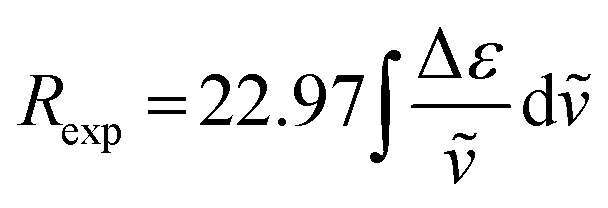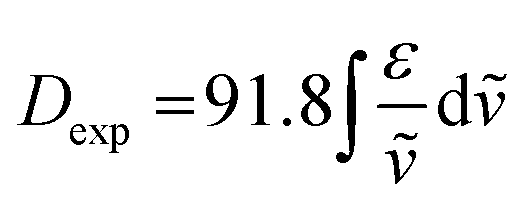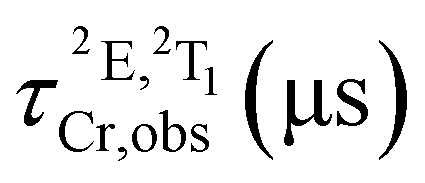DOI:
10.1039/D2TC05344F
(Paper)
J. Mater. Chem. C, 2023,
11, 2582-2590
EuIII functionalized silica nanoparticles encapsulating chiral CrIII complexes with simultaneous unpolarized red and polarized NIR-I luminescence†
Received
14th December 2022
, Accepted 27th January 2023
First published on 27th January 2023
Abstract
In this contribution we report on the synthesis and characterization of NIR-I luminescent silica nanoparticles doped with the chiral luminescent CrIIIN6 complex [Cr(dqp)2]3+ (dqp = 2,6-di(quinolin-8-yl)pyridine), Cr@SiO2NPs, as well as on their post-functionalization with the highly luminescent Eu(tta)3(phen-Si) (tta = 2-Thenoyltrifluoroacetonate; phen-Si = phenanthroline silica precursor), EuCr@SiO2NPs. These hybrid materials were prepared via reverse micelle techniques. Electronic microscopy analyses determined an average particle size of 94 nm with the Cr complex randomly distributed into the silica matrix. The EuCr@SiO2NPs material displays the characteristic ruby-like emission arising from the metal-centered spin-flip transitions within the 730–750 nm range (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–12700 cm−1) together with the Eu-centered 5D0 → 7FJ (J = 0–4) transitions in the red region of the electromagnetic spectrum. CPL measurements showed two mirror-image polarized bands ascribed to the Cr(2E → 4A2) and Cr(2T1 → 4A2) with dissymetry factor (glum) in the range of 0.1–0.2. The encapsulated CrIII complex showed larger quantum yield (4%) and longer excited state lifetime (336–1500 μs) compared to the non-encapsulated complex, providing an enhanced CPL brightness of 80 M−1 cm−1 in aerated media. Thus, these hybrid materials display an efficient and bright long-lived dual simultaneous non-polarized red and polarized NIR-I luminescence.
500–12700 cm−1) together with the Eu-centered 5D0 → 7FJ (J = 0–4) transitions in the red region of the electromagnetic spectrum. CPL measurements showed two mirror-image polarized bands ascribed to the Cr(2E → 4A2) and Cr(2T1 → 4A2) with dissymetry factor (glum) in the range of 0.1–0.2. The encapsulated CrIII complex showed larger quantum yield (4%) and longer excited state lifetime (336–1500 μs) compared to the non-encapsulated complex, providing an enhanced CPL brightness of 80 M−1 cm−1 in aerated media. Thus, these hybrid materials display an efficient and bright long-lived dual simultaneous non-polarized red and polarized NIR-I luminescence.
Introduction
Chiral materials displaying circularly polarized luminescence (CPL), which is the emission of polarized light with a certain degree of handedness, are attracting systems not only for fundamental purposes but also for potential applications in many different fields of biology, physics and chemistry.1–6 Thus, many CPL-active (nano)materials have been developed and they are mainly based on chiral organic molecules (discrete or supramolecular), chiral metal complexes, chiral polymers or chiral inorganic solids.7–21 However, silica platforms are overlooked materials in this field, particularly, silica nanoparticles. These nanomaterials are well-known to be cheap and appealing candidates for many applications since they have been proved to be biocompatible, stable, inert and their surface can be easily functionalized with active species leading to multifunctional materials.22–26 In consequence, these systems can show great potential in several biological and biomedical applications such as time-gated detection when combined with chiral long-lived emissive chromophores.27 Within this context, our group is interested in exploiting chiral Cr(III) polypyridine complexes displaying strong luminescence on the field of CPL as well as improving their stability and ease of handling by introduction into hybrid materials.28 In this sense, earth abundant metal-based hybrid materials can replace the costly Ru(II)- and Ir(III)-based nanoparticles which have been largely exploited in this field.29,30
Generally, promising photoactive emissive chiral materials should fulfill at least: (i) good chemical stability, (ii) large dissymmetry factor (glum), which quantifies the degree of handedness of a CP emission, and (iii) high emission brightness.31 Combining these properties in a solely system is still quite challenging since, generally, enhanced brightness implies low dissymmetry factors.32 The theoretical description of the dissymmetry factor reveals that maximized values of glum can be expected for those transitions on which the electric and magnetic dipole transitions moments |μ| and |m|, are similar in magnitude and their vectors are collinear.33–35 Interestingly, the spin-flip transition on Cr(III), which is the rearrangement of one electron spin in the d(t2g) orbitals, is an electric dipole forbidden and magnetic dipole allowed transition leading to |μ| ≈ |m|.36 Furthermore, it has been demonstrated that Cr(III) complexes embedded in a strong ligand field with nearly octahedral geometry show efficient quantum yields, ϕ, of up to 30%,37,38 and long-lived (millisecond range) ruby-like metal centered (MC) emissions although deoxygenation was required and the ϕ decreases to <1% under air conditions.39 Therefore, combining large glum and high quantum yields on Cr(III) complexes is achievable and allow high CPL brightness (BCPL = ελ × ϕλ × glum/2) which is key for the different applications employing chiral photoactive materials, where having good responses under ambient conditions is also advisable.31 Based on the inertness,40 inexpensive character of chromium,41,42 and the interesting photophysical properties of Cr(III) complexes,39,43,44 these systems have been recently used for molecular upconversion,45–49 molecular thermometry,50 pressure sensors,51 photocalysis,52–56 NIR-II luminescence,57,58 and, remarkably, as CPL emitters.48,59,60 Aiming at transferring the latter application to the nanoscale, herein we present amorphous silica nanoparticles which encapsulate photoactive chiral Cr(III) chromophores. We envisioned that the encapsulation of photoactive chiral Cr(III) chromophores could enhance both stability and emission under ambient conditions, hence expanding their possible applications. Altogether, in this work, we will discuss the synthetic approach, the morphology and photophysical properties of silica nanoparticles doped with the chiral photoactive [Cr(dqp)2]3+ (dqp = 2,6-di(quinolin-8-yl)pyridine) complex, as well as their Eu(III) functionalized analogs. We have demonstrated that both the stability and CPL brightness can be improved through encapsulation of chiral active Cr(III) complexes into silica nanoparticles and the successful nanoparticle functionalization with a second chromophore.
Results
Synthesis and chiral resolution of the chiral Cr(III) chromophore.
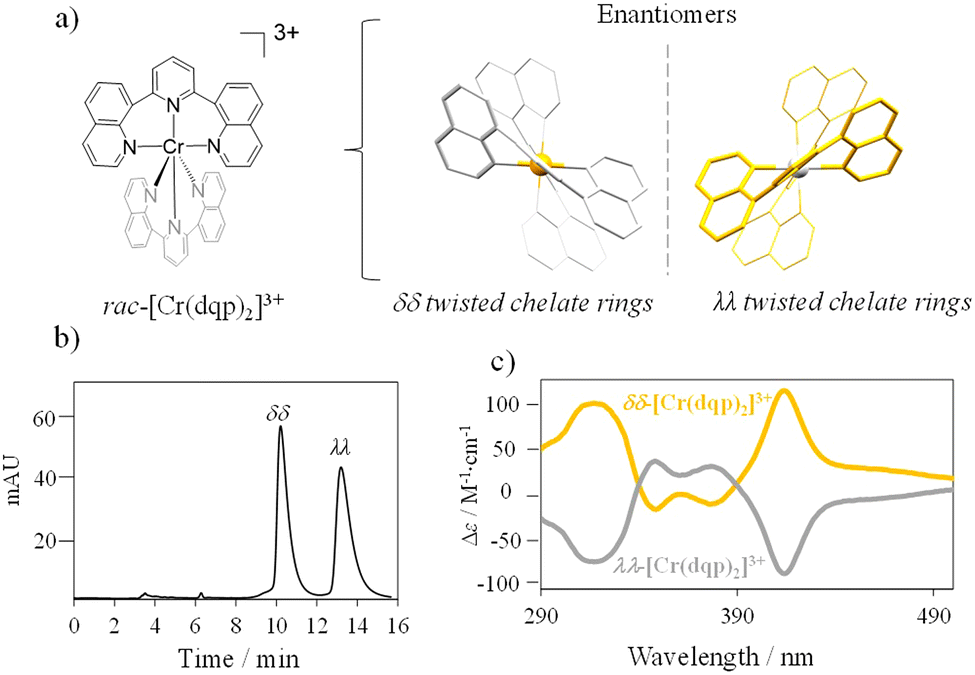
The complex rac-[Cr(dqp)2]3+ (Fig. 1(a) left) was prepared according to the reported procedure.60 The dqp ligand coordinates to the CrIII in a meridional fashion and the two quinoline branches distort from the planarity generating a pair of helical enantiomers (λλ/δδ), with λ (left-handed) and δ (right-handed) being the descriptors for the twisted conformation of the dqp ligand (Fig. 1(a) right).
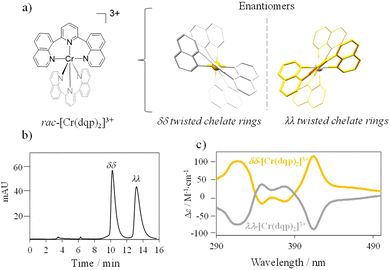 |
| | Fig. 1 (a) Picture of the [Cr(dqp)2]3+ complex highlighting the two helical conformations of the two enantiomers. (b) Chromatogram with separation conditions (inset) for the resolution of the rac-[Cr(dqp)2]3+. (c) CD spectra of the two eluted fractions in CH3CN. | |
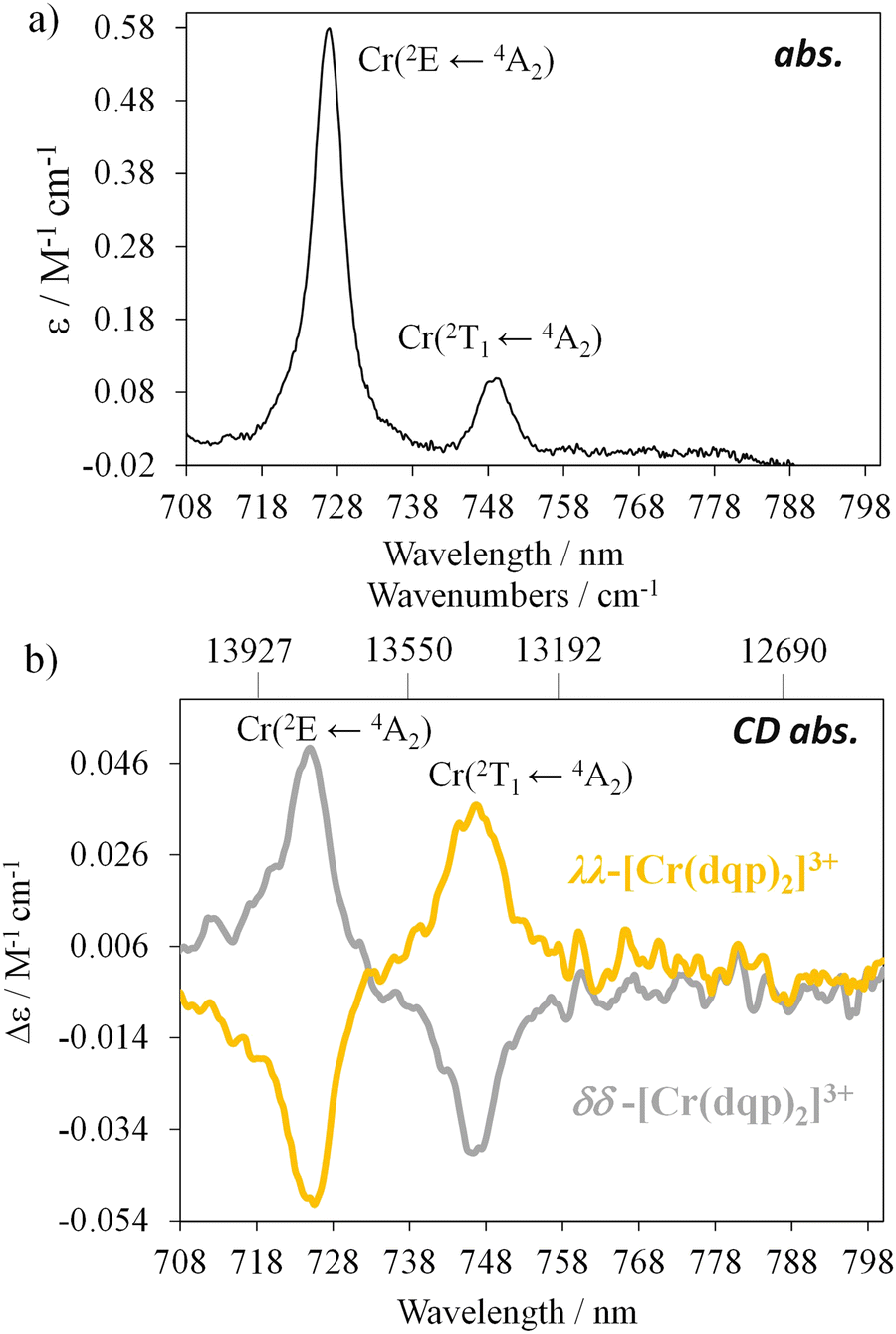
Chiral resolution of the two enantiomers was achieved by using chiral stationary phase high performance liquid chromatography (CSP-HPLC) instead of column chromatography as previously reported (Fig. 1(b)).60 Upon injection of 1 mL of an ethanolic concentrated solution (10 mM), we were able to separate approximately 15 mg of each enantiomer. The two collected fractions containing the target complex, [Cr(dqp)2](CF3COO)3, and a huge excess of Et4N+CF3COO− salt coming from the mobile phase, were purified by anion exchange using KPF6 in a mixture of water/acetone (ESI†). This step enabled the isolation of the pure enantiomers. The HPLC analysis demonstrated the chiral purity of the two fractions (Fig. S1, ESI†). The circular dichroism (CD) spectra in the 290–500 nm range for the less and more retained fractions displayed mirror images which proved their enantiomeric relationship (Fig. 1(c)). Our previous structural analysis by means of single crystal X-ray diffraction on the λλ enantiomer together with its CD spectrum,60 allowed us to determine that the more retained fraction, showing a negative cotton effect at 414 nm and 318 nm in the CD spectrum, corresponds to the λλ enantiomer. In consequence the less retained fraction corresponds to the δδ configuration. The CD spectrum recorded on a concentrated solution (1 mM) in the 800–700 nm region (showed two bands with opposite sign which are ascribed to the spin-flip Cr(2T1 ← 4A2) and Cr(2E ← 4A2) transitions assuming octahedral symmetry for the [CrN6] chromophore (Fig. 2(a) and (b)). The gabs, which is the absorption dissymmetry factor, gabs = 2(εL − εR)/(εL + εR), where εL and εR are the molar absorption coefficients for left and right circularly polarized light of these two transitions are 0.017 and 0.02, respectively. These values are roughly two orders of magnitude larger than those usually exhibited by electronic transitions induced in organic molecules.36
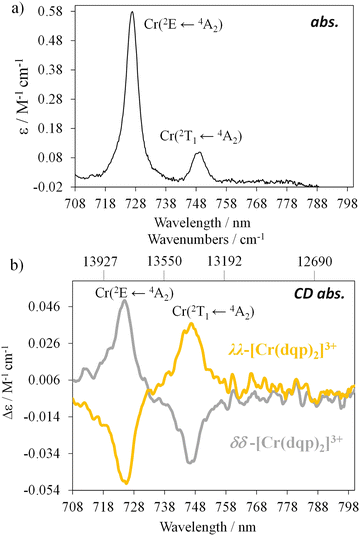 |
| | Fig. 2 (a) Absorption spectrum of the rac-[Cr(dqp)2]3+ and (b) CD spectra in the spin-flip region for the two enantiomers (CH3CN; c = 1 mM). | |
The integral of a CD band derived from a single electronic transition is related to the rotational strength (R) through the simplified eqn (1). Likewise, the dipole strength (D) of the spin-flip transitions can be calculated by integration of the absorption spectrum (Fig. 2(b)) using the simplified eqn (2).61
| | 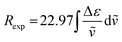 | (1) |
| | 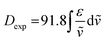 | (2) |
The rotatory strength (R), which is the probability of absorbing (or emitting) a circularly polarized photon from an initial state to a final state, is usually expressed in 10−40 cgs units.62,63 The computed values for R are 229 × 10−40 cgs and 107 × 10−40 cgs (in absolute values) for the respective transitions. Those R values, are four to five orders of magnitude larger compared to the spin-forbidden transitions on chiral Co(III) and Cr(III) complexes.61,64,65 Finally, the experimentally obtained dipole strength values are 7340 × 10−40 cgs and 2050 × 10−40 cgs for the lowest and highest energy transitions (eqn (2)).
Synthesis and morphology of the chiral silica nanoparticles
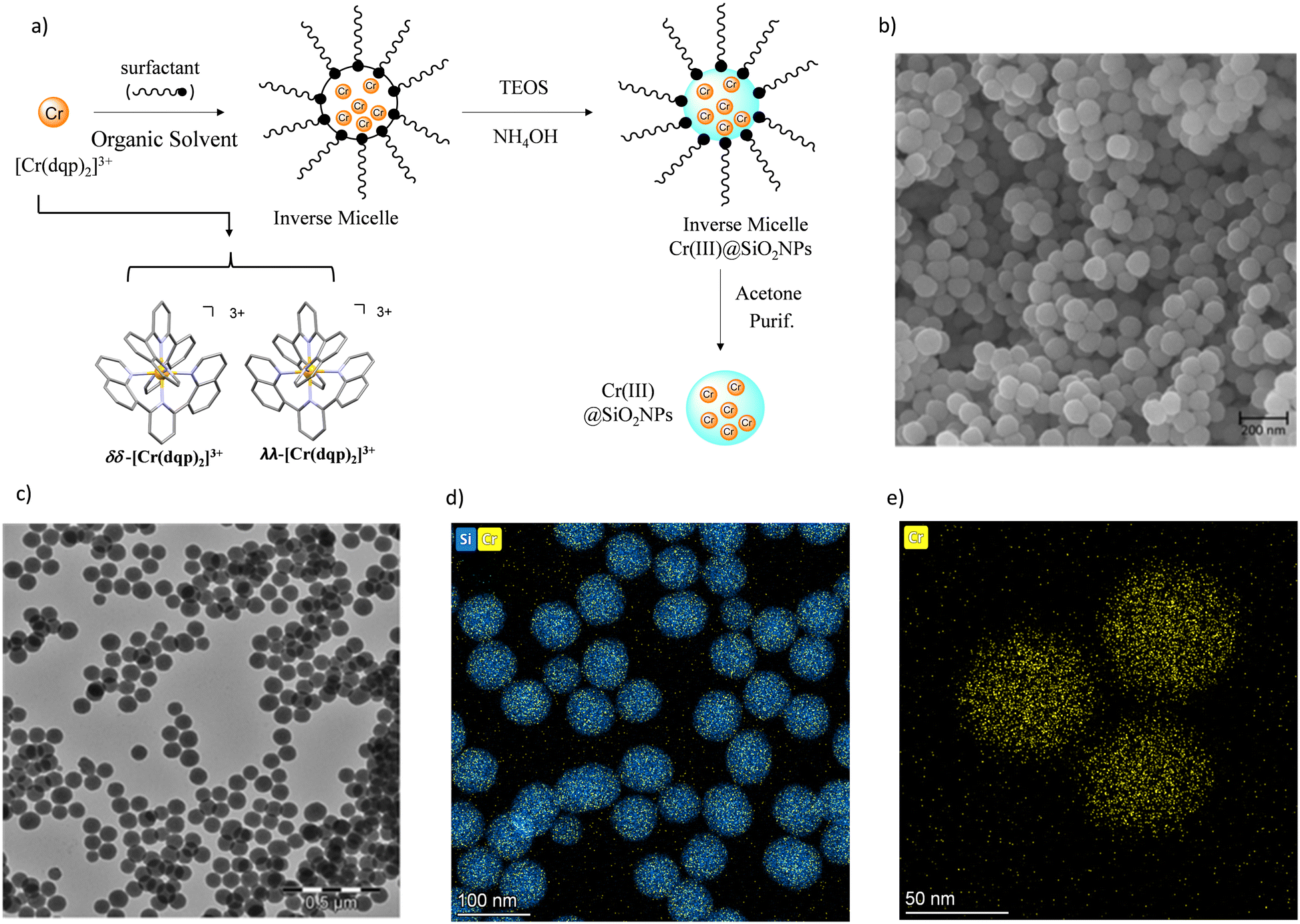
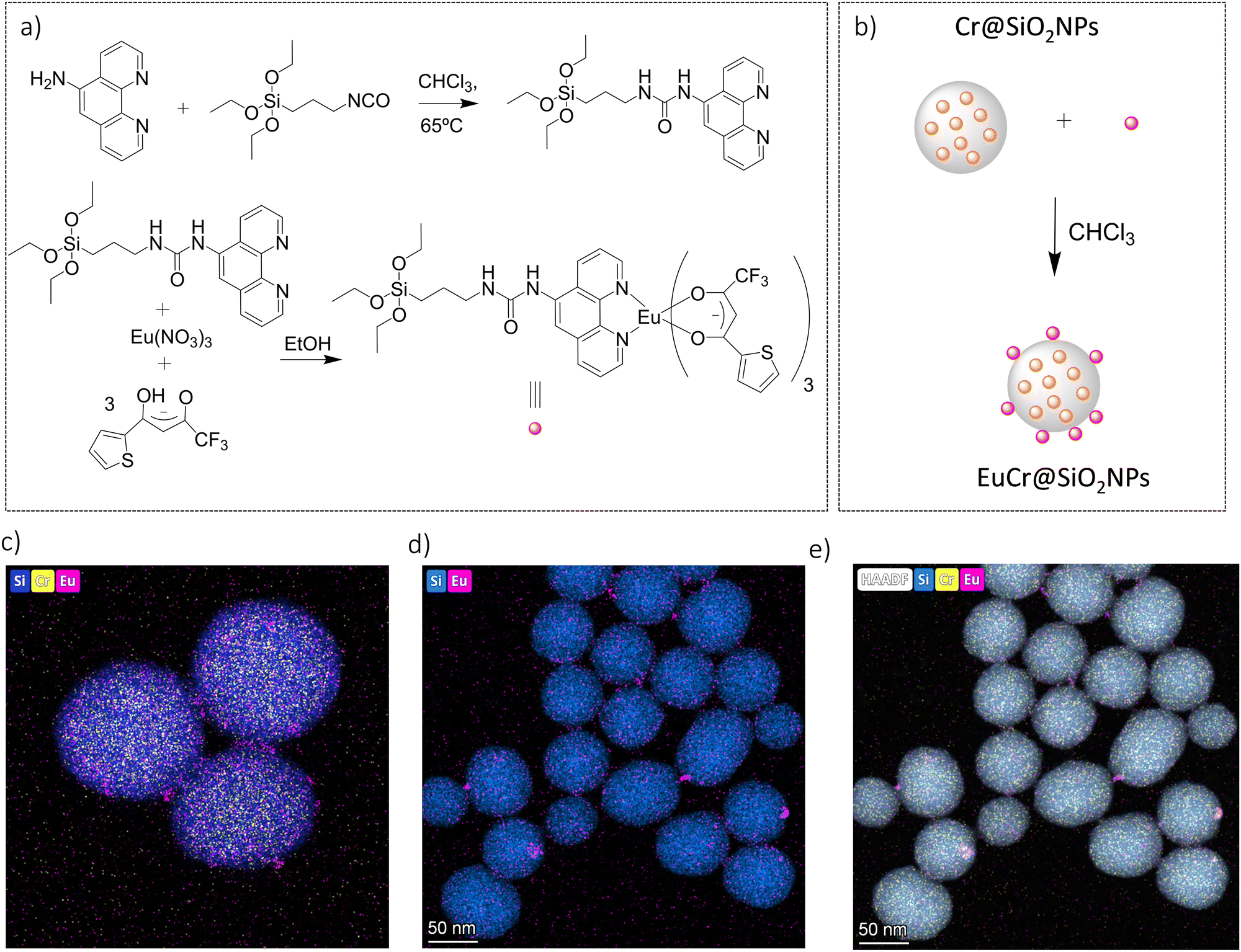
The preparation of the doped nanoparticles has been adapted from a literature method (Fig. 3(a) and ESI†).22,66 An aqueous solution of rac-[Cr(dqp)2](Cl)3, λλ-[Cr(dqp)2](Cl)3 or δδ-[Cr(dqp)2](Cl)3 (1 mL, ∼15 mM) and 200 μL of tetraethyl orthosilicate (TEOS) were added to a solution of Triton X-100 (1.8 mL), hexanol (1.8 mL) and cyclohexane (7.5 mL). The resulting mixture was stirred for 10 min before the addition of 60 μL of concentrated ammonia. The formed microemulsion was stirred for 24 h in the dark. Addition of acetone induces the precipitation of the formed nanoparticles which were recovered by centrifugation, washed several times with water, ethanol and acetone and finally dried at 50 °C for 12 h leading to fine orange-yellow powder. For the next, rac-Cr@SiO2NPs, λλ-Cr@SiO2NPs and δδ-Cr@SiO2NPs stand for the nanoparticles encapsulating the racemic [Cr(dqp)2]3+ complex and the λλ and δδ enantiopure complexes respectively. The method for preparing these hybrid silica nanoparticles is based on the well-established reverse micelle technique using water soluble complexes as doping agents.67,68 The reaction takes place in a reverse microemulsion made of a small amount of water solution of the Cr(III) complex and a surfactant (Triton X-10), co-surfactant (hexanol) in an organic solvent (cyclohexane). The formation of the silica matrix takes place within the reverse micelle which serves as a nano reactor, where the hydrophilic precursor TEOS diffuses, hydrolizes and condensates (polymerizes) in the presence of a base catalyst (ammonia). The cationic chromium complex (3+) rests inmobilized within the silica matrix through electrostatic interactions with the negatively charged silanol groups located at the surface of the nanoparticles (Scheme S1 in ESI†). The CD spectra of the λλ-Cr@SiO2NPs and δδ-Cr@SiO2NPs could be recorded and it demonstrates their enantiomeric relationship (Fig. S2, ESI†). The solid material is stable in air and does not leach the metal complex after at least 2 months (Fig. S3, ESI†). The morphology, size and chemical composition of the samples were determined by electron microscopy techniques. Fig. 3 shows representative Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM), and High-Angle Annular Dark-Field-STEM (HAADF-STEM) of rac-Cr@SiO2NPs (Fig. 3(b) and Fig. S4, ESI†) and λλ-Cr@SiO2NPs (Fig. 3(c), (d) and (e)). The nanoparticles show a well-defined spherical topology with a mean diameter of 94.1 ± 7.0 nm (Fig. S5, ESI†). EDX-compositional mapping images confirm that the Cr(III) complex is embedded and randomly distributed within the SiO2 nanoparticles (Fig. 3(d) and (e) and Fig. S6, ESI†). The amount of Cr(III) complex encapsulated into the silica matrix was determined by Inductive Coupled Plasma (ICP) emission spectroscopy showing a weight percentage of 0.7.
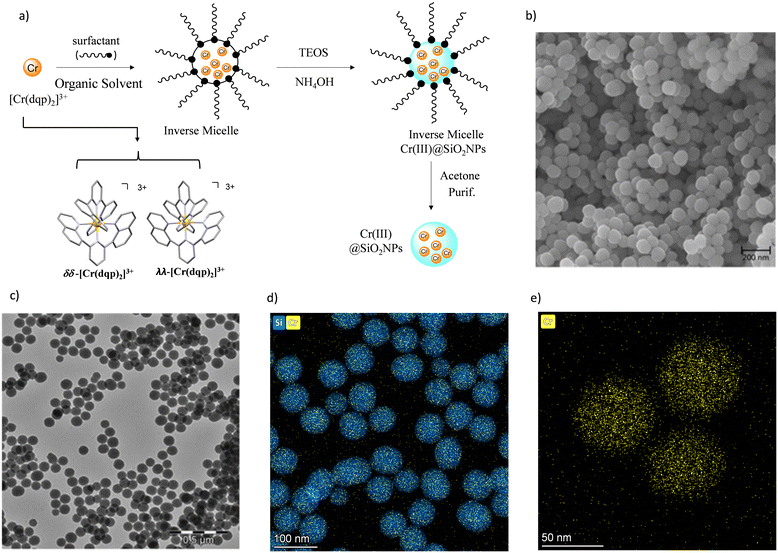 |
| | Fig. 3 (a) Schematic synthetic approach for preparing the silica nanoparticles (b) SEM image of the prepared rac-@SiO2NPs nanoparticles (c) TEM image of the prepared λλ-Cr@SiO2NPs; (d) and (e) HAADF-STEM compositional mapping image of λλ-Cr@SiO2NPs. | |
Photophysical properties of the Cr(III) silica nanoparticles
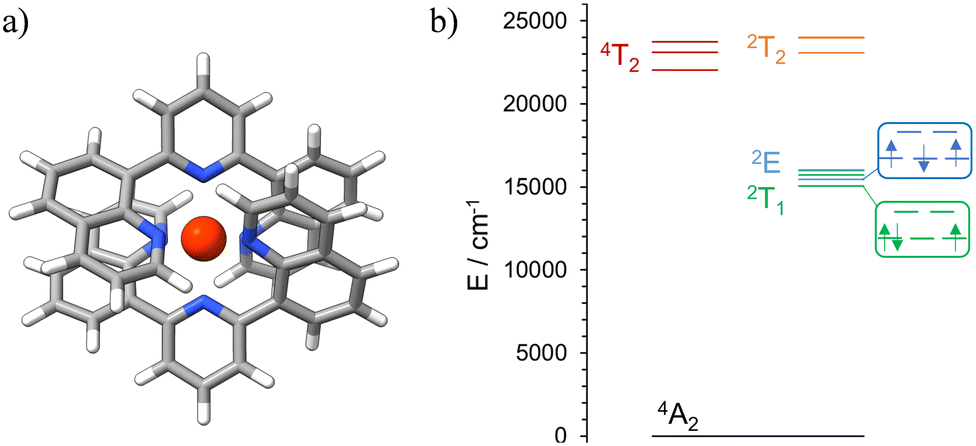
Initial ab initio ligand field theory (AILFT, CASSCF(3,5)-FIC-NEVPT2) yielded the Racah parameters B and C and the ligand field splitting Δ0 (Table S2, ESI†) for the [Cr(dqp)2]3+ optimized geometry (UB3LYP/def2TZVPP/GD3BJ/RIJCOSX/ZORA). Contrary to our predicted spin-flip energy level ordering based on the Tanabe-Sugano diagram for a d3 electronic configuration,69 refined CASSCF(7,12)-FIC-NEVPT2 calculations of the quartet and doublet excited states yielded the spin-flip 2T1 microstate below the 2E microstate (Fig. 4, Fig. S9 and Tables S3, S4, ESI†). Similar results have been observed for the [Cr(ddpd)2]3+ analog, using the same level of theory.70 In addition, the calculated energy values of the 4T2 and 2T2 microstates are very similar (Fig. 4(b) and Table S3, ESI†), favoring the intersystem crossing (ISC) process towards the doublet microstate.
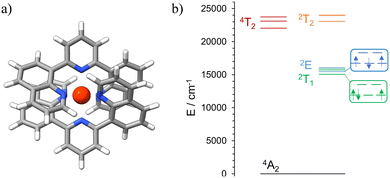 |
| | Fig. 4 (a) DFT optimized geometry of the quartet ground state of [Cr(dqp)2]3+. Spin density at the Cr center: 3.151647. (b) Schematic representation of the energy levels for the calculated (CASSCF(7,12)-FIC-NEVPT2) excited states of [Cr(dqp)2]3+. | |
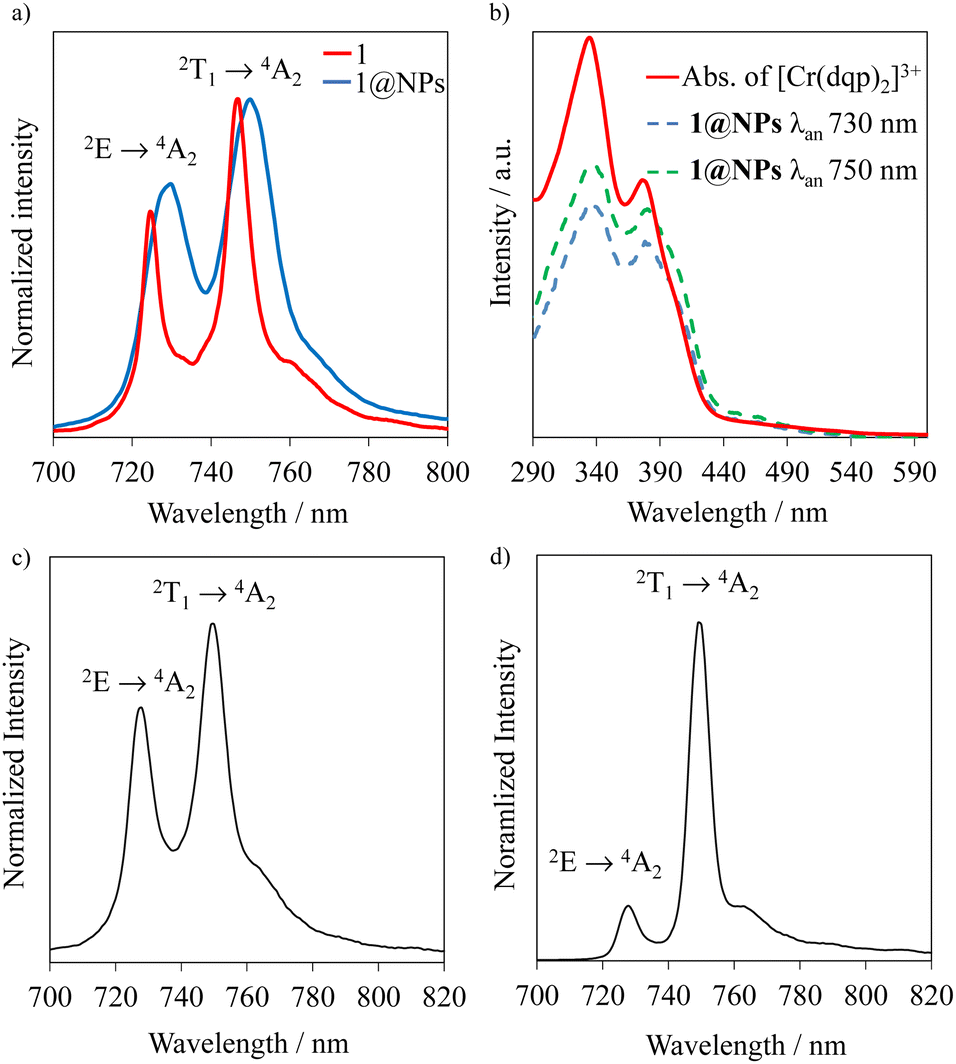
Upon excitation of rac-Cr@SiO2NPs at 410 nm (4A2 → 4T2/LMCT manifold) with a Xe lamp at room temperature and in a water suspension, two emission bands in the NIR-I window (700–800 nm) were detected. The observed transitions are ascribed to the spin-flip metal-centered Cr(2T1 → 4A2) (λmax = 750 nm) and Cr(2E → 4A2) (λmax = 730 nm) bands. This emission spectrum shows a similar profile to that exhibited by the non-encapsulated [Cr(dqp)2]3+ complex in aqueous solution (Fig. 5(a)). The emission maxima for the encapsulated complexes (blue spectra in Fig. 5(a)) are slightly red shifted (∼4 nm) and a bit broader compared to the isolated complexes (red spectra in Fig. 5(a)). This feature is also observed in the solid-state spectrum of the [Cr(dqp)2]3+ and can be ascribed to the reduced motion and vibration of the complex upon encapsulation.38,60 The excitation spectra recorded upon monitoring the emission bands at 750 and 730 nm for rac-Cr@SiO2NPs (Fig. 5(b); blue and green dashed lines) closely match the absorption spectra of the non-encapsulated [Cr(dqp)2]3+ complex (Fig. 5(b); red line) demonstrating the participation of ππ*, LMCT, and LMCT/MC excited states for feeding the emissive Cr-centered doublet states.
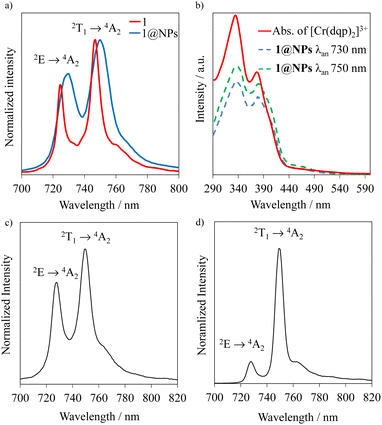 |
| | Fig. 5 (a) Emission spectra of the rac-Cr@SiO2NPs (blue trace) compared to the non-encapsulated rac-[Cr(dqp)2]3+ complex (red trace) λexc = 410 nm; (b) excitation spectra of the rac-Cr@SiO2NPs (dashed lines, λan = analysis wavelength) with the absorption spectra of the non-encapsulated [Cr(dqp)2]3+ (solid line); (c) solid state emission spectrum at 293 K of rac-Cr@SiO2NPs and (d) emission spectrum at 77 K of rac-Cr@SiO2NPs. | |
Time-resolved experiments on rac-Cr@SiO2NPs in aqueous solution and solid state, at the emission maximum, showed biexponential decays probably due to the different chemical environments around the encapsulated Cr(III) complexes (Fig. S10 and S11, ESI†). The obtained exponential decays displayed two contributions of: (i) 336–420 μs and (ii) 1500 μs in aerated solutions (Table 1). The same experiments were repeated after 35 minutes bubbling nitrogen obtaining the same biexponential decay profile with the same excited-state lifetimes. At cryogenic temperature (77 K) and in a H2O/DMSO solvent mixture, the nanoparticles display narrower emissions with slightly longer excited state lifetimes (Fig. 5(d), Table 1 and Fig. S12, ESI†). The room temperature overall quantum yields in deaerated and aerated water solutions amount to 4.0%. This value represents a 8-fold increase compared to the non-encapsulated complex measured in the same experimental conditions. This also indicates that the silica matrix shields and inhibits the detrimental Cr(III) interactions with the oxygen quencher (Table 1).71 Assuming that the radiative rate constant (kr) of the CrIII complex was not significantly affected by encapsulation, the larger value of τobs and ϕ at 298 K can be attributed to smaller non-radiative rate constants (knr) within the nanoparticles. Indeed, the rigid silica matrix restricts the vibrational motions of the complexes and penalizes non-radiative relaxation pathways of the excited state. For the sake of completeness, we have studied the photophysical properties of the λλ-Cr@SiO2NPs and δδ-Cr@SiO2NPs resulting in the same patterns and parameters as for the rac-Cr@SiO2NPs.
Table 1 Photophysical properties of isolated [Cr(dqp)2]3+ and rac-Cr@SiO2NPs under different experimental conditions. Lifetimes and quantum yields: estimated relative uncertainty ±10%
| Emitter |
Conditions |
ϕ
(%) |
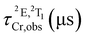
|
|
Room temperature.
In H2O/DMSO (50/50).
Excited state lifetime at λanalysis = 750 nm.
|
| [Cr(dqp)2]3+ 60 |
Aerated H2Oa |
0.5 |
89 |
| Powder |
— |
τ
1 = 32; τ2 = 6 |
| 77 Kb |
— |
3000 |
|
Cr@SiO2NPs
|
Aerated H2Oa |
4.0 |
τ
1= 420; τ2 = 1570 |
| Powder |
— |
τ
1 = 336; τ2 = 1500 |
| 77 Kb |
— |
τ
1 = 337; τ2 = 2160 |
Synthesis and morphology of the functionalized chiral silica nanoparticle
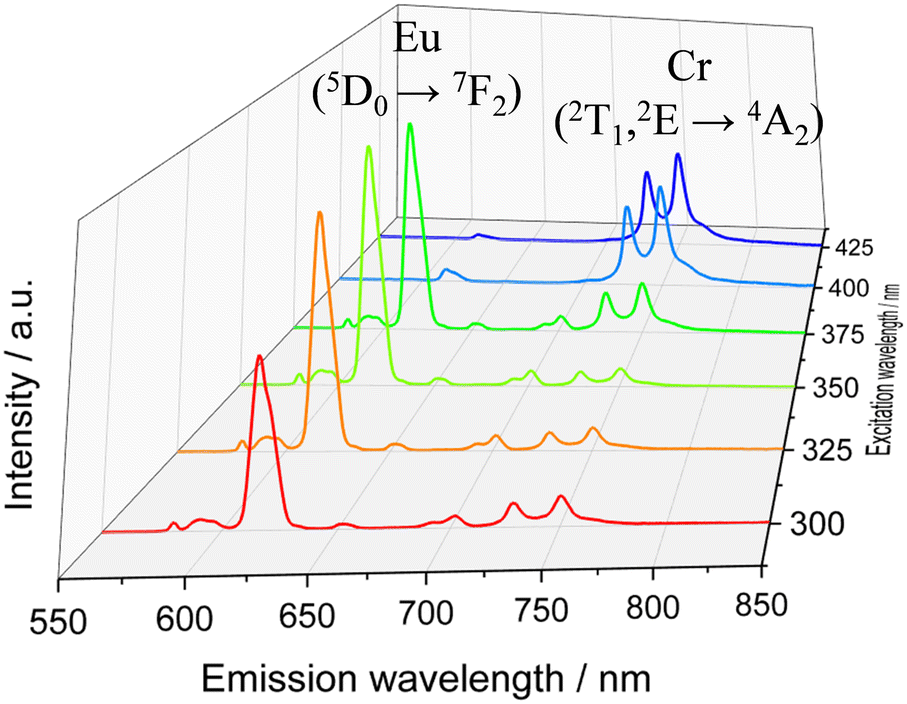
The surface of the Cr@SiO2NPs can be post-functionalized with luminescent metal complexes conjugated with alcoxysilane groups, leading to multi-coloured luminescent silica nanoparticles. In our case, the alkoxysilane conjugate [Eu(tta)3(phen-Si)] was prepared by reaction of 1 equiv. of 5-amino-1,10-phenanthroline with 1 equiv. of 3-(triethoxysilyl)propyl isocyanate in CHCl3 at 65 °C, followed by reaction with 1 equiv. of Eu(tta)3 (Fig. 6(a) and (b)).72 The mixture was stirred for 3 h at 50 °C under argon. The grafting reaction to prepare the (λλ/δδ)-EuCr@SiO2NPs nanoparticles was carried out in dichloromethane during 48 h under inert atmosphere. TEM microscopy confirms the same size and morphology as the non-functionalized nanoparticles (Fig. 6(c)). To probe the fixation of the EuIII complex to the silica matrix, EDX-FESEM (Energy Dispersive X-ray spectrometry and Field Emission Scanning Electron Microscopy) were performed, leading to an image that maps the spatial distribution of the Cr and Eu ions within the sample and confirms the grafting of the Eu(III) complex to the Cr@SiO2NPs surface (Fig. 6(d) and (e) and Fig. S7, ESI†). X-ray Photoelectron Spectroscopy (XPS) was applied in order to analyze the elements constituting the sample surface. Distinct peaks of Si(2p), C(1s), N(1s), O(1s), F(1s), Cr(2p) and Eu(3d) have been detected (Fig. S8a, ESI†). In Fig. S8b (ESI†), the peaks at 1132 eV and 1163 eV correspond to Eu(III) 3d5/2 and 3d3/2. The peaks at 575 eV and 586 eV were assigned to Cr(III) 2p3/2 and 2p1/2 (Fig. S8c, ESI†). Quantitative analysis on the surface demonstrated a greater amount of Eu(III) than Cr(III) on the surface of the nanoparticle which in agreement with the electronic microscope images (Table S1, ESI†). The emission spectrum of the hybrid nanoparticles at room temperature and upon excitation at 375 nm showed narrow bands corresponding to the Eu-centered 5D0 → 7FJ (J = 0–4) transitions as well as the Cr-centered 2E/2T1 → 4A2 transitions (Fig. 7). Time resolved experiments showed on the one hand a multi-exponential decay with two contributions, 300 μs (70%) and 1.5 ms (30%) for the Cr(2T1 → 4A2) transition, and a mono-exponential decay yielding 300 μs for the Eu(5D0 → 7F2) at 620 nm (Fig. S13, ESI†).
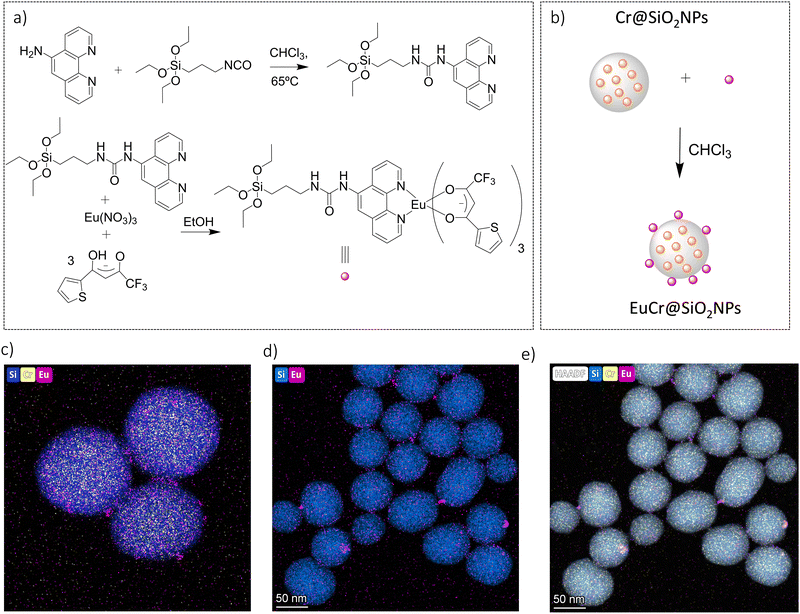 |
| | Fig. 6 (a) Synthetic steps for the preparation of the EuIII complex. (b) Schematic synthesis of (λλ/δδ)-EuCr@SiO2NPs (c) TEM image of the functionalized nanoparticle. (d) and (e) HAADF-STEM compositional mapping image of λλ-EuCr@SiO2NPs. | |
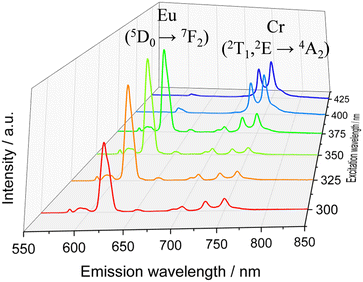 |
| | Fig. 7 Emission spectra for the (λλ/δδ)-EuCr@SiO2NPs under different excitation wavelength in ethanolic solution at room temperature. | |
It is worth mentioning that the excited state lifetime of the isolated Eu complex in solution at room temperature amounts to 300 μs as well. This indicates that downshifting Eu (donor) → Cr (acceptor) energy transfer does not occur in this system contrary to that observed in molecular compounds where efficient intramolecular energy transfer (f → d) has been detected.73 In the present nanoparticles, the large separation expected between the two metallic centers together with their poor spectral overlap prevent the energy transfer phenomenon. Thus, luminescence from Cr(III) and Eu(III) can be simultaneously induced and can be tuned as a function of the excitation wavelength (Fig. 7 and Fig. S14, ESI†). For instance, upon excitation at λ > 400 nm, mainly the spin-flip emission in NIR-I region arising from the Cr(III) is observed due to the efficient energy transfer from the dqp → Cr(III). In the range of excitation 390–375 nm, both emissions, arising from Cr(III) and Eu(III) are significantly intense as a consequence of the overlap of their excitation spectra. As the excitation wavelength moves to higher energies the red Eu(III)-based emission becomes more intense. Thus, the (λλ/δδ)-EuCr@SiO2NPs behaves as a non-polarized and polarized colour-tunable material in which the colour emission can be conveniently modified by selecting a specific excitation wavelength.
CPL of the nanoparticles
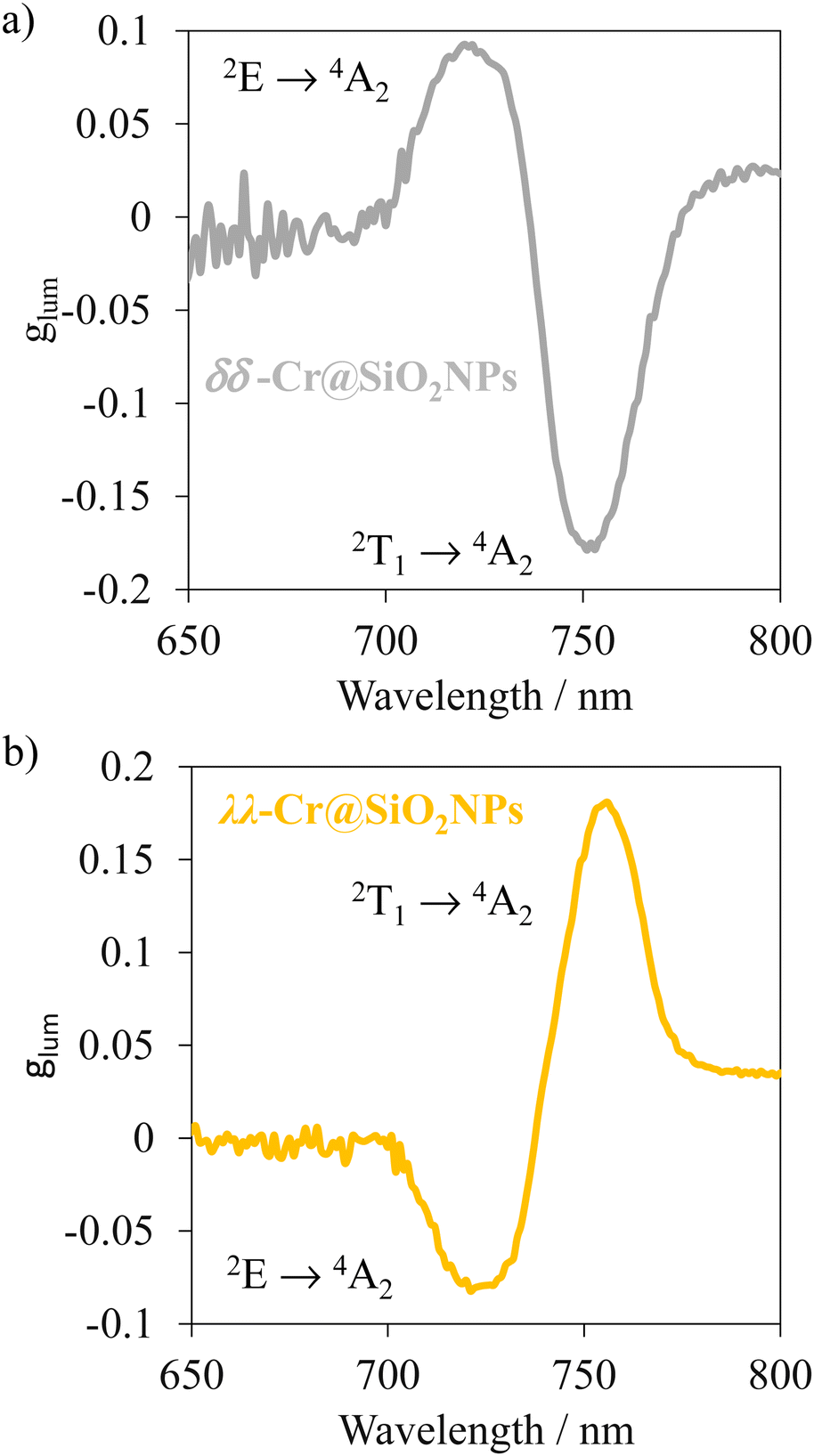
Upon non-polarized excitation at 370 nm, λλ-Cr@SiO2NPs and δδ-Cr@SiO2NPs showed two CP emission bands at 749 and 729 nm corresponding to Cr(2T1 → 4A2) and Cr(2T1 → 4A2) transitions, respectively (Fig. 8). The calculated glum which is 2(IL − IR)/(IL + IR), where IL and IR, represent the left and right circularly polarized emission intensities, respectively, yielded 0.18 and 0.08 (absolute values). These values of dissymmetry factor are taken from the maxima emission wavelength of the Cr(2T1 → 4A2) and Cr(2E → 4A2) transitions respectively and are comparable to the non-encapsulated enantiomers.60 This indicates that enantiomers encapsulation into the silica-defined matrix does not impact the glum value, thus the chiral Cr(III) chromophores behave just as isolated entities. The dissymmetry factor (glum) is also defined in terms of the rotatory and dipole strength (R and D) by the equation glum = 4R/D.74 By computing the obtained values of R and D into that equation (i.e. Rexp(2T1 ← 4A2) = 229 × 10−40 cgs and Rexp(2E ← 4A2) = 107 × 10−40 cgs; Dexp(2T1 ← 4A2) = 7340 × 10−40 cgs and Dexp(2E ← 4A2) = 2056 × 10−40 cgs), absolute values of 0.2 and 0.11 for the glum are obtained. These values closely match the experimental results obtained through the CPL experiments. Although the glum of the chiral Cr(III) complex is not tuned upon encapsulation in the silica nanoparticles, more importantly, upon excitation at 370 nm (ελ = 20.000 M−1 cm−1)59 the CPL brightness, which allows quantifying the overall efficiency of circularly polarized emitters, is enhanced significantly, in going from 1 M−1 cm−1 to 80 M−1 cm−1 in aerated media. The silica matrix protection of the chiral chromophores from the triplet oxygen and solvent interactions enhances this factor. Thus, these values of glum and brightness remain among the highest reported up to date.31 Future investigations will be devoted to decrease the nanoparticle size in order to study possible second order interactions between chromophores and that affecting the CPL properties.
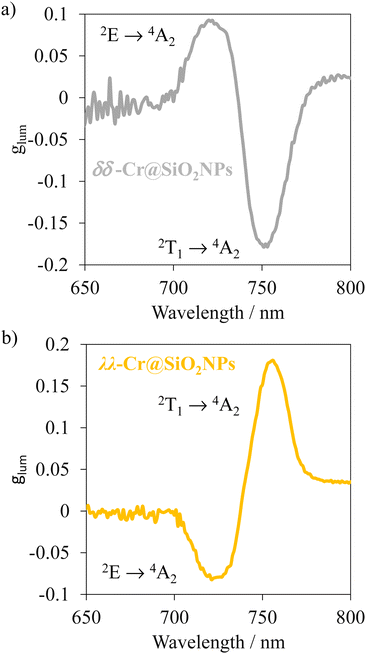 |
| | Fig. 8 CPL spectra of λλ-Cr@SiO2NPs and δδ-Cr@SiO2NPs in aerated diluted ethanolic solutions (λexc = 370 nm). | |
Conclusions
In summary, following a water-in-oil synthetic approach, hybrid silica nanoparticles have been prepared using the chiral [Cr(dqp)2]3+ ruby as doping agent. The silica matrix protects the chromophores from the triplet oxygen quencher and since the complexes have a limited mobility in the silica matrix, the vibrational motions are reduced and so the non-radiative deactivation pathways are also reduced. Altogether, the excited state lifetimes and quantum yields are few orders of magnitude larger than that of the non-encapsulated complexes under aerobic conditions. The CPL emission of the silica nanoparticles (glum ∼ 0.2) encapsulating the chiral rubies is very similar compared to the non-encapsulated complexes which indicates not only the lack of interaction between chiral chromophores but also the absence of electronic tuning upon encapsulation. Functionalization of the chiral nanoparticles with achiral Eu(III) complexes provided a bright and tunable long-lived dual simultaneous non-polarized red and polarized NIR-I luminescence where the Cr(III) and the Eu(III) behave independently. Importantly, the use of the earth abundant chromium and silicon make these cheap materials appealing candidates for many applications.22
Author contributions
J. R. J., S. M. L., A. G. C. and J. M. H. supervised the project and provided financial funds. J. R. J., Y. Y., J. M. H synthesized the luminescent complexes and the doped SiO2 nanoparticles. J. R. J, M. P. and C. P. have carried out the chiral resolution of the materials. S. M. L. and A. G. C. have performed the CPL experiments. C. M. performed theoretical calculations. C. L. and J. R. J. performed the photophysical characterization. J. R. J. and S. M. L. supervised the writing of the manuscript and all the authors have given the approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the funding received from the Special Research Programme from the University of Granada, PPJIA2021.11., grant TED2021.129598A.I00 funded by MCIN/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR and Junta de Andalucía. This work was financially supported by Ministerio de Ciencia e Innovación (PGC2018 102052-B-C21) MCIN/AEI/10.13039/501100011033/FEDER “Una manera de hacer Europa”, Junta de Andalucía (FQM-195, projects I + D + i A-FQM-172-UGR18 and B.FQM.328.UGR20) and the University of Granada. J. R. J. and J. M. H. acknowledge the Juan de la Cierva Incorporation grant IJC2020-044040-I funded by MCIN/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR.
Notes and references
- W. R. Kitzmann and K. Heinze, Angew. Chem., Int. Ed., 2022, 17, 955 Search PubMed.
- F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef.
- K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Chem. Sci., 2021, 12, 5522–5533 RSC.
- L. E. Mackenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS.
- F. Zinna, L. Arrico, T. Funaioli, D. Bari, M. Pasini, C. Botta and U. Giovannella, J. Mater. Chem. C, 2022, 10, 463–468 RSC.
- J. Bin Xiong, H. T. Feng, J. P. Sun, W. Z. Xie, D. Yang, M. Liu and Y. S. Zheng, J. Am. Chem. Soc., 2016, 138, 11469–11472 CrossRef PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1–33 CrossRef PubMed.
- J. Roose, B. Z. Tang and K. S. Wong, Small, 2016, 12, 6495–6512 CrossRef CAS PubMed.
- Y. Shi, P. Duan, S. Huo, Y. Li and M. Liu, Adv. Mater., 2018, 30, 1–7 Search PubMed.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed.
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfold and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC.
- J. Wade, J. Brandt, D. Reger, F. Zinna, K. Amsharov, N. Jux, D. Andrews and M. J. Fuchter, Angew. Chem., Int. Ed., 2020, 60, 222–227 CrossRef PubMed.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- X. Yang, J. Han, Y. Wang and P. Duan, Chem. Sci., 2019, 10, 172–178 RSC.
- C. M. Cruz, I. R. Márquez, I. F. A. Mariz, V. Blanco, C. Sánchez-Sánchez, J. M. Sobrado, J. A. Martín-Gago, J. M. Cuerva, E. Maçôas and A. G. Campaña, Chem. Sci., 2018, 9, 3917–3924 RSC.
- J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef CAS PubMed.
- S. Titos-Padilla, E. Colacio, S. J. A. Pope, J. J. Delgado, M. Melgosa and J. M. Herrera, J. Mater. Chem. C, 2013, 1, 3808–3815 RSC.
- L. Armelao, D. B. Dell’Amico, L. Bellucci, G. Bottaro, L. Di Bari, L. Labella, F. Marchetti, S. Samaritani and F. Zinna, Inorg. Chem., 2017, 56, 7010–7018 CrossRef CAS PubMed.
- S. Zhou, Q. Zhong, Y. Wang, P. Hu, W. Zhong, C. B. Huang, Z. Q. Yu, C. Di Ding, H. Liu and J. Fu, Coord. Chem. Rev., 2022, 452, 214309 CrossRef CAS.
- H. Li, X. Chen, D. Shen, F. Wu and R. Pleixats, Nanoscale, 2021, 13, 15998–16016 RSC.
- A. Sardarian, H. Eslahi and M. Esmaeilpour, ChemistrySelect, 2018, 3, 1499–1511 CrossRef CAS.
- J. C. G. Bünzli, Trends Chem., 2019, 1, 751–762 CrossRef.
- M. Poncet, A. Benchohra, J. R. Jiménez and C. Piguet, ChemPhotoChem, 2021, 5, 880–892 CrossRef CAS.
- Y. Li, W. Liu and X. Zheng, LANL, 2015, 48, 1907–1917 CrossRef CAS.
- Y. Jin, A. Li, S. G. Hazelton, S. Liang, C. L. John, P. D. Selid, D. T. Pierce and J. X. Zhao, Coord. Chem. Rev., 2009, 253, 2998–3014 CrossRef CAS.
- L. Arrico, L. Di Bari and F. Zinna, Chem. – Eur. J., 2020, 27, 2920–2934 CrossRef PubMed.
- Y. Nagata and T. Mori, Front. Chem., 2020, 8, 1–6 CrossRef PubMed.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS.
- F. S. Richardson, Chem. Rev., 1979, 79, 17–36 CrossRef CAS.
- G. L. Hilmes, H. G. Brittain and F. S. Richardson, Inorg. Chem., 1977, 16, 528–533 CrossRef CAS.
- B. Doistau, J. R. Jiménez and C. Piguet, Front. Chem., 2020, 8, 1–27 CrossRef PubMed.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed.
- T. H. Maiman, Nature, 1960, 187, 493–494 CrossRef.
- J. R. Jiménez, B. Doistau, M. Poncet and C. Piguet, Coord. Chem. Rev., 2021, 434, 213750 CrossRef.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- L. S. Forster, Chem. Rev., 1990, 90, 331–353 CrossRef CAS.
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J. C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed.
- Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, J. Phys. Chem. C, 2013, 117, 26957–26963 CrossRef CAS.
- B. Golesorkhi, H. Bolvin and C. Piguet, Dalton Trans., 2021, 50, 7955–7968 RSC.
- C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC.
- J. Kalmbach, C. Wang, Y. You, C. Förster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 59, 18804–18808 CrossRef CAS PubMed.
- S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Chem. – Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed.
- S. Otto, J. P. Harris, K. Heinze and C. Reber, Angew. Chem., Int. Ed., 2018, 57, 11069–11073 CrossRef CAS PubMed.
- T. H. Bürgin, F. Glaser and O. S. Wenger, J. Am. Chem. Soc., 2022, 144, 14181–14194 CrossRef PubMed.
- S. Sittel, R. Naumann and K. Heinze, Front. Chem., 2022, 10, 340 Search PubMed.
- R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappé and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451–5464 CrossRef CAS PubMed.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed.
- S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- N. Sawicka, C. J. Craze, P. N. Horton, S. J. Coles, E. Richards and S. J. A. Pope, Chem. Commun., 2022, 58, 5733 RSC.
- N. Sinha, J. R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef CAS PubMed.
- J.-R. Jiménez, M. Poncet, S. Miguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef PubMed.
- J. R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed.
- A. J. McCaffery and S. F. Mason, Trans. Faraday Soc., 1963, 59, 1–11 RSC.
-
F. Gendron, B. Moore, O. Cador, F. Pointillart, J. Autschbach and B. Le Guennic, ChemRxiv, 2019, 1–33.
- M. Scott, D. R. Rehn, S. Coriani, P. Norman and A. Dreuw, J. Chem. Phys., 2021, 154, 024111 CrossRef PubMed.
- S. F. Mason and B. J. Peart, J. Chem. Soc., Dalton Trans., 1977, 937–941 RSC.
- J. Fan, M. Seth, J. Autschbach and T. Ziegler, Inorg. Chem., 2008, 47, 11656–11668 CrossRef CAS PubMed.
- I. F. Díaz-Ortega, E. L. Fernández-Barbosa, S. Titos-Padilla, S. J. A. Pope, J. R. Jiménez, E. Colacio and J. M. Herrera, Dalton Trans., 2021, 50, 16176–16184 RSC.
- R. P. Bagwe, C. Yang, L. R. Hilliard and W. Tan, Langmuir, 2004, 20, 8336–8342 CrossRef CAS PubMed.
- F. J. Arriagada and K. Osseo-Asare, J. Colloid Interface Sci., 1999, 211, 210–220 CrossRef CAS PubMed.
- Y. Tanabe and S. Sugano, J. Phys. Soc. Jpn., 1954, 9, 766–779 CrossRef CAS.
- W. R. Kitzmann, C. Ramanan, R. Naumann and K. Heinze, Dalton Trans., 2022, 51, 6519–6525 RSC.
- C. Wang, W. R. Kitzmann, F. Weigert, C. Förster, X. Wang, K. Heinze and U. Resch-Genger, ChemPhotoChem, 2022, 6, 1–9 Search PubMed.
- S. Cousinié, M. Gressier, C. Reber, J. Dexpert-Ghys and M. J. Menu, Langmuir, 2008, 24, 6208–6214 CrossRef PubMed.
- M. Cantuel, G. Bernardinelli, D. Imbert, J. C. G. Bünzli, G. Hopfgartnerd and C. Piguet, J. Chem. Soc., Dalton Trans., 2002, 1929–1940 RSC.
- F. S. Richardson, Inorg. Chem., 1980, 19, 2806–2812 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Sandra
Míguez-Lago
*a,
Sandra
Míguez-Lago