
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of a Ti layer on the growth of binder-free carbon nanotubes on Cu foil and their performance as lithium ion battery anodes
Ganesh Sainadh
Gudavalli
ab,
Mahesh
Nepal
ab,
Joshua
Young
de,
Manuel
Smeu
 cd and
Tara P.
Dhakal
*abc
cd and
Tara P.
Dhakal
*abc
aCenter for Autonomous Solar Power (CASP), Binghamton University, NY 13902, USA. E-mail: tdhakal@binghamton.edu
bDepartment of Electrical and Computer Engineering, Binghamton University, Binghamton, NY 13902, USA
cMaterials Science and Engineering Program, Binghamton University, Binghamton, NY 13902, USA
dDepartment of Physics, Binghamton University, Binghamton, NY 13902, USA
eDepartment of Chemical and Materials Engineering, New Jersey Institute of Technology, Newark, NJ 07102, USA
First published on 12th June 2023
Abstract
We report a unique approach to fabricate lithium ion battery anodes based on multiwalled carbon nanotubes (MWCNTs) grown directly on copper foils via chemical vapor deposition. This process eliminates the use of binders for lithium ion battery anodes. The intermediate conductive Ti and alumina blocking layers have been optimized to seamlessly connect the CNTs with the Cu substrate, providing robust structural integrity that facilitates charge transfer. This anode material showed a high capacity of 448 mA h g−1 at 50 mA g−1, superior rate capability and no capacity degradation up to 70 cycles at different current densities (100 mA g−1 to 500 mA g−1). Within a small voltage window of 0.8 V vs. Li/Li+, this anode offers the usable capacity of 300 mA h g−1. The observed electrochemical performance of this anode can be attributed to the high theoretical capacity of carbon nanotubes, faster Li-ion insertion into the walls of CNTs and better conductivity at the point of contact between the substrate and the CNTs enhanced by the inclusion of Ti at the substrate–CNT interface.
Introduction
Decades of usage of fossil fuels have created problematic situations such as an increase in CO2 emissions leading to global warming. To satisfy the urgent environmental demands, lithium ion batteries (LIBs) are among the most promising electrochemical cells and are expected to replace traditional fossil fuels in transportation as well as being employed to buffer the intermittent and fluctuating green energy supplies, such as solar and wind power.1,2 Although LIBs have achieved the dominant position in cell markets compared to lead–acid and nickel metal hydride (Ni–MH) batteries, there is still a need for improvement to be used in a larger distribution of the energy supply, such as powering electric and hybrid vehicles and large scale grid storage. In particular, electric vehicles require next-generation Li-ion batteries with high power density, high energy density, high charging rate, long cycle life and improved safety. The most dominant area of LIBs is in portable electronic devices, particularly cell phones and laptop computers.3 As the performance of these devices depends on the performance of energy storage media, i.e., the battery, significant efforts have been made towards developing new and high-performance materials for fabricating the battery components.Several strategies have been employed over the years to improve the performance of Li-ion batteries. For example, (1) there is an effort towards the utilization of high-voltage cathode materials; the main hindrance with achieving this is the electrolyte decomposition voltage (∼4.2 V vs. Li/Li+ for organic solid electrolytes).4 Hence, a lot of research is being focused on engineering the electrolyte to be stable at higher voltages. (2) Utilization of high-energy and high-power density anodes is another way of improving the overall performance of a battery. (3) Nanostructures have various advantages over their bulk counterparts, which could be exploited, especially in the case of anodes for Li-ion batteries. Nanostructures exhibit improved damage tolerance, thereby accommodating volumetric changes by preserving the cycling capacity. The high surface area of nanostructures, e.g. nanoparticles, helps rapid lithiation and delithiation due to reduced lithium ion diffusion distance, thereby improving the power density.5 In this paper, we focused on a strategy to improve the anode of LIBs.
The graphite used in conventional Li-ion battery anodes has both advantages and disadvantages. The advantages are low working potential vs. Li/Li+, low cost, good cycling life and lower material expansion during cycling. The disadvantages include low theoretical capacity (372 mA h g−1), and lower diffusion rate of Li (10−9–10−7 cm2 S−1) leading to low power density.6–8 Additionally, the conventional methods of fabricating carbonaceous anodes use polymer binders mixed with active material that are then cast onto conductive substrates such as copper, nickel, etc. The contact resistance between the active material particles and polymer binder might inhibit the performance of the anode. Therefore, a major challenge in fabricating an advanced LIB is the selection of suitable anodes which can provide high capacity, good cycling stability and ease of Li ion diffusion. Several studies have been done in investigating both carbon and non-carbon based materials. Carbonaceous materials include carbon nanotubes (1100 mA h g−1),9 carbon nanofibers (450 mA h g−1),10 graphene (960 mA h g−1),11 and porous carbon (800–1100 mA h g−1).12 Non-carbon materials include silicon oxide (2400 mA h g−1),13 silicon (3587 mA h g−1),14 germanium (1600 mA h g−1),15 tin (994 mA h g−1)16 and transition metal oxides (500–1000 mA h g−1).17–19 The values in the parentheses are the theoretical capacities. Even though the non-carbonaceous materials have higher capacity, they suffer from high volume expansion, poor electron conductivity, capacity fading after a few cycles, etc.
Carbon nanotubes (CNTs) have been studied as promising anode materials to replace graphite in LIBs.20 Their superior properties include (1) high theoretical specific capacity (1116 mA h g−1) for SWCNTs,21 and (2) shorter diffusion length of Li ions due to controlled nanostructure. Simulation results of Li diffusion in CNTs have shown that the Li+ ion could enter the tube via topographical defects during intercalation.22,23 In addition, they have faster electron transport due to their high electron conductivity of 106 S m−1 at 300 K for SWCNTs and >105 S m−1 for MWCNTs.24,25 CNTs also help with the reduction in volumetric expansion while preserving the structure of the electrode after cycling (intercalation/de-intercalation) due to their porous structures.26 Recent experiments have shown a higher Li intercalation capacity of CNTs over the well-known LiC6 of graphite; this higher capacity of Li implies a lower weight in battery applications.27,28
Commercial LIBs use polymeric binders such as poly(vinylidene fluoride) (PVDF), polyacrylonitrile (PAN), etc., during the electrode fabrication process along with the active material for better adhesion to the current collector. The usage of these binders poses several disadvantages. (1) At elevated temperatures binders such as PVDF react with lithiated graphite to form undesirable stable LiF.29 (2) PVDF reacts with metal lithium leading to exothermic reactions ∼7180 J g−1 of PVDF.29,30 These two might be the cause of thermal runaway in batteries.31 Binders are also known to add unnecessary weight leading to reduced capacity. In this work we report the growth of CNTs directly on copper substrates, thus eliminating the use of polymeric binders completely. Various blocking and buffer layers such as Ti, Al2O3, TiN, Cr, and Au are employed to optimize the growth in different studies using similar CNT growth techniques.32–34 In this study, alumina is used as the blocking layer to protect the copper substrate and prevent the diffusion of the catalyst into the copper substrate. A Ti layer is used as a buffer layer to increase the conductivity and reduce the contact resistance between the CNTs and the substrate. Sputtered iron (Fe) is used as the catalyst for CNT growth.35
Experimental
Materials and chemicals
9 μm thick polished pure copper foils (99.99%) were purchased from MTI corporation to be used as both the current collectors and substrates for growing CNTs. For the growth of alumina (Al2O3), trimethyl aluminium and water are used as precursors for the atomic layer deposition (ALD) system. 99.995% pure Ti pellets purchased from Kurt J. Lesker were evaporated following the alumina buffer layer, as shown in Fig. 1.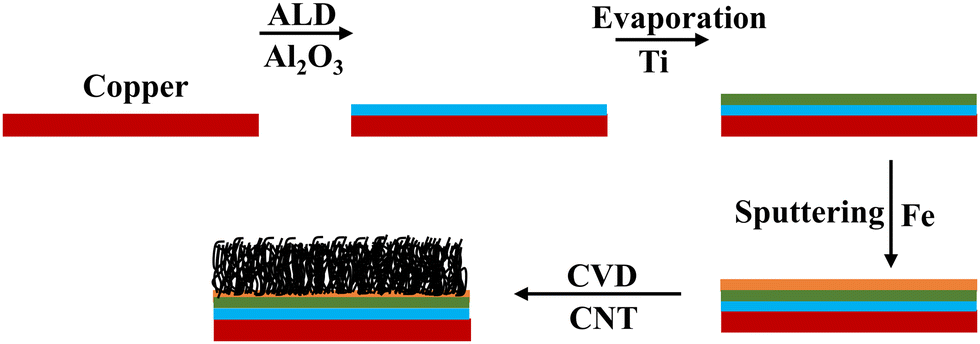 | ||
| Fig. 1 Schematic of the step-by-step fabrication process for CNT growth in a CVD reactor. | ||
Electrode synthesis
Copper substrates were first ultrasonically cleaned in acetone and then ethanol for 15 minutes each. Then they were annealed to make sure that the surface was free of contamination and to ensure flattening of the surface. The blocking layer alumina was deposited on the cleaned substrates using atomic layer deposition (ALD) at a deposition rate of 1 Å per cycle at a temperature of 200 °C.36 A thin film of Fe as a catalyst layer was deposited on the alumina coated copper using magnetron sputtering at a deposition rate of approximately 0.5 Å s−1. It has been shown that the thickness of the Fe layer has a significant effect on the morphology of the CNTs; hence, 5 nm was chosen.35,37 Then, the substrates were transferred into a chemical vapor deposition (CVD) chamber for CNT growth. Vertically aligned carbon nanotubes were grown via ambient pressure CVD using ethylene as the carbon source and a 5% hydrogen/95% argon mixture as the carrier gas. The detailed procedure and experimental setup are described elsewhere.38 The carbon-containing gasses, at their specific decomposition temperature, decompose at the catalyst sites to form CNTs. To deposit the Ti buffer layer, the substrate is transferred into an evaporation chamber where Ti is evaporated at 1 Å s−1. The schematic of the process flow is shown in Fig. 1.Electrochemical measurements and material characterization tools
The microstructure and morphology of the samples were examined by a Zeiss field-emission scanning electron microscopy (FE-SEM) Supra-55 VP equipped with X-ray Energy-Dispersive Spectroscopy (EDS) capability for compositional measurement. Raman spectra were taken from a DXR Raman imaging microscope (532 nm laser) from Thermo Scientific. The electrochemical properties of half cells were measured using a VMP2 multichannel potentiostat (Biologic) for cycling and galvanostatic charge–discharge (GCD) tests. The tests were conducted at a fixed voltage window between 0 V to 3 V (vs. Li/Li+).Fabrication and testing of lithium battery coin cells
The half cells were assembled in 2325-type coin cells under helium gas ambient in the glove box with moisture and oxygen levels below 1 ppm. Copper foils with CNTs and pure lithium metal were used as the anode and counter electrode, respectively. WHA1822021 glass fiber was used as a separator and 1 M LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio) solution as an electrolyte. The coin cells were fabricated with CNT electrodes, where the configurations of the electrode with order of layers deposited are “Cu/50 nm Alumina/25 nm Ti/5 nm Fe/30 min CNT growth by CVD” (left to right).
:1 mass ratio) solution as an electrolyte. The coin cells were fabricated with CNT electrodes, where the configurations of the electrode with order of layers deposited are “Cu/50 nm Alumina/25 nm Ti/5 nm Fe/30 min CNT growth by CVD” (left to right).
Density functional theory calculations
Density functional theory calculations were performed using the Vienna Ab initio Simulation Package (VASP).39,40 We utilized projector augmented wave (PAW) potentials and the PBE exchange-correlation functional.41 We generated 100 atom amorphous MxAl2O3 simulation cells (M = Ti or Fe; x = 0, 1, 4) using the PACKMOL software to randomly distribute the M, Al, and O atoms. We then performed a series of ab initio molecular dynamics (AIMD) trajectories to equilibrate the cells and extract formation energies; this approach, proposed by Jung et al., is called “computational quenching”.42 All runs utilized a Nosé thermostat, a 1 fs time step, a 450 eV planewave cutoff, and the Γ k-point, and all heating and cooling procedures were done at a rate of 1 K s−1. We first heated each simulation cell from 300 to 3000 K, followed by a 5 ps trajectory at 3000 K. We then cooled each system back from 3000 to 300 K and performed another 5 ps trajectory at 300 K. We then randomly selected 5 structures from this last trajectory and performed full DFT relaxations of the lattice parameters and atomic positions to determine the volume of the simulation cell at each M concentration. We then fixed each cell to the new volume and performed a final 5 ps trajectory at 300 K. Finally, we selected 10 structures from this trajectory and self-consistently computed their total energy. The density of states was computed using an increased 4 × 4 × 4 k-point mesh and tetrahedral smearing.Results and discussion
The growth time of the CVD process dictates the thickness of the as-grown CNTs. Fig. 2(a) shows the vertically aligned CNTs on Ti@Al2O3@Cu with 7–8 μm thickness and 30–40 nm diameter for the 30 min CVD growth time. The CNTs show a forest-like structure with very high surface area potentially contributing towards more lithiation. As the growth time is increased, the thickness of the CNTs increases up to a certain time and then remains the same from then on, which could be due to the fact that the amount of catalyst available tends to decrease or due to deactivation of the catalyst when too much carbon accumulates on them.34,43 For electrodes with prolonged CVD time, the adhesion of CNTs to the substrate was poor beyond 2-hour growth time as shown in Fig. 2(c); hence, the optimum growth time was fixed at 30 minutes. Fig. 2(b) is the cross-sectional zoomed out picture of the CNTs showing uniformity in thickness and Fig. 2(d) shows the SEM image of the tilted sample. It has been shown that at higher temperatures the iron catalysts convert into iron nanoparticles and assemble on clouds of alumina, which act as starting points for carbon supersaturation, leading to tubular CNT growth.44 The Ti buffer layer has been used earlier for many reasons such as formation of TiC helping to bind the CNTs with the substrate and thereby preventing breakage.45 The introduction of a Ti buffer layer over alumina has been found to have many advantages. (i) During the CVD process, the intermediate Ti forms highly conductive TiC, which helps with lowering the contact resistance at the Cu–CNT interface.45 (ii) The presence of Ti over alumina helps with protecting the copper substrate from oxidation and electrochemical degradation. (iii) It helps with strong bonding between the substrate and CNTs.46 The use of the catalyst, precursor, and growth temperature in our experiment favoured the formation of multi-walled carbon nanotubes (MWCNTs), as demonstrated by our previous research group.47 They took high-resolution transmission electron microscopy (HR-TEM) images, which confirmed the presence of multi-walled (∼11 walls per tube) carbon nanotubes. | ||
| Fig. 2 SEM images representing (a) 7–8 μm thick vertically aligned CNTs on Cu after 30 min CVD growth. (b) Zoomed out picture of panel (a) showing uniformity in thickness of the CNTs. (c) Poor adhesion of CNTs to the substrate after prolonged growth time and (d) tilted image showing forest-like growth. | ||
In order to confirm the presence of TiC at the interface between the CNTs and the substrate, a CVD experiment was run with the same parameters as discussed in the electrode synthesis section but excluding the Fe catalyst. This absence of catalyst layer prevented the growth of CNTs during CVD but served the purpose of TiC growth. Raman spectroscopy has been performed on the as-grown TiC. Raman spectra shown by the red curve in Fig. 3 show two characteristic D and G bands at 1345 cm−1 and 1574 cm−1, associated to A1g and E2g vibrational modes of the graphitic structure in TiC.48,49 The inset shows the high resolution peaks of TiC at 242 cm−1, 450 cm−1, and 613 cm−1 corresponding to the acoustic mode and optical mode spectral characteristics of titanium compounds, which are comparable to those reported by Iqcin et al.50,51 The black curve in Fig. 3 shows the Raman spectra over the electrode with CNTs grown. The 2D band at 2683 cm−1 mainly originates due to the defects on the MWCNTs.52,53 This peak also appeared in TiC, which can be attributed to defects in the graphitic layer as a CNT is a sheet of graphene rolled into a cylinder.
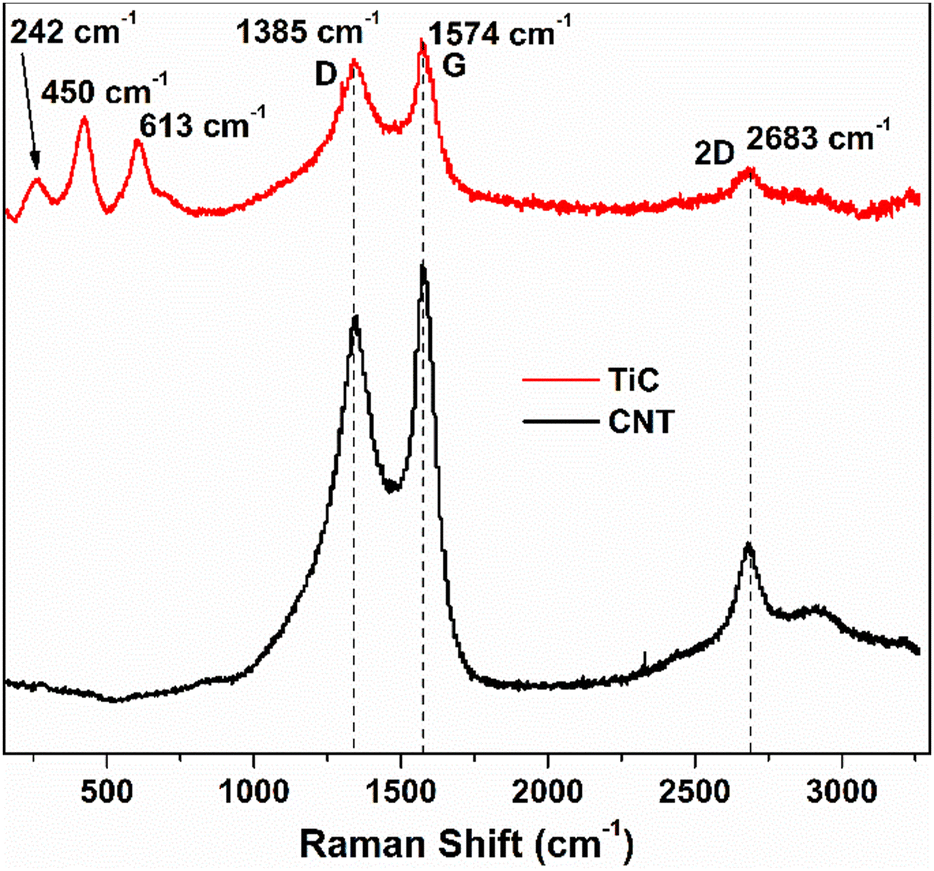 | ||
| Fig. 3 Raman spectra collected from two samples at a laser excitation wavelength of 532 nm: (black) standard CNT/Fe/Al2O3/Cu sample where CNTs were grown by CVD and (red) the sample without the Fe catalyst resulting in no CNT growth but verifying the presence of the TiC interface. | ||
The electrochemical characteristics of CNTs on Cu are shown in Fig. 4. The first three charge–discharge curves at a current density of 50 mA g−1 are shown in Fig. 4(a). The very first discharge cycle reached a capacity of 849 mA h g−1 and is characterized by a voltage plateau between voltages 1.2–1.5 V (vs. Li/Li+). This large irreversible capacity in the first discharge process is attributed to the formation of the solid electrolyte interphase (SEI) layer.54–57 The SEI layer is a passivation layer that forms on the anode surface of LIB batteries comprising both inorganic and organic components during early charge cycles because of electrolyte decomposition. The components on the SEI layer are the products of irreversible reactions and can vary depending on the kind of salts, solvents, and electrodes used. Based on previous studies, EC: DMC + LiPF6 electrolyte with a carbon-based anode tends to form products such as LiF, (CH2OCO2Li)2, Li2CO3, etc. Some of the irreversible reactions are given below.58
| EC: 2(CH2O)2CO + 2e− + 2Li+ → (CH2OCO2Li)2 + 2CH2(g) | (i) |
| (CH2O)2CO + 2e− + 2Li+ → Li2CO3 + C2H4(g) | (ii) |
| DMC: CH3OCO2CH3 + 2e− + 2Li+ → Li2CO3 + C2H6(g) | (iii) |
| LiPF6: LiPF6 + Li2CO3 → 3LiF + POF3 + CO2(g) | (iv) |
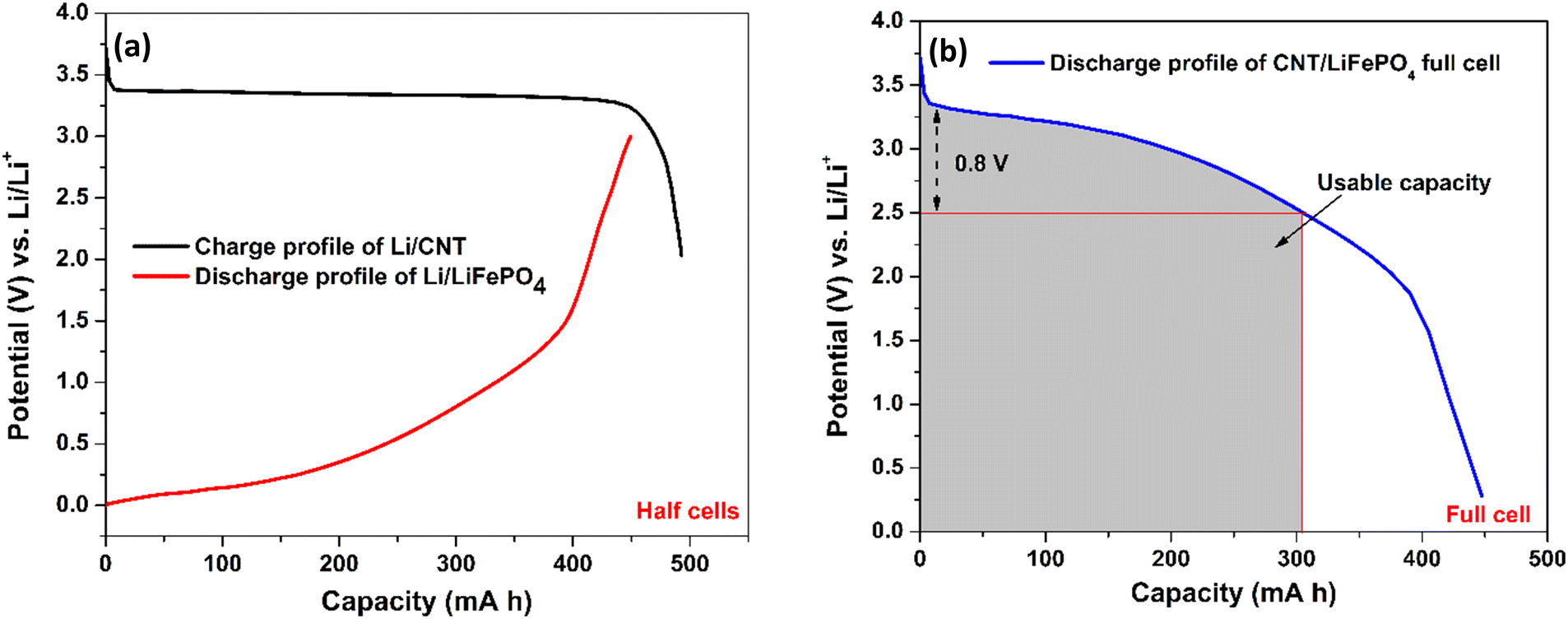 | ||
| Fig. 4 Illustration of a practical capacity of the CNT anode by forming a full cell: (a) charge and discharge profile of our Li/CNT half-cell and Li/LiFePO4 cell from the published literature,65 respectively. 1 gram of CNTs and 3.5 gram of LiFePO4 have been taken to balance the capacity in each electrode. (b) A full cell CNT‖LiFePO4 is modelled by merging the charge–discharge profile of the Li/CNT and Li/LiFePO4 cells. As the discharge curve of the full cell has a broad voltage profile, capacity only within a small voltage fluctuation (∼0.8 V) is useful, which is around 300 mA h g−1 for the CNT anode. | ||
The 2nd and 3rd cycles have reached a nearly identical capacity of 458 mA h g−1, which is higher than previously reported for carbon-based anodes.59–61 This high capacity could be because of defective CNTs, which are known to have better conductivity and lithiation capability than graphite.62,63Fig. 4(b) shows the charge–discharge profiles at various current densities of 50 mA g−1, 100 mA g−1, 250 mA g−1 and 500 mA g−1. It is observed that as the current density increases, the capacity decreases which is the standard behaviour of an LIB. At 50 mA g−1 the capacity is 458 mA h g−1 and even at 500 mA g−1 (10 times the initial current density), 73% of the capacity is retained and reached 336 mA h g−1, which clearly shows the improved rate capability of the anode structure. Fig. 4(c) shows the charge and discharge capacity of the LIB, which is subjected to increasing current densities in step 1 (50–500 mA g−1) followed by decreasing current densities in reverse order (500–50 mA g−1) in step 2 with 10 cycles for each current density. This is done in order to observe the reversible capabilities and stability after long-term cycling when combined with different current densities. The charging and discharging capacity at a particular current density almost overlap indicating good coulombic efficiency and the stability of the device structure.
Interestingly, an enhancement in the capacity with an increase in the cycle number is observed at low charge/discharge current, i.e., 50 mA g−1. Similar behaviour was observed by Kant et al.64 in 2016 in free-standing carbon nanotubes as an anode for a flexible lithium-ion battery. Masarapu et al.62 in 2009 claimed to be the first to observe this anomalous behaviour with MWCNTs synthesized on a stainless-steel substrate. They argued that enhanced capacity came from the gradual structural damage in MWCNTs, which led to the increase in the intercalation ability of the nanotubes. This argument only applies partially to our case because the capacity enhancement only occurred during the slow charge/discharge, and the substrate is copper which cannot intercalate Li ions, unlike stainless-steel as mentioned by Masarapu et al.62 Here we argue that when MWCNT anodes are charged slowly, Li+ ions get enough time to get intercalated first into some outer layers of the nanotubes (which are basically graphene sheets) through surface defects. Diffusion of more and more Li+ ions puts a lot of stress on the graphene layers, thereby expanding the inter-graphene space and easing ion diffusion. As cycling progresses, these ions create more defects in the nanotubes, which helped the Li+ ions go deeper into the layers increasing the energy density. However, when cycling is performed faster, the ions would not get enough time to go deeper, and the capacity remains almost constant until the electrode starts to degrade. Fig. 4(d) illustrates the coulombic efficiency and discharge capacity of the CNT electrode as a function of cycle number at the current density of 200 mA g−1. The Coulombic efficiency is observed to remain at 99.4% after 70 cycles, indicating the stability of the SEI layer and the integrity of the electrode. However, it is important to note that the Coulombic efficiency obtained in the half-cell may not be reflected in the actual device (full cell) due to the limited inventory of lithium ions in the anode.
To illustrate the capacity of our CNT electrode in a full cell (Fig. 5(b)), we have combined our CNT anode capacity (Fig. 5(a)) with the published discharge cycle capacity of a LiFePO4 cathode paired against a Li anode (Fig. 5(a)).65 In order to balance the capacity in the anode and cathode, the capacity for 1 g of CNTs and 3.5 g of LiFePO4 is taken. The plot in Fig. 5(b) was created by subtracting the voltage of Li/CNT (Fig. 5(a)) by the voltage of the Li/LiFePO4 because Li is the counter electrode to both devices. This way, the value shown in the y-axis of Fig. 5(b) shows the voltage achievable in a full cell for our CNT electrode. From Fig. 5(b), it is thus clear that a practical capacity of ∼300 mA h g−1 at a rated voltage of 2.5 V is possible with our CNTs if employed in a full cell configuration. As a lithium ion capacitor, the full capacity of 458 mA h g−1 can be utilized.
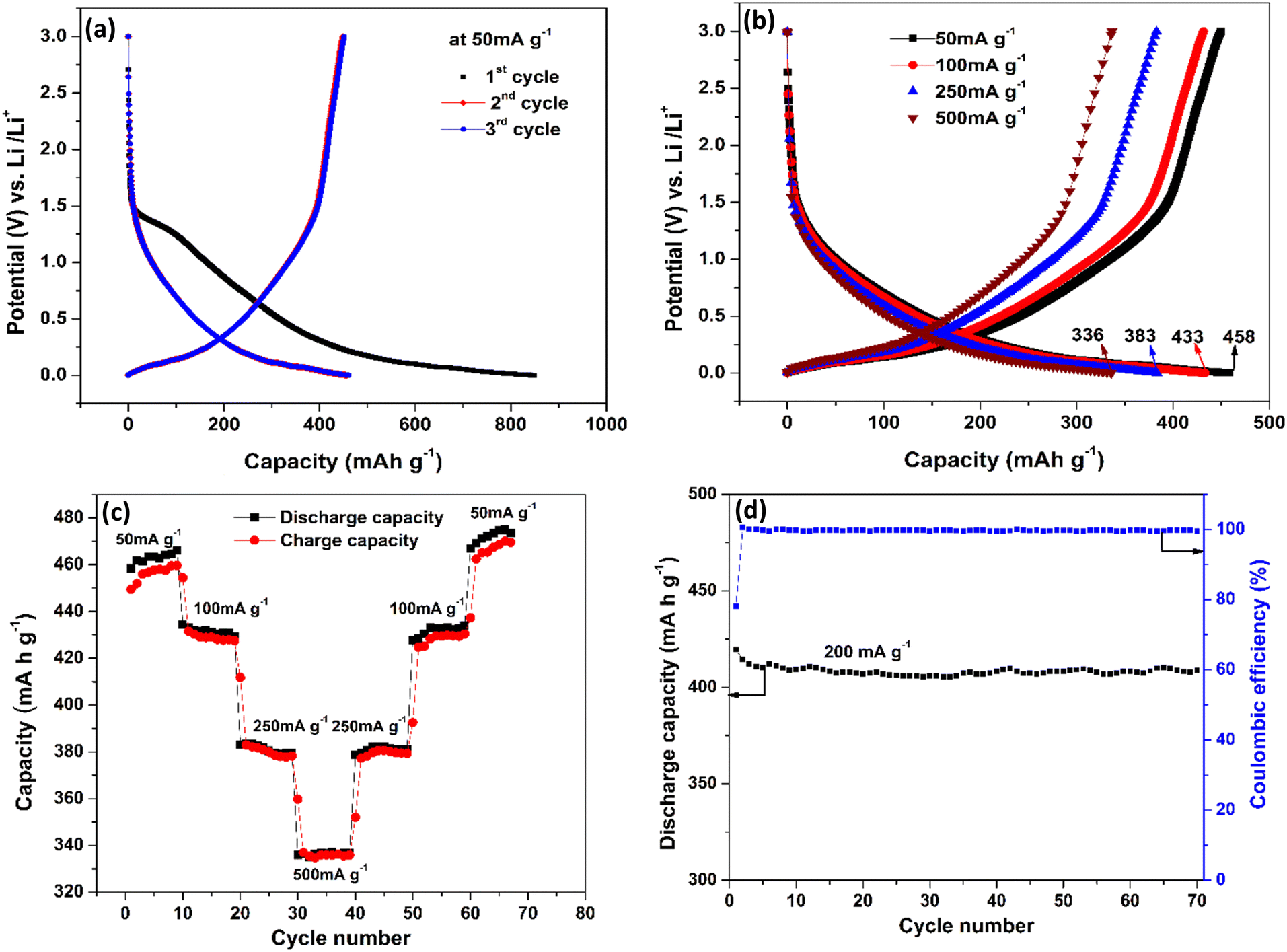 | ||
| Fig. 5 Galvanostatic charge discharge profiles of CNTs on the Cu substrate. (a) First three charge–discharge cycles at 50 mA g−1. (b) Electrode subjected to various current densities from 50–500 mA g−1. (c) High-rate cycling performance. (d) Discharge capacity and coulombic efficiency vs. cycle number at the current density of 200 mA g−1. | ||
Calculation of density of states
In order to confirm the advantage of the Ti buffer layer, which increases the conductivity at the alumina/CNT interface, density of states calculations were carried out on alumina with various amounts of Ti to investigate changes in the electronic structure. Pure alumina with two concentrations of Ti (0.2 and 0.5 per Al2O3 formula unit) were used for the calculations. The density of states for the compounds Al2O3, Ti0.2Al2O3, and Ti0.5Al2O3 are shown in Fig. 6(a). At both concentrations, it was found that it is favourable for the Ti to diffuse into amorphous alumina due to the negative formation energy Eform. Therefore, the introduction of Ti causes states to appear at the Fermi level (set to 0 eV here), resulting in metallic behaviour. Fig. 6(b) shows the density of states with small amounts of Ti and Fe atoms in alumina. First, both Ti and Fe intercalation cause the system to turn metallic. As the concentration of both increase, more states appear at the Fermi level. Second, at low concentration, it is found that it is energetically unfavorable for Ti or Fe to intercalate into Al2O3. However, it is 50% more unfavorable for Fe to intercalate (true across the concentrations). Comparing the results of the density of states with the experimental results, the following is derived. (1) In the case of Fe alone, it is unfavorable for Fe to diffuse into the Al2O3 layer, which then remains insulating. This is important because the Fe nanoparticles act as a catalyst for CNT growth, and any diffusion into alumina would not result in CNT growth. (2) In the case of Ti, it is more favorable for small amounts to get into the Al2O3 layer and make it metallic, leading to enhanced conductivity.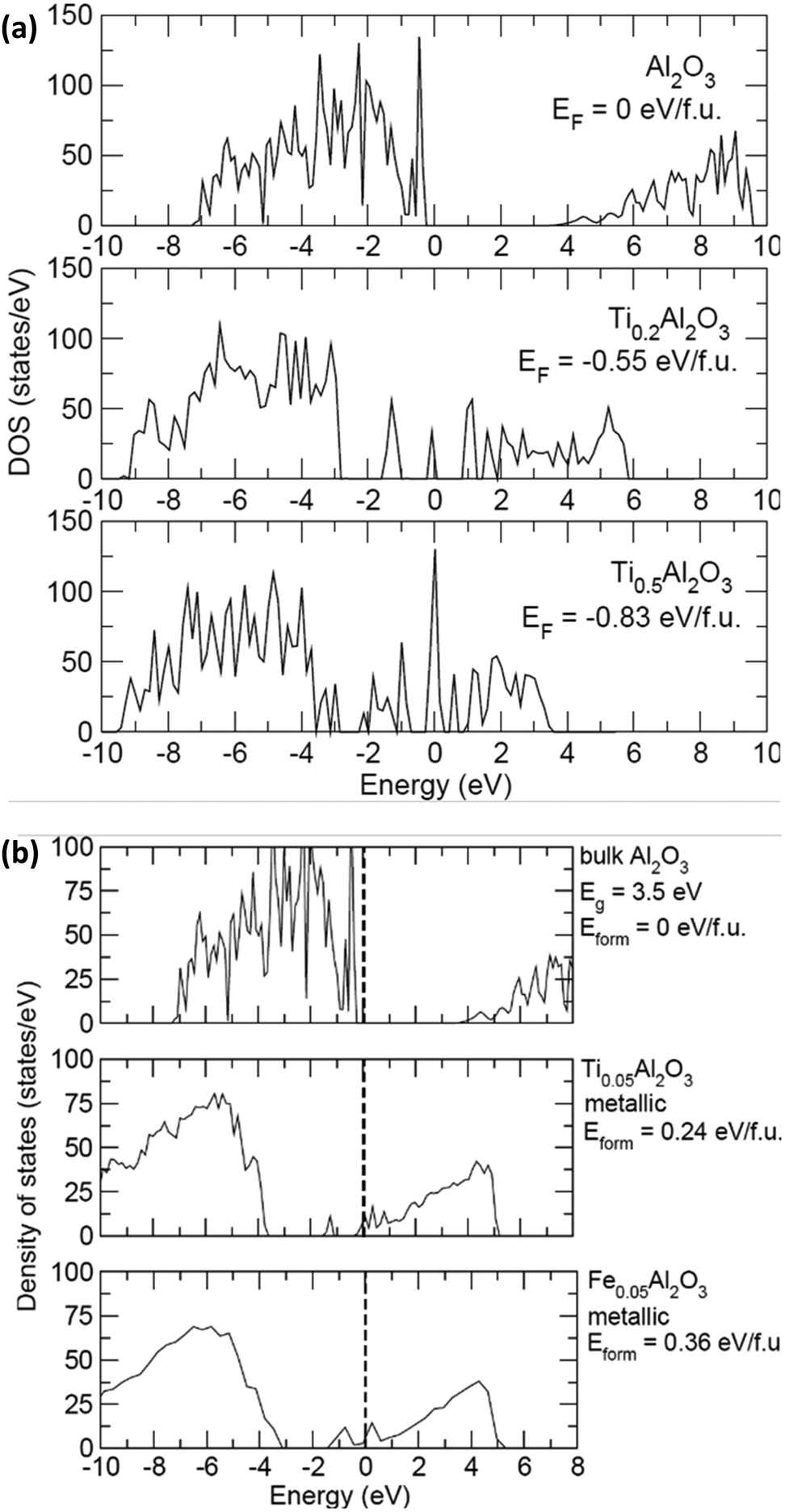 | ||
| Fig. 6 Density of states (states per eV) vs. energy (eV) at (a) 0, 0.2 and 0.5 Ti per Al2O3 formula unit showing the appearance of states across the Fermi level (set at 0 eV) and (b) at 0, 0.05 Ti and 0.05 Fe per Al2O3 formula unit showing the lower Eform of Ti compared to Fe to intercalate into Al2O3 to increase the electronic conductivity at the interface. | ||
Conclusions
The viability of high surface area carbon nanotubes (CNTs) as an anode for LIBs, grown on copper foils using chemical vapor deposition has been studied. CNTs with their high theoretical capacity and superior material properties could be used as a replacement for graphite. The effect of the Ti buffer layer in the growth on CNTs via CVD technique has also been studied by making an electrode with structure Cu/Al2O3/Ti/Fe/CNT. Coin cells were made from this electrode with Li metal as the counter electrode and 1 M LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 mass ratio) solution as an electrolyte. A high capacity of 300 mA h g−1 (0.8 V vs. Li/Li+) and 458 mA h g−1 (3 V vs. Li/Li+) at 50 mA g−1 has been achieved. Raman spectroscopy confirmed the formation of TiC at the interface between the CNTs and the substrate. The high rate capacity on the LIB could be attributed to the conductive TiC, which helped with better contact between the substrate and CNTs, thereby reducing the interface contact resistance and the high surface area CNTs contributing towards higher lithiation capability. The capacity of this CNT anode can be enhanced by the incorporation of active anode material such as Si, which can help achieve the target of energy density required for electric vehicles.
Author contributions
Ganesh S. Gudavalli fabricated the device, performed experiments, analysed the data and wrote the original draft of the manuscript. Mahesh Nepal performed analysis of the observed results and edited the manuscript accordingly. Manuel Smeu and Joshua Young performed the DFT calculations and provided the corresponding analysis. Tara P. Dhakal conceptualized the project, designed the experiment and reviewed the findings and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported work was supported in part by Dhakal's start-up funds provided by the Department of Electrical and Computer Engineering at Binghamton University. The authors also would like to acknowledge the support from Northeast Center for Chemical Energy Storage (NECCES) for charge–discharge cycle measurements. In addition, we would like to thank Prof. Nobel Laurate Stan Whittingham for reading our manuscript and providing us with valuable comments.References
- A. G. Olabi and M. A. Abdelkareem, Renewable Sustainable Energy Rev., 2022, 158, 112111 CrossRef CAS.
- M. S. Whittingham, Nano Lett., 2020, 20, 8435 CrossRef CAS PubMed.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292 CrossRef.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176 CrossRef CAS.
- C. Wang, C. Yang and Z. Zheng, Adv. Sci., 2022, 9, 2105213 CrossRef CAS PubMed.
- R. Yazami and P. Touzain, J. Power Sources, 1983, 9, 365 CrossRef CAS.
- K. Persson, V. A. Sethuraman, L. J. Hardwick, Y. Hinuma, Y. S. Meng, A. Van Der Ven, V. Srinivasan, R. Kostecki and G. Ceder, J. Phys. Chem. Lett., 2010, 1, 26 Search PubMed.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664 CrossRef CAS PubMed.
- A. Abdollahi, A. Abnavi, S. Ghasemi, S. Mohajerzadeh and Z. Sanaee, Electrochim. Acta, 2019, 320, 134598 CrossRef CAS.
- L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang, Y.-H. Huang, L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047 CrossRef PubMed.
- J. Hou, Y. Shao, M. W. Ellis, R. B. Moore and B. Yi, Phys. Chem. Chem. Phys., 2011, 13, 15384 RSC.
- Y. Dou, X. Liu, K. Yu, X. Wang, W. Liu, J. Liang and C. Liang, Diamond Relat. Mater., 2019, 98, 107514 CrossRef CAS.
- C. Ying Kuo, H. Ping Hsu and C. Wen Lan, J. Power Sources, 2023, 558, 232599 CrossRef.
- H. Su, X. Li, C. Liu, Y. Shang and H. Liu, Chem. Eng. J., 2023, 451, 138394 CrossRef CAS.
- W. Lang, C. Yue, M. Dang, G. Wang, Y. Chen, F. Hu, Z. Liu and J. Shu, J. Power Sources, 2023, 560, 232706 CrossRef CAS.
- R. Mo, X. Tan, F. Li, R. Tao, J. Xu, D. Kong, Z. Wang, B. Xu, X. Wang, C. Wang, J. Li, Y. Peng and Y. Lu, Nat. Commun., 2020, 11, 1374 CrossRef CAS PubMed.
- K. Zhuo, M.-G. Jeong and C.-H. Chung, J. Power Sources, 2013, 244, 601 CrossRef CAS.
- Z. Wang, L. Zhou and X. W. David Lou, Adv. Mater., 2012, 24, 1903 CrossRef CAS PubMed.
- P. P. Prosini, M. Carewska, S. Loreti, C. Minarini and S. Passerini, Int. J. Inorg. Mater., 2000, 2, 365 CrossRef CAS.
- J. E. Fischer, Acc. Chem. Res., 2002, 35, 1079 CrossRef CAS PubMed.
- C. M. Schauerman, M. J. Ganter, G. Gaustad, C. W. Babbitt, R. P. Raffaelle and B. J. Landi, J. Mater. Chem., 2012, 22, 12008 RSC.
- V. Meunier, J. Kephart, C. Roland and J. Bernholc, Phys. Rev. Lett., 2002, 88, 075506 CrossRef PubMed.
- K. Nishidate and M. Hasegawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 245418 CrossRef.
- Y. Ando, X. Zhao, H. Shimoyama, G. Sakai and K. Kaneto, Int. J. Inorg. Mater., 1999, 1, 77 CrossRef CAS.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. H. Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. E. Scuseria, D. Tománek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483 CrossRef CAS PubMed.
- H. Dai, A. Javey, E. Pop, D. Mann, W. Kim and Y. Lu, Nano, 2006, 1, 1 CrossRef CAS.
- V. A. Sugiawati, F. Vacandio, N. Yitzhack, Y. Ein-Eli and T. Djenizian, Polymers, 2020, 12, 406 CrossRef CAS PubMed.
- Y. Yan, C. Li, C. Liu, Z. Mutlu, B. Dong, J. Liu, C. S. Ozkan and M. Ozkan, Carbon, 2019, 142, 238 CrossRef CAS.
- S. S. Zhang and T. R. Jow, J. Power Sources, 2002, 109, 422 CrossRef CAS.
- A. Du Pasquier, F. Disma, S. A. S. Gozdz and J. Tarascon, J. Electrochem. Soc., 1998, 145, 472 CrossRef.
- H. Maleki, G. Deng, A. Anani and J. Howard, J. Electrochem. Soc., 1999, 146, 3224 CrossRef CAS.
- G. Li, S. Chakrabarti, M. Schulz and V. Shanov, J. Mater. Res., 2009, 24, 2813 CrossRef CAS.
- J. García-Céspedes, S. Thomasson, K. B. K. Teo, I. A. Kinloch, W. I. Milne, E. Pascual and E. Bertran, Carbon, 2009, 47, 613 CrossRef.
- H. Wang, J. Feng, X. Hu and K. M. Ng, J. Phys. Chem. C, 2007, 111, 12617 CrossRef CAS.
- G. S. Gudavalli, J. Turner and T. P. Dhakal, MRS Adv., 2017, 2, 3263 CrossRef CAS.
- T. Dhakal, D. Vanhart, R. Christian, A. Nandur, A. Sharma and C. R. Westgate, J. Vac. Sci. Technol., A, 2012, 30, 021202 CrossRef.
- Y. Gao, G. P. Pandey, J. Turner, C. Westgate and B. Sammakia, Phys. Scr., 2012, 86, 065603 CrossRef CAS.
- Y. Gao, G. G. P. Pandey, J. Turner, C. R. Westgate and B. Sammakia, Nanoscale Res. Lett., 2012, 7, 651 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- S. C. Jung and Y. K. Han, J. Phys. Chem. Lett., 2013, 4, 2681 CrossRef CAS.
- J. Zhu, J. Jia, F. Kwong and D. H. L. Ng, Carbon N. Y., 2012, 50, 2504 CrossRef CAS.
- M. Kumar and Y. Ando, J. Nanosci. Nanotechnol., 2010, 10, 3739 CrossRef CAS PubMed.
- I. Lahiri, S.-W. Oh, J. Y. Hwang, S. Cho, Y.-K. Sun, R. Banerjee and W. Choi, ACS Nano, 2010, 4, 3440 CrossRef CAS PubMed.
- J. Zhang, X. Wang, W. Yang, W. Yu, T. Feng, Q. Li, X. Liu and C. Yang, Carbon N. Y., 2006, 44, 418 CrossRef CAS.
- S. P. Adusumilli and C. R. Westgate, J. Nanosci. Nanotechnol., 2015, 15, 6609 CrossRef CAS PubMed.
- B. H. Lohse, A. Calka and D. Wexler, J. Alloys Compd., 2007, 434–435, 405 CrossRef CAS.
- M. Amer, M. W. Barsoum, T. El-Raghy, I. Weiss, S. Leclair and D. Liptak, J. Appl. Phys., 1998, 84, 5817 CrossRef CAS.
- M. V. Iqcin, J. A. Holy and W. S. Williams, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 17, 1546 CrossRef.
- N. Saoula, K. Henda, R. Kesri, S. Shrivastava, R. M. Erasmus and J. D. Comins, Acta Phys. Pol., A, 2012, 121, 172 CrossRef CAS.
- B. P. Vinayan, R. I. Jafri, R. Nagar, N. Rajalakshmi, K. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2012, 37, 412 CrossRef CAS.
- A. Kaniyoor, R. Imran Jafri, T. Arockiadoss and S. Ramaprabhu, Nanoscale, 2009, 1, 382 RSC.
- P. Sehrawat, C. Julien and S. S. Islam, Mater. Sci. Eng., B, 2016, 213, 12 CrossRef CAS.
- R. Maheswaran and B. P. Shanmugavel, J. Electron. Mater., 2022, 51, 2786 CrossRef CAS PubMed.
- X. Jiao, Y. Liu, B. Li, W. Zhang, C. He, C. Zhang, Z. Yu, T. Gao and J. Song, Carbon N. Y., 2019, 148, 518 CrossRef CAS.
- J. Ren, Z. Wang, F. Yang, R. P. Ren and Y. K. Lv, Electrochim. Acta, 2018, 267, 133 CrossRef CAS.
- V. A. Agubra and J. W. Fergus, J. Power Sources, 2014, 268, 153 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-S. Zhou, T. Kudo and I. Honma, Nano Lett., 2009, 9, 72 CrossRef PubMed.
- F. Ji, Y.-L. Li, J.-M. Feng, D. Su, Y.-Y. Wen, Y. Feng and F. Hou, J. Mater. Chem., 2009, 19, 9063 RSC.
- M. Liang and L. Zhi, Artic. J. Mater. Chem., 2009, 19, 5871 RSC.
- C. Masarapu, V. Subramanian, H. Zhu and B. Wei, Adv. Funct. Mater., 2009, 19, 1008 CrossRef CAS.
- P. C. P. Watts, W.-K. Hsu, H. W. Kroto and D. R. M. Walton, Nano Lett., 2003, 3, 549 CrossRef CAS.
- C. Kang, E. Cha, R. Baskaran and W. Choi, Nanotechnology, 2016, 27, 105402 CrossRef PubMed.
- L. Liao, P. Zuo, Y. Ma, X. Chen, Y. An, Y. Gao and G. Yin, Electrochim. Acta, 2012, 60, 269 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |