
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon atom doping in heterotrimetallic sulfides for non-noble metal alkaline water electrolysis†
Mohamed Barakat Zakaria
Hegazy
 *ab,
Leila
Bahri
c,
David
Tetzlaff
ad,
Sebastian A.
Sanden
a and
Ulf-Peter
Apfel
*ad
*ab,
Leila
Bahri
c,
David
Tetzlaff
ad,
Sebastian A.
Sanden
a and
Ulf-Peter
Apfel
*ad
aInorganic Chemistry I-Technical Electrochemistry, Faculty for Chemistry and Biochemistry, Ruhr University Bochum, 44801 Bochum, Germany. E-mail: mohamed.barakat@rub.de; ulf.apfel@rub.de
bDepartment of Chemistry, Faculty of Science, Tanta University, 31527 Tanta, Egypt
cDepartment of Chemistry, College of Sciences, University of Hafar Al-Batin, 39524 Hafar Al-Batin, Kingdom of Saudi Arabia
dFraunhofer Institute for Environmental, Department for Electrosynthesis, Safety and Energy Technology UMSICHT, 46047 Oberhausen, Germany
First published on 23rd June 2023
Abstract
This study investigates the modification of materials by doping with foreign elements to enhance electrocatalytic activity and focuses on the engineering of an inorganic material composed of transition heterometal-rich pentlandite (Fe3Co3Ni3S8, FCNS) doped with silicon (FCNSSi) as a bifunctional catalyst for the overall electrochemical water splitting process. The FCNSSi electrode exhibits remarkable catalytic activity for oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). The OER performance of FCNSSi was evaluated in a 1.0 M KOH solution, achieving an overpotential of 313 mV at 10 mA cm−2. The FCNSSi electrode exhibits a current density of −10 mA cm−2 at a remarkably low overpotential of 164 mV with a Tafel slope of 80.7 mV dec−1 in HER. Density functional theory (DFT) calculation suggests that Si doping adjusts the binding energies of intermediates on the surface, which weakened the *OH, *O, and *OOH adsorption energies, resulting in enhanced activity for both OER and HER. Moreover, Si doping enhances the hydrogen adsorption activity of all sites. Finally, a two-electrode zero-gap cell assembly was used to investigate the durability of FCNSSi catalyst towards efficient and durable alkaline water electrolysis, demonstrating the promising potential of this catalyst for practical applications at 500 mA cm−2.
Introduction
Climate change has emerged as a pressing global concern due to the significant changes it causes in Earth's atmosphere, temperature, and air quality, primarily resulting from the emission of CO2 generated by the combustion of fossil fuels and industrial steam reforming for hydrogen production.1,2 The development of renewable energy sources capable of replacing these environmentally damaging technologies has become an urgent requirement. Among the promising candidates for clean hydrogen production with zero CO2 emission, water electrolysis has recently gained significant attention.3 Hydrogen generated through water electrolysis is highly demanded for sustainable economic and societal development.4 However, this process is energetically demanding and requires a minimum voltage of 1.23 V versus RHE under standard conditions,5 which is only achievable at a much higher potential of at least 1.6 to 2.0 V vs. RHE in practice due to cathodic and anodic overpotentials.6 Additional energy losses occur due to multiple proton and electron transfer reactions resulting in pH changes and irreversible redox reactions.7To minimize the required energy and maintain continuous and efficient electrochemical water splitting for sustainable hydrogen production, the development of stable and efficient electrocatalysts is critical.8 However, the current industrial use of scarce noble metal-based electrocatalysts, such as Pt/C and IrO2, in proton exchange membrane (PEM) technology incurs high H2 production costs.9 Similarly, the developed Ni and Co-based materials, alloys, hydroxides, etc. catalysts used in alkaline water electrolyzers still lack in efficiency compared to commercial PEM.10
Recent research efforts have focused on using earth-abundant elements as non-precious catalysts for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).11–18 Along this line, transition metal chalcogenides have emerged as promising catalysts for water electrolysis due to their abundance, cost-effectiveness, high conductivity, efficient redox chemistry, and stability.19–23 A variety of nickel, iron, and cobalt-based transition metal-based catalysts have been developed for electrocatalytic hydrogen production to date.11,12,24–26 Among them, metal-rich pentlandite-type catalysts have been suggested as a promising material class.25,26 For instance, a Fe4.5Ni4.5S8 electrode from the natural ore pentlandite achieved a current density of 10 mA cm−2 at 280 mV for catalytic hydrogen evolution under acidic conditions.27 Similarly, Co9S8 nanoparticles supported by carbon nanosheets catalyst exhibited an overpotential of 294 mV at 10 mA cm−2 for OER in alkaline electrolyte.28 Our group recently adapted a mechanochemical method for making various compositions of nanosized pentlandites which work efficiently as cathodic electrocatalysts in a zero-gap PEM electrolyzer for water splitting.29 Additionally, more insights into the electrocatalytic activity of pentlandites upon variation of the metal content were reported.30 To further improve the HER and OER performance of these materials, doping with non-metallic foreign elements such as nitrogen and phosphorus heteroatoms was shown as a viable strategy.24,31
In addition to transition metal chalcogenides, silicon has been extensively investigated as an electrode material in electrochemical semiconductors due to its high theoretical gravimetric hydrogen storage efficiency of 14 wt%, suggesting its excellent potential for hydrogen generation.32 Yang and colleagues have reported that Si prepared by molten salt electrolysis has potential applications in large-scale H2 production.33 Recent studies have also shown that electrolyzed silicon can generate hydrogen when exposed to pure water.34 This hydrogen liberation can occur in both ionic and atomic forms of silicon in a spontaneous, exothermic reaction.34 In addition, silicon possesses an anodic polarization in alkaline solutions, and hydrogen evolution is the dominant reaction at silicon electrodes in alkaline aqueous solutions.35,36 However, pure silicon electrodes are not preferrable in water splitting applications due to the formation of a silicon dioxide passivation layer, which limits its electrochemical activity.35,36
To connect the promising electrochemical properties of pentlandite materials and silicon for overall electrochemical water splitting, we investigated the doping of silicon into the pentlandite crystal structure. Herein, the discrepancy in atomic radius between S and Si atoms is expected to modify the overall electronic structure of the active site due to the change in bond length, similar to previous reports of metal substitutions.37 Thus, in this article, we propose a synthetic protocol for Si incorporation into pentlandite to regulate and investigate their electrocatalytic activity towards the overall water splitting. Moreover, we studied the effect of silicon adsorption at the pentlandite surface on the electrochemical performance compared to the doped material. The developed silicon-doped pentlandite (FCNSSi) material showed significantly improved HER and OER performances than the trimetallic pristine FCNS material. Furthermore, FCNSSi demonstrated a significantly improved catalytic HER performance after activation for 24 hours in 0.5 M H2SO4 during long-term chronoamperometry test at −0.1 V vs. RHE. Finally, a promising performance and durability of FCNSSi towards alkaline water electrolysis at elevated current densities was shown in a membrane electrode assembly.
Experimental details
Materials
Iron powder (≥99.0%, reduced), cobalt powder (2 μm particle size, 99.8%), nickel powder (99.9%, 3N), sulfur powder (99.5–100.5%), silicon tetrachloride (99.0%), Nafion perfluorinated resin 5% in alcohol, potassium hydroxide, and sulfuric acid were purchased from Sigma-Aldrich, Merck, Germany. Sustainion® XA-9 Alkaline Ionomer 5% in ethanol was purchased from Dioxide Materials, USA. SiLibeads Ceramic beads Type ZY-S were purchased from Sigmund Lindner GmbH, Germany. 0.05 μm polishing alumina suspension for electrode polishing were purchased from Gamry.Synthesis of Fe3Co3Ni3S8 (FCNS)
FCNS was synthesized via a modified mechanochemical synthesis recently published by our group.24 A reaction mixture (mtotal = 25 g) composed of stoichiometric amounts of the elements iron, cobalt, nickel, and sulfur were milled employing a planetary ball mill (Fritsch Pulverisette 7, premium line) with ZrO2 milling containers (V = 80 mL) and ZrO2 milling balls (100 g, d = 5 mm). The reaction mixture was prepared inside a glovebox to assure an inert argon atmosphere inside the milling vessel. Ball milling was performed at a constant rotation speed of 900 rpm for 4 × 60 min, with a 60 min break after each cycle.Synthesis of Fe3Co3Ni3S8−xSix (FCNSSi) for silicon atom doping in pentlandite structure
FCNSSi was prepared through annealing of FCNS at 800 °C in an inert nitrogen gas atmosphere with a flow rate of 80 mL min−1 in presence of SiCl4 as Si source (Fig. 1, right side). FCNS powder (1.0 g) and SiCl4 liquid (3.0 g) were placed and mixed in a separate quartz-boat. A large excess of SiCl4 was used due to anticipated losses of Si e.g. by evaporation. After aging for 1 h, the annealing process was initiated by heating from room temperature up to 57 °C, the boiling point of SiCl4, within 1 h. After continuous annealing at 57 °C for 1 h, the temperature was raised up to 800 °C within 2 h. After continuous annealing at 800 °C min−1 for 4 h, the powder was cooled inside the furnace at a rate of 10 °C min−1. The obtained FCNSSi powder was collected for characterization and electrochemical testing.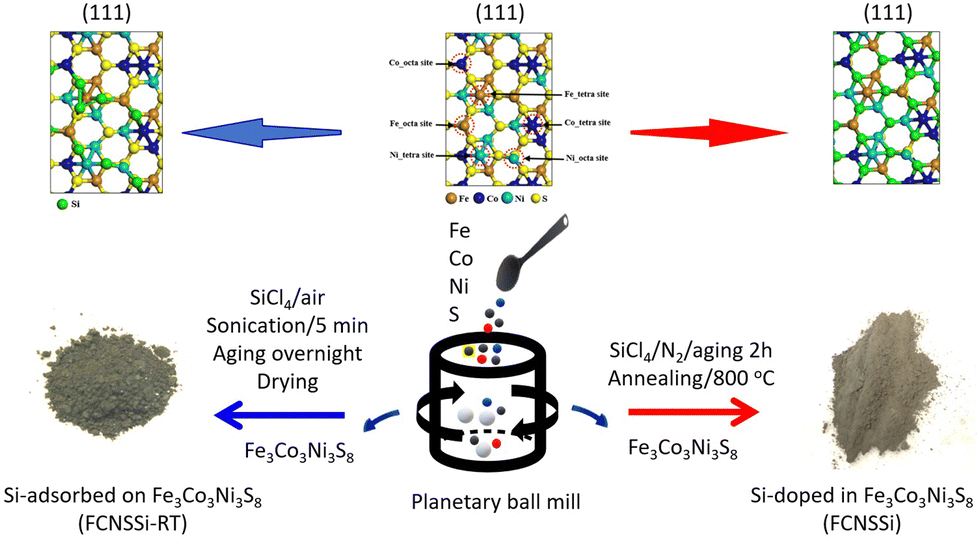 | ||
| Fig. 1 Schematic illustration of the synthetic pathway of Si-doped in trimetallic pentlandite (FCNSSi) and Si-adsorbed at trimetallic pentlandite (FCNSSi-RT) surface. | ||
Synthesis of Fe3Co3Ni3S8−xSix (FCNSSi-RT) for silicon adsorption at pentlandite surface
FCNSSi-RT was prepared through mixing FCNS and SiCl4 at room temperature in an inert atmosphere (Fig. 1, left side). FCNS and SiCl4 (wt/wt = 1/3) were mixed in a small vial in an inert atmosphere. The vial was aged tightly closed in a fume hood overnight. Afterwards the vial was opened and aged in the fume hood until a dry powder is obtained. The obtained FCNSSi powder was collected for characterization and electrochemical testing.Electrochemical measurements
A conventional H-type three-electrode electrochemical cell or an in-house built zero-gap membrane reactor (see below) was used for OER and HER measurements. All electrochemical tests were performed on a GAMRY 1010B interface/potentiostat. A glassy carbon electrode (GCE) of a geometric surface area of 0.071 cm2 and carbon paper electrode (CPE) with a geometric surface area of 0.16 cm2 coated with our catalysts were used as working electrodes, saturated calomel electrode (SCE) as a reference electrode, and a platinum mesh as a counter electrode. The reported potential versus the reversible hydrogen electrode (RHE) was estimated using the following equation: ERHE = ESCE + 0.241 + 0.059 pH. To prepare the ink for the working electrode, 5 mg of our catalyst was well-dispersed in a mixture of water and ethanol (950 μL, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) until obtaining a homogenous solution. 50 μL of Nafion (5 wt%) perfluorinated resin solution was added into the prepared suspension while keeping the sonication continuous for one additional hour until affording a homogeneous ink (N.B. Sustainion® XA-9 Alkaline Ionomer 5% in ethanol is used as a binder for making ink for zero gap cell/membrane electrode assembly measurements (Fig. 8)). Finally, 5 μL of prepared ink was drop-casted at GCE surface and 10 μL of prepared ink was drop-casted at CPE surface and then dried at room temperature. Linear sweep voltammetry (LSV) measurements were performed at a potential range between 1.0 V to 2.0 V vs. RHE for OER testing in KOH solution and 0.2 V and −0.8 V vs. RHE in H2SO4 solution for HER with a scan rate of 50 mV s−1. Tafel plots were derived from LSV curves around the onset potential region. Cyclic voltammetry (CV) measurements were performed at different scan rates to estimate the electrochemical active surface area (ECSA). The calculation of ECSA was performed according to the formula, ECSA = RfS, in which S represents the geometric surface area of the smooth electrode, equivalent to the geometric area of the working electrode. The roughness factor (Rf) was obtained from the formula,
:1 v/v) until obtaining a homogenous solution. 50 μL of Nafion (5 wt%) perfluorinated resin solution was added into the prepared suspension while keeping the sonication continuous for one additional hour until affording a homogeneous ink (N.B. Sustainion® XA-9 Alkaline Ionomer 5% in ethanol is used as a binder for making ink for zero gap cell/membrane electrode assembly measurements (Fig. 8)). Finally, 5 μL of prepared ink was drop-casted at GCE surface and 10 μL of prepared ink was drop-casted at CPE surface and then dried at room temperature. Linear sweep voltammetry (LSV) measurements were performed at a potential range between 1.0 V to 2.0 V vs. RHE for OER testing in KOH solution and 0.2 V and −0.8 V vs. RHE in H2SO4 solution for HER with a scan rate of 50 mV s−1. Tafel plots were derived from LSV curves around the onset potential region. Cyclic voltammetry (CV) measurements were performed at different scan rates to estimate the electrochemical active surface area (ECSA). The calculation of ECSA was performed according to the formula, ECSA = RfS, in which S represents the geometric surface area of the smooth electrode, equivalent to the geometric area of the working electrode. The roughness factor (Rf) was obtained from the formula, 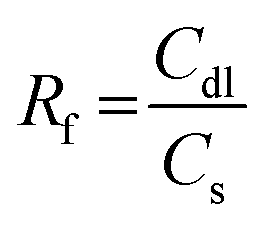 , in which the double layer capacitance (Cdl) was equal to the slope of the double layer charging current versus the scan rate slope using this formula i = vCdl. The general specific capacitance Cs corresponded to the average double layer capacitance of a smooth surface about 20–40 μF cm−2.38 Electrochemical impedance spectroscopy (EIS) measurements were performed to determine the charge transfer speed. Finally, chronoamperometry and chronopotentiometry tests were performed for 24 h to determine the catalyst durability and stability for long-term OER and HER performance.
, in which the double layer capacitance (Cdl) was equal to the slope of the double layer charging current versus the scan rate slope using this formula i = vCdl. The general specific capacitance Cs corresponded to the average double layer capacitance of a smooth surface about 20–40 μF cm−2.38 Electrochemical impedance spectroscopy (EIS) measurements were performed to determine the charge transfer speed. Finally, chronoamperometry and chronopotentiometry tests were performed for 24 h to determine the catalyst durability and stability for long-term OER and HER performance.
Zero-gap cell assembly
An in-house made zero-gap cell with an active area of 2 cm2 was employed for all experiments at high current densities.39,40 The compression and position of the porous transport electrodes were adjusted by polytetrafluoroethylene (PTFE) gaskets. We focused our investigation on AEM-based zero-gap electrolyzers, for which chalcogenide materials hold greater promise for both the anodic and cathodic side. The employed Fumasep FAA-3-PK-130 membrane was conditioned in 1.0 M KOH for one day prior the electrochemical measurements. KOH (1.0 M) was used as an electrolyte for both the anode and the cathode and were flooded through the used titanium parallel flow-fields with a flow rate of 10 mL min−1.Characterizations
Powder X-ray diffraction (PXRD) measurements were performed on a Bruker D2 Phaser diffractometer equipped with a Lynx Eye XE-T detector operating at 30 kV acceleration voltage and 10 mA emission current using Cu K-α radiation (λ = 1.54184 Å). The data was recorded in a range from 10–70° 2θ. The thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed using Netzsch STA 449 F3 Jupiter equipped with nitrogen-purged SiC-Oven (<1550 °C), DSC-Sample holder. The device was properly calibrated using the pure elements In, Zn, Al, Ag and Au. The FTIR spectra were collected using Shimadzu IR Tracer-100 with a Pike miracle ATR unit. Scanning electron microscopy (SEM) was performed on a ZEISS Gemini2 Merlin HR-FESEM equipped with an OXFORD Aztec Energy X-ray microanalysis system for energy dispersive X-ray spectroscopy (EDX). The SEM images were recorded at an acceleration voltage of 4 kV while EDX mappings were performed from 0–20 kV. The high-resolution TEM (HRTEM) image was collected using a JEOL JEM-2800 operated at 200 keV. XPS measurements were carried out in an ultra-high-vacuum (UHV) setup equipped with a polychromatic Al Kα X-ray source (1486.6 eV) or Mg Kα X-ray source (1253.6 eV) and a hemispherical analyzer (type CLAM2, VG, Scientific, Thermo Fischer Scientific). The base pressure in the measurement chamber was maintained at about 10−9 mbar. All spectra were recorded with a pass energy of 100 eV at beam current of 13 mA and a high voltage of 14 kV, which equals a Power of 13 × 14 = 182 W. ICP was performed to measure the elements Fe, Ni and Co after microwave digestion on an Analytik Jena Model ContrAA800 AAS. The nitrogen gas adsorption–desorption isotherms were obtained using a Autosorb-1, Quantachrome Instruments. The pore size distribution was calculated based on the DFT model. Prior the measurements the materials have undergone preheating treatments at 100 °C for 24 h.Online gas chromatography (GC) measurements
An Agilent 7820A GC system for gas analytics was used for qualitative and quantitative analysis of O2 and H2 gases and for faradaic efficiency (FE%) calculations. The gas chromatography system is equipped with two columns (HaySep Q and molesieve 5A), a flame ionization detector (FID) and a thermal conductivity detector (TCD). Argon was used as carrier gas for standard measurements.Computational methods
The spin-unpolarized DFT calculations with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional were performed using the Vienna ab initio simulation package (VASP).41 The projector augmented wave method (PAW),42 with a plane-wave kinetic energy cutoff of 400 eV, was used, with the Gaussian smearing of 0.05 eV. The Brillouin zone was sampled by only the 3 × 3 × 1 K-point. The pentlandite Fe3Co3Ni3S8 (111) surface was modeled by a (2 × 3 × 2) supercell, and a vacuum layer of 15 Å was added to eliminate artificial interactions between periodic images for modeling surface chemistry. We substituted the surface sulfur atoms with silicone for doped pentlandite systems (FCNSSi sample) to evaluate the dopant effects. All atoms were allowed to relax during geometry optimization, and the atomic positions were optimized until the forces were less than 0.02 eV Å−1. The effects of van der Waals corrections were modeled using Grimme's method, with Becke–Johnson damping.43 The adsorption energy of hydrogen atom (EHad) is defined as the energy difference before and after the adsorption with respect to the gas-phase H2 molecule as shown in the following equation: EHad = Etotal − Esurafce − 1/2EH2, where EHad, EH2, and Etotal are the energies for the clean surface, H2 molecule in the gas phase, and hydrogen atom adsorbed on the surface, respectively. The Gibbs-free energy of H adsorption (ΔGH) is obtained by applying the entropy correction as shown in the equation ΔGH = ΔEH + ΔEZPE − ΔSH.The HER takes place on the surface of the cathode via multi-step electrochemical process. Specifically, in acidic conditions the multi-step electrochemical process occurs via the following reactions.44,45
| H3O+ + e− + M → M–H + H2O (Volmer) |
| M–H + H3O+ + e− → H2 + H2O + M (Heyrovsky) |
| 2M–H → 2M + H2 (Tafel) |
| Step I: OH− + * → *OH + e− |
| Step II: *OH + OH− → *O + H2O + e− |
| Step III: *O + OH− → *OOH + e− |
| Step IV: *OOH → * + O2 + e− |
Under ideal conditions, the OER reaction with a total energy change of 4.92 eV can be driven at 1.23 V, whereas the free energy of each elementary reaction would be equally divided into 1.23 eV. Thus, the overpotential (η) is introduced to represent the additional required potential and rationalize the catalytic performance of the catalyst, which is defined in theoretical calculations as: η(PLS) = max(ΔG(1,2,3,4))/e − 1.23 eV.
Results and discussion
Materials characterization
Using a combinatorial chemical synthesis approach that involves planetary ball milling followed by monitored annealing in an inert atmosphere and in the presence of SiCl4, we were able to successfully obtain Si-doped pentlandite powder (as depicted on the right side of Fig. 1). Likewise, to prepare Si adsorbed at the surface of pentlandite, we conducted an additional control experiment at room temperature as shown on the left side of Fig. 1. The sample obtained through annealing is denoted as FCNSSi, while the sample obtained at room temperature is labeled as FCNSSi-RT.Using SEM and HRTEM images, we determined the particle size of FCNSSi to be about 300 nm, as shown by SEM pictures (Fig. 2a and b). The crystalline fringes at d-spacings of 0.28, 0.22, and 0.20, correspond to (222), (420), and (422) planes respectively and thus, features of the pentlandites phase are reflected in the HRTEM image (Fig. 2c). We then analyzed the crystal structure and phase purity of the materials using PXRD diffraction (Fig. S1, ESI†). All materials exhibited diffraction peaks consistent with the trimetallic pentlandite phase (PDF card no. 30-0444).47 The dominant diffraction peaks of FCNSSi also matched the pentlandite phase, indicating that the doping of Si atoms into FCNS had no significant impact on the pristine crystal structure (Fig. S2, ESI†). Additionally, the peaks' intensity and sharpness increased, suggesting an improvement in the overall crystallinity of the material. While some peaks associated with NiS and Ni31Si12 phases were detected (Fig. S1, ESI†),48 they were minor compared to the main pentlandite phase in terms of peak intensity, likely due to phase preferential crystallization.
 | ||
| Fig. 2 (a and b) Low and high magnifications SEM images, and (c) HRTEM image of FCNSSi powder. | ||
To examine the morphology and composition of FCNSSi on carbon paper electrodes, we used SEM in combination with an energy-dispersion X-ray system (EDX). The microscale particles of FCNSSi (1–10 μm) were distributed across the electrode surface (as displayed in Fig. 3a). The EDX images (Fig. 3) and spectra (Fig. S3, ESI†) revealed the presence and overlapping distribution of Fe, Co, Ni, S, and Si atoms throughout the entire FCNSSi electrode surface.
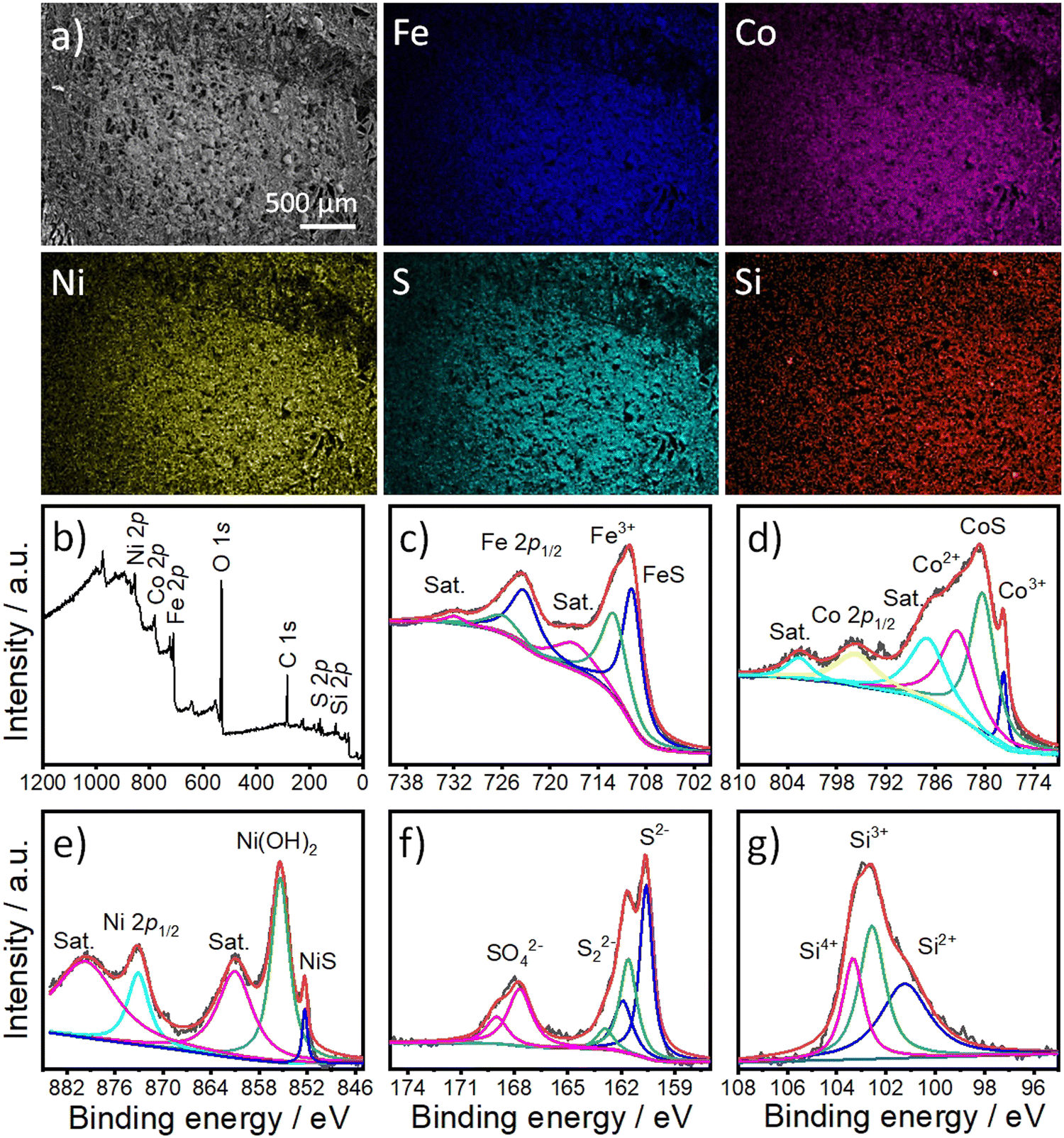 | ||
| Fig. 3 (a) Top-view SEM and the corresponding atoms (Fe, Co, Ni, S, and Si) mapping images of FCNSSi on carbon paper electrode. (b) Wide scan XPS survey of FCNSSi powder and high-resolution XPS spectra of (c) Fe 2p, (d) Co 2p, (e) Ni 2p, (f) S 2p, and (g) Si 2p orbitals. | ||
X-Ray photoelectron spectroscopy (XPS) analysis was then conducted to determine the oxidation state of the elemental and material composition of FCNSSi powder (Fig. 3b). The photopeaks corresponding to Fe 2p, Co 2p3/2, Ni 2p3/2, S 2p, and Si 2p orbitals were clearly observed in the XPS survey scan (Fig. 3b). The Fe 2p orbital photopeak was deconvoluted into two splits that are assignable to the Fe 2p3/2 and Fe 2p1/2 orbitals (Fig. 3c).24 The peak at 709.8 eV for FCNSSi is attributed to the Fe–S bond in pentlandite phase.49 The Co 2p3/2 photopeaks were then deconvoluted into four peaks which indicate not only presence of Co3+ and Co2+ species at tetrahedron and octahedron positions in pentlandite, but high intensity photopeak at 780.2 eV is assigned to Co–S phase.50 Similarly, Ni 2p3/2 is deconvoluted into three peaks at 852.4 eV, 855.35 eV, and 860.85 eV attributed to NiS, Ni(OH)2, and a shake-up satellite, respectively.51 The S 2p photopeak was deconvoluted into two peaks at 160.6 eV and 161.9 eV, characteristic of S 2p3/2 and S 2p1/2 orbitals in metal–sulfur bond in pentlandite phase, respectively.52 An additional doublet at 161.7 eV and 163.0 eV can be assigned to a disulfide phase.53 The signals at 167.6 eV and 168.9 eV corresponding to S 2p3/2 and S 2p1/2 orbitals can be assigned to sulfate species.53 Finally, the S 2p signal is deconvoluted to one dominant peak at 101.22 eV and two additional peaks at 102.5 eV and 103.4 eV characteristics of Si2+, Si3+, and Si4+, respectively.54,55 We are assuming that the peak observed at 101.22 eV originates from the silicon–metal bond due to silicon doping as proposed by the DFT calculations (see below) and/or Ni31Si12 alloy.56 The additional peaks at 102.5 eV and 103.4 eV are assigned to oxidized silicon.57
To further confirm the successful doping of the pentlandite materials, the chemical composition of the materials was determined using Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES). The compositional analysis confirmed the presence of Si in pentlandite as a part of the material's composition (Table S1, ESI†). The chemical composition of the FCNS sample was determined as Fe2.97Co2.98Ni3.01S8, while that of FCNSSi was Fe2.82Co2.84Ni2.8S7.7Si0.3, which is almost typical of the composition of the pentlandite phase. Notably, a low content of silicon doping was preferred to avoid passivation caused by silicon dioxide formation intentionally. At the same time, incorporation of a large amount of Si may disrupt the crystal structure of pentlandite phase as a common behavior of sulfur boned atom.37 On the other hand, the Si-adsorbed FCNSSi-RT sample had a chemical composition of Fe3.1Co2.94Ni2.95S8.61Si2.21, which is not in accord with the chemical composition of the pentlandite phase and provided evidence of Si adsorption and/or SiO2 formation at the surface of FCNS at room temperature. This observation is further evidenced in the corresponding XPS spectra showing surface modification of the FCNS sample by removal of sulfide after aging in SiCl4 at room temperature (Fig. S4, ESI†). The Si 2p orbital peak was deconvoluted into several peaks, revealing the presence of Si with different oxidation states, as expected from SiCl4. Additionally, deconvolution of the high-resolution XPS scan of the Si 2p orbital shows a peak at around 103.5 eV that could be attributed to oxidized silicon at FCNS surface.57
To investigate the effect of silicon on the porous nature of FCNS and FCNSSi powders, we performed nitrogen gas adsorption–desorption isotherms (Fig. S5a, ESI†). An obvious change in sorption behavior is observed, which could be attributed to the incorporation of Si atoms and resulted in a remarkable increase in the BET surface area of FCNSSi (120.19 m g−1) compared to FCNS, which had a BET of 93.49 m g−1 (Fig. S5a, ESI†). A likely factor contributing to the increase in specific surface area is the etching of the material's surface due evolution of hydrochloric acid during Si incorporation. Another aspect is the development of Si–O bond with improved adsorptive characteristics. Surprisingly, FCNSSi-RT shows a significant improvement in the specific surface area (230.08 m g−1) reflecting the significant changes in the surface due to Si adsorption, but without benefits to the electrocatalytic activity (Fig. S5a, ESI†). The average pore size distribution curves are depicted in Fig. S5b (ESI†). The three samples each showed an average pore size distribution of 16.97 nm within the range of mesoporous materials according to IUPAC classifications.58 In addition, FCNSSi-RT shows one more different average pore size distribution of 7.04 nm within the mesoporous range as well, which might result from etching of FCNS surface due to treatment with SiCl4.
OER performance of FCNS and FCNSSi
To assess the OER performance of our catalysts, we subsequently analyzed the materials using LSV both with and without iR compensation and Fig. 3a and b illustrate the obtained results. To precisely evaluate the catalyst performance, we performed the measurements before and after iR compensation (between 4 to 7 Ω) for the whole electrodes. We found that FCNSSi exhibited superior OER performance, with an overpotential of 315 mV at 10 mA cm−2, outperforming pristine FCNS, which showed an overpotential of 350 mV (Fig. 4a). At elevated current density of 100 mA cm−2, still FCNSSi shows superior performance (465 mV) compared to pristine FCNS (549 mV) (Fig. 4a). In particular, FCNSSi showed an overpotential of 313 mV at 10 mA cm−2, compared to FCNS, which showed an overpotential of 353 mV (Fig. 4b). The improvement is significant at higher current density (100 mA cm−2), whereas FCNSSi displays an overpotential of 400 mV, which is significantly improved compared to pristine FCNS (452 mV) at identical conditions (Fig. 4b).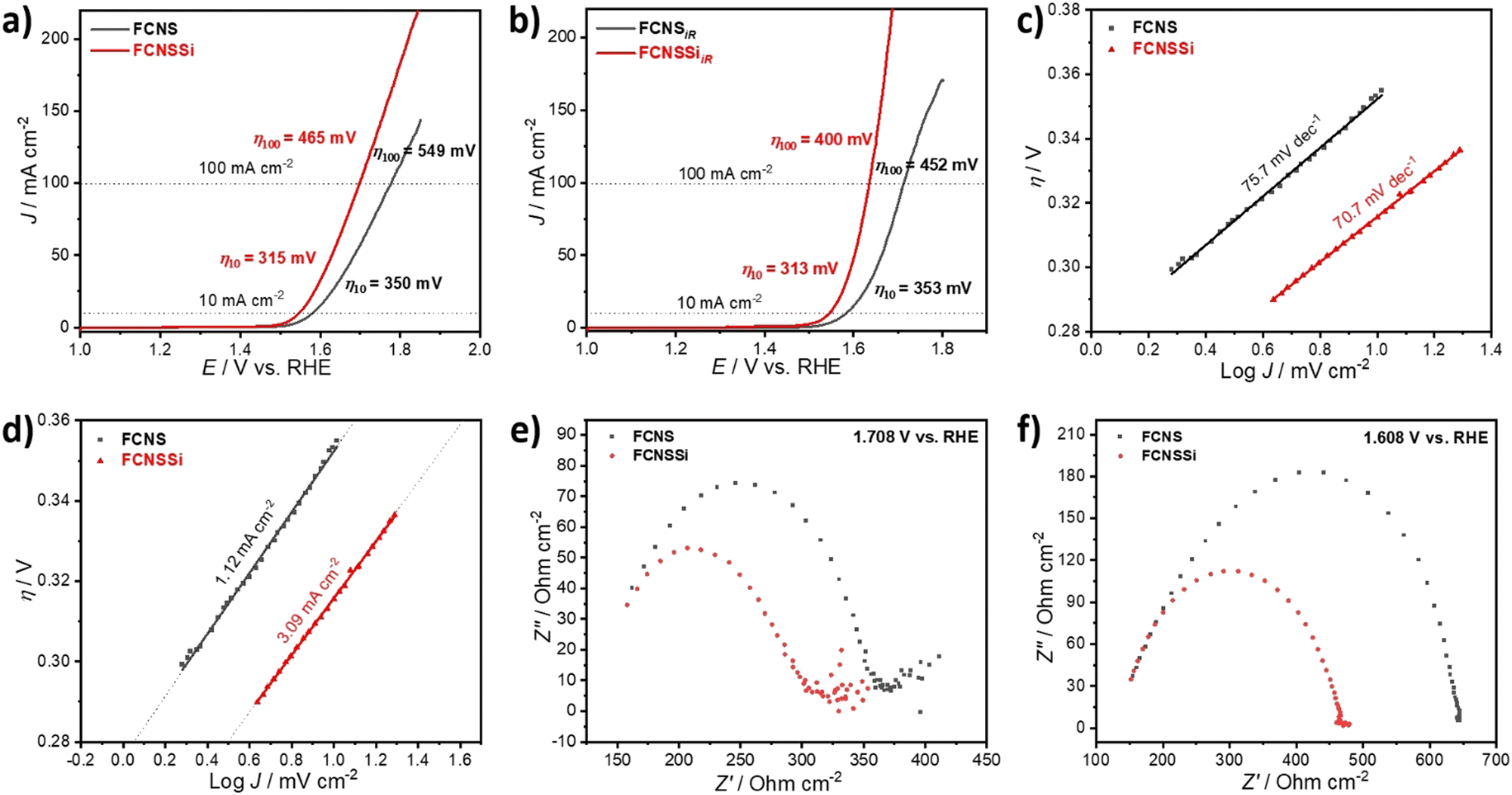 | ||
| Fig. 4 LSV curves (a) before and (b) after iR compensation in 1.0 M KOH solution at scan rate of 10 mV s−1, (c) derived Tafel plots from LSV curves, (d) the estimated exchange current density (J0), and EIS at (e) 1.708 V vs. RHE and (f) 1.608 V vs. RHE of our materials on glassy carbon electrode. | ||
To gain additional insight into the kinetics of oxygen formation on our catalysts, we calculated the Tafel slopes and exchange current densities (J0) (Fig. 4c and d). Our results indicate that the FCNSSi sample exhibits fast oxygen formation kinetics with the lowest Tafel slope of 70.7 mV dec−1 (Fig. 4c) and a high J0 of 3.09 mA cm−2 (Fig. 4d). These findings represent a significant improvement over previously published results (see Table S2, ESI†).
To elucidate the charge transfer speed, we conducted Electrochemical Impedance Spectroscopy (EIS) at different potentials at 1.71 V and 1.61 V versus RHE to enable polarization at the catalyst surface (refer to Fig. 4e and f). It is widely recognized that pentlandites have high conductivity due to their high metal-to-sulfide ratio.59 The Nyquist plots show a linear behavior at a low frequency region and a semicircle at high frequency one (Fig. 4e). The FCNSSi sample displayed a very narrow Nyquist arc (around 138 Ω cm−1 at 1.71 V vs. RHE and 313 Ω cm−1 at 1.61 V vs. RHE), indicating a fast charge transfer at the interface between electrode and electrolyte, supporting its excellent OER performance. In the case of the FCNSSi sample, the linear region simultaneously displays fast mass transfer (Fig. 4e). Since the series resistance (Rs) originates from the solution and electrode substrate, it is expected that both samples will have almost identical Rs values as shown in Fig. 4e and f. We calculated the electrochemical surface-active area (ECSA) by measuring the dielectric charging current in a non-faradaic region using cyclic voltammetry at various scan rates (refer to Fig. S6, ESI†). The FCNSSi material demonstrated a higher Cdl value (2.66 mF cm−2), implying a high ECSA (6.39 cm−2), which represents a twofold increase over pristine FCNS (1.28 mF cm−2 and 3.07 cm−2). This result, combined with an improved specific surface area/BET, supports our initial hypothesis that Si doping can increase the number of active sites for better OER performance.
To identify the gases produced during the oxidation process, we analyzed the gases obtained in an inert argon gas carrier using gas chromatography (GC) at 20 mA cm−2 for 1 h (refer to Fig. S7, ESI†). It is evident that the primary product is O2. Catalyst stability is a crucial factor for its potential industrial applications; therefore, we performed a long-term stability test of our materials for 24 hours at a high current density of 100 mA cm−2 (Fig. S8a, ESI†). FCNSSi exhibited an increase in performance in the first few hours followed by almost stable catalytic OER performance, reflecting its high durability compared to pristine FCNS, which continuously loses its OER activity over time. At the same time, we examined the surface structure, morphology, and composition of the FCNSSi sample on carbon paper electrode after the stability test using Scanning Electron Microscopy (SEM) (Fig. 5a), XPS (Fig. 5b and c), Energy-Dispersive X-ray Spectroscopy (EDX) (Fig. S9, ESI†), PXRD (Fig. S8b, ESI†). The SEM and atom mapping images clearly depict the homogenous distribution of Fe, Co, Ni, S, and Si atoms on the surface of carbon paper, even after the long-term stability test (Fig. 5).
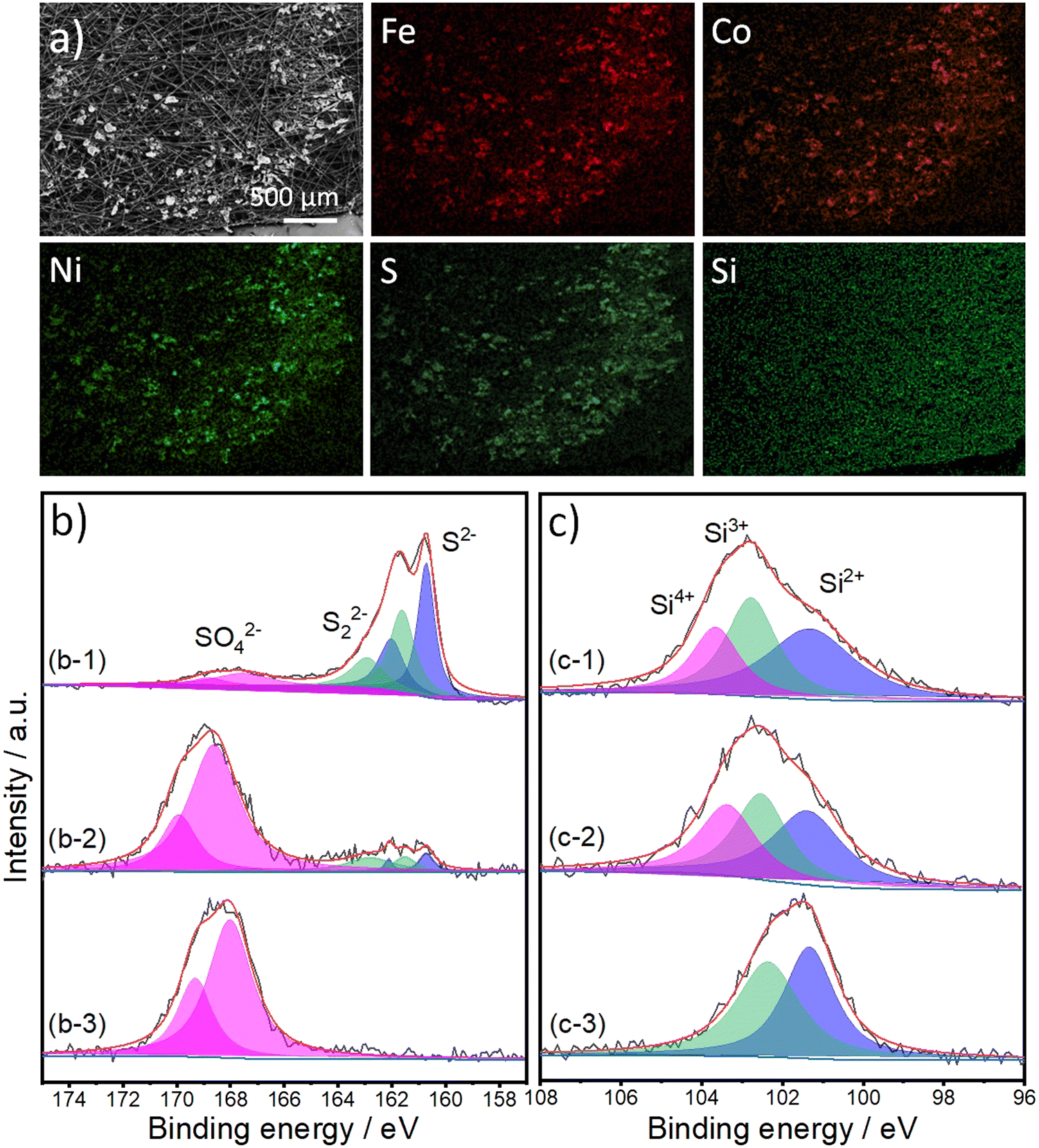 | ||
| Fig. 5 (a) Top-view SEM and the corresponding atoms mapping (Fe, Co, Ni, S, and Si) images of FCNSSi on carbon paper electrode after chronopotentiometry test at 100 mA cm−2 for 24 h. High resolution XPS spectra of (b) S 2p and (c) Si 2p orbitals of (b-1 and c-1) the prepared electrode, and after (b-2 and c-2) 1 h and (b-3 and c-3) 24 h OER. | ||
The high resolution XPS spectra of S 2p orbital collected from FCNSSi/CPE are similar to those of the as-prepared powder (Fig. 5b-1). The three doublets can be assigned to sulfides, disulfides, and sulfate species. After one hour of electrolysis, the doublet corresponding to sulfides and disulfides species are decreased in intensity and, at the same time, the doublet corresponding to sulfate species are remarkably increased (Fig. 5b-2). After 24 hours of electrolysis, the whole surface is oxidized ((Fig. 5b-3), which agrees with the XRD patterns (Fig. S8, ESI†) reflecting that the surface tends to oxidize during OER performance, forming a more thermodynamically stable transition metal oxyhydroxide shell, which is in line with our previous work.24 The XRD peaks are assigned to carbon paper,60 iron and cobalt oxyhydroxide,61 nickel oxyhydroxide,62 and iron oxides.63 This phenomenon has been extensively studied previously, as transition metal oxides and hydroxides (oxyhydroxides) are more thermodynamically stable than transition metal chalcogenides, particularly sulfides.64–66 On the other hand, XPS spectra of the Si 2p orbital collected from the as-prepared FCNSSi/CPE and after one-hour OER are similar to the spectra collected from the as-prepared powder (Fig. 5c-1 and c-2). The peak at 103.4 eV disappeared, which suggests the dissolution of some oxidized silicon species after long term OER performance (Fig. 5c-3).57 The decrease in sulfur content herein was determined by EDX after long-term OER (24 h at 100 mA cm−2) on FCNSSi (Fe3.01Co3.078Ni2.93S4.98Si0.95) (Fig. S9, ESI†). All previous results indicate that sulfur from the surface is oxidized during OER, and the surface is dominantly composed of Si, SO4, and O atoms, which we anticipate is significant contribution to OER catalytic performance.
Due to its low boiling point of 57.6 °C and tendency to evaporate at room temperature, we also tested the doping process of SiCl4 on pentlandite (Fe4.5Ni4.5S8) at ambient conditions (Fig. 1, left side). To characterize the morphology and composition, we performed SEM and EDX measurements on FCNSSi-RT powder (Fig. S10, ESI†). The SEM images suggest that either a phase segregation occurred, or that more than one phase is produced (Fig. S10a, ESI†). The element mapping images reveal that the overlapping of Fe, Co, Ni, S, O, and Cl, while the Si signals are not overlapping. These results, in addition to presence of chloride ions, may likewise indicate an unsuccessful Si doping at room temperature, which in turn suggests that the Si atoms may have been only adsorbed on the surface (Fig. S10b, ESI†). Notably, XRD analysis showed typical diffraction peaks characteristic of pentlandite, indicating that the FCNS crystal structure was retained (Fig. S11a, ESI†). Despite this observation, the FCNSSi-RT sample exhibited poor OER and HER performances compared to FCNSSi obtained at 800 °C (Fig. S11d and e, ESI†), supporting the idea that Si doping in FCNS is preferable to Si adsorption on FCNS for OER. It is expected that Si adsorption may block the active centers on the metal surface, leading to a decrease in OER catalytic activity.
Theoretical study of FCNSSi on OER
To understand the observed enhancement of OER activity for FCNSSi, DFT calculations were conducted to analyze the whole process of the four-electron OER reaction on different metal site moieties embedding in FCNSSi matrix. Considering the conditions of OER in our previous work the surface of catalyst was oxidized as is the case here the surface would oxidize to be covered by 7/4 ML of O*.24 The calculations were performed on the optimized (111) surface. As expected, the oxidation of the surface lead to the formation of self-assembled amorphous metal oxide (Fig. S12, ESI†), where similar observations were noted for pentlandite doped by nitrogen and phosphorus in our previous work by combining systematic experimental and theoretical studies.24 These findings may explain the outstanding OER activity of the pentlandite catalyst compared to other crystalline catalysts since the amorphous catalysts are reported to show better activities of water splitting reaction than their crystalline counterparts.67To quantify further OER activity of the FCNSSi, we investigated the atomic-scale mechanism of OER on the clean FCNSSi and FCNSSi oxidized surface, as depicted in Fig. 6. We found that the potential limiting step for the unoxidized FCNSSi surface corresponds to the oxidation of *OOH intermediate to form the O2 product. The calculated overpotential on clean FCNSSi surface was 2.08, 2.26 and 2.30 V for Ni, Fe, and Co metal sites, respectively. The high overpotential can be ascribed to the strong binding between *OOH and metal atoms. However, as described in Fig. 6a, oxidation of FCNSSi surface significantly adjusts binding of the intermediates on the surface due a decrease in electron density in the octahedral metal site through additional M–O bond formation. The oxidation of the surface is also evidenced by PXRD of the catalyst surface after OER (Fig. S8b, ESI†) and the increased coordination with oxygen decreases the *OH, *O and *OOH adsorption energies compared with those on clean FCNSSi surface. Therefore, the calculated theoretical overpotential significantly decreased to 0.20, 0.26 and 0.48 V on Ni, Fe, and Co metal site, respectively in oxidized FCNSSi. By comparing the OER energy profile of Si with the N and P dopant in our previous publication,24 it is clear that Si dopant leads to moderate adsorption energy of intermediates closer to the ideal energy profile showing the advantage of Si doping over the pristine FCNS and N and P dopants as well.
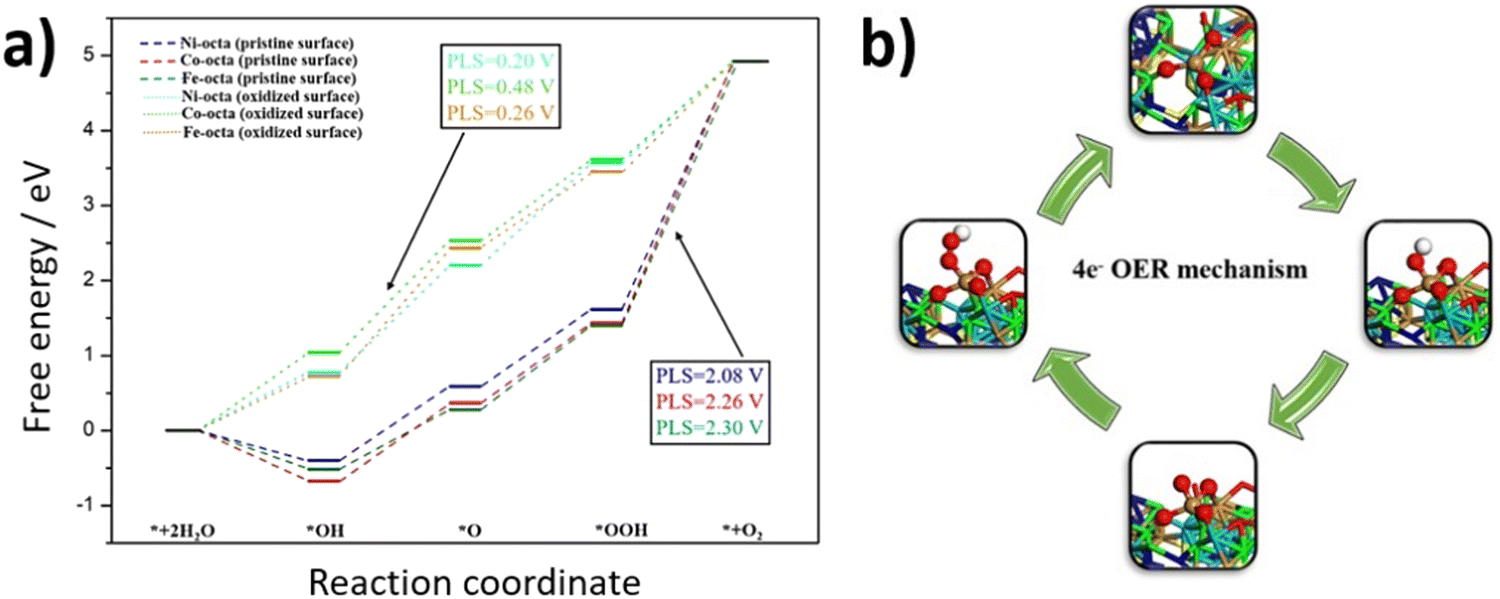 | ||
| Fig. 6 (a) Quantum-theoretical calculations at the level of density functional theory (DFT) on the electrocatalytic OER performance in FCNSSi, and (b) the computational optimized OER intermediates on Fe in FCNSSi. | ||
HER performance of FCNS and FCNSSi
As mentioned in the introductory part, both ionic and metallic forms of silicon show promise for hydrogen production. Therefore, it is necessary to also evaluate the potential of FCNSSi for HER. The catalytic HER performance of the FCNSSi sample was thus subsequently assessed using LSV measurements on a glassy carbon electrode (GCE). While the HER performance of the as-prepared FCNSSi electrode was significantly improved compared to the pristine FCNS, achieving a current density of only −10 mA cm−2 at a high overpotential of 425 mV (Fig. 7a).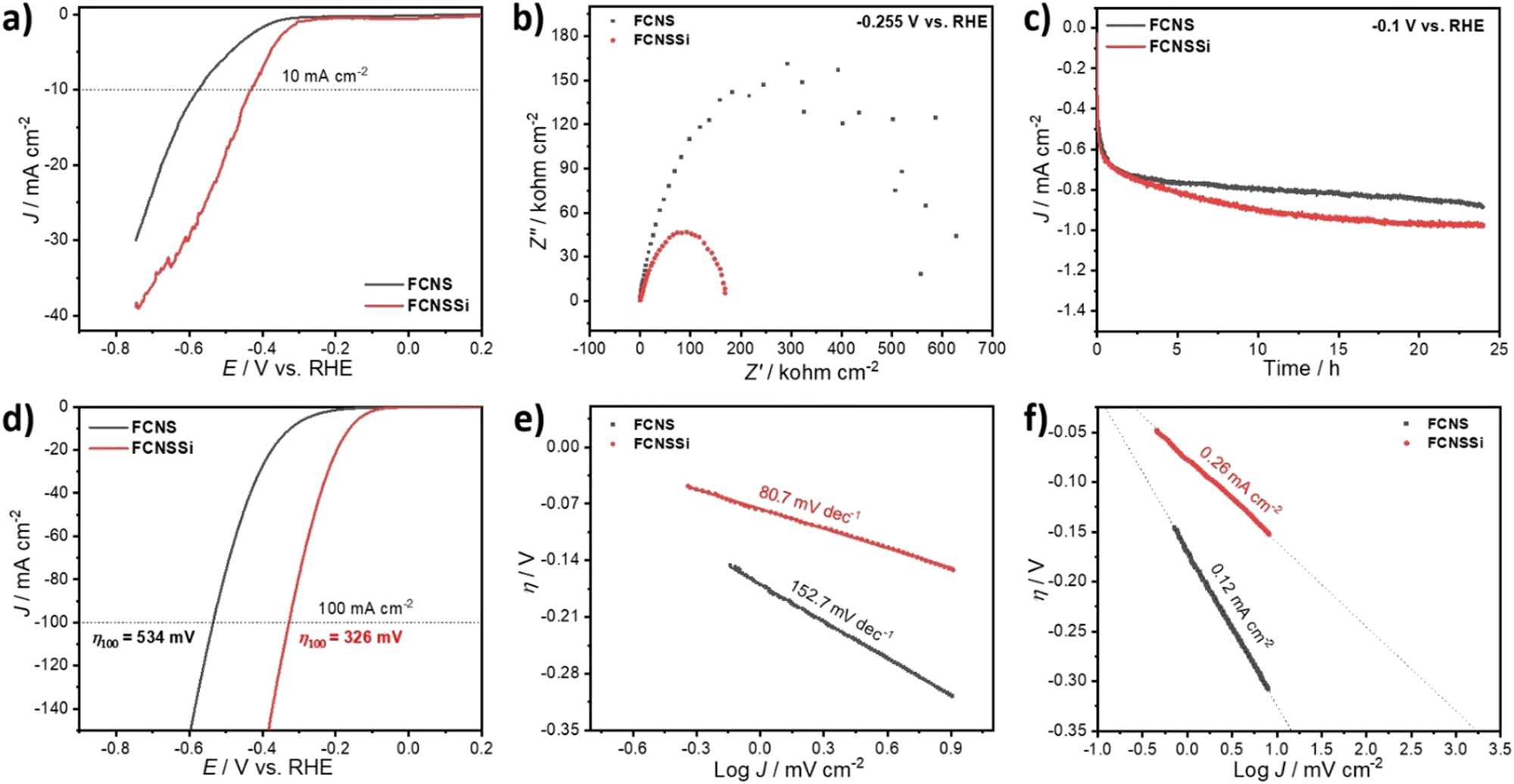 | ||
| Fig. 7 (a) LSV curves of the as-prepared FCNS and FCNSSi in 0.5 M H2SO4 solution at scan rate of 50 mV s−1, (b) EIS at −0.255 vs. RHE, (c) chronoamperometry test at −0.1 vs. RHE, (d) LSV curves after HER performance for 24 h, (e and f) the derived Tafel plots and the estimated exchange current density (J0), respectively, of FCNS and FCNSSi samples on glassy carbon electrode. | ||
However, the FCNSSi electrode showed a much smaller Nyquist arc around 169.0 Ω cm−2, compared to FCNS (629.0 Ω cm−2) (Fig. 7b), indicating a faster charge transfer between the electrode–electrolyte interface. After 24 hours of FCNSSi electrolysis, a significant HER improvement was observed (Fig. 7c) and the activated FCNSSi electrode achieved a current density of −10 mA cm−2 at an overpotential of 164 mV and a low Tafel slope of 80.7 mV dec−1, which was significantly better than the pristine FCNS materials (316 mV and 152.7 mV dec−1, respectively) at identical conditions (Fig. 7d and e). These results are also improved from those reported for previously published pentlandite materials in terms of energy required for water reduction and kinetics of hydrogen formation (Table S3, ESI†). Additionally FCNSSi (η100 = 326 mV) significantly displays HER performance better than pristine FCNS (η100 = 534 mV) at −100 mA cm−2 (Fig. 7c). To obtain further information concerning the HER rate and kinetics, we estimated the exchange current density (J0) from the intercept between the equilibrium potential at an overpotential of zero (Fig. 7f). The calculated J0 for FCNSSi was 0.26 mA cm−2, which is almost twice the value for FCNS reflecting the faster hydrogen formation rate in case of FCNSSi electrocatalyst (0.12 mA cm−2).
Furthermore, to identify and estimate the amount of H2 produced through hydrogen evolution in acid solution, we performed gas chromatography (GC) (Fig. S13, ESI†). We applied a current density of −20.0 mA cm−2 for 10 hours and GC injections in one-hour intervals. The chromatograms showed that the main product was H2 gas, with negligible traces of O2 and N2 that might have been due to residual air (Fig. S14a, ESI†). The faradaic efficiency was calculated to be 109 ± 10% for FCNSSi and 94 ± 10% for FCNS on a carbon paper electrode at identical conditions (Fig. 7b) (Table S4, ESI†). To receive post-mortem information on a FCNSSi on a carbon paper electrode before and after chronopotentiometry tests at −20.0 mA cm−2 for 10 hours, we performed PXRD analysis (Fig. S14, ESI†). The pentlandite crystal structure was well-preserved, indicating high structural stability and durability of FCNSSi during H2 evolution in acidic solution.
The HER results in combination with the OER clearly demonstrate that the FCNSSi electrode is a highly active and stable bifunctional electrocatalyst for both HER and OER. To investigate its potential and durability for overall water splitting, we designed a two-electrode cell in which FCNSSi served as both anode and cathode in an alkaline solution (Fig. S15 and S16, ESI†).
Theoretical study of FCNSSi on HER
To further understand the HER activity and nature of active sites on the hypothetical silicon doped FCNS, the adsorption free energy of hydrogen for the possible sites were calculated to estimate their catalytic activity toward HER (Fig. S17a, ESI†). The hydrogen adsorption free energy is calculated at a potential U = 0 relative to the standard hydrogen electrode (SHE) at pH = 0. Although our synthesis strategy envisioned doping of FCNS with Si, we considered Si adsorption as an alternative possibility for the modification of the material in the form of Si adsorbed and atomically dispersed on the surface (Fig. S17b and c, ESI†). After optimization of the structure, we found that the adsorption of a single Si atom on the surface is unstable. The DFT calculation suggests that adsorbed Si atoms tend to aggregate, forming small clusters Si3 or Si4 atoms on the metal site (Fe, Co and Ni) on the surface, leading to blocking hydrogen adsorption active sites, which agrees with our experimental results (Fig. 8 and Fig. S11b, ESI†). In this case the Si sites were supposed to be the only possible site for HER. The calculated free energy illustrated in Fig. 8a shows that the Si site exhibit a high ΔG*H, indicating its limited HER activity due to excessive binding strength.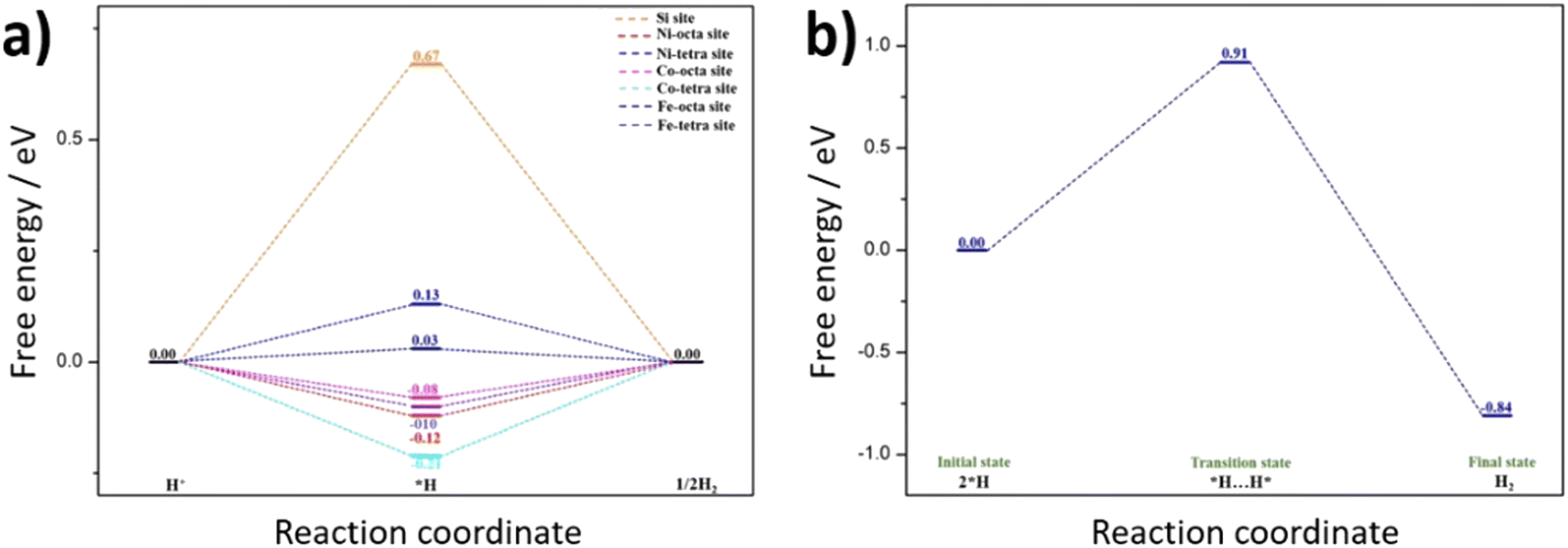 | ||
| Fig. 8 Quantum-theoretical calculations at the level of density functional theory (DFT) on the electrocatalytic HER performance in FCNSSi via (a) Volmer–Heyrovsky and (b) Volmer–Tafel mechanisms. | ||
On the other hand, compared to Si adsorbed at FCNS (FCNSS-RT), Si doping in FCNS (FCNSSi) enhances the hydrogen adsorption activity of all sites. We found the global minimum energy of hydrogen adsorption on each type of metal site tends to be much closer to zero, where the ΔG*H in the range of (0.10, −0.2) eV, as depicted in Fig. 8a. Moreover, DFT calculations have shown that H* adsorbs on the surface as bridging two metal atoms explaining the improvement of H adsorption activity over the sites following Heyrovsky mechanism (Fig. S15c and d, ESI†).
Alternatively, hydrogen evolution could proceed via the Tafel reaction. Therefore, we studied the Tafel reaction energetics on Fe octahedral site as an example (Fig. 8b). The results show that the Tafel reaction requires an energy barrier of 0.91 eV. The overall reaction energy from 2H* to H2(g) is exothermic with −0.84 eV. This result indicates that the Tafel reaction seems to be kinetically unfavorable compared to Heyrovsky reaction in our study. This result is in accordance with our previous work, whereas a Volmer–Heyrovsky mechanism is favorable to the pristine FCNS.25 Smialkowski et al. found that the content of Co plays a significant role in manipulating the HER mechanism and kinetics.25 The Co-rich FCNS adapts a Volmer–Heyrovsky mechanism, while the mechanism shifts to a Volmer–Tafel mechanism with increasing Fe or Ni content.
Zero-gap cell experiments
To assess the effectiveness of the materials presented here on an industrial scale, we constructed a zero-gap cell in-house using FCNSSi deposited through thermal drop casting at 90 °C on Ni-foam on the anodic side (FCNSSi/Ni foam), and Pt/C(40%) on CPE on the cathodic side (Pt-C/CPE) for alkaline water electrolysis (Fig. 9a). The results of LSV measurements showed that FCNSSi had better performance towards water oxidation compared to the pristine FCNS electrode (Fig. 9b). The overpotential estimated for the FCNSSi/Ni foam electrode at 500 mA cm−2 is 2.28 V, outperforming the pristine FCNS/Ni foam electrode (2.56 V). The long-term chronopotentiometry test at an elevated current density of 500 mA cm−2 further confirmed the high performance and durability of FCNSSi/Ni foam, with an overall cell potential of approximately 2.31 V for 16 h (Fig. 9c). There is a potential increase of around 23.0 mV after 16 h of consecutive performance at 500 mA cm−2, which represents a decay of 3 mV h−1 in the overall cell voltage (Fig. 9c). It is currently unclear if this drop stems from an overall material decomposition or mechanical faults of the electrode assembly/membrane. In contrast, the pristine FCNS/Ni foam electrode demonstrated lower performance, with a higher cell potential of 2.58 V.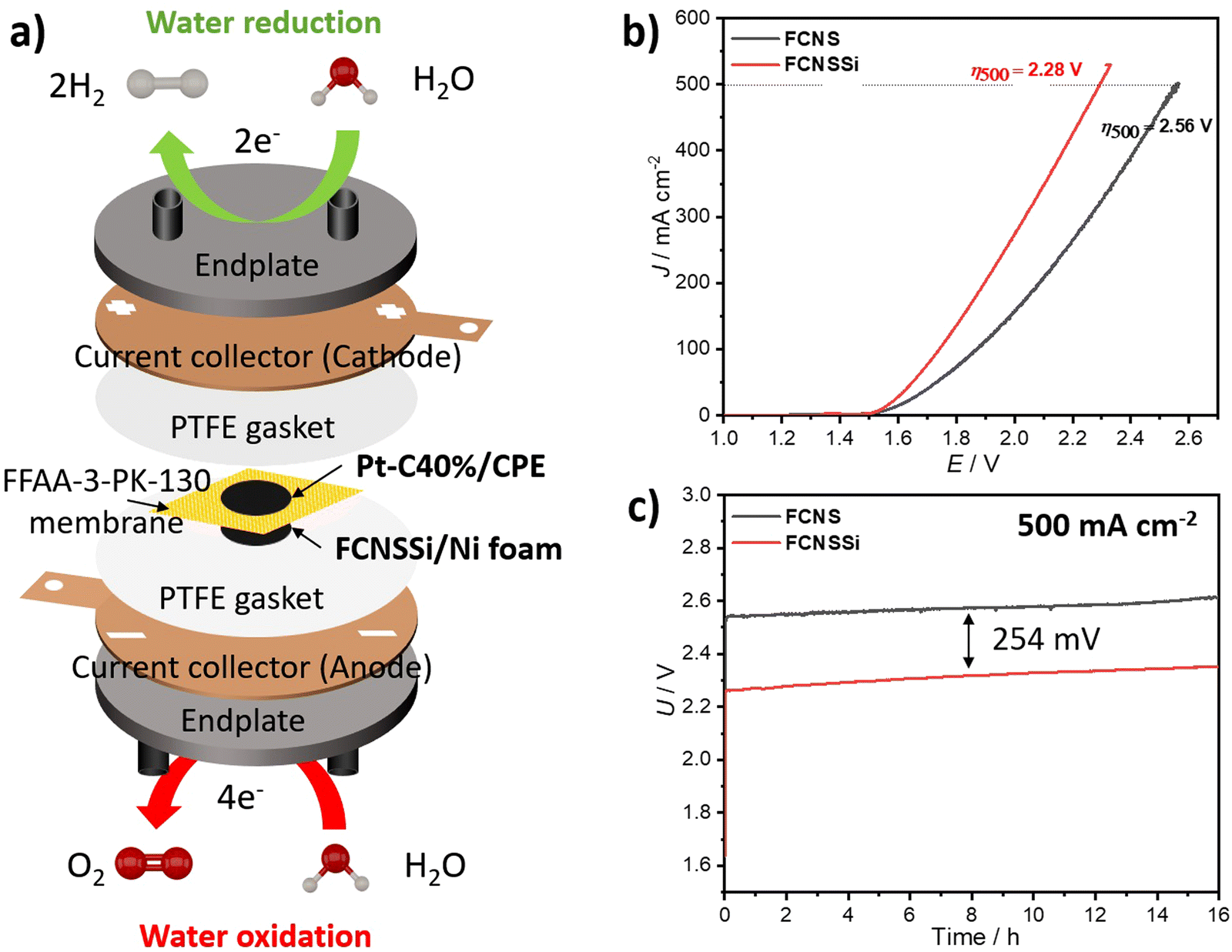 | ||
| Fig. 9 (a) Zero-gap cell assembly using FCNSSi on Ni foam at anodic side against Pt/C (40%) on CPE at cathodic side, and (b) LSV measurements, and (c) chronopotentiometry test at 500 mA cm−2 for 16 h of both pristine FCNS and FCNSSi on Ni foam electrodes. | ||
As a reference cell, we constructed a zero-gap cell using bare CPE as the cathode and FCNSSi/Ni foam and bare Ni foam as anodes. The chronopotentiometry test at 500 mA cm−2 showed an improvement in the FCNSSi/Ni foam electrode compared to bare Ni foam by 679 mV, reflecting the significant contribution of the FCNSSi catalyst towards the water oxidation activity (Fig. S18, ESI†).
Likewise, to evaluate the HER catalytic performance in 1.0 M KOH, we constructed a zero-gap cell in which FCNSSi/CPE acted as the cathode against bare Ni foam as the anode (Fig. S19a, ESI†). The cell achieved a cell voltage of 2.56 V at −100 mA cm−2, with an average steady FE% of 104 ± 5% within 10 hours of performance (Fig. S19b, ESI†), which agrees with the three-electrode set up results (Fig. S13b, ESI†).
Conclusion
In this study, we successfully doped heterotrimetallic pentlandite (FCNS) with silicon (Si) atoms through controlled annealing at 800 °C in an inert atmosphere of pristine FCNS and SiCl4 mixture. The obtained FCNSSi exhibited superior performance and durability towards electrochemical OER in alkaline solution with an overpotential 313 mV at 10 mA cm−2 and a Tafel slope of 70.7 mV dec−1 and HER in acid solution with an overpotential 164 mV at −10 mA cm−2 and a Tafel slope of 80.7 mV dec−1 for overall electrochemical water splitting, outperforming pristine FCNS. Moreover, the FCNSSi electrode exhibited exceptional durability and robustness for long-term HER and OER. For comparison, the pristine FCNS was treated with SiCl4 at room temperature to obtain FCNSSi-RT. We found that the FCNSSi-RT has neither improved OER nor HER implying that heating is necessary for Si atom doping. DFT calculations suggest that the surface oxidation of FCNSSi during OER improved its performance by weakening the adsorption energy of *OH, *O and *OOH. Additionally, Si doping in FCNS structure leads to moderate adsorption energy of intermediates closer to the ideal energy profile, showing the advantage of Si doping over the pristine FCNS. Furthermore, Si doping in FCNS structure enhanced the hydrogen adsorption activity of all sites, where the global minimum energy of hydrogen adsorption on each type of metal site tended to be much closer to zero, thereby improving the HER performance.Our results suggest competitive performance of FCNSSi with previously published materials towards overall electrochemical water splitting. The zero-gap cell testing further confirmed the excellent FCNSSi activity and robustness for long-term alkaline water electrolysis. Si doping of metal sulfides is therefore an effective strategy to improve catalytic activity by adsorption of hydrogen on unoxidized pentlandite and *OOH on oxidized pentlandites and possibly other metal chalcogenides.
Author contributions
M. B. Z. H. synthesized and characterized materials, tested their electrochemical performance, performed analyses, prepared figures and schemes, wrote the first and last draft of the article, and revised final submission. L. B. performed DFT analysis and discussions. D. T. helped in XRD analysis and FCNS synthesis. S. A. S. cooperated in XPS and SEM measurements, and revised last drafts. U. P. A. proposed the concept, guided the team, helped in data interpretation, revised the last draft of the manuscript, and did submission.Conflicts of interest
The authors have no conflict to declare.Acknowledgements
The authors acknowledge the financial support received from Alexander von Humboldt Foundation, 53173 Bonn, Germany (ref. 3.4-1211515-EGY-HFST-P) to complete this project and are grateful for the financial support from the Deutsche Forschungsgemeinschaft (AP242/6-1; under Germany's Excellence Strategy EXC-2033, Project number 390677874, RESOLV) and the Fraunhofer Internal Programs under Grant No. Attract 097-602175 and the Fraunhofer Cluster of Excellence CINES.References
- S. Solomona, G.-K. Plattnerb, R. Knuttic and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef PubMed.
- P. Friedlingstein, M. W. Jones, M. O'Sullivan, R. M. Andrew, D. C. E. Bakker, J. Hauck, C. Le Quéré, G. P. Peters, W. Peters, J. Pongratz, S. Sitch, J. G. Canadell, P. Ciais, R. B. Jackson, S. R. Alin, P. Anthoni, N. R. Bates, M. Becker, N. Bellouin, L. Bopp, T. T. T. Chau, F. Chevallier, L. P. Chini, M. Cronin, K. I. Currie, B. Decharme, L. M. Djeutchouang, X. Dou, W. Evans, R. A. Feely, L. Feng, T. Gasser, D. Gilfillan, T. Gkritzalis, G. Grassi, L. Gregor, N. Gruber, Ö. Gürses, I. Harris, R. A. Houghton, G. C. Hurtt, Y. Iida, T. Ilyina, I. T. Luijkx, A. Jain, S. D. Jones, E. Kato, D. Kennedy, K. Klein Goldewijk, J. Knauer, J. I. Korsbakken, A. Körtzinger, P. Landschützer, S. K. Lauvset, N. Lefèvre, S. Lienert, J. Liu, G. Marland, P. C. McGuire, J. R. Melton, D. R. Munro, J. E. M. S. Nabel, S.-I. Nakaoka, Y. Niwa, T. Ono, D. Pierrot, B. Poulter, G. Rehder, L. Resplandy, E. Robertson, C. Rödenbeck, T. M. Rosan, J. Schwinger, C. Schwingshackl, R. Séférian, A. J. Sutton, C. Sweeney, T. Tanhua, P. P. Tans, H. Tian, B. Tilbrook, F. Tubiello, G. R. van der Werf, N. Vuichard, C. Wada, R. Wanninkhof, A. J. Watson, D. Willis, A. J. Wiltshire, W. Yuan, C. Yue, X. Yue, S. Zaehle and J. Zeng, Earth Syst. Sci. Data, 2022, 14, 1917–2005 CrossRef.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schafer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- B. H. R. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Nat. Commun., 2019, 10, 5599 CrossRef CAS PubMed.
- Z. N. Zahran, E. A. Mohamed, Y. Tsubonouchi, M. Ishizaki, T. Togashi, M. Kurihara, K. Saito, T. Yui and M. Yagi, Energy Environ. Sci., 2021, 14, 5358–5365 RSC.
- M. T. M. Koper, Chem. Sci., 2013, 4, 2710–2723 RSC.
- X. Bu, Y. Li and J. C. Ho, MRS Bull., 2020, 45, 531–538 CrossRef CAS.
- E. Amores, M. Sánchez, N. Rojas and M. Sánchez-Molina, Renewable hydrogen production by water electrolysis, 2021, pp. 271–313 Search PubMed.
- Y. Li, L. Zhou and S. Guo, EnergyChem, 2021, 3, 100053 CrossRef CAS.
- M. B. Z. Hegazy, M. R. Berber, Y. Yamauchi, A. Pakdel, R. Cao and U. P. Apfel, ACS Appl. Mater. Interfaces, 2021, 13, 34043–34052 CrossRef CAS PubMed.
- M. B. Zakaria, D. Zheng, U. P. Apfel, T. Nagata, E. S. Kenawy and J. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 40186–40193 CrossRef CAS PubMed.
- X. Gao, Y. Chen, T. Sun, J. Huang, W. Zhang, Q. Wang and R. Cao, Energy Environ. Sci., 2020, 13, 174–182 RSC.
- X. Jin, X. Li, H. Lei, K. Guo, B. Lv, H. Guo, D. Chen, W. Zhang and R. Cao, J. Energy Chem., 2021, 63, 659–666 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, Y. Guo, Y. Ide, B. Yuliarto, X. Jiang, Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 3035–3047 RSC.
- P. Bhanja, Y. Kim, B. Paul, Y. V. Kaneti, A. A. Alothman, A. Bhaumik and Y. Yamauchi, Chem. Eng. J., 2021, 405 Search PubMed.
- S. Hu, S. Wang, C. Feng, H. Wu, J. Zhang and H. Mei, ACS Sustain. Chem. Eng., 2020, 8, 7414–7422 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, Y. Guo, B. Yuliarto, X. Jiang, Y. Ide, N. Nugraha, H. K. Dipojono, A. Yu, Y. Sugahara, D. Golberg and Y. Yamauchi, ChemSusChem, 2020, 13, 1645–1655 CrossRef CAS PubMed.
- S. Dou, X. Wang and S. Wang, Small Methods, 2019, 3, 1800211 CrossRef.
- D. Siegmund, N. Blanc, M. Smialkowski, K. Tschulik and U. P. Apfel, ChemElectroChem, 2020, 7, 1514–1527 CrossRef CAS.
- P. S. Adarakatti, M. Mahanthappa, J. P. Hughes, S. J. Rowley-Neale, G. C. Smith, S. Ashoka and C. E. Banks, Int. J. Hydrog. Energy, 2019, 44, 16069–16078 CrossRef CAS.
- K. Ao, Q. Wei and W. A. Daoud, ACS Appl. Mater. Interfaces, 2020, 12, 33595–33602 CrossRef CAS PubMed.
- Y. Guo, C. Zhang, J. Zhang, K. Dastafkan, K. Wang, C. Zhao and Z. Shi, ACS Sustain. Chem. Eng., 2021, 9, 2047–2056 CrossRef CAS.
- M. B. Z. Hegazy, K. Harrath, D. Tetzlaff, M. Smialkowski, D. Siegmund, J. Li, R. Cao and U.-P. Apfel, iScience, 2022, 25, 105148 CrossRef CAS PubMed.
- M. Smialkowski, D. Siegmund, K. Stier, L. Hensgen, M. P. Checinski and U.-P. Apfel, ACS Mater. Au, 2022, 2, 474–481 CrossRef CAS PubMed.
- M. Smialkowski, D. Tetzlaff, L. Hensgen, D. Siegmund and U.-P. Apfel, Chin. J. Catal., 2021, 42, 1360–1369 CrossRef CAS.
- B. Konkena, K. Junge Puring, I. Sinev, S. Piontek, O. Khavryuchenko, J. P. Durholt, R. Schmid, H. Tuysuz, M. Muhler, W. Schuhmann and U. P. Apfel, Nat. Commun., 2016, 7, 12269 CrossRef CAS PubMed.
- M. Al-Mamun, Y. Wang, P. Liu, Y. L. Zhong, H. Yin, X. Su, H. Zhang, H. Yang, D. Wang, Z. Tang and H. Zhao, J. Mater. Chem. A, 2016, 4, 18314–18321 RSC.
- D. Tetzlaff, K. Pellumbi, D. M. Baier, L. Hoof, H. Shastry Barkur, M. Smialkowski, H. M. A. Amin, S. Gratz, D. Siegmund, L. Borchardt and U. P. Apfel, Chem. Sci., 2020, 11, 12835–12842 RSC.
- D. Tetzlaff, K. Pellumbi, K. J. Puring, D. Siegmund, W. S. K. Polet, M. P. Checinski and U. P. Apfel, ChemElectroChem, 2021, 8, 3161–3167 CrossRef CAS.
- M. Smialkowski, D. Siegmund, K. Pellumbi, L. Hensgen, H. Antoni, M. Muhler and U. P. Apfel, Chem. Commun., 2019, 55, 8792–8795 RSC.
- P. Brack, S. E. Dann, K. G. U. Wijayantha, P. Adcock and S. Foster, Int. J. Energy Res., 2017, 41, 220–228 CrossRef CAS.
- X. Yang, T. Nohira and I. Sohn, J. Electrochem. Soc., 2020, 167 Search PubMed.
- M. N. P. E. A. Tutov, I. V. Protasova and V. M. Kashkarov, Tech. Phys. Lett., 2002, 28, 729–731 CrossRef.
- X. G. Zhang, Anodic Oxide, Electrochemistry of Silicon and Its Oxide, Springer, Boston, 2004, pp. 91–130 Search PubMed.
- X. G. Zhang, Electrochemistry of Silicon and Its Oxide, Springer, Boston, 2004, pp. 237–278 Search PubMed.
- G. V. Gibbs, N. L. Ross and D. F. Cox, Phys. Chem. Miner., 2017, 44, 561–566 CrossRef CAS.
- A. S. B. Mohd Najib, M. Iqbal, M. B. Zakaria, S. Shoji, Y. Cho, X. Peng, S. Ueda, A. Hashimoto, T. Fujita, M. Miyauchi, Y. Yamauchi and H. Abe, J. Mater. Chem. A, 2020, 8, 19788–19792 RSC.
- K. Pellumbi, L. Wickert, J. T. Kleinhaus, J. Wolf, K. Junge Puring, D. Siegmund and U.-P. Apfel, ECS Meeting Abstracts, 2022, MA2022-02, 2376 CrossRef.
- K. Pellumbi, L. Wickert, J. T. Kleinhaus, J. Wolf, A. Leonard, D. Tetzlaff, R. Goy, J. A. Medlock, K. Junge Puring, R. Cao, D. Siegmund and U. P. Apfel, Chem. Sci., 2022, 13, 12461–12468 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, e1808066 CrossRef PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- X. Zeng, Q. Luo, J. Li, Y. Li, W. Wang, Y. Li, R. Wu, D. Pan, G. Song, J. Li, Z. Guo and N. Wang, Adv. Compos. Hybrid. Mater., 2021, 4, 392–400 CrossRef CAS.
- K. Frank and K. Schubert, Acta. Crystallogr. B. Struct. Sci. Cryst. Eng. Mater., 1971, 27, 916–920 CrossRef CAS.
- D. L. Legrand, Am. Mineral., 2005, 90, 1042–1054 CrossRef CAS.
- Y. Xi, N. Angulakshmi, B. Zhang, X. Tian, Z. Tang, P. Xie, G. Z. Chen and Y. Zhou, J. Alloys Compd., 2020, 826 Search PubMed.
- S. Piontek, K. Junge Puring, D. Siegmund, M. Smialkowski, I. Sinev, D. Tetzlaff, B. Roldan Cuenya and U. P. Apfel, Chem. Sci., 2019, 10, 1075–1081 RSC.
- M. B. Zakaria, Y. Guo, J. Na, R. Tahawy, T. Chikyow, W. A. El-Said, D. A. El-Hady, W. Alshitari, Y. Yamauchi and J. Lin, ChemSusChem, 2020, 13, 3269–3276 CrossRef CAS PubMed.
- C. Simon, J. Zander, T. Kottakkat, M. Weiss, J. Timm, C. Roth and R. Marschall, ACS Appl. Energy Mater., 2021, 4, 8702–8708 CrossRef CAS.
- S. Meskinis, A. Vasiliauskas, M. Andrulevicius, D. Peckus, S. Tamulevicius and K. Viskontas, Materials, 2020, 13 Search PubMed.
- A. A. Sokolov, E. O. Filatova, V. V. Afanas’ev, E. Y. Taracheva, M. M. Brzhezinskaya and A. A. Ovchinnikov, J. Phys. D: Appl. Phys., 2009, 42, 035308 CrossRef.
- D. A. Zatsepin, D. W. Boukhvalov, A. F. Zatsepin, A. N. Mikhaylov, N. N. Gerasimenko and O. A. Zaporozhan, J. Mater. Sci., 2020, 56, 2103–2112 CrossRef.
- R. Lukose, M. Lisker, F. Akhtar, M. Fraschke, T. Grabolla, A. Mai and M. Lukosius, Sci. Rep., 2021, 11, 13111 CrossRef CAS PubMed.
- M. Thommes and C. Schlumberger, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 137–162 CrossRef CAS PubMed.
- H. M. A. Amin and U. P. Apfel, Eur. J. Inorg. Chem., 2020, 2679–2690 CrossRef CAS.
- H. Zhang, J. Zheng, Y. Chao, K. Zhang and Z. Zhu, New J. Chem., 2018, 42, 7254–7261 RSC.
- R. Duan, Y. Li, S. Gong, Y. Tong, Z. Li and W. Qi, Electrochimi. Acta, 2020, 360 Search PubMed.
- R. Thimmasandra Narayan, Indian J. Mater. Sci., 2015, 2015, 1–7 CrossRef.
- P. L. Suryawanshi, S. H. Sonawane, B. A. Bhanvase, M. Ashokkumar, M. S. Pimplapure and P. R. Gogate, Green Process. Synth., 2018, 7, 1–11 CAS.
- Y. X. Yang, B. L. Li, Q. Zhang, W. H. Guo, X. H. Wang, L. J. Li, H. Q. Luo and N. B. Li, Energy Technol., 2020, 9, 2000178 CrossRef.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- Z. J. Xu, Sci. China Mater., 2019, 63, 3–7 CrossRef.
- W. Cai, R. Chen, H. Yang, H. B. Tao, H. Y. Wang, J. Gao, W. Liu, S. Liu, S. F. Hung and B. Liu, Nano Lett., 2020, 20, 4278–4285 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00218g |
| This journal is © The Royal Society of Chemistry 2023 |