
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent ReaxFF MD studies on pyrolysis and combustion mechanisms of aviation/aerospace fuels and energetic additives
Menghui
Chen†
a,
Wei
Li†
b,
Houjun
Zhang
a,
Menghui
Liu
a,
Jinli
Zhang
 a,
Xiangyuan
Li
c and
You
Han
*a
a,
Xiangyuan
Li
c and
You
Han
*a
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: yhan@tju.edu.cn
bScience and Technology on Aerospace Chemical Power Laboratory, Hubei Institute of Aerospace Chemotechnology, Xiangyang 441003, China
cSchool of Chemical Engineering, Sichuan University, Chengdu 610065, China
First published on 30th November 2022
Abstract
Development of aviation and aerospace fuels requires deep insight into the pyrolysis and combustion mechanisms. However, rapid and complex reactions during fuel combustion make it difficult to accurately describe the free radicals, reaction paths and other relevant details in large-scale reaction systems through experiments or quantum mechanics. The molecular dynamics (MD) simulation based on reaction force-fields (ReaxFF) is an effective method to solve the above problems, which can reveal the overall reaction mechanism in the fuel combustion process by dynamically describing the breakage and formation of bonds. Here in, the recent progress of ReaxFF MD in exploring the pyrolysis and combustion mechanism of aviation fuels, aerospace fuels and energetic additives is summarized, especially for aviation kerosene model substitutes, high-density aviation fuels, aerospace fuels and energetic additives under various conditions. Finally, the shortcomings of ReaxFF MD simulation in describing fuel pyrolysis or combustion systems are discussed, and the application prospect of ReaxFF MD simulation in the fuel field is prospected.
1 Introduction
The construction of a reasonable physical model and chemical kinetic mechanism of aviation and aerospace fuels will help researchers to deepen their understanding of the pyrolysis and combustion process of aerospace fuel, and then play a guiding role in engine design and optimization. Researchers have proposed a large number of physical replacement models1–3 and related chemical mechanisms for aviation fuel through experimental studies.4,5 These studies enable people to understand the thermophysical and thermochemical properties of aviation fuel, but there is still a lack of details about the fuel reaction path and free radicals in the combustion process. At this extreme temperature and pressure, chemical reactions take place in picoseconds and the reaction paths are coupled, which brings great difficulties to the detection of free radicals, intermediates and reaction paths; even with the latest instruments, it is difficult to capture them completely.6 However, the development of computational chemistry makes up for the shortcomings of experiments.Quantum chemical (QC) simulation is a widely used molecular simulation method based on quantum mechanics (QM). It obtains a high-precision solution at the electronic scale by solving the Schrödinger equation, which allows it to accurately predict the potential barrier of the reaction.7,8 However, the high computational cost limits the quantum chemical simulation to the simple system of small molecules and picosecond time scale. In addition, the defect of the preset reaction path makes this simulation method unsuitable for complex macromolecular systems such as coal and biomass. The molecular dynamics (MD) simulation method can calculate the trajectory of atoms by solving Newtonian equations, which makes it possible to apply it in large-scale systems9 with lower cost and shorter time than quantum chemical simulation methods. However, it is worth noting that molecular dynamics simulation is lacking in the study of chemical reaction processes, which limits its further application.
Quantum chemical simulation and molecular dynamics simulation have their strengths and weaknesses, and the urgent need to study large-scale reaction systems promotes their integration, thus developing a new simulation method – reaction molecular dynamics simulation method. The ReaxFF reaction force field developed by van Duin et al.10 expresses bond length in terms of bond order. Based on this method, ReaxFF can dynamically describe the breakage and formation of bonds in the reaction process. And in this process, a smooth and continuous transition can be achieved between non-bonded states and bond states without the existence of energy or force discontinuities. The ReaxFF potential parameters are optimized by the data obtained from experiments and quantum chemistry calculations, which makes the ReaxFF MD simulation close to the accuracy of quantum chemistry calculations. The same calculation method as molecular dynamics simulation makes ReaxFF MD simulation several orders of magnitude faster than quantum chemistry calculation, which greatly reduces the cost. More importantly, ReaxFF MD simulation does not rely on prior chemical knowledge, which makes it widely used in complex reaction systems such as coal,11 biomass,12 and fuel.13
The application of ReaxFF is closely related to the development and improvement of its reaction force field. As one of the earliest branches of ReaxFF, the reaction force field related to the combustion branch14 has been continuously updated and expanded in recent years. In order to study the initial reaction of the hydrocarbon gas phase at high temperature, Chenoweth et al.15 developed the CHO-2008 force field by adding the transition state information and relevant quantum mechanical data during hydrocarbon oxidation to the initial training set of hydrocarbons. And the CHO-2008 force field was used for methane, propylene, o-xylene and benzene oxidation systems, and achieved satisfactory results. So far, the CHO-2008 force field is still one of the most widely used reaction force fields.16–19 Ashraf et al.20 developed the CHO-2016 force field based on the CHO-2008 force field, improving the range of applications and simulation accuracy of ReaxFF. Compared to the CHO-2008 force field, the CHO-2016 force field maintains the prediction accuracy of the CHO-2008 force field for large hydrocarbons. On this basis, the CHO-2016 force field provides a richer set of radicals and reaction pathways, systematically improving on the lack of prediction accuracy of the CHO-2008 force field in describing the oxidation of small hydrocarbons to carbon dioxide. In addition, the shortcomings of the CHO-2008 force field regarding the overprediction of the rate of hydrogen extraction by oxygen and thus the underestimation of the initial temperature of hydrocarbon oxidation have been effectively addressed. In order to explore the combustion of hydrogen at low temperature and high pressure, Cheng et al.21 developed the OH-2014 force field, which improved the lack of prediction of H3O and other intermediates in the CHO-2008 force field, and provided a feasible tool for studying the reaction mechanism of H2 at real temperature. In addition, they have developed adaptive Accelerated ReaxFF Reaction Dynamics by combining reaction molecular dynamics with acceleration dynamics and have tested a hydrogen combustion system using the HO-2014 force field to achieve satisfactory results at a low cost. They showed that this approach may have significant advantages in the future for describing longer reaction systems at lower temperatures. Kowalik et al.22 have added a parametric description of the N atom to the CHO-2016 force field, resulting in the development of a CHON-2019 force field that is more suitable for probing the specific details of the transformation of polymers into carbon fiber structures. They explored the formation kinetics of N2 and its interaction with related free radicals during the carbonization of polymers using the improved CHON-2019 force field, determining the formation path of all-carbon rings, and put forward new insights into understanding how polymers evolve into graphene structures. ReaxFF has absorbed a large amount of metal23–25 and non-metallic elements26,27 in the process of continuous application to new fields, and developed a large number of force fields, which makes it applicable in most scientific research fields.
During the simulation of ReaxFF MD, specific parameter settings such as temperature, pressure and time step affect the results of the simulation. In view of the cost of the simulation, ReaxFF MD is generally set at a much higher temperature than the experimental temperature, but numerous studies have shown that an increase in temperature only accelerates the reaction and has a small effect on the reaction process, and the simulation results are in qualitative agreement with the experimental values. The effect of pressure on the reaction process is not as significant as that of temperature, but an increase in pressure increases the probability of molecular collisions, which also has an accelerating effect on the reaction process. However, given the stability of the system, the pressure is generally only set when simulating larger reaction systems. The choice of time step determines the accuracy of the simulation results. Jensen et al.28 explored the effect of time step setting on the reaction by testing six carbon-based molecules using different time steps. The results showed that smaller time steps were required at higher temperatures to obtain accurate simulation results. The Chenoweth parameter is sufficient for accurate simulations at temperatures below 3000 K with a time step of 0.1 fs. At present, most of the studies on the molecular dynamics of fuel pyrolysis and combustion use this time step (0.1 fs).
Recent articles14,29–31 provide a comprehensive review of the development of ReaxFF and its applications in various fields. However, these articles are too sketchy in describing the reaction mechanism of the pyrolysis and combustion of aviation fuel. Moreover, there are few studies on the pyrolysis and combustion mechanism of aerospace fuels and their energetic additives. Therefore, this review intends to sort out the studies on the application of ReaxFF in exploring the reaction mechanism of pyrolysis or combustion of aviation fuels, aerospace fuels and energetic additives. In the second part, the progress of using ReaxFF to study the pyrolysis and combustion mechanism of aviation kerosene model substitutes and high-density aviation fuels is introduced. In the third part, the use of ReaxFF in exploring the pyrolysis and combustion mechanism of aerospace fuels and energetic additives is introduced. Finally, the content of the full text is summarized, the shortcomings of ReaxFF are discussed and the development of ReaxFF is prospected.
2 Pyrolysis and combustion mechanism of aviation fuels
The study of single component pyrolysis systems with ReaxFF MD provides comprehensive and unobtrusive information on the mechanism of component pyrolysis and reaction products. These chain, cycloalkane and aromatic hydrocarbon components are the basis for the model compounds of aviation paraffin. The study of these single components has provided a wealth of information that has led to a better understanding of the overall mechanism of aviation paraffin combustion. In contrast to single-component pyrolysis, radicals, intermediates and other species formed by the first component to be pyrolysed in multi-component pyrolysis may attack other reactant molecules, causing changes in the reaction pathway and thus affecting the reaction mechanism and products. A comparison of multi-component and single-component pyrolysis systems can demonstrate the differences between the two and thus reveal the influence of the interaction of the different components on the reaction process and improve the understanding of the overall mechanism of aerospace paraffin combustion. Overall, the combination of the single- and multi-component models of ReaxFF MD provides a clearer and more comprehensive picture of the overall reaction mechanism of aviation kerosene and provides a basis and guidance for the design and optimisation of engines.2.1 One-component pyrolysis and combustion mechanism of aviation fuels
The general fuels currently used in domestic and foreign aviation fields are RP-3 and Jet A/Jet A-1 series kerosene. The research on the combustion mechanism of real aviation kerosene at high temperature can provide a reference for the optimization and improvement of the engine. However, considering the complexity of the composition of real aviation fuel, this is an almost impossible task. Therefore, the alternative model selected based on the physical and chemical properties of aviation kerosene has become an acceptable solution for the research of aviation kerosene. A large number of researchers have proposed different multi-component aviation kerosene substitution models, such as the four-component RP-3 aviation kerosene substitution model proposed by Yu et al.,32 the three-component Jet A-1 aviation kerosene substitution model proposed by Dagaut et al.,33 the three-component Jet A aviation kerosene substitution model proposed by Dooley et al.34 and so on. Although there are a variety of alternative models for aviation kerosene, most of them include alkanes, naphthenes and aromatic hydrocarbons. And undisturbed kinetics and reaction mechanism can be obtained by simulating these components separately. These works, combined with the research of multi-component models, can provide a clearer and more comprehensive understanding of the reaction mechanism of aviation kerosene.Liu et al.35 explored the thermal decomposition mechanism of n-octane in the liquid state, the effect of temperature and the formation mechanism of three main thermal decomposition products through reaction molecular dynamics and density functional theory calculation. Eight thermal decomposition paths of n-octane were determined and it was found that the end of the n-octane molecule was not easy to react, and the increase of temperature greatly promoted the reaction. The major products, H2 and CH4, are mainly from the attack of H and CH3 free radicals and the binding reaction between free radicals. On the other hand, ethylene mainly comes from β-scission reactions of macromolecules. Wang et al.36 applied the optimized reaction force field to the pyrolysis of isooctane, and they used a time step of 0.1 fs in the simulation, which showed that the cleavage path of the C–C bond connected to quaternary carbon occupies the highest proportion in the pyrolysis path of isooctane. And it can be observed from the number of isooctane pyrolysis products and intermediates that C2 species play an important role in the pyrolysis of isooctane. Liu et al.37 investigated and compared the combustion mechanism, reaction performance and coking status of six octane isomers. The results show that both n-octane and isooctane undergo the stage of isomerization during combustion, but the activity of isooctane decreases due to the existence of a branched chain compared with n-octane, and the trend of coking is obvious with the increase of the branched chain.
Liu et al.38 simulated the pyrolysis process of n-decane and focused on the formation of soot nanoparticles during the pyrolysis of n-decane. The results show that the dissociation energy of the C2–C3 bond of the n-decane molecule is the lowest, and the cleavage of the C2–C3 bond is the most likely path for the decomposition of the n-decane molecule. In addition, it is also found that the formation of soot nanoparticles needs to go through four stages: initial carbon ring formation, polycyclic aromatic hydrocarbon (PAH) growth, PAH aggregation nucleation and carbon core surface growth, as shown in Fig. 1(a). Zhou et al.39 explored the decomposition process of n-decane under the action of an electric field. The decomposition pathway and proportion distribution of n-decane are shown in Fig. 1(b). The study found that the electric field enhanced the polarization of n-decane at low temperature. It also significantly promoted the decomposition of n-decane, and the number of PAHs was significantly reduced during the decomposition of n-decane, as shown in Fig. 1(c). These works provide a reference for the development of anti-coking methods in the combustion process of aviation fuel. Wang et al.40 tested the effects of several additives on the pyrolysis of n-decane and it was found that 1-nitropropane greatly reduces the decomposition and activation energy of reaction molecules during the pyrolysis of n-decane, and thus plays the highest role in promoting the pyrolysis of n-decane. This work provides a reference precedent for the screening of fuel additives.
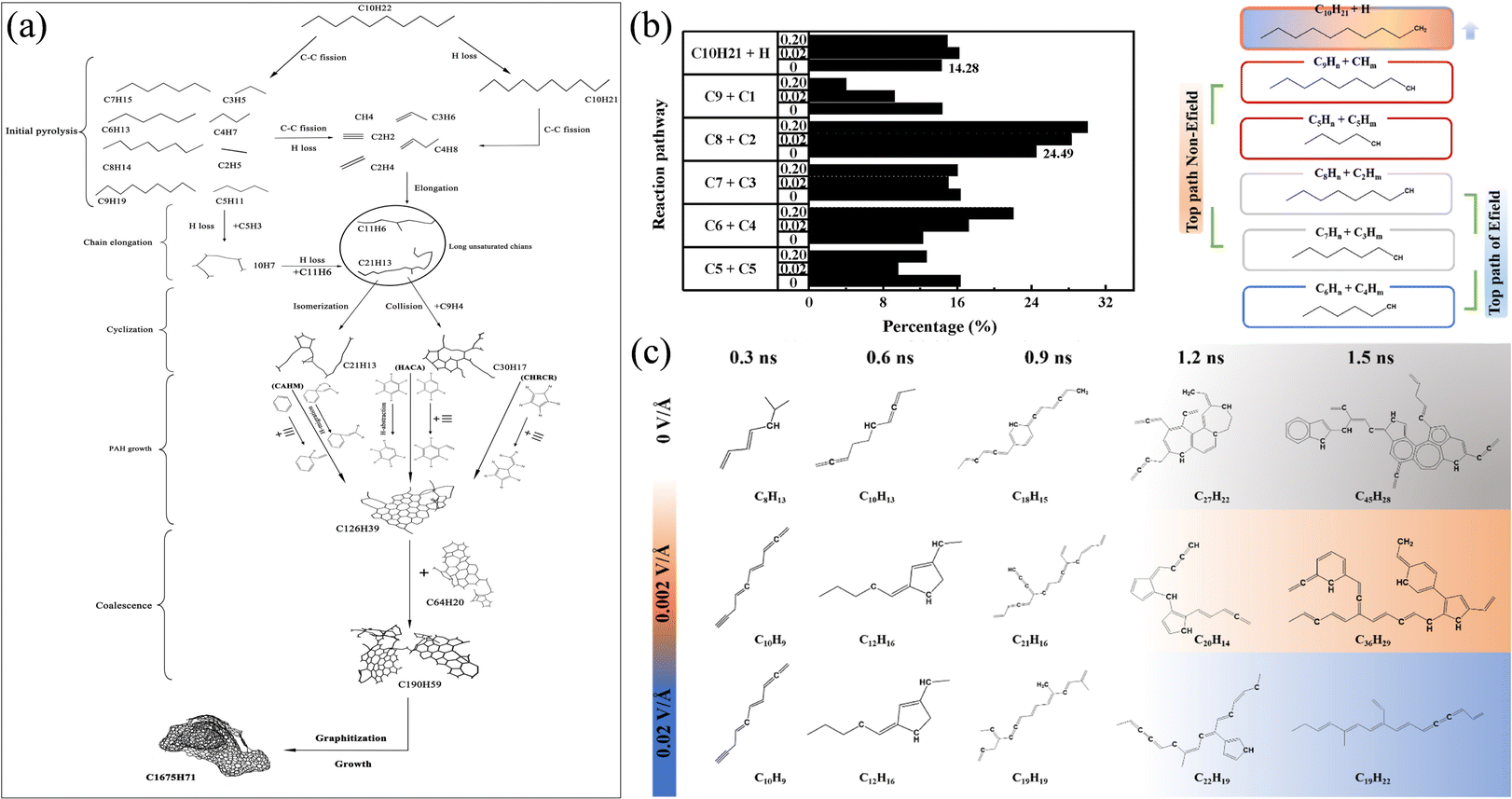 | ||
| Fig. 1 (a) Mechanism of soot formation by pyrolysis of n-heptane at 3000 K;38 (b) initial pyrolysis path and proportion of n-decane under an electric field;39 and (c) the largest molecule structure evolution during the reaction process.39 | ||
Wang et al.18 simulated the pyrolysis and combustion process of n-dodecane. The results show that the pyrolysis path of n-dodecane is C–C bond breakage, dehydrogenation and H extraction of small molecular free radicals. In addition, it is also found that the effect of density/pressure on the pyrolysis of n-dodecane is not as significant as that of temperature and there is a threshold for the effect of density/pressure on pyrolysis. The importance of formaldehyde molecules can be observed from the oxidation simulation process of n-dodecane, which is consistent with the existing kinetic models. The common reactive molecular dynamics studies of fuel combustion properties are carried out at high temperatures, but these temperatures deviate from the temperature for practical industrial applications. To obtain atomic-scale information of fuel combustion processes at industrially relevant temperature and time scales, Bal et al.41 studied the pyrolysis and oxidation of n-dodecane at low temperature to 700 K and ultra-long time to 39 seconds by collective variable driven super dynamics (CVHD42). The simulation results are consistent with the previous studies. This work provides a reference for exploring the reaction mechanism of reaction molecular dynamics under experimental conditions.
Chen et al.43 studied the pyrolysis process of n-hexadecane at high temperature, which showed that the eight pyrolysis paths of n-hexadecane involved C–C bond breakage, and the existence of double free radicals accelerated the reaction during the pyrolysis of n-hexadecane. In addition, the optimum conditions for the pyrolysis of n-hexadecane to ethylene and hydrogen were determined by exploring the effects of temperature and pressure on the pyrolysis process. Subsequently, the research group44 further studied the pyrolysis process of n-hexadecane under the action of three kinds of catalysts. The results showed that the introduction of hydroxyl groups on the surface of the catalyst was beneficial to the increase of ethylene production. On the other hand, the addition of aluminum stabilized the structure of the catalyst, promoting the dehydrogenation of reactant molecules and inhibiting the formation of the C–O bond. These works provide a reference for the application of heavy hydrocarbons in industry.
As the most basic component of aviation fuels, the research work on the pyrolysis and oxidation mechanism of endothermic hydrocarbon fuel saturated alkanes provides an opportunity to deeply understand the ignition and combustion properties of aviation fuel, further achieve full combustion and avoid coking of aviation fuel in the engine.
Liu et al.47 studied the detailed pyrolysis process of methylcyclohexane and found that there are four initial decomposition paths of methylcyclohexane, as shown in Fig. 2(a). It can be observed that the first type of reaction C7H14 → ˙C7H14˙ has the highest proportion. It can be seen from Fig. 2(b) that the main product (i.e., C7H14 double radical) is further decomposed to produce ethylene and propylene, which is different from the previous study. Another work50 studied the pyrolysis mechanism of methylcyclohexane in the presence of suspended functional fossil graphene tablets (FGS). As shown in Fig. 2(c), the functional groups on FGS significantly reduce the potential barrier of methylcyclohexane dehydrogenation, accelerate the decomposition of reaction molecules and produce more small molecular free radicals, thus increasing the conversion of methylcyclohexane to low-carbon species. Liu et al.53 further studied the oxidation process of methylcyclohexane and clarified the oxidation mechanism of methylcyclohexane. Through the research, they found a new fuel consumption route in which methylcyclohexane is isomerized first and then decomposed into small molecular olefins. In addition, they also determined the important role of intermediates CH2O and H2O2 in the product formation process. Another study52 reveals the growth of polycyclic aromatic hydrocarbons (PAHs) during n-hexane combustion. The research results show that the formation of PAHs involves the process of entanglement of long carbon chains into rings and growth by molecular chain attachment. In addition, the optimal temperature range for the growth of PAHs has also been found through research. These works enrich the data of high temperature cracking or oxidation of cycloalkanes and provide support for the establishment of the overall reaction mechanism and kinetic model of cycloalkanes.
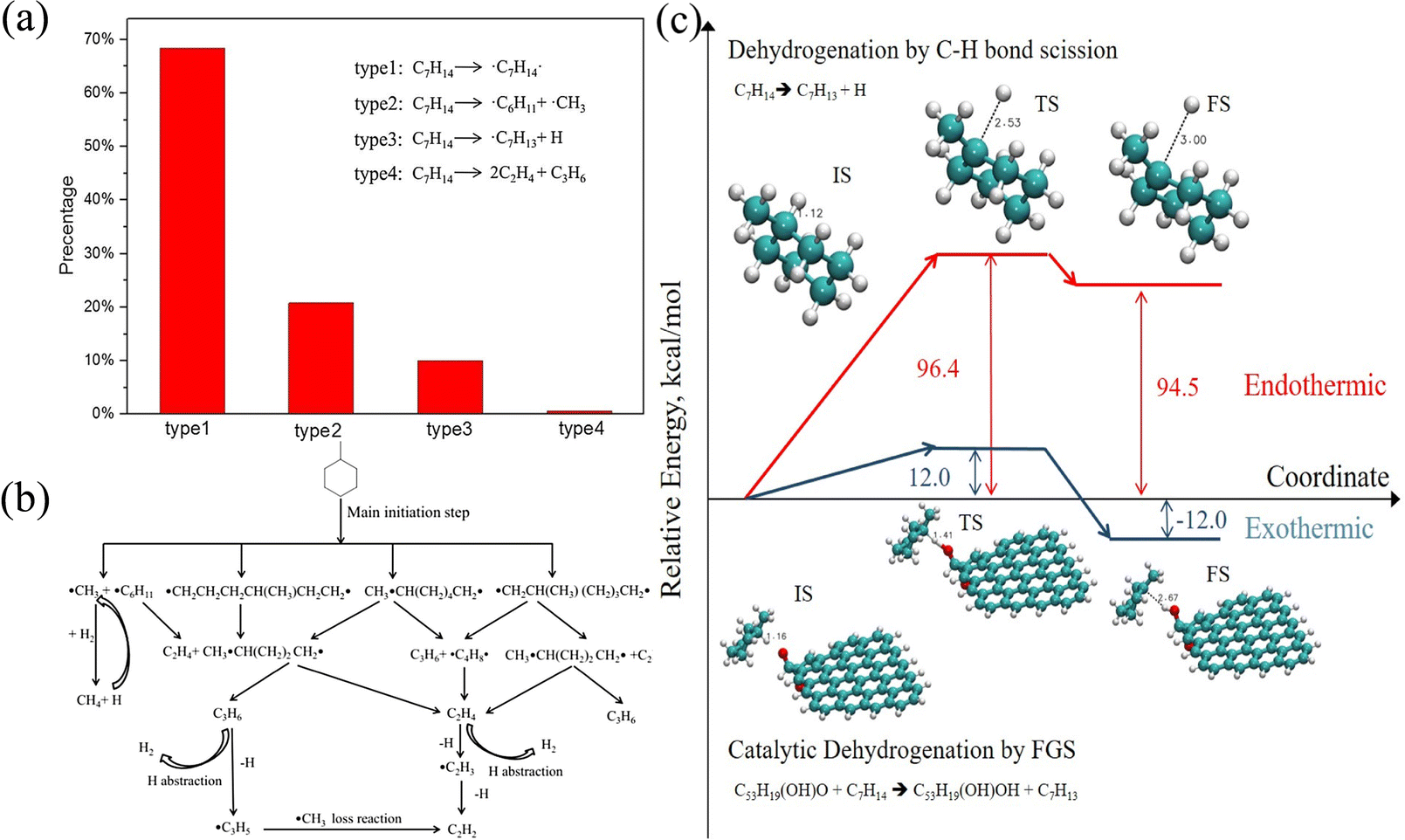
Cheng et al.17 explored the oxidation process of toluene and found three paths for the initial reaction of toluene, namely, (1) dehydrogenation of toluene induced by oxygen and other small molecular free radicals, (2) the cleavage of the C–H bond of methyl to benzyl, and (3) the cleavage of the C–C bond of methyl to benzene to form phenyl. In these three paths, the first reaction mainly occurs when oxygen is sufficient, and the proportion of the other two reactions increases when oxygen is insufficient. Tan et al.58 explored the effect of an applied electric field on the oxidation of toluene, and the results show that the electric field significantly promoted the oxidation process of toluene, so that toluene reacted at 2100 K where no reaction had occurred before. And the oxidation rate of toluene at 2100 K is consistent with the oxidation rate at 2900 K without an electric field. In addition, the authors also studied the effect of the electric field on the initial oxidation path of toluene. The results show that although the electric field promoted the formation of initial radicals and made toluene decompose faster, the effect of the electric field on the two reaction paths seemed to be indistinguishable. Finally, the authors further show that the electric field does not take into account the possible effect of the reaction molecular polarization, which may cause the simulation results to deviate from the correct value. The authors suggest that researchers take note of this in subsequent studies. Fu et al.59 then evaluated the effect of an external electric field on the oxidation and pyrolysis of toluene. The results showed that the electric field promoted the oxidation of toluene but did not significantly promote the pyrolysis of toluene. The internal reason was that the electric field increased the entropy change of the reactants and transition state, but the pyrolysis of toluene was driven by heat, so the effect was not obvious. These works are helpful to deepen people's understanding of the mechanism of pyrolysis and oxidation of toluene.
Wei et al.54 studied the oxidation of benzene molecules on palladium nanoparticle catalysts. Fig. 3(a) shows the detailed process of benzene molecules being oxidized by palladium nanoparticles. As shown in Fig. 3, benzene molecules are first adsorbed on palladium nanoparticles, and then benzene molecules get rid of hydrogen atoms through several steps to form C6H, which is oxidized to form an important intermediate C6HO. C6HO is further oxidized to form small molecular species, and the path is shown in Fig. 3(b). Statistical analysis of the ring-opening path shows that the ring-opening reaction of C6HO occurs at oxygen-containing sites in 88% of cases. Further study revealed the detailed path of C6HO oxidation, and demonstrated that the core substance of the C6HO oxidation process is C2 species, and the final oxidation product is CO2. This work provides an idea for the study of the oxidation mechanism of benzene molecules in the presence of catalysts.
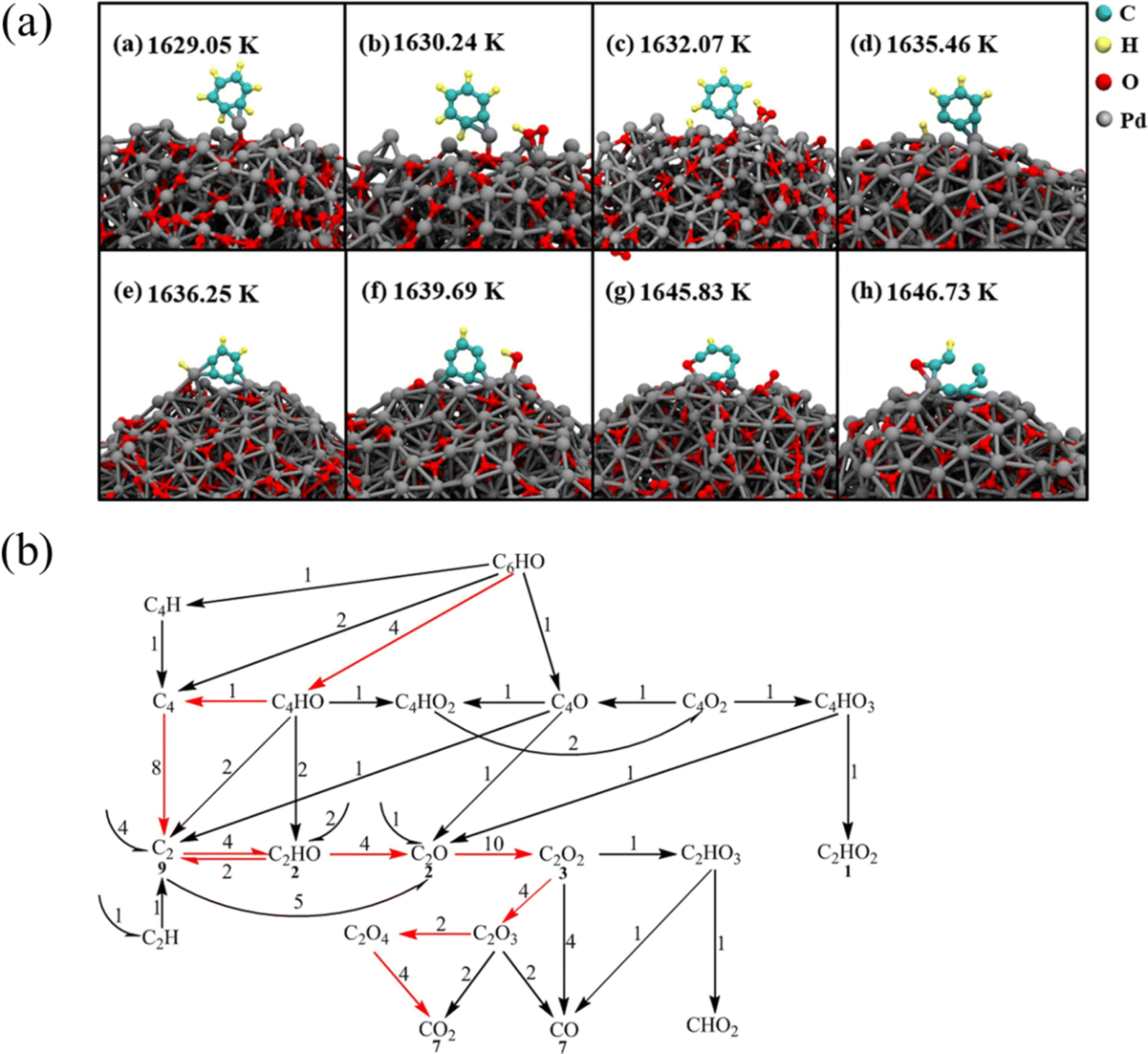 | ||
| Fig. 3 (a) The oxidation process of C6H6 on Pd nanoparticles and (b) further oxidation of C6HO to small molecules.54 | ||
Another study conducted by Qian et al.55 explored the cause of explosion caused by the mixing of hydrogen peroxide and1,3,5-trimethybenzene (TMB). The study shows that the explosion process can be divided into two parts. First, H2O2 due to its instability decomposes into OH and OOH free radicals, and the hydrogen extraction reaction and binding reaction caused by the attack of the OH radical on TMB molecules lead to the cracking of the benzene ring. Subsequently, the small molecular species formed by TMB decomposition undergo further oxidation reaction. A huge amount of heat is released, causing the risk of explosion. This study confirmed that the existence of OH free radicals determines the speed of the whole reaction process, and controlling the number of OH free radicals can effectively reduce the risk of explosion. This work provides inspiration for the development of further explosion suppression methods in similar systems.
Chaudret et al.62 studied the initial reaction paths of naphthalene and mono naphthalene and dimethyl naphthalene and the formation of coke molecules. It is revealed that the initial reaction paths of these substances are hydrogen dissociation and the reversed radical disproportionation mutation, and the position and number of methyl substituents will affect the kinetics and thermodynamics of the reaction. These results are supported by quantum mechanics. In addition, the formation of coke molecules involves the activation of the naphthalene derivative, dimerization or trimerization to form low molecular weight coke molecules, and condensation to form high molecular weight coke molecules. The information about the pyrolysis mechanism of complex aromatic hydrocarbon molecules, the effect of the number and position of substituents on pyrolysis, and the formation process of coke molecules obtained in this work provide a reference for the study of complex aromatic hydrocarbon molecules.
2.2 Pyrolysis and combustion mechanism of multi-components of aviation fuels
The study63–70 of the aviation kerosene multi-component or alternative model can provide information on the influence of multi-component interaction on the overall fuel combustion, and then help improve the combustion kinetic model and mechanism of aviation kerosene.Song et al.69 investigated the effect of mixing ratio on the mixed oxidation of toluene and n-decane. Compared with the single-molecule oxidation simulation, the initial reaction pathway of n-decane was not affected in the mixed oxidation simulation, while the oxidation pathway of toluene was changed. This is mainly because compared with toluene, n-decane is more likely to react when heated, and then a large number of highly reactive radicals are generated, which changes the oxidation path of toluene. The effect of H/C ratio on the representative intermediate HCHO was further discussed. The results show that the existence time of HCHO decreased with the increase of H/C ratio at low temperature. However, at high temperature, the H/C ratio has almost no effect on HCHO. This phenomenon is primarily because the H/C ratio affects the generation of OH radicals, while the consumption of HCHO is dominated by OH and O2. This work shows the important influence of the H/C ratio on fuel oxidation, and provides guidance for the selection of various components in aviation kerosene. Liu et al.66 simulated the combustion process of the model substitute of aviation kerosene JP8-1 composed of n-dodecane, n-tetradecane, isooctane, methylcyclohexane, m-xylene and tetralin, and explored the combustion mechanism and kinetics of JP8-1 aviation fuel. Table 1 lists the details of the alternative components and the path and combustion rate of the initial reaction. As shown in Table 1, the initial reaction paths of most components are consistent with the single-component simulation, mainly the C–C bond cleavage reaction, the C–H bond cleavage reaction, and the decomposition reaction caused by the attack of active free radicals. The kinetic analysis showed that the reaction rate of n-alkanes was the highest, followed by that of isooctane and methylcyclohexane, while the reaction rate of aromatics was the lowest, and the reaction rate of similar components decreased with the increase of molecular weight. The analysis of the formation path of coke molecules shows that ternary rings, five-membered rings and six-membered rings may be the precursors for the formation of coke molecules. This work provides insights for further understanding the combustion properties of JP8-1 and designing new and more reasonable model substitutes.
| Name | Reaction | Snapshot | Type | k′ |
|---|---|---|---|---|
| n-Dodecane | C12H26 →˙C6H13 + ˙C6H13 |
|
Normal-paraffin | 3.81 × 105 |
| n-Tetradecane | C14H30 → ˙C7H15 + ˙C7H15 |
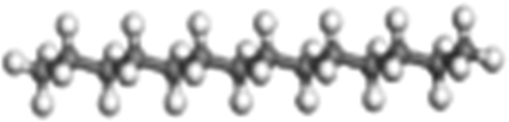
|
Normal-paraffin | 3.5 × 105 |
| Isooctane | C8H18 → ˙CH2CH(CH3)2 + ˙C(CH3)3 |
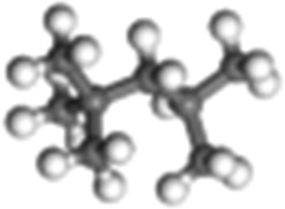
|
Branched-paraffin | 2.96 × 105 |
| Methylcyclohexane |
|
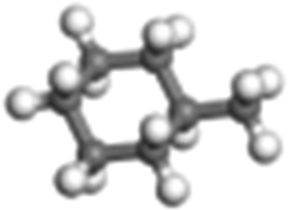
|
Cyclo-paraffin | 1.01 × 105 |
| m-Xylene |
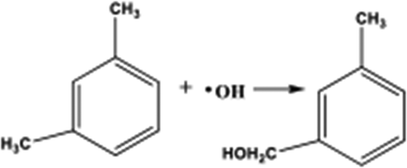
|
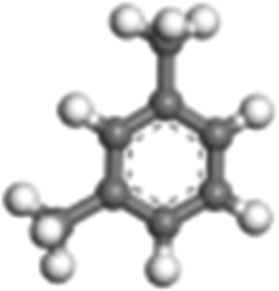
|
Monocyclic-aromatic | 5.32 × 103 |
| Tetralin |
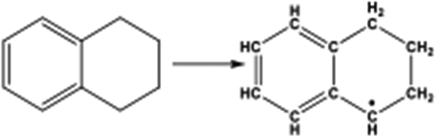
|
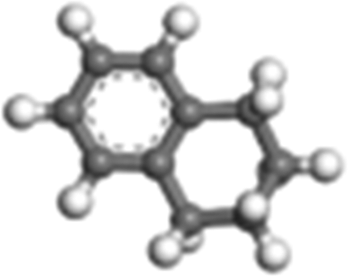
|
Bicyclic-aromatic | 2.32 × 102 |
Zhao et al.63 used reaction molecular dynamics to evaluate the difference in combustion between RP-3 aviation kerosene 3-component and 4-component model substitutes and the 45-component real model. A time step of 0.1 fs was used in the simulation. The results show that in terms of combustion kinetics, the 3-component and 4-component models have a relatively high pre-exponential factor and reaction activation energy than the 45-component models, which explains the higher reaction rate of the 3-component and 4-component models at high temperature. In terms of the formation of the main products, the three-component and four-component model substitutes showed a higher ethylene production rate, and more propylene was produced in the combustion products of the three-component model substitutes. This is caused by the higher content of n-alkanes in the two model substitutes and the large amount of 1,3,5-methylcyclohexane in the three-component model substitutes. This work shows the influence of the difference in fuel molecular structure and component content on the combustion mechanism and kinetics of fuel, and further deepens the understanding of the internal reasons for the reaction difference of model substitutes. It provides a reference for obtaining a more reasonable aviation kerosene substitution model through design optimization.
A study by Han et al.68 used reactive molecular dynamics combined with machine learning to take the chemical composition content in the RP-3 model surrogate as the independent variable and the chemical reactivity data as the dependent variable. The RP-3 alternative model with higher reactivity can be obtained by optimizing the content of each component in the RP-3 alternative model. Through the error evaluation results, the best LightGBM machine learning model was selected among the three machine learning models of LR, SVR and LightGBM, and the component content of the substitute model compound was optimized by this model, and an improved three-component model was finally obtained. The rationality of the improved model is confirmed by the experimental results of ignition delay time. This study provides guidance for fine-tuning component fractions in aviation kerosene model compounds to obtain more reasonable surrogate models.
2.3 Pyrolysis and combustion mechanism of high-density aviation fuels
High-density aviation fuels used in scramjets have recently been extensively studied based on the need for higher Mach-number flight speeds for aircraft. There has been a lot of work on high-energy density aviation fuel synthesis,71 experimental research72–74 and quantum mechanics research.74–76 At the same time, research on the pyrolysis or oxidation mechanisms of high-density aviation fuels by using reactive molecular dynamics is also steadily progressing.JP-10 is a kind of high-density hydrocarbon propellant with exo-tetrahydrodicyclopentadiene (exo-THDCPD) as the main body. JP-10 is the standard fuel of supersonic cruise missile because of its high density, high energy and high thermal stability. The small-scale molecular dynamics simulation of JP-10 has deepened people's understanding of the pyrolysis and combustion mechanism of JP-10. Chenoweth et al.13 identified two paths for the initial decomposition of JP-10 by single molecule simulation, that is, (1) JP-10 molecule loses ethylene to form cyclooctadiene, and (2) JP-10 undergoes cleavage to produce 1,4-pentadiene and cyclopentene. To obtain the complete reaction kinetics and mechanism of JP-10, Liu et al.19 performed a large-scale simulation of JP-10 using a time step of 0.1 fs. The results show that the pyrolysis of JP-10 mainly has three stages, as shown in Fig. 4(a). JP-10 first undergoes ring-opening reaction in the first stage to form four kinds of double radicals, and then these four kinds of double radicals are further decomposed to form eight kinds of products. In the second stage, the products formed in the previous stage mainly undergo β-scission reactions to form free radicals. Then, the free radicals are further propagated through β-scission reactions or C–H bond cleavage reaction, and finally the chain termination reaction occurs in the third stage to form stable small molecular products. C5H7 and C3H5 radicals play an important role in the whole reaction stage. This work shows that ReaxFF can be used to explore large-scale reaction systems and obtain complete reaction mechanisms. Guo et al.77 further discussed the combustion mechanism of JP-10 molecules. The results show that JP-10 first burns to form free radical pools, and then the resulting free radicals attack JP-10 molecules causing it to decompose and oxidize to form alcohols, ketones and other species. Finally, these species further react with small molecular free radicals to form final products such as CO2 and H2O.
 | ||
| Fig. 4 (a) Three stages of pyrolysis of JP-10 molecules.19 (b) Pyrolysis and oxidation of JP-10 molecules on functionalized graphene sheets (FGS).78 | ||
Feng et al.78 studied the reaction process of pyrolysis and oxidation of JP-10 on functionalized graphene sheets (FGS) and found the reaction path of pyrolysis and oxidation of JP-10 on FGS. As shown in Fig. 4(b), OH on FGS is separated from FGS at first, and then OH participates in the pyrolysis or oxidation of JP-10. Finally, more OH and H radicals are further involved in the reaction to form stable small molecules. The calculation shows that this path greatly reduces the energy barrier of the reaction and accelerates the reaction.
For the sake of environmental protection, renewable biojet fuels have been widely studied recently. Kwon et al.79 and Lele et al.80 have studied the pyrolysis processes of a series of head-to-head (HtH) polycyclic hydrocarbon molecules using a time step of 0.1 fs. The structures of the six molecules are shown in Fig. 5(a). These six molecules can be divided into two categories: one is connected by a C–C bond to two cyclic molecules, and the other is connected by a central four-membered ring to two cyclic molecules. The calculation of kinetic parameters shows that the fuel molecules with central four-membered rings show higher reaction activity. The pyrolysis of the reaction molecules shows that the initial decomposition paths can be divided into two types: (1) central bond breakage and (2) molecular ring-opening reaction. The analysis of the pyrolysis path of HtH polycyclic hydrocarbon molecules shows that small molecular olefins and alkynes constitute the main body of the product, which is different from the pyrolysis products of other fuel molecules. This gives people a more comprehensive understanding of the mechanism of fuel initial pyrolysis. These efforts provide a reference for people to explore the pyrolysis and combustion mechanism of a wider range of new aviation fuels.
 | ||
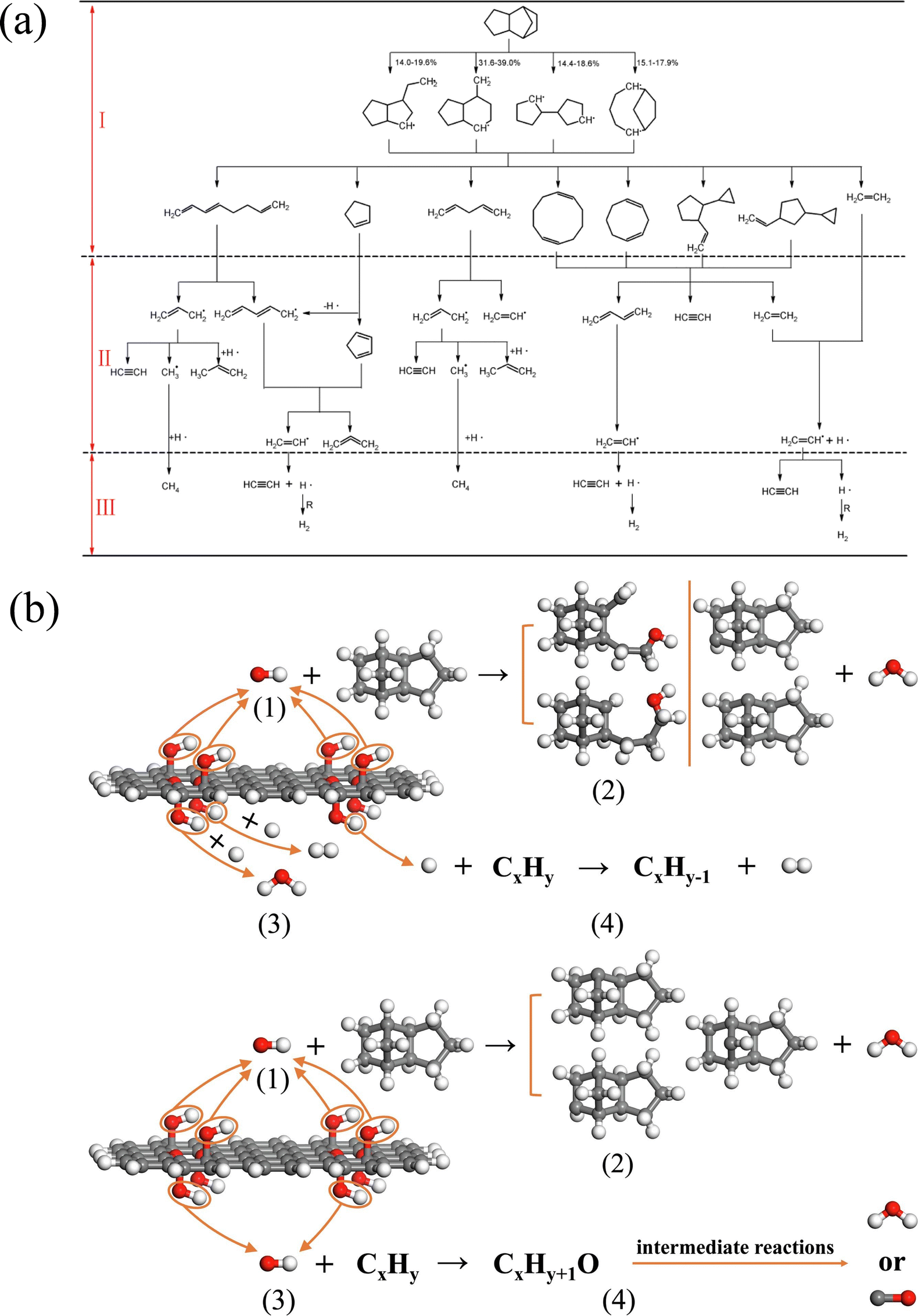
| Fig. 5 (a) Molecular structure of six HTH polycyclic alkanes;80 (b) molecular structure of farnesane, α-farnesene, and β-farnesene;81 and (c) the initial decomposition path and its proportion of PMT molecules.82 | ||
Goncalves et al.81 explored the pyrolysis and combustion kinetics and mechanism of three substances, farnesane, α-farnesene and β-farnesene, which can be used as biojet fuels. The molecular structures of the three species are shown in Fig. 5(b). The analysis shows that the initial path is mainly C–H bond cleavage, accompanied by a small amount of C–C bond breakage. But compared with farnesane, the existence of double bonds in α-farnesene and β-farnesene increases the molecular reaction activity and accelerates the reaction. The strength of the C–C bond is enhanced by the presence of the double bond, and C–C bond breakage is reduced. In addition, the calculated results of kinetic parameters are verified by experimental data. Wang et al.82 studied the pyrolysis process of p-menthane (1-isopropyl-4-methylcyclohexane, PMT) and analyzed the initial decomposition path of PMT. It can be seen from Fig. 5(c) that the cleavage reaction of the isopropyl group and the ring-opening reaction caused by the cleavage of the C–C bond near the isopropyl group are the main reaction pathways. Further analysis shows that all kinds of ring-opening products continue to decompose through β-scission reactions and finally produce a large number of ethylene, propylene and other products. These works can further deepen our understanding of the pyrolysis and combustion mechanisms of high-density aviation fuels.
3 Pyrolysis and combustion mechanism of aerospace fuels and energetic additives
Although information about the macroscopic combustion properties of aerospace fuels has been obtained through experiments, the limitations of the experiments themselves prevent people from gaining insight into the atomic-level details of the combustion process of aerospace fuels. However, the emergence of ReaxFF10 enables people to study the overall mechanism of pyrolysis and combustion of space fuels more intuitively at the micro level. Energetic materials as additives can greatly improve the specific impulse of rocket fuels. The study of their pyrolysis and combustion mechanism can deepen people's understanding of the interaction between energetic materials and aerospace fuels, and further improve the selection of energetic materials and fuels, providing guidance on improving the combustion efficiency of aerospace fuels.3.1 Pyrolysis and combustion mechanism of aerospace fuels
At present, the widely used aerospace fuels are refined kerosene RP-1, hydrazine, unsymmetrical dimethylhydrazine, nitromethane, liquid hydrogen, triple-base solid propellants and so on. There have been some studies on the pyrolysis or combustion mechanism of these fuels by reactive molecular dynamics.Han et al.83 studied the pyrolysis process of the RP-1 aerospace kerosene 3-component model and the 24-component alternative model using a time step of 0.1 fs. The results show that there are some differences in the pyrolysis characteristics of these two alternative models. The pyrolysis curve shows that the 24-component substitution model has higher reactivity than the three-component substitution model. The analysis of the reactivity of the components of the two substitution models shows that the high stability and high content of cycloalkanes in the 3-component substitution model are the reasons for the difference. Further analysis of the two substitution models revealed the differences in reaction paths and products. Compared with the 24-component substitution model, the existence of quaternary carbon in isohexadecane molecules in the 3-component substitution model led to the formation of a large amount of 2-methpropene. The diversity of cyclic species in the 24-component substitution model made the ring-opening reaction more abundant, resulting in more diolefins and cyclic olefins. Another study84 on the pyrolysis of the RP-1 aerospace kerosene 24-component substitution model provides information on soot nanoparticle generation. The formation of a soot nanoparticle can be divided into three stages: PAH formation, aggregation nucleation and graphitization to form a soot nanoparticle. The evolution process is shown in Fig. 6. In the first stage, the long carbon chain containing polyalkyne groups forms a large ring, then a bridge is formed within the molecule, and some small rings are formed within the large ring to form a PAH molecule. In the second stage, the side chain of PAH is closed, a bridge bond is generated inside, and the molecules are aggregated into nuclei. In the last stage, the C5/C7 ring is transformed into a six-membered ring to form a soot nanoparticle. These studies have made an effort to evaluate the overall mechanism of the RP-1 fuel substitution model and prevent coking during combustion.
 | ||
| Fig. 6 Soot nanoparticle nucleation evolution process.84 | ||
As clean energy, liquid hydrogen is often mixed with liquid oxygen for aerospace fuel. The HO-2011 reaction force field developed by Agrawalla et al.,85 the CHO-200815 reaction force field and the HO-201421 and CHO-201620 reaction force fields improved on the basis of the CHO-2008 reaction force field are all suitable for hydrogen combustion or pyrolysis systems. The pyrolysis process of liquid hydrogen at ultra-high temperature was studied by Kudinov et al.,86 and the results showed that the pyrolysis products and pyrolysis activation energies were consistent with previous studies. Palacios et al.87 studied the combustion of hydrogen in a high-pressure system. The simulation results showed that H2 + O2 → H + HO2 was the most important initial reaction, but further calculations show that collisions between three molecules frequently occur at ultra-high pressures, and the trimolecular reaction 2O2 + H2 → 2HO2 and 2O2 + H2 → H + HO2 + O2 may be the new initiation mechanism of H2 oxidation. Sun et al.88 studied the mixed combustion of H2 and CH4 under the action of an electric field. The results show that the consumption rate of H2 increases obviously under the action of an external electric field, and the initial reaction time changes. In addition, the addition of an electric field reduces the diversity of free radicals but produces unique species. This study confirms the broad application prospect of ReaxFF in exploring the mechanism of fuel combustion under the action of an external electric field. Bertels et al.89 evaluated the above four reaction force fields that can be used in hydrogen combustion systems based on quantum mechanical methods. The results show that ReaxFF using these reaction force fields is not accurate enough to describe hydrogen combustion systems, and further optimization is needed to better conform to the results of quantum mechanical calculations.
Hydrazine has become a widely used aerospace fuel due to its good energy properties and long-term storage at low temperatures. Zhang et al.90 studied the pyrolysis of bulk hydrazine (N2H4) and the decomposition of bulk hydrazine over Pt catalysts, and explored the effect of temperature on the pyrolysis process. The results show that bulk hydrazine decomposes to produce NH3 at lower temperatures, but the initial products change to N2 and H2 at higher temperatures. Compared with bulk hydrazine, the catalytic effect of the Pt catalyst reduced the initial decomposition temperature of hydrazine by half. This study further affirms the applicability of ReaxFF. Liu et al.91 studied the combustion process of spontaneous combustion system monomethylhydrazine (MMH) and dinitrogen tetroxide (NTO). Compared with non-spontaneous combustion system ethanol (EtOH) and NTO, the MMH-NTO system showed a higher reaction rate. The initial path analysis of the MMH-NTO system shows that the initial path of pyrolysis of the MMH-NTO system is N–N bond breakage and hydrogen extraction reaction, and further analysis of the product shows that NO2 is diffused into the fuel to react. This study shows that the fuel decomposition path in the spontaneous combustion system should have a lower reaction energy barrier to achieve rapid reaction, and the oxidant should have a larger diffusion coefficient to achieve more uniform mixing and higher combustion efficiency.
Nitromethane exhibits fine combustion performance and detonation performance. It is often used as aerospace fuel. There have been some studies92–94 on its pyrolysis or combustion mechanism using ReaxFF MD. Xu et al.93 studied the pyrolysis of liquid nitromethane in different 2D nanocarbon materials, and the results showed that hybrid carbon atoms and hydroxyl groups participated in the decomposition of nitromethane and promoted the reaction. This work provides insights into the selection of 2D nanocarbon material catalysts for specific fuel pyrolysis or combustion systems. Guo et al.94 studied the improvement of detonation performance of nitromethane by mixing nitromethane and NCNO2. The results showed that the Chapman–Jouguet (CJ) temperature, CJ pressure and detonation velocity of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed system were greatly improved. Product analysis showed that the addition of NCNO2 increased the production of gaseous molecules while reducing the mass of carbon clusters, which caused this change. This work provides a reference for the improvement of detonation performance of nitromethane or other fuels.
:1 mixed system were greatly improved. Product analysis showed that the addition of NCNO2 increased the production of gaseous molecules while reducing the mass of carbon clusters, which caused this change. This work provides a reference for the improvement of detonation performance of nitromethane or other fuels.
Yi et al.95 evaluated the pyrolysis of an RDX modified triple-base solid propellant. Analysis of pyrolysis products shows that carbon clusters react with H2O at high temperature to generate more H2, and the pyrolysis products are more abundant at high temperature. These results show that the complete combustion of the propellant at higher temperature can be achieved by adding nano-metals and increasing the content of the oxidant, so as to further improve the energy output.
3.2 Pyrolysis and combustion mechanism of energetic additives
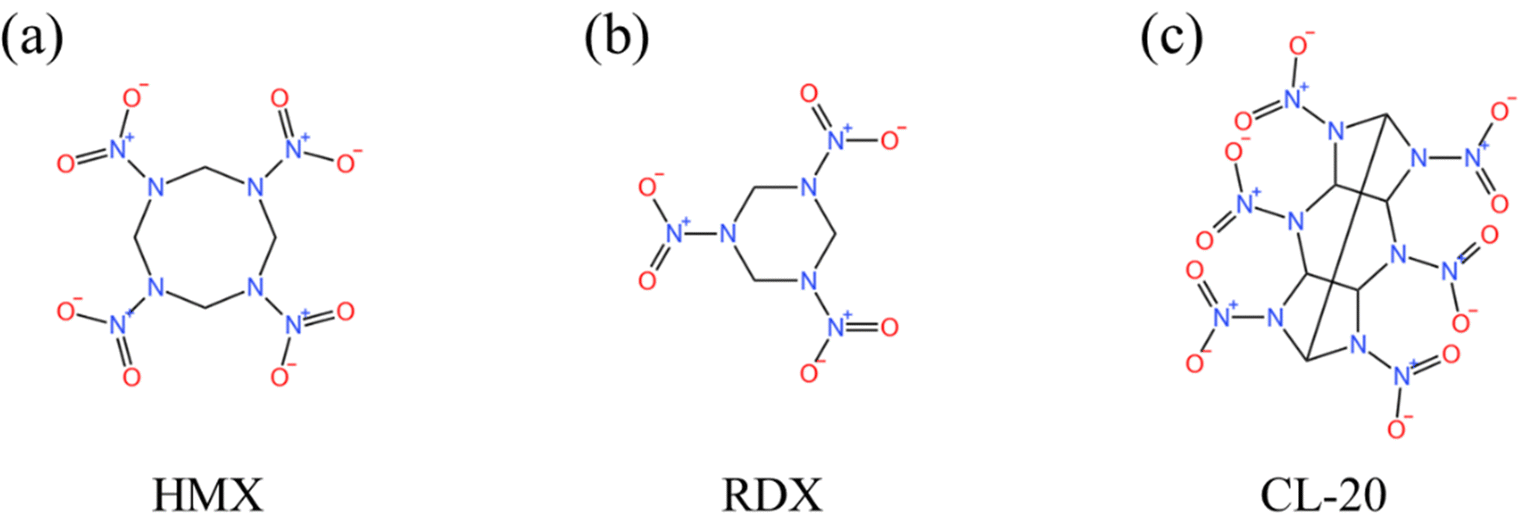 | ||
| Fig. 7 Molecular structure of (a) HMX, (b) HMX and (c) CL-20. | ||
3.2.1.1 Pyrolysis and combustion mechanism of HMX. HMX has an eight-membered ring nitramine structure, which has good stability and strong explosive power, and can be used as a component of solid rocket propellants. Zhou et al.96 investigated the pyrolysis mechanism of β-HMX crystals at different densities. Studies have shown that β-HMX mainly undergoes intramolecular decomposition at low density, and the initial decomposition paths are N–N bond breakage, C–N bond breakage and HONO elimination. However, at high density, molecular collisions are frequent, and there are more intermolecular retroactive reactions, resulting in O2, HO and macromolecular clusters. Zhu et al.97 studied the physicochemical properties and reaction mechanism of HMX nanocrystals at low temperature. The results show that the change of the position and direction of NO2 groups in HMX will seriously affect the phase transition of HMX. Furthermore, the transition temperature of the β crystal form and δ crystal form of HMX nanocrystals is determined by analysis. Meanwhile, it is found that the initial reaction path of HMX nanocrystals at low temperature is the breaking of the C–H bond, and the product at low temperature is the same as that at high temperature. Zhao et al.98 studied the pyrolysis process of nano-AlH3 and HMX composites. The results show that HMX adsorbs on the particle surface by forming Al–O and Al–N bonds. During the reaction, the cleavage of C–N and N–O bonds is the main pyrolysis path. Due to the attraction of Al to O and N atoms, the initial decomposition path is barrier-free. Further studies show that the temperature and specific impulse of the composite system are higher than those of the HMX system. In another study,99 the triaminoguanidine glyoxal energetic polymer was added to HMX crystals to obtain QY-HMX. The results showed that the stability of QY-HMX was improved and it did not decompose at low temperature. This study provides a reference for improving the stability of energetic materials.
Zhou et al.100 studied the decomposition process of HMX crystals induced by thermal shock. The results show that the initial decomposition path of HMX crystals is mainly the fracture of the N–N bond, HONO detachment and ring breaking reaction. In addition, the analysis shows that compression promotes heat conduction, which leads to earlier initial reaction and more intense propagation reaction. Huang et al.101 studied the pyrolysis process of defective β-HMX crystals under thermal shock. The results show that compared with the β-HMX crystal, the impact sensitivity of the defective β-HMX crystal is improved, and there is a high temperature region in the crystal, which leads to the occurrence of the reaction. In addition, the initial reaction paths of defective crystals are N–N bond cleavage and N–O bond cleavage, and the final products are more abundant. He et al.102 studied the catalysis of water in the process of shock explosion of HMX. The results show that water molecules accelerate the oxidation of C atoms by promoting the transfer of oxygen atoms from N to C, as shown in Fig. 8. As a result, the energy barrier for C–N bond breakage is reduced, resulting in rapid cleavage of HMX.
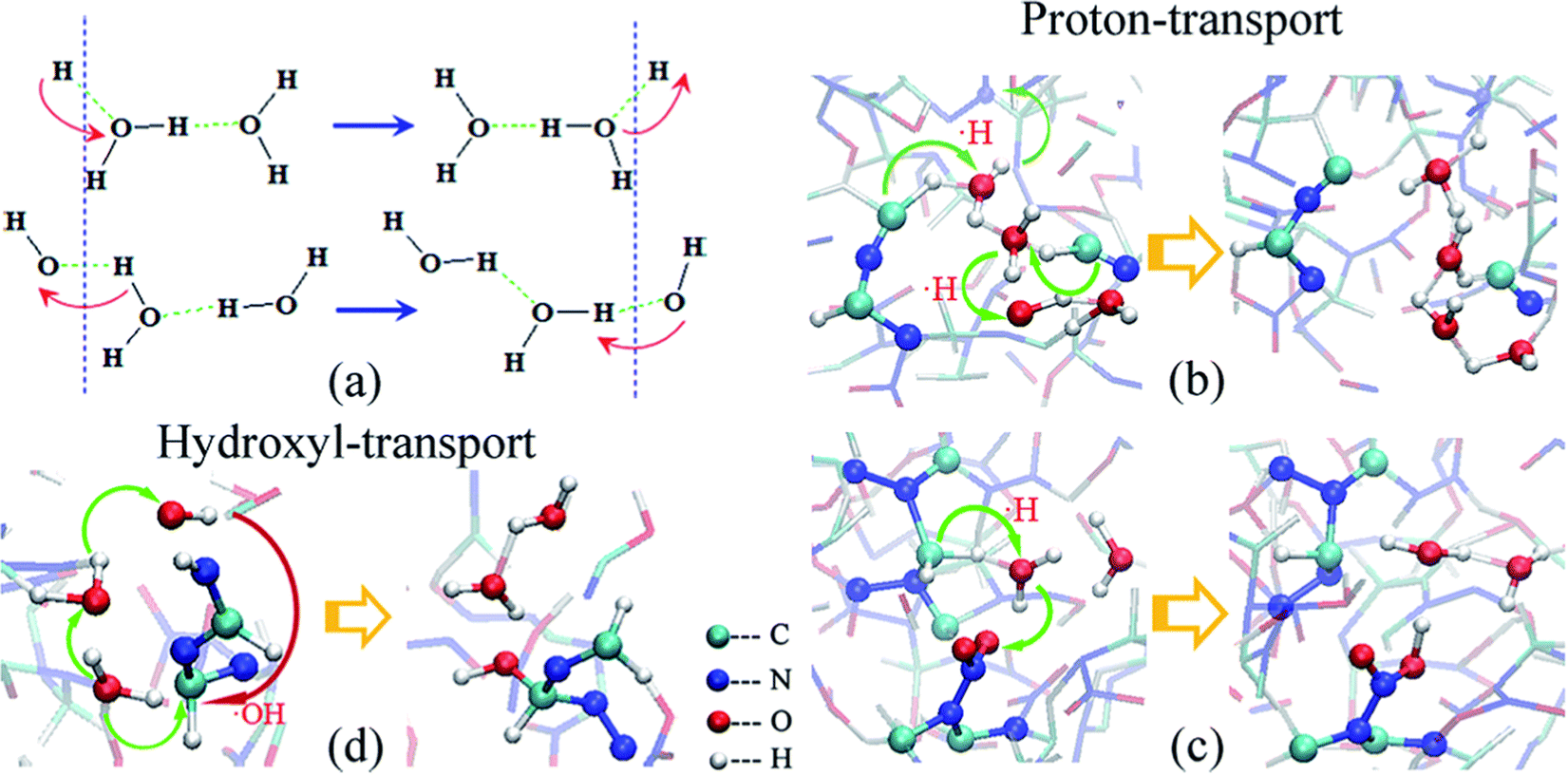 | ||
| Fig. 8 H and OH transport of water in β-HMX:102 schematic of (a) H and OH transport, (b and c) H transport, and (d) OH transport. | ||
3.2.1.2 Pyrolysis and combustion mechanism of RDX. RDX is a kind of energetic additive with good comprehensive performance because of its stable chemical property and great explosive power. In order to understand the combustion properties of RDX, related research studies103–108 are being carried out. Peng et al.108 studied the pyrolysis process of α-RDX and its derivatives. The initial decomposition path shows that the first step of the reaction is the breaking of the N–N bond. The difference is that the ring-opening reaction occurs only after the substituents of α-RDX derivatives are completely removed, while the ring-opening reaction of α-RDX occurs when there is a nitro group. Further analysis shows that a smaller C/N ratio is beneficial to improve the pyrolysis performance of energetic materials. Zheng et al.107 investigated the effects of solid-phase pyrolysis of RDX and nanosize effects. The results show that the solid-phase pyrolysis of RDX is divided into two stages: solid-dominated and non-condensed. The decomposition reaction randomly occurred inside the RDX during the RDX pyrolysis, and the smaller size nanocrystals exhibited higher decomposition rates. This study furthers our understanding of the complex interactions of the pyrolysis process of energetic crystals. Peng et al.106 have studied the pyrolysis process of the perfect RDX crystal and the RDX crystal with a molecular vacancy under shock. The initial decomposition path shows that the N–N bond breakage occurs at first, then the C–N bond breakage occurs in both crystals, and the possible C–H bond breaks to form HONO. Further comparative analysis showed that the existence of vacancies enhanced the activity of the N–N bond and accelerated the reaction.
The presence of aluminum can increase the temperature of the system. Researchers often add aluminum to energetic materials to achieve more complete combustion. The pyrolysis of aluminum-containing RDX109–112 is widely studied. Hao et al.109 studied the promoting effect of Al and Al2O3 nanoparticles on the pyrolysis of RDX. The analysis shows that the participation of Al changes the initial path from the unimolecular reaction of N–N bond cleavage to the bimolecular reaction without an energy barrier or with a low energy barrier, which leads to the early decomposition of RDX. Zhao et al.110 studied the decomposition of RDX in the presence of pure aluminum, surface AlN and surface AlO. The results show that RDX adsorbs on the particle surface by forming N–Al and O–Al bonds. In these systems, the initial decomposition path of RDX is the fracture of the N–O bond. In addition, the calculation of the energy barrier shows that the existence of the AlO surface weakens the attraction of the Al atoms in the bulk phase to the O and N atoms in RDX, resulting in the decrease of the catalytic effect of surface AlO on RDX. The study of Li et al.24 shows that the smaller size of AlH3 can promote the pyrolysis of RDX more thoroughly. These studies provide insights for the further application of active nano-metals in energetic materials.
3.2.1.3 Pyrolysis and combustion mechanism of CL-20. CL-20 is a kind of energetic compound with a cage-like polycyclic nitramine structure, which is often used in additives to improve the specific impulse because of its excellent properties. Through some research studies, the pyrolysis and combustion mechanism of CL-20113–120 have been preliminarily understood.
Ren et al.120 have studied the pyrolysis process of β-CL-20. The results show that the initial decomposition paths of β-CL-20 are (1) the fracture of the N–NO2 bond and (2) the fracture of the C–N bond. High temperature is beneficial to the first path, but has little effect on the second path. In addition, during the low-temperature simulation, a bimolecular reaction in which NO2 molecules take away oxygen occurs (PI-3), and the overall reaction path is shown in Fig. 9. At the same time, further analysis shows that the simulated intermediates and final products are consistent with the experimental results. Wang et al.114,119 discussed the effect of temperature and density on the pyrolysis of ε-CL-20. It is found that temperature only affects the pyrolysis rate of ε-CL-20. The increase of temperature leads to the destruction of the ε-CL-20 structure in advance, but does not affect the reaction path of ε-CL-20 pyrolysis. The increase of density decreases the frequency of N–NO2 bond breakage, although the initial reaction rate increases at high density, the activation energy does not change. And large clusters are easy to be formed at high density, thus reducing the number of small molecular products. Han et al.116 explored the effect of particle size on the pyrolysis of ε-CL-20. It is found that the decrease of particle size promotes the breaking of only the N–NO2 bond, increases the reaction rate, and promotes the formation of some products at different reaction stages, but does not form a new ε-CL-20 decomposition path.
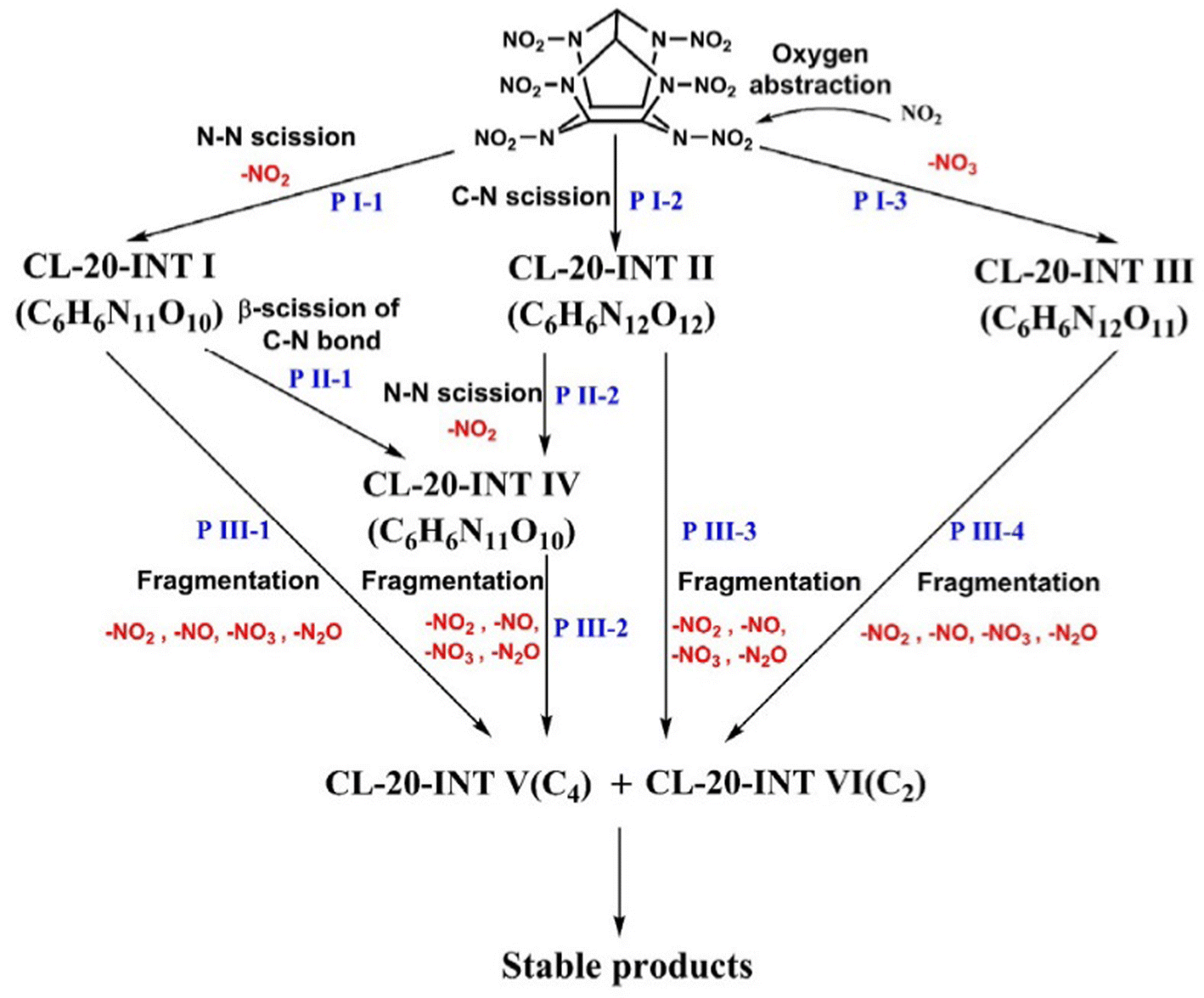 | ||
| Fig. 9 Initial pyrolysis path of the CL-20 molecule.119 | ||
Hu et al.121 studied the pyrolysis process of defective ε-CL-20 at high temperature. They used a time step of 0.1 fs in the simulation. The results show that the main initial path of ε-CL-20 pyrolysis is the breaking of the N–NO2 bond. However, the introduction of vacancy defects reduces the proportion of this path and increases the number of ring-opening reactions, and the reactions first occur around the vacancy defects. In addition, vacancy defects decrease the activation energy of the reaction and increase the reaction rate of ε-CL-20 pyrolysis, which is more obvious at low temperature. Wang et al.117 studied the pyrolysis of ε-CL-20 under different conditions. The results show that the initial pyrolysis path of ε-CL-20 at high temperature is mainly the fracture of N–NO2, while under high pressure and impact, ε-CL-20 first causes the separation of NO2 by forming intramolecular or intermolecular N–O bonds. Mei et al.118 studied the decomposition of CL-20 in the presence of AlH3. The analysis shows that CL-20 adsorbs on the surface of AlH3 particles by forming Al–O bonds, and then completes the fracture of N–NO2 bonds. In addition, further results show that the existence of AlH3 reduces the activation energy of CL-20 decomposition, increases the release of energy, and increases the formation of gas products. This work provides a reference for exploring the properties of new composite energetic materials.
3.2.2.1 Pyrolysis and combustion mechanism of Al. Zeng et al.125 studied the evolution behavior of core–shell Al/Al2O3 nanoparticles at high temperature. The results show that the diffusion of O atoms in the shell causes the reaction, and the AlO generated at the core is positively charged and migrates to the shell under the action of the electric field formed by the migration of Al and O until the melting electric field of the shell is zero. Chu et al.124 studied the oxidation process of core-shell Al/Al2O3 nanoparticles in an oxygen atmosphere. The results show that the whole oxidation process can be divided into four stages: preheating, melting, Al nuclear oxidation and moderate shell oxidation. In the second stage, the Al core melts from the shell junction. At this stage, the core Al diffuses outward, and the core Al diffuses to the oxide layer to be oxidized, releasing heat, which further promotes the melting of the Al core until it melts completely. In the third stage, the inward diffusion of O atoms in the oxide layer and the outward diffusion of Al atoms lead to the rapid oxidation of the Al core. These studies further deepen people's understanding of the ignition and combustion mechanism of Al nanoparticles.
Hao et al.130 studied the evolution behavior of Al nanoparticles (ANPs) in a high temperature environment in the presence of hot 2,4,6-trinitro-1,3,5-triaminobenzene (TATB). The results show that the smaller size of ANPs is beneficial to the occurrence of micro-explosion, which leads to the more complete decomposition of TATB. Moreover, as shown in Fig. 10, the destruction of the core–shell structure is observed in ANPs with an oxide layer, the core Al atoms gushing out achieve rapid oxidation, and rigid Al–O bonds lead to the formation of central voids. On the other hand, in ANPs without an oxide layer, O atoms diffuse inward and gather in ANPs, while C–Al bonds are formed on the surface, which is mainly caused by hypoxia. In addition, it was also found that although the addition of ANPs increased the heat release of the system and played a catalytic role in the pyrolysis of TATB, it also reduced the yield of gas molecules and increased the number of C/Al clusters. Zhao et al.131 have similar conclusions on the pyrolysis of pentaerythritol tetranitrate (PETN) in the presence of ANPs. In this system, the hot spots in PETN originate from the exothermic oxidation of ANPs. ANPs inhibit the production of CO2 but promote the formation of H2O, which is beneficial to the increase of specific impulse. The difference in the yield of small molecules between the two systems is mainly caused by the difference in energetic substances. These studies have made efforts to further understand the evolution mechanism of ANPs during the pyrolysis of energetic materials.
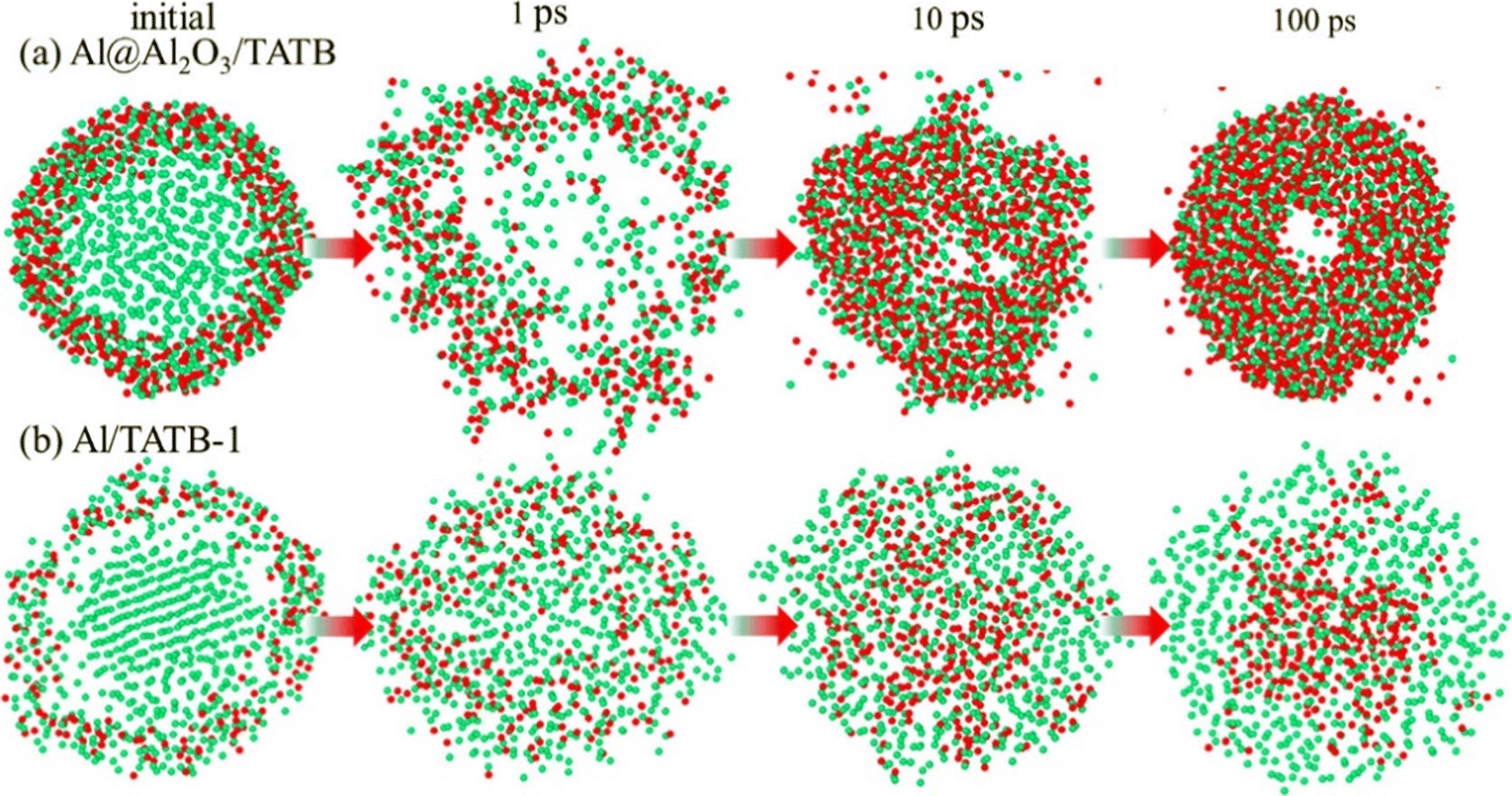 | ||
| Fig. 10 Morphological evolution of Al atoms in the (a) Al@Al2O3/TATB and (b) Al/TATB-1 systems at 3000 K.130 | ||
Dong et al.139 studied the effects of the size of ANPs and the existence of the oxide layer on the reaction of ANPs with water molecules. The results show that when the size of ANPs decreases, the consumption of H2O increases, but ANPs are easy to accumulate at high temperature, which will counteract this effect. The existence of the Al oxide layer will reduce the energy release of the system and reduce the production of H2, which can be eliminated by partially replacing the oxide layer with AlH3. In addition, it is also found that in the pure Al/H2O system, the reaction 3Al + 2H2O → 2AlO + AlH3 + H occurs on the outer surface of the particles, while in the Al/H2O system with an oxide layer, the reaction 4Al + Al2O3 →3Al2O occurs in the interior of the particles. Chu et al.123 studied the physical and chemical interaction between ANPs and small molecular oxidants. The results show that from melting to oxidation, the chemical adsorption and reaction diffusion of small molecular oxidants are dominant, and the duration of these two stages varies with different types of oxidants. The diffusion analysis of several small molecular oxidants shows that the reaction diffusion rate is positively related to the molecular diffusion coefficient of the oxidant, but negatively related to the binding energy of Al, which leads to the difference of ANP structure in different oxidants. This study reveals the complex interaction between oxidants and ANPs, which provides guidance for the proportion design of different oxidants in the presence of ANPs.
3.2.2.2 Pyrolysis and combustion mechanism of AlH3. AlH3 has excellent hydrogen storage performance and a large combustion calorific value. Compared with Al, AlH3 has lower combustion temperature and higher gas production. It is a high-energy additive with great potential to replace Al.
Song et al.128 studied the morphology evolution of aluminum hydride nanoparticles (AHNPs) during dehydrogenation and oxidation. They used a time step of 0.1 fs in the simulation. The results show that the evolution of AHNPs involves four stages: sphere, notched sphere, large branch and small string. It is found that the initial reaction originates from the surface dehydrogenation of AHNPs, and then the oxidation reaction takes place. The decomposition reaction caused by heat transfer into the particles leads to the formation and accumulation of hydrogen. When the temperature rises to 1600 K, the oxide layer breaks and the hydrogen overflows, and the morphology evolves into the second stage. With the temperature rising to 3000 K, large branches appear in the further oxidation of the AHNP, and the morphology enters the third stage. When the temperature rises to 3500 K, the AHNP explodes and the morphology evolves into the fourth stage. Zhao et al.127 studied the evolution behavior of pure and core-shell AHNPs in an O2 atmosphere. The results show that the oxidation of pure AHNPs begins on the surface of the particles and shows a non-uniform distribution. The further oxidation of pure AHNPs is carried out through the outward diffusion of Al and the inward diffusion of O. The core–shell AHNP analysis shows that the oxidation process is caused by the outward diffusion of Al atoms driven by an electric field and then their reaction with O. On the other hand, the phenomenon of micro-explosion appeared in the oxidation process of AHNPs with smaller size, which accelerated the oxidation process. At lower oxygen concentration, the inner diffusion of O atoms and the outer diffusion of core Al result in the formation of a homogeneous Al–O phase. On the other hand, at higher oxygen concentration, the diffusion rate of core Al increases and the center forms a hollow structure.
In-depth understanding of AHNP oxidation in small molecule systems is of great significance for improving propellant combustion. Wang et al.134 studied the pyrolysis process of the nitromethane/nano-aluminum hydride (NM/AHNP) composite system. The results show that the addition of AHNPs reduces the energy barrier of pyrolysis, promotes the reaction and increases the release of energy. Further analysis shows that the AHNP oxidizes by capturing O atoms in NM, followed by explosion in the small size AHNP to achieve rapid hydrogen release and oxidation, while further oxidation in the large size AHNP depends on the external diffusion of Al and the internal diffusion of O. Song et al.141 studied the oxidation process of AHNPs in the presence of H2O. Studies have shown that AHNPs at low temperature are dominated by dehydrogenation reactions inside and on the surface of the particles. The dehydrogenation on the surface causes the surface rearrangement of Al atoms, while the internal dehydrogenation causes the formation of hydrogen bubbles. When the temperature increases, the internal pressure causes the micro-explosion of AHNPs, which accelerates the reaction. Moreover, the research also reveals the oxidation process of AHNPs, that is, H2O is decomposed to form the Al–OH bond, and then O atoms move inward under the action of three Al atoms, resulting in O–H bond cleavage, which leads to the oxidation of Al. This study deepens people's understanding of the overall mechanism of metal hydrides.
4. Challenges for ReaxFF application
ReaxFF MD can dynamically capture the overall morphology of the reaction system and the evolution of different species over time, and then clarify the detailed mechanism of pyrolysis or combustion of the reaction system through analysis, which provides theoretical support for further research. However, a large number of studies have exposed some problems about ReaxFF, such as the deficiency of the QEQ method in describing molecular polarization and the setting of the ground state of high-energy materials by ReaxFF. The QEQ142 method is insufficient to predict the charge distribution of molecules far from equilibrium; it overestimates the charge flow in the molecule, resulting in the QEQ method describing a much larger molecular polarizability than the true situation. The QTPIE142 method more comprehensive treatment of the charges inside and outside the molecule compared to the QEQ method allows the method to obtain a lower net charge and a more accurate molecular polarizability, and the addition of such methods is expected to improve the accuracy of the ReaxFF MD calculations. Recently, O'Hearn et al.143 proposed a new charge model, namely ACKS2. Compared with the ReaxFF bond description currently used, this method estimates the Coulomb interaction energy term better, and this improvement is more significant in the ion system. Through the simulation of lithium oxides, they showed that this method can describe the breakage of the molecular covalent bond and ionic bond and the transfer of charge more correctly than the QEQ method. They suggest that this method is more suitable for describing materials similar to ceramics or with high ion bonding. ReaxFF's assumptions about the ground state of energetic materials lead to inaccuracies in ReaxFF's description of explosive or shock processes in energetic materials. At high thermal temperatures and in the shock state, the electrons of the energetic material may be in an excited state, which is different from the ground state assumption, and this has an impact on the results. This should be noted and improved in subsequent improvements to ReaxFF in relation to excited state chemistry.In addition, the simulation of ReaxFF in a larger system can fully avoid the influence of accidental factors, and reaction molecules can produce more kinds of intermediates and free radicals through richer reaction pathways. Large-scale19 reaction molecular dynamics simulation has unique advantages in revealing the overall pyrolysis mechanism of fuel and obtaining overall kinetic information. However, the reaction network in the large-scale reaction system is very complex, and the overall reaction mechanism is difficult to obtain by general methods. The visualization platform VARxMD developed by Liu et al.144 has great advantages in dealing with the details and information of the reaction system. VARxMD completely describes the whole reaction process by combining visual data and visual molecular structure. At present, VARxMD has been applied in many studies19,145 on fuel pyrolysis. In addition, the large-scale verification of the results of reaction molecular dynamics simulation and experiments is also one of the problems that limit the development of ReaxFF. Due to the limitation of reaction molecular dynamics, most of the current related simulations are carried out at a temperature higher than the experimental temperature. Although the simulation results are qualitatively consistent with the experimental results, they can’t be quantitatively compared with the experimental values, especially in terms of kinetic data. The solution of this problem may require the combination of accelerated dynamics and reaction molecular dynamics (such as Accelerated ReaxFF Reactive Dynamics (aARRDyn21), Collective Variable-driven Hyperdynamics (CVHD146) and ChemTraYzer-TAD147), so that the reaction molecular dynamics simulation can reach the same time scale as the experiment at close to the experimental temperature.
5. Conclusion
In this work, the research of ReaxFF MD in the pyrolysis and combustion of aviation/aerospace fuels and energetic additives has been introduced, especially for aviation kerosene model substitutes, high-density aviation fuels, aerospace fuels and energetic additives under various conditions. In addition, we discuss the limitations of ReaxFF applications, particularly in terms of charge, excited state chemistry, verification of simulation results against experiments, and extraction of reaction mechanisms, and point out possible solutions accordingly. In conclusion, ReaxFF has considerable potential to explore the pyrolysis or combustion mechanisms of fuels and energetic materials under extreme conditions.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Open Research Fund Program of Science and Technology on Aerospace Chemical Power Laboratory (STACPL220221B03), National Natural Science Foundation of China (Grant no. 21978210 and U20A20151) and the National Key R & D Program of China (Grant no. 2018YFA0702403).References
- W. Yao, Acta Mech. Sin., 2019, 35, 1155–1177 CrossRef CAS.
- K. L. Tay, W. B. Yu, F. Y. Zhao and W. M. Yang, Proc IMechE, Part D: J Automobile Engineering, 2020, 234, 303–333 CrossRef CAS.
- P. Dagaut and M. Cathonnet, Prog. Energy Combust. Sci., 2006, 32, 48–92 CrossRef CAS.
- S. Honnet, K. Seshadri, U. Niemann and N. Peters, Proc. Combust. Inst., 2009, 32, 485–492 CrossRef CAS.
- A. Violi, S. Yan, E. G. Eddings, A. F. Sarofim, S. Granata, T. Faravelli and E. Ranzi, Combust. Sci. Technol., 2002, 174, 399–417 CrossRef CAS.
- S. Wang, G. Dai, H. Yang and Z. Luo, Prog. Energy Combust. Sci., 2017, 62, 33–86 CrossRef.
- E. Huo, S. Zhang, L. Xin, S. Wang, S. Cai, L. Zhang and M. Bai, Comput. Theor. Chem., 2022, 1211, 113696 CrossRef CAS.
- E. Huo, L. Xin, S. Zhang, C. Liu, S. Wang and L. Zhang, Process Saf. Environ. Prot., 2022, 161, 603–610 CrossRef CAS.
- Z. Chen, J. Pei, R. Li and F. Xiao, Constr. Build. Mater., 2018, 189, 695–710 CrossRef.
- A. C. T. van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- F. Castro-Marcano, A. M. Kamat, M. F. Russo, A. C. T. van Duin and J. P. Mathews, Combust. Flame, 2012, 159, 1272–1285 CrossRef CAS.
- C. Chen, L. Zhao, J. Wang and S. Lin, Ind. Eng. Chem. Res., 2017, 56, 12276–12288 CrossRef CAS.
- K. Chenoweth, A. C. T. van Duin, S. Dasgupta and W. A. Goddard Iii, J. Phys. Chem. A, 2009, 113, 1740–1746 CrossRef CAS PubMed.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, npj Comput. Mater., 2016, 2, 15011 CrossRef CAS.
- K. Chenoweth, A. C. T. van Duin and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS PubMed.
- L. Liu, C. Bai, H. Sun and W. A. Goddard, J. Phys. Chem. A, 2011, 115, 4941–4950 CrossRef CAS PubMed.
- X.-M. Cheng, Q.-D. Wang, J.-Q. Li, J.-B. Wang and X.-Y. Li, J. Phys. Chem. A, 2012, 116, 9811–9818 CrossRef CAS PubMed.
- Q.-D. Wang, J.-B. Wang, J.-Q. Li, N.-X. Tan and X.-Y. Li, Combust. Flame, 2011, 158, 217–226 CrossRef CAS.
- H. Liu, J. Liang, R. He, X. Li, M. Zheng, C. Ren, G. An, X. Xu and Z. Zheng, Combust. Flame, 2022, 237, 111865 CrossRef CAS.
- C. Ashraf and A. C. T. van Duin, J. Phys. Chem. A, 2017, 121, 1051–1068 CrossRef CAS PubMed.
- T. Cheng, A. Jaramillo-Botero, W. A. Goddard and H. Sun, J. Am. Chem. Soc., 2014, 136, 13467 CrossRef CAS.
- M. Kowalik, C. Ashraf, B. Damirchi, D. Akbarian, S. Rajabpour and A. C. T. van Duin, J. Phys. Chem. B, 2019, 123, 5357–5367 CrossRef CAS PubMed.
- S. Cheung, W.-Q. Deng, A. C. T. van Duin and W. A. Goddard, J. Phys. Chem. A, 2005, 109, 851–859 CrossRef CAS PubMed.
- C.-F. Li, Z. Mei, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Phys. Chem. Chem. Phys., 2018, 20, 14192–14199 RSC.
- H. Zhang, F. Chen, J. Xu, J. Zhang and Y. Han, Front. Chem. Sci. Eng., 2022, 16, 886–896 CrossRef CAS.
- G. Psofogiannakis and A. C. T. van Duin, Surf. Sci., 2016, 646, 253–260 CrossRef CAS.
- D. H. Kim, S. J. Kwak, J. H. Jeong, S. Yoo, S. K. Nam, Y. Kim and W. B. Lee, ACS Omega, 2021, 6, 16009–16015 CrossRef CAS PubMed.
- B. D. Jensen, A. Bandyopadhyay, K. E. Wise and G. M. Odegard, J. Chem. Theory Comput., 2012, 8, 3003–3008 CrossRef CAS PubMed.
- Y. Han, D. Jiang, J. Zhang, W. Li, Z. Gan and J. Gu, Front. Chem. Sci. Eng., 2016, 10, 16–38 CrossRef CAS.
- X. Li, M. Zheng, C. Ren and L. Guo, Energy Fuels, 2021, 35, 11707–11739 CrossRef CAS.
- G. Li, F. Zheng, Q. Huang, J. Wang, B. Niu, Y. Zhang and D. Long, J. Anal. Appl. Pyrolysis, 2022, 166, 105620 CrossRef CAS.
- J. Yu, B. Yu and J. Yu, JASP, 2020, 35, 673–681 Search PubMed.
- P. Dagaut, A. El Bakali and A. Ristori, Fuel, 2006, 85, 944–956 CrossRef CAS.
- S. Dooley, S. H. Won, M. Chaos, J. Heyne, Y. Ju, F. L. Dryer, K. Kumar, C.-J. Sung, H. Wang, M. A. Oehlschlaeger, R. J. Santoro and T. A. Litzinger, Combust. Flame, 2010, 157, 2333–2339 CrossRef CAS.
- C. Liu, S. Qiu, H. Huang, P. Guo and E. Huo, Mater. Rev., 2019, 33, 1251–1256 Search PubMed.
- E. Wang, J. Ding, Z. Qu and K. Han, Energy Fuels, 2018, 32, 901–907 CrossRef CAS.
- Y. Liu, X. Wei, W. Sun and L. Zhao, Energy Fuels, 2021, 35, 16778–16790 CrossRef CAS.
- L. Liu, H. Xu, Q. Zhu, H. Ren and X. Li, Chem. Phys. Lett., 2020, 760, 137983 CrossRef CAS.
- W. Zhou, X. Zhang, W. Zhou, L. Yang and Z. Jia, J. Energy Inst., 2022, 102, 82–91 CrossRef CAS.
- Q.-D. Wang, X.-X. Hua, X.-M. Cheng, J.-Q. Li and X.-Y. Li, J. Phys. Chem. A, 2012, 116, 3794–3801 CrossRef CAS PubMed.
- K. M. Bal and E. C. Neyts, Chem. Sci., 2016, 7, 5280–5286 RSC.
- K. M. Bal and E. C. Neyts, J. Chem. Theory Comput., 2015, 11, 4545–4554 CrossRef CAS PubMed.
- Z. Chen, W. Sun and L. Zhao, J. Phys. Chem. A, 2017, 121, 2069–2078 CrossRef CAS PubMed.
- Z. Chen, P. Zhao, L. Zhao and W. Sun, Energy Fuels, 2017, 31, 10515–10524 CrossRef CAS.
- D. Kim, J. Martz and A. Violi, Combust. Flame, 2014, 161, 1489–1498 CrossRef CAS.
- K. Narayanaswamy, H. Pitsch and P. Pepiot, Combust. Flame, 2016, 165, 288–309 CrossRef CAS.
- Y. Liu, J. Ding and K.-L. Han, Fuel, 2018, 217, 185–192 CrossRef CAS.
- Y. Dai, W. Zhao, H. Xie, Y. Guo and W. Fang, J. Anal. Appl. Pyrolysis, 2020, 145, 104723 CrossRef CAS.
- Y. Guan, J. Lou, R. Liu, H. Ma and J. Song, Fuel, 2020, 261, 116447 CrossRef CAS.
- H. S. Sim, R. A. Yetter, S. Hong, A. C. T. van Duin, D. M. Dabbs and I. A. Aksay, Combust. Flame, 2020, 217, 212–221 CrossRef CAS.
- L. Xin, C. Liu, Y. Liu, E. Huo, Q. Li, X. Wang and Q. Cheng, Fuel, 2020, 275, 117885 CrossRef CAS.
- H. Hirai, Chem. Phys., 2021, 548, 111225 CrossRef CAS.
- Y. Liu, G. Li and J. Ding, Fuel, 2019, 241, 273–282 CrossRef CAS.
- M. Wei, S. Wu, Q. Mao, Y. Wang, G. Guo and D. Zhang, Fuel, 2020, 275, 117989 CrossRef CAS.
- Y. Qian, W. Xu, J.-H. Zhan, X. Jia and F. Zhang, Process Saf. Environ. Prot., 2021, 147, 578–588 CrossRef CAS.
- T. Shou, N. Xu, Y. Li, G. Sun, M. T. Bernards, Y. Shi and Y. He, Plasma Process., 2019, 39, 863–876 CrossRef CAS.
- M. Duplančić, V. Gomzi, A. Pintar, S. Kurajica and V. Tomašić, Ceram. Int., 2021, 47, 3108–3121 CrossRef.
- S. Tan, T. Xia, Y. Shi, J. Pfaendtner, S. Zhao and Y. He, Sci. Rep., 2017, 7, 1710 CrossRef PubMed.
- C. D. Fu, Y. He and J. Pfaendtner, J. Phys. Chem. A, 2019, 123, 3080–3089 CrossRef CAS PubMed.
- H. Kwon, S. Shabnam, A. C. T. van Duin and Y. Xuan, Fuel, 2020, 262, 116545 CrossRef CAS.
- P. K. Naik, S. Paul and T. Banerjee, ChemistrySelect, 2019, 4, 12996–13005 CrossRef CAS.
- R. Chaudret, A. Bick and X. Krokidis, Energy Fuels, 2016, 30, 6817–6821 CrossRef.
- P. Zhao, S. Han, X. Li, T. Zhu, X. Tao and L. Guo, Energy Fuels, 2019, 33, 7176–7187 CrossRef CAS.
- Y. Wen, C. Jiang, W. Li, Y. Xie, G. Wang and Y. Hou, CIESC J., 2021, 72, 1100–1106 CAS.
- Y. Wen, W. Li, Y. Xie, Z. Qin, M. Gu, T. Wang and Y. Hou, Environ. Technol., 2022, 43, 2002–2016 CrossRef CAS PubMed.
- Y. Liu, X. Wei, W. Sun and L. Zhao, Ind. Eng. Chem. Res., 2021, 60, 14674–14684 CrossRef CAS.
- C. Ashraf, S. Shabnam, A. Jain, Y. Xuan and A. C. T. van Duin, Fuel, 2019, 235, 194–207 CrossRef CAS.
- S. Han, X. Li, L. Guo, H. Sun, M. Zheng and W. Ge, Energy Fuels, 2020, 34, 11381–11394 CrossRef CAS.
- K.-F. Song, G.-F. Ji, K. M. Kumari and D.-Q. Wei, Mol. Simul., 2018, 44, 21–33 CrossRef CAS.
- J. Zeng, L. Cao, C.-H. Chin, H. Ren, J. Z. H. Zhang and T. Zhu, Phys. Chem. Chem. Phys., 2020, 22, 683–691 RSC.
- X. Zhang, L. Pan, L. Wang and J.-J. Zou, Chem. Eng. Sci., 2018, 180, 95–125 CrossRef CAS.
- H. Wang, S. Gong, L. Wang, X. Zhang and G. Liu, Fuel, 2019, 239, 935–945 CrossRef CAS.
- E. Xiu-tian-feng, L. Pan, X. Zhang and J.-J. Zou, Fuel, 2020, 276, 118047 CrossRef.
- H. Wang, B. Zhang, S. Gong, L. Wang, X. Zhang and G. Liu, Combust. Flame, 2021, 226, 163–181 CrossRef CAS.
- W.-H. Kong, Y.-W. Li, J.-B. Wang and X.-Y. Li, Comput. Theor. Chem., 2021, 1200, 113232 CrossRef CAS.
- S. Wang, Z. Cui, C. Yu and T. Tan, J. Phys. Chem. A, 2020, 124, 6660–6666 CrossRef CAS PubMed.
- F. Guo, X. Cheng and H. Zhang, Combust. Sci. Technol., 2012, 184, 1233–1243 CrossRef CAS.
- M. Feng, X. Z. Jiang, Q. Mao, K. H. Luo and P. Hellier, Fuel, 2019, 254, 115643 CrossRef CAS.
- H. Kwon, A. Lele, J. Zhu, C. S. McEnally, L. D. Pfefferle, Y. Xuan and A. C. T. van Duin, Fuel, 2020, 279, 118548 CrossRef CAS.
- A. Lele, H. Kwon, K. Ganeshan, Y. Xuan and A. C. T. van Duin, Fuel, 2021, 297, 120724 CrossRef CAS.
- R. F. B. Goncalves, B. K. V. Iha, J. A. F. F. Rocco and A. E. Kuznetsov, Fuel, 2022, 310, 122157 CrossRef CAS.
- Y. Wang, S. Gong, H. Wang, L. Li and G. Liu, J. Anal. Appl. Pyrolysis, 2021, 155, 105045 CrossRef CAS.
- S. Han, X. Li, M. Zheng and L. Guo, Fuel, 2018, 222, 753–765 CrossRef CAS.
- S. Han, X. Li, F. Nie, M. Zheng, X. Liu and L. Guo, Energy Fuels, 2017, 31, 8434–8444 CrossRef CAS.
- S. Agrawalla and A. C. T. van Duin, J. Phys. Chem. A, 2011, 115, 960–972 CrossRef CAS PubMed.
- A. V. Kudinov, Y. A. Bogdanova and S. A. Gubin, Phys. At. Nucl., 2019, 82, 1486–1489 CrossRef CAS.
- M. Monge-Palacios and H. Rafatijo, Phys. Chem. Chem. Phys., 2017, 19, 2175–2185 RSC.
- F. Sun and W. Zeng, Int. J. Hydrogen Energy, 2020, 45, 20194–20199 CrossRef CAS.
- L. W. Bertels, L. B. Newcomb, M. Alaghemandi, J. R. Green and M. Head-Gordon, J. Phys. Chem. A, 2020, 124, 5631–5645 CrossRef CAS PubMed.
- L. Zhang, A. C. T. v Duin, S. V. Zybin and W. A. Goddard Iii, J. Phys. Chem. B, 2009, 113, 10770–10778 CrossRef CAS PubMed.
- Y. Liu, S. V. Zybin, J. Guo, A. C. T. van Duin and W. A. Goddard, J. Phys. Chem. B, 2012, 116, 14136–14145 CrossRef CAS PubMed.
- L. Song, F.-Q. Zhao, S.-Y. Xu, X.-H. Ju and C.-C. Ye, Fuel, 2021, 303, 121221 CrossRef CAS.
- J. Xu, Y. Bian, Y. Liu and D. Zhai, Comput. Mater. Sci., 2017, 131, 126–131 CrossRef CAS.
- D. Guo, S. V. Zybin, W. A. Goddard and Q. An, J. Phys. Chem. C, 2020, 124, 9787–9794 CrossRef CAS.
- J. Yi, Z. Qin, H. Li, F. Zhao, H. Ma and Z. Guo, J. Mol. Model., 2022, 28, 216 CrossRef CAS PubMed.
- T.-T. Zhou, Y.-D. Shi and F.-L. Huang, Acta Phys.-Chim. Sin., 2012, 28, 2605–2615 CAS.
- S. Zhu and W. Zhu, J. Nanopart. Res., 2020, 22, 362 CrossRef CAS.
- Y. Zhao, Z. Mei, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, ACS Omega, 2020, 5, 23193–23200 CrossRef CAS PubMed.
- H.-R. Zhang, Z.-H. Xue, X. Fu, J.-P. Liu, X. Qi and Q.-L. Yan, Fuel, 2022, 324, 124646 CrossRef CAS.
- T. Zhou, H. Song, Y. Liu and F. Huang, Phys. Chem. Chem. Phys., 2014, 16, 13914–13931 RSC.
- X. Huang, Z. Qiao, X. Dai, K. Zhang, M. Li, G. Pei and Y. Wen, J. Appl. Phys., 2019, 125, 195101 CrossRef.
- Z.-H. He, J. Chen, Q. Wu and G.-F. Ji, RSC Adv., 2016, 6, 93103–93110 RSC.
- Y. Zhang, H. Liu, Z. Yang, Q. Li and Y. He, ACS Omega, 2019, 4, 8031–8038 CrossRef CAS PubMed.
- J. Meng, S. Zhang, R. Gou, Y. Chen, Y. Li, M. Chen and Z. Li, J. Mol. Model., 2020, 26, 245 CrossRef CAS PubMed.
- C. Deng, J. Liu, X. Xue, X. Long and C. Zhang, J. Phys. Chem. C, 2018, 122, 27875–27884 CrossRef CAS.
- Y. J. Peng, S. Sun, W. N. Liu and Y. H. Liu, Acta Phys. Sin., 2021, 70, 158202 CrossRef.
- K. Zheng, Y. Wen, B. Huang, J. Wang, J. Chen, G. Xie, G. Lv, J. Liu, Z. Qiao and G. Yang, Phys. Chem. Chem. Phys., 2019, 21, 17240–17252 RSC.
- Q. Y. Li-Juan Peng, J.-B. Wang, Z.-R. Li, Q. Zhu and X.-Y. Li, Acta Phys.-Chim. Sin., 2017, 33, 745–754 Search PubMed.
- W. Hao, L. Niu, R. Gou and C. Zhang, J. Phys. Chem. C, 2019, 123, 14067–14080 CrossRef CAS.
- Y. Zhao, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Comput. Mater. Sci., 2020, 177, 109556 CrossRef CAS.
- N. Wang, J. Peng, A. Pang, T. He, F. Du and A. Jaramillo-Botero, J. Phys. Chem. C, 2017, 121, 14597–14610 CrossRef CAS.
- Z. Mei, Q. An, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Phys. Chem. Chem. Phys., 2018, 20, 29341–29350 RSC.
- F. Wang, L. Chen, D. Geng, J. Lu and J. Wu, J. Phys. Chem. C, 2019, 123, 23845–23852 CrossRef CAS.
- F. Wang, L. Chen, D. Geng, J. Wu, J. Lu and C. Wang, J. Phys. Chem. A, 2018, 122, 3971–3979 CrossRef CAS PubMed.
- J. Zhang and W. Guo, J. Phys. Chem. A, 2022, 126, 286–295 CrossRef CAS PubMed.
- Q. Han and W. Zhu, Chem. Phys. Lett., 2020, 761, 138067 CrossRef CAS.
- F. Wang, L. Chen, D. Geng, J. Lu and J. Wu, Phys. Chem. Chem. Phys., 2020, 22, 23323–23332 RSC.
- Z. Mei, C.-F. Li, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, J. Mater. Sci., 2019, 54, 7016–7027 CrossRef CAS.
- F. Wang, L. Chen, D. Geng, J. Lu and J. Wu, Phys. Chem. Chem. Phys., 2018, 20, 22600–22609 RSC.
- C.-X. Ren, X.-X. Li and L. Guo, Acta Phys. -Chim. Sin., 2018, 34, 1151–1162 CAS.
- J. Hu, Q. Gan, C. Feng, C. Li, S. Zhu and N. Cheng, J. Mater. Eng., 2021, 29, 482–491 Search PubMed.
- B. Wu, F. C. Wu, Y. B. Zhu, A. M. He and H. A. Wu, J. Appl. Phys., 2019, 126, 144305 CrossRef.
- Q. Chu, B. Shi, L. Liao, X. Zou, K. H. Luo and N. Wang, J. Phys. Chem. C, 2020, 124, 3886–3894 CrossRef CAS.
- Q. Chu, B. Shi, L. Liao, K. H. Luo, N. Wang and C. Huang, J. Phys. Chem. C, 2018, 122, 29620–29627 CrossRef CAS.
- H. Zeng, X. Cheng, C. Zhang and Z. Lu, J. Phys. Chem. C, 2018, 122, 9191–9197 CrossRef CAS.
- G. Li, L. Niu, W. Hao, Y. Liu and C. Zhang, Combust. Flame, 2020, 214, 238–250 CrossRef CAS.
- Y. Zhao, D. X. Ma, Z. Mei, F. Q. Zhao, S. Y. Xu and X. H. Ju, Int. J. Energy Res., 2022, 46, 11079–11091 CrossRef CAS.
- L. Song, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Appl. Surf. Sci., 2020, 519, 146249 CrossRef CAS.
- Q. Chu, X. Chang and D. Chen, Combust. Flame, 2022, 237, 111739 CrossRef CAS.
- W. Hao, G. Li, L. Niu, R. Gou and C. Zhang, J. Phys. Chem. C, 2020, 124, 10783–10792 CrossRef CAS.
- Y. Zhao, Z. Mei, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Appl. Surf. Sci., 2021, 563, 150296 CrossRef CAS.
- Y. Jiang, S. Deng, S. Hong, S. Tiwari, H. Chen, K.-I. Nomura, R. K. Kalia, A. Nakano, P. Vashishta, M. R. Zachariah and X. Zheng, ACS Appl. Mater. Interfaces, 2020, 12, 7451–7458 CrossRef CAS PubMed.
- S. Huang, S. Hong, Y. Su, Y. Jiang, S. Fukushima, T. M. Gill, N. E. D. Yilmaz, S. Tiwari, K.-I. Nomura, R. K. Kalia, A. Nakano, F. Shimojo, P. Vashishta, M. Chen and X. Zheng, Combust. Flame, 2020, 219, 467–477 CrossRef CAS.
- X.-K. Wang, Y. Zhao, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, J. Mol. Graphics Modell., 2021, 108, 107987 CrossRef CAS PubMed.
- L. Song, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Phys. Chem. Chem. Phys., 2021, 23, 11886–11892 RSC.
- R.-K. Dong, Z. Mei, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Int. J. Hydrogen Energy, 2021, 46, 1234–1245 CrossRef CAS.
- R.-K. Dong, Z. Mei, S.-Y. Xu, F.-Q. Zhao, X.-H. Ju and C.-C. Ye, Int. J. Hydrogen Energy, 2019, 44, 19474–19483 CrossRef CAS.
- Y. Zhao, D.-X. Ma, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, Appl. Surf. Sci., 2022, 586, 152777 CrossRef CAS.
- R.-K. Dong, Z. Mei, F.-Q. Zhao, S.-Y. Xu and X.-H. Ju, RSC Adv., 2019, 9, 41918–41926 RSC.
- L. Song, Z. Mei, S.-Y. Xu, F.-Q. Zhao and X.-H. Ju, J. Am. Ceram. Soc., 2022, 105, 1343–1357 CrossRef CAS.
- L. Song, F.-Q. Zhao, S.-Y. Xu, C.-C. Ye and X.-H. Ju, Mater. Today Commun., 2021, 26, 101804 CrossRef CAS.
- J. Chen and T. J. Martínez, Chem. Phys. Lett., 2007, 438, 315–320 CrossRef CAS.
- K. A. O'Hearn, M. W. Swift, J. L. Liu, I. Magoulas, P. Piecuch, A. C. T. van Duin, H. M. Aktulga and Y. Qi, J. Chem. Phys., 2020, 153 Search PubMed.
- J. Liu, X. Li, L. Guo, M. Zheng, J. Han, X. Yuan, F. Nie and X. Liu, Comput. Appl. Chem., 2014, 31, 641–647 CAS.
- M. Zheng, Z. Wang, X. Li, X. Qiao, W. Song and L. Guo, Fuel, 2016, 177, 130–141 CrossRef CAS.
- S. Fukuhara, K. M. Bal, E. C. Neyts and Y. Shibuta, Comput. Mater. Sci., 2020, 177, 109581 CrossRef CAS.
- L. Krep, I. S. Roy, W. Kopp, F. Schmalz, C. Huang and K. Leonhard, J. Chem. Inf. Model., 2022, 62, 890–902 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |