out under a large current density of 2.0 A g−1 within 0.01–3 V (vs. Li/Li+) for the Li-SHEO and SHEO anodes. As shown in Fig. 4(f), the Li-SHEO anode exhibits better cyclic properties with a 200th discharge capacity of 850.7 mA h g−1 with a capacity retention of 122.0%, while the SHEO anode can only deliver a low 200th discharge capacity of 460.6 mA h g−1 with a capacity retention of 73.9%. This excellent cycle stability can be attributed to the unique high-entropy effect of Li-SHEO. Notably, an increase in capacity can be observed. In the first 150th cycles, both Li-SHEO and SHEO anodes exhibit an increasing trend in specific capacity. This phenomenon of capacity enhancement is commonly observed in metal oxide anodes. The capacity rise can be attributed to the enhanced utilization of conversion reactions, the optimization of the electrolyte surface layer, and the changes in morphology.36 Specifically, the active material particles undergo reversible lithiation/de-lithiation through conversion reactions, leading to smaller particles. The reduction in particle size generates new active sites for lithium storage, resulting in a capacity increase. Moreover, the decomposition of the electrolyte and solid electrolyte interphase (SEI) under the catalytic effect of nano transition metal generated from the conversion reaction also contributes to the capacity increase.37 The difference can be attributed to the higher Sconfig of Li-SHEO. The stabilization effect of entropy ensures that the Li-SHEO anode remains stable under a high current density of 2.0 A g−1, while SHEO experiences a rapid capacity decrease due to phase structure degradation. Moreover, as displayed in Fig. 4(f), Li-SHEO shows higher Coulombic efficiency than the SHEO anode, indicating the fast electrochemical kinetics process caused by the successful introduction of Li2O, which is consistent with the former enhanced rate capability.
To investigate the phase transition behaviour during the lithiation process in the SHEO anode and Li-SHEO anode, ex situ XRD analysis was conducted on electrodes under different states of charge (SOC). As shown in Fig. 5(b), when first discharged to 1.0 V, Li-SHEO exhibited the characteristics of a spinel phase corresponding well to the Fe1.145Mn1.148Li0.706O4 phase (ICCD:01-089-7827). When further discharged to 0.8–0.5 V, the spinel phase still existed and it is noteworthy that a change had taken place in the peak intensity of the (311) and (222) planes, corresponding to the Fe2.74Li1.26O4 phase (ICCD:01-085-1992). It can be observed that there is no phase transition behavior in the SHEO anode as found in the Li-SHEO anode within the voltage range of 1.0 V–0.5 V. The phase transition from Fe1.145Mn1.148Li0.706O4 to Fe2.74Li1.26O4 indicates that the first discharge plateau belongs to the Li+ ion intercalation process of the Li-SHEO anode, while the SHEO anode exhibits only a conversion reaction process. During the late discharge and early charge stages, no distinct peaks can be observed in the Li-SHEO and SHEO anodes. When recharged to 3 V, the characteristic peak of the spinel phase of the (311) and (222) peaks can be restored in the Li-SHEO anode. However, no significant peaks can be observed in the recharged SHEO anode.
 | ||
| Fig. 5 The ex situ XRD of the Li-SHEO and SHEO anodes recorded during the initial cycle: (a) and (c) the related discharge–charge profile and (b) and (d) ex situ XRD patterns. D1–D4 represent discharged to 1.0 V, 0.8 V, 0.5 V, 0.1 V, respectively; C1–C4 represent charged to 0.5 V, 0.8 V, 1.0 V, 3.0 V, respectively. | ||
The disappearance of the spinel phase may be related to the conversion reaction of highly disordered atoms in the Li-SHEO and SHEO anodes. The special high-entropy effects (lattice distortion effect, slow diffusion effect) suppress the formation of the secondary phase of the conversion reaction, resulting in an amorphous state with a long-range disorder, which cannot be observed with distinct peaks in the XRD patterns. As the charging proceeds, some metal cations return to their original lattice sites, and the spinel structure is partially restored in the Li-SHEO anode.
Based on the experimental results, we propose the lithiation mechanism in SHEO and the reasons for its increased reversible capacity. The lithiation process of the Li-SHEO anode consists of two parts, which is the former fast Li+ ion intercalation process at high working potential and the subsequent conversion reaction at low working potential. The increase in the reversible capacity of the Li-SHEO anode results from the joint contribution of Li+ ion intercalation into the crystal lattice of the Li-SHEO anode and subsequent more complete transformation reactions for lithium storage. It was illustrated that the addition of Li2O can induce the formation of more oxygen vacancies, which can provide additional active sites for lithium storage.38 In particular, the higher plateau voltage and the more distinct plateau in the initial charge–discharge curve, and the higher reduction peak potential and intensity in the CV curve corresponding to the lithiation process also indicate that more Li+ ions are embedded into the crystal structure of Li-SHEO with a fast charge transfer rate. Upon comparing the charge–discharge curves and CV curves of the Li-SHEO and SHEO anodes, it was found that the voltage platform and reduction peak corresponding to the conversion reaction in the Li-SHEO anode are raised and widened. These observations provide evidence that the conversion reaction can occur over a broader voltage range in the Li-SHEO anode. Thus, the introduction of Li2O reduces the conversion reaction barrier and leads to a more complete conversion reaction in the Li-SHEO anode.
Conclusions
In this work, Li2O-doped spinel high-entropy oxide (FeMgNiCrMn)3O4 was successfully synthesized via a modified solution combustion synthesis with a ball-milling method. The obtained Li-SHEO anode shows an improved discharge capacity, long cycle life, and good rate capability. A series of experimental analyses shows that the spinel structure phase can be reversibly recovered during the cycling process, ensuring the cycling stability of the Li-SHEO anode. It was also indicated that the introduction of Li2O increased the valence state of Mn and the content of oxygen vacancies in the Li-SHEO material, resulting in additional sites for lithium storage to improve the discharge capacity. Furthermore, it was illustrated that the lithiation process of the Li-SHEO anode consists of two parts: the former intercalation process of Li+ ions and subsequent conversion reaction of transition metals. The increase in lithium-ion insertion and subsequent more complete conversion reaction during cycling jointly promoted the dramatic increase in the capacity of the Li2O-doped SHEO materials. Our work discussed in detail the lithiation mechanism of the SHEO anodes and revealed the feasibility and effectiveness of Li doping, which can provide a new insight for the development of HEO anode materials for Li-ion batteries.Author contributions
G. Ma: conceptualization, investigation, writing – original draft. Y. Zheng: investigation, methodology. F. Meng: formal analysis, writing – original draft. R. Hu: conceptualization, supervision, funding administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (52071144, 52231009, and 51831009), Guangdong Basic and Applied Basic Research Foundation (2023B1515040011), Guangzhou Key Research and Development Program (No. 202103040001), and TCL Science and Technology Innovation Fund.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- Y. Lu, L. Yu and X. W. Lou, Chem, 2018, 4, 972–996 CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- X. Zuo, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Nano Energy, 2017, 31, 113–143 CrossRef CAS.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed.
- A. Sarkar, B. Breitung and H. Hahn, Scr. Mater., 2020, 187, 43–48 CrossRef CAS.
- J. W. Yeh, S. K. Chen, S. J. Lin, J. Y. Gan, T. S. Chin, T. T. Shun, C. H. Tsau and S. Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef PubMed.
- Y. Ma, Y. Chen, M. Sun and Y. Zhang, Chem. Rec., 2023, 23, e202200195 CrossRef CAS PubMed.
- Y. Chen, H. Fu, Y. Huang, L. Huang, X. Zheng, Y. Dai, Y. Huang and W. Luo, ACS Mater. Lett., 2021, 3, 160–170 CrossRef CAS.
- K. Y. Tsai, M. H. Tsai and J. W. Yeh, Acta Mater., 2013, 61, 4887–4897 CrossRef CAS.
- B. L. Musicó, D. Gilbert, T. Z. Ward, K. Page, E. George, J. Yan, D. Mandrus and V. Keppens, APL Mater., 2020, 8, 040912 CrossRef.
- S. S. Aamlid, M. Oudah, J. Rottler and A. M. Hallas, J. Am. Chem. Soc., 2023, 145, 5991–6006 CrossRef CAS PubMed.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- X. Wang, G. Liu, C. Tang, H. Tang, W. Zhang, Z. Ju, J. Jiang, Q. Zhuang and Y. Cui, J. Alloys Compd., 2023, 934, 167889 CrossRef CAS.
- T. X. Nguyen, J. Patra, J.-K. Chang and J.-M. Ting, J. Mater. Chem. A, 2020, 8, 18963–18973 RSC.
- P. Ghigna, L. Airoldi, M. Fracchia, D. Callegari, U. Anselmi-Tamburini, P. D’Angelo, N. Pianta, R. Ruffo, G. Cibin, D. O. de Souza and E. Quartarone, ACS Appl. Mater. Interfaces, 2020, 12, 50344–50354 CrossRef CAS PubMed.
- D. Bérardan, S. Franger, D. Dragoe, A. K. Meena and N. Dragoe, Phys. Status Solidi RRL, 2016, 10, 328–333 CrossRef.
- N. Osenciat, D. Bérardan, D. Dragoe, B. Léridon, S. Holé, A. K. Meena, S. Franger and N. Dragoe, J. Am. Ceram. Soc., 2019, 102, 6156–6162 CrossRef CAS.
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC.
- L. Su, J. Ren, T. Lu, K. Chen, J. Ouyang, Y. Zhang, X. Zhu, L. Wang, H. Min, W. Luo, Z. Sun, Q. Zhang, Y. Wu, L. Sun, L. Mai and F. Xu, Adv. Mater., 2023, 35, e2205751 CrossRef PubMed.
- E. Lökçü, Ç. Toparli and M. Anik, ACS Appl. Mater. Interfaces, 2020, 12, 23860–23866 CrossRef PubMed.
- Y. Gao, Y. Liu, H. Yu and D. Zou, Appl. Catal., A, 2022, 631, 118478 CrossRef CAS.
- Y. Zhang, Z. Ding, C. W. Foster, C. E. Banks, X. Qiu and X. Ji, Adv. Funct. Mater., 2017, 27, 1700856 CrossRef.
- C. Hou, Y. Hou, Y. Fan, Y. Zhai, Y. Wang, Z. Sun, R. Fan, F. Dang and J. Wang, J. Mater. Chem. A, 2018, 6, 6967–6976 RSC.
- T. Meng, B. Li, L. Hu, H. Yang, W. Fan, S. Zhang, P. Liu, M. Li, F. L. Gu and Y. Tong, Small Methods, 2019, 3, 1900185 CrossRef CAS.
- A. Mao, H.-Z. Xiang, Z.-G. Zhang, K. Kuramoto, H. Yu and S. Ran, J. Magn. Magn. Mater., 2019, 484, 245–252 CrossRef CAS.
- D. Wang, S. Jiang, C. Duan, J. Mao, Y. Dong, K. Dong, Z. Wang, S. Luo, Y. Liu and X. Qi, J. Alloys Compd., 2020, 844, 156158 CrossRef CAS.
- A. Sarkar and G. G. Khan, Nanoscale, 2019, 11, 3414–3444 RSC.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- K. Cao, T. Jin, L. Yang and L. Jiao, Mater. Chem. Front., 2017, 1, 2213–2242 RSC.
- H. Kim, W. Choi, J. Yoon, J. H. Um, W. Lee, J. Kim, J. Cabana and W.-S. Yoon, Chem. Rev., 2020, 120, 6934–6976 CrossRef CAS PubMed.
- Y. Zheng, X. Wu, X. Lan and R. Hu, Processes, 2022, 10(1), 49 CrossRef CAS.
- Y.-Y. Hu, Z. Liu, K.-W. Nam, O. J. Borkiewicz, J. Cheng, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L.-S. Du, K. W. Chapman, P. J. Chupas, X.-Q. Yang and C. P. Grey, Nat. Mater., 2013, 12, 1130–1136 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |