
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable one-pot method to transform seashell waste calcium carbonate to osteoinductive hydroxyapatite micro-nanoparticles†
Raquel
Fernández-Penas
 a,
Cristóbal
Verdugo-Escamilla
a,
Carla
Triunfo
bc,
Stefanie
Gärtner
d,
Annarita
D'Urso
e,
Francesca
Oltolina
e,
Antonia
Follenzi
e,
Gabriele
Maoloni
f,
Helmut
Cölfen
d,
Giuseppe
Falini
b and
Jaime
Gómez-Morales
*a
a,
Cristóbal
Verdugo-Escamilla
a,
Carla
Triunfo
bc,
Stefanie
Gärtner
d,
Annarita
D'Urso
e,
Francesca
Oltolina
e,
Antonia
Follenzi
e,
Gabriele
Maoloni
f,
Helmut
Cölfen
d,
Giuseppe
Falini
b and
Jaime
Gómez-Morales
*a
aLaboratorio de Estudios Cristalográficos, IACT (CSIC-UGR), Avda. Las Palmeras, no 4, 18100 Armilla, Spain. E-mail: jaime@lec.csic.es
bDepartment of Chemistry “Giacomo Ciamician”, University of Bologna, via F. Selmi 2, 40126 Bologna, Italy
cFano Marine Center, The Inter-Institute Center for Research on Marine Biodiversity, Resources and Biotechnologies, viale Adriatico 1/N, 61032 Fano, Italy
dPhysical Chemistry, Department of Chemistry, University of Konstanz, Universitätsstrasse 10, Box 714, D-78457 Konstanz, Germany
eDipartimento di Scienze della Salute, Università del Piemonte Orientale, A. Avogadro” Via Solaroli, 17, 28100 Novara, Italy
fPlant Ascoli Piceno, Finproject S.p.A., 3100 Ascoli Piceno, Italy
First published on 21st July 2023
Abstract
We have developed a straightforward, one-pot, low-temperature hydrothermal method to transform oyster shell waste particles (bCCP) from the species Crassostrea gigas (Mg-calcite, 5 wt% Mg) into hydroxyapatite (HA) micro/nanoparticles. The influence of the P reagents (H3PO4, KH2PO4, and K2HPO4), P/bCCP molar ratios (0.24, 0.6, and 0.96), digestion temperatures (25–200 °C), and digestion times (1 week–2 months) on the transformation process was thoroughly investigated. At 1 week, the minimum temperature to yield the full transformation significantly reduced from 160 °C to 120 °C when using K2HPO4 instead of KH2PO4 at a P/bCCP ratio of 0.6, and even to 80 °C at a P/bCCP ratio of 0.96. The transformation took place via a dissolution–reprecipitation mechanism driven by the favorable balance between HA precipitation and bCCP dissolution, due to the lower solubility product of HA than that of calcite at any of the tested temperatures. Both the bCCP and the derived HA particles were cytocompatible for MG-63 human osteosarcoma cells and m17.ASC murine mesenchymal stem cells, and additionally, they promoted the osteogenic differentiation of m17.ASC, especially the HA particles. Because of their physicochemical features and biological compatibility, both particles could be useful osteoinductive platforms for translational applications in bone tissue engineering.
1. Introduction
Seashells represent a waste by-product from fishery industry and marine aquaculture.1 In 2018, world production of shelled molluscs reached 17.3 million tonnes representing 56% of the production of marine and coastal aquaculture.2 Waste seashells are often just dumped in the sea causing damage to the marine ecosystem3 or disposed in a landfill affecting the quality of life for people living in proximity because of the unpleasant smell and causing health issues due to the microbial decomposition.4,5 On the other hand, their disposal represents an environmental and economic issue since it involves the use of incineration or burial.6 A way to overcome these issues may involve turning cast-off waste seashells into new materials by observing the main circular economy targets, i.e., efficiency, recycling, recovering and reducing.7 Mollusc shells are hierarchical organic/mineral composites made of CaCO3 embedded within an organic framework (1–5 wt%).8 So far, this waste by-product has been used as a source of calcium carbonate in different fields.9 Many studies report the use of CaCO3 from the shells as calcium supplements for livestock feeding,10,11 agricultural liming agents for soil acidity treatment12 and as aggregate substitutes in concrete preparation.13,14 In recent years, several papers have been published on the transformation of biogenic CaCO3 particles (bCCP) to calcium phosphates of the hydroxyapatite phase (HA).HA [Ca10(PO4)6(OH)2] is the model phase representing the bone and teeth mineral component. Synthetic HA is the most largely studied calcium phosphate for biomedical applications such as coatings for dental implants, drug delivery systems, bioimaging and bone filling, substitution, replacement and regeneration in orthopaedics.15–21 The global HA market, including nano-, micron- and larger sizes, is expected to grow to around USD 3,086.05 million by 2027.22 The interest in HA is due to its excellent features i.e., biocompatibility, bioactivity, no-toxicity and osteointegration.23 Recently, HA nanoparticles, in particular, have widely drawn attention due to their large surface-to-volume ratio, which mimics the dimensions of calcified tissue constituents allowing a higher biocompatibility and cell affinity and proliferation.15,24,25
Different biogenic CaCO3 sources have been employed as a source of calcium to produce HA such as eggshells,26 mussel shells,27 clam shells,28 cockle shells,29 oyster shells,30 snail shells,31 corals32 and cuttlefish bones.33 Among them, oyster shells are constituted by CaCO3 in the form of calcite with an isomorphic substitution of magnesium to calcium of about 5 mol%.34 Both valves comprise different microstructures of calcite that are layers of sheet-like foliae alternated with discontinuous lense-like chalk layers and a thin external layer of prisms 5–25 μm wide and 20–50 μm high which are in turn constituted by an internal laminated structure.35,36 HA particles obtained from oyster shells having biocompatibility and bioactivity properties have been already produced. However, nothing about the osteoinductive properties of these HA particles was reported.37,38
Several methods have been developed to convert bCCP in HA so far. Most of them are two-step processes involving the calcination of CaCO3 to CaO at 900–1200 °C followed by the reaction with a phosphate reagent (typically H3PO4) with Ca(OH)2 produced by the hydration of CaO.39–42 Calcium precursors for the wet chemical precipitation of HA can also be obtained by dissolving CaCO3 in an acidic solution such as HCl or lactic acid.43,44 The hydrothermal method was used in a three-step procedure consisting of calcination of bCCP, hydration and carbonation of resulting CaO to produce precipitated CaCO3 particles, which were then used for the hydrothermal reaction with (NH4)2HPO4 at 160 °C in an autoclave to yield HA nanoparticles.45 Rod and flower-like HA nanoparticles were synthesized by a microwave irradiation method with the aid of EDTA as a calcium chelating agent.46–48 Agalya et al. performed one-pot ultrasound-assisted synthesis to produce Fe-doped HA nanoparticles using mussel shells as the calcium source with the aid of trisodium citrate as a complexing agent.49 HA nanoparticles were also obtained via wet mechanosynthesis by the use of ball milling but for the full conversion of bCCP to be achieved, thermal treatment had to be performed after the milling process.50 Balu et al. introduced an oil-bath-mediated precipitation method at 80 °C for the synthesis of HA nanorods from cuttlefish bone powder using basic pH.51
In this work, we investigate the transformation of bCCP from oyster shell waste of the species Crassostrea gigas into HA micro/nanoparticles with suitable cytocompatible and osteoinductive properties for osteogenic applications using a one-step hydrothermal process. Compared to most of the methods reported in the literature so far, the method we suggest here is straightforward, one-pot and involves the full transformation of bCCP into HA nanoparticles. Moreover, it is low-cost since it can avoid the use of expensive hydrothermal autoclaves, additives, and high-purity calcium reagents, and importantly, it can be performed at relatively low hydrothermal temperatures without any pH adjustment. This method will benefit the environment since it proposes an efficient and low-cost production of HA particles with applications in medicine and therapeutics using waste seashells as the starting material, thus contributing to reducing the waste material derived from the fishery industry and marine aquaculture.
2. Materials and methods
2.1. Preparation of bCCP
Oyster shells of the species Crassostrea gigas were purchased from F.lli Terzi (Palosco, BG, Italy). The shells were carefully washed with tap water to eliminate the meat residues and mineral debris. Then they were treated with 5% v/v sodium hypochlorite solution for 24 hours to remove the organic residues from the surface, washed with tap water and air-dried. The dry shells were finally crushed by a hammer mill and sieved at Ø < 45 μm by analytical sieving in order to get biogenic calcium carbonate particles (bCCP).2.2. Reagents
Potassium phosphate monobasic (KH2PO4, ACS Reagent, >99%), potassium phosphate dibasic (K2HPO4, ACS Reagent, ≥ 98%), and phosphoric acid (H3PO4, ACS reagent, 85 wt% in H2O) were provided by Sigma-Aldrich. All solutions were prepared with deionized water (0.22 μS, 25 °C, Milli-Q, Millipore).2.3. Experimental method
The experiments were carried out in a furnace with circulated forced air, using 10 mL glass Pyrex test tubes with PBT screw caps and a rubber disc coated with PTFE at temperatures in the range 25 °C ≤ T ≤ 120 °C, with ΔT = 20 °C. For higher temperatures (120 °C < T ≤ 200 °C, ΔT = 20 °C), an aluminium box was designed to contain four 10 mL PTFE tubes. The box is provided with an aluminium cap coated with PTFE to close the ensemble. Alternatively, we used the Berghoff BR100 model hydrothermal reactor equipped with a 100 mL PTFE liner (see in Fig. S1 of ESI† a sketch of the experimental set-up and the screened processing parameters). All tubes were filled at 70% volume with the bCCP aqueous dispersions. The dispersions were prepared with the bCCP by varying the phosphate reagent (H3PO4, KH2PO4, K2HPO4) and the P/bCCP mol ratio to 0.24 (deficiency of phosphate), 0.6 (stoichiometric with respect to HA), and 0.96 (excess of phosphate). The initial concentrations of phosphate and bCCP were as follows: For P/bCCP = 0.24, 120 mM P/500 mM bCCP, while for P/bCCP = 0.6 and 0.96, 300 mM P/500 mM bCCP and 480 mM P/500 mM bCCP, respectively. The maturation time (t) varied depending on the P/bCCP mol ratio and the setting temperature. Thus, for P/CaCO3 = 0.24 in the range 25 °C ≤ T ≤ 80 °C, the experiments lasted 1 and 2 months, while for the range 100 °C ≤ T ≤ 200 °C, they lasted 1 week. This set of preliminary experiments allowed us to fix the maturation time in further experiments with P/CaCO3 = 0.6 and 0.96 in 1 week. The initial pH was not adjusted but measured at the beginning and at the end of the experiments in the residual solutions by using a Sension + + CAT pH electrode connected to a Hach Sension+ pH-meter with an accuracy ≤ 0.02 pH. Upon completion, the precipitates were subjected to three consecutive cycles of washing by centrifugation with deionized water to remove unreacted species or salts such as NaCl. Afterward, they were freeze-dried overnight at −50 °C under a vacuum (3 mbar).2.4. Particle characterization
Different techniques were used for the characterization of the precipitates including X-ray diffraction (XRD), thermogravimetry, FTIR and Raman spectroscopies, scanning and transmission electron microscopies (SEM, TEM and HRTEM), energy dispersive X-ray spectroscopy (EDS) analysis, and thermogravimetry. The selected samples were additionally analysed by dynamic light scattering (DLS) and ζ-potential against pH.XRD data were collected using a Bruker D8 Advance Vario diffractometer with a Bragg–Brentano parafocusing geometry and Cu Kα1 radiation (1.5406 Å). Data processing of most matured samples was carried out using software TOPAS 6.0 (Bruker AXS, Karlsruhe).52 The contribution of the isotropic peak broadening due to domain size was modeled using the TOPAS macro based on the Scherrer approximation and considering the instrumental contribution from a measurement of LaB6 standard (NIST SRM 660c).
Thermogravimetric analyses of some selected samples were performed using a Shimadzu TGA-50H calorimeter at a flow rate of 100 mL min−1 air in an alumina crucible. The measurements were carried out from 25 to 950 °C at a rate of 20 °C min−1.
FESEM observations and EDS were both performed using a field emission high-resolution microscope GEMINI LEO 1500 (Zeiss, Jenna, Germany) equipped with an energy dispersive X-ray (EDX) spectroscope by Oxford Instruments.
Transmission electron microscopy (TEM) was performed using a Carl Zeiss Libra 120 Plus instrument at 80 kV. High-resolution transmission electron microscopy (HR-TEM) analyses were performed using a TITAN G2 60-300 FEI Instrument (FEI, Hillsboro, OR, USA) operating at 300 kV. The instrument is equipped with an EDX Super X detector to perform microanalysis, and a STEM type HAADF detector. For both techniques, the powder samples were ultrasonically dispersed in H2O (MilliQ), and then a few droplets of the slurry were deposited on copper microgrids coated with a FORMVAR carbon film prior to observation.
Fourier transform infrared spectra were recorded using an Invenio R FTIR spectrometer (Bruker) equipped with an attenuated total reflectance (ATR) accessory of diamond crystals. The FTIR spectra in a transmittance mode were recorded within the wavenumber range from 4000 cm−1 to 400 cm−1 at a resolution of 4 cm−1. Raman spectra were recorded with a LabRAMHR spectrometer (Jobin–Yvon, Horiba, Japan). The excitation line was provided by a diode laser emitting at a wavelength of 532 nm. A Peltier cooled charge-coupled device (1064 × 256 pixels) was used as the detector.
The analysis of both the crystal size distribution by dynamic light scattering (DLS) and the electrophoretic mobility (ζ-potential) of the HA samples was performed with a Zetasizer Nano ZS analyzer with a 633 nm laser (Malvern, UK) using disposable polystyrene cuvettes containing the particles suspended in deionized water (0.5 mg mL−1) at 25 °C. For the measurements of the ζ-potential versus pH, the MPT-2 autotitrator (Malvern, UK) connected to the analyser was employed. Diluted HCl and NaOH solutions (0.25 and 0.1 M, respectively) were used as titration agents. No additional electrolytes were added. For oyster shell bCCP, the analysis was performed by laser diffraction with a Mastersizer 2000 instrument (Malvern, UK) coupled with a Hydro 2000SM system.
2.5. Biological tests
3. Results and discussion
3.1. Influence of P/bCCP molar ratio, P reagent, and temperature on bCCP to apatite transformation
Fig. 1 summarizes the main results of XRD and spectroscopic characterization (FTIR and Raman) of the precipitates resulting in experiments with initial molar ratios of P/bCCP = 0.24 at 1 month (Fig. 1(a)–(c)), P/bCCP = 0.60 at 1 week (Fig. 1(d)–(f)), and P/bCCP = 0.96 at 1 week (Fig. 2(g)–(i)). For the sake of simplicity, the figure shows the results at 25 °C and at the minimum temperature yielding the highest transformation (Fig. 1(a)–(c)) or at temperatures yielding full transformation (Fig. 1(d)–(f) and (g)–(i)).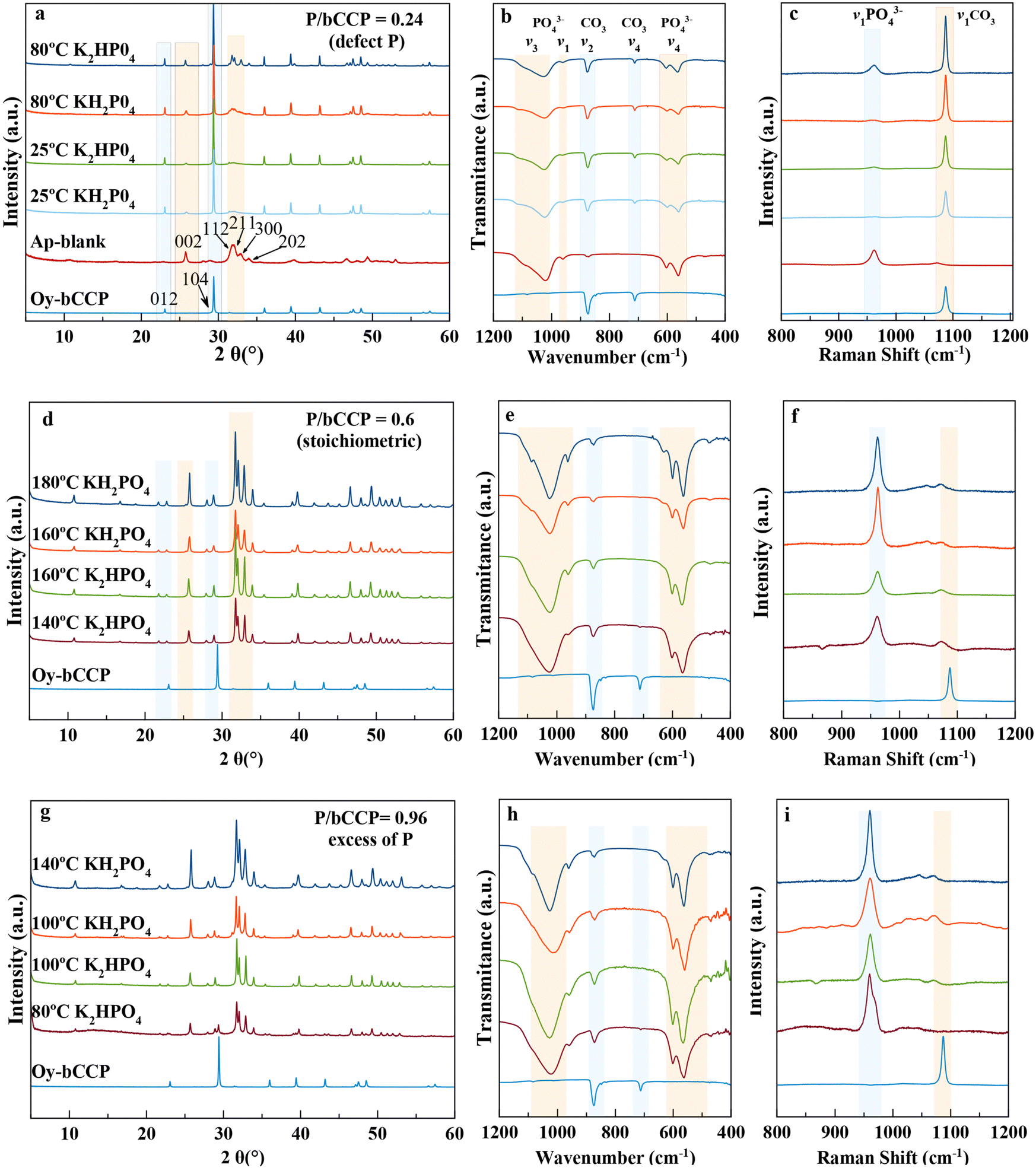 | ||
| Fig. 1 XRD patterns (a), (d) and (g), FTIR (b), (e) and (h) and Raman (c), (f) and (i) spectra of precipitates obtained from aqueous suspensions with initial P/bCCP ratios of 0.24 (defect of P) at 1 month, 0.6 (stoichiometric composition with respect to HA) at 1 week, and 0.96 (excess of P) at 1 week at 25 °C and at the minimum temperatures allowing the maximum or full transformation percentage. | ||
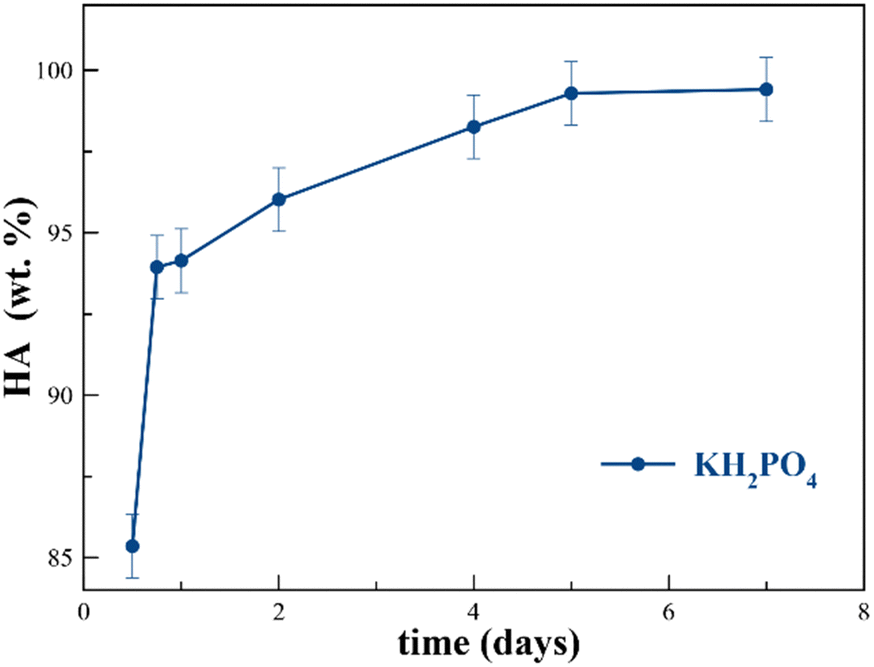 | ||
| Fig. 2 Evolution of HA wt% with time using KH2PO4 at a P/bCCP molar ratio 0.6. | ||
In these plots, the starting material bCCP presents the characteristic XRD pattern of calcite (PDF 00-005-0586), with reflections at 2θ = 29.3°(104) and 23.03° (012) marked in the diffractogram.56 The XRD pattern of a carbonated-hydroxyapatite blank57 shows the main reflections at 2θ = 25.87° (002) and the quadruplet 31.77° (211), 32.19° (112), 32.90° (300), and 33.97 (202) (PDF 01-1008) (Fig. 1(a)). The FTIR spectrum of bCCP in the range of 400–1200 cm−1 shows the υ2(CO3) and υ4(CO3) vibration modes at ∼875 cm−1 and 713 cm−1,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 58 while for HA, it shows the characteristic asymmetric stretching υ3 (PO4) at 1000–1100 cm−1, symmetric stretching υ1(PO4) at ∼960 cm−1, and the bending mode υ4(PO4) at 562 cm−1 and 603 cm−1. The presence of a small band in the υ2(CO3) region is due to the CO32− ions substituting both OH− (A-type) and PO43−(B-type) (Fig. 1(b)).20,57 Concerning the Raman spectra in the range 800 cm−1 to 1200 cm−1 (Fig. 1(c)), we find the bands of HA υ1(PO4) at ∼960 cm−159 and calcite υ1(CO3) at ∼1086 cm−160 as the most representative ones.
58 while for HA, it shows the characteristic asymmetric stretching υ3 (PO4) at 1000–1100 cm−1, symmetric stretching υ1(PO4) at ∼960 cm−1, and the bending mode υ4(PO4) at 562 cm−1 and 603 cm−1. The presence of a small band in the υ2(CO3) region is due to the CO32− ions substituting both OH− (A-type) and PO43−(B-type) (Fig. 1(b)).20,57 Concerning the Raman spectra in the range 800 cm−1 to 1200 cm−1 (Fig. 1(c)), we find the bands of HA υ1(PO4) at ∼960 cm−159 and calcite υ1(CO3) at ∼1086 cm−160 as the most representative ones.
In the experiments with a P/bCCP ratio of 0.24 (Fig. 1(a)–(c) and Fig. S2 of ESI†), we obtained mixtures of bCCP and HA. This P/bCCP ratio allowed us a preliminary screening of the influences of time (t), temperature (T) and P reagent on the evolution of the precipitation. The effect of these parameters on HA wt% was quantified by Rietveld analysis of the XRD patterns using TOPAS 6.0 software (ESI,† Fig. S3). The plot shows an increase of transformation with T within the range from 25 °C to 80 °C (Fig. S3a, ESI†), with a maximum percentage at 80 °C of 39.5 wt%, which was attained with 120 mM KH2PO4. The conversion percentages when using K2HPO4 were worse (34.4 wt% and 30.4 wt% for 120 and 30 mM, respectively) but improved when the maturation time doubled to 2 months (37.1 wt%) (Fig. S3b, ESI†). Only when increasing the temperature to 180 °C and 200 °C, the HA wt% increased to around 43.6 wt%, even reducing the time to 1 week. The experiments using H3PO4 (at 25 °C) did not produce any transformation, so this reagent was ruled out in further experiments at higher P/bCCP molar ratios using 1 week as the experimental time.
At P/bCCP molar ratios of 0.6 (stoichiometric) and 0.96 (excess of P), the study shows the influences of P reagent (KH2PO4 or K2HPO4) and T (25 °C to 200 °C) at t = 1 week. A detailed examination of Fig. 1(d)–(f) and Fig. S4a,b (ESI†) for the P/bCCP ratio of 0.6 and Fig. 1(g)–(i) and Fig. S5a,b (ESI†) for the ratio of 0.96 allowed us to determine the minimum temperature to yield the full conversion (Tmin). The examination considered the disappearance of the calcite reflection C(104) in the XRD pattern and the lack of both the C(υ4CO3) vibration mode in the FTIR spectrum and C(υ1CO3) in the Raman spectrum at the same T. It was observed that for the P/bCCP ratio of 0.6, the Tmin is 160 °C when using KH2PO4 and 120 °C for K2HPO4. However, for the P/bCCP ratio of 0.96, the Tmin was 120 °C when using KH2PO4 and 80 °C when using K2HPO4. The evolution of HA wt% with time (from 12 h to 7 days) at the molar ratio of 0.6, and T = Tmin, shows a growing transformation trend with both reagents yielding >99.5 wt% HA at 7 days (Fig. 2). The decrease of Tmin ≤ 120 °C allows the process to be scaled up using technology already established in the chemical industry based on glass reactors, thus avoiding the use of expensive hydrothermal autoclaves.
Thermogravimetric analyses (TGA) of HA particles obtained at a molar ratio of 0.6 using both reagents (ESI,† Fig. S6) revealed the presence of three main regions. The first one between 25 °C and 230 °C is associated to adsorbed surface water, the second one up to 500–600 °C is due to loss of internal or structural water, and the third one between 600 and 950 °C is due to loss of CO2 by decomposition of CO32−.61 The latter region is divided into two sub-regions in the CO3-apatite obtained with KH2PO4 (Fig. S6a, ESI†), with losses of 3.4 wt% and 2.3 wt%, and the associated first derivative (DTG) peaks centered at 708 °C and 795 °C. These losses can correspond to A- and B-type CO32− substituted groups in AB-type CO3-apatites.62 The presence of both types of substitutions is reflected in the υ2(CO3) region of the FTIR spectrum, which is decomposed into two small peaks at 879–880 cm−1 and 870–872 cm−1 attributable to A-type and B-type CO32− substitutions, respectively.18,63 In the CO3-apatite prepared with K2HPO4 (Fig. S6b, ESI†), only one temperature region was found with a loss of 6.8 wt% and a DTG peak centered at 796 °C. The υ2(CO3) of the FTIR spectrum shows a main peak at ∼872 cm−1 attributable to B-type CO3.18,63 When increasing the conversion temperature to 200 °C, the CO2 region shifted to around 750–950 °C in both apatites, with losses of 1.6 and 1.7 wt%, respectively, evidencing the minor content of carbonate of these samples. The three mentioned thermal regions of HA compare with those of Oy-CaCO3. The first from 25–260 °C is due to the loss of adsorbed water, the second one to the loss of internal structural water and organics, and the third one between 650 and 790 °C, with a DTG peak at 760 °C, to the loss of CO2 yielding the residue of CaO.
The type of P reagent regulated the initial pH of the suspensions, i.e., 5.6–5.8, when using KH2PO4 and 8.5–8.8 in the case of K2HPO4 (Table S1 of ESI†), while the conversion process was responsible for the higher pH at the end of the experiment, reaching in some cases values higher than 10.0. This increase of pH is explained by the progressive dissolution of bCCP that favors the increase of activities of Ca2+, CO32−, HPO43−, PO43−, and OH−, as well as of neutral (CaHPO4°) and ion pair (CaPO4+) species up to the critical supersaturation for the nucleation of the HA phase. The favorable balance between HA precipitation and CaCO3 dissolution, due to the lower-solubility product of HA than that of calcite, was responsible for the whole dissolution of CaCO3. For a better understanding of the process, Fig. S7 of the ESI† shows the results of a simulated experiment using K2HPO4 and P/CaCO3 of 0.96 in which the full conversion was attained at 80 °C. The decrease of activities of the aqueous species Ca2+ and CaHPO4° with an increase in pH and a slow decrease of HPO42− is observed (Fig. S7a and c, ESI†), which indicates that these are the species involved in the nucleation process. Recently, some authors have shown pieces of evidence that the non-classical calcium phosphate precipitation pathway involves prenucleation clusters (PNC) identified as Ca2(HPO4)32−, which are constituted by a molecule of CaHPO4° plus HPO42− and an extra Ca2+ ion,64 or ion associations of the type Ca(HPO4)34−.65 Going back to the simulation, the decrease of the activities of Ca2+, CaHPO4°, and HPO42− also agrees with the formation of these types of PNCs. The crucial role of PNCs in the formation of the solid phase could explain the decrease in Tmin to attain the full CaCO3 to HA transformation when using K2HPO4.
3.2. Morphological and microstructural characteristics of the precipitated HA particles
Following the precipitation processes at different temperatures, the FESEM micrographs (Fig. 3) show that the initial micrometer-sized Oy-bCCP layered rods (Fig. 3(a)) were coated with HA nanoparticles in the experiments at 25 °C, irrespective of the P/bCCP molar ratio and P reagent used (Fig. 2(b), (e), (f), (i) and (j)). In experiments with a P/bCCP ratio of 0.24, at the temperatures for maximum transformation (Fig. 3(c) and (d)), the HA nanoparticles displayed mostly isometric morphologies. At P/bCCP molar ratios of 0.6 and 0.96, the obtained nanoparticles at the Tmin displayed predominant morphologies depending on the P reagent type, i.e., thin needle- and plate-like anisometric morphologies when using KH2PO4 (Fig. 3(g) and (k)) or more isometric quasi-spherical rods when using K2HPO4 (Fig. 3(h) and (l)). The two morphologies were found at the different processing temperatures (Fig. S8, ESI†). | ||
| Fig. 3 FESEM micrographs of bCCP and apatites obtained at 25 °C and at the minimum temperature allowing full transformation using KH2PO4 and K2HPO4 at P/bCCP molar ratios of 0.24 (a)–(d), 0.6 (e)–(h), and 0.96 (i)–(l). | ||
The results are confirmed by HRTEM characterization of samples transformed at 200 °C. Fig. 4(a) and (i) show the high-angle annular dark field (HAADF) images of samples prepared at a P/bCCP molar ratio of 0.6 using KH2PO4 and K2HPO4, respectively. While the former particles are mostly needle-like, the latter are more isometric. In both cases, the EDX elemental mappings (Fig. 4(b)–(d) and (j)–(l)) reveal the presence of Ca, P, and to a minor extent Mg with 0.61 ± 0.06 wt% and a molar ratio (Ca + Mg)/P of 1.73 ± 0.03 when using KH2PO4 and 0.49 ± 0.11 wt% with a molar ratio of 1.72 ± 0.08 or 0.71± 0.11 wt% with a molar ratio of 1.81 ± 0.08, when using K2HPO4, as revealed in 2 different zones of this sample. The amount of Mg is similar to that reported in bone HA of 0.72 wt%.66 The role of Mg2+ is essential in bone formation since a decrease in Mg2+ level adversely affects all stages of skeletal metabolism and increases bone fragility and the risk of osteoporosis.67 The preparation of Mg-HA is therefore considered of great interest in the development of artificial bone substitutes.68,69
 | ||
| Fig. 4 (a) and (i) HAADF-STEM micrographs of samples prepared with KH2PO4 and K2HPO4, respectively; (b)–(d) and (j)–(l) EDX element mapping analysis of Ca, P, and Mg of particles of images (a) and (i), respectively. (e) and (g) SAED patterns of samples prepared with KH2PO4 and K2HPO4, respectively, showing the planes of the apatite phase. (f) and (h) HR-TEM images showing the lattice fringes of particles prepared with KH2PO4 and KH2PO4, respectively. Note than in the latter image, lattice fringes correspond to the hydroxyapatite planes (112), (211) and (300) and calcite planes (104) and (006). | ||
The indexed selected area electron diffraction (SAED) pattern of the first sample (Fig. 3I) shows the characteristic crystallographic planes of HA, i.e., (002), (211), (310), and (152), while the HRTEM image (Fig. 4(f)) shows lattice fringes of the elongated particles structured as single crystal domains whose d-spacing of 8.15 Å corresponds to planes (100).
On the other hand, the indexed SAED pattern of the second sample (Fig. 3(g)) shows different crystallographic planes of the HA phase, i.e., (101), (112), (212), and (312), but the HRTEM image (Fig. 4(h)) displays in addition lattice fringes corresponding to the (104) and (006) planes of calcite. This finding indicates that even after increasing the digestion temperature, a small amount of calcite, not detected by XRD, is present in the samples. Whether it is untransformed bCCP or precipitated crystals in the experiment is not possible to be distinguished by microscopy. Most likely, they correspond to freshly precipitated calcite due to oscillations in the solution saturation index at the stationary state (dynamic) once the full transformation is attained. Note in the simulated experiment of Fig. S7 (ESI†) that after full transformation, the solution remains only slightly undersaturated with respect to calcite.
3.3. Crystal size distribution and electrophoretic mobility (ζ-potential) of the HA particles
The crystal size distribution (CSD) and electrophoretic mobility (ζ-potential) against pH of aqueous suspensions of both Oy-bCCP and HA-derived particles are relevant properties to assess their potential biomedical applications as carriers for drug delivery or as osteoinductive implantable bone filling materials. The trend of a colloidal suspension to disperse in simulated physiological conditions of the blood (pH∼7.4), in a tumor microenvironment, or in an inflamed tissue (pH 5–6) depends on the size and surface charge of their particles. While the dispersion of the colloidal particles is essential for their use as carriers of chemotherapeutic or anti-inflammatory drugs,18,70 it seems a priori not a crucial feature for their use as an osteoinductive implantable material.The CSD is here plotted as both volume-based (Fig. 5(a) and (d)) and cumulative volume-based distribution (Fig. 5(b) and (e)) because these latter plots allow measuring the percentiles D10, D50 and D90 of the distribution. The percentiles characterize the percentage of cumulative volume undersize distribution. D10 is close to the individual particle size observed microscopically, while D50 (the median of the population) and D90 are influenced by the aggregation of particles. We can observe the big reduction of the particle size experienced when transforming the Oy-bCCP particles to HA, with D10 reducing from 2270 nm to 99 nm (ratio P/bCCP 0.6) or to 10 nm (ratio P/bCCP 0.96) in both experiments using KH2PO4 at 200 °C. The influence of P/bCCP ratio, P reagent, and T on the percentiles is evident. The experiments with a P/bCCP ratio of 0.96 at 200 °C yielded a lower particle size with negligible aggregation, with D10, D50 and D90 percentiles equal to 10, 61 and 67 nm, respectively, when using KH2PO4 and 41, 67 and 70 nm when using K2HPO4. However, by decreasing the temperature to 120 °C and 80 °C, the percentiles increased greatly. In experiments with a P/bCCP ratio of 0.6, there is not a clear trend. Using this molar ratio, both the particle size and aggregation tendency are higher. Following the discussion, the particles prepared at a P/bCCP ratio of 0.96 at 200 °C could find applications as drug carriers, while those prepared at a P/bCCP ratio of 0.6 could find applications as implantable materials. For these particles, we have studied their osteoinductive properties.
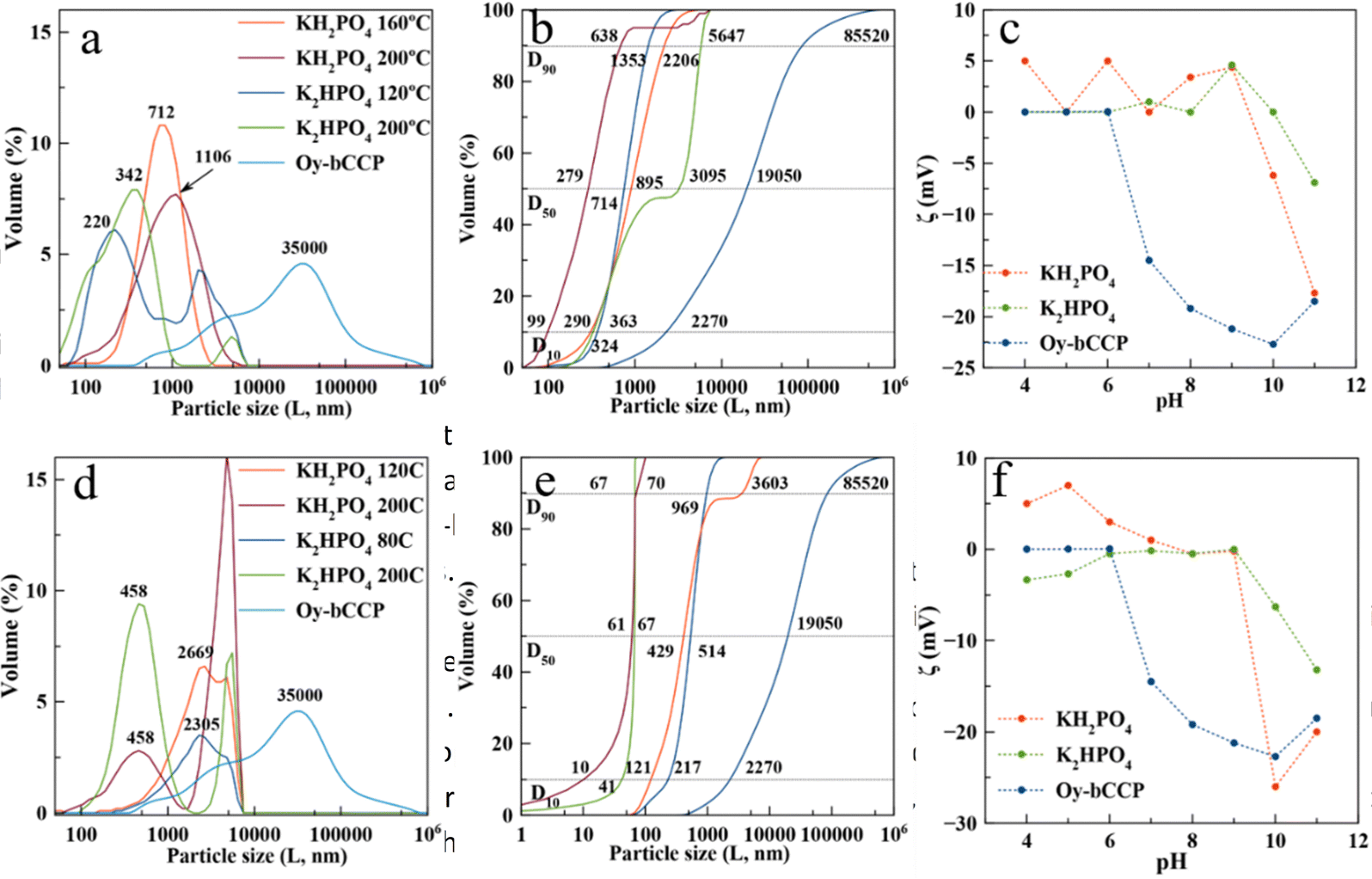 | ||
| Fig. 5 Volume-based CSD (a) and (d), cumulative volume-based CSD (b) and (e), and ζ-potential versus pH (c)–(f) of Oy-bCCP and HA particles prepared at different temperatures using KH2PO4 and K2HPO4 and P/bCCP molar ratios of 0.6 (a)–(c), and 0.96 (d)–(f). Note that CSDs of HA and Oy-bCCP particles, though plotted in the same figures, were measured by different techniques, i.e., DLS and laser diffraction, respectively. | ||
4. Biological tests
4.1. Cytocompatibility of the Oy-bCCP and HA-derived nanoparticles
The cytocompatibility of Oy-bCCP and HA-derived NPs was tested in an MTT assay on the MG-63 human osteosarcoma cells and on the m17.ASC murine mesenchymal stem cells after 3-day incubation at different concentrations ranging from 100 to 0.1 μg mL−1. No significant toxicity was observed on MG-63 cells at any particle concentration (Fig. 6(A)) since in all cases, the cell viability is higher than 85%.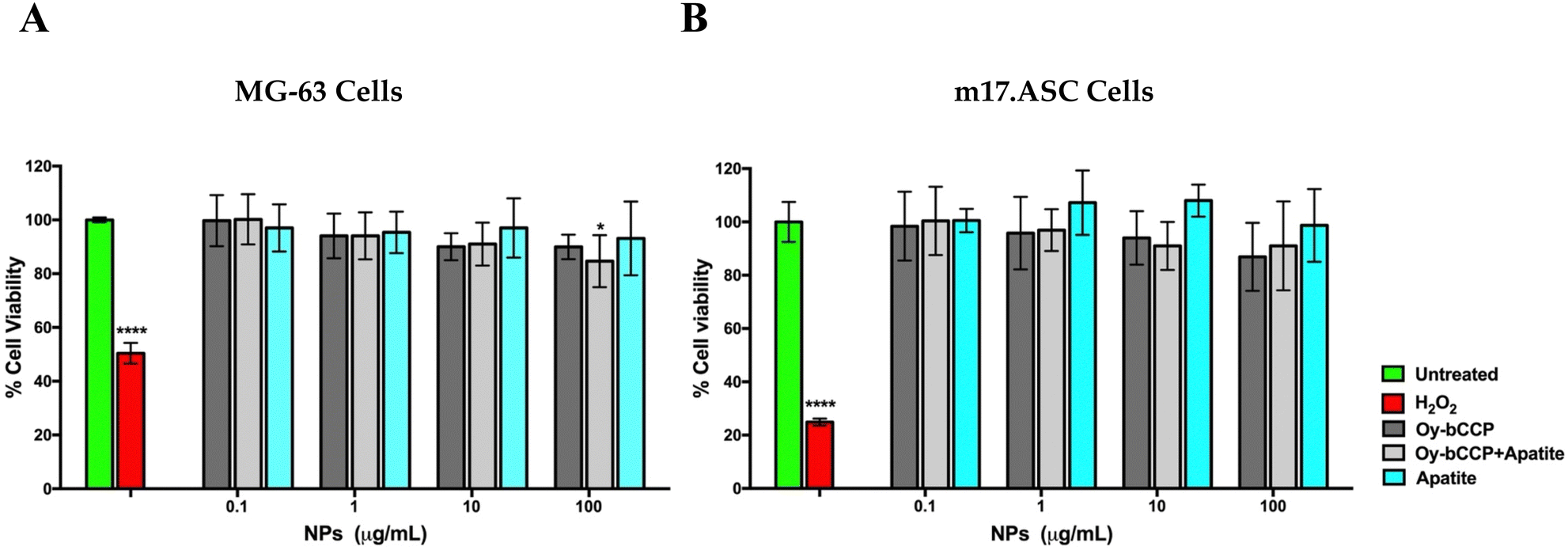 | ||
| Fig. 6 Viability of MG-63 (A) and of m17.ASC cells (B) incubated for three days with different nanoparticles derived from the oyster shell and with different concentrations. Viability was assessed in MTT assays. Data represent means ± SD of three independent experiments performed in triplicate, and statistical analyses were carried out using one-way ANOVA, with Bonferroni comparison test. For statistical analysis, all data were compared to untreated samples (*p < 0.05, ****p < 0.0005). | ||
On the other hand, the mesenchymal stem cells are more sensitive to the inorganic particles than the osteosarcoma cell line, especially when they were incubated with the highest particle concentration of 100 μg mL−1 (Fig. 6(B)). Nevertheless, in all cases, the cell viability was always higher than 80%, which is above the cut-off of 70% indicated by ISO 10993-5:2009.71 As expected, all the cells were affected by the addition of 1 μM hydrogen peroxide, which reduced their viability to less than 50%. Thus, these data show the good cytocompatibility of the different Ca-based materials that will be used in this study focused on the osteogenic differentiation of the mesenchymal stem cells.
4.2. HA particles derived from oyster shells induce osteogenesis and mineralization in m17.ASC cells
Since calcium-based nanomaterials play a critical role in the osteogenesis process,72,73 we analyzed the ability of Oy-bCCP and apatite-derived nanoparticles to induce the osteogenic differentiation of the mesenchymal cell line m17.ASC. It has been reported that the increase of ALP enzyme activity is a pivotal event during the early stage of the osteogenesis and generally reaches the maximum level at 2 weeks.74 Hence, after m17.ASC cells were incubated with 25 μg mL−1 (minimum effective dose reported in the literature) of particles for 14 days in the absence of osteogenic stimulators, we evaluated the ALP activity by the staining assay.As shown in Fig. 7(A), a strong violet signal, typical of ALP staining, was visualized in both particle-treated groups, in contrast to untreated cells. In fact, the quantitative analysis (Fig. 7(B)) showed that the intensity of violet staining associated with the particles (7000 and 9000 a.u. for Oy-bCCP and apatite, respectively) is nearly comparable to the signal detected (12000 a.u.) in the case of differentiated cells with the osteogenic medium. Thus, these findings suggest that the tested particles could play a role at early stages of the osteogenesis process. As a matter of fact, mineralization is another hallmark of the osteogenesis contributing to the final stage of the differentiation process, which is characterized by the calcium salt deposits forming the bone matrix.75 Therefore, we evaluated whether Oy-bCCP and apatite particles were also able to induce the mineralization in cell line m17.ASC performing the Alizarin Red staining after the cells underwent the same treatments without or with the particles as described above. The formation of the mineralized matrix, which was visualized as a red nodule-like staining, was not only detectable in differentiated cells with osteogenic factors but also detectable when cells were incubated with both types of NPs (Fig. 8(A)). For quantitative analysis, we measured the intensity of ARS extracted from the stained wells and expressed it as the optical density (O.D.). It is clear from Fig. 7(B) that particle-treated groups attained O.D. values similar to the positive control. Thus, the data obtained after 14 days of incubation illustrate that both Oy-bCCP and apatite particles significantly increased the mineralized matrix depositions in cells compared to the untreated group (negative control). Taken together, these results suggest that these calcium-based materials derived from oyster shells benefit the osteogenic capacity even in the absence of osteogenesis stimulators. Two recent studies have tested the osteoinductive properties of calcium-based materials derived from oyster shells, indicating the interest in reusing this waste material in the biomaterials field. In the first one, Coringa et al.76 tested the internal layer of the shell (i.e., nacre) of the species Crassostrea rhizophora from northeastern Brazil as a bone substitute material and concluded that nacre presents good biocompatibility and promoted the stimulation for bone-forming cells. In the second one, Ho et al.77 produced biphasic calcium phosphate (HA/β-TCP) granules from oyster shells and found that this material was able to promote the differentiation of induced pluripotent stem cells.77 Based on the results of the current work, the bCCP recovered from Crassostrea gigas oyster shells and the HA-derived particles can be useful osteoinductive platforms for translational applications in bone tissue engineering.
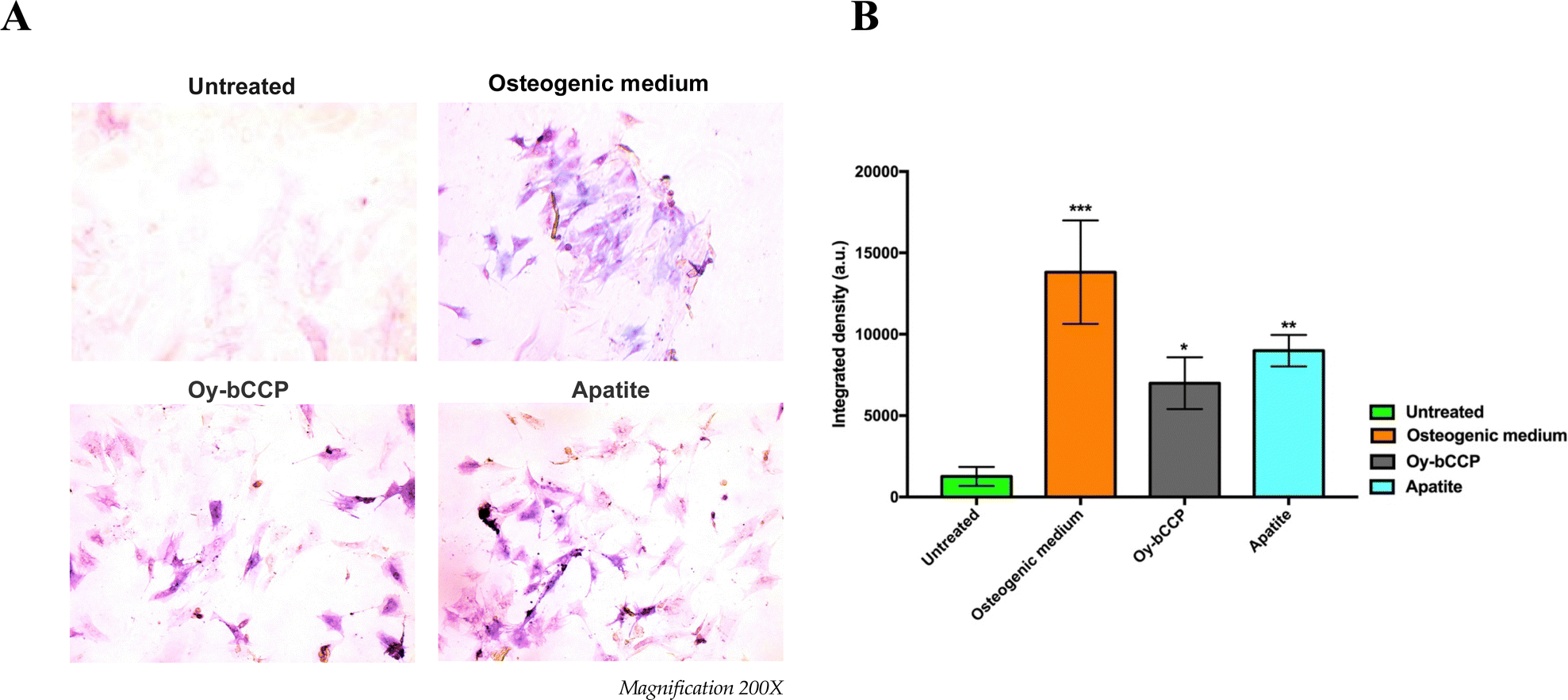 | ||
| Fig. 7 Effects of Oy-bCCP and apatite particles on ALP activity of m17.ASC cells after 14 days incubation with 25 μg mL−1 particles or with the osteogenic induction medium used as a positive control. (A) Representative images of ALP staining assay obtained at optical microscopy. Magnification 200X. (B) Quantitative analysis of ALP activity obtained by counting ALP spots with ImageJ software. Data representing three independent experiments are reported in the histogram as an average (A.U. ± SD), and statistical analyses were carried out using one-way ANOVA, with Bonferroni comparison test. Significance was considered as follows: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
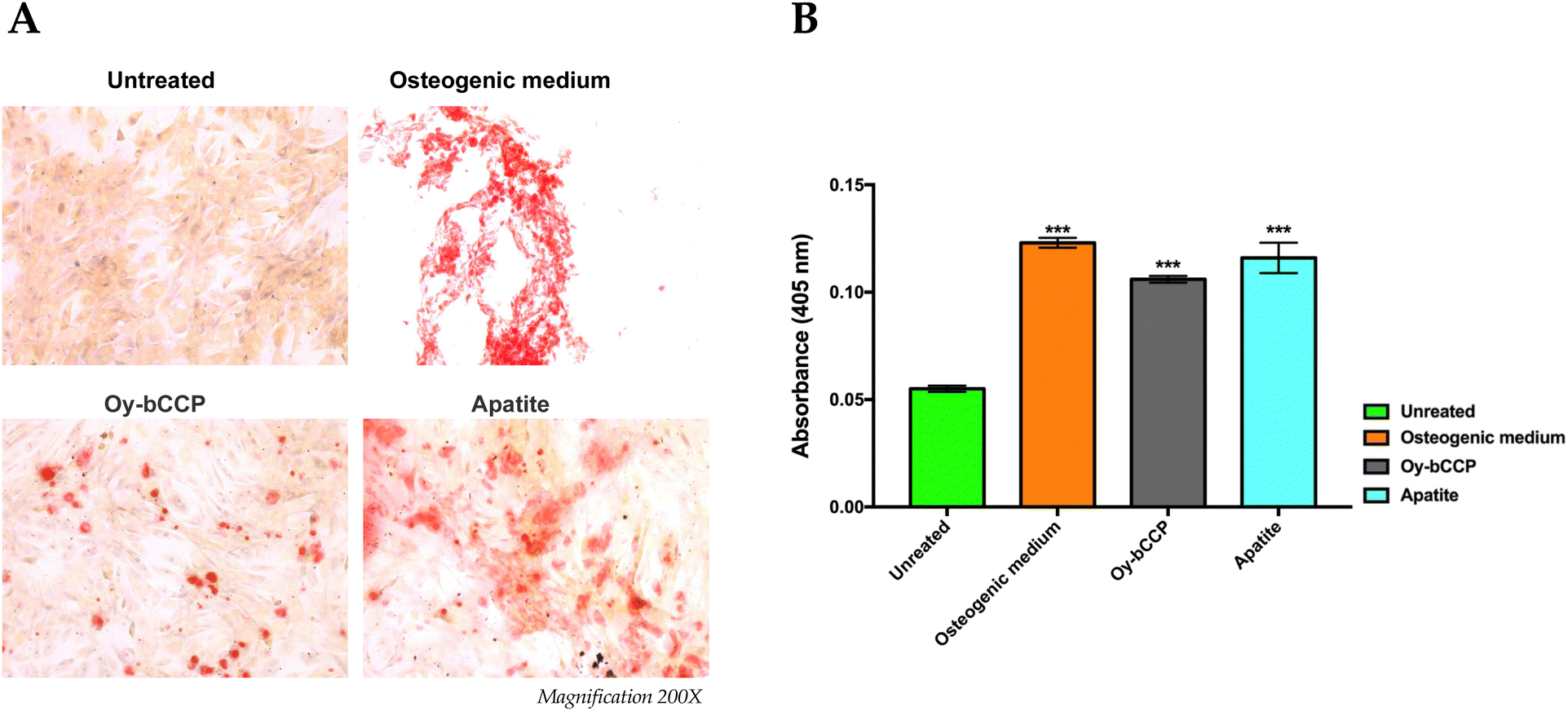 | ||
| Fig. 8 Effects of Oy-bCCP and apatite nanoparticles on the mineralization process of m17.ASC cells. (A) The extracellular calcium deposition was visualized by Alizarin Red S staining after cells were cultured with 25 μg mL−1 particles or with the osteogenic induction medium used as a positive control. Magnification 200x. (B) Mineralization was quantified following the colorimetric analysis of Alizarin Red S solution from calcium deposition. Results were obtained in three independent experiments made in triplicates, and they were reported in the histogram as an average ± SD of the optical density measured in a multiwell reader at 405 nm. For statistical analysis, all data were compared to untreated samples using one-way ANOVA, with Bonferroni comparison test (***p < 0.001). | ||
5. Conclusions
We have set up a one-pot low-temperature hydrothermal method to transform oyster shell bCCP particles into HA (Mg-doped) micro-nanoparticles. The following crystallization parameters were screened to find the minimum temperature allowing complete transformation: P reagent (H3PO4, KH2PO4, and K2HPO4), P/bCCP molar ratio (0.24, 0.6, and 0.96), temperature (25–200 °C) and time (1 week–2 months) in the absence of any additive and without pH adjustment. The digestion temperature significantly reduced from 160 °C to 120 °C by using K2HPO4 instead of KH2PO4 in experiments with a P/bCCP molar ratio of 0.6 and even to 80 °C when increasing the P/bCCP molar ratio to 0.96. The P reagent also influenced the HA morphology and its carbonation type. The transformation process followed a dissolution–reprecipitation mechanism driven by the favorable balance between HA precipitation and CaCO3 dissolution due to the lower-solubility product of HA than that of calcite at any of the temperatures tested. Both the bCCP and the derived HA particles were cytocompatible when incubated with MG-63 human osteosarcoma cells and with the m17.ASC murine mesenchymal stem cells. In addition, both materials promoted the osteogenic differentiation of the m17.ASC cells in the absence of osteogenic differentiation media, especially the HA particles. We conclude that due to their biological features, both particles can be employed as osteoinductive platforms with applications in bone tissue engineering.Author contributions
Conceptualization: J. G.-M., G. F., H. C., and G. M.; investigation: R. F.-P., C. T., C. V.-E., S. G., A. D., and F. O.; methodology: R. F.-P., C. T., C. V.-E., S. G., A. D., F. O., and A. F.; supervision: J. G.-M. and A. F.; writing – original draft preparation: J. G.-M.; writing – review and editing: J. G.-M., C. T., G. F., and H. C.; funding acquisition: J. G.-M., G. F., H. C., and G. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by project PCI2020-112108 funded by MCI/AEI/10.13039/501100011033 (Spain) and the European Union “NextGenerationEU”/PRTR”. PCI2020-112108 is part of the CASEAWA project of the ERA-NET Cofund BlueBio Programme, supported by the European Union (H2020), (Grant Agreement ERA-NET no. 817992). The authors acknowledge the Scientific Instrumentation Centre (CIC) of the University of Granada for FESEM, TEM, HRTEM and TGA characterization. This article is dedicated to the memory of Maria Prat, who promoted the realization of biological tests.References
- J. P. Morris, T. Backeljau and G. Chapelle, Rev. Aquacul., 2019, 11, 42 CrossRef.
- Anonymous, The State of World Fisheries and Aquaculture, 2020, 1-206, I-XIII.
- Y. Hou, A. Shavandi, A. Carne, A. A. Bekhit, T. B. Ng, R. C. F. Cheung and A. E. A. Bekhit, Crit. Rev. Environ. Sci. Technol., 2016, 46, 1047 CrossRef CAS.
- K. H. Mo, U. J. Alengaram, M. Z. Jumaat, S. C. Lee, W. I. Goh and C. W. Yuen, Constr. Build. Mater., 2018, 162, 751–764 CrossRef CAS.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS.
- D. Suteu, D. Bilba, M. Aflori, F. Doroftei, G. Lisa, M. Badeanu and T. Malutan, Clean: Soil, Air, Water, 2012, 40, 198–205 CAS.
- P. Morseletto, Resour., Conserv. Recycl., 2020, 153, 104553 CrossRef.
- D. E. Jacob, A. L. Soldati, R. Wirth, J. Huth, U. Wehrmeister and W. Hofmeister, Geochim. Cosmochim. Acta, 2008, 72, 5401 CrossRef CAS.
- A. Hart, Waste Manage. Res., 2020, 38, 514 CrossRef.
- C. McLaughlan, P. Rose and D. C. Aldridge, Environ. Manage., 2014, 54, 1102 CrossRef.
- A. O. Oso, A. A. Idowu and O. T. Niameh, J. Anim. Physiol. Anim. Nutr., 2011, 95, 461 CrossRef CAS.
- C. H. Lee, D. K. Lee, M. A. Ali and P. J. Kim, Waste Manage., 2008, 28, 2702 CrossRef CAS PubMed.
- E.-I. Yang, S.-T. Yi and Y.-M. Leem, Cem. Concr. Res., 2005, 35, 2175 CrossRef CAS.
- W.-T. Kuo, H.-Y. Wang, C.-Y. Shu and D.-S. Su, Constr. Build. Mater., 2013, 46, 128 CrossRef.
- J. Gómez-Morales, M. Iafisco, J. M. Delgado-López, S. Sarda and C. Drouet, Prog. Cryst. Growth Charact. Mater., 2013, 59, 1 CrossRef.
- A. Ressler, A. Žužić, I. Ivanišević, N. Kamboj and H. Ivanković, Open Ceram., 2021, 6, 100122 CrossRef CAS.
- M. Okada and T. Furuzono, Sci. Technol. Adv. Mater., 2012, 13, 064103 CrossRef CAS.
- S. M. Cano Plá, A. D’Urso, J. F. Fernández-Sánchez, D. Colangelo, D. Choquesillo-Lazarte, R. Ferracini, M. Bosetti, M. Prat and J. Gómez-Morales, Nanomaterials, 2022, 12, 562 CrossRef.
- J. Gómez-Morales, C. Verdugo-Escamilla, R. Fernández-Penas, C. M. Parra-Milla, C. Drouet, F. Maube-Bosc, F. Oltolina, M. Prat and J. F. Fernández-Sánchez, RSC Adv., 2018, 8, 2385 RSC.
- J. Gómez-Morales, R. Fernández-Penas, F. J. Acebedo-Martínez, I. Romero-Castillo, C. Verdugo-Escamilla, D. Choquesillo-Lazarte, L. D. Esposti, Y. Jiménez-Martínez, J. F. Fernández-Sánchez and M. Iafisco, Nanomaterials, 2022, 12, 1257 CrossRef.
- J. Knaus, D. Schaffarczyk and H. Cölfen, Macromol. Biosci., 2020, 20, 1900239 CrossRef CAS.
- Proficient Market Insights.
- A. Szcześ, L. Hołysz and E. Chibowski, Adv. Colloid Interface Sci., 2017, 249, 321 CrossRef.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 715 CrossRef CAS PubMed.
- Y. Cai, Y. Liu, W. Yan, Q. Hu, J. Tao, M. Zhang, Z. Shi and R. Tang, J. Mater. Chem., 2007, 17, 3780 RSC.
- N. K. Nga, N. T. Thuy Chau and P. H. Viet, Colloids Surf., B, 2018, 172, 769–778 CrossRef CAS.
- S. Scialla, F. Carella, M. Dapporto, S. Sprio, A. Piancastelli, B. Palazzo, A. Adamiano, L. Degli Esposti, M. Iafisco and C. Piccirillo, Mar. Drugs, 2020, 18, 309 CrossRef CAS.
- A. Pal, S. Maity, S. Chabri, S. Bera, A. R. Chowdhury, M. Das and A. Sinha, Biomed. Phys. Eng. Express, 2017, 3, 015010 CrossRef.
- S. Hajar Saharudin, J. Haslinda Shariffuddin, N. Ida Amalina Ahamad Nordin and A. Ismail, Mater. Today: Proc., 2019, 19, 1208 CrossRef CAS.
- S. Rujitanapanich, P. Kumpapan and P. Wanjanoi, Energy Procedia, 2014, 56, 112 CrossRef CAS.
- A. Shavandi, A. E.-D. A. Bekhit, A. Ali and Z. Sun, Mater. Chem. Phys., 2015, 149–150, 607 CrossRef CAS.
- Y. Xu, D. Wang, L. Yang and H. Tang, Mater. Charact., 2001, 47, 83–87 CrossRef CAS.
- H. Ivankovic, E. Tkalcec, S. Orlic, G. Gallego Ferrer and Z. Schauperl, J. Mater. Sci.: Mater. Med., 2010, 21, 2711 CrossRef CAS.
- G. Magnabosco, D. Giuri, A. P. Di Bisceglie, F. Scarpino, S. Fermani, C. Tomasini and G. Falini, ACS Sustainable Chem. Eng., 2021, 9, 6203 CrossRef CAS.
- A. G. Checa, E. M. Harper and A. González-Segura, Sci. Rep., 2018, 8, 7507 CrossRef PubMed.
- R. Higuera-Ruiz and J. Elorza, Estuarine, Coastal Shelf Sci., 2009, 82, 201 CrossRef CAS.
- S.-C. Wu, H.-C. Hsu, S.-K. Hsu, C.-P. Tseng and W.-F. Ho, Adv. Powder Technol., 2017, 28, 1154 CrossRef CAS.
- Z. Wang, S. Jiang, Y. Zhao and M. Zeng, Mater. Sci. Eng., C, 2019, 105, 110102 CrossRef CAS.
- S. Santhosh and S. Balasivanandha Prabu, Mater. Lett., 2013, 97, 121 CrossRef CAS.
- B. N. Alhussary, G. A. Taqa and A. A. A. Taqa, J. Appl. Vet. Sci., 2020, 5, 25 Search PubMed.
- K. Salma-Ancane, L. Stipniece and Z. Irbe, Ceram. Int., 2016, 42, 9504 CrossRef CAS.
- D. L. Goloshchapov, A. S. Lenshin, D. V. Savchenko and P. V. Seredin, Results Phys., 2019, 13, 102158 CrossRef.
- M. A. M. Castro, T. O. Portela, G. S. Correa, M. M. Oliveira, J. H. G. Rangel, S. F. Rodrigues and J. M. R. Mercury, Bol. Soc. Esp. Ceram. Vidrio, 2022, 61, 35 CrossRef CAS.
- J. R. M. Ferreira, L. H. L. Louro, A. M. Costa, J. B. De Campos and M. H. Silva, Cerâmica, 2016, 62, 386 CrossRef CAS.
- D. F. Fitriyana, R. Ismail, Y. I. Santosa, S. Nugroho, A. J. Hakim and M. Syahreza Al Mulqi, in 2019 International Biomedical Instrumentation and Technology Conference (IBITeC), 2019, 1, 7.
- G. S. Kumar, L. Sathish, R. Govindan and E. K. Girija, RSC Adv., 2015, 5, 39544 RSC.
- G. S. Kumar, A. Thamizhavel and E. K. Girija, Matter. Lett., 2012, 76, 198 CrossRef CAS.
- S.-C. Wu, Y.-L. Kao, Y.-C. Lu, H.-C. Hsu and W.-F. Ho, J. Aust. Ceram. Soc., 2021, 57, 1541–1551 CrossRef CAS.
- P. Agalya, G. Suresh Kumar, K. M. Prabu, S. Cholan, G. Karunakaran, J. Hakami, M. Shkir and S. Ramalingam, Ceram. Int., 2022, 48, 28299–28307 CrossRef CAS.
- F. Cestari, F. Agostinacchio, A. Galotta, G. Chemello, A. Motta and V. M. Sglavo, Nanomaterials, 2021, 11, 264 CrossRef CAS PubMed.
- S. Balu, M. V. Sundaradoss, S. Andra and J. Jeevanandam, Beilstein J. Nanotechnol., 2020, 11, 285 CrossRef CAS PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210 CrossRef CAS.
- A. Zamperone, S. Pietronave, S. Merlin, D. Colangelo, G. Ranaldo, E. Medico, F. Di Scipio, G. N. Berta, A. Follenzi and M. Prat, Stem Cells Dev., 2013, 22, 2873–2884 CrossRef CAS.
- J. Gómez-Morales, R. Fernández-Penas, I. Romero-Castillo, C. Verdugo-Escamilla, D. Choquesillo-Lazarte, A. D’Urso, M. Prat and J. F. Fernández-Sánchez, Nanomaterials, 2021, 11, 322 CrossRef PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato and S. Bicciato, Nature, 2011, 474, 179 CrossRef CAS PubMed.
- C. Triunfo, S. Gärtner, C. Marchini, S. Fermani, G. Maoloni, S. Goffredo, J. Gomez Morales, H. Cölfen and G. Falini, ACS Omega, 2022, 7, 43992 CrossRef CAS PubMed.
- J. M. Delgado-López, M. Iafisco, I. Rodríguez, A. Tampieri, M. Prat and J. Gómez-Morales, Acta Biomater., 2012, 8, 3491 CrossRef.
- F. Marchegiani, E. Cibej, P. Vergni, G. Tosi, S. Fermani and G. Falini, J. Cryst. Growth, 2009, 311, 4219 CrossRef CAS.
- G. B. Ramírez-Rodríguez, J. M. Delgado-López and J. Gómez-Morales, CrystEngComm, 2013, 15, 2206 RSC.
- M. De La Pierre, C. Carteret, L. Maschio, E. André, R. Orlando and R. Dovesi, J. Chem. Phys., 2014, 140, 164509 CrossRef.
- Z. Zyman and M. Tkachenko, J. Eur. Ceram. Soc., 2011, 31, 241 CrossRef CAS.
- S. Barinov, J. Rau, S. N. Cesaro, J. Urisin, I. Fadeeva, D. Ferro, L. Medvecky and G. Trionfetti, J. Mater. Sci.: Mater. Med., 2006, 17, 597 CrossRef CAS.
- P. Ducheyne, K. E. Healy, D. W. Hutmacher, D. W. Grainger and C. J. Kirkpatrick, Comprehensive Biomaterials, Elsevier, 1st edn, 2011, vol. 1 Search PubMed.
- W. J. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. Bomans and P. M. Frederik, Nat. Commun., 2013, 4, 1507 CrossRef PubMed.
- X. Yang, C. Zhang, X. Yang and Z. Xu, J. Mol. Liq., 2023, 121585 CrossRef CAS.
- R. Z. LeGeros, Calcium phosphates in oral biology and medicine, Karger AG, Switzerland, Basel, 1991 Search PubMed.
- E. Landi, G. Logroscino, L. Proietti, A. Tampieri, M. Sandri and S. Sprio, J. Mater. Sci.: Mater. Med., 2008, 19, 239–247 CrossRef CAS PubMed.
- D. Predoi, S. L. Iconaru, M. V. Predoi, G. E. Stan and N. Buton, Nanomaterials, 2019, 9, 1295 CrossRef CAS.
- X. Yuan, B. Zhu, G. Tong, Y. Su and X. Zhu, J. Matter. Chem. B, 2013, 1, 6551–6559 RSC.
- M. Iafisco, J. M. Delgado-Lopez, E. M. Varoni, A. Tampieri, L. Rimondini, J. Gomez-Morales and M. Prat, Small, 2013, 9, 3834 CrossRef PubMed.
- ISO 10993-5 Biological Evaluation of Medical Devices Part 5: Tests for In Vitro Cytotoxicity, International Standard Organization, Geneva, Switzerland, 2009 Search PubMed.
- X. Yang, Y. Li, X. Liu, W. He, Q. Huang and Q. Feng, Nanoparticles and their effects on differentiation of mesenchymal stem cells, Biomater. Transl., 2020, 1, 58–68, DOI:10.3877/cma.j.issn.2096-112X.2020.01.006.
- W. Liang, P. Ding, G. Li, E. Lu and Z. Zhao, Drug Des., Dev. Ther., 2021, 15, 905 CrossRef.
- X. Yang, Y. Li, X. Liu, R. Zhang and Q. Feng, Stem Cells Int., 2018, 2036176 Search PubMed.
- C. Yi, D. Liu, C.-C. Fong, J. Zhang and M. Yang, ACS Nano, 2010, 4, 6439–6448 CrossRef CAS.
- R. Coringa, E. M. de Sousa, J. N. Botelho, R. S. Diniz, J. C. de Sá, M. C. F. N. da Cruz, M. A. B. Paschoal and L. M. Gonçalves, PLoS One, 2018, 13, e0198697 CrossRef.
- W.-F. Ho, M.-H. Lee, J. L. Thomas, J.-A. Li, S.-C. Wu, H.-C. Hsu and H.-Y. Lin, Int. J. Mol. Sci., 2021, 22, 9444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00856h |
| This journal is © The Royal Society of Chemistry 2023 |