
Injectable spontaneously formed asymmetric adhesive hydrogel with controllable removal for wound healing†
Lei
Liang
a,
Xi
Li
a,
Zhouying
Tan
a,
Min
Liu
a,
Yuwei
Qiu
a,
Qingyu
Yu
a,
Chaojie
Yu
a,
Mengmeng
Yao
a,
Bingyan
Guo
a,
Fanglian
Yao
 ab,
Pengcheng
Che
*c,
Hong
Zhang
*a and
Junjie
Li
*ab
ab,
Pengcheng
Che
*c,
Hong
Zhang
*a and
Junjie
Li
*ab
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, China
bFrontiers Science Center for Synthetic Biology and Key Laboratory of Systems Bioengineering (Ministry of Education), Tianjin University, Tianjin 300350, China
cSchool of Nursing and Rehabilitation, North China University of Science and Technology, Tangshan 063210, China
First published on 26th October 2023
Abstract
Healing large-scale wounds has been a long-standing challenge in the field of biomedicine. Herein, we propose an injectable oxidated sodium alginate/gelatin/3,3′-dithiobis(propionic hydrazide)-aurum (Alg-CHO/gelatin/DTPH-Au) hydrogel filler with asymmetric adhesion ability and removability, which is formed by the Schiff-base reaction between aldehyde-based sodium alginate and multi-amino crosslinkers (gelatin and DTPH), combined with the coordination interaction between Au nanoparticles and disulfide bond of DTPH. Consequently, the prepared Alg-CHO/gelatin/DTPH-Au hydrogel exhibits high mechanical properties and injectable behaviors owing to its multiple-crosslinked interactions. Moreover, because various types of interaction bonding form on the contact side with the tissue, denser crosslinking of the upper layer relative to the lower layer occurs. Combined with the temperature difference between the upper and lower surfaces, this results in asymmetric adhesive properties. Owing to the photothermal effect, the reversible coordination interaction between Au nanoparticles and DTPH and the change in the triple helix structure of gelatin to a coil structure impart the filler-phased removability and antibacterial ability. The choice of all natural polymers also allows for favorable degradability of the wound filler and outstanding biocompatibility. Based on these features, this versatile wound filler can achieve a wide range of applications in the field of all-skin wound repair.
1. Introduction
For decades, cutaneous wound healing has attracted considerable attention in the emergency field owing to its high morbidity and mortality.1,2 The process of wound healing involves four stages: coagulation, inflammation, repair, and regeneration.3,4 Generally, small size wounds can be repaired by the body itself, while severe skin damage requires fillers to assist in all stages of wound healing and overcome the external environment. Among the medical wound fillers, wound dressings must not only have the ability to promote the repair and regeneration of damaged skin but also have the wound binding capacity. Previous studies have developed a variety of natural polymer/synthetic polymer dressings to achieve antibacterial5,6 and anti-fouling3,7 functions. Nowadays, increasing attention is being paid to the adaptability of wounds as a key to enhancing their restorative treatment outcomes.8,9 According to the findings of the studies, the adaptability of the wound is of increasing interest and has become crucial for improving the effectiveness of its repair treatment. Adaptability includes injectability, adhesion properties, and removability. Injectability is a viable alternative for improving the adaptability of wound fillers. It enables the wound dressing to cover the wound more easily and completely, promoting better adhesion properties and allowing the wound dressing to contact the wound more closely and absorb exudate,10 whereas removability prevents secondary injuries caused by dressing changes.11 Hence, the development of an injectable removable adhesive hydrogel is of vital importance in the field of wound healing medical devices.Many approaches have been developed to achieve the injectable property, such as the shear thinning property,12,13 Schiff-base reaction,14 thiol–ene click reaction, and photo-crosslinking method.15 Among them, the shear thinning property is one of the most common methods. Zhang et al.16 prepared a shear-thinning zwitterionic granular hydrogel through the reversible binding of physical interactions to achieve the repair of articular cartilage injury, which could be easily broken under external force and recombined after the force was removed. In addition, injectable capability can also be achieved as in situ forming hydrogels. As a flowing liquid, the pre-gel solution can be easily injected and adapted to the wound site, and the resulting hydrogel can be firmly filled and adhered to the skin wound. The pre-gel solution can also absorb fluid around the wound and hydrogel after breaking the hydration layer, thus promoting a stable wet adhesion capability. A et al.17 fabricated an injectable multi-responsive hydrogel with the Schiff-base reaction between the amino group of 3,3′-dithiobis(propionic hydrazide) (DTPH) and aldehyde group from oxidized hyaluronic acid. In addition to Schiff-base bonds, other physical interactions and dynamic bonds, such as hydrogen bond18 and electronic interaction,3 can also be designed to achieve injectable hydrogels.
Hydrogel adhesion properties can be further improved via the modification of specific groups or the introduction of hydrophobic interactions.19–22 However, these methods result in the hydrogel having high adhesion properties on both sides, which may accidentally cause adhesion to the surrounding tissues or devices.23–25 Achieving asymmetric adhesion is also one of the hot research topics and solutions of double-side adhesion in the field of hydrogel adhesion and wound healing.26–29 Owing to the in vivo application, bilateral adhesion may lead to many operational difficulties or subsequent side effects, such as postoperative tissue adhesion or dressing surface contamination. However, existing methods for the preparation of asymmetric hydrogels often require processing in a subsequent step30 and have limitations in their preparation method, and most of them cannot be applied in the form of an injectable hydrogel.31,32 In recent years, the use of post-crosslinking techniques has been proposed for the design of injectable asymmetric adhesive hydrogels. Su et al.33 fabricated the gelatin/tea polyphenols/urea injectable hydrogel with triggered underwater adhesion through molecular diffusion. The urea diffuses underwater, and the gelatin-tea polyphenol combination that achieves post-crosslinking is stronger. Wu et al.34 also proposed an injectable catechol-grafted hyaluronic acid hydrogel with double bond groups that could be photo-crosslinked after being injected into the wound to achieve antiadhesive capability but still needs post-irradiation. Despite the existence of partial studies, the preparation method of post-crosslinking into hydrogels still needs to be optimized. Consequently, the development of injectable asymmetric adhesion hydrogels is one of the most important research directions for future wound dressings, which can promote the binding ability by close contact with injectable hydrogels and utilize the selection of multi-scale crosslinking to achieve asymmetric adhesion hydrogels.
Meanwhile, traditional adhesion hydrogels have a strong adhesion ability with tissues, and difficult dressing changes can easily cause secondary damage, which is an important cause of late wound healing efficiency. Therefore, removability is the key to enhancing clinical outcomes. Existing means of removal often achieve dressing removal by reducing wound dressing adhesion, using an appropriate removal solution, or spontaneously degrading the hydrogel network.35–39 However, problems still exist, including the difficulty for the hydrogel network to adequately contact the degradation solution, further leading to low removal efficiency. Consequently, the phase transition through hydrogels is an ideal way to achieve their non-destructive removal.36 Aurum nanoparticles (AuNPs) can form coordination interactions with compounds containing disulfide bonds (e.g., DTPH), and this in coordination interactions can detach controllably under near-infrared (NIR) light irradiation at 808 nm,40 thus causing the macroscopic or microscopic phase transition of the hydrogel. Therefore, AuNPs combined with DTPH (DTPH-AuNPs) are used in injectable hydrogels for gradient crosslinking with a Schiff-base reaction.17 Additionally, AuNPs have a marvellous photothermal property under NIR irradiation at 808 nm, which can modulate the formation and dissociation of the temperature responsive crosslinked structure. For example, the photothermal may result in a change in the triple helix structure of gelatin to coil structure.11 Thus, the development of phase transition hydrogels based on gold-disulfide bond crosslinking is an important way to enhance their removability.
In recognition of its low cost and favourable biocompatibility, sodium alginate (Alg) is widely used in the production of biomedical materials.41,42 Gelatin also has predominant biocompatibility and the ability to promote wound healing.43 Accordingly, we fabricate a kind of injectable hydrogel with excellent asymmetric adhesion and controlled removal using a Schiff-base reaction between Alg–CHO and multi-scale crosslinkers (DTPH/DTPH-AuNPs/gelatin). As shown in Fig. 1, the oxidated sodium alginate (Alg–CHO) and gelatin were used to fabricate the basic structure of the hydrogel as the first scale crosslinking network, the coordination interaction between AuNPs and DTPH was the second scale crosslinking network, and DTPH crosslinked Alg–CHO was the third scale crosslinking network. In this multi-scale crosslinking system, the Schiff-base reaction between Alg–CHO and gelatin achieves the injectable property, while the Aurum-disulfide bond promotes the phase transition hydrogel. Different molecular scales also have different crosslinking times, resulting in gradient crosslinking degrees and asymmetric adhesion. Concurrently, the introduction of DTPH-AuNPs and DTPH contributes to the controlled removal of injectable hydrogels. In addition to facilitating the application process, the phased removability in the later stage of wound healing allows them to reach a softened state for easy replenishment of the pre-gel solution or absorption of the degradation solution before refilling or removal. The soft state also makes it easier to clean surface contamination under NIR light irradiation, which endows the injectable hydrogel filler with a wonderful bacteria clearance ability.
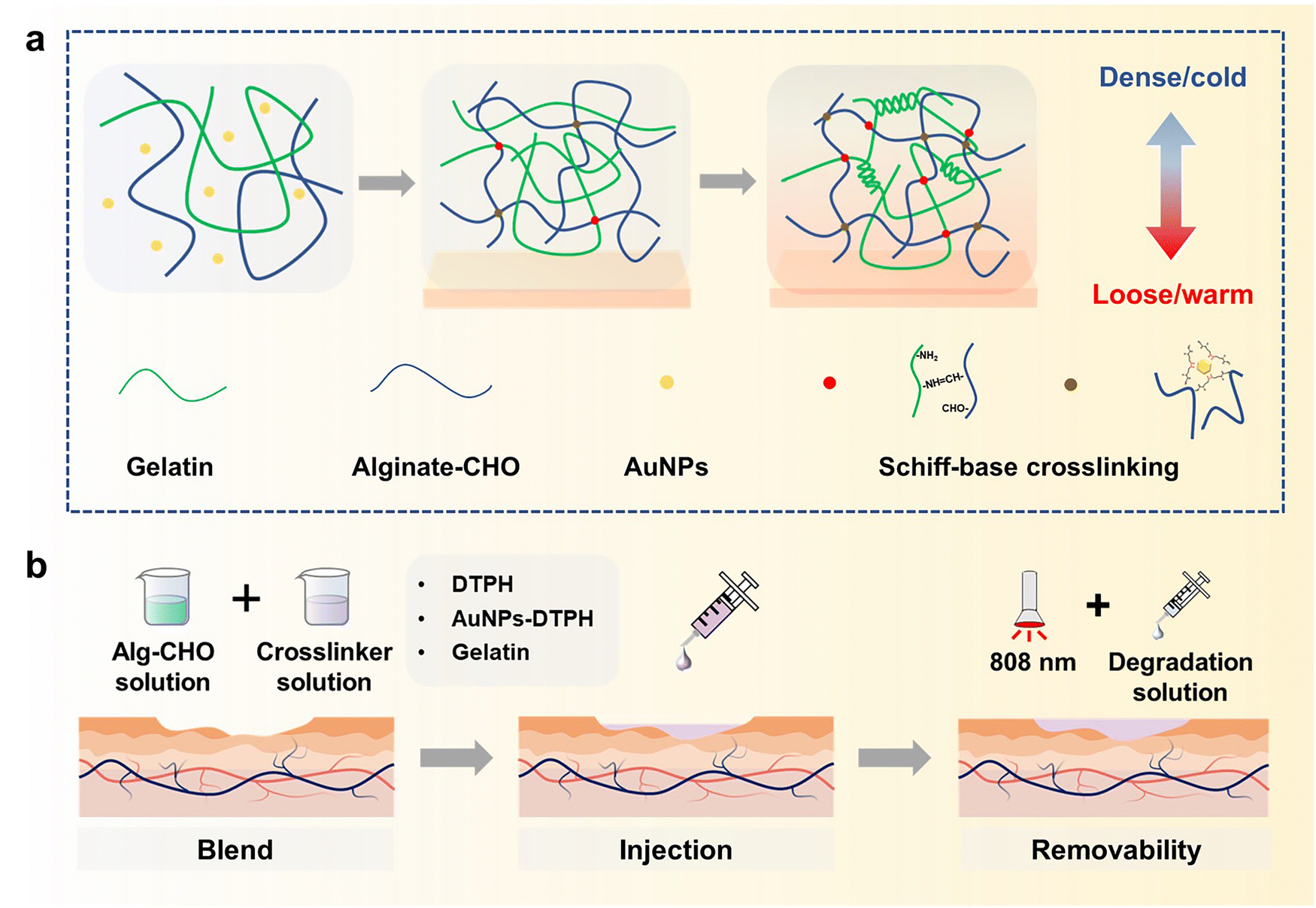 | ||
| Fig. 1 (a) Formation mechanism of injectable gradient adhesive hydrogels injected on skin tissue. (b) Application and removal process of injectable hydrogel on skin wound. | ||
2. Materials and methods
2.1. Materials
Chloroauric acid (HAuCl4) was purchased from Macklin reagent. 3,3′-dimethyl dithiodipropionate, hydrazine hydrate, sodium citrate solution, gelatin, sodium alginate (Alg), and sodium periodate (NaIO4) were obtained from Sigma-Aldrich. A CCK-8 cell proliferation and cytotoxicity assay kit (CA1210, Solarbio) and acridine orange/propidium iodide staining (HR0462, Biolabs) were used for the cytotoxicity assay. All the above chemicals and solvents, unless otherwise mentioned, were used without further purification.2.2. Preparation of Aurum nanoparticles
Monodisperse aurum nanoparticles (AuNPs) with an average diameter of 16 nm were prepared according to a previous report.40 First, HAuCl4 solution (0.3 mmol L−1, 0.102 g L−1) and sodium citrate solution (38.7 mmol L−1, 9.99 g L−1) were prepared. Then, 10 mL of HAuCl4 solution was heated under stirring until boiling in the flask, and 3 mL of sodium citrate solution was added. After 2 min, the mixed solution was bright red, indicating that the monodisperse AuNPs were formed. The particle morphology and size of the prepared AuNPs were measured using a Transmission Electron Microscope (TEM), and the mapping mode was used to determine the combination of DTPH and AuNPs.2.3. Preparation of 3,3′-dithiobis(propionic hydrazide)
First, 12 mL of dimethyl 3,3′-dimethyl dithiodipropionate and 6 mL of hydrazine hydrate were added sequentially to the methanol solvent and stirred for 12 h. Then, the reaction products were washed sequentially with water and methanol at least three times. The final filtration products were dried under vacuum conditions to obtain pure 3,3′-dithiobis(propionic hydrazide) (DTPH).2.4. Synthesis of aldehyde sodium alginate
5 g of sodium alginate (Alg) was dissolved in 25 mL of ethanol to form a suspension solution, and then a certain quality of NaIO4 (the molar ratio of NaIO4 to Alg is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) was dissolved in 25 mL of water. Then, the NaIO4 solution was added into the Alg suspension under stirring in the dark. After 4 h, ethylene glycol with the same molar amount as NaIO4 was added to terminate the reaction. The obtained mixed solution was poured into absolute ethanol to precipitate white precipitates under strong stirring. The mixture was filtered by suction, dissolved with deionized water, separated out with absolute ethanol, filtered by suction, and repeated 3 times. Finally, the white product was put into a glassware and dried under vacuum at 40 °C for 24 h to obtain aldehyde sodium alginate (Alg–CHO).
:5) was dissolved in 25 mL of water. Then, the NaIO4 solution was added into the Alg suspension under stirring in the dark. After 4 h, ethylene glycol with the same molar amount as NaIO4 was added to terminate the reaction. The obtained mixed solution was poured into absolute ethanol to precipitate white precipitates under strong stirring. The mixture was filtered by suction, dissolved with deionized water, separated out with absolute ethanol, filtered by suction, and repeated 3 times. Finally, the white product was put into a glassware and dried under vacuum at 40 °C for 24 h to obtain aldehyde sodium alginate (Alg–CHO).
The oxidation degree (OD) of Alg–CHO was determined using the hydroxylamine hydrochloride titration method. Briefly, 0.1 g of Alg–CHO was dissolved in 25 mL of hydroxylamine methyl orange hydrochloride solution. After being left for 12 h, titration was performed with a standard NaOH solution (10 mM). The amount of standard solution added each time, and the corresponding pH value were recorded. Then, the amount of all NaOH added at the end point of titration was measured and calculated using the potentiometric method, and then the OD of the oxidized sodium alginate prepared in this experiment was calculated using the following equation:
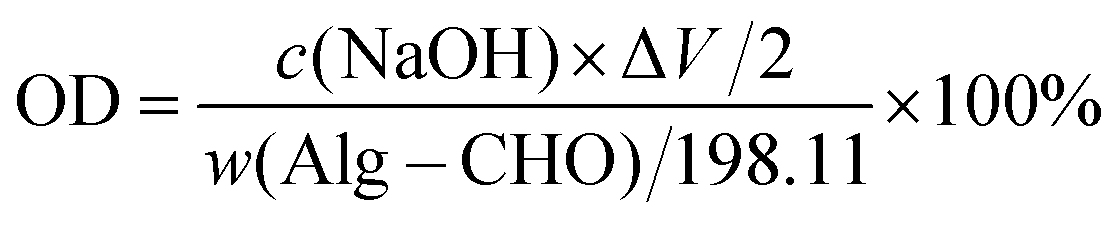 | (1) |
2.5. Preparation of injectable gradient asymmetric hydrogels
The obtained Alg–CHO was dissolved in deionized water to obtain the Alg–CHO solution (10% (w/v)). Next, 1.2 g of gelatin was dissolved in 10 mL deionized water at 50 °C. Then, different amounts of DTPH (15%, 25%, and 35% mass ratio to Alg–CHO) and AuNPs solution (10% volume ratio to gelatin solution) were added to the gelatin solution. An equal volume of Alg–CHO solution and gelatin/DTPH solution was mixed at 37 °C. Finally, the mixed solution was pumped into a syringe and injected into a specific mold to form injectable gradient asymmetric hydrogels. The prepared injectable gradient asymmetric hydrogels with different proportions were named AG, AD15, AD25, AD35, AGD15, AGD25, AGD35, and AGD15-Au, where A, G, and D represent Alg–CHO, gelatin, and DTPH, respectively. The lower corner marker refers to the amount of DTPH.The chemical structures of the prepared hydrogel were investigated using a Fourier Transform Infrared Spectrometer (iS50 FT-IR). Moreover, their morphology was investigated using a scanning electron microscope (SEM) after freeze drying and brittle fracture operations.
2.6. Swelling behaviours test
The swelling ratio of the prepared injectable gradient asymmetric hydrogels was determined through weight changing under a liquid environment. Different hydrogels were cut into about 8.5 mm in diameter and 3 mm in height. First, all hydrogel samples were immersed in 5 mL of PBS solution (pH = 7.4). The weight of the samples was tested after removing the surface liquid. The initial and weight at different times of injectable hydrogels were recorded as w0 and wt, respectively. The equilibrium swelling ratio (ESR) was calculated using the following equation: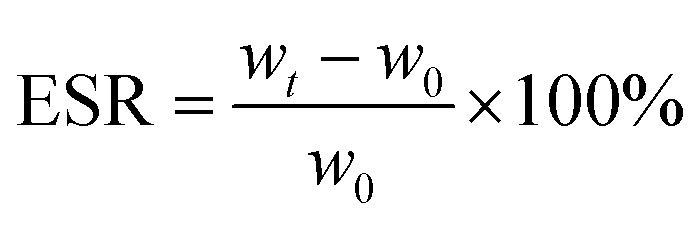 | (2) |
2.7. Mechanical property test
The rheological properties of the prepared injectable gradient asymmetric hydrogels were characterized by applying time scanning curves using a rotational rheometer. The pre-gel liquid of the hydrogels was kept for 3 min before being injected into the test table as preparation time. Then, the rheological properties of different injectable hydrogels were tested at 37 °C. The frequency was set to 1 Hz, and the strain was set to 1%. After mixing and injection, the gradient asymmetric hydrogel was tested for 3000 s to monitor its mechanical properties during the gradual and complete curing process. In addition, the change in rheological properties under light was tested. To explore the mechanism more obviously, the pre-prepared AD25-Au and AGD15-Au hydrogels were cut into a diameter of 2 cm and a thickness of 2 mm to perform the time scanning test at 25 °C, 37 °C, and 37 °C after 808 nm illumination (AD25-Au for 6 min and AGD15-Au for 10 min). The test conditions were consistent with the above measurement.The compressive properties of the prepared injectable gradient asymmetric hydrogels were also characterized by a universal testing machine equipped with a 500 N load cell. The hydrogels were prepared as cylindrical samples with a diameter of 8.5 mm and a height of about 6 mm. The compressive rate was set at 5 mm min−1.
2.8. Lap shear measurement
The lap shear measurement was performed using a universal testing machine equipped with a 500 N load cell. Glass (2.5 cm × 6 cm) was used as the substance of measurement by default. First, the glass was successively washed with water and ethanol. The AG, AGD and AGD-Au gradient asymmetric hydrogels were injected into one side of the glass surface with a 2.5 cm × 2.5 cm contact area and kept at 37 °C at different times (between 0 min and 10 min). After the crosslinking process, the other side substrate was covered to the upper side of the injectable hydrogel to test the bonding strength for non-adhesive side adhesion. The test samples were kept at 37 °C for 1 h to completely cure. The lap shear strength of the gradient asymmetric hydrogel on the glass was tested at a stretching rate of 20 mm min−1. In addition to stainless steel, aluminum and pigskin were also used as substrates to evaluate the adhesive capacity of the prepared injectable gradient asymmetric hydrogel using the same method.2.9. Photothermal property test
The photothermal property of the prepared injectable gradient asymmetric hydrogel was performed at body temperature (37 °C), and the test samples were set as AGD15, AGD15-Au, AD25, and AD25-Au groups. These hydrogels were cut into about 8.5 mm in diameter and 5 mm in height. Then, the temperature of the preheated samples was exposed to near-infrared light irradiation at 808 nm, the near-infrared light power was set as 2 W, and the distance between the light source and the sample was about 10 cm. At different times (every minute below 10 min, and every 5 min below 30 min), the temperature of the hydrogel was also measured using an infrared measuring instrument, and infrared images were obtained, which corresponded to the optical pictures.2.10. Anti-bacterial adhesion test
Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) were used to investigate the anti-bacterial adhesion of the prepared gradient asymmetric hydrogel. First, the prepared hydrogel was in situ formed in a 48-well plate. Then, 200 μL of bacterium solution (1 × 107) was added to the hydrogel surface and cultured at 37 °C for 2 h in a shaker. After the bacterium solution was removed, the specimens were washed with PBS solution, and a certain amount of SDS solution (1% (wt/v) with PBS) was added into each well. After 1 min ultrasound, 100 μL of solution was drawn into the culture dishes and cultured at 37 °C for 12 h. Then, the colony count was measured to calculate the bacterial clearance ratio. The washed PBS solution can also be dyed through the bacteria Live/Dead straining kit by adding 100 dye solution. Furthermore, 10 μL of liquid was added to the slide and observed using a fluorescent microscope.2.11. Hemocompatibility test
The blood compatibility of the injectable gradient asymmetric hydrogel was verified by performing a hemolysis test. First, the rabbit plasma (sodium citrate) was centrifugated at 2000 rpm. Then, the supernatant was removed, and the precipitate was resuspended with PBS solution. The above steps were repeated at least 5 times to obtain red blood cells, which were diluted to a concentration of 10% (v/v). 0.5 g of the hydrogels were added to 5 mL of PBS solution and incubated at 37 °C for 24 h to obtain the hydrogel extract. The extract was mixed with the prepared red blood cell solution and shaken at 37 °C for 4 h. The mixed solution was centrifuged at 2000 rpm for 10 min to collect the supernatant. The absorbance was measured at 545 nm using an orifice plate, and the hemolysis rate was calculated using the following equation: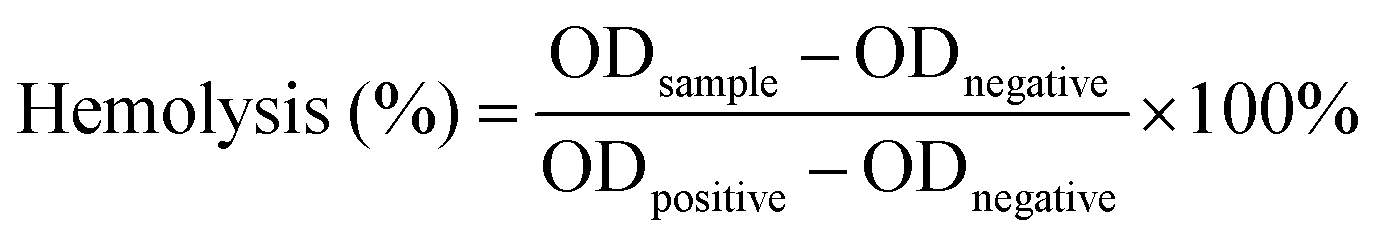 | (3) |
2.12. Cytotoxicity assay
The cytotoxicity of the injectable gradient asymmetric hydrogel was assessed by CCK-8 assay (CCK-8 cell proliferation and cytotoxicity assay) using L929 cells. An amount of AGD15 and AGD15-Au hydrogel was immersed in a cell culture medium for 24 h to prepare the extract (0.05 g mL−1). Then, L929 cells were seeded in 96-well plates (1 × 104 per well) and cultured for 12 h. After being washed with PBS solution, the culture medium was replaced with hydrogel extract and cultured for another 24 h. Additionally, L929 cells were cultured in a normal medium as a control. The dye solution was configured according to the instructions of the CCK-8 kit, and the appropriate amount was added to each well at a 24 h time. After being cultured at 37 °C for 2 h, the absorbance of each well was measured at 450 nm using a microplate reader. The cell viability was also calculated according to the kit instruction.In addition, the cell viability was determined by acridine orange (AO)/propidium iodide (PI) staining. First, the L929 cells were incubated in 96-well plates (2 × 104 per well) for 12 h to achieve the attachment and continued to cultivate for 24 h using hydrogel extract as the cell culture medium. The dye solution was configured according to the AO/PI kit instruction, and then 100 μL dye solution was added to each well to observe the cell survival status for 24 h after washing with PBS solution. After culturing at 37 °C for 20 min, the cell morphology and viability were detected through fluorescence observation after removal of the dye solution and washing with PBS solution. The morphology of the cells was also observed through the images of bright fields.
2.13. Evaluation of the wound healing effect in vivo
To evaluate the effect of the prepared gradient asymmetric hydrogel on wound repair, we established a large full skin defect model in mice. All animal studies were approved by the Animal Ethics Committee of the North China University of Science and Technology (Tangshan, China), and the animal experiments were performed at the North China University of Science and Technology (Approval No. LAEC-NCST-2020177, Tangshan, China). Briefly, the mice were anesthetized by intraperitoneal injection of chloral hydrate. Then, the back was shaved, and a full skin circular wound of about 20 mm in diameter was performed. The ADG15-Au pre-gel (about 0.5 mL) was injected directly to cover the wound surface. After several minutes, the hydrogel was formed on the wound surfaces. The wounds were observed at different times (0 day, 3 days, 7 days, and 10 days), and optical pictures were taken for preliminary quantitative statistics of wound healing. The relative wound percentage (%) was calculated using eqn (4), where “R” is an abbreviation for relative wound area and the lower corner marker “t” represents the date, i.e., 0, 3, 7 and 10 days: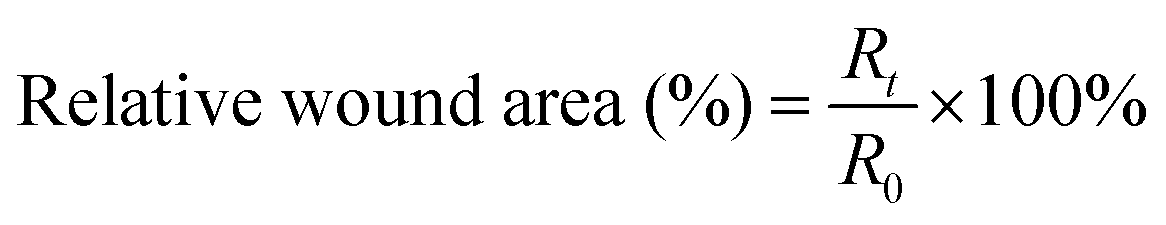 | (4) |
2.14. Histological analysis
Skin tissues from the back of the mice were taken at specific time points and fixed with 4% paraformaldehyde solution. The fixed tissue subsequently underwent an embedding and sectioning process, followed by Hematoxylin and Eosin (H&E) staining (nucleus and cytoplasm of cells) and Masson Trichrome (MT) staining (collagen and muscle fibers) for follow-up analysis. Immunofluorescence staining was similarly tested to determine the status of wound healing in the late stage. Specific markers of α-SMA and CD34 were used to determine the status of neovascularization and endothelial cells.2.15. Statistical analysis
All experiments had at least three parallel samples (n ≥ 3), and the mean with error was calculated. Differences were considered significant at p < 0.05. Microsoft excel was used for the statistical analysis.3. Results and discussion
3.1. Formation of an injectable gradient asymmetric hydrogel
To fabricate the injectable gradient asymmetric hydrogels, DTPH was synthesized via a nucleophilic addition reaction (Fig. S1a, ESI†). The 1H NMR (500 MHz, D2O, δ) (Fig. S1c, ESI†) demonstrated the characteristic chemical shift of DTPH at 2.83–2.90 (t, 2H; CH2; H1) and 2.50–2.57 (t, 2H; CH2; H2). The coordination interactions between DTPH and AuNPs were one of the main driving forces for the formation and removal of the hydrogel. As shown in Fig. 2a, the DTPH-Au coordination interaction could achieve reversible conversion under 808 nm NIR irradiation. TEM images discovered that the prepared AuNPs had an average of 15–25 nm particle size. The mapping images of the Au and S elements almost overlapped in the DTPH-AuNP solution, implying that DTPH was adsorbed on the surface of the AuNPs (Fig. 2a).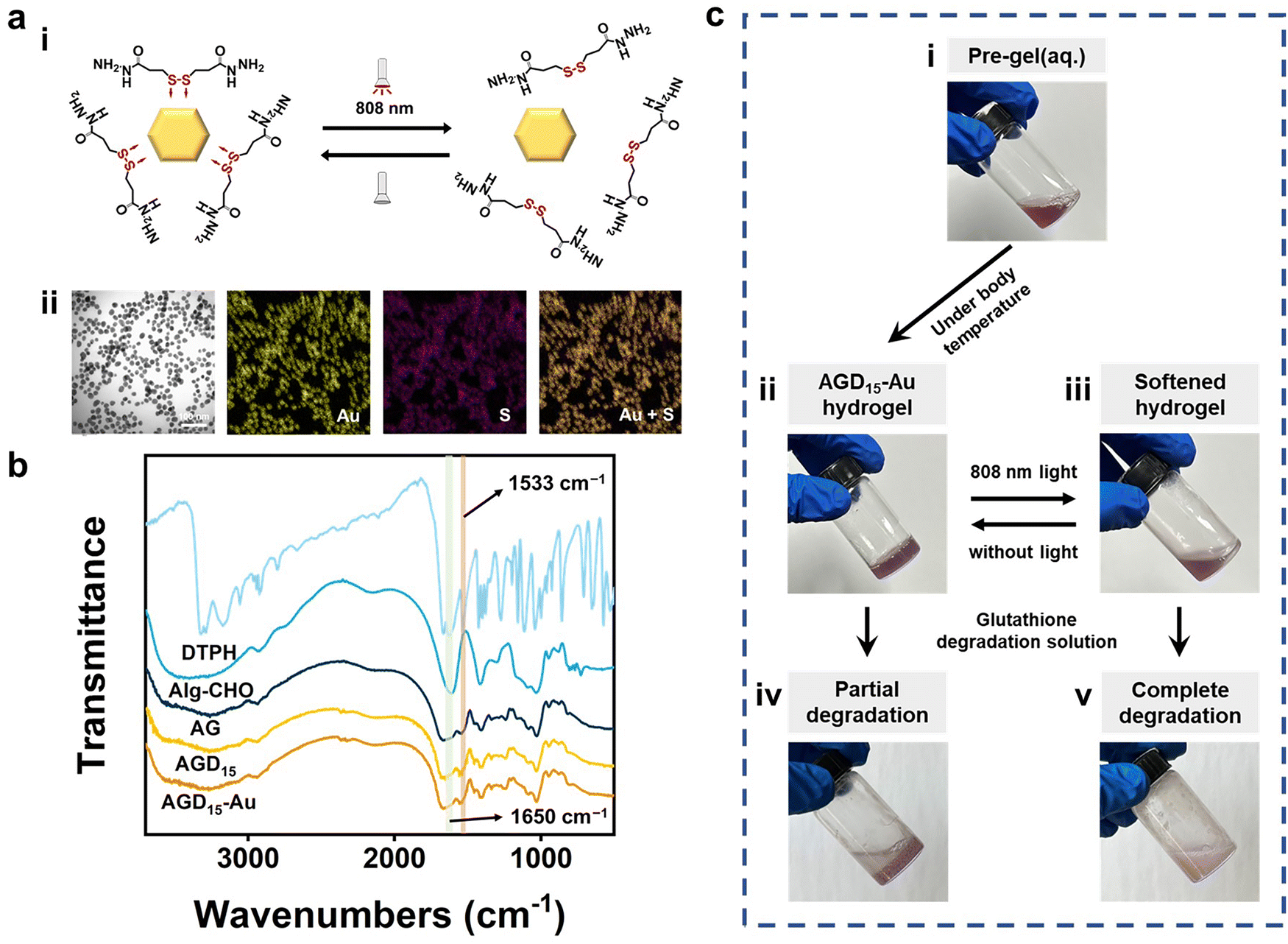 | ||
| Fig. 2 (a) (i) Reversible interaction of DTPH-Au coordination under 808 nm near-infrared light irradiation. (ii) The transmission electron microscopy and mapping images of DTPH-AuNPs. (b) FT-IR spectroscopy of DTPH, oxidated sodium alginate, AG, AGD15, and AGD15-Au hydrogels. (c) Formation and phased-transition process of injectable gradient asymmetric hydrogel (AGD15-Au). | ||
To prepare the injectable gradient asymmetric hydrogel, the oxidated sodium alginate (Alg–CHO) was first synthesized with a 24.76% oxidation degree using the sodium periodate method (Fig. S2, ESI†).44 Compared with sodium alginate, new chemical shifts at 3.97–5.20 ppm (m, 2H; CH; peaks 1 and 2) were observed in Alg–CHO (Fig. S1d, ESI†). In addition, the chemical shifts at 5.4 and 5.6 indicated a partially formed hemiacetal structure. The pre-gel solution consisting of an equal volume of Alg–CHO solution and crosslinker solution was mixed homogeneously and then injected into the mold. After 3 min, the mixed solution was gradually transformed to gradient asymmetric Alg/gelatin/DTPH-Au hydrogels without any external stimuli. The injectable hydrogel with different components was prepared by adjusting the gelatin/AuNPs/DTPH ratio. The obtained injectable gradient asymmetric hydrogels were named AG, ADx, AGDx, and AGDx-Au, where A, G, and D represent Alg–CHO, gelatin, and DTPH, respectively. The lower corner marker “x” represents the mass percentage of DTPH to Alg–CHO.
The formation of injectable gradient asymmetric hydrogels involves gradient crosslinking interactions (Fig. 1): (i) the Schiff-base reaction between Alg–CHO and gelatin, (ii) the formation of gelatin trihelix structure via the hydrogen bond, (iii) the combination of DTPH-Au coordination interaction between disulfide bonds and AuNPs, and (iv) the Schiff-base bond between DTPH-Au/DTPH aggregate and Alg–CHO.17,40 For different crosslink interactions, the crosslinking time also have differences. The amino groups of the gelatin chain could rapidly react with the aldehyde groups on Alg–CHO to format the Schiff base crosslinked Alg/Gel network. Similarly, DTPH can act as a crosslinker to induce the formation of the Schiff-base crosslinked Alg-CHO network. The AuNPs enhanced crosslinking interaction via the coordination interactions between the disulfide bonds in DTPH and AuNPs. Ultraviolet (UV) absorbance tests (Fig. S3, ESI†) were also used to validate DTPH-Au interactions. The absorption peak of Au was red-shifted from 528 to 758 nm with the introduction of DTPH.40 Meanwhile, the formation of a trihelix structure among gelatin was associated with temperature. At physiological temperatures, the gelatin chains moved freely, and the network structure was relatively loose, while the compact network structure was formed at room temperature owing to the formation of a trihelix structure. Hence, when the pre-gel solution was injected into the wound surface, the gradient crosslink structure was gradually formed with time owing to the different crosslinking mechanisms (Fig. 1a). And only a chemical crosslinked structure can be formed on the skin surface. Subsequently, a physical crosslinked structure of the gelatin triple helix was formed with the increases in thickness owing to the decrease in temperature.
Owing to the photothermal effect of AuNPs, the Au-disulfide bond dissociated when exposed to NIR irradiation at 808 nm. Moreover, the triple helix structure of gelatin dissociated owing to the increase in temperature, leading to the relaxation of the network structure of the hydrogel. Meanwhile, the prepared hydrogel exhibited different adhesive behaviors owing to the different crosslinking times. It can adhere to the skin surface via hydrogen bonds and Schiff-base interaction, while the adhesive capacity of its top surface was low due to the consumption of crosslinking reaction between the aldehyde and amino groups. Therefore, during the healing process of a large area wound, the injectable gradient asymmetric hydrogels could act as natural human skin to protect damaged skin. Simultaneously, the cleavage of partial crosslinking interaction under the photothermal effect provided the possibility of removing the hydrogel and avoiding secondary damage to the defected tissue (Fig. 1b).
Fourier transform infrared spectroscopy (FTIR) was further used to investigate the interactions in the injectable gradient asymmetric hydrogels. As shown in Fig. 2b, the DTPH had an absorption band at 1533 cm−1, which belonged to the –N–H bond bending vibration of the primary amino group (−NH2). Only weak amino absorption peaks remained in the formed hydrogel, which laterally confirmed the occurrence of amino effect depletion. The characteristic peak at 1650 cm−1 appearing in AGD15 and AGD15-Au hydrogel groups was attributed to the newly formed Schiff-base bond from the amine groups of DTPH/gelatin and the aldehyde groups of Alg–CHO. To elucidate the formation and distribution of DTPH-Au, mapping images of C/N/Au/S elements were investigated. Fig. S4 (ESI†) showed that the constituent elements were uniformly distributed in the pore walls, implying that the crosslinking of different species achieved a stable crosslinked structure for hydrogel formation. Moreover, the phase transition behaviors of the prepared hydrogel were depicted in Fig. 2c. AGD15-Au could maintain a stable hydrogel state at 37 °C (Fig. 2c(ii)), and the AGD15-Au hydrogel could become a soft flowing state (Fig. 2c(iii)) under NIR radiation. In the presence of the degradation solution (glutathione solution), the AGD15-Au hydrogel can be completely transformed into a flowable liquid after a few seconds under stirring (Fig. 2c(v)), while it can only be partially degraded in the absence of softening. The phase inversion process of hydrogel in solution with different glutathione concentrations was shown in Fig. S5 (ESI†), implying that disulfide bonds played an important connecting role in the internal crosslinking network of injectable hydrogels, and the breakage of disulfide bonds by sulfhydryl exchange can lead to the complete degradation of hydrogels. Thus, it can be concluded from this trend that the time required for complete degradation can be reduced with an increase in the concentration of the degradation solution or the use of other oxidant solutions, such as hydrogen peroxide solution,45 which could break disulfide bonds through oxidation interaction, thereby enabling a rapid removal effect.
3.2. Time-induced mechanical compatibility
The rheological behaviors and compressive strength of the injectable gradient asymmetric hydrogels were investigated to verify the compatibility of the hydrogels with skin tissue. As shown in Fig. 3a, the compressive strength of the hydrogel decreased with an increase in DTPH. The compressive strengths of the AGD15, AGD25, and AGD35 hydrogels were 123 kPa, 95.4 kPa, and 45.9 kPa, respectively (Fig. 3b), which was mainly attributed to that the AGD15 group had the most appropriate aldehyde group to amino group ratio. Consequently, the excessive DTPH of AGD25 and AGD35 resulted in more monodentate crosslinking, which corresponded to lower compressive strength. As expected, the introduction of AuNPs obviously improved the mechanical strength, and the compressive strengths of hydrogel were largely improved to 465 kPa (AGD15-Au), 354 kPa (AGD25-Au), and 378 kPa (AGD35-Au). The AuNPs can act as crosslinking sites to enhance the density of the polymer networks via the coordination interactions between disulfide and AuNPs. Owing to the multiscale crosslinking, a difference in microstructure can also be observed when the hydrogels contain different amounts of crosslinkers. As shown in Fig. S6 (ESI†), the scanning electron microscope (SEM) images of AGD15, AGD25, and AGD35 hydrogel had the structure of coexisting large and small pores, which originated from the Schiff-base crosslinking of gelatin/Alg, DTPH-Au/Alg and DTPH/Alg. The small pores of AGD15, AGD25, and AGD35 also had a gradual tendency of increasement for the changing dosage of DTPH. Notably, the AGD15-Au hydrogel demonstrated both denser small pore and large pore structures compared to the AGD hydrogels without AuNPs. The incorporation of AuNPs formed DTPH-Au conjugates, the aggregate crosslinking agents, to further tighten the pores. Besides, the swelling ratio implied a similar phenomenon (Fig. S7, ESI†). The swelling rate of all hydrogels increased continuously with time until equilibrium. It can be observed that the swelling rate of AGD15, AGD25 and AGD35 gradually increased with the increase in DTPH content, and the AGD35 hydrogel started to degrade slightly after 2 days. This further confirmed that the crosslinking ratio of AGD15 was more suitable. The AGD15-Au group showed a slight increase in the dissolution rate relative to the AGD15 group. It may be inferred that the adsorption of Au on DTPH led to an increase in the local crosslinking degree, while the remaining crosslinking degree slightly decreased, which was consistent with the SEM image results.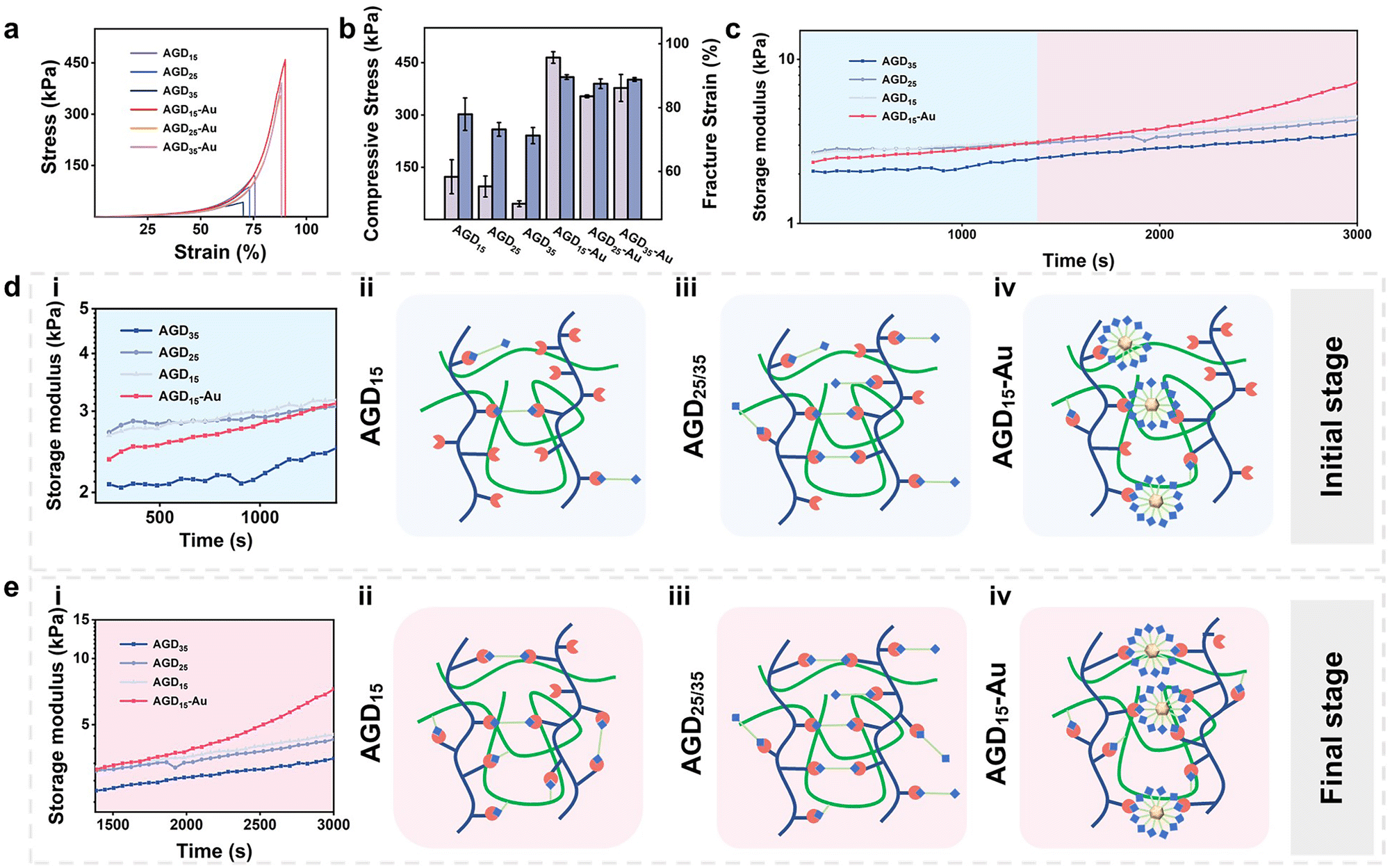 | ||
| Fig. 3 (a) Compressive curves, (b) stress and fracture strain of AGD/AGD-Au hydrogels (AGD15/AGD25/AGD35/AGD15-Au/AGD25-Au/AGD35-Au), (c) the storage modulus of the prepare hydrogel vs. the time (AGD15-Au/AGD25-Au/AGD35-Au), and (d) rheological property at the initial and final stages and (ii)–(iv) gelation mechanism of AGD-Au hydrogels. | ||
A rheological test was also conducted to examine the mechanical properties and time-induced multiscale crosslinking of the injectable gradient hydrogels (Fig. 3c). It was noteworthy that the storage modulus (G′) of AGD35 hydrogel was lowest during the whole test time, which was due to the large amount of monodentate crosslinking caused by the excessive DTPH (Fig. 3d(iii)). Consequently, there were insufficient aldehyde sites to form bidentate crosslinking (Fig. 3e(iii)). Accordingly, the AGD15 hydrogel could achieve bidentate crosslinking during the curing progress (Fig. 3e(ii)) and had a higher G′. Surprisingly, the introduction of AuNPs did not enhance the mechanical strength of the AGD hydrogel during the initial period. For example, the G′ of AGD15-Au hydrogel was lower than that of AGD15 and AGD25 hydrogels within 20 min because the DTPH-Au aggregate was hard to achieve complete crosslinking in a short time (Fig. 3d(iv)), while the DTPH could quickly achieve single tooth crosslinking or double tooth crosslinking (Fig. 3d(ii)). DTPH adsorbed on the surface of AuNPs enabled them to have more Schiff base cross-linking sites. However, although the DTPH-Au aggregate crosslinker had more amino groups on the AuNPs surface, as shown in the diagram, the time taken to form a multi-tooth coordination also became significantly longer. Hence, the mechanical properties of AGD15-Au were relatively low at the initial stage (Fig. 3d(i)), making it easier to adapt to the wound shape. The G′ of all hydrogels also increased as time increased owing to the crosslinking process. In particular, the G′ of the AGD15-Au hydrogel increased significantly after 1380 s to the extent that the modulus was higher than the other groups. During this period, complete crosslinking continued to form, including a Schiff-base bond between DTPH-Au aggregate and Alg–CHO (Fig. 3e(iv)). Finally, despite the overall increase in the G′ of all hydrogels, the AGD15-Au hydrogel showed a greater increase in G′ than the other hydrogel groups (Fig. 3e). All the injectable gradient asymmetric hydrogels had the appropriate mechanical performance to act as artificial skin for wound filling or external foreign matter resistance.
The rheological behaviors of injectable gradient asymmetric hydrogels without gelatin were also investigated to verify the above results (Fig. S8, ESI†). The G′ of AD15, AD25, AD35, and AD45 increased and then decreased with an increase in DTPH content, proving the most suitable composition of AD25. After the addition of AuNPs, the adsorption of AuNPs to DTPH led to a concentration of crosslinking points and a slight decrease in their strength. Thus, in combination with the above experimental results, the AD25-Au and AGD15-Au hydrogels were used to investigate the mechanism related to the injectable hydrogels.
3.3. Asymmetric adhesion property and mechanism
For wound filling applications, the injectable hydrogels need predominant stable and adhesive performance with the wound surface. However, the strong adhesion ability of normal double-sided adhesive hydrogels was often accompanied by operational difficulties or unpredictable side effects. To resist external foreign matters or convenient operation, asymmetric adhesion performance was urgently needed in the biomedical field. Here, the gradient crosslinking approach was used to achieve asymmetric adhesion. During the mixing process of AGD15-Au pre-gel solution, the Schiff-base bond was first formed between Alg–CHO and gelatin, and the DTPH or DTPH-Au formed monodentate and partial multi-site crosslinking with Alg–CHO. In this stage, the AGD15-Au system presented a flowing state and could be easily injected into the large area wound to fill or adapt to the skin defect. After being injected into the wound site, the Schiff-base reaction continuously occurred between the unilateral crosslinked DTPH and the aldehyde groups of Alg–CHO, which resulted in complete small molecule crosslinking. The multi-scale crosslinking inferred excellent injectability (Fig. S9, ESI†). The cured hydrogel could stably combine with the damaged skin depending on its self-fixation and partial interactions with skin tissue. The partially gel-forming state also allowed the AGD-Au hydrogel to absorb fluid from the wound for stable binding. Although the hydrogel was immobilized at the wound surface, the crosslinking process was still in progress. The complete crosslinking of the DTPH-Au aggregate crosslinker and the gradient modulus change caused by the temperature sensitivity of gelatin finally achieved the asymmetry of the AGD-Au hydrogel. Because of the partial combination of hydrogel and skin tissue, slow crosslinking between DTPH-Au and Alg–CHO mostly occurred in the upper zone of the hydrogel. Taking advantage of the different gelatin conformations at room temperature and physiological temperature, the gelatin in the AGD-Au hydrogel formed a gradient triple helix structure. Together with gradient crosslinking and more intensive hydrogen bonding between gelatins, the hydrogel network in the upper region was tighter internally, while the polymer chains in the lower region can move more freely. Therefore, the final AGD-Au hydrogel had a gradient structure induced by multi-scale crosslinking, which further led to gradient performance.In order to confirm the asymmetric adhesion property of the AGD-Au hydrogel, a single-side lap shear adhesive test on glass was operated using a universal testing machine (Fig. 4a). To eliminate the influence of gelatin temperature sensitivity and explore only the effect of time-induced multi-scale crosslinking, this experiment was conducted at 37 °C. The adhesive strengths of the injectable hydrogels were shown in Fig. 4b. Compared with the AGD hydrogel, all ADx hydrogels had a lower adhesive strength. The lab shear strengths of AD15/25/35 were 26.2 kPa, 32.5 kPa and 12.4 kPa, respectively. For comparative analysis, the bonding strength of AGDx hydrogels showed a trend of increasing first and then decreasing with an increase in DTPH contents. The AGD15 hydrogel showed the highest adhesive strength (67.0 kPa), and the adhesive strength of AGD35 hydrogel decreased to 37.8 kPa. Notably, the introduction of AuNPs increased the adhesive strength of the hydrogel. The lab shear strengths of AGD15-Au, AGD25-Au, and AGD35-Au hydrogels were 85.0 kPa, 55.5 kPa, and 39.3 kPa, respectively. The injectable gradient hydrogel could withstand water flushing for 1 min (Fig. 4d(i)) as a consequence of its high bonding ability. Considering the above-mentioned prominent mechanical compatibility, the hydrogel could also experience twisting and bending processes without falling off (Fig. 4d(ii)). It can also be combined with different substrates stably at room temperature (Fig. 4d(iii) and Fig. S10, ESI†). These results indicated that the AGD-Au-based injectable hydrogel could adhere to the wound surface at the initial stage. However, the adhesive strength of the upper surface of the hydrogel obviously decreased after hydrogel formation. After 10 min, the adhesive strength of the AG, AGD15, AD15, and AGD15-Au hydrogels decreased to 32.1 kPa, 10.4 kPa, 9.27 kPa, and 20.5 kPa, respectively (Fig. 4c). In addition, considering the formation of the triple helix structure of gelatin, its upper layer viscosity almost achieved zero at room temperature and further decreased the adhesive strength. Thus, the prepared injectable AGD-Au-based hydrogel showed gradient asymmetric adhesion performance owing to the time-induced and multiscale crosslinking network.
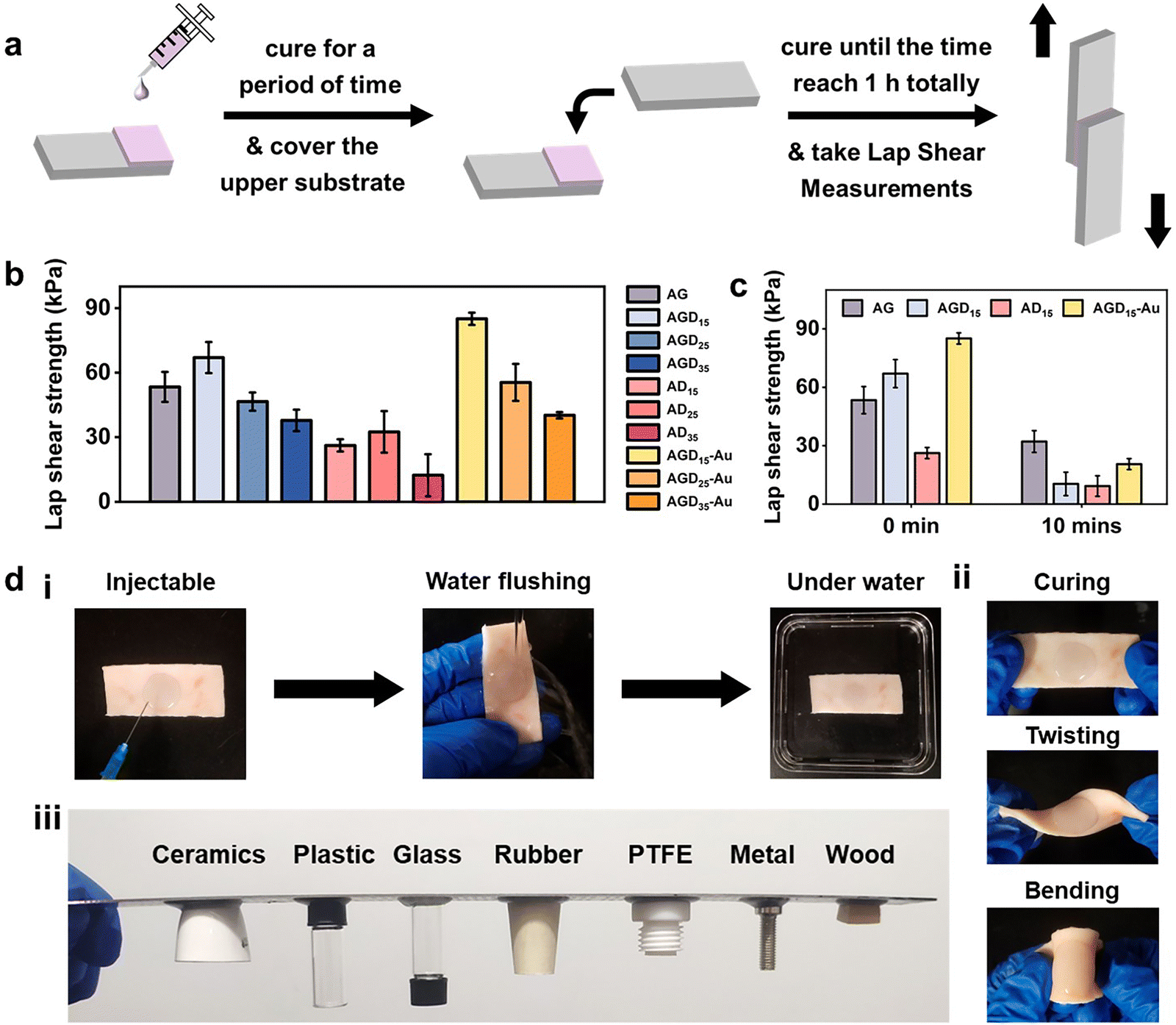 | ||
| Fig. 4 (a) Schematic diagram of unilateral adhesion performance characterization. (b) Lap shear adhesive strength of injectable asymmetric hydrogels (AG/AGG/AD/AGD-Au). (c) Unilateral lap shear adhesive strength of injectable asymmetric hydrogels (AG/AGD15/AD15/AGD15-Au) with different curing times. (d)(i) and (ii) Stability, and (iii) adhesive ability of AGD15-Au hydrogel. | ||
3.4. Phased removal behaviors and bacterial clearance ability
Both the replaceability and removal characteristics of the hydrogel are challenges facing the biomedical hydrogel dressing field. After a period of wound healing, the hydrogel may experience varying degrees of contamination or degradation. Under this condition, the hydrogel filler needs to be complemented or further removed (Fig. 5a). To solve this problem, various hydrogel structures have been proposed. The design of a cleavable crosslinking agent is one of the most popular approaches. In this study, we took advantage of multi-scale crosslinkers to achieve phased removal capability. The DTPH-Au coordination interaction and trihelix structure of gelatin can be reversed via the photothermal efficiency of the AuNPs and the cleaved solution of disulfide bonds (Fig. 1b).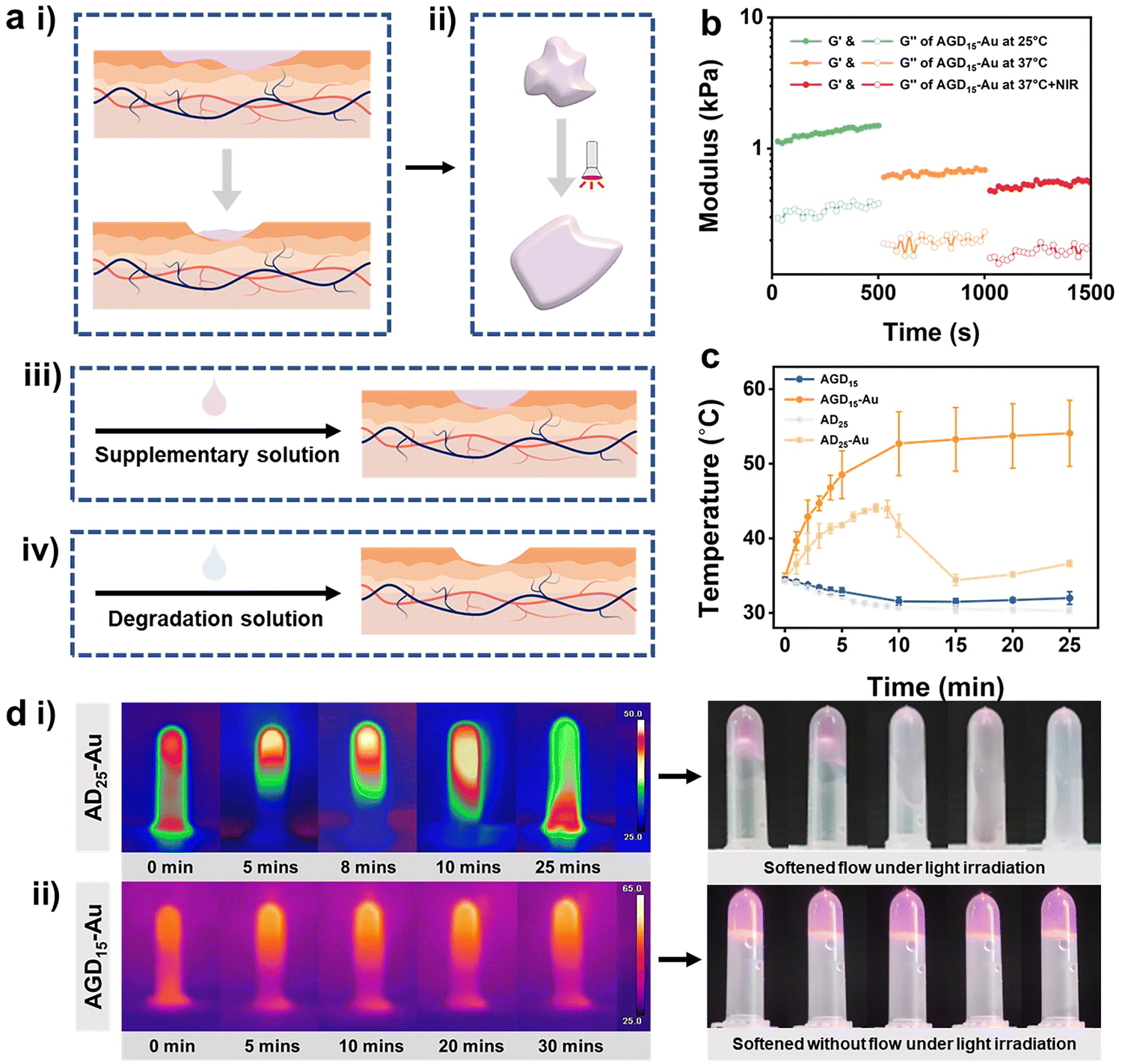 | ||
| Fig. 5 (a) Mechanism of phased removal ability of injectable hydrogels, (i) natural dressing attrition, (ii) near-infrared illumination softening, addition of (iii) supplementary solution and (iv) degradation solution. (b) Controlled softening process of AGD15-Au hydrogel at 25 °C, 37 °C and 37 °C under light exposure. (c) Temperature journeys of the AGD15, AGD15-Au, AD25, and AD25-Au hydrogels over time (d) infrared and optical images of near-infrared illumination softening of (i) AD25-Au and (ii) AGD15-Au groups. | ||
To validate the de-association of DTPH-Au coordination interaction, the rheological behavior of the AGD15-Au and AD25-Au hydrogel was investigated after NIR irradiation. Fig. 5b showed the variation of the rheological properties of AGD15-Au at 25 °C, 37 °C and 37 °C under light exposure. Both G′ and loss modulus (G′′) at body temperature decreased significantly relative to G′ and G′′ at room temperature. After further NIR irradiation, the G′ and G′′ decreased again, verifying the unraveling of the gelatin triple helix structure and the DTPH-Au interaction, respectively. However, the G′ value was still higher than the G′′ value, suggesting that the hydrogel was still gel stated and could softened owing to the photothermal effects. To observe the change in DTPH-Au interaction more obviously, rheological behaviors of AD25-Au were performed before and after light irradiation (Fig. S11, ESI†). Owing to the decrease in the overall crosslinking degree, a significant decrease in the energy storage modulus after light exposure was observed. In contrast, the modulus gradually increased over time after the withdrawal of light, indicating a slow recovery of internal interactions.
The photothermal behavior of the prepared AGD/AD-based hydrogel was investigated under NIR irradiation at 808 nm. Fig. 5c showed the temperature journeys of the AGD- and AD-based hydrogels over time. AGD15 and AD25 hydrogels maintained a constant temperature of 31–35 °C after 25 minutes with no variations. As expected, the introduction of AuNPs can obviously increase the temperature of the prepared hydrogel. Under 808 nm irradiation, AD25-Au hydrogel exhibited an obvious phenomenon of phase transition, and it gradually softened and became a flowable sol (Fig. 5d(i)). The time-temperature curve demonstrated a trend of rapid warming. The temperature of the AD25-Au hydrogel initially increased to 44.1 °C, then began to rapidly decrease at 8 min and decreased to 34.4 °C at 15 min (Fig. 5d(i)). This was mainly because it was difficult to keep AD25-Au in its stable form under NIR, which led to flow and erroneous temperature readings. Conversely, because of the presence of gelatin, AGD15-Au only softened without macroscopic flow when it rose rapidly to 52.7 °C within 10 min, and the maximum temperature value was greater than that of AD25-Au (Fig. 5d(ii)). After the extra addition of gelatin, the free movement of the Alg–CHO chain in the hydrogel network was restrained even after NIR irradiation, while the ADG15-Au system could still maintain the hydrogel state instead of the loosened network, which was conducive to combining with the newly added supplementary liquid or absorbing the degradation liquid for subsequent removal (Fig. 5a(iii) and (iv)).
The softening state also facilitated the removal of bacteria by rinsing with PBS, as some of the surface hydrogels could be shed, allowing the surface bacteria to be cleared. To verify the ability of bacterial clearance, the anti-bacterial adhesion behaviors of AGD15-Au hydrogel were investigated with/without NIR irradiation (Fig. 6a). It was observed that the control group had a higher number of bacteria colonies, representing high bacterial adsorption. The AGD15-Au hydrogel had a prominent anti-bacterial adhesion effect for S. aureus and E. coli owing to the action of AuNPs in the hydrogel and the presence of a large number of hydrophilic groups. When NIR light irradiation was applied, the hydrogel softened as a whole due to the unraveling of the internal DTPH-AuNPs interaction. The part of the outer layer adsorbed with bacteria can be partially shed, thus achieving a favorable bacterial removal effect. The temperature increase after light illumination also had a killing effect on the bacteria.43,46 The colony count results of different groups were shown in Fig. 6b(i). Compared with the control group, the AGD15-Au hydrogel presented outstanding anti-adhesive properties against bacteria, and the AGD15-Au group with NIR light irradiation possessed better bacteria clearance ability for both E. coli and S. aureus (more than 98%, Fig. 6b(ii)). Live/Dead staining also showed similar results (Fig. 6c) compared with the control group. Only a small number of bacteria were detected in both the AGD15-Au hydrogel and the AGD15-Au + NIR group, and some of the bacteria were killed. Based on these findings, the AGD-Au hydrogels were partially softened by NIR irradiation, allowing them to be partially exfoliated by PBS or other solutions and any bacteria or contamination attached to them to be removed.
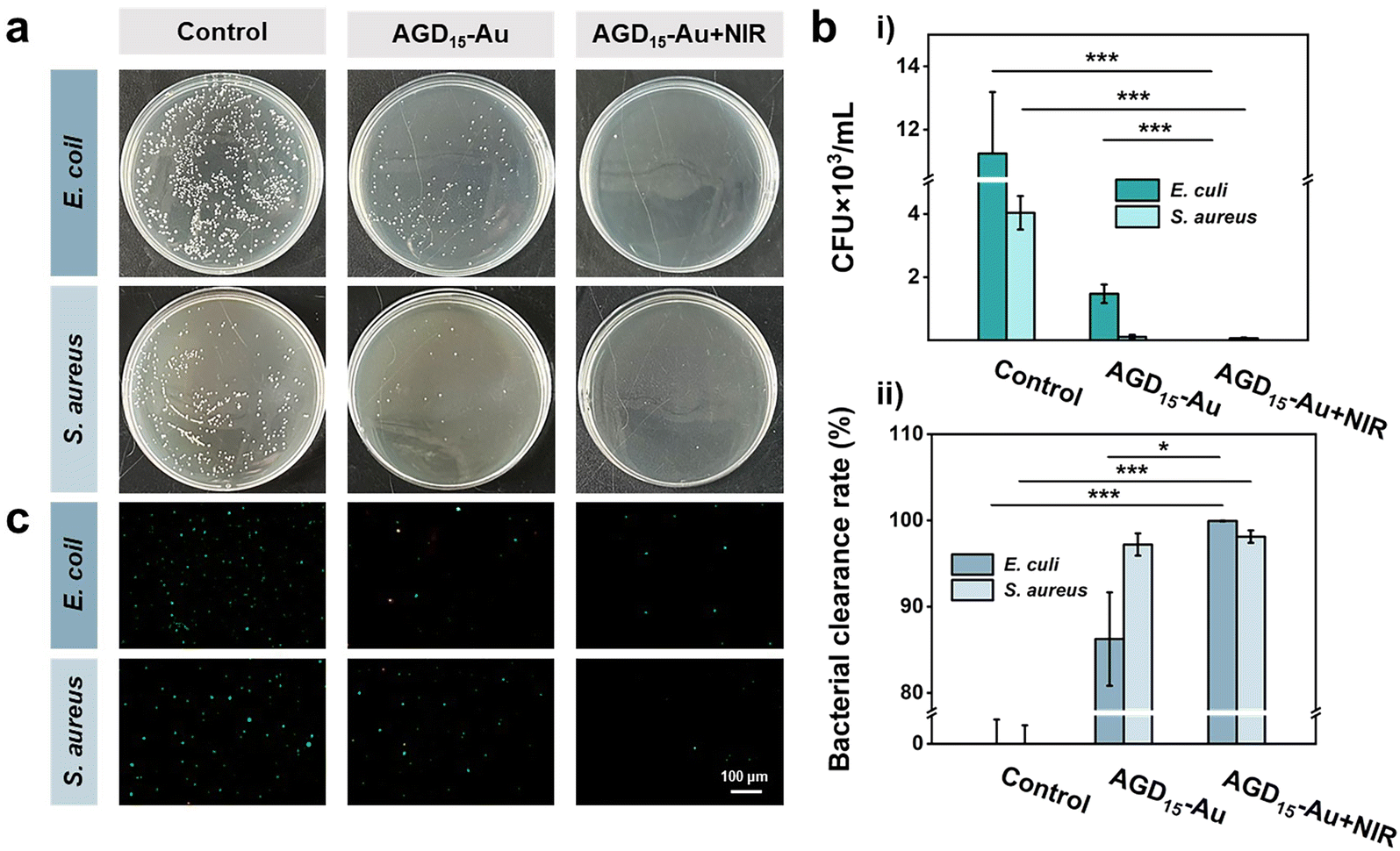 | ||
| Fig. 6 (a) Anti-bacterial adhesion property of AGD15-Au hydrogel with and without near-infrared illumination. (b) Anti-bacterial adhesion test results of (i) colony count results and (ii) bacterial clearance ratio (*, p < 0.05, **, p < 0.01, ***, and p < 0.001). (c) Bacterial clearance fluorescence images of different groups (control/AGD15-Au/AGD15-Au + NIR). | ||
3.5. Healing effects of hydrogel on large area full-thickness skin defect in vivo
Biocompatibility is the premise and basis of the AGD-Au hydrogel in wound tissue repair. Thus, blood compatibility and cytocompatibility tests were performed to investigate the biocompatibility of the AGD-Au hydrogel. As shown in Fig. S12 (ESI†), all AGD and AGD-Au hydrogels had predominant blood compatibility compared with the negative control group (deionized water) and had the same level as the positive group (PBS). Afterwards, L929 mouse fibroblasts were used to investigate the cytocompatibility of AGD15-Au hydrogels using the CCK-8 assay. Compared with the control group, the cell viabilities of AGD15 and AGD15-Au hydrogel were 113% and 95.6%, respectively (Fig. S13a, ESI†), corresponding to the cell number results derived from the bright field images (Fig. S13b, ESI†). Furthermore, L929 cells co-cultured with AGD15 and AGD15-Au hydrogels were spindle-shaped with little morphological damage, suggesting that L929 grew well over the entire experiment. Live/Dead staining images also showed that most of the cells were alive (green), and few dead cells (red) were observed (Fig. S14, ESI†). These findings confirmed that the injectable asymmetric adhesive hydrogel had excellent biocompatibility, and the incorporation of AuNPs had no effect on the hydrogel's biocompatibility.Following the bio-safe experiments, an in vivo wound experiment in mice was performed to demonstrate the feasibility of injectable gradient hydrogel as a wound filler. An infected full-thickness skin defect model was prepared on the back of the mice. Subsequently, the AGD15-Au hydrogel was injected into the wounds. Actual pictures of full layer wound healing on the back of mice on different days determined the effect of injectable hydrogel to promote wound closure and hair growth (Fig. 7a). As shown in Fig. 7b(i), the area of the wounds in both the control and AGD15-Au hydrogel groups shrank with time. An obvious suppuration phenomenon could be observed in the control group at day 3, whereas it did not appear in AGD15-Au hydrogel. Moreover, the wound was completely healed and covered by hair on days 7 and 10 in the AGD15-Au group. In contrast, in the control group, obvious wound could still be observed even on day 10, and there were few hairs on the wound. Notably, the AGD15-Au group had a better wound closure and skin regeneration performance compared to the control group, which were 85.8% and 96.2% at 7 days (Fig. 7b(ii)).
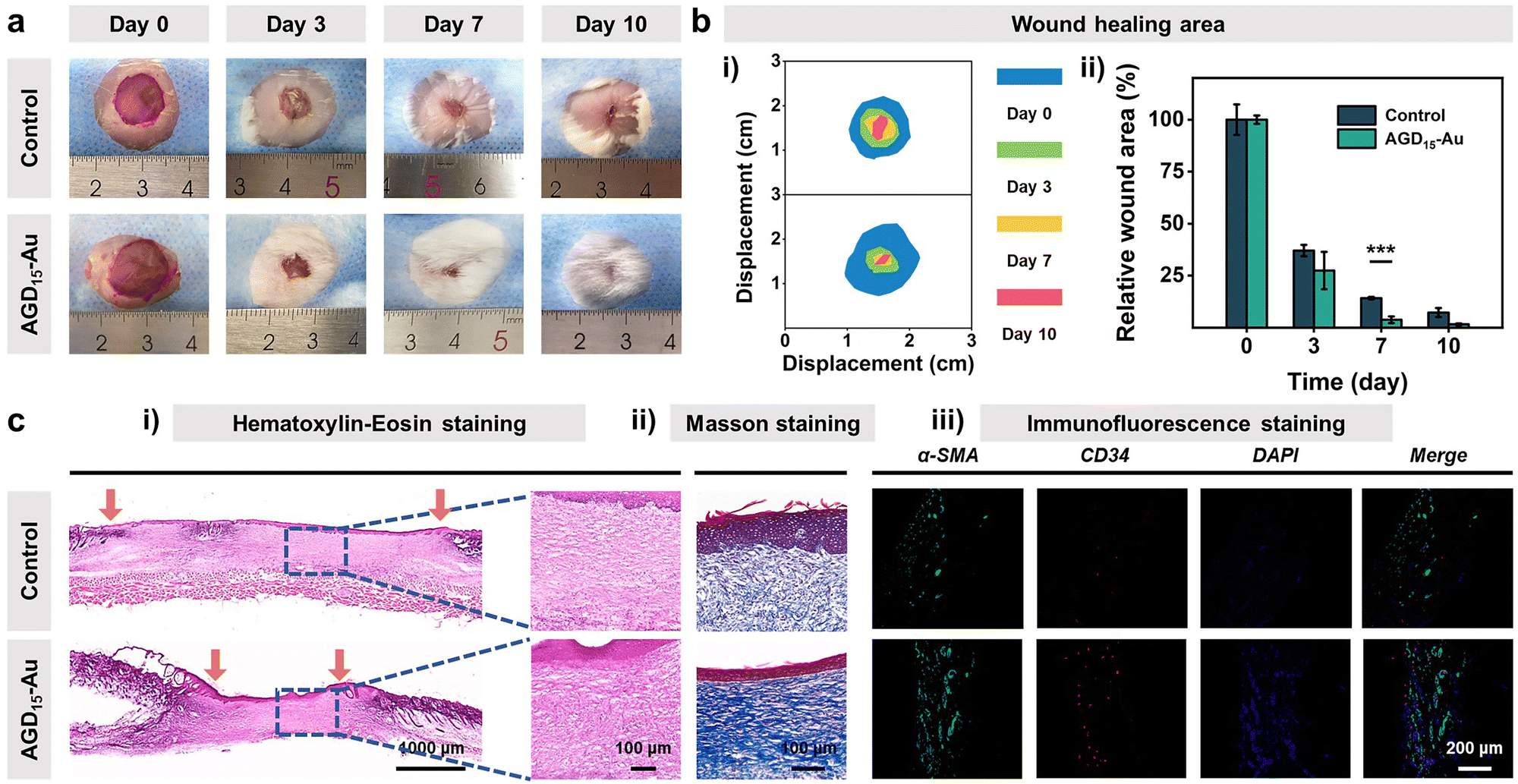 | ||
| Fig. 7 (a) Optical images of the healed skin wound at different times of the control and AGD15-Au groups. (b)(i) Schematic diagram of wound shrinkage and (ii) relative wound area of the control and AGD15-Au groups (***, p < 0.001). (c) (i) H&E staining images, (ii) Masson tricolor staining images, and (iii) double immunofluorescence staining images of α-SMA (green)/CD34 (red) at day 10 of the control and AGD15-Au groups. | ||
To further determine the effects of AGD15-Au hydrogel on wound healing, the quality of the regenerated skin tissue was investigated through Hematoxylin–Eosin (H&E) staining, Masson tricolor staining, and immunofluorescence staining (Fig. 7c). As can be observed in Fig. 7c(i), the AGD15-Au hydrogel had a more remarkable would repair effect and epidermal regeneration compared with the control group. In comparison with the control group, where a larger wound gap still existed, a better wound healing promotion effect existed in the experimental group owing to the selection of all natural polymers. This resulted in a prominent healing effect with a relatively smaller wound gap. In addition, lots of nuclei (blue mark) could be observed in the control group, which was significantly higher than that in AGD15-Au hydrogel, proving that AGD15-Au hydrogel can obviously reduce the inflammatory reaction. Meanwhile, Masson staining (Fig. 7c(ii)) showed that the collagen deposition level (blue) in the AGD15-Au hydrogel was significantly higher than that in the control group, indicating more collagen deposition. Additionally, there were some strip marks in the blue area with a more regular texture. The collagen deposition was oriented, and the wound healing process was more complete. α-SMA and CD34 immunofluorescence staining (Fig. 7c(iii)) suggested that the number of neovascular endothelial cells in AGD15-Au hydrogel was significantly greater than that in the control group. The experimental group not only promoted the initial closure and healing of a large whole-layer wound on the back of the animal but also positively affected its eventual recovery.
4. Conclusions
In conclusion, an injectable gradient asymmetric hydrogel was designed and prepared. The fabricated hydrogel was proposed to fill and promote the healing of large area skin defects. The different sacles of crosslinking with different crosslinking times promoted the gradient combination of hydrogel. Because of the choice of AuNP/DTPH crosslinking point and reversible coordination interactions between the Au and disulfide bonds, the injectable AuNP hydrogel could achieve easier operation of change and removal of the wound filler, which also promoted bacterial clearance ability. In addition, the hydrogel has sealing and healing effects on full-thickness skin defects. The hydrogel filler will have a broad application prospect in the medical field of large tissue defects.Author contributions
All the authors participated in the collection and analysis of the literature; L. L., X. L., Z. Y. T., M. L., Y. W. Q., Q. Y. Y., C. J. Y., M. M. Y., and B. Y. G. assisted in investigation; writing – original draft was finished by L. L.; J. J. L., H. Z., and F. L. Y. participated in writing – review & editing.Conflicts of interest
These authors declare no conflict of interest.Acknowledgements
This work was supported by National Natural Science Foundation of China [No. U20A20261, 52073205 and 32271390], and Tianjin Research Innovation Project for Postgraduate Students [No. 2022BKY091].Notes and references
- Y. Li, P. Yu, J. Wen, H. Sun, D. Wang, J. Liu, J. Li and H. Chu, Adv. Funct. Mater., 2021, 32, 2110720, DOI:10.1002/adfm.202110720.
- Z. Guo, Z. Zhang, N. Zhang, W. Gao, J. Li, Y. Pu, B. He and J. Xie, Bioact. Mater., 2021, 15, 203–213, DOI:10.1016/j.bioactmat.2021.11.036.
- X. Dong, F. Yao, L. Jiang, L. Liang, H. Sun, S. He, M. Shi, Z. Guo, Q. Yu, M. Yao, P. Che, H. Zhang and J. Li, J. Mater. Chem. B, 2022, 10, 2215–2229, 10.1039/d2tb00027j.
- Q. Yu, H. Sun, Z. Yue, C. Yu, L. Jiang, X. Dong, M. Yao, M. Shi, L. Liang, Y. Wan, H. Zhang, F. Yao and J. Li, Adv. Healthcare Mater., 2023, 12(7), 2202309, DOI:10.1002/adhm.202202309.
- D. Liu, L. Li, B. L. Shi, B. Shi, M. D. Li, Y. Qiu, D. Zhao, Q. D. Shen and Z. Z. Zhu, Bioact. Mater., 2023, 24, 96–111, DOI:10.1016/j.bioactmat.2022.11.023.
- X. Xie, X. Jin, B. He, Y. Zou, J. Yang, C. Liu, X. Kong, W. Liu and W. Wang, Appl. Mater. Today, 2022, 27, 101477, DOI:10.1016/j.apmt.2022.101477.
- N. Zhao and W. Yuan, Composites, Part B, 2022, 230, 109525, DOI:10.1016/j.compositesb.2021.109525.
- K. Chen, Z. Wu, Y. Liu, Y. Yuan and C. Liu, Adv. Funct. Mater., 2021, 32(12), 2109687, DOI:10.1002/adfm.202109687.
- G. Cha, W. Lee, S. Sunwoo, D. Kang, T. Kang, K. Cho, M. Kim, O. Park, D. Jung, J. Lee, S. Choi, T. Hyeon and D. Kim, ACS Nano, 2022, 16(1), 554–567, DOI:10.1021/acsnano.1c07649.
- C. Chen, X. Yang, S. Li, C. Zhang, Y. Ma, Y. Ma, P. Gao, S. Gao and X. Huang, Green Chem., 2021, 23(4), 1794–1804, 10.1039/d0gc02909b.
- L. Zhou, C. Dai, L. Fan, Y. Jiang, C. Liu, Z. Zhou, P. Guan, Y. Tian, J. Xing, X. Li, Y. Luo, P. Yu, C. Ning and G. Tan, Adv. Funct. Mater., 2021, 31(14), 2007457, DOI:10.1002/adfm.202007457.
- C. Yu, Z. Yue, M. Shi, L. Jiang, S. Chen, M. Yao, Q. Yu, X. Wu, H. Zhang, F. Yao, C. Wang, H. Sun and J. Li, ACS Nano, 2022, 16(10), 16234–16248, DOI:10.1021/acsnano.2c05168.
- C. Yu, Z. Yue, H. Zhang, M. Shi, M. Yao, Q. Yu, M. Liu, B. Guo, H. Zhang, L. Tian, H. Sun, F. Yao and J. Li, Adv. Funct. Mater., 2023, 33(15), 2211023, DOI:10.1002/adfm.202211023.
- X. Zhao, Y. Yang, J. Yu, R. Ding, D. Pei, Y. Zhang, G. He, Y. Cheng and A. Li, Biomaterials, 2022, 282, 121387, DOI:10.1016/j.biomaterials.2022.121387.
- Z. Bao, M. Gao, X. Fan, Y. Cui, J. Yang, X. Peng, M. Xian, Y. Sun and R. Nian, Int. J. Biol. Macromol., 2020, 155, 163–173, DOI:10.1016/j.ijbiomac.2020.03.210.
- J. Zhang, W. Xin, Y. Qin, Y. Hong, Z. Xiahou, K. Zhang, P. Fu and J. Yin, Chem. Eng. J., 2022, 430, 132713, DOI:10.1016/j.cej.2021.132713.
- A. Sigen, Q. Xu, M. Johnson, J. Creagh-Flynn, M. Venet and D. Zhou, Appl. Mater. Today, 2021, 22, 100967, DOI:10.1016/j.apmt.2021.100967.
- Z. Tan, X. Li, C. Yu, M. Yao, Z. Zhao, B. Guo, L. Liang, Y. Wei, F. Yao, H. Zhang and J. Li, Int. J. Biol. Macromol., 2023, 232, 123449, DOI:10.1016/j.ijbiomac.2023.123449.
- A. Cholewinski, F. Yang and B. Zhao, Mater. Horiz., 2019, 6, 285–293 RSC.
- H. Wang, X. Su, Z. Chai, Z. Tian, W. Xie, Y. Wang, Z. Wan, M. Deng, Z. Yuan and J. Huang, Chem. Eng. J., 2022, 428, 131049, DOI:10.1016/j.cej.2021.131049.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174, DOI:10.1038/s41586-019-1710-5.
- S. He, B. Guo, X. Sun, M. Shi, H. Zhang, F. Yao, H. Sun and J. Li, ACS Appl. Mater. Interfaces, 2022, 14(40), 45869–45879, DOI:10.1021/acsami.2c13371.
- X. Su, Y. Luo, Z. Tian, Z. Yuan, Y. Han, R. Dong, L. Xu, Y. Feng, X. Liu and J. Huang, Mater. Horiz., 2020, 7(10), 2651–2661, 10.1039/d0mh01344g.
- B. Guo, S. He, M. Yao, Z. Tan, X. Li, M. Liu, C. Yu, L. Liang, Z. Zhao, Z. Guo, M. Shi, Y. Wei, H. Zhang, F. Yao and J. Li, Chem. Eng. J., 2023, 461, 142099, DOI:10.1016/j.cej.2023.142099.
- B. Guo, Y. Wu, S. He, C. Wang, M. Yao, Q. Yu, X. Wu, C. Yu, M. Liu, L. Liang, Z. Zhao, Y. Qiu, F. Yao, H. Zhang and J. Li, J. Mater. Chem. A, 2023, 11, 8038–8047, 10.1039/d2ta09973j.
- W. Liang, W. He, R. Huang, Y. Tang, S. Li, B. Zheng, Y. Lin, Y. Lu, H. Wang and D. Wu, Adv. Mater., 2022, 34(15), 2108992, DOI:10.1002/adma.202108992.
- Y. Wang, D. Xiao, H. Yu, R. Ke, S. Shi, Y. Tang, Y. Zhong, L. Zhang, X. Sui, B. Wang, X. Feng, H. Xu and Z. Mao, Composites, Part B, 2022, 235, 109762, DOI:10.1016/j.compositesb.2022.109762.
- J. Yu, Y. Qin, Y. Yang, X. Zhao, Z. Zhang, Q. Zhang, Y. Su, Y. Zhang and Y. Cheng, Bioact. Mater., 2023, 19, 703–716, DOI:10.1016/j.bioactmat.2022.05.010.
- Y. Jia, J. Feng, Z. Feng, J. Liu, Y. Yang, X. Li, M. Lei, H. Guo, Z. Wei, Y. Lv and F. Xu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(6), 2219024120, DOI:10.1073/pnas.2219024120.
- C. Cui, T. Wu, X. Chen, Y. Liu, Y. Li, Z. Xu, C. Fan and W. Liu, Adv. Funct. Mater., 2020, 30(49), 2005689, DOI:10.1002/adfm.202005689.
- H. Wang, X. Yi, T. Liu, J. Liu, Q. Wu, Y. Ding, Z. Liu and Q. Wang, Adv. Mater., 2023, 35, 2300394, DOI:10.1002/adma.202300394.
- K. Chen, C. Liu, J. Huang, L. Che, Y. Yuan and C. Liu, Adv. Funct. Mater., 2023, 33(41), 2303836, DOI:10.1002/adfm.202303836.
- X. Su, W. Xie, P. Wang, Z. Tian, H. Wang, Z. Yuan, X. Liu and J. Huang, Mater. Horiz., 2021, 8, 2199–2207, 10.1039/d1mh00533b.
- X. Wu, W. Guo, L. Wang, Y. Xu, Z. Wang, Y. Yang, L. Yu, J. Huang, Y. Li, H. Zhang, Y. Wu, G. Li and W. Huang, Adv. Funct. Mater., 2021, 32(9), 2110066, DOI:10.1002/adfm.202110066.
- T. Xie, J. Ding, X. Han, H. Jia, Y. Yang, S. Liang, W. Wang, W. Liu and W. Wang, Mater. Horiz., 2020, 7(2), 605–614, 10.1039/c9mh01255a.
- J. Shi, D. Wang, H. Wang, X. Yang, S. Gu, Y. Wang, Z. Chen, Y. Chen, J. Gao, L. Yu and J. Ding, Acta Biomater., 2022, 145, 106–121, DOI:10.1016/j.actbio.2022.04.020.
- B. Freedman, O. Uzun, N. Luna, A. Rock, C. Clifford, E. Stoler, G. Ostlund-Sholars, C. Johnson and D. Mooney, Adv. Mater., 2021, 33(17), 2008553, DOI:10.1002/adma.202008553.
- G. Tian, D. Yang, C. Liang, Y. Liu, J. Chen, Q. Zhao, S. Tang, J. Huang, P. Xu, Z. Liu and D. Qi, Adv. Mater., 2023, 35(18), 2212302, DOI:10.1002/adma.202212302.
- L. Liang, Z. Qin, X. Dong, S. He, M. Yao, Q. Yu, C. Yu, M. Liu, B. Guo, H. Zhang, F. Yao and J. Li, Macromol. Rapid Commun., 2022, 43(17), 2200182, DOI:10.1002/marc.202200182.
- H. Qin, T. Zhang, H.-N. Li, H.-P. Cong, M. Antonietti and S.-H. Yu, Chem, 2017, 3(4), 691–705, DOI:10.1016/j.chempr.2017.07.017.
- Q. Liu, A. Chiu, L. Wang, D. An, M. Zhong, A. Smink, B. de Haan, P. de Vos, K. Keane, A. Vegge, E. Chen, W. Song, W. Liu, J. Flanders, C. Rescan, L. Grunnet, X. Wang and M. Ma, Nat. Commun., 2019, 10(1), 5262, DOI:10.1038/s41467-019-13238-7.
- S. Choi, J. R. Moon, N. Park, J. Im, Y. E. Kim, J. H. Kim and J. Kim, Adv. Mater., 2022, 35(3), 2206207, DOI:10.1002/adma.202206207.
- X. Li, Z. Tan, B. Guo, C. Yu, M. Yao, L. Liang, X. Wu, Z. Zhao, F. Yao, H. Zhang, S. Lyu, C. Yuan and J. Li, Chem. Eng. J., 2023, 463, 142387, DOI:10.1016/j.cej.2023.142387.
- Q. Wang, Y. Liao, W. Teng, Z. Liao, S. He, J. Xu and H. Ye, Chin. J. Anal. Lab., 2008, 27, 4, DOI:10.3969/j.issn.1000-0720.2008.z1.026.
- Y. Yang, H. Xu, M. Li, Z. Li, H. Zhang, B. Guo and J. Zhang, ACS Appl. Mater. Interfaces, 2022, 14(37), 41726–41741, DOI:10.1021/acsami.2c10490.
- Y. Wu, X. Zhang, B. Tan, Y. Shan, X. Zhao and J. Liao, Biomater. Adv., 2022, 133, 112641, DOI:10.1016/j.msec.2022.112641.
Footnote |
| † Electronic supplementary information (ESI) available: Basic material characterization, scanning electron microscopy and mapping images of AGD15-Au surface, swelling and degradation properties of injectable asymmetric hydrogels, rheological properties and biocompatibility test of different groups. See DOI: https://doi.org/10.1039/d3tb02014b |
| This journal is © The Royal Society of Chemistry 2023 |