
Highly modular hepatitis B virus-like nanocarriers for therapeutic protein encapsulation and targeted delivery to triple negative breast cancer cells†
Daniel
Yur
 ,
Millicent O.
Sullivan
* and
Wilfred
Chen
*
,
Millicent O.
Sullivan
* and
Wilfred
Chen
*
Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE, USA. E-mail: msullivan@udel.edu; wilfred@udel.edu
First published on 5th April 2023
Abstract
Protein therapeutics offer enormous clinical impact in treating a variety of diseases by offering high selectivity with limited off-target effects. However, delivery challenges severely hinder functional proteins from reaching their target cells and necessitate frequent administration. To address these problems, nanocarrier encapsulation can provide protease protection and enhanced targeted transportation of functional proteins to their intended disease site. Inspired by their viral analogues, virus-like particles (VLPs) are non-infectious viral capsids that have potential for drug delivery applications because of their shared structural characteristics, such as high loading capacity, particle stability, and structural uniformity. Here, we describe a modular hepatitis B virus (HBV) VLP delivery platform offering tunable modifications of both the exterior and interior viral capsid surfaces via SpyCatcher–SpyTag bioconjugation and a multi-expression system, respectively. This new platform facilitates modification with epidermal growth factor receptor (EGFR)-targeting proteins and encapsulation with both model green fluorescent protein (GFP) and prodrug-converting yeast cytosine deaminase (yCD) enzyme. The resultant targeted VLPs demonstrated enhanced uptake and toxicity in EGFR-overexpressing triple negative breast cancer (TNBC) cells in contrast to non-malignant breast epithelial cells.
Introduction
Protein therapeutics is one of the fastest growing markets in the healthcare sector due to the capacity of proteins to perform diverse functions and recognize biological targets with high specificity.1–3 Because many diseases result in global phenotypic anomalies, these therapies represent ideal candidates for treatment of cancers,4 genetic disorders,5,6 and autoimmune disorders.7 Despite the rapid development of protein biologics, delivery challenges remain a critical limitation.8,9 Most proteins exhibit limited membrane permeability, resulting in few protein drug therapies and necessitating the need for targeting moieties to aid in cellular internalization.10,11 Enhancing delivery could usher in the increased utilization of untapped intracellular drug targets in treating diseases.Nanoparticle delivery platforms improve drug biodistribution and pharmacokinetics and can be modified with targeting moieties for cell-specific uptake.12–15 Nanoparticle encapsulation provides the simultaneous delivery of multiple copies of a target protein cargo, which enhances the effective concentration at the delivery site. However, conventional liposomal and polymeric nanoparticles typically require non-aqueous synthesis conditions that may affect cargo protein stability, and they are often polydisperse in size and potentially toxic, yielding slow and non-uniform drug release and off-target cytotoxicity.16 Furthermore, current strategies for exterior and interior modifications with cell-surface binders and protein cargos, respectively, typically exhibit poor loading and site- and orientation-specific control, which limit the targeted delivery of therapeutics.17,18
In nature, viruses have developed an efficient delivery system because of their regular, multifunctional architecture and their desirable size for phagocytic evasion.19–21 Virus-like particles (VLPs) are non-infectious viral capsids that are ideal for drug delivery applications because of their shared structural characteristics and biocompatibility.22 Because of these advantages, VLPs are an emerging class of nanocarriers used to deliver many therapeutics, ranging from small molecule drugs to biologics, such as proteins and gene therapies.23–26 The well-characterized hepatitis B virus (HBV) VLP has been widely explored for protein delivery because of its regular solvent exposed features for incorporating surface modifications, a large interior cavity amenable for loading various cargos, and a diameter of 36 nm for favorable pharmacokinetics.27–35
While small proteins and peptides can be inserted into the surface loop of HBV,32,35–37 this strategy is limited by the physical dimensions of the cargos31,38 and by the correct assembly of the resultant VLPs.39 Chemical conjugation is an alternative for protein modification and typically occurs by exploiting reactive side groups of amino acids, such as cysteine or lysine,40–42 but lacks site- and orientation-specific control to retain the high-binding affinity with the corresponding target.43 Similarly, internal loading of smaller peptides and proteins is possible by direct genetic fusion to the HBV monomers;44 however, encapsulation of larger proteins typically requires re-assembly after urea denaturaion.33,45 The need for refolding is tedious and often limits the type of protein cargos that can be loaded.46
Herein, we developed a highly modular HBV VLP-based delivery platform capable of simultaneous interior and exterior modifications (Fig. 1). We exploited the robust SpyCatcher–SpyTag bio-click chemistry strategy to modify the exterior with binding moieties, resulting in a simple plug-and-play nanocarrier platform for specific cell targeting and delivery.47–50 We demonstrated that the exterior could be decorated with different SpyCatcher fusion proteins for VLP purification and cell targeting via a SpyTag inserted within the exposed exterior loop on the HBV monomer. Interior loading of multiple protein cargos was made possible by using a multi-expression system, wherein we tuned the expression levels of different HBV monomer–cargo protein fusions to alleviate the steric effects on VLP assembly. This new design was demonstrated for the simultaneous loading of green fluorescent protein (GFP) and the therapeutic protein yeast cytosine deaminase (yCD), which can convert a non-toxic prodrug 5-fluorocytosine (5-FC) into the chemotherapeutic 5-fluorouracil (5-FU).24,51 To enable cell-selective delivery to epidermal growth factor receptor (EGFR)-overexpressing triple negative breast cancer (TNBC) cells, an EGFR-binding designed ankyrin repeat protein (DARPin)52,53 was conjugated to the exterior of the VLP. The resultant targeted VLPs demonstrated up to 20-fold increase in uptake compared to their untargeted counterparts as well as non-malignant breast epithelial cells (MCF10A). Selective uptake of yCD-loaded VLPs resulted in significantly greater cell toxicity in TNBC cells when treated with 5-FC compared to the level of cell toxicity when the non-targeted VLP and 5-FC were co-delivered. Toxicity was also significantly greater using the targeted VLP/5-FC combination in TNBC cells vs. non-malignant control MCF10A cells. In the long term, the ability to tune both the exterior and interior decorations allows customizable control of cellular uptake and cancer cell killing using a range of therapeutic agents.
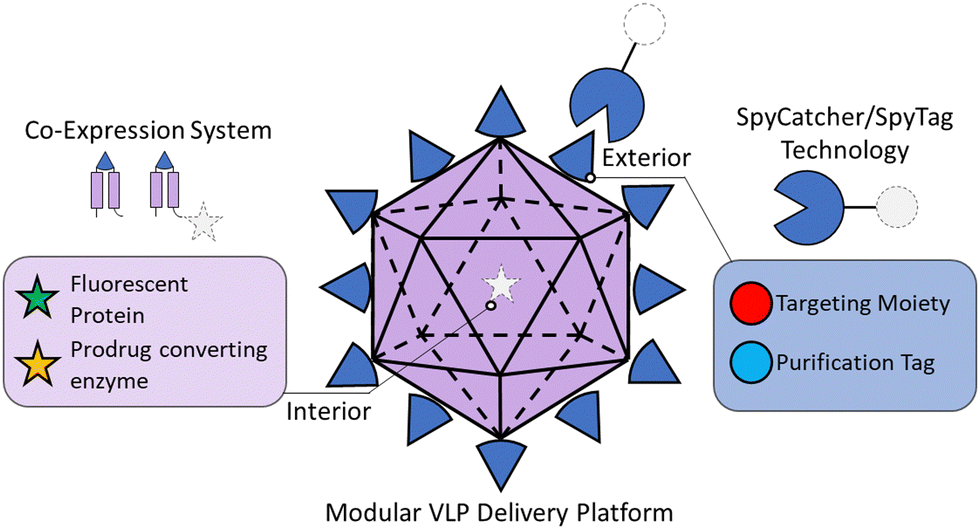 | ||
| Fig. 1 Schematic of modular virus-like particle nanocarrier. The VLP can be tunably modified on the interior with the multi-expression system and on the exterior with SpyCatcher/SpyTag technology. | ||
Materials and methods
Materials
DNA oligonucleotides used in polymerase chain reaction (PCR) were purchased from Integrated DNA Technologies (Coralville, IA, USA). Restriction enzymes, T4 DNA ligase, and Q5 DNA polymerase were purchased from New England Biolabs (Ipswich, MA, USA). Plasmid preparation and DNA clean-up kits were purchased from Zymo Research (Irvine, CA, USA) following DNA digestion and gel electrophoresis. All genetic manipulation was performed in the Escherichia coli strain NEB5α (New England Biolabs, Ipswitch, MA, USA) [fhuA2Δ(argF-lacZ)U169 phoA gln V44Φ80Δ(lacZ)M15 gyrA96 recA1 relA1 endA1thi-1 hsdR17] and protein expression was performed in BL21 (DE3) [F-ompT hsdSB(rB-mB-) gal dcm (DE3)] (EMD Millipore, Madison, WI, USA). All bacterial culture medium ingredients were purchased from Fisher Scientific (Pittsburgh, PA, USA). Antibiotics, isopropyl-β-D-1-thiogalactopyranoside (IPTG), L-arabinose, and anhydrotetracycline (aTc) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and reagents for SDS-PAGE were purchased from Bio-Rad (Hercules, CA). Dulbecco's Phosphate Buffered Saline (DPBS, 1×), Ham's F-12, Dulbecco's Modified Eagle's Medium/Ham's F12 50/50 Mix, and Leibovitz's L-15 Medium were purchased from Thermo Fisher Scientific (Grand Island, NY, USA).Construction of expression plasmids
All plasmid constructs were prepared using standard molecular cloning techniques and the DNA oligonucleotide sequences that were used are listed in Table S1 (ESI†). To clone HBVSpyTag, the HBV core capsid protein was obtained from the previously cloned HBV_SpyCatcher construct.25 HBV_SpyCatcher was cloned from a pET24a vector containing restriction enzymes sites, KpNI and BamHI, upstream and downstream of the SpyCatcher, inserted at the c/e1 immunodominant loop of the HBV capsid protein. SpyTag with N- and C-terminal flexible linkers was synthesized via annealing DNA oligos, KpNI-GGGSGGG-SpyTag-GG-BamHI (+) and KpNI-GGGSGGG-SpyTag-GG-BamHI (−) and the product was then phosphorylated with T4 polynucleotide kinase. The SpyCatcher was removed from the HBV capsid protein by digesting with KpNI and BamHI and the purified product was then ligated to the phosphorylated KpNI-GGGSGGG-SpyTag-GG-BamHI fragment.The ELP[AV-60]-SpyCatcher (ELP60-SpyCatcher) and ELP[KV8F-40]-SpyCatcher (ELP40-SpyCatcher) constructs were previously cloned.24 To clone DARPinEGFR-ELP[AV-60]-SpyCatcher (DARPin-ELP60-SpyCatcher), a previously cloned GE11-ELP[AV-60]-SpyCatcher was first digested with NcoI and NdeI to yield ELP60-SpyCatcher. The DARPinEGFR gene was amplified from a pET29a(+) plasmid containing Bim-DARPinEGFR (a gift from Professor David Baker52), with primers NcoI-DARPinEGFR_Fw and NdeI-S-(G4S)2-GGSA-DARPinEGFR_Rev. The purified product was then digested with NcoI and NdeI and ligated with ELP60-SpyCatcher.
To clone the dual-expression plasmid, HBVSpyTag-GFP was first cloned from HBVSpyTag. The entire pET24a-HBVSpyTag plasmid was amplified with SacI-HBVSpyTag_Fw and XhoI-HBVSpyTag_Rev and the purified PCR product was digested with XhoI and SacI to yield pET24a-HBVSpyTag-XhoI-SacI. A linker sequence (XhoI-A-(G3S)3-G4T-AgeI) was synthesized by annealing DNA oligos and GFP was then amplified with XhoI-GFP_Fw and SacI-His6-GFP_Rev and the purified product was digested with XhoI and SacI and ligated with XhoI-A-(G3S)3-G4T-AgeI to the digested pET24a-HBVSpyTag-XhoI-SacI vector, yielding HBVSpyTag-GFP. The dual expression plasmid was then synthesized via Gibson assembly, which stitched together the following purified DNA fragments generated by primers listed in Table S2 (ESI†): pET24a-HBVSpyTag vector, the araC expression cassette and pBAD promoter sequence, HBVSpyTag-GFP, and the rrn1 and rrn2 terminator sequences.
To develop the tri-expression system, a second plasmid containing HBVSpyTag-yCD2 was cloned by inserting the gene into a pLtetO1 plasmid with a p15A origin of replication. HBVSpyTag was digested from the dual expression system with SphI and AgeI and two yCD fragments were generated with AgeI-yCD_Fw and BamHI-G3-yCD_Rev, and BamHI-G4S-yCD_Fw and AvrII-His6-yCD_Rev. The yCD fragments were then digested by the pairs of restriction enzymes, AgeI/BamHI and BamHI/AvrII, respectively. The purified products were ligated together with the digested HBVSpyTag into the modified pLtetO1 vector with enzyme sites SphI and AvrII at the upstream and downstream ends of the multiple cloning site. For the alternative tri-expression system, HBVSpyTag-GFP was first cloned into the second plasmid in the tri-expression system by amplification with SphI-HBVSpyTag-GFP-h6 Fw and HBV-SpyTag-GFP-h6-AvrII Rev primers. The SphI-HBVSpyTag-GFP-h6-AvrII fragment and tri-expression pLtetO1 vector were digested with SphI and AvrII and then ligated to form the alterative pLtetO1_HBVSpyTag-GFP-h6 plasmid. To incorporate HBVSpyTag-yCD2 into the alternative dual expression plasmid used in the tri-expression system, AgeI-yCD2-AvrII was first generated by digesting the original pLtetO1_HBVSpyTag-yCD2-h6 plasmid with AgeI and AvrII and then ligated into the AgeI- and AvrII-digested HBVSpyTag(GFP) dual expression plasmid.
Protein expression
HBVSpyTag, HBVSpyTag(GFP), ELP60-SpyCatcher, ELP40-SpyCatcher, and DARPinEGFR-ELP60-SpyCatcher constructs were grown in Luria-Bertani Broth (LB) supplemented with 50 μg mL−1 kanamycin. HBVSpyTag(GFP,yCD2) constructs were grown in LB supplemented with 50 μg mL−1 kanamycin and 35 μg mL−1 chloramphenicol. All cultures were grown at 37 °C at 250 rpm to an OD600 of 0.7–1.0, after which HBVSpyTag, ELP40-SpyCatcher, ELP60-SpyCatcher, and DARPinEGFR-ELP60-SpyCatcher were expressed overnight (12–16 h) at 20 °C using 100 μM IPTG. Dual-expressed HBVSpyTag(GFP) was expressed overnight (12–16 h) at 20 °C using 100 μM IPTG, 0.2% L-arabinose, or 100 μM IPTG and 0.2% L-arabinose. HBVSpyTag(GFP,yCD2) and HBVSpyTag(yCD2,GFP) were expressed overnight (12–16 h) with0.2% L-arabinose and 1 mM IPTG or 0.2% L-arabinose, 1 mM IPTG, and 0.1 μg mL−1 aTc. After expression, cells were harvested by centrifugation at 3000g at 4 °C and resuspended in PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4 at pH 7.4) at an optical density (OD600) of 30. Cell lysis was performed with a Fisher Scientific sonicator (Pittsburgh, PA, USA) and the resulting lysate was clarified via centrifugation at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 10 min.
000g at 4 °C for 10 min.
Protein purification
ELP60-SpyCatcher and ELP40-SpyCatcher were purified via inverse transition cycling (ITC). Ammonium sulfate stock solution (3 M) was added to a final concentration of 0.5 M to precipitate ELPs. The proteins were centrifuged at 16000 g at 30 °C for 10 min. The supernatant was removed, and the pellet was incubated in cold PBS buffer for 1 h on ice and resuspended gently by pipetting. The protein was then centrifuged at 16000 g at 4 °C for 10 min to remove residual insoluble contaminants. The process of ITC was repeated a second time to further improve purity. Concentrations were measured using A280 via theoretical extinction coefficients.
DARPinEGFR-ELP60-SpyCatcher was purified via Ni-NTA immobilized metal ion chromatography (Thermo Scientific, Rockford, IL, USA). The column was equilibrated with 10 mM imidazole and lysate was bound to the column in PBS buffer with 10 mM imidazole. The column was washed with PBS buffer with 25 mM imidazole and then eluted with PBS buffer with 250 mM imidazole. The protein was then dialyzed into PBS buffer and the final concentration was quantified with A280 with theoretical extinction coefficients.
HBVSpyTag, HBVSpyTag(GFP), HBVSpyTag(GFP,yCD2), and HBVSpyTag(yCD2,GFP) samples were purified by ligating pure ELP60-SpyCatcher or ELP40-SpyCatcher and performing ITC on the reacted VLP products. The concentration of HBV proteins in the lysate was estimated by using Bradford assay (BioRad) and densitometry. The Bradford assay was performed based on the manufacturer's instruction through the construction of a BSA standard curve. Densitometry was performed with Commassie Blue-stained SDS-PAGE gels and using the Image Lab 5.1 software (BioRad), specifically the “Lands and Bands” tool that calculated the percentage of each protein band in the sample. The concentration of each HBV protein was quantified by multiplying the lysate protein concentration obtained from the Bradford assay with the band percentage determined from densitometry. For dual- and tri-expressed HBV samples, the total HBV protein was a summation of the concentration from each HBV species. SpyTagged HBV proteins in lysate were reacted with ELP40-SpyCatcher at a 1:2 or with ELP60-SpyCatcher at a 20:1 mole ratio at 4 °C overnight. Inverse transition cycling was performed by first adding 0.4 M ammonium sulfate to the reacted HBV proteins, followed by centrifugation at 10000g at 30 °C for 10 min. The supernatant was removed and the pellet was incubated in cold PBS buffer overnight at 4 °C. The pellet was gently resuspended and the protein was centrifuged at 16000g at 4 °C for 10 min and ITC was repeated a second time. Concentrations of the purified samples were assessed with Bradford assay and densitometry.
Sucrose gradient sedimentation was performed on VLP lysate samples using 10–60% sucrose. Equal volumes of 10%, 20%, 30%, 40%, 50%, and 60% sucrose and protein lysate were added into polypropylene tubes (Beckman-Coulter, Indianapolis, IN, USA) and centrifuged at 60000 rpm at 20 °C for 40 min in a SW60-Ti rotor (Beckman-Coulter). Fourteen fractions were then removed from the top of each tube and loaded into SDS-PAGE gel for analysis. Fractions that contained VLPs were dialyzed in PBS buffer and further assessed with TEM.
Transmission electron microscopy
HBVSpyTag, HBVSpyTag(GFP), HBVSpyTag(GFP,yCD2), and HBVSpyTag(yCD2,GFP) were imaged with TEM. Particle samples at 0.1 mg mL−1 total protein were added to carbon-coated copper grids that were glow discharged with a PELCO easiGlow (Ted Pella Inc., Redding, CA, USA). The grids were washed three times and stained with 2% uranyl acetate and then imaged with a Zeiss Libra 102 transmission electron microscope (Oberkochen, Germany) at 120 V.Targeted VLP assembly
SpyCatcher/SpyTag reactions were performed overnight at 4 °C at the described molar ratio of purified HBV and SpyCatcher proteins in PBS at pH 7.4. After assembly, densitometry was performed to assess the average ligation density of each decoration species. The band percentages were converted into molar percentages by dividing each by its respective molecular weight and then dividing the resultant number by the sum of the total molecular weight-normalized band percentages.E. coli growth retardation assay
E. coli strain GIA39 (F-thr-1 leuB6(Am) fhuA21 codA1 lacY1 tsx-95 glnX44(AS) λ-dadX3 pyrF101 his-108 argG6 ilvA634 thiE1 deoC1 glt-15) was used for the growth retardation assay.54 Briefly, 500 nM yCD protein samples were incubated with 1 mg mL−1 5-FC in a 50 μL reaction for 2 h at room temperature. Reaction samples were then added to GIA39 cultures at an OD600 of 0.05 and grown in a 37 °C shaker at 250 rpm for 4 hours. OD600 was measured at 4 h, subtracted from the initial OD600 to measure total E coli growth, and divided by the total E coli growth of the LB only control to yield the normalized E coli growth.Cell culture
MDA-MB-468 cells were purchased from ATCC (Manassas, Virginia, USA) and were cultured in Leibovitz's L-15 medium, supplemented with 10% FBS and 1% (v/v) penicillin/streptomycin. IBC SUM149 cells (a gift from Kenneth van Golen55) were grown in Ham's F12 medium, supplemented with 5% FBS, 1% (v/v) penicillin/streptomycin, 1% (v/v) glutamine, 5 μg mL−1 insulin, 2.5 μg mL−1 transferrin, 200 ng mL−1 selenium, and 1 μg mL−1 hydrocortisone according to previously established methods.51,56 MCF10A cells were purchased from ATCC and were cultured in 50/50 DMEM/Ham's F12 medium supplemented with 5% FBS, 1% (v/v) penicillin/streptomycin, 50 μg mL−1 bovine pituitary extract, 10 μg mL−1 insulin, 0.5 μg mL−1 hydrocortisone, 100 ng mL−1 cholera toxin, and 20 ng mL−1 epidermal growth factor.Uptake of HBV VLPs
Flow cytometry was used to quantify VLP uptake in MDA-MB-468, SUM149, and MCF10A cells. Briefly, cells were seeded in tissue culture treated 24-well plates (Corning Inc., Corning, NY, USA) at a density of 4 × 104 cells per well and incubated overnight at 37 °C. The medium was removed and cells were incubated with 50 nM, 1 μM, or 6 μM HBV proteins in OptiMEM for 1 or 2 h. Cells were washed three times with 1× DPBS and trypsinized. Following trypsinization, cells were neutralized with their respective cell media and centrifuged at room temperature for 4 min at 120 g. Cells were then resuspended in cold 1× DPBS and assessed by flow cytometry (NovoCyte, ACEA Biosciences Inc., San Diego, CA, USA). A 488 nm laser and 530 nm filter were used to measure the fluorescence intensity from each cell sample. The mean fluorescence intensity reported was calculated from three replicates.Prodrug treatment and cytotoxicity assays
Propidium iodide staining was used to assess the cytotoxicity of yCD delivery. In each well, 4 × 104 MDA-MB-468 cells were seeded in a tissue culture treated 24-well plate (Fisher Scientific, Pittsburgh, PA, USA) and incubated overnight. The medium was replaced and cells were incubated with 1 μM HBVSpyTag(GFP,yCD2) or HBVSpyTag(yCD2,GFP) protein in OptiMEM for 2 h at 37 °C. Cells were washed three times with 1× DPBS and trypsinized at 37 °C for 15 min. They were then neutralized with medium and pelleted at 120 g for 4 min at room temperature. Cells were resuspended in medium alone or medium supplemented with 1 mg mL−1 5-FC or 5-FU, seeded in a fresh well in a 24-well plate, and incubated for 48 h at 37 °C. Following incubation, cells were washed with 1× DPBS and trypsinized at 37 °C for 15 min. After neutralization with medium, cells were pelleted at 120 g for 4 min at room temperature and then stained with propidium iodide (Invitrogen, Waltham, MA, USA) using the manufacturer's protocol. Afterwards, mean cell fluorescence was assessed across three replicates with flow cytometry using the 488 nm laser and 675 nm filter.Results and discussion
SpyTag incorporation into the surface-exposed loop of HBV VLPs for site-specific conjugation
The HBV VLP is a commonly used carrier for protein delivery, biosensing,24,57 and vaccines,58 owing to its robust structure across a wide range of pH and temperature.59 The truncated form of the monomer at residue 149 (denoted here as HBV) has been shown to maintain the ability to self-assemble60,61 and was used to generate the VLPs in this study. While a SpyCatcher was successfully inserted into the surface exposed c/e1 loop for bioconjugation,24 we chose to replace it with a SpyTag to produce less steric strain during protein folding and capsid assembly (Fig. 1). The HBVSpyTag protein was expressed in E. coli and the soluble lysate was assessed with SDS-PAGE, which confirmed soluble expression with a band at 20 kDa (Fig. 2(A)). Sucrose gradient analysis of the soluble lysate demonstrated localization of HBVSpyTag proteins in fractions 7–10 (Fig. 2(B)), which suggests the presence of intact HBV capsids.45,62 To confirm that HBVSpyTag maintained the ability to self-assembly, fractions 7–10 were pooled and imaged with transmission electron microscopy (TEM) (Fig. 2(C)). Detection of intact HBVSpyTag particles of around 34 nm (Fig. S1A, ESI†) confirmed their assembly into VLPs.30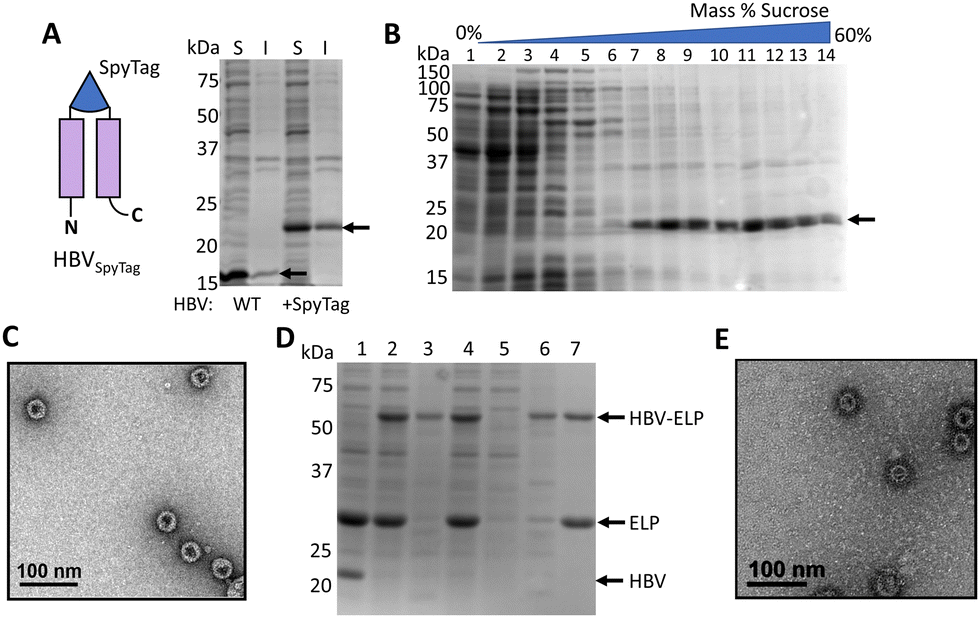 | ||
| Fig. 2 Incorporation of SpyTag into HBV capsid. (A) SDS-PAGE analysis of soluble (S) and insoluble (I) cell lysates, where full length proteins are denoted with an arrow (B) SDS-PAGE analysis after sucrose gradient analysis of soluble cell lysates; HBVSpyTag is denoted by an arrow. (C) TEM image of HBVSpyTag, pooled from fractions 7–10. (D) SDS-PAGE analysis of HBVSpyTag and ELP40-SpyCatcher ligation. (1) HBVSpyTag lysate and purified ELP40-SpyCatcher were mixed at a 1:2 molar ratio (t = 0). (2) Post-reaction protein sample. (3) Insoluble proteins after reaction. (4) Soluble proteins after reaction. (5) Soluble contaminants after ITC. (6) Insoluble contaminants after ITC. (7) Purified conjugates. (E) TEM image of purified conjugates after one cycle of ITC. | ||
To demonstrate that the HBVSpyTag VLPs were capable of bioconjugation, cell lysates were ligated with a ELP40-SpyCatcher fusion protein.24 Elastin-like polypeptides (ELP) were chosen because they can be used as a purification tag for rapid VLP purification due to the well-characterized inverse transition cycling (ITC) behavior.63–67 ELP40-SpyCatcher was mixed with HBVSpyTag cell lysate in a 2:1 molar ratio (Fig. 2(D)). Complete conversion of the HBVSpyTag protein into the corresponding conjugated (HBVSpyTag-ELP40) product was observed, confirming the activity of the inserted SpyTags. The protein products were subjected to one cycle of ITC, yielding only HBVSpyTag-ELP40 products and unreacted ELP40-SpyCatcher (Fig. 2(D)). Neither the SpyCatcher–SpyTag reaction nor ITC had any effect on capsid integrity as intact nanoparticles were detected with the purified VLPs (Fig. 2(E) and Fig. S1B, ESI†).
Protein encapsulation within HBV VLPs
While cargo proteins have been loaded within HBV VLPs by direct genetic fusion to the C-terminus of the HBV monomer,35,44 not all proteins can be incorporated successfully using this strategy due to size restrictions.68 To test the limitations of the C-terminal loading strategy, a small RNA-binding protein (19 kDa) from Carnation Italian Ringspot Virus, p19,69,70 was initially selected as a model cargo protein and genetically fused to the C-terminus of the HBV monomer. To assess encapsulation, HBV-p19 was expressed and soluble protein was analyzed by sucrose gradient (Fig. S2A and B, ESI†), which suggested assembly of some HBV-p19 proteins into intact nanostructures. However, upon imaging the pooled gradient fractions 8 and 9 with TEM, predominantly non-capsid aggregates were observed (Fig. S2C, ESI†), demonstrating that incorporation of 240 copies of even a small protein cargo within the VLP drastically affected capsid assembly. This is most likely due to steric effects of encapsulating a large number of protein cargoes simultaneously, which prevents efficient assembly of complete capsid structures.45,71To address the steric constraints of cargo encapsulation, we developed a multi-expression strategy to facilitate interior loading of multiple protein cargos based on co-assembly of both unmodified HBV monomers and modified HBV monomers fused with a cargo protein at the C-terminus. To implement this approach, we began with a dual-expression system to encapsulate GFP inside the HBVSpyTag VLP. This was accomplished by placing expression of HBVSpyTag and HBVSpyTag-GFP under the control of two different orthogonal inducible promoters, such that the expression of HBVSpyTag and HBVSpyTag-GFP could be tuned separately by the addition of arabinose and IPTG, respectively (Fig. 3(A)). By tuning the amount of IPTG and arabinose added, up to an average of 25% GFP per VLP was encapsulated as quantified by densitometry without overcrowding the VLP lumen (Fig. 3(B)). Intact assembly was confirmed after ligating the VLPs with ELP40-SpyCatcher (Fig. S3, ESI†) and undergoing one cycle of ITC before imaging the purified capsids with TEM (Fig. 3(C)). More importantly, the encapsulated GFP was functional as determined by measuring the fluorescence of the purified VLPs. Fluorescence was observed in all HBVSpyTag(GFP) samples, and the fluorescence signal was directly proportional to GFP loading (Fig. 3(D)), with signal from the non-arabinose induced VLPs likely resulting from leaky expression of HBVSpyTag-GFP.
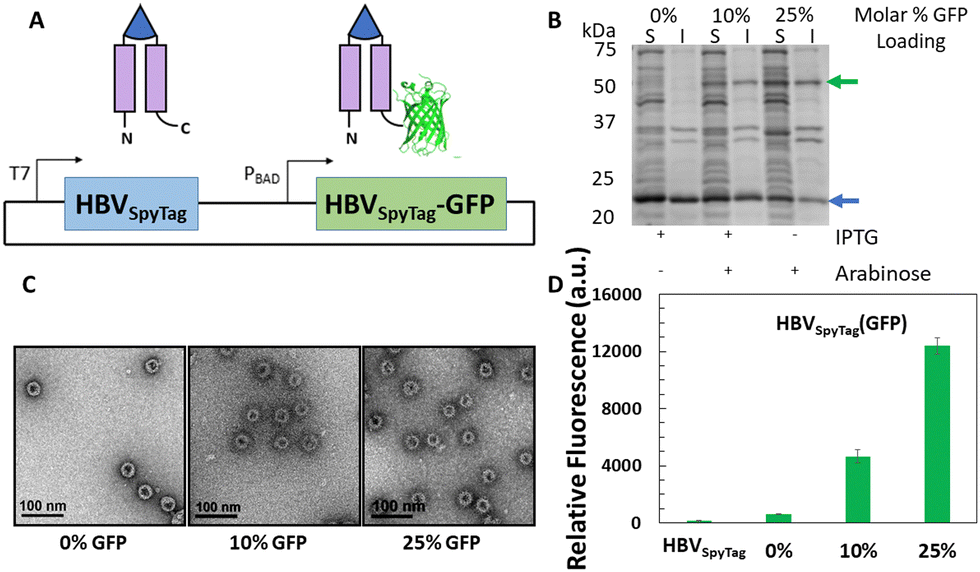 | ||
| Fig. 3 A dual-expression system for encapsulation of GFP in HBVSpyTag. (A) Schematic of the dual expression strategy. (B) SDS-PAGE analysis of dual expression in the soluble (S) and insoluble (I) cell lysates. By varying the expression conditions, up to 25% GFP was encapsulated within HBVSpyTag VLPs (green arrow - HBVSpyTag-GFP, blue arrow - HBVSpyTag). The percentages are the average per VLP. (C) ELP40-SpyCatcher ligated HBVSpyTag(GFP) samples were purified with one cycle of ITC and imaged with TEM. (D) Fluorescence levels of purified HBVSpyTag(GFP) VLPs. Data represent the mean values and error bars represent the standard deviation across three individual replicates for each sample. | ||
Simultaneous loading of multiple protein cargos within HBV VLPs
After demonstrating the tunable loading of GFP within the HBVSpyTag VLPs, we next extended the framework to simultaneous loading of GFP and a therapeutic protein. yCD, which can convert the non-toxic prodrug 5-fluorocytosine (5-FU) into the FDA-approved chemotherapeutic 5-fluorouracil (5-FU), was chosen.51,72 To encapsulate two proteins, we expanded the dual-expression system into a tri-expression system. Since yCD is a dimer, it was incorporated as a tandem fusion to HBVSpyTag to facilitate better folding within the VLP. To co-express HBVSpyTag-yCD2, it was placed under a tetracycline-inducible promoter in a second plasmid and co-transformed with the dual-expression plasmid (Fig. 4(A)). Expression of all proteins was confirmed with SDS-PAGE and Western blotting (Fig. 4(B)). From densitometry, the resulting VLPs (HBVSpyTag(GFP,yCD2)) were loaded with an average of 25% GFP and 3% yCD2. To confirm intact loading of both GFP and yCD2, HBVSpyTag(GFP,yCD2) VLPs were first purified via reaction with ELP40-SpyCatcher and one cycle of ITC (Fig. S4A, ESI†). TEM analysis (Fig. S4B, ESI†) confirmed the formation of intact capsid structures even after co-encapsulation of both GFP and yCD2. Similar fluorescence intensities were detected for HBVSpyTag(GFP) and HBVSpyTag(GFP,yCD2) with similar GFP loading, confirming that simultaneous loading of GFP and yCD2 had a negligible effect on GFP fluorescence (Fig. 4(C)). yCD activity was assessed using an E. coli growth retardation assay. Co-incubation of HBVSpyTag(GFP,yCD2) and 5-FC resulted in significant growth reduction, similar to that of 5-FU alone, indicating that 100% of the 5-FC was converted into 5-FU (Fig. 4(D)). In contrast, negligible effects were observed when incubating E. coli cells with HBVSpyTag(GFP) and 5-FC, protein only, and 5-FC only.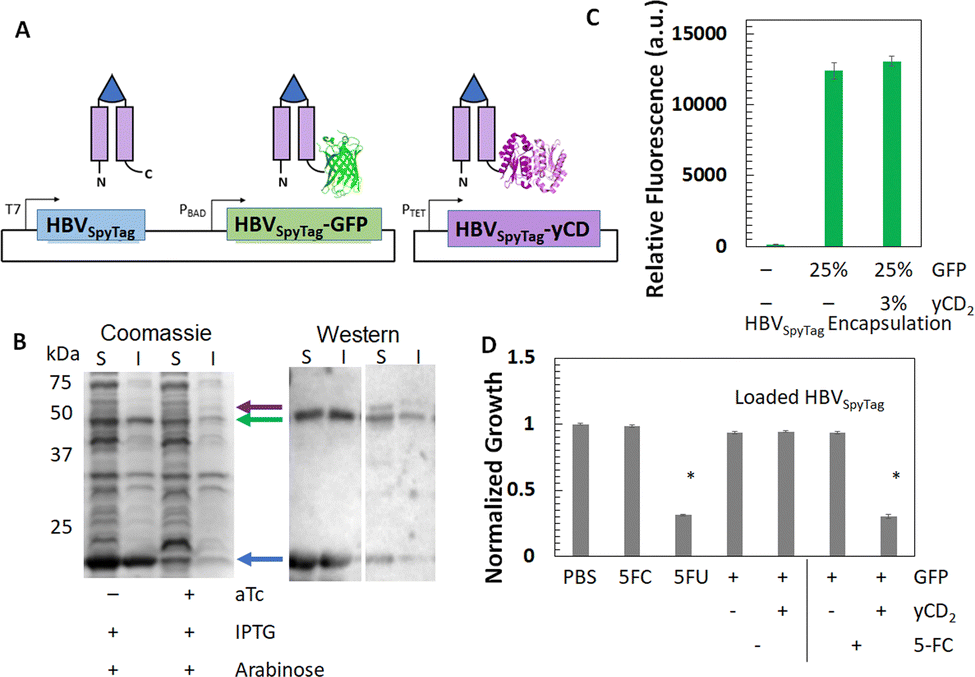 | ||
| Fig. 4 A tri-expression system for encapsulation of both GFP and yCD in HBVSpyTag VLPs. (A) Schematic of the tri-expression system. (B) Controlled expression of three VLP proteins depending on the induction conditions as visualized by SDS-PAGE (S – soluble lysate, I – Insoluble lysate) and Western blotting with an anti-his6 antibody for detection of his tags at the C-terminus of each HBV protein. (C) Fluorescence levels of purified HBVSpyTag(GFP,yCD2) VLPs compared to HBVSpyTag(GFP) VLPs with a similar GFP loading. The percentages are the average per VLP. (D) Normalized growth of E. coli when incubated with dual-expressed and tri-expressed VLPs with or without 5-FC. Controls using PBS, 5-FC and 5-FU were used for comparison where * denotes significance relative to other samples (*p < 10−4). Data represent the mean values and error bars represent the standard deviation across three individual replicates for each sample. | ||
Surface modification of VLPs with an EGFR-targeting DARPin for cancer cell-specific uptake
EGFR is over-expressed in many cancer cell types, such as TNBC cells, and is typically linked with poor prognosis.73–76 To minimize untargeted delivery, ELP60-SpyCatcher was ligated to VLPs at 40% density, which demonstrated the greatest reduction in non-specific uptake in SUM149 TNBC cells (Fig. S5, ESI†). To use EGFR for specific cancer cell targeting, we decorated VLPs with an antibody-mimetic DARPin, which was previously developed with phage display to target EGFR with a sub nanomolar affinity (Kd ∼ 0.5 nM).52,53 DARPinEGFR was first fused to the N-terminus of ELP60-SpyCatcher to improve solvent accessibility. Purified DARPinEGFR-ELP60-SpyCatcher and ELP60-SpyCatcher proteins (Fig. S6, ESI†) were then ligated to ELP-purified HBVSpyTag(GFP) and HBVSpyTag(GFP,yCD2) VLPs at a 10% and 25% conjugation density, respectively, as assessed by densitometry (Fig. S6, ESI†).Targeted and non-targeted HBVSpyTag(GFP) VLPs were delivered to two types of EGFR-overexpressing TNBC cell lines, MDA-MB-468 and SUM149, as well as a control non-malignant breast epithelial cell line, MCF10A. Significantly higher levels of uptake were detected using flow cytometry (Fig. 5(A)) for targeted HBVSpyTag(GFP) VLPs in both TNBC cell lines in contrast to their untargeted VLP counterparts. The higher level of uptake observed in MDA-MB-468 cells is consistent with the reported higher level of EGFR expression.74 Low levels of uptake were observed for both types of VLPs in MCF10A cells, demonstrating the effectiveness of DARPinEGFR to confer cell specificity. Similar delivery results were observed with the targeted HBVSpyTag(GFP,yCD2) VLPs with significantly greater levels of uptake to TNBC cells according to flow cytometry (Fig. 5(B)) than their untargeted counterparts. Once again, a negligible level of uptake was detected in MCF10A cells.
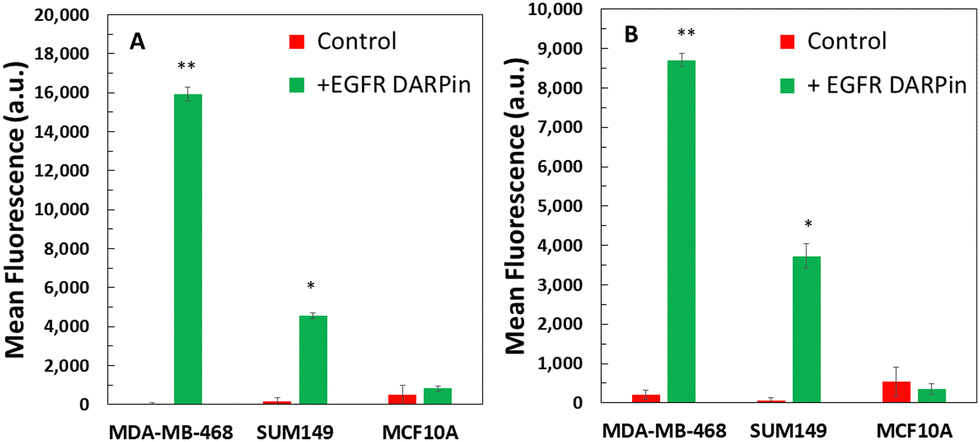 | ||
| Fig. 5 Targeted delivery to TNBC cells using DARPin-decorated VLPs. (A) Mean fluorescence intensities of untargeted (Control) and targeted (+EGFR DARPin) HBVSpyTag(GFP) uptake as measured with flow cytometry (*p < 0.01, **p < 0.001). (B) Mean fluorescence intensities of untargeted (control) and targeted (+EGFR DARPin) HBVSpyTag(GFP,yCD2) uptake as measured with flow cytometry (*p < 0.01, **p < 0.001). Data represent the mean values and error bars represent the standard deviation across three individual replicates for each sample. | ||
Selective delivery of DARPinEGFR-decorated HBVSpyTag(yCD2,GFP) VLPs leads to targeted cytotoxicity in MDA-MB-468 cells
We next investigated whether the delivery of bioactive yCD inside HBVSpyTag(GFP,yCD2) to MDA-MB-468 cells could elicit targeted toxicity. DARPinEGFR-decorated VLPs were first delivered to MDA-MB-468 cells to confirm biocompatibility with negligible effects in cell viability (Fig. 6(B)). When both targeted and untargeted VLPs were delivered to MDA-MB-468 cells, only the targeted VLPs exhibited a slight but insignificant cytotoxicity in MDA-MB-468 cells upon 5-FC addition. This lack of significant cytotoxicity even with the increased uptake of targeted VLPs can be attributed to insufficient conversion of 5-FC to 5-FU by the low level of encapsulated yCD (3%) to elicit a desirable therapeutic effect.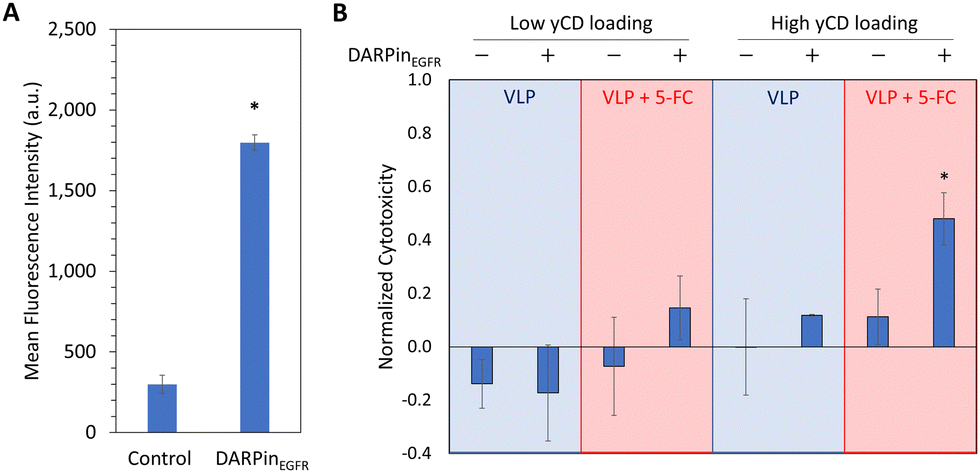 | ||
| Fig. 6 Targeted cytotoxicity in MDA-MB-468 cells achieved using DARPinEGFR-decorated HBVSpyTag(yCD2,GFP) VLPs. (A) Mean fluorescence intensities of untargeted (Control) and targeted (DARPinEGFR) HBVSpyTag(yCD2,GFP) uptake as measured with flow cytometry. Error bars are the standard deviation across three individual replicates. (*p < 10−5) (B) Normalized cytotoxicity was assessed by staining treated cells with propidium iodide after delivering VLP samples with or without 5-FC incubation. Normalized cytotoxicity is defined as the relative fluorescence intensity from propidium iodide staining with respective to PBS treatment (value of 0) and 5-FU treatment (value of 1). Error bars are the standard deviation from three individual replicates. * denotes statistical significance compared to all other samples (p < 0.05). | ||
One of the key benefits of the modular expression platform is the ability to tune the levels of encapsulated cargos. To improve the conversion of 5-FC to 5-FU, we increased the yCD2 loading within the VLP by exchanging the E. coli promoters controlling HBVSpyTag-GFP and HBVSpyTag-yCD2 expression (Fig. S7A, ESI†). Using a stronger pBAD promoter to control HBVSpyTag-yCD2 expression resulted in greater yCD loading in the VLP: an average of 40% yCD2 and 2% GFP from densitometry (Fig. S7B, ESI†). Because the low expression of HBVSpyTag-GFP was difficult to observe with SDS-PAGE, it was confirmed with the fluorescence assay as described above (Fig. S7C, ESI†). To prepare the high-yCD loaded VLP for targeted delivery, VLPs were purified by ITC after ligation with ELP60-SpyCatcher (Fig. S8A, ESI†). After confirming particle assembly with DLS (Fig. S8B, ESI†), purified VLPs were similarly modified with 10% DARPinEGFR-ELP60-SpyCatcher and 25% ELP60-SpyCatcher (Fig. S8C, ESI†). To investigate targeted cytotoxicity in TNBC cells, we first delivered high-yCD loaded VLPs to MDA-MB-468 cells and observed significantly greater uptake with flow cytometry for the targeted version (Fig. 6(A)). The increased yCD loading also improved the conversion of 5-FC to 5-FU, resulting in enhanced cytotoxicity in MDA-MB-468 cells when compared to cells treated with the non-targeted counterpart and 5-FC, or with the targeted construct in the absence of 5-FC (Fig. 6(B)). The ability to fine-tune the therapeutic effect without affecting specific cancer targeting is the most powerful feature of this modular HBV VLP platform.
Conclusions
In summary, we developed a modular HBV VLP nanocarrier platform for tunable cargo protein loading and surface functionalization. Interior modification was achieved using a multi-expression system to modulate expression of different VLP monomers fused to a desired cargo protein. Exterior modification was performed by inserting a SpyTag to an external loop of the VLP monomer for SpyCatcher–SpyTag ligation of desired decorations. The new approach was used to demonstrate the modular decoration of several functional cargoes to both the exterior and the interior of the engineered VLPs. Surface decoration with an ELP tag offered a simple approach for VLP purification, while decoration with an EGFR-specific DARPin provided highly selective intracellular delivery to TNBC cells. Dual interior decoration with GFP and yCD enabled simple quantification of VLP delivery and prodrug activation for TNBC cell treatment. The versatility of the design offered a high degree of tunability in modulating the interior cargo loading, allowing optimization of yCD content for targeted cell killing. Furthermore, exterior decorations with site- and orientation-specific control will ultimately facilitate the ability to tailor the surface properties to promote delivery of larger encapsulated proteins to any desired target. While we only demonstrated the use of this platform for directed enzyme prodrug cancer therapy,77 the same strategy can be easily tailored to other applications by changing the loaded cargo and corresponding targeting domain. Furthermore, this VLP could be further modified with hydrophilic stealth biopolymers and endosomolytic peptides to enhance in vivo circulation and cytosolic delivery of bioactive cargos, respectively.Author contributions
D. Y., M. O. S. and W. C. conceived the project and designed the experiments. D. Y. performed the experiments. D. Y., M. O. S. and W. C. analyzed the data. D. Y., M. O. S. and W. C. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the funding and support from NSF (DMR1609621). D. Y. was also partially supported by an IGERT training grant (NSF 1144726). We thank Shannon Modla in the Bio-Imaging Center at the Delaware Biotechnology Institute for assistance with TEM, and we thank NIH-NIGMS (P20 GM103445), the NSF (IIA-1301765), and the State of Delaware for support for microscopy access at DBI. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation, the National Institute of General Medical Sciences, or the National Institutes of Health.Notes and references
- H. A. D. Lagassé, A. Alexaki, V. L. Simhadri, N. H. Katagiri, W. Jankowski, Z. E. Sauna and C. Kimchi-Sarfaty, F1000Research, 2017, 6, 113 Search PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- N. Skalko-Basnet, Biol.: Targets Ther., 2014, 107, DOI:10.2147/btt.s38387.
- N. Serna, L. Sanchez-Garcia, U. Unzueta, R. Diaz, E. Vazquez, R. Mangues and A. Villaverde, Trends Biotechnol., 2018, 36, 318–335 CrossRef CAS PubMed.
- J. A. Gorzelany and M. P. de Souza, Sci. Transl. Med., 2013, 5, 178fs10 Search PubMed.
- P. J. Koch and M. I. Koster, Front. Genet., 2021, 12, 714764 CrossRef CAS PubMed.
- J. M. Thurman and R. Yapa, Front. Immunol., 2019, 10, 672 CrossRef CAS PubMed.
- A. L. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602–1608 CrossRef CAS PubMed.
- F. Moncalvo, M. I. M. Espinoza and F. Cellesi, Front. Bioeng. Biotechnol., 2020, 8, 89 CrossRef PubMed.
- S. Miersch and S. S. Sidhu, F1000Research, 2016, 5, 1947 Search PubMed.
- G. L. Verdine and L. D. Walensky, Clin. Cancer Res., 2007, 13, 7264–7270 CrossRef CAS PubMed.
- D. Yur, R. M. Lieser, M. O. Sullivan and W. Chen, Curr. Opin. Biotechnol, 2021, 71, 41–48 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- R. M. Lieser, E. J. Hartzell, D. Yur, M. O. Sullivan and W. Chen, Bioconjugate Chem., 2022, 33, 452–462 CrossRef CAS PubMed.
- A. K. Jain and S. Thareja, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 524–539 CrossRef CAS PubMed.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed.
- R. M. Lieser, D. Yur, M. O. Sullivan and W. Chen, Bioconjugate Chem., 2020, 31, 2272–2282 CrossRef CAS PubMed.
- J. T. Bulcha, Y. Wang, H. Ma, P. W. L. Tai and G. P. Gao, Signal Transduction Targeted Ther., 2021, 6, 53 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- A. Alcami and U. H. Koszinowski, Immunol. Today, 2000, 21, 447–455 CrossRef CAS PubMed.
- S. Nooraei, H. Bahrulolum, Z. S. Hoseini, C. Katalani, A. Hajizade, A. J. Easton and G. Ahmadian, J. Nanobiotechnol., 2021, 19, 59 CrossRef CAS PubMed.
- W. J. Shan, D. L. Zhang, Y. L. Wu, X. L. Lv, B. Hu, X. Zhou, S. F. Ye, S. L. Bi, L. Ren and X. Z. Zhang, Nanomedicine, 2018, 14, 725–734 CrossRef CAS PubMed.
- E. J. Hartzell, R. M. Lieser, M. O. Sullivan and W. Chen, ACS Nano, 2020, 14, 12642–12651 CrossRef CAS PubMed.
- M. Segel, B. Lash, J. W. Song, A. Ladha, C. C. Liu, X. Jin, S. L. Mekhedov, R. K. Macrae, E. V. Koonin and F. Zhang, Science, 2021, 373, 882–889 CrossRef CAS PubMed.
- S. Banskota, A. Raguram, S. Suh, S. W. Du, J. R. Davis, E. H. Choi, X. Wang, S. C. Nielsen, G. A. Newby, P. B. Randolph, M. J. Osborn, K. Musunuru, K. Palczewski and D. R. Liu, Cell, 2022, 185, 250–265 CrossRef CAS PubMed.
- M. J. Rohovie, M. Nagasawa and J. R. Swartz, Bioeng. Transl. Med., 2017, 2, 43–57 CrossRef CAS PubMed.
- I. Suffian and K. T. Al-Jamal, Adv. Drug Delivery Rev., 2022, 180, 114030 CrossRef CAS PubMed.
- P. Pumpens and E. Grens, FEBS Lett., 1999, 442, 1–6 CrossRef CAS PubMed.
- S. A. Wynne, R. A. Crowther and A. G. W. Leslie, Mol. Cell, 1999, 3, 771–780 CrossRef CAS PubMed.
- X. K. Yu, L. Jin, J. Jih, C. H. Shih and Z. H. Zhou, PLoS One, 2013, 8, e69729 CrossRef CAS PubMed.
- H. Peyret, A. Gehin, E. C. Thuenemann, D. Blond, A. El Turabi, L. Beales, D. Clarke, R. J. C. Gilbert, E. E. Fry, D. I. Stuart, K. Holmes, N. J. Stonehouse, M. Whelan, W. Rosenberg, G. P. Lomonossoff and D. J. Rowlands, PLoS One, 2015, 10, 0120751 CrossRef PubMed.
- K. W. Lee and W. S. Tan, J. Virol. Methods, 2008, 151, 172–180 CrossRef CAS PubMed.
- L. H. Shen, J. Zhou, Y. X. Wang, N. Kang, X. B. Ke, S. L. Bi and L. Ren, Small, 2015, 11, 1190–1196 CrossRef CAS PubMed.
- K. M. Choi, S. H. Choi, H. Jeon, I. S. Kim and H. J. Ahn, ACS Nano, 2011, 5, 8690–8699 CrossRef CAS PubMed.
- B. Schwarz, P. Madden, J. Avera, B. Gordon, K. Larson, H. M. Miettinen, M. Uchida, B. LaFrance, G. Basu, A. Rynda-Apple and T. Douglas, ACS Nano, 2015, 9, 9134–9147 CrossRef CAS PubMed.
- J. K. Pokorski, M. L. Hovlid and M. G. Finn, ChemBioChem, 2011, 12, 2441–2447 CrossRef CAS PubMed.
- A. Walker, C. Skamel and M. Nassal, Sci. Rep., 2011, 1, 5 CrossRef PubMed.
- S. Costa, A. Almeida, A. Castro and L. Domingues, Front. Microbiol., 2014, 5, 63 Search PubMed.
- A. Chatterji, W. F. Ochoa, M. Paine, B. R. Ratna, J. E. Johnson and T. W. Lin, Chem. Biol., 2004, 11, 855–863 CrossRef CAS PubMed.
- A. Chatterji, W. Ochoa, L. Shamieh, S. P. Salakian, S. M. Wong, G. Clinton, P. Ghosh, T. W. Lin and J. E. Johnson, Bioconjugate Chem., 2004, 15, 807–813 CrossRef CAS PubMed.
- E. Gillitzer, D. Willits, M. Young and T. Douglas, Chem. Commun., 2002, 2390–2391, 10.1039/b207853h.
- K. W. Yong, D. Yuen, M. Z. Chen, C. J. H. Porter and A. P. R. Johnston, Nano Lett., 2019, 19, 1827–1831 CrossRef CAS PubMed.
- G. Beterams, B. Bottcher and M. Nassal, FEBS Lett., 2000, 481, 169–176 CrossRef CAS PubMed.
- M. Vogel, M. Diez, J. Eisfeld and M. Nassal, FEBS Lett., 2005, 579, 5211–5216 CrossRef CAS PubMed.
- H. Yamaguchi and M. Miyazaki, Biomolecules, 2014, 4, 235–251 CrossRef PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- G. Veggiani, B. Zakeri and M. Howarth, Trends Biotechnol., 2014, 32, 506–512 CrossRef CAS PubMed.
- S. C. Reddington and M. Howarth, Curr. Opin. Chem. Biol., 2015, 29, 94–99 CrossRef CAS PubMed.
- L. Li, J. O. Fierer, T. A. Rapoport and M. Howarth, J. Mol. Biol., 2014, 426, 309–317 CrossRef CAS PubMed.
- R. M. Lieser, W. Chen and M. O. Sullivan, Bioconjugate Chem., 2019, 30, 432–442 CrossRef CAS PubMed.
- M. J. Lajoie, S. E. Boyken, A. I. Salter, J. Bruffey, A. Rajan, R. A. Langan, A. Olshefsky, V. Muhunthan, M. J. Bick, M. Gewe, A. Quijano-Rubio, J. Johnson, G. Lenz, A. Nguyen, S. Pun, C. E. Correnti, S. R. Riddell and D. Baker, Science, 2020, 369, 1637–1643 CrossRef CAS PubMed.
- D. Steiner, P. Forrer and A. Pluckthun, J. Mol. Biol., 2008, 382, 1211–1227 CrossRef CAS PubMed.
- R. H. J. Beelen, A. M. Feldmann and H. J. W. Wijsman, Mol. Gen. Genet., 1973, 121, 369–374 CrossRef CAS PubMed.
- H. L. Lehman, S. J. Van Laere, C. M. van Golen, P. B. Vermeulen, L. Y. Dirix and K. L. van Golen, Mol. Cancer Res., 2012, 10, 1306–1318 CrossRef CAS PubMed.
- N. L. Ross and M. O. Sullivan, Biotechnol. Bioeng., 2016, 113, 2686–2697 CrossRef CAS PubMed.
- M. Raeeszadeh-Sarmazdeh, E. Hartzell, J. V. Price and W. Chen, Curr. Opin. Chem. Eng., 2016, 13, 109–118 CrossRef PubMed.
- K. D. Brune, D. B. Leneghan, I. J. Brian, A. S. Ishizuka, M. F. Bachmann, S. J. Draper, S. Biswas and M. Howarth, Sci. Rep., 2016, 6, 19234 CrossRef CAS PubMed.
- M. Newman, F. M. Suk, M. Cajimat, P. M. Chua and C. Shih, J. Virol., 2003, 77, 12950–12960 CrossRef CAS PubMed.
- I. Sominskaya, D. Skrastina, I. Petrovskis, A. Dishlers, I. Berza, M. Mihailova, J. Jansons, I. Akopjana, I. Stahovska, D. Dreilina, V. Ose and P. Pumpens, PLoS One, 2013, 8, e75938 CrossRef CAS PubMed.
- I. Sominskaya, D. Skrastina, A. Dislers, D. Vasiljev, M. Mihailova, V. Ose, D. Dreilina and P. Pumpens, Clin. Vaccine Immunol., 2010, 17, 1027–1033 CrossRef CAS PubMed.
- M. Vogel, J. Vorreiter and M. Nassal, Proteins: Struct., Funct., Bioinf., 2005, 58, 478–488 CrossRef CAS PubMed.
- W. Hassouneh, T. Christensen and A. Chilkoti, Curr. Protoc. Protein Sci., 2010, 61, 6.11.1–6.11.16 Search PubMed.
- A. R. Swartz, Q. Sun and W. Chen, Biomacromolecules, 2017, 18, 1654–1659 CrossRef CAS PubMed.
- H. Kim and W. Chen, J. Biotechnol., 2016, 234, 27–34 CrossRef CAS PubMed.
- J. Y. Kim, A. Mulchandani and W. Chen, Biotechnol. Bioeng., 2005, 90, 373–379 CrossRef CAS PubMed.
- F. Liu and W. Chen, Biotechnol. Prog., 2013, 29, 968–971 CrossRef CAS PubMed.
- M. Vogel, M. Diez, J. Eisfeld and M. Nassal, FEBS Lett., 2005, 579, 5211–5216 CrossRef CAS PubMed.
- D. C. Danielson, N. Sachrajda, W. Wang, R. Filip and J. P. Pezacki, Mol. Ther.–Nucleic Acids, 2016, 5, e303 CrossRef CAS PubMed.
- J. Cheng, R. Koukiekolo, K. Kieliszkiewicz, S. M. Sagan and J. P. Pezacki, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 1197–1203 CrossRef CAS PubMed.
- D. McNeale, N. Dashti, L. C. Cheah and F. Sainsbury, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, e1869, DOI:10.1002/wnan.1869.
- A. S. Gaynor and W. Chen, ACS Synth. Biol., 2020, 9, 2639–2647 CrossRef CAS PubMed.
- N. Normanno, A. De Luca, C. Bianco, L. Strizzi, M. Mancino, M. R. Maiello, A. Carotenuto, G. De Feo, F. Caponigro and D. S. Salomon, Gene, 2006, 366, 2–16 CrossRef CAS PubMed.
- K. L. Mueller, Z.-Q. Yang, R. Haddad, S. P. Ethier and J. L. Boerner, J. Mol. Signaling, 2010, 5, 8 CrossRef PubMed.
- R. I. Nicholson, J. M. W. Gee and M. E. Harper, Eur. J. Cancer, 2001, 37, S9–S15 CrossRef CAS PubMed.
- H. S. Park, M. H. Jang, E. J. Kim, H. J. Kim, H. J. Lee, Y. J. Kim, J. J. Kim, E. Kang, S.-W. Kim, I. A. Kim and S. Y. Park, Mod. Pathol., 2014, 27, 1212–1222 CrossRef CAS PubMed.
- J. Zhang, V. Kale and M. Chen, AAPS J., 2015, 17, 102–110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00445g |
| This journal is © The Royal Society of Chemistry 2023 |