
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHard carbons: potential anode materials for potassium ion batteries and their current bottleneck
Xiaoyi
Lu
 a,
Handong
Peng
a,
Guoping
Liu
a,
Fangya
Qi
a,
Chenglong
Shi
a,
Sheng
Wu
a,
Yanxue
Wu
b,
Huanping
Yang
*d,
Jie
Shan
*e and
Zhipeng
Sun
*ac
a,
Handong
Peng
a,
Guoping
Liu
a,
Fangya
Qi
a,
Chenglong
Shi
a,
Sheng
Wu
a,
Yanxue
Wu
b,
Huanping
Yang
*d,
Jie
Shan
*e and
Zhipeng
Sun
*ac
aSchool of Materials and Energy, Guangdong University of Technology, Guangzhou, 510006, Guangdong, P. R. China. E-mail: zpsunxj@163.com
bAnalysis and Test Center, Guangdong University of Technology, Guangzhou, 510006, Guangdong, P. R. China
cKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin, 300071, P. R. China
dSchool of Science, Zhejiang University of Science & Technology, Hangzhou, 310023, Zhejiang, P. R. China
eDepartment of Chemistry and Chemical Engineering, Changji University, Changji, 831100, Xinjiang Autonomous Region, P. R. China
First published on 19th July 2023
Abstract
Owing to the desire for fulfilled energy density and competitively low production costs of lithium-ion battery supplements, potassium ion batteries with the advantages of abundant raw material sources, high safety and excellent electrochemical performance have been a strong contender. In practical applications, hard carbon materials are considered promising anodes owing to their wide range of sources, remarkable economic benefits, environmental friendliness and excellent performance. At present, researchers focus on the study of structure–activity relationship and energy storage mechanism, aiming to obtain universal rules and guide the real commercialization of hard carbon anodes. For this purpose, we herein summarize the current mainstream research views on the K+ storage mechanism, list the common hard carbon anodes and discuss the effects of various modification techniques. In addition, we notice the bottleneck of hard carbon anode development and propose the future direction for technical improvement.
 Xiaoyi Lu | Xiaoyi Lu is a graduate student pursuing a PhD with Prof. Zhipeng Sun, at the School of Materials and Energy, Guangdong University of Technology, China. Currently, she is focused on energy storage devices with special interest in potassium ion batteries. She is devoting herself to develop hard carbon anodes with excellent cycle life and satisfactory fast-charging ability. |
 Guoping Liu | Guoping Liu received his bachelor's degree in Materials Chemistry from the East China University of Technology in 2016 and master's degree in Materials Engineering from Guangdong University of Technology in 2019. He is currently a PhD candidate under the supervision of Prof. Zhipeng Sun at the School of Materials and Energy, Guangdong University of Technology. His current research is focused on rechargeable alkali metal-ion batteries. |
 Huanping Yang | Huanping Yang obtained her PhD from Nanyang Technological University. She was appointed as a Postdoctoral Research fellow at Agency for Science, Technology and Research, Singapore. She is currently a Professor at the Department of Science at the Zhejiang University of Science and Technology, China. Her research interests include lithium-ion batteries and optical and electrical properties of graphenes. |
 Jie Shan | Jie Shan is a lecture at the Department of Chemistry and Chemical Engineering in Changji University, China. She obtained her master's degree in chemical engineering from the Department of Chemistry and Chemical Engineering, Zhejiang University in 2015. Her research interests include the synthesis and application of complex materials such as magnetic materials and lithium-ion batteries. |
 Zhipeng Sun | Zhipeng Sun is a professor in the School of Materials and Energy at Guangdong, University of Technology, China. He obtained his PhD from the Nanjing University of Aeronautics and Astronautics, China in 2010. He then joined the National University of Singapore as a Postdoctoral Research Fellow. In 2012, he moved to the Nanyang Technological University as a Postdoctoral Research Fellow. His research interests include energy storage materials, mesoporous functional composites and gas sensors. In his research career, he has published over 120 publications, including 110 papers in peer-reviewed journals and 8 patents, and managed 15 projects as chief investigator. |
1 Introduction
With the wide-ranging application of green energy, there is an urgent need for stable large-scale energy storage equipment in order to realize centralized energy storage and off-peak utilization. Given the limited lithium resources and the high cost of technology, it is necessary to search for novel secondary batteries. Potassium, the first major group of elements with more affluent crustal abundance (2.09%),1–4 exhibits quite similar chemical properties and electrochemical behaviour to lithium. In propylene carbonate electrolytes, the standard redox potential of K+/K (−2.88 V vs. SHE) is proved to be even lower than that of Li+/Li (−2.79 V vs. SHE), allowing an expanded voltage window to improve the overall energy density.5–8 Meanwhile, despite the larger ionic radius, potassium ions showed a smaller Stokes radius and desolvation energy in ester-based electrolytes,9–12 displaying a greater ionic conductivity and mobility. With regard to actual safety, the potassium metal texture is too soft to form long and sharp dendrites;13 and its low melting point ensures that the short circuit can be cut off by dendrite melting before the whole battery becomes seriously thermal runaway.14 Therefore, potassium ion batteries (PIBs) are considered potential secondary batteries for the lithium-ion battery supplements.To date, the practical application of PIBs is hampered by the lack of anode materials that can effectively mitigate against the serious volume change caused by potassiation/depotassiation.15–19 For this reason, hard carbons, already widely used in lithium-ion batteries,20–22 have attracted the attention of researchers owing to their unique turbine-like graphite domain structure. The hard carbon usually refers to a kind of amorphous carbon that cannot be completely graphitized even at ultra-high pyrolysis temperatures (above 2000 °C).23–25 It is usually derived from thermosetting resins or polymers, biomass and small organic molecules with weak aromatic properties.26–28 The hard carbon contains curly and scattered graphene sheets that cannot be completely unrolled or flattened (Fig. 1(a) and (d)), making it difficult to stack further into ordered graphite.29–31 Different from the soft carbon and graphite (Fig. 1(a)–(c)), the curling short-range graphite-like structure can effectively buffer the mechanical deformation caused by K+ intercalation.32,33 In general, there are numerous doped heteroatoms in the hard carbon structure, especially C–O–C bonds, which are the key to the existence of abundant defects.34 For energy storage, these defects allow it to exceed the theoretical capacity of graphite.35,36 However, this discontinuous structure, to a certain extent, can block the conductive network, hindering the speed of electronic transmission (Fig. 1(g)).37,38 Relatively speaking, the long-range ordered structure of soft carbon and graphite endows them with excellent conductivity, which contributes to rate performance. However, the accompanying narrow carbon layer spacing cannot provide sufficient buffer for K+ intercalation, jeopardizing long-cycle life (shown in Fig. 1(e) and (f)).
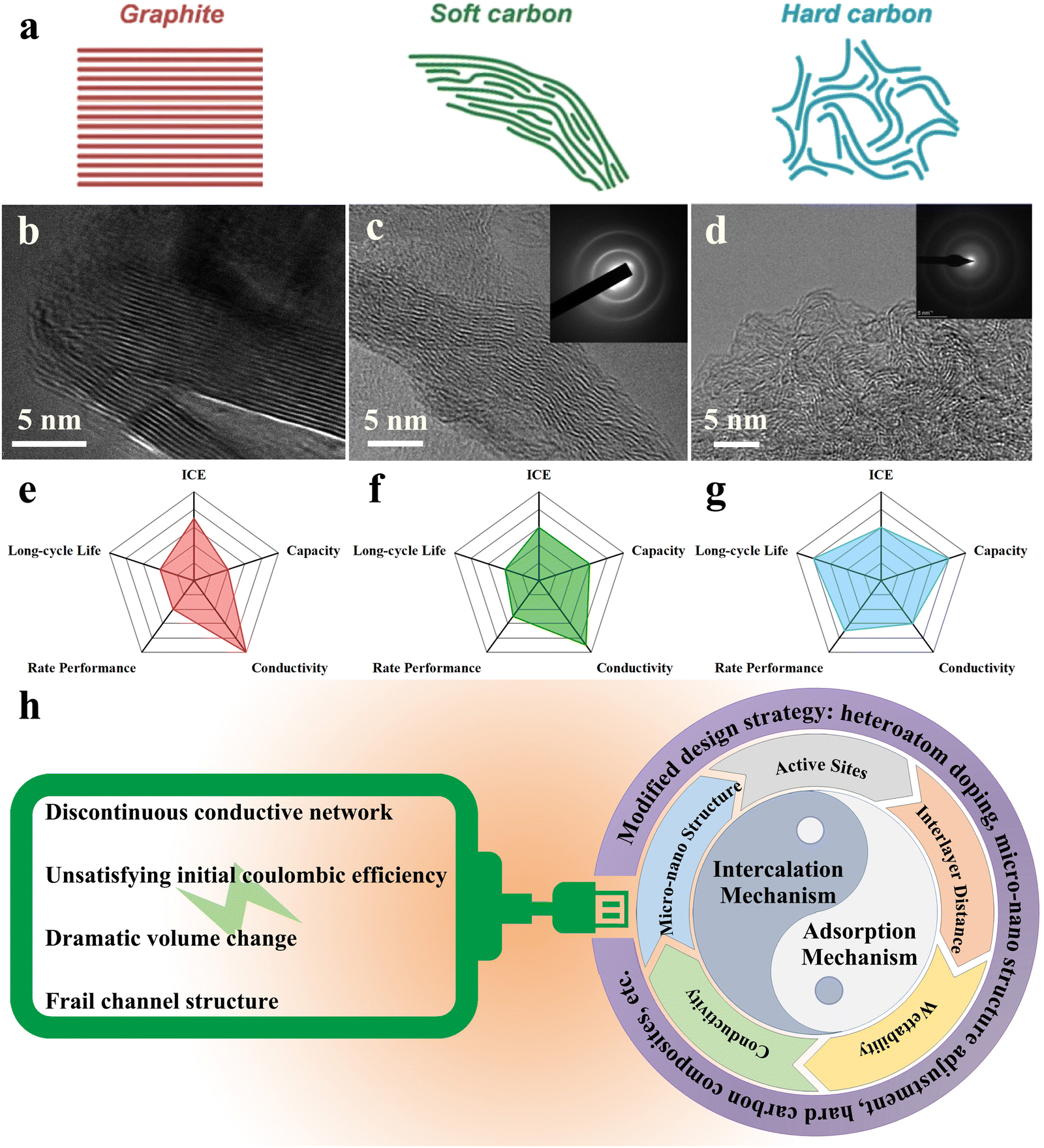 | ||
| Fig. 1 Classification, microstructure and performance comparison of mainstream carbon anodes in PIBs: (a) Illustration of the typical structure of graphite, soft carbon, and hard carbon (Reprinted with permission from ref. 50. Copyright 2018 Wiley). HR-TEM images of (b) graphite (Reprinted with permission from ref. 51. Copyright 2008 American Chemical Society), (c) soft carbon (Reprinted with permission from ref. 52. Copyright 2020 Elsevier) and (d) hard carbon (Reprinted with permission from ref. 53. Copyright 2022 Elsevier); comparison of the comprehensive electrochemical performance of (e) graphite, (f) soft carbon and (g) hard carbon. (h) Schematic diagram of two main energy storage mechanisms in hard carbon, major dilemmas and their solutions. | ||
The electrochemical behavior of hard carbons, common anode materials, in lithium-ion batteries has been studied in detail.22,39 Part of the experience can be used as a guide to explain the storage behavior of potassium ions in hard carbons, but cannot be completely appropriated. The slope region similar to that of lithium ion batteries can be observed in the charge–discharge curve of PIBs, but there is no obvious platform.41,42 According to current typical studies, there are two conventional K+ storage mechanisms: one is diffusion-controlled intercalation mechanism40,43 and the other is surface-driven adsorption mechanism.44,45 These two mechanisms are not opposed to each other, but often co-exist to contribute to energy storage, the “bulk insertion and surface adsorption” mechanism.46 Among them, the pseudocapacitance behavior generated by the adsorption mechanism is the main reason that hard carbon materials exhibit storage capacity exceeding the theoretical capacity. Usually, the storage mechanism puts forward strict requirements to the material micro-nano structure, and in turn, the material structure becomes the precondition to decide the storage path (shown in Fig. 1). In other words, the expansion of the inner carbon layer spacing is conducive to the stable diffusion of K+ within the material, and the contribution of pseudocapacitance is also due to the abundant defects formed by heteroatom doping.47–49
Unfortunately, the application of hard carbons in PIBs is still restricted by the unsatisfying initial coulombic efficiency (ICE), poor conductivity and so on.54,55 In order to deal with the above-mentioned dilemma, researchers have made various attempts to modify hard carbon materials, including micro-nano structure control and intrinsic property optimization. In this review, we summarize the current research on the K+ storage mechanism in hard carbons, and list the main preparation strategies of high-performance anodes. It is expected to explore the future direction of potassium ions and make strategic planning for overall performance.
2 Potassium storage mechanism for hard carbon anode materials
It is well known that the electrochemical performance of hard carbon anodes is closely related to their K+ storage mechanism. Therefore, it is of great significance to discuss the mechanism of potassium storage for the optimization of hard carbon anodes. As shown in Fig. 2, according to the difference of K+ storage sites, the present research shows that the storage mechanism can be divided into two kinds: intercalation mechanism and adsorption mechanism. In detail, the former stores K+ in the gap of carbon layers, while the latter absorbs K+ in the defects, surface and nanovoids of hard carbon materials to provide capacity. In reality, energy storage is mostly achieved by the cooperation of the these two.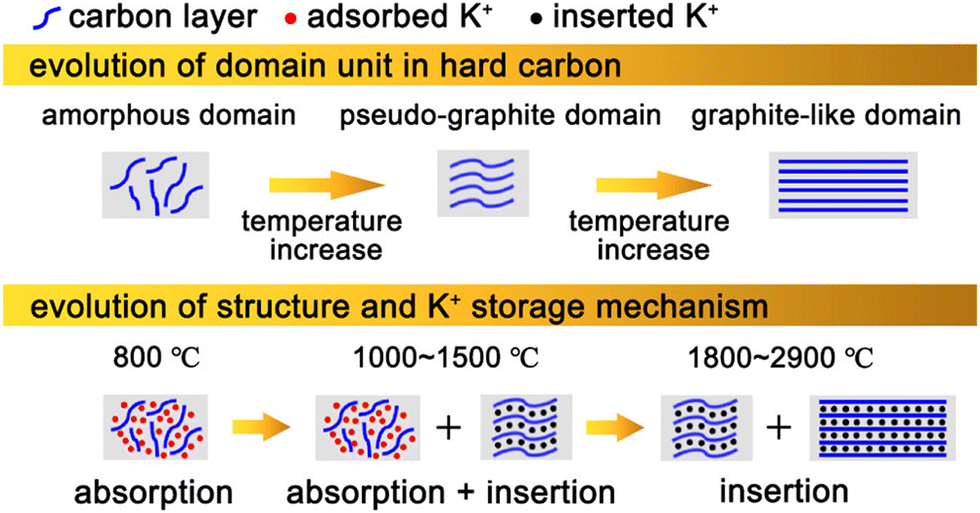 | ||
| Fig. 2 Illustration of hard carbon with different microstructures (above) and its corresponding K+ storage mechanism (below). (Reprinted with permission from ref. 56. Copyright 2022 American Chemical Society). | ||
2.1 Intercalation mechanism
Traditional graphite exhibits a typical layered structure, which consists of several graphite layers stacked by ABAB stacking sequence,57–59 and K+ can be inserted into the graphite layers to form a series of graphite intercalation compounds (GICs). However, there is no large-scale regular graphite lamellar structure in the hard carbon, which is mainly composed of curly short-range ordered graphite domains.60–62 The K+ intercalation mechanism of hard carbons is different from that of traditional graphite due to the special micromorphology. In the potassiation process, the local intercalation reaction takes place between the turbine-like quasigraphitic domain layers of hard carbons.63–65In the process of potassiation, volume expansion of hard carbons (about 160%) occurs by gradually intercalating K+ with a large ionic radius, and the most direct result is the gradual enlargement of spacing between hard carbon layers. Therefore, it is possible to observe the change in layer spacing to reflect the K+ intercalating process indirectly. Some researchers used in situ XRD to trace the process of K+ intercalation.66 In Fig. 3(a), an interesting result was found that as the degree of potassiation increased, the broad peak of amorphous graphite (002) split into two parts: one part remained stationary at 22.80° and the other part shifted to 22.4°. This fully indicates that the spacing of carbon lattices expands. After the removal of K+, the position and intensity of the corresponding diffraction peaks gradually returned to the original state, indicating that the process is reversible.
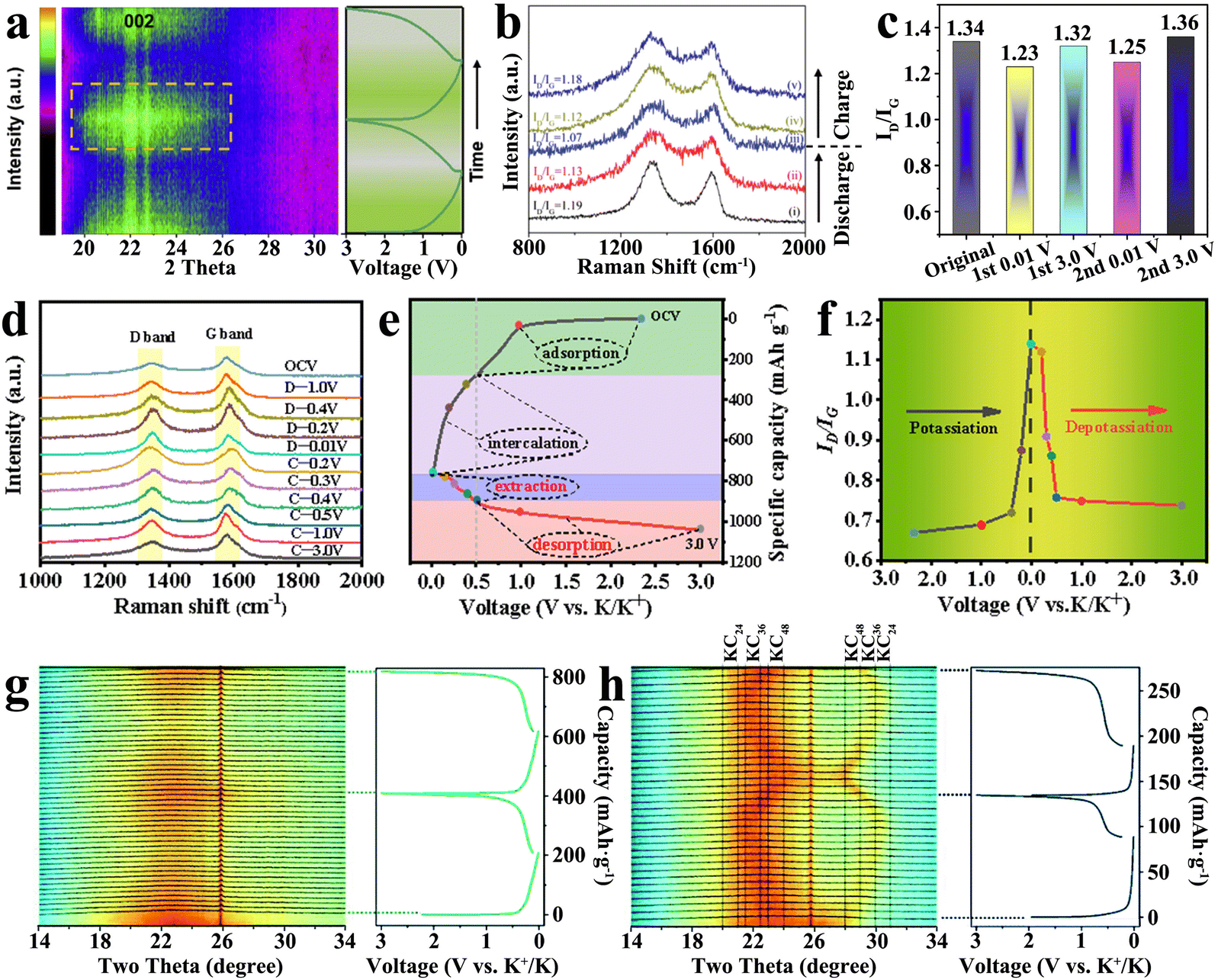 | ||
| Fig. 3 K+ storage intercalation mechanism. (a) In situ XRD patterns during the charge and discharge process (Reprinted with permission from ref. 66. Copyright 2020 Elsevier). (b) Ex situ Raman spectra in the potassiation/depotassiation process (Reprinted with permission from ref. 67. Copyright 2022 Wiley). (c) ID/IG ratios in the first two cycles (Reprinted with permission from ref. 68. Copyright 2020 Elsevier). (d) Ex situ Raman spectra, (e) discharge–charge curve and (f) the corresponding ID/IG value changes at the first cycle (Reprinted with permission from ref. 69. Copyright 2020 Elsevier). In situ XRD patterns of (g) DGC-1600 and (h) DGC-2800 with galvanostatic discharge/charge profiles (Reprinted with permission from ref. 71. Copyright 2022 Royal Society of Chemistry). | ||
In general, the structure of hard carbon materials changes along with K+ intercalation. Some studies found that the intercalation of K+ can cause the renormalization of hard carbon materials and increase the graphitization degree, which can be observed by the decrease in the ID/IG value in Raman spectra (Fig. 3(b)).67 As presented in Fig. 3(c), this internal change is reversible and inheritable, and the researchers found similar results in the follow-up charge–discharge monitoring.68 On the contrary, it has been observed in other studies that the ID/IG value gradually increased with the deepening of the degree of potassium (as shown in Fig. 3(d)–(f)),69 which may be due to the decrease in the intensity of the G peak caused by the weakening of resonance induced by K+ intercalation in the graphite domain.70 The results clearly indicated that the change rate of the ID/IG value was related to the degree of charge and discharge, changing sharply in the low-potential region, which also reflected the difference between intercalation and adsorption energy storage.
In the graphite anode, the intercalation process of K+ is often accompanied by the formation of GICs. However, this experience cannot be applied perfectly to hard carbon materials. Some studies have shown that stable GICs may not be formed, and the GICs are directly related to the degree of graphitization of hard carbon materials.71 In the trace of highly graphitized sample (DGC-2000), significant formation of KC8(stage I) was observed at the end of complete potassiation. Surprisingly, two-phase intercalation products, KC48 (stage IV), KC36 (Stage III) and KC24 (Stage II), were separated using the in situ XRD pattern signal during the electrochemical cycle of DGC-2800 (Fig. 3(h)). However, the high-capacity final-phase product KC8 was not found, which may be caused by the restricted intercalation kinetics. In the potassiation/depotassiation process of local ordered DGC-1600 with lower graphitization, it was found that the broad peak of amorphous graphite (002) became wider and more asymmetric with K+ intercalation, and then recovered (shown in Fig. 3(g)). The results indicated that the single-phase solid-solution intercalation reaction occurred. The intercalation behavior of single-phase K+ is significantly different from that of graphite-based materials reported previously. In the process of repeated K+ insertion/deinsertion, there is no formation or disappearance of new phases.
Although the intercalation mechanism of hard carbons is still controversial, there is no doubt that the storage of K+ involves solid-phase diffusion and causes drastic volume change in hard carbon materials during the electrochemical process. Obviously, it is significant to shorten the diffusion path and enlarge the interlayer spacing for ameliorating the electrochemical properties of intercalated-energy-storage hard carbon anodes.
2.2 Adsorption mechanism
For the K+ storage of hard carbon anodes, another important mechanism is the surface-driven adsorption, that is, K+ is absorbed in the defects, surfaces and nanoholes of hard carbon materials, which is a pseudocapacitance behavior. In contrast to the intercalation mechanism, this way is feasible to avoid K+ intercalation into carbon lattices, maintaining the integrity of the materials while providing a faster chemical kinetics path.72In practice, there is no complete surface adsorption-controlled K+ storage. The adsorption mechanism also includes the diffusion of K+ into hard carbon materials and its storage in the bulk phase, which is mainly controlled by diffusion. In order to clarify the electrochemical behavior induced by different storage modes, cyclic voltammetry is generally used to determine the control steps of K+ storage by using the power-law relationship between peak current (i) and scanning rate (v). The linear curve of![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(|i|) tolog(v) is obtained by mathematical treatment of the formula i = avb, and the slope of the curve obtained is the b-value.73 When the b-value is 0.5, K+ is stored by a diffuse-controlled intercalation process, while b = 1 is expressed as a surface-driven pseudocapacitance process, like Fig. 4(a). In addition, the capacity contribution of diffusion and pseudocapacitance can be calculated by the equation i = k1v + k2v1/2, where k1v and/or k2v1/2 signify the pseudocapacitance control and diffusion control, respectively (Fig. 4(b) and (c)).
log(|i|) tolog(v) is obtained by mathematical treatment of the formula i = avb, and the slope of the curve obtained is the b-value.73 When the b-value is 0.5, K+ is stored by a diffuse-controlled intercalation process, while b = 1 is expressed as a surface-driven pseudocapacitance process, like Fig. 4(a). In addition, the capacity contribution of diffusion and pseudocapacitance can be calculated by the equation i = k1v + k2v1/2, where k1v and/or k2v1/2 signify the pseudocapacitance control and diffusion control, respectively (Fig. 4(b) and (c)).
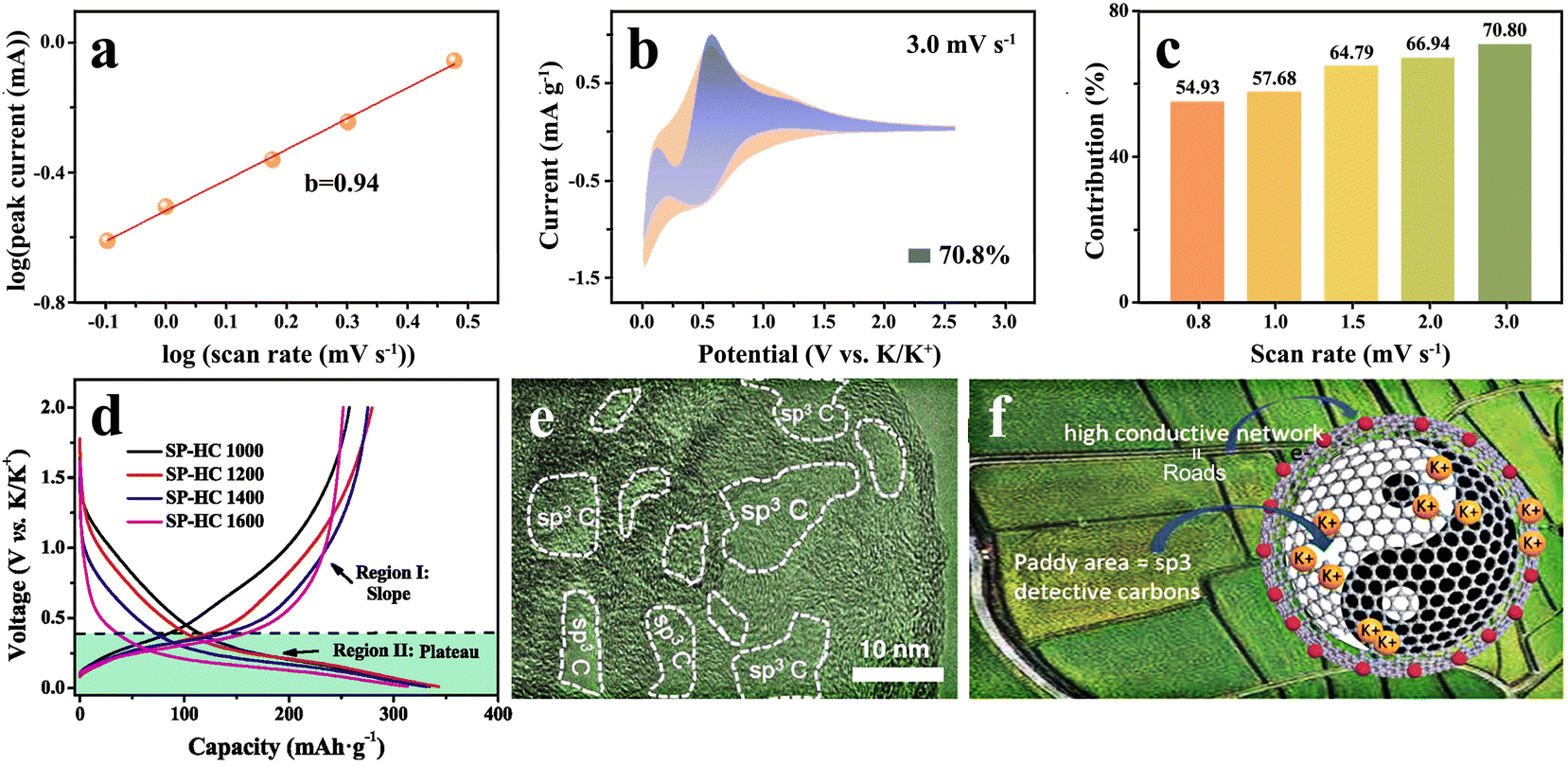 | ||
| Fig. 4 K+ storage adsorption mechanism. (a) Calculation of the b-value, (b) the fitting of capacitance contribution and (c) the percentage of capacitance contribution at different scan rates (Reprinted with permission from ref. 73. Copyright 2021 Elsevier). (d) Charge–discharge curves of hard carbon materials synthesized at different temperatures (Reprinted with permission from ref. 78. Copyright 2019 Elsevier). (e) Several sp3 defective carbon areas in the continuous sp2 graphitic carbon conductive network. (f) Schematic sketch of the K+ transport and storage mechanism in the hard carbon containing defects (Reprinted with permission from ref. 80. Copyright 2021 Wiley). | ||
It was found that the distinction between intercalation and absorption can be clearly indicated by the charge–discharge cycling profiles. The pseudocapacitance behavior exhibits a sloping charge–discharge curve, rather than forming an obvious voltage plateau.74,75 Its capacity is mainly contributed by the sloping region, and it shows a higher average potential. Correspondingly, the intercalation mechanism often presents a voltage plateau in the low voltage range, providing most of the capacity and a lower average voltage.71,76,77 The storage mechanism is closely bound up with the micro-nano structure of the hard carbon anodes themselves. As displayed in Fig. 4(d), the increase in temperature is conducive to the local ordering of hard carbons, and K+ is more likely to be intercalated into the graphite-like turbine lattice, which means that the voltage plateau shows a gradual lengthening.78
The surface adsorption-controlled pseudocapacitance behavior shows remarkable ability to effectively avoid the repeated intercalation of K+ in the carbon lattice, which can not only endow hard carbon anodes with the ability to exceed the theoretical capacity, but also achieve superior diffusion kinetic and excellent cyclic performance.
It is a common method to introduce more non-carbon intrinsic defects into hard carbon materials by doping with heteroatoms. These heteroatoms induce the existence of external defects, which can change the microstructure of hard carbons, effectively improving the K+ adsorption site and optimizing its electrochemical performance. There is a lack of widely systematic understanding of the relationship between defects and synthesis methods, but it is certain that doped heteroatoms are positively correlated with the types and content of defects. The doping of heteroatoms can destroy the integrity of the carbon plane and form new topological-structure-rich defects. Meanwhile, heteroatom doping can cause a change in the micro-nano structure of carbon materials, form more voids and introduce a large number of edge defects.79 Further studies show that inclusion of heteroatoms in the carbon structure is not always favorable, and excessive heteroatom doping will produce much more sp3 defects, as can be seen in Fig. 4(e), and distort the π conjugated structure in the carbon layer, which is not conducive to rapid transfer of electrons (Fig. 4(f)).80,81 Therefore, it is very crucial for improving the overall properties of hard carbon materials to correctly examine and control the balance of heteroatom doping.
3 Recent development of hard carbon materials
As a very important branch of PIB anode materials, hard carbon materials have been developed rapidly in recent years due to their prominent electrochemical performance and potential economic benefits. Their special “pseudo-graphite” nanostructure, which exhibits turbo-enhanced effects, can resist structural degradation in the reduplicative charge/discharge process to a certain extent, showing satisfactory electrochemical cycling performance.82,83As early as 2016, hard carbon microspheres (HCSs) with sucrose as the carbon source were introduced as the fresh anode of PIBs, whose charge–discharge curve presents a gradient slope in the high potential region and a quasi-plateau shape in the low potential region.84 Compared with the performance of sodium ion batteries, the HCS, as a K+ storage host, shows better rate performance, which may be related to a higher K+ diffusion coefficient in the hard carbon structure. Meanwhile, the intercalation potential of K+ in the HCS is much higher than that of potassium plating, which reduces the risk of pernicious dendrite formation. Therefore, in this aspect, hard carbon anodes are preferred for widespread application in PIBs over sodium ion batteries.
Inevitably, for practical applications of K+ storage, the diffusion-controlled intercalation mechanism is limited by energy density,85,86 while the surface-driven pseudocapacitance process is subject to low power densities and low ICE to some extent.87,88 In one word, the traditional hard carbon materials cannot fully exhibit their structural advantages. At present, the research focuses on the modification, composite and micro-nano structure adjustment of traditional hard carbon materials, aiming to improve its capacity and enlarge its economic advantages. We also summarized some technical parameters of hard carbon anodes in recent years, listing in Table 1 to help explain various modification methods.
| Anode | Source | Method | Morphology | Electrolyte | Cycle life [capacity (mA h g−1) (cycle number) @current density (A g−1)] | Rate performance [capacity (mA h g−1) @ current density (A g−1)] | ICE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| NCS | Chitin | Sol–gel process | Sphere-shaped | — | 180(4000)@0.504 | 154@20.16 | — | 93 |
| N-SHC | Glucose | Hydrothermal reaction | Sphere-shaped | 0.8 M KPF6 in EC:PC(1:1) |
200(600)@0.2 | 93@2 | 53 | 94 |
| NPC | Sodium citrate & urea | Carbonization process | Network-shaped | 0.8 M KPF6 in EC:DEC(1:1) |
144.4(1000)@5 | 185@10 | 43.1 | 99 |
| NHCFs | Pyrrole & CS2 | Carbonization process | Fiber-shaped | 1.0 M KPF6 in DME | 264.2(500)@2 | 261@5 | 80.7 | 100 |
| NOHPHC | MOFs | Carbonization process | Flower-like microsphere | 0.8 M KPF6 in EC:DEC(1:1) |
130(1100)@1.05 | 118@3 | 25 | 91 |
| NSHCF | Aspergillus niger | Sn-catalyzing & carbonization process | Fiber-shaped | 0.8 M KPF6 in EC:DEC(1:1) |
179.3(500)@1 | 124.5@2 | 51 | 101 |
| NPHC | Cellulose | Carbonization process | Network-shaped | 0.8 M KPF6 in EC:DEC(1:1) |
172(600)@2 | 76@20 | — | 105 |
| NSO-HCN | MPUA resin | Carbonization process | Network-shaped | 0.8 M KPF6 in EC:DEC(1:1) |
100.3(1000)@0.5 |
126.4@0.5 | 31.6 | 106 |
| SNOC | Polybenzoxazine | Vulcanization process | Sphere-shaped | 1 M KFSI in the EMC | 218.9(7300)@2 | 174.5@2 | 60.8 | 107 |
| HCS-SC | Sugar & PTCDA | Hydrothermal reaction & balled milled mixture | Hard–soft composite carbon | 0.8 M KPF6 in EC:DEC(1:1) |
200(200)@ 279 | 190@558 | 67 | 108 |
| QLGC | Alkali lignin & GO | Polymerization reaction & pyrolysis | Thin carbon layer | 0.8 M KPF6 in EC:DEC(1:1) |
200(100)@0.1 | 63@2 | 45.4 | 112 |
| WS | Water chestnut | Carbonization process | Irregular-shaped | 1.5 M KFSI in EC:DEC(1:1) |
220.5(1000)@0.1 | 134.8@1 | — | 41 |
| NOCNBs | Dragon shrimp shells | Carbonization process | Fiber-shaped | 0.8 M KPF6 in EC:DEC(1:1) |
277(1600)@1 | 200@3.2 | 49 | 66 |
| CPC | Ganoderma lucidum spore | Carbonization process | Cage-shaped | 1 M KPF6 in EC:DEC(1:1) |
124.6(700)@1 | 133@1 | 38.2 | 114 |
3.1 Heteroatomic-doped hard carbon materials
The introduction of heteroatom doping is the most common hard carbon modification method, which is expected to improve the electrochemical performance by regulating the defects. In general, heteroatomic doping has effects on K+ storage in several ways such as ameliorating the intrinsic conductivity,89,90 increasing the number of active sites,66 enhancing surface wettability,91 and expanding the interlayer spacing to reduce diffusion barriers.92 According to the classification of doping, the common heteroatomic doping includes single-atom doping, dual-atom doping and triple-atom doping.Among them, as an element adjacent to C in the periodic table, N is very similar to C in physical and chemical properties, facilitating the entry and modification of the carbon skeleton. Furthermore, N atoms are incorporated into the honeycomb-like carbon network via sp2 hybridization, which can not only provide additional activity defects with lone electron pairs, but also ameliorate the electrical conductivity by adjusting the intrinsic electron local structure.96 Different doping species of N atoms in the carbon matrix play different roles in improving the actual energy storage ability, as shown in Fig. 5(a), which can be mainly divided into three types, namely, graphitic-N (N-Q), pyrrolic-N (N-5) and pyridinic-N (N-6), among which the latter two are usually referred to as edge N defects.97 The theoretical calculation shows that N-Q is not the active site of K+ adsorption, and its adsorption energy is −0.024 eV (Fig. 5(b)), higher than that of the undoped pristine carbon (−0.229 eV), while N-6 shows a stronger K+ adsorption.93 Both N-Q and N-6 promote the density of states (DOS) around the Fermi level, leading to an excellent electronic conductivity. With the progress of research, the role of N doping morphology in the K+ storage process has been further understood and enriched. Abundant edge-N doping can not only broaden the interlayer spacing, but also elevate the contribution of pseudocapacitance, representing the better rate performance, capacity and cycle performance of hard carbon materials.98 For N-5 and N-6, which can introduce vacancy defects in the carbon layer, there is a slight difference in their contribution to the storage of K+. As displayed in Fig. 5(c), the adsorption energy (Eads) of vacancy defects caused by N-5 and N-6 in the carbon layer is −2.38 eV and −2.68 eV, respectively, indicating that N-6 is more favorable for the adsorption and storage of K+.99 The introduction of abundant edge N doping defects, as highly chemically active sites, delivered exceptional electrochemical performance of hard carbon, retaining a competitive reversible specific capacity of 384.2 mA h g−1 after 500 cycles at 0.1 A g−1. Meanwhile, the presence of N-Q deep in the carbon skeleton made an indelible contribution to enhancing the electrical conductivity, exhibiting a superior rate performance (capacity of 185.0 mA h g−1 at 10 A g−1). The selection of precursor and reaction conditions is an effective way to control the content and morphology of doped N and to directly regulate the physical and chemical properties of hard carbons.
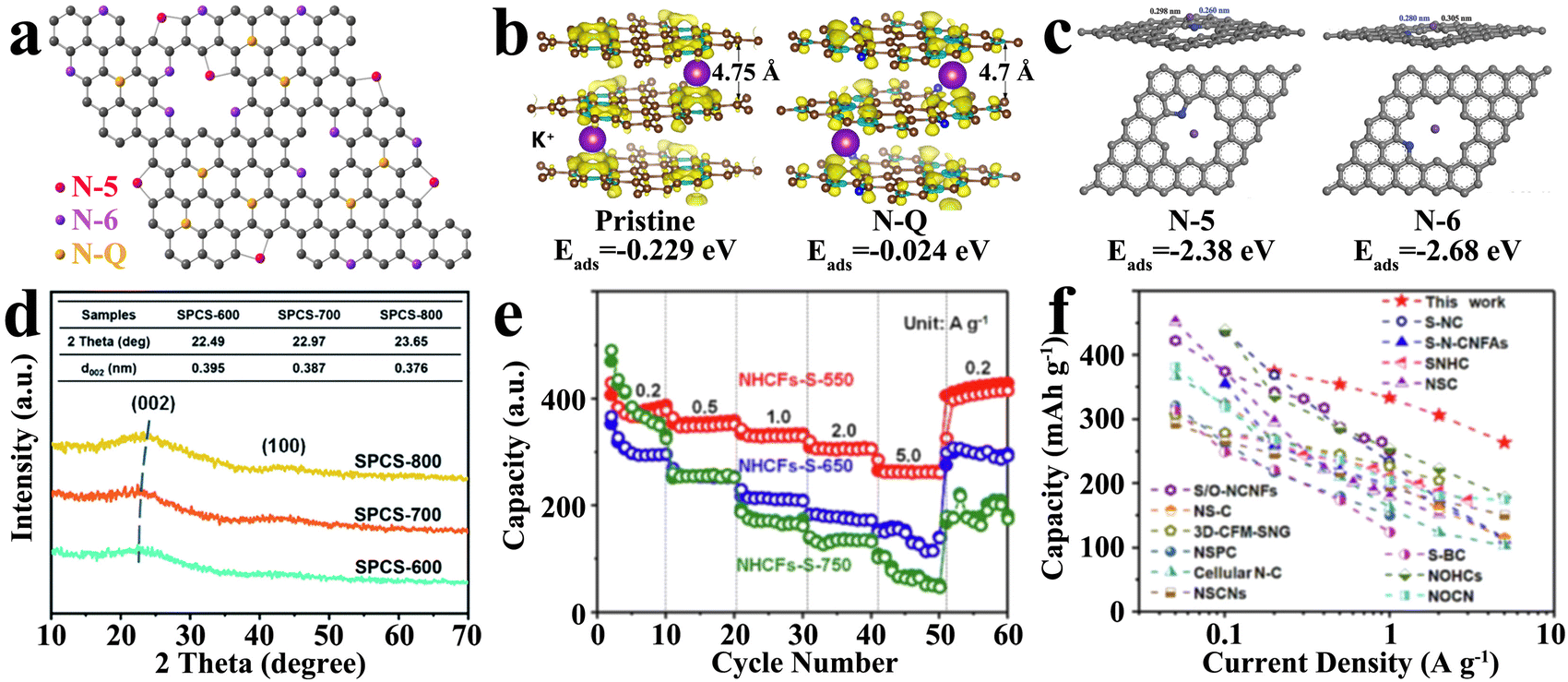 | ||
| Fig. 5 Single atom-doped hard carbon materials as PIB anodes: (a) schematic illustration of N-doping species (Reprinted with permission from ref. 97. Copyright 2018 Springer Nature); (b) adsorption energy (Eads) of K+ adsorbed on pristine carbon and graphitic N-doped (N-Q) carbon (Reprinted with permission from ref. 93. Copyright 2017 Elsevier); (c) adsorption energy (Eads) of K+ adsorbed on pyrrolic-N (N-5) and pyridinic-N (N-6) (Reprinted with permission from ref. 99. Copyright 2018 Wiley); (d) XRD patterns of various hard carbon materials synthesized at different temperatures (Reprinted with permission from ref. 79. Copyright 2021 Royal Society of Chemistry); (e) rate performances of S-doped hard carbon materials and (f) its comparison with previous work (Reprinted with permission from ref. 93. Copyright 2023 Elsevier). | ||
An important factor restricting the cycling performance of unmodified hard carbon anodes is the limited layer spacing relative to a larger K+ radius. In the process of repeated charge and/or discharge, the materials undergo serious irreversible deformation, which seriously affects the cycling life. Unfortunately, N-doping is exploited to supply plentiful defects, while it seems to be difficult to expand interlayer spacing. Some large-sized or highly electronegative heteroatom doping can cause graphitic lattice distortion of carbon and expand the layer spacing. Meanwhile, some heteroatoms may also bond between carbon layers to achieve the purpose of layer spacing expansion. Commonly, S, as a large radius doping element, can provide plenty of structural defects after entering into the carbon layer, while extending the layer spacing to 0.387 nm (Fig. 5(d)).79 Larger interlayer spacings are beneficial to alleviate the volume expansion, exhibiting an extremely low capacity-decay rate of 0.009% per cycle and retaining a satisfying specific capacity of 188.9 mA h g−1 after 1000 cycles. Besides, S-doping could establish more enduring active sites to break the limitations of K+ storage capacitance by absorption mechanism, achieving a desirable rate performance of 261 mA h·g−1 even at 5.0 A·g−1 (shown in Fig. 5(e) and (f)).100
In order to fit the actual application requirements, we usually expect to be able to achieve a variety of performance improvements in the simplest way. It is an effective and economic way to choose precursors containing different heteroatoms as reactants, such as amino acids and thermosetting resins. It is well known that N-doping can effectively promote the conductivity of hard carbon anodes, and the synergistic effect with other elements can make this improvement more obvious. The results indicated that N/O co-doping can effectively reduce the diffusion energy barrier of K+, so as to create a fast channel (Fig. 6(a)–(c)).103 Meanwhile, the DFT calculation gave strong evidence (Fig. 6(d)) that the N/O co-doped structures exhibit enhanced DOS around the Fermi level in comparison to only the N-doped carbon matrix, disclosing that the increase in electron transport efficiency. The rich vacancy defect, torn out by N/O co-doping, can effectively alleviate the drastic expansion of layer distance to relieve the volume variation after K+ intercalation.104 Besides, the widespread intrinsic carbon vacancy defects and N/O co-doping together induce the spontaneous migration of K+ in the bulk phase and improve the availability of active sites.
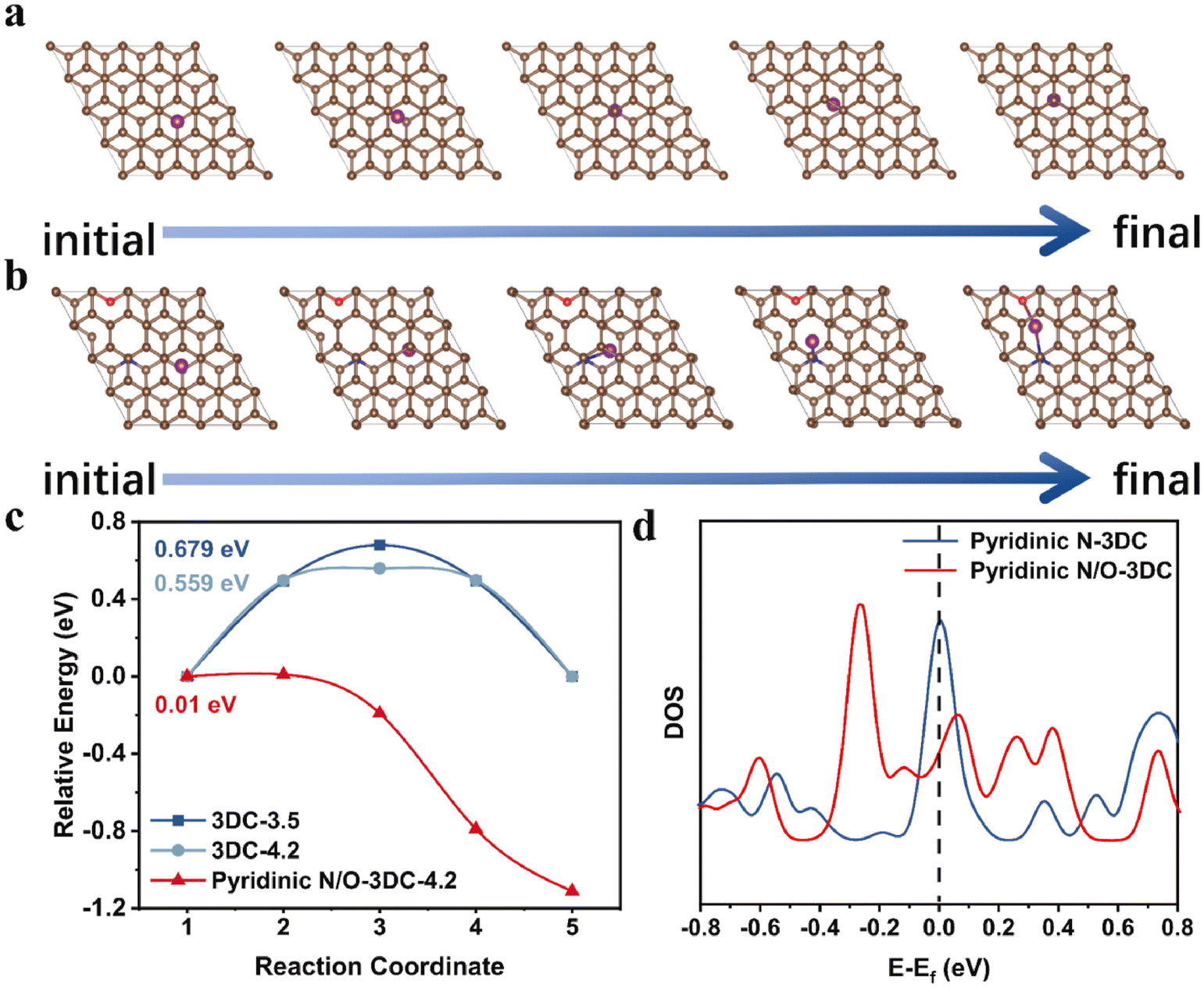 | ||
| Fig. 6 Model diagram of the minimum diffusion barrier path for K+ transport from one active site to another active site in (a) pristine graphite (3DC-3.5 and 3DC-4.2 with different interlayer distances), and (b) N/O co-doped hard carbon materials (pyridinic N/O-3DC with interlayer distance of 4.2 Å). (c) Comparison of diffusion energy barriers. (d) Comparison of DOS (Reprinted with permission from ref. 103. Copyright 2021 Elsevier). | ||
Expanding the interlayer spacing of hard carbons is the most direct and effective method to expedite K+ diffusion and maintain structural stability. Encouragingly, the enlarged graphitic interlayer spacing can also introduce affluent edge defects, which is conducive to achieve rapid absorption capacity.79 For example, single large-radius P doping can effectively enlarge the layer spacing, and the synergistic effect of N/P-co-doping is beneficial to reduce the adsorption energy of hard carbons for K+.105 As a result, the specific capacity rose from 140 to 253 mA h g−1, and the rate performance was also improved significantly (from 16 to 76 mA h g−1 at a current density of 20 A g−1).
Building on the previous work, a N/O/S-tri-doped hard carbon nanosphere born from polybenzoxazine showed excellent rate performance (capacity retention of 39.8% at 2000 mA g−1) and surprising ultra-long cycling life (218.9 mA h g−1 after 7300 cycles at 2000 mA g−1).107 Based on the results of DFT calculations, N/O/S-tri-doping can not only promote the ability of K+ adsorption, but also improve the electronic conductivity.
It is a pity that the current works of triple atom-doped hard carbons are still focused on the improvement of electrochemical properties. The complex structure makes it extremely difficult to study the synergistic mechanism among the three types of heteroatoms. Thus, there is a long way to explain the electrochemical behavior in complex systems.
3.2 Hard carbon composites
In order to overcome the limitation of single hard carbons, it is undoubtedly a new development trend to make up the deficiency by multiphase composites. At present, the focus of research is on the design of reasonable hard carbon composites to integrate the characteristics of different materials and improve their comprehensive properties.As another member of amorphous carbon materials, the soft carbon shows unique advantages in conductivity, but its cycle life, as the anode of PIBs, is restricted by the limited layer spacing. In the hard carbon, however, discontinuous graphite domains block the conducting network at the microscopic level. The researchers found that, when the hard and soft carbon phases were only in close proximity to each other on the same active particles (shown in Fig. 7(a)), the composite (HCS-SC) could combine the advantages of the soft carbon in terms of high rate performance with the hard carbon in terms of long cycle life.108 Brightly, the HCS-SC still exhibited an excellent capacity of up to 190 mA h g−1 even at a current density of 558 mA g−1 (2 C, shown in Fig. 7(b)), and retained an impressive capacity retention of 93% after 200 cycles at a current density of 279 mA g−1 (1 C). Similarly, another prepared composite (PI-700-P28) with only 20 wt% hard carbon achieved a high reversible specific capacity of 376.8 mA h g−1 and a competitive ICE of 71.04% (Fig. 7(c)),109 giving prominence to the prospect of hard–soft carbon composites.
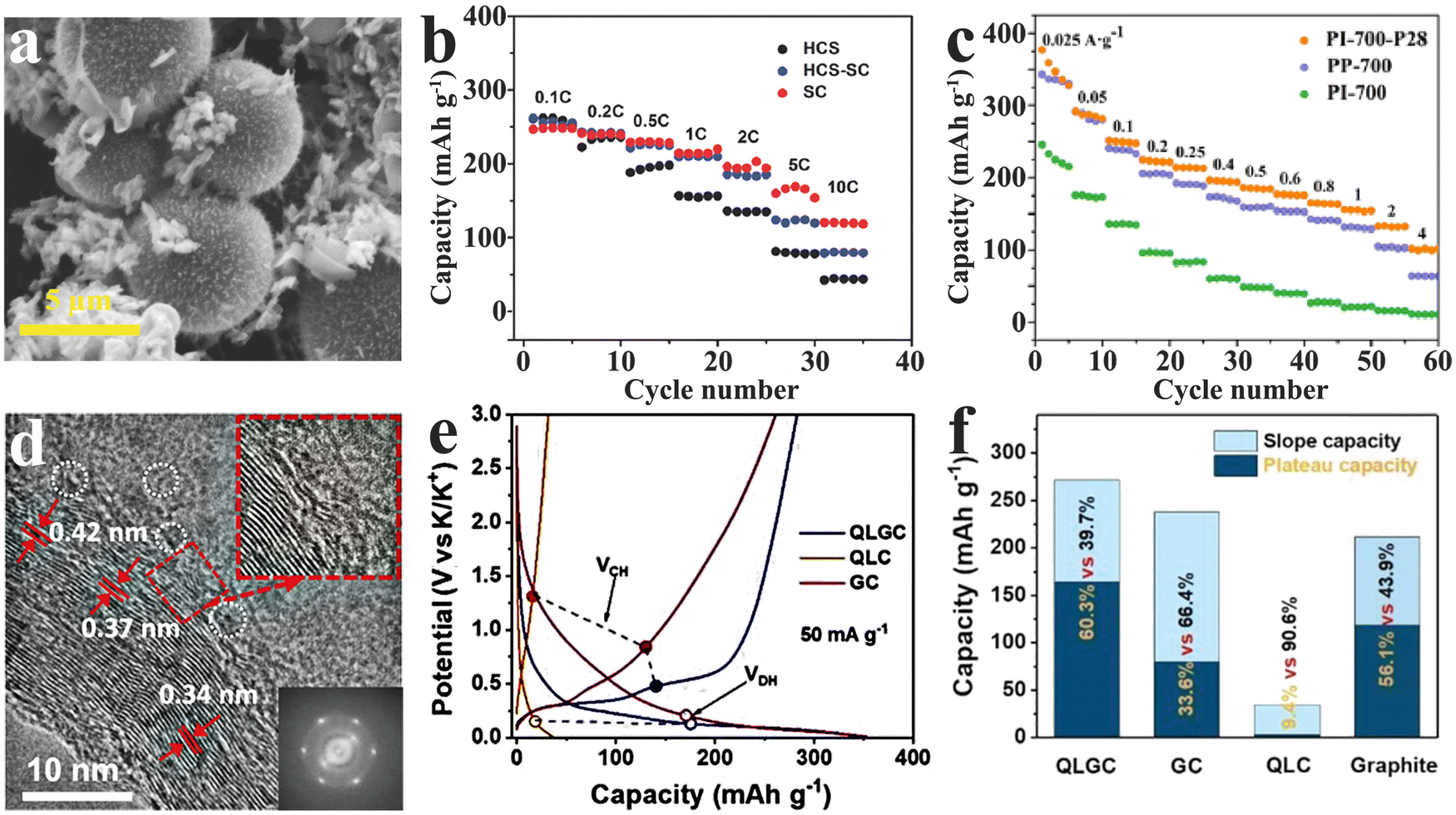 | ||
| Fig. 7 Hard carbon composites as PIB anodes: SEM image(a) and rate performance (b) of hard carbon spheres and soft carbon composites (HCS-SC) (Reprinted with permission from ref. 108. Copyright 2017 Wiley). (c) Rate performance of hard–soft carbon composites (PI-700-P28) (Reprinted with permission from ref. 109. Copyright 2017 Wiley). HR-TEM image (d), potential hysteresis (e) and capacity contribution distribution upon potential (f) of modified hard carbons (QLGC) (Reprinted with permission from ref. 112. Copyright 2022 Wiley). | ||
The special turbine-like graphite domain structure in the hard carbon endows a buffer zone to endure the volume expansion induced by K+ intercalation/deintercalation. Inevitably, these short-range ordered structures lead to a large voltage hysteresis (high polarization) at high rates due to insufficient conductivity.110 Previous studies have shown that an appropriate improvement of local graphitization degree in the hard carbon phase is conducive for improving the overall conductivity, thereby contributing to the efficiency of K+ storage.71,111 To achieve local graphitization, a modified hard carbon (QLGC) was developed by simple graphene-induced quaternized lignin carbonization.112 The results indicated that a little GO incorporation can efficaciously induce in situ partial graphitization of the hard carbon phase while preserving a wide enough K+-admission layer spacing (4.2 nm) in the amorphous region (Fig. 7(d)). As shown in Fig. 7(e) and (f), the treated hard carbon displayed a suppressive potential hysteresis and a remarkable low-potential-platform capacity with 164 mA h g−1 (stable plateau capacity contributions of 60.3%).
Troubled by the low theoretical capacity, the introduction of high-capacity non-carbon materials provides a new way to solve the problem. A porous carbon-loaded Sn4P3 composite (Sn4P3@C) was used for K+ storage, delivering a splendid capacity of 473.3 mA h g−1 at 50 mA g−1 and a superior rate performance of 183.6 mA h g−1 even at 2.0 A g−1.113 The Sn4P3@C composite also showed strong strength in the aspect of cycling stability, maintaining a reversible specific capacity of 181.5 mA h g−1 after 800 cycles at 500 mA g−1. The porous hard carbon made contribution to alleviate the volume change, enhance the electrical conductivity, and prevent the agglomeration of Sn4P3 particles during the whole charge/discharge process.
3.3 Biomass-based hard carbon materials
As an important source of hard carbon, biomass-based materials stand out because of their unique sustainable development and structural advantages. The inherent special and ordered biological architectures such as spherality, network and fiber of the biomass precursor are preserved after carbonization, which bodes well for the possibility of the self-template method. Moreover, the heteroatoms in biomass can be partially retained to form active sites for K+ adsorption. These intrinsic advantages of structures ensure the mechanical stability of high-strength electrodes during K+ repeated insertion/extraction and facilitate the expansion of electrolyte contact area.As a typical internal structure of biomass, the fiber is widely found in the biological world, such as bamboo, cotton, and animal hair. The derived fiber-shaped carbons, a one-dimensional (1D) material, have been recognized as promising candidates for high-performance anode materials. In nature, crustaceans such as dragon shrimp shells possess abundant chitin and CaCO3. As shown in Fig. 8(a), During the pyrolysis process of dragon shrimp shells, the carbon layer can grow along the CaCO3 nanoparticles and retain large amounts of N and O doping, forming a series of N/O-co-doped porous hard carbon nanoribbons (NOCNBs).66 When used in K+ storage, NOCNBs delivered a conspicuous reversible capacity of 468 mA h g−1 at 50 mA g−1 and an excellent cycling stability with a capacity of 277 mA h g−1 over 1600 cycles at 1000 mA g−1. The interconnected nanobelt-like morphology of NOCNBs, formed by vertically stacked nanofibers (Fig. 8(b)–(d)), ensures rapid K+ diffusion and electron transfer, exhibiting a glorious rate performance of 200 mA h g−1 even at a high current of 3200 mA g−1. In situ N/O-co-doping introduced additional active sites in the carbon skeleton and extended the layer spacing to 0.4 nm.
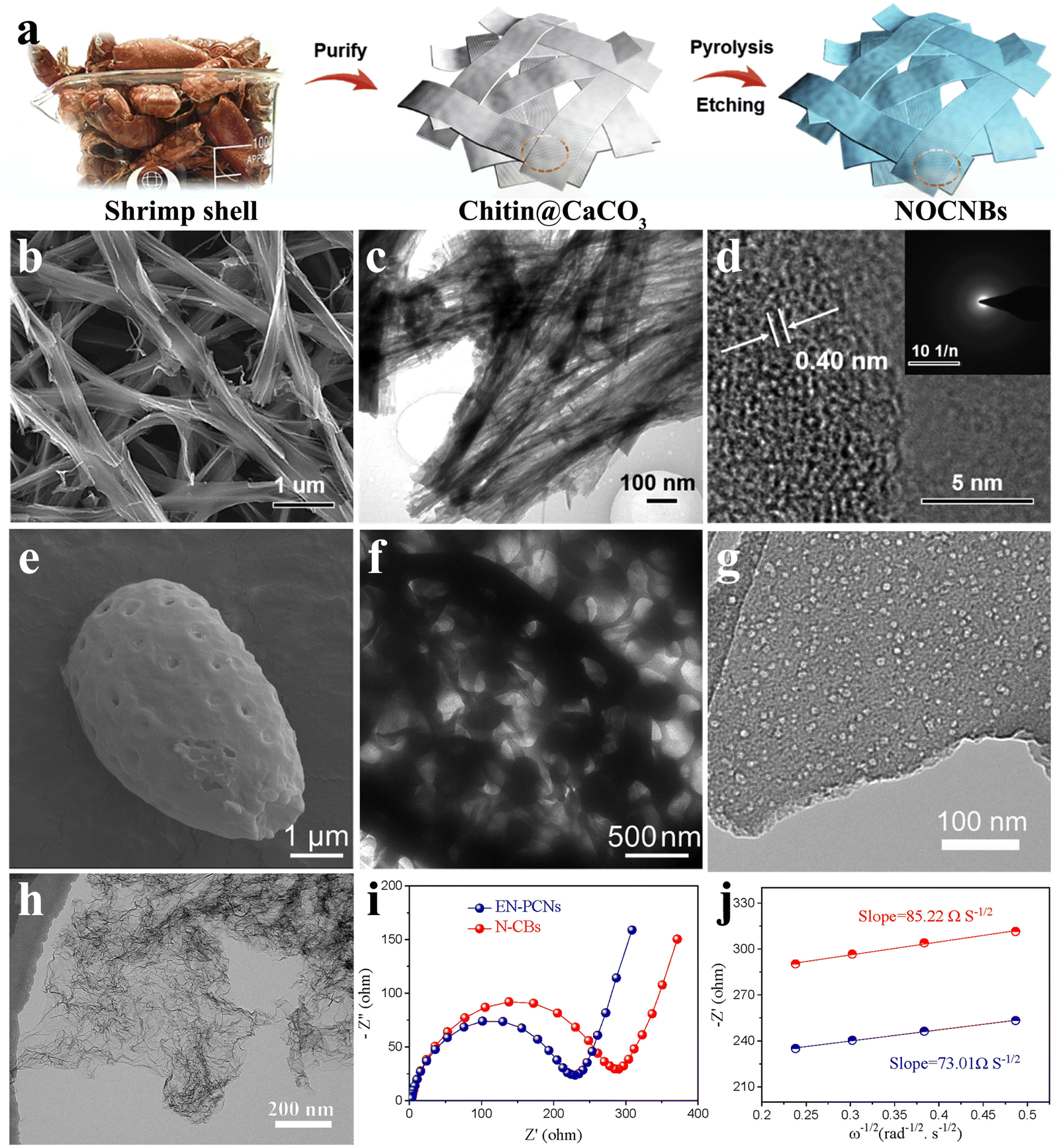 | ||
| Fig. 8 Biomass-based hard carbon materials as PIB anodes: (a) schematic illustration of the fabrication of taenioid biomass-based N/O-co-doped porous hard carbon nanoribbons (NOCNBs). SEM image (b), TEM image (c) and HRTEM image (d, inset is selected-area electron-diffraction patterns) of NOCNBs (Reprinted with permission from ref. 66. Copyright 2020 Elsevier). SEM image (e) and TEM image (f) of biomass-based hollow cage-like hard carbon (Reprinted with permission from ref. 114. Copyright 2020 Elsevier). (g) TEM image of potato-derived porous hard carbon (Reprinted with permission from ref. 115. Copyright 2019 Elsevier). (h) TEM image of porous carbon nanosheets (EN-PCNs). (i) Initial Nyquist plots of EN-PCNs and the control group. (j) Linear relation of ω−1/2versus Z′ of EN-PCNs and the control group (Reprinted with permission from ref. 117. Copyright 2022 Elsevier). | ||
In the electrochemical cycling process, the existence of the microscopic pore structure is helpful to relieve the pressure caused by volume expansion and sustain the stability of carbon structure. Moreover, the open channel corresponds to a fuller specific surface, favoring the exposure of internal active sites. For this purpose, researchers turned their attention to some special biomass materials with an internal hierarchical pore structure. The natural hollow cage-like double-wall structure of Ganoderma lucidum spore could be preserved completely after carbonization, which endowed hard carbons with enough specific surface area and relatively stable skeleton structure (Fig. 8(e) and (f)).114 The prepared cage-like porous carbon (CPC) showed a remarkable reversible capacity of 407 mA h g−1 at 50 mA g−1 after 50 cycles. Due to the excellent microstructure, the stability of CPC was particularly outstanding, exhibiting an extremely long cycling stability with 124.6 mA h g−1 after 700 cycles at a high current density of 1 A g−1. Commonly, some biomass materials containing monosaccharides or polysaccharides may undergo partial carbon chain decomposition during heat treatment, resulting in gas leakage and leaving abundant pores in the material. As shown in Fig. 8(g), potato-derived materials (PBPC-1000), for example, exhibit abundant smooth and crack-free internal mesoporous pores (specific surface area around 531.67 m2 g−1 and mean pore size around 10 nm).115 It showed excellent structural stability for K+ storage, with a teeny capacity decay of 0.0312% per cycle. The abundant open surface gives K+ the possibility of rapid transmission, presenting a rapid diffusion coefficient of 1.732 × 10−11 cm2 s−1. The structure-oriented rapid K+ transport corresponded to excellent rate performance, which retained a reversible capacity of 152 mA h g−1 at 1000 mA g−1.
In terms of shortening the K+ transport distance and improving the electrical conductivity, the carbon network structure shows unique advantages. This architecture makes it easier to transfer large amounts of charge near the interface between the materials and the electrolyte, that is, a lower diffusion impedance. An animal bone-derived 3D hierarchically porous carbon scaffold (3D-HPCS) was fabricated by one-step pyrolysis of chicken bones.116 When applied to K+ batteries, it showed that the charge-transfer resistance was around 1300 Ω at the initial cycle. Profiting from its preeminent surface chemistry, the prepared 3D-HPCS attained a splendid rate behavior (around 113 mA h g−1 at 580 mA g−1) and cycling stability (95% capacity retention after 2000 cycles). Recently, a kind of porous carbon nanosheet with high edge-nitrogen-doping (EN-PCNs) was introduced for potassium storage (Fig. 8(h)), with a promising charge-transfer resistance of 197.5 Ω (Fig. 8(i) and (j)).117 Moreover, the EN-PCN electrode delivered a high rate performance of 226.3 mA h g−1 at 502.2 mA h g−1 and revealed a stable capacity of 255.4 mA h g−1 over 100 cycles at 195.3 mA h g−1. It has to be admitted that cheap and easily available biomass-based hard carbon materials have innate advantages in practical applications, but the electrochemical properties need to be further optimized.
4 Conclusion and outlook
Hard carbons, as commercialized anodes, shine in the field of energy storage. With the development of PIBs, the hard carbon as an anode with strong competitiveness has attracted widespread attention. However, the inevitable large radius of K+ presents a new challenge to the electrochemical stability of hard carbon materials.For the energy storage behavior of the hard carbon, its structure controls the mechanism of K+ storage, and in turn, the mechanism puts forward various requirements for microstructures. Technically, there are two mechanisms for K+ storage in hard carbons: one by intercalation between turbine-like quasi-graphite domains, and the other by adsorption onto the surface, defects, and nano-pores of materials. In general, interestingly, these two mechanisms tend not to emerge independently, but rather in tandem. The reversible intercalation of K+ in the quasi-graphite domains is often accompanied by the process of adsorption, and the contribution of adsorption-driven pseudocapacitance is indispensable in the intercalation energy-harvesting. It has been widely proved that a larger interlayer spacing and more defects are beneficial to obtain more stable cycling properties and better storage capacity (even higher than the theoretical capacity of GICs).
However, until date, hard carbons as anodes for PIBs have fallen far short of commercial requirements, and their further development still faces several challenges:
(1) Extensive research has shown that heteroatom doping can effectively promote the electrochemical performance of the hard carbon, but it has to be admitted that excessive heteroatom doping will block the continuous conductive network, resulting in a decrease in the conductivity and the impact of rate performance. In order to achieve the compromise between the contribution of pseudocapacitance and the conductivity, it is necessary to adjust the loading content and type of doped heteroatoms. Meanwhile, it is a cost-effective means to combine hard carbons with other materials with salient electrical conductivity.
(2) Sufficient specific surface is conducive to shorten the diffusion path and accelerate the transfer rate of K+, which plays a positive role in realizing the rapid energy storage at high current density. However, inevitably, too much open surface leads to irreversible capacity and electrolyte consumption. At the same time, the large specific surface is associated with abundant pore structures. When the pore volume reaches a certain level, the carbon network may collapse, which is not conducive to the stability of the material. Therefore, it is the focus of future research to precisely regulate the micro-nano structure of hard carbon and form reasonable hierarchical pores.
(3) Current research shows that the unsatisfactory ICE is an inevitable pain point for PIBs. In laboratory half-cell evaluation, this effect is usually shielded by an unlimited supply of K+, but fatal in a full cell. This irreversible loss can be partially recovered by pre-potassium treatment, and more stable interface control is also a feasible approach.
At present, the application of hard carbon anodes in PIBs is developed vigorously, but some deep-seated mechanisms need to be explored to guide the direction for high-energy density and long-lifespan anodes. The researchers have noted the positive effects of interface on hard carbons, and expected to improve the ICE by regulating the electrolyte. In short, the hard carbon is a kind of promising and controllable anode, and researchers hope to make full use of its properties, making it outstanding compared with the rest of the anodes.
Author contributions
Xiaoyi Lu: conceptualization, formal analysis, writing–draft, creating the graphics, investigation, reviewing and editing; Handong Peng: investigation, analysis, writing–review and editing; Guoping Liu: investigation, analysis, writing–review and editing; Fangya Qi: investigation and analysis; Chenglong Shi: investigation and analysis; Sheng Wu: investigation and analysis; Yanxue Wu: methodology, supervision, reviewing and editing; Huanping Yang: investigation, supervision, writing–review and editing; Jie Shan: resources, supervision, writing–review and editing; Zhipeng Sun: conceptualization, methodology, investigation, resources, supervision, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Guangdong University of Technology Hundred Talents Program (No. 220418136), Macao Young Scholars Program (AM2021009) and Basic Research Expenses Program of Universities in Xinjiang autonomous region (XJEDU2022P107).Notes and references
- Q. Yang, H. Li, C. Feng, Q. Ma, L. Zhang, R. Wang, J. Liu, S. Zhang, T. Zhou, Z. Guo and C. Zhang, Nanoscale, 2022, 14, 5814–5823 RSC.
- J. Xie, J. Li, X. Li, H. lei, W. Zhuo, X. Li, G. Hong, K. N. Hui, L. Pan and W. Mai, CCS Chem., 2021, 3, 791–799 CrossRef CAS.
- J. Zhang, T. Liu, X. Cheng, M. Xia, R. Zheng, N. Peng, H. Yu, M. Shui and J. Shu, Nano Energy, 2019, 60, 340–361 CrossRef CAS.
- B. Wang, Y. Peng, F. Yuan, Q. Liu, L. Sun, P. Zhang, Q. Wang, Z. Li and Y. A. Wu, J. Power Sources, 2021, 484, 229244 CrossRef CAS.
- H. Lei, J. Li, X. Zhang, L. Ma, Z. Ji, Z. Wang, L. Pan, S. Tan and W. Mai, InfoMat, 2022, 4, e12272 CrossRef CAS.
- W. Zhang, J. Yin, W. Wang, Z. Bayhan and H. N. Alshareef, Nano Energy, 2021, 83, 105792 CrossRef CAS.
- X. Min, J. Xiao, M. Fang, W. Wang, Y. Zhao, Y. Liu, A. M. Abdelkader, K. Xi, R. V. Kumar and Z. Huang, Energy Environ. Sci., 2021, 14, 2186–2243 RSC.
- Z. Wu, J. Zou, S. Chen, X. Niu, J. Liu and L. Wang, J. Power Sources, 2021, 484, 229307 CrossRef CAS.
- P. Liu and D. Mitlin, Acc. Chem. Res., 2020, 53, 1161–1175 CrossRef CAS PubMed.
- Z. Feng, R. Chen, R. Huang, F. Zhang, W. Liu and S. Liu, Metals, 2023, 13, 658 CrossRef CAS.
- Y. Liu, C. Gao, L. Dai, Q. Deng, L. Wang, J. Luo, S. Liu and N. Hu, Small, 2020, 16, 2004096 CrossRef CAS PubMed.
- X. Zhang, J. Meng, X. Wang, Z. Xiao, P. Wu and L. Mai, Energy Storage Mater., 2021, 38, 30–49 CrossRef.
- C. Zhang, H. Pan, L. Sun, F. Xu, Y. Ouyang and F. Rosei, Energy Storage Mater., 2021, 38, 354–378 CrossRef.
- P. Hunderkar, S. Basu, X. Fan, L. Li, A. Yoshimura, T. Gupta, V. Sarbada, A. Lakhnot, R. Jain, S. Narayanan, Y. Shi, C. Wang and N. Koratkar, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5588–5594 CrossRef PubMed.
- S. Wang, P. Xiong, X. Guo, J. Zhang, X. Gao, F. Zhang, X. Tang, P. H. L. Notten and G. Wang, Adv. Funct. Mater., 2020, 30, 2001588 CrossRef CAS.
- Y. Feng, M. Xu, T. He, B. Chen, F. Gu, L. Zu, R. Meng and J. Yang, Adv. Mater., 2021, 33, 2007262 CrossRef CAS.
- Y. Wu, J. Zheng, Y. Tong, X. Liu, Y. Sun, L. Niu and H. Li, ACS Appl. Mater. Interfaces, 2021, 13, 51066–51077 CrossRef CAS PubMed.
- S. Imtiaz, N. Kapuria, I. S. Amiinu, A. Sankaran, S. Singh, H. Geaney, T. Kennedy and K. M. Ryan, Adv. Funct. Mater., 2023, 33, 2209566 CrossRef CAS.
- W. Xiao, X. Li, B. Cao, G. Huang, C. Xie, J. Qin, H. Yang, J. Wang and X. Sun, Nano Energy, 2021, 83, 105772 CrossRef CAS.
- Y. Shen, J. Qian, H. Yang, F. Zhong and X. Ai, Small, 2020, 16, 1907602 CrossRef CAS.
- H. Zheng, Q. Qu, L. Zhang, G. Liu and V. S. Battaglia, RSC Adv., 2012, 2, 4904–4912 RSC.
- L. Xie, C. Tang, Z. Bi, M. Song, Y. Fan, C. Yan, X. Li, F. Su, Q. Zhang and C. Chen, Adv. Energy Mater., 2021, 11, 2101650 CrossRef CAS.
- D. Cheng, X. Zhou, H. Hu, Z. Li, J. Chen, L. Mao, X. Ye and H. Zhang, Carbon, 2021, 182, 758–769 CrossRef CAS.
- F. Yuan, D. Zhang, Z. Li, H. Sun, Q. Yu, Q. Wang, J. Zhang, Y. Wu, K. Xi and B. Wang, Small, 2020, 18, 2107113 CrossRef PubMed.
- H. Zhang, C. Luo, H. He, H. Wu, L. Zhang, Q. Zhang, H. Wang and M. Wang, Nanoscale Horiz., 2020, 5, 895–903 RSC.
- A. Beda, P. Taberna, P. Simon and C. M. Ghimbeu, Carbon, 2018, 139, 248–257 CrossRef CAS.
- P. Yu, W. Tang, F. Wu, C. Zhang, H. Luo, H. Liu and Z. Wang, Rare Met., 2020, 39, 1019–1033 CrossRef CAS.
- Z. Yu, B. Qin, Z. Ma, J. Huang, S. Li, H. Zhao, H. Li, Y. Zhu, H. Wu and S. Yu, Adv. Mater., 2019, 31, 1900651 CrossRef PubMed.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- M. Dahbi, M. Kiso, K. Kubota, T. Horiba, T. Chafik, K. Hida, T. Matsuyama and S. Komaba, J. Mater. Chem. A, 2017, 5, 9917–9928 RSC.
- J. M. Stratford, A. K. Kleppe, D. S. Keeble, P. A. Chater, S. S. Meysami, C. J. Wright, J. Barker, M. Titirici, P. K. Allen and C. P. Grey, J. Am. Chem. Soc., 2021, 143, 14274–14286 CrossRef CAS PubMed.
- Y. Xu, S. Duan, Y. Sun, D. Bin, X. Tao, D. Zhang, Y. Liu, A. Cao and L. Wan, J. Mater. Chem. A, 2019, 7, 4334–4352 RSC.
- Y. Zhu, Y. Wang, Y. Wang, T. Xu and P. Chang, Carbon Energy, 2022, 4, 1182–1213 CrossRef CAS.
- N. Li, Z. Jiang, X. Wu, R. Cai, Z. Mo, C. Wu, S. Wu and S. Liu, Chem. Eng. J., 2022, 431, 133736 CrossRef CAS.
- G. Xia, C. Wang, P. Jiang, J. Lu, J. Diao and Q. Chen, J. Mater. Chem. A, 2019, 7, 12317–12324 RSC.
- L. Zhang, W. Wang, S. Lu and Y. Xiang, Adv. Energy Mater., 2021, 11, 2003640 CrossRef CAS.
- N. Sun, Q. Zhu, B. Anasori, P. Zhang, U. Liu, Y. Gogotsi and B. Xu, Adv. Funct. Mater., 2019, 29, 1906282 CrossRef CAS.
- Q. Jin, K. Wang, P. Feng, Z. Zhang, S. Cheng and K. Jiang, Energy Storage Mater., 2020, 27, 43–50 CrossRef.
- L. Xie, C. Tang, M. Song, X. Guo, X. Li, J. Li, C. Yan, Q. Kong, G. Sun, Q. Zhang, F. Su and C. Chen, J. Energy Chem., 2022, 72, 554–569 CrossRef CAS.
- S. Alvin, H. S. Cahyadi, J. Hwang, W. Chang, S. K. Kwak and J. Kim, Adv. Energy Mater., 2020, 10, 2000283 CrossRef CAS.
- Z. Xu, S. Du, Z. Yi, J. Han, C. Lai, Y. Xu and X. Zhou, ACS Appl. Energy Mater., 2020, 3, 11410–11417 CrossRef CAS.
- D. Wang, G. Du, D. Han, Q. Su, S. Ding, M. Zhang, W. Zhao and B. Xu, Carbon, 2021, 181, 1–8 CrossRef CAS.
- Z. Zhu, W. Zhong, Y. Zhang, P. Dong, S. Sun, Y. Zhang and X. Li, Carbon Energy, 2021, 3, 541–553 CrossRef CAS.
- Z. Liu, S. Wu, Y. Song, T. Yang, Z. Ma, X. Tian and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 47674–47684 CrossRef CAS.
- M. Chen, W. Wang, X. Liang, S. Gong, J. Liu, Q. Wang, S. Guo and H. Yang, Adv. Energy Mater., 2018, 8, 1800171 CrossRef.
- Z. Wu, J. Zou, Y. Zhang, X. Lin, D. Fry, L. Wang and J. Liu, Chem. Eng. J., 2022, 427, 131547 CrossRef CAS.
- Q. Wang, C. Gao, W. Zhang, S. Luo, M. Zhou, Y. Liu, R. Liu, Y. Zhang, Z. Wang and A. Hao, Electrochim. Acta, 2019, 324, 134902 CrossRef CAS.
- M. Hu, J. Song, H. Fan, L. Bai, Y. Wang, S. Liu, Y. Jin, Y. Cui and W. Liu, Chem. Eng. J., 2023, 451, 138452 CrossRef CAS.
- X. Chen, X. Cheng and Z. Liu, J. Energy Chem., 2022, 68, 688–698 CrossRef CAS.
- D. Saurel, B. Orayech, B. Xiao, D. Carriazo, X. Li and T. Rojo, Adv. Energy Mater., 2018, 8, 1703268 CrossRef.
- J. Chattopadhyay, A. Mukherjee, C. E. Hamilton, J. Kang, S. Chakraborty, W. Guo, K. F. Kelly, A. R. Barron and E. Billups, J. Am. Chem. Soc., 2008, 130, 5414–5415 CrossRef CAS PubMed.
- Y. Qi, Y. Lu, L. Liu, X. Qi, F. Ding, H. Li, X. Huang, L. Chen and Y. Hu, Energy Storage Mater., 2020, 26, 577–584 CrossRef.
- S. Guo, Y. Chen, L. Tong, Y. Cao, H. Jiao, Z. Long and X. Qiu, Electrochim. Acta, 2022, 410, 140017 CrossRef CAS.
- L. Ma, J. Li, T. Wu, P. Sun, S. Tan, H. Wang, W. Xie, L. Pan, Y. Yamauchi and W. Mai, Nano Energy, 2021, 87, 106150 CrossRef CAS.
- Y. Qian, S. Jiang, Y. Li, Z. Yi, J. Zhou, T. Li, Y. Han, Y. Wang, J. Tian, N. Lin and Y. Qian, Adv. Energy Mater., 2019, 9, 1901676 CrossRef.
- J. Xu, C. Fan, M. Ou, S. Sun, Y. Xu, Y. Liu, X. Wang, Q. Li, C. Fang and J. Han, Chem. Mater., 2022, 34, 4202–4211 CrossRef CAS.
- Z. Zhou, W. G. Bouwman, H. Schut and C. Pappas, Carbon, 2014, 69, 17–24 CrossRef CAS.
- H. Onuma, K. Kubota, S. Muratsubaki, W. Ota, M. Shishkin, H. Sato, K. Yamashita, S. Yasuno and S. Komaba, J. Mater. Chem. A, 2021, 9, 11187–11200 RSC.
- M. Rykner and M. Chandesris, J. Phys. Chem. C, 2022, 126, 5457–5472 CrossRef CAS.
- J. Zhao, X. He., W. Lai, Z. Yang, X. Liu, L. Li, Y. Qiao, Y. Xiao, L. Li, X. Wu and S. Chou, Adv. Energy Mater, 2023, 13, 2300444 CrossRef CAS.
- X. Zhao, Y. Ding, Q. Xu, X. Yu, Y. Liu and H. Shen, Adv. Energy Mater., 2019, 9, 1803648 CrossRef.
- C. Cai, Y. Chen, P. Hu, T. Zhu, X. Li, Q. Yu, L. Zhou, X. Yang and L. Mai, Small, 2022, 18, 2105303 CrossRef CAS.
- S. Alvin, C. Chandra and J. Kim, Chem. Eng. J., 2021, 411, 128490 CrossRef CAS.
- Z. Yu, C. Chen, Q. Lin, J. Liu, M. Tang, Y. Zhu and B. Zhang, Energy Storage Mater., 2023, 60, 102805 CrossRef.
- X. Han, T. Chen, P. Zhang, Y. Qi, P. Yang, Y. Zhao, M. Shao, J. Wu, J. Weng, S. Li and F. Huo, Adv. Funct. Mater., 2022, 32, 2109672 CrossRef CAS.
- K. Zhang, Q. He, F. Xiong, J. Zhou, Y. Zhao, L. Mai and L. Zhang, Nano Energy, 2020, 77, 105018 CrossRef CAS.
- S. Chong, L. Yuan, T. Li, C. Shu, S. Qiao, S. Dong, Z. Liu, J. Yang, H. K. Liu, S. X. Dou and W. Huang, Small, 2022, 18, 2104296 CrossRef CAS.
- J. Du, S. Gao, P. Shi, J. Fan, Q. Xu and Y. Min, J. Power Sources, 2020, 451, 227727 CrossRef CAS.
- J. Hu, Y. Xie, M. Yin and Z. Zhang, J. Energy Chem., 2020, 49, 327–334 CrossRef.
- S. Huang, Z. Li, B. Wang, J. Zhang, Z. Peng, R. Qi, J. Wang and Y. Zhao, Adv. Funct. Mater., 2018, 28, 1706294 CrossRef.
- H. Kim, J. C. Hyun, J. I. Jung, J. B. Lee, J. Choi, S. Y. Cho, H. Jin and Y. S. Yun, J. Mater. Chem. A, 2022, 10, 2055–2063 RSC.
- H. Wang, A. Artemova, G. Yang, H. Wang, L. Zhang, X. Cao, E. Akhipova, J. Liu, Y. Huang, J. Lin and Z. Shen, J. Power Sources, 2020, 466, 228303 CrossRef CAS.
- M. Yang, Q. Kong, W. Feng and W. Yao, Carbon, 2021, 176, 71–82 CrossRef CAS.
- X. Lin, J. Huang and B. Zhang, Carbon, 2019, 143, 138–146 CrossRef CAS.
- X. Wu, C. W. K. Lam, N. Wu, S. Pang, Z. Xing, W. Zhang and Z. Ju, Mater. Today Energy, 2019, 11, 182–191 CrossRef CAS.
- S. Mo, L. Liu, H. Zhao and Y. Lei, Carbon Energy, 2020, 2, 350–369 CrossRef.
- S. Zeng, X. Chen, R. Xu., X. Wu, Y. Feng, H. Zhang, S. Peng and Y. Yu, Nano Energy, 2020, 73, 104807 CrossRef CAS.
- C. Chen, M. Wu, Y. Wang and K. Zaghib, J. Power Sources, 2019, 444, 227310 CrossRef CAS.
- R. Huang, X. Zhang, Z. Qu, X. Zhang, J. Lin, F. Wu, R. Chen and L. Li, J. Mater. Chem. A, 2022, 10, 682–689 RSC.
- W. Feng, N. Feng, W. Liu, Y. Cui, C. Chen, T. Dong, S. Liu, W. Deng, H. Wang and Y. Jin, Adv. Energy Mater., 2021, 11, 2003215 CrossRef CAS.
- B. Cao, Q. Zhang, H. Liu, B. Xu, S. Zhang, T. Zhou, J. Mao, W. K. Pang, Z. Guo, A. Li, J. Zhou, X. Chen and H. Song, Adv. Energy Mater., 2018, 8, 1801149 CrossRef.
- J. Sun, L. Ma, H. Sun, Y. Xu, J. Li, W. Mai and B. Liu, Chem. Eng. J., 2023, 455, 140902 CrossRef CAS.
- Z. Wu, L. Wang, J. Huang, J. Zou, S. Chen, H. Cheng, C. Jiang, P. Gao and X. Niu, Electrochim. Acta, 2019, 306, 446–453 CrossRef CAS.
- Z. Jian, Z. Xing, C. Bommier, Z. Li and X. Ji, Adv. Energy Mater., 2016, 6, 1501874 CrossRef.
- H. Tan, R. Zhou and B. Zhang, J. Power Sources, 2021, 506, 230179 CrossRef CAS.
- Q. Li, Y. Zhang, Z. Chen, J. Zhang, Y. Tao and Q. Yang, Adv. Energy Mater., 2022, 12, 2201574 CrossRef CAS.
- H. Deng, L. Wang, S. Li, M. Zhang, T. Wang, J. Zhou, M. Chen, S. Chen, J. Cao, Q. Zhang, J. Zhu and B. Lu, Adv. Funct. Mater., 2021, 31, 2107246 CrossRef CAS.
- Y. Xu, X. Sun, Z. Li, L. Wei, G. Yao, H. Niu, Y. Yang, F. Zheng and Q. Chen, Nanoscale, 2021, 13, 19634–19641 RSC.
- R. C. Cui, B. Xu, H. J. Dong, C. C. Yang and Q. Jiang, Adv. Sci., 2020, 7, 1902547 CrossRef CAS.
- C. Gao, Q. Wang, S. Luo, Z. Wang, Y. Zhang, Y. Liu, A. Hao and R. Guo, J. Power Sources, 2019, 415, 165–171 CrossRef CAS.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef.
- Y. Li, W. Zhong, C. Yang, F. Zheng, Q. Pan, Y. Liu, G. Wang, X. Xiong and M. Liu, Chem. Eng. J., 2019, 358, 1147–1154 CrossRef CAS.
- C. Chen, Z. Wang, B. Zhang, L. Miao, J. Cai, L. Peng, Y. Huang, J. Jiang, Y. Huang, L. Zhang and J. Xie, Energy Storage Mater., 2017, 8, 161–168 CrossRef.
- C. Chen, K. Zhao, M. La and C. Yang, Materials, 2022, 15, 4249 CrossRef CAS PubMed.
- Y. Feng, K. Wu, S. Wu, M. He, X. Xu and M. Xue, ACS Appl. Energy Mater., 2022, 5, 12966–12976 CrossRef CAS.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Adv. Energy Mater., 2020, 10, 2000927 CrossRef CAS.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- B. Wang, F. Yuan, Q. Yu, W. Li, H. Sun, L. Zhang, D. Zhang, Q. Wang, F. Lai and W. Wang, Energy Storage Mater., 2021, 38, 329–337 CrossRef.
- D. Li, X. Ren, Q. Ai, Q. Sun, L. Zhu, Y. liu, Z. Liang, R. Peng, P. Si, J. Lou, J. Feng and L. Ci, Adv. Energy Mater., 2018, 8, 1802386 CrossRef.
- F. Wang, D. Li, G. Zhang, J. Li, C. Zhang, D. Wei, J. Yang, C. Ye, J. Tan and J. Liu, Appl. Surf. Sci., 2023, 614, 156149 CrossRef CAS.
- J. Li, J. Cao, X. Li, J. Hu, Y. Zhang, H. M. K. Sari, C. Lv, L. V. Zatovsky and W. Han, J. Energy Chem., 2021, 55, 420–427 CrossRef CAS.
- X. Ma, N. Xiao, J. Xiao, X. Song, H. Guo, Y. Wang, S. Zhao, Y. Zhong and J. Qiu, Carbon, 2021, 179, 33–41 CrossRef CAS.
- Y. Sun, Q. Wu, Y. Wang, C. Li, X. Liang and H. Xiang, J. Power Sources, 2021, 512, 230530 CrossRef CAS.
- J. Wang, Y. Qin, L. Lin, S. Zhang, X. Pei, Z. Niu, X. Zheng and D. Li, Chem. Eng. J., 2023, 457, 141253 CrossRef CAS.
- Q. Wang, Y. Wang, J. Zeng, C. Xu, P. Liu, Y. Meng, C. Zhang, Y. Wang, L. Gao, R. Ding, J. Liu, X. Jiang, Y. Zhang, J. Tang and X. Wang, FlatChem, 2022, 34, 100398 CrossRef CAS.
- M. Chen, Y. Cao, C. Ma and H. Yang, Nano Energy, 2021, 81, 105640 CrossRef CAS.
- X. Chen, W. Zhou, J. Liu, Y. Wu and Z. Liu, J. Energy Chem., 2023, 77, 338–347 CrossRef CAS.
- Z. Jian, S. Hwang, Z. Li, A. S. Hernandez, X. Wang, Z. Xing, D. Su and X. Ji, Adv. Funct. Mater., 2017, 27, 1700324 CrossRef.
- M. Wang, Y. Zhu, Y. Zhang, T. Yang, J. Duan and C. Wang, Electrochim. Acta, 2021, 368, 137649 CrossRef CAS.
- L. Zhao, Z. Hu, W. Lai, Y. Tao, J. Peng, Z. Miao, Y. Wang, S. Chou, H. Liu and S. Dou, Adv. Energy Mater., 2021, 11, 2002704 CrossRef CAS.
- Z. Wang, X. Wang, Y. Bai, H. Yang, Y. Li, S. Guo, G. Chen, Y. Li, H. Xu and C. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 2481–2489 CrossRef CAS PubMed.
- L. Zhong, W. Zhang, S. Sun, L. Zhao, W. Jian, X. He, Z. Xing, Z. Shi, Y. Chen, H. N. Alshareef and X. Qiu, Adv. Funct. Mater., 2023, 33, 2211872 CrossRef CAS.
- D. Li, Y. Zhang, Q. Sun, S. Zhang, Z. Wang, Z. Liang, P. Si and L. Ci, Energy Storage Mater., 2019, 23, 367–374 CrossRef.
- M. Yang, J. Dai, M. He, T. Duan and W. Yao, J. Colloid Interface Sci., 2020, 567, 256–263 CrossRef CAS.
- W. Cao, E. Zhang, J. Wang, Z. Liu, J. Ge, X. Yu, H. Yang and B. Lu, Electrochim. Acta, 2019, 293, 364–370 CrossRef CAS.
- X. Yuan, B. Zhu, J. Feng, C. Wang, X. Cai and R. Qin, Mater. Res. Bull., 2021, 139, 111282 CrossRef CAS.
- L. Zhu, Y. Wang, M. Wang, M. Huang, Y. Huang, Z. Zhang, J. Yu, Y. Qu, C. Li and Z. Yang, Carbon, 2022, 187, 302–309 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |