
Inflammation-targeting polyamine nanomedicines for the treatment of ulcerative colitis
Akihiro
Nishiguchi
 * and
Tetsushi
Taguchi
* and
Tetsushi
Taguchi
Polymers and Biomaterials Field, Research Center for Functional Materials, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: NISHIGUCHI.Akihiro@nims.co.jp
First published on 5th April 2023
Abstract
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder characterized by immune system dysfunction. Despite the availability of various anti-inflammatory drugs, they exhibit low therapeutic efficacy with systemic side effects. In this study, we developed oral anti-inflammatory polyamine-based nanomedicines for the treatment of ulcerative colitis. Polyamine-bearing nanoparticles were prepared by the self-assembly of hyaluronic acid in organic solvents and crosslinking with branched oligoethyleneimine. Polyamine nanoparticles were found to suppress excessive inflammatory responses by scavenging the reactive oxygen species (ROS). Moreover, these nanoparticles inhibited enzymatic degradation and targeting of inflamed intestinal tissues. Additionally, they suppressed the inflammatory responses and recovered the pathological disorders in the colon of an ulcerative colitis mouse model. Therefore, polyamine-based nanomedicines exhibit great potential as biocompatible ROS-scavenging drugs for the treatment of IBD.
 Akihiro Nishiguchi | Dr Akihiro Nishiguchi is a biomaterial scientist working on functional polymeric hydrogels in the field of medical materials, tissue engineering, regenerative medicine, and drug delivery system. He is currently a Principal Researcher in Research Center for Macromolecules and Biomaterials, National Institute for Materials Science (NIMS). He received his PhD from Osaka University in 2015 under the supervision of Prof. Mitsuru Akashi. Between 2015–2017, he worked as a Postdoctoral Researcher with Prof. Martin Moeller at the DWI Leibniz-Institute for Interactive Materials. In 2017, he moved to NIMS (polymeric biomaterials group). He currently focuses on tissue regeneration, tissue adhesion, supramolecular chemistry, and tissue reconstruction using advanced medical hydrogels. |
1. Introduction
Inflammatory bowel disease (IBD) is a serious inflammatory disorder of the intestinal tract caused by mucosal immune abnormalities.1 Loss of barrier function and translocation of commensal microorganisms further increase the inflammation, resulting in diarrhea, bloody stools, and tumorigenesis.2 As IBD repeats the active and remission phases, affected patients require lifelong treatment. Although mesalazine or 5-aminosalicylic acid (5-ASA), steroids, biologics, and immunosuppressive agents are used for treatment depending on the disease severity, many patients do not respond well to these therapies.3 In addition, these drugs often cause systemic side effects, such as opportunistic infections, cancer, adrenal cortex dysfunction, and hepatotoxicity.4As one of the most important mediators of inflammatory responses, reactive oxygen species (ROS), such as hydroxyl radicals and hydrogen peroxides, have attracted increasing attention.5 In normal physiological conditions, the endogenous production of ROS is essential for the body to regulate intracellular and intercellular signal transduction. However, under inflammatory conditions, inflammatory cells, such as macrophages, produce excessive ROS, causing oxidative stress via DNA damage, lipid peroxidation, and protein oxidation.6 Clinical studies have reported that excessive ROS production is associated with accelerated inflammatory responses in the intestinal mucosa and pathogenesis of IBD.7,8 Therefore, ROS may act as therapeutic targets, with ROS scavenging a potential treatment strategy for IBD. However, some ROS scavengers, such as beta-carotene and vitamin A, have been reported to increase patient mortality owing to cytotoxicity.9 In particular, these ROS scavengers can act on the mitochondrial electron transfer system and inhibit the membrane potential functions. Therefore, there is a need to identify novel therapeutic agents that can eliminate excess ROS to suppress inflammation. Biomaterial-based nanomedicines have been developed as ROS-scavenging reagents for the treatment of inflammatory diseases.10 Previous studies have developed anti-inflammatory nanoparticles (NPs), including antioxidants,11 bilirubin,12 and cyclodextrin NPs.13 However, use of these biomaterials is limited due to several issues, such as the in vivo kinetics of non-biodegradable materials, retention of radicals, and long-term safety.14
In this study, we report the use of orally administered anti-inflammatory polyamine (PA)-based NPs (PANPs) for the treatment of ulcerative colitis (Fig. 1). Previously, we screened various PA species with different structural characteristics. PA is an effective anti-inflammatory compound with ROS-scavenging function,15 but it exhibits poor biocompatibility. We previously found that branched oligoethyleneimine (bOEI), with a relatively low molecular weight (Mw = 300 and 600 Da), possessed potent ROS-scavenging functions and high biocompatibility.16 Bioconjugation of bOEI to hyaluronic acid (HA), which has anti-inflammatory properties, via HA-CD44 signaling17 enhances its biocompatibility. However, the ability of bOEI–HA conjugates to target inflamed tissues is limited. To improve their targeting ability, we prepared NPs composed of PA and HA by developing globule structures of HA and crosslinking with PA via carbodiimide chemistry. Oral PANPs can accumulate in the submucosal tissue of the large intestine, where the barrier integrity is reduced due to inflammation. We also investigated whether ROS scavenging by PANPs can suppress the amplification of inflammatory responses in the intestinal mucosa in an ulcerative colitis mouse model. Inflammation-targeting, ROS-scavenging, and biocompatible PANPs may serve as therapeutic drugs for IBD.
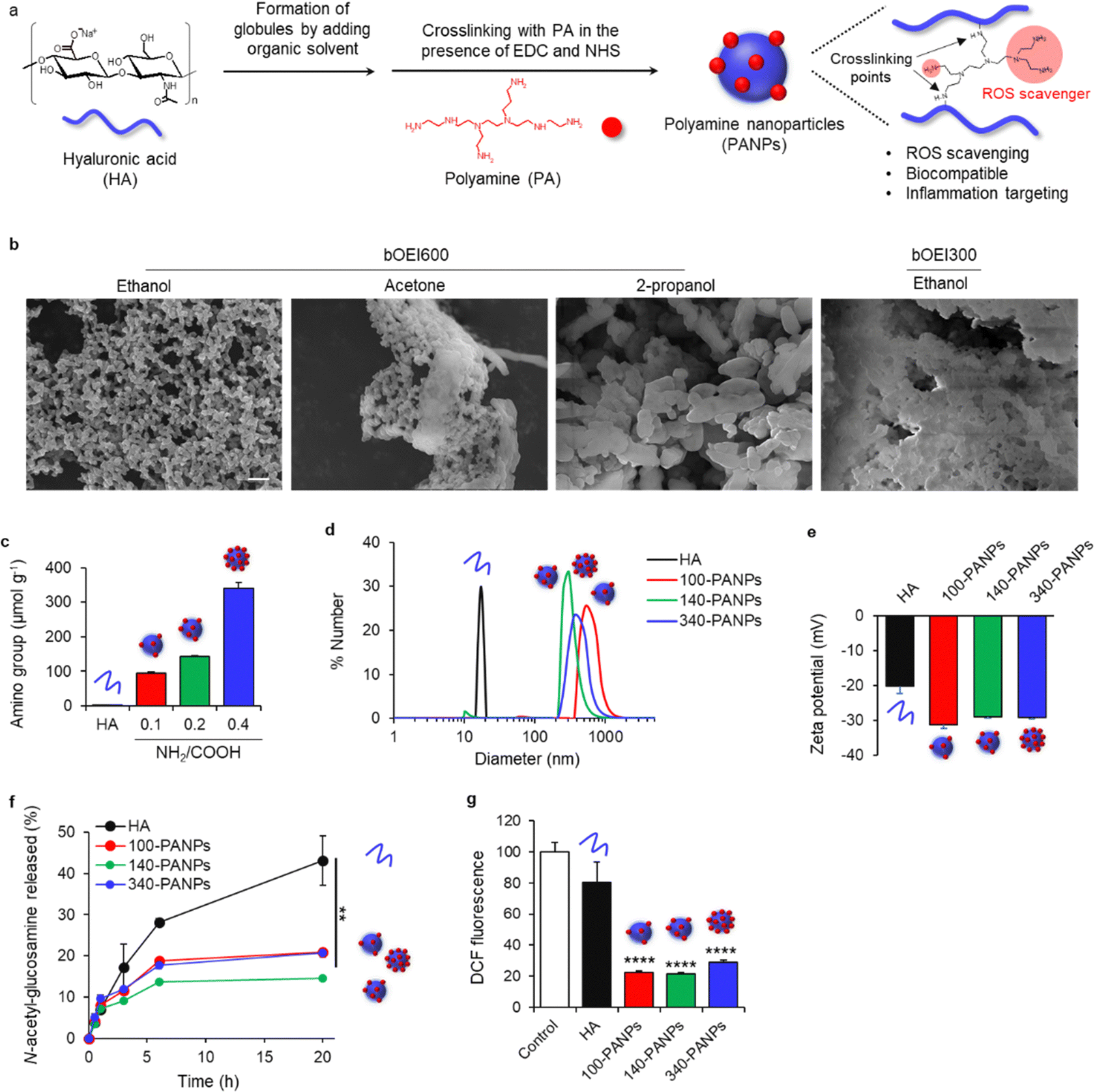 | ||
| Fig. 1 (a) Schematic illustration of the preparation of polyamine nanoparticles (PANPs). Organic solvent was added to the hyaluronic acid (HA) globule structure and crosslinking with PA (bOEI600) in the presence of carbodiimide led to the formation of PANPs. PA functioned as a crosslinker and ROS scavenger. (b) Scanning electron microscopy (SEM) images of PANPs prepared using different organic solvents (ethanol, acetone, and 2-propanol) and molecular weights (300 and 600 Da) of bOEI. (c) Quantification of amino group content in HA and PANPs prepared with different feeding ratios of PA. Amino group content was measured using the 2,4,6-trinitrobenzensulfonic acid (TNBS) method. (d and e) Dynamic light scattering (DLS) and zeta potential measurement of HA and 100-, 140-, and 340-PANPs. (f) Release of N-acetyl-glucosamine from HA and PANPs in the presence of hyaluronidase. (g) Reactive oxygen species (ROS) scavenging assay of PA, HA, and PANPs (n = 3). DCF fluorescence of control was set to 100%. Data are presented as the mean ± standard deviation (SD) from one representative experiment. **P < 0.01, ****P < 0.0001 analyzed via one-way analysis of variance (ANOVA) with Tukey's multiple comparison post-hoc test. Scale bar, 1 μm. | ||
2. Experimental
2.1 Preparation of PANPs
PANPs were prepared by the condensation of HA with bOEI-600 using water-soluble carbodiimide. HA (100 mg, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00000 Da; Seikagaku Corporation, Japan) was dissolved in 100 mL of 0.1 M 2-(N-morpholino) ethanesulfonic acid (Sigma-Aldrich, USA) buffer (pH = 4.8) and stirred for 3 h at 25 °C. Ethanol (100 mL) was added to obtain the HA globule form. Then, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, 25 mg, 0.16 mmol; Fujifilm Wako Pure Chemical Corporation, Japan) and 10 mM N-hydroxysuccinimide (NHS; Fujifilm Wako Pure Chemical Corporation, Japan) were added to the solution. Next, 2.5, 5, and 10 mg of bOEI-600 (Mw = 600 Da; Junsei Kagaku, Japan) were added (NH2/COOH = 0.1, 0.2, and 0.4). The condensation reaction proceeded for 1 h at 25 °C. The product was dialyzed using a dialysis membrane (molecular weight cut-off value: 12000–14000 Da; Repligen, USA) in deionized water for three days and dried via freeze-drying for three days. The number of amino groups in PANPs was estimated by determining the residual amino groups using a 2,4,6-trinitrobenzensulfonic acid (TNBS; Tokyo Chemical Industry Co., Ltd, Japan) assay.18 The morphology of PANPs was observed via scanning electron microscopy (SEM; S-4800 ultrahigh-resolution SEM; HITACHI, Japan). Platinum was sputtered onto the PANPs for 30 s to form a 10 nm-thick coated layer. The accelerating voltage and working distance were set as 10 kV and 8 mm, respectively.
00000 Da; Seikagaku Corporation, Japan) was dissolved in 100 mL of 0.1 M 2-(N-morpholino) ethanesulfonic acid (Sigma-Aldrich, USA) buffer (pH = 4.8) and stirred for 3 h at 25 °C. Ethanol (100 mL) was added to obtain the HA globule form. Then, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, 25 mg, 0.16 mmol; Fujifilm Wako Pure Chemical Corporation, Japan) and 10 mM N-hydroxysuccinimide (NHS; Fujifilm Wako Pure Chemical Corporation, Japan) were added to the solution. Next, 2.5, 5, and 10 mg of bOEI-600 (Mw = 600 Da; Junsei Kagaku, Japan) were added (NH2/COOH = 0.1, 0.2, and 0.4). The condensation reaction proceeded for 1 h at 25 °C. The product was dialyzed using a dialysis membrane (molecular weight cut-off value: 12000–14000 Da; Repligen, USA) in deionized water for three days and dried via freeze-drying for three days. The number of amino groups in PANPs was estimated by determining the residual amino groups using a 2,4,6-trinitrobenzensulfonic acid (TNBS; Tokyo Chemical Industry Co., Ltd, Japan) assay.18 The morphology of PANPs was observed via scanning electron microscopy (SEM; S-4800 ultrahigh-resolution SEM; HITACHI, Japan). Platinum was sputtered onto the PANPs for 30 s to form a 10 nm-thick coated layer. The accelerating voltage and working distance were set as 10 kV and 8 mm, respectively.
2.2 Dynamic light scattering (DLS) and zeta potential measurement
HA and 100-, 140-, and 340-PANPs were dissolved in phosphate-buffered saline (PBS; Nacalai Tesque, Japan) for DLS measurement and in 10 mM NaCl solution for zeta potential measurement, respectively. The samples were incubated at 25 °C for 24 h prior to measurement for complete dispersion. The sizes and zeta potentials of the samples were measured using a DLS spectrophotometer (DLS-8000HAL; Otsuka Electronics, Japan) and zeta-potential analyzer (ELSZ-1000Z Otsuka Electronics, Japan), respectively.2.3 Degradability test
Degradation of HA and PANPs was assessed by measuring the concentration of terminal N-acetyl-D-glucosamine, a hyaluronan degradation product, according to a previously described colorimetric method.19 HA (1 mg mL−1) and 100-, 140-, and 340-PANPs (1 mg mL−1) were dispersed in PBS (pH = 7.4) and incubated with 1000 U mL−1 of hyaluronidase II (Sigma-Aldrich) at 37 °C. N-Acetyl-D-glucosamine (1 mg mL−1) was used as the control. After incubation for 0.5, 1, 3, 6, and 24 h, 100 μL of the sample was collected, and the mixture was diluted with 500 μL of PBS. The enzymatic reaction was quenched by heating for 5 min at 100 °C, after which 100 μL of 0.8 M potassium tetraborate (pH = 9.0) was added and heated for another 3 min at 100 °C. Then, 3 mL of 4-dimethylaminobenzaldehyde (DMAB; Sigma-Aldrich) reagent, comprising 1 g of DMAB dissolved in 10 mL of glacial acetic acid containing 20% (v/v) of 6 M HCl, was added to the solution and incubated in a water bath at 37 °C for 20 min. Absorbance was measured at 544 nm using a microplate reader (Spark10M; TECAN, Switzerland).2.4 ROS scavenging test
ROS scavenging was evaluated using 2′,7′-dichlorodihydrofluorescin diacetate (DCF-DA; OxiSelect; Cell Biolabs, Inc., USA), a fluorescence probe that detects hydroxyl radicals. DCF-DA was dissolved in ethanol and diluted in PBS. DCF-DA solution was added to PBS containing H2O2 (100 μM) in a 96-well plate. PA (bOEI-600, 100 μg mL−1), HA (1 mg mL−1), and PANPs (1 mg mL−1) were then added to the DCF-DA solution. The final DCF-DA concentration was 50 μM. The solution was incubated at 37 °C for 2 h and the fluorescence intensity (ex = 485 nm, em = 525 nm) was measured using a microplate reader (Spark10M).2.5 Preparation of bone marrow-derived macrophages (BMDMs)
All animal experiments were approved by the Animal Care and Use Committee of the National Institute for Materials Science (66-2021-1). Mice (female, C57BL/6J, 7 weeks-old) were euthanized via cervical dislocation, and the femur and tibia were removed and cleaned with 70% ethanol for 1 min. After washing the bones with basal media (RPMI-1640 medium; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin streptomycin (P/S; Thermo Fisher Scientific, USA), both ends of the bones were cut, and bone marrow cells were collected by flushing the media using a 25-gauge syringe. The obtained cells were passed through a 70 μm filter, and ACK lysing buffer (Thermo Fisher Scientific, USA) was added to the tube for hemolysis. Cells were collected via centrifugation at 1300 rpm for 5 min, washed with the medium, and resuspended in the differentiation medium (RPMI-1640 medium supplemented with 10% FBS, 1% P/S, and 40 ng mL−1 of mouse macrophage colony-stimulating factor [M-CSF; Miltenyi Biotec, Germany]). Cells were cultured at 37 °C in a 5% CO2 incubator for three days. Fresh differentiation medium was added and the cells were cultured for another two days. Differentiated cells were harvested using a scraper and used in further inflammation assays.2.6 Cell viability and inflammation assays
RAW-Blue cells (3 × 104 cells well−1; InvivoGen, USA) were seeded in a 96-well culture plate and cultured in the basal medium at 37 °C in a 5% CO2 incubator for 24 h. RAW-Blue cells stably express the secreted embryonic alkaline phosphatase (SEAP) gene induced by nuclear factor (NF)-κB/AP-1 were used as an inflammation assay model. In response to LPS exposure, RAW-Blue cells were activated to produce SEAP and the levels of SEAP were monitored using QUANTI-Blue™ solution. Basal medium containing PA or PANPs and lipopolysaccharide (LPS; Escherichia coli, 100 ng mL−1; Fujifilm Wako Pure Chemical Corporation, Japan) was added to the cells and incubated for 24 h. Cell morphology was observed using an optical microscope (EVOS XL Cell Imaging System; Thermo Fisher Scientific, USA), and cell numbers were quantified using a cell counting kit (WST-8 assay; DOJINDO, Japan). Briefly, 10 μL of WST-8 reagent was added to each well and incubated for 2 h. Absorbance at 450 nm was recorded using a microplate reader (Spark10M). To evaluate NF-κB activity, the RAW-Blue assay kit (Invitrogen, USA) was used, according to the manufacturer's instructions. After culturing RAW-Blue cells with PA or PANPs and LPS, supernatants containing SEAP were collected and mixed with alkaline phosphatase detection medium. Absorbance at 655 nm was measured using a microplate reader (Spark10M).To measure the secretion of inflammatory cytokines from BMDMs, BMDMs (3 × 104 cells well−1) were seeded in a 96-well culture plate and cultured in the differentiation medium at 37 °C in a 5% CO2 incubator for 24 h. Differentiation medium containing PANPs (1 mg mL−1) and LPS (100 ng mL−1) was added to the cells and incubated for 24 h. The supernatants were collected and the levels of inflammatory cytokines were quantified using an enzyme-linked immunosorbent assay (ELISA) kit (Mouse Quantikine ELISA kit for tumor necrosis factor-α [TNF]-α; R&D Systems, USA) according to the manufacturer's protocols. Absorbance at 450 nm was measured using a microplate reader (Spark10M; TECAN), and the concentration of TNF-α was calculated from a standard curve.
2.7 Polymerase chain reaction (PCR) analysis
Reverse transcription PCR analysis was performed using a PCR array kit (RT2 Profiler PCR Array Mouse Wound Healing; Qiagen, USA). BMDMs (3 × 105 cell well−1) were seeded in a 12-well culture plate and cultured in the differentiation medium at 37 °C in a 5% CO2 incubator for 24 h. Differentiation medium containing LPS (100 ng mL−1) with or without 100-PANPs (1 mg mL−1) was added to the cells and incubated for 24 h. Total RNA was extracted using an RNA micro-scale kit (NucleoSpin, TAKARA, Japan) according to the manufacturer's protocol. cDNA was prepared from 200 ng total RNA using a cDNA synthesis kit. Reverse transcription-PCR (RT-PCR) was performed using SYBER green gene expression assays. Glyceraldehyde 3-phosphate dehydrogenase was used as a housekeeping gene. Gene expression was quantified using GeneGlobe analysis (Qiagen, USA).2.8 Fluorescence imaging
To check whether PANPs accumulated in the inflamed colon, fluorescently labeled PANPs were administered, and the tissues were analyzed by histological observation and an in vivo imaging system. To conjugate Cy5.5 to HA, HA was dialyzed against 0.1 M HCl overnight. HA was then dialyzed against distilled water three times for 1 d and freeze-dried to obtain the acidic form of HA. The acidic form of HA (800 μmol) was dissolved in 64 mL of DMSO overnight. Then, 160 μmol EDC and 160 μmol NHS were added to the mixture. After mixing for 10 min, 8 μmol Cy5.5-NH2 was added to the reaction mixture. After stirring overnight at 25 °C, the products were dialyzed against 0.01 M NaOH three times for 1 d. After dialysis against distilled water twice for two days, the products were freeze-dried to obtain HA-Cy5.5. Cy5.5-labbeled PANPs were prepared using HA-Cy5.5 in the same manner as described above. Mice were administered with water or water containing 3% dextran sulfate sodium (DSS) by free drinking from days 0 to 5. On day 5, DSS-containing water was changed to water and the mice were housed without food for 16 h. On day 6, the mice were orally administered HA-Cy5.5 and PANPs-Cy5.5 (10 mg kg−1). After 6 h, the mice were euthanized via cervical dislocation, and organs including the heart, kidney, lung, spleen, liver, stomach, small intestine, and colon were collected. The organs were then observed using an in vivo imaging system (IVIS Lumina; PerkinElmer, USA) with a Cy5.5 filter channel. For histological observation, colon was fixed with 10% formalin, and the cross-sections of colon stained with 4′,6-diamidino-2-phenylindole were observed using confocal laser scanning microscope (Leica TCS SPE; Leica, Germany).2.9 In vivo biocompatibility test
Mice (female, C56BL/6J, 7 weeks-old) were administered PBS and 100-PANPs (10 and 100 mg kg−1) on days 0, 2, 4, and 6. Body weight was monitored during sample administration. On day 9, mice were euthanized via cervical dislocation, and the heart, lungs, liver, kidneys, spleen, colon, and blood were collected for histological observation and blood tests. For histological observation, tissues were collected and fixed with 10% formalin, and the cross-sections of HE-stained tissues were observed using a digital slide scanner (NanoZoomer S210; Hamamatsu Photonics, Japan). The collected blood samples were analyzed using blood chemistry to evaluate systemic toxicity.2.10 Ulcerative colitis models
Mice were administered with water or water containing 3% DSS (Mw = 36000–50000, colitis grade; Fujifilm Wako Pure Chemical Corporation) via free drinking from days 0 to 5. On days 5, 7, and 9, mice were administered PBS, 100-, 140-, 340-PANPs (10 mg kg−1), and 5-ASA (10 mg kg−1). Body weight was monitored during sample administration. On day 10, mice were euthanized via cervical dislocation, and colons were collected for measurement of length, histological observation, and quantification of pro-inflammatory cytokines. For histological observation, colons were fixed with 10% formalin, and the cross-sections of HE-stained tissues were observed using a digital slide scanner (NanoZoomer S210; Hamamatsu Photonics). The colonic damage was scored according to previous reports.12 To measure inflammatory cytokines, interleukin-6, colon sections in radioimmunoprecipitation buffer (Fujifilm Wako Pure Chemical Corporation, Japan) was homogenized using a homogenizer and centrifuged at 10000 × g for 10 min. The supernatants were used to determine the levels of IL-6 using an ELISA kit (Mouse Quantikine ELISA kit for IL-6; R&D Systems), according to the manufacturer's instructions. The protein concentration in the supernatant was measured using a Micro BCA Protein Assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Absorbance was measured using a microplate reader (Spark10M) and the concentrations of IL-6 and proteins were calculated using a standard curve.
2.11 Statistical analysis
Data are expressed as the mean ± standard deviation or standard error of the mean. One-way analysis of variance, followed by Tukey's multiple comparison post-hoc test, was used to determine the statistical differences. Independent experiments were performed multiple times. The data shown in each figure are a complete dataset from one representative independent experiment. None of the samples were excluded from the analysis. Statistical significance is indicated by *P < 0.05, **P < 0.01, and ****P < 0.0001. Statistical analyses were performed using GraphPad Prism v.8.0 (GraphPad Software, USA).3. Results and discussion
3.1 Preparation and characterization of PANPs
PANPs were prepared by self-assembly of HA in organic solvents and crosslinking with PA (Fig. 1a). The addition of organic solvents to the HA aqueous solution resulted in the formation of globular structures.20 This process is useful for initiating the nucleation and facilitating the formation of NPs. HA was then cross-linked with PA (bOEI600) in the presence of EDC and NHS. PA functioned not only as a ROS scavenger but also as a crosslinker. Several organic solvents were screened to optimize the preparation conditions for the PANPs. The use of ethanol resulted in the formation of monodispersed PANPs, while acetone induced the aggregation of PANPs, and 2-propanol formed micrometer-sized particles (Fig. 1b). Since it has been reported that nanometer-sized particles effectively target ulcerative colitis,21 ethanol was used as an organic solvent. Moreover, the relatively low molecular weight PA (bOEI300) showed aggregation, and bOEI600 was suitable for nanoparticle formation. The amounts of amino groups in the PANPs were tuned by varying the feeding ratio of bOEI600 in the range of 0.1 0.4 (NH2/COOH) (Fig. 1c). TNBS measurements showed the formation of PANPs with 100, 140, and 340 μmol g−1 amino groups (100, 140, and 340-PANPs), indicating successful amination of HA with bOEI600. DLS measurements revealed that the average diameters of the 100-, 140-, and 340-PANPs were 620, 300, and 430 nm, respectively (Fig. 1d). We expected that nanoparticulation of bOEI-bearing HA would improve the targeting ability to inflamed tissues compared with the previously reported bOEI–HA polymer.15 Each PANPs possesses a negative charge under physiological conditions (Fig. 1e). Although the introduction of amino groups to HA increases the zeta potential, the amounts of amino groups introduced into NPs were 0.34 mmol g−1 at the maximum, which was much lower than carboxylic groups in original HA (approximately 2.4 mmol g−1).22 Therefore, the net charge of the PANPs was negative, which contributed to the high dispersity of the PANPs originating from electrostatic repulsion. Stability against enzymatic treatment with hyaluronidase was improved in PANPs compared to HA (Fig. 1f). The crosslinking of HA by bOEI changes the molecular structures of HA, and the recognition of HA degradation moieties by hyaluronidase may be suppressed. An increase in enzyme stability is beneficial for oral administration. ROS scavenging of PANPs was evaluated by DCF assay, which can detect residual hydroxyl radicals. All PANPs suppressed the formation of hydroxyl radicals and showed high ROS-scavenging ability (Fig. 1g). The primary amino groups of PA play a role in ROS-scavenging.15,16 Although some amino groups in PA formed amide bonds with HA, remaining amino groups functioned as a ROS scavenger.3.2 Anti-inflammatory effects of PANPs
Biological functions of PANPs in immune cells were evaluated using cultured RAW-Blue cells and BMDMs. RAW-Blue cells were exposed to 100, 140, and 340-PANPs for 24 h, and cytocompatibility was assessed using the WST-8 assay. All PANPs were highly cytocompatible at less than 2.5 mg mL−1 (Fig. 2a). To evaluate the anti-inflammatory functions of PANPs, LPS-activated RAW-Blue cells were used as an inflammation model and the nuclear translocation of NF-κB was quantified. The mammalian NF-κB family, including p50, p52, RelA (p65), RelB, and c-Rel, forms an inactivated complex with I kappa B (IκB) and functions as a key regulator of immune responses.23 In response to inflammatory signals via toll-like receptors, the I kappa B kinase (IKK) complex initiates the phosphorylation of IκB and undergoes nuclear translocation of NF-κB, leading to the transcription of inflammation-related genes. PANPs substantially suppressed the activation and nuclear translocation of NF-κB in RAW-Blue cells (Fig. 2b). Treatment with approximately 1 mg mL−1 of PANPs effectively suppressed inflammatory responses, although higher concentrations of PANPs were not cytotoxic. This result indicates that PANPs prevent the nuclear translocation of NF-κB through ROS scavenging. Inflammatory responses were evaluated in primary macrophages and BMDMs. Multiple gene expression assays revealed that PANPs downregulated the expression of a broad range of inflammatory cytokines, such as interleukin-1β (IL-1β) and TNF-α (Fig. 2c). Several genes related to cellular adhesion, such as integrin, were upregulated. The production of TNF-α secreted from BMDMs was evaluated using ELISA. The 100- and 140-PANPs showed lower levels of TNF-α secretion than the control and 340-PANPs (Fig. 2d). LPS binds to TLR4 to activate the gene expression of inflammatory cytokines, such as IL-1β and TNF-α, through the nuclear translocation of NF-κB.24,25 Since NF-κB is highly expressed in inflamed tissues and plays crucial roles in the inflammation signaling, it can be a therapeutic target for IBD.26 We previously reported that bOEI suppressed the nuclear translocation of NF-κB and inhibited the production of inflammatory cytokines.16 bOEI-bearing PANPs may attenuate the nuclear translocation of NF-κB by inhibiting IKK functions, IκBα degradation, nuclear translocation of NF-κB, and binding to DNA. These anti-inflammatory processes may rely on ROS scavenging property of bOEI. The results of these experiments correlated with those of the bOEI-conjugated polymer, indicating that the anti-inflammatory function of bOEI was maintained in PANPs. Moreover, when BMDMs were exposed to Cy5.5-labeled PANPs, cellular uptake was confirmed (Fig. 2e). This result Indicates that PANPs were localized in cells and possibly scavenged excessive intra/extracellular ROS.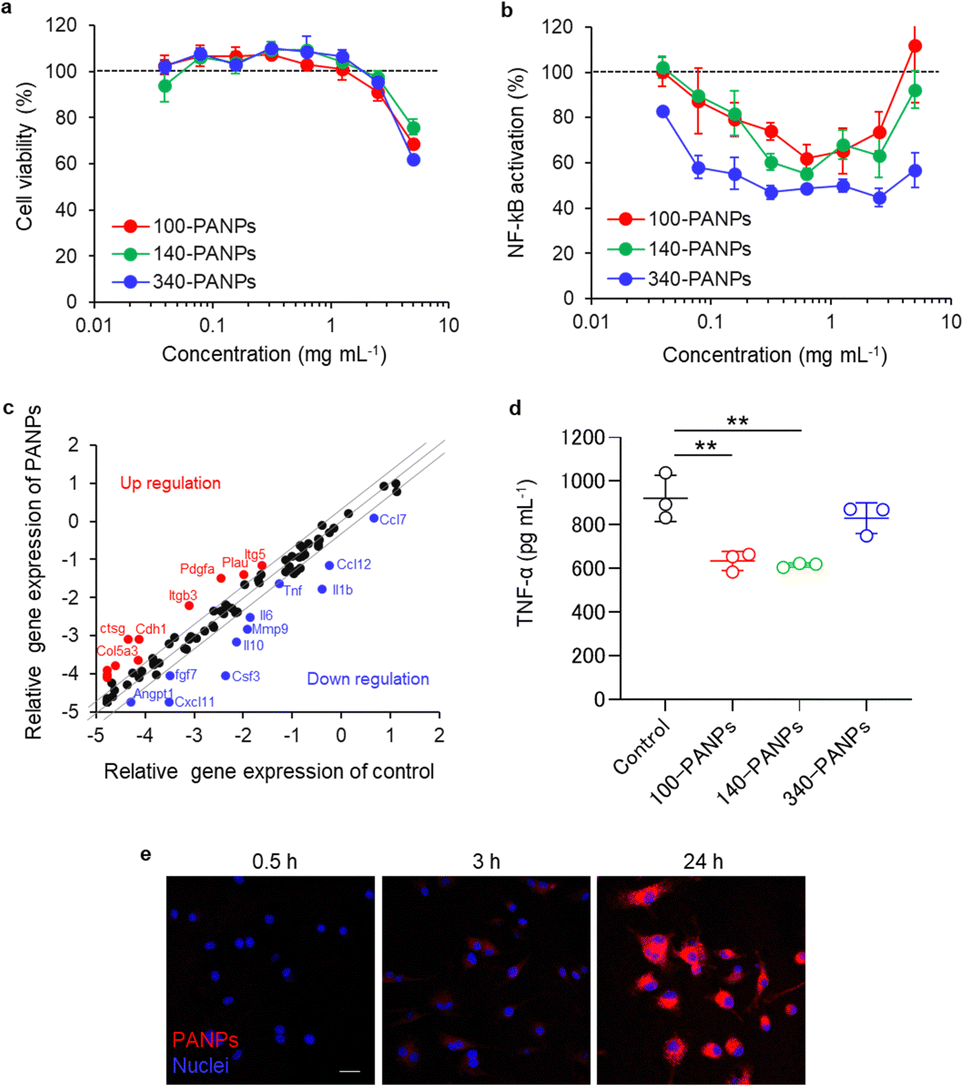 | ||
| Fig. 2 (a and b) Cell viability and nuclear factor (NF)-κB activation of RAW-blue cells exposed to lipopolysaccharides (LPS; 100 ng mL−1) or LPS and PA or PANPs. The value of control (exposed to only LPS) was set to 100% (n = 3). (c) Scatter plot of the expression levels of inflammation-related genes in bone marrow-derived macrophages (BMDMs) exposed to LPS (100 ng mL−1) or LPS and PANPs (1 mg mL−1). (d) Secretion of tumor necrosis factor (TNF)-α from BMDMs exposed to LPS (100 ng mL−1) or LPS and PANPs (1 mg mL−1). TNF-α levels were quantified using an enzyme-linked immunosorbent assay (ELISA; n = 3). (e) Uptake of cy5.5-labeled 100-PANPs by BMDMs. Data are presented as the mean ± SD from one representative experiment except (d). **P < 0.01 analyzed via one-way ANOVA with Tukey's multiple comparison post-hoc test. Scale bar represents 20 μm. | ||
3.3 Targeting ability of PANPs
To assess the targeting ability of 100-PANPs to inflamed colons, histological observations and in vivo fluorescence imaging were performed. Mice were treated with 3% DSS for five days to induce ulcerative colitis. After drinking water for 1 day, Cy5.5-labeled 100-PANPs were orally administered and their distribution in the colon was imaged. Treatment with DSS causes bloody diarrhea, ulcerations, and granulocyte infiltration through strong inflammatory responses.27 Fluorescence images of sectioned colon tissues showed the accumulation of 100-PANPs on the surface of the mucosa, although there was no fluorescence on the mucosa when Cy5.5-HA was administered due to rapid clearance (Fig. 3a). In contrast, 100-PANPs did not accumulate in the normal colon of mice that were not treated with DSS. Size-dependent accumulation in the colon was in agreement with a previous report using nanomedicines.28 Moreover, in vivo fluorescence imaging revealed that 100-PANPs overcame the degradation in the stomach and accumulated in the colon owing to the increased stability of the enzyme (Fig. 3b and c). In ulcerative colitis, intestinal barriers are disrupted and the permeation of orally administered drugs across the barriers is enhanced depending on the size.29 It has been reported that NPs effectively accumulate in inflamed tissues compared to small molecules.21 Nanoparticulation of HA and PA is effective in enhancing the accumulation of drugs. Moreover, it has been reported that inflamed mucosa is covered with positively charged proteins, such as transferrin,30 and electrostatic interactions can be used as a driving force for active drug delivery.31 Therefore, negatively charged PANPs can target damaged mucosal layers via electrostatic interactions to improve the delivery efficacy, although PA itself may not be associated with targeting. These results indicate that the nanoparticulation of bOEI and HA from polymers can improve the targeting ability, and 100-PANPs can target the inflamed tissues, where barrier integrity is disrupted, but not the normal tissues.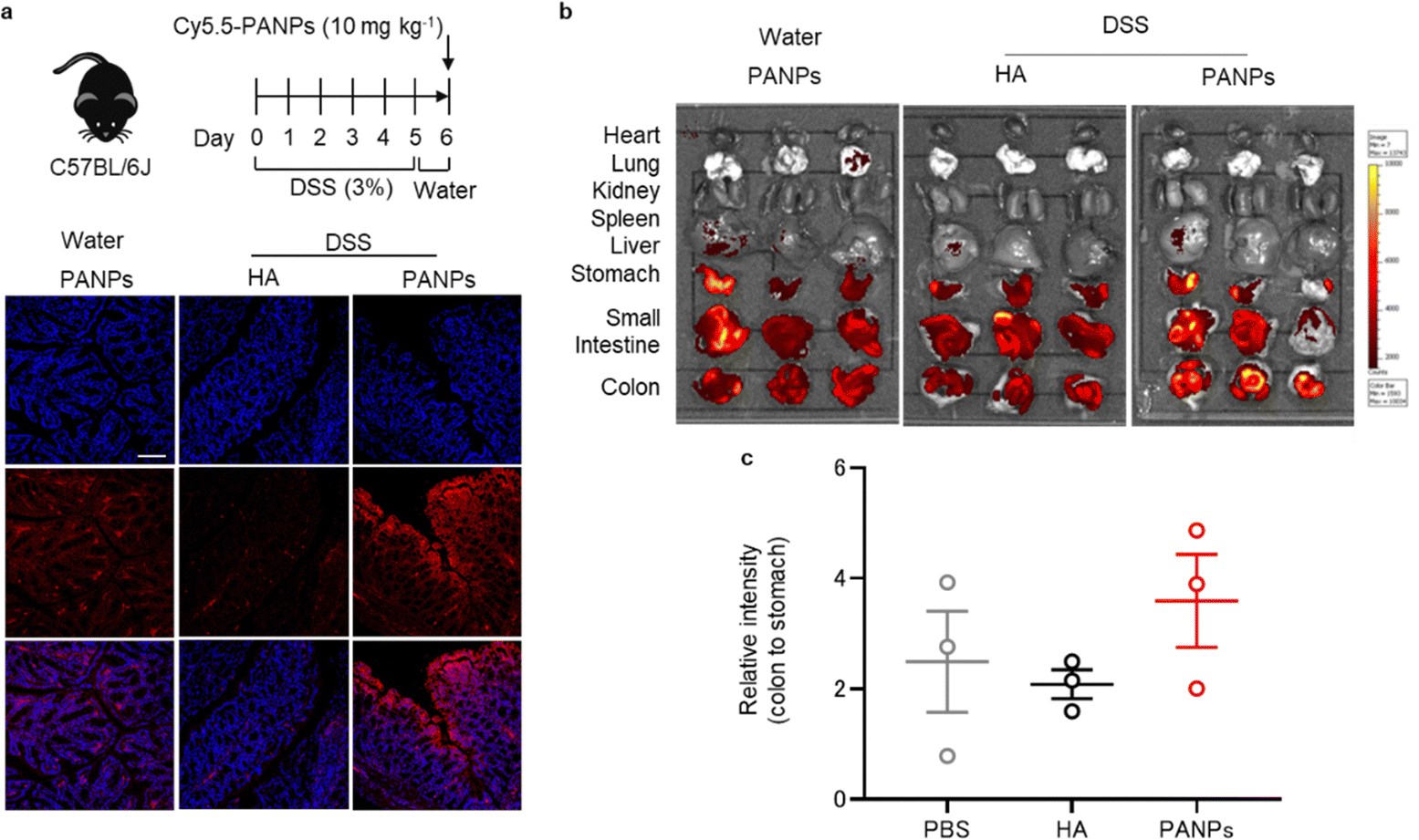 | ||
| Fig. 3 (a) Mice (C57BL/6J) were provided water or 3% dextran sulfate sodium (DSS)-containing water for 5 d. After 1 d of providing with water, mice were orally administered Cy5.5-HA (10 mg kg−1) and Cy5.5-labeled 100-PANPs (10 mg kg−1). Tissues were collected 6 h after administration. Fluorescence images of histological sections of colons stained with 4′,6-diamidino-2-phenylindole (DAPI) were observed. HA and 100-PANPs were fluorescently-labeled (red) and nuclei were stained with DAPI (blue). (b) Fluorescence observation of the localization of Cy5.5-labeled HA and 100-PANPs in the heart, lungs, kidneys, spleen, liver, stomach, small intestine, and colon tissues using an in vivo imaging system (IVIS). Scale bar, 100 μm. Data are presented as the mean ± standard error of the mean (SEM) from one representative experiment (biologically independent samples). | ||
3.4 Treatment of DSS-induced ulcerative colitis
Finally, we evaluated the therapeutic efficacy of 100-PANPs against ulcerative colitis models in mice. Prior to the experiments of ulcerative colitis, the biocompatibility of 100-PANPs was evaluated in mice. Oral administration of 100-PANPs did not change the weight of mice, even at a higher dose (10 times the dose for the therapeutic experiment) (Fig. 4a). Histological observations of the heart, lungs, liver, kidneys, spleen, and colon tissues showed no severe damage, hemorrhage, or necrosis (Fig. 4b). Blood tests revealed that 100-PANPs did not cause changes in the biological components associated with white cells, red cells, liver, and kidney functions (Fig. 4c). Biodegradable HA-based nanomedicines are expected to be absorbed through enzymatic degradation and eliminated. These results indicate that there was no severe systemic toxicity in orally administered 100-PANPs. Compared to the original PA with a positive charge, negatively charged PANPs may avoid nonspecific protein adsorption and cation-induced toxicity,32 which potentially suppresses the off-target effects and poor targeting efficacy of drugs. Moreover, negatively charged PANPs may reduce the interaction with the cell membrane and cellular uptake, which can aid in scavenging ROS in extracellular microenvironments in inflamed tissues and avoid disruption of the mitochondrial activity.6 100-PANPs were orally administered to DSS-induced ulcerative colitis models (Fig. 5a). Note that PANPs were administered after DSS treatment for 5 days, although previous reports often used prophylactic inflammation models treated with DSS and drugs simultaneously.21 DSS-treated mice showed drastic drop in body weight around 80% on day 10 (Fig. 5b). The body weight of mice administered with all PANPs recovered after day 7 and 100-PANPs-treated samples reached over 90% on day 10. These PANPs showed higher recovery of body weight than 5-ASA, which is a commercially available drug for ulcerative colitis. Although 5-ASA has been widely used for ulcerative colitis, its therapeutic effects are often limited and cause off-target side effects, including abdominal pain, hair loss, and allergic reactions.33 In PBS-treated ulcerative colitis mice, the colon was shortened due to thickening of the intestinal tract through severe inflammation (Fig. 5c). Moreover, 100-PANPs suppressed the shortening of colon length induced by DSS and showed the longest length among the samples. ELISA of the collected colon tissues showed that the secretion of the inflammatory cytokine, IL-6, was suppressed in 100-PANPs-treated mice (Fig. 5d). These results indicate that treatment with anti-inflammatory PANPs suppresses colon damage by inhibiting the inflammatory responses. Histological observation of HE-stained cross-sectional tissues revealed disruption of intestinal barrier structures and infiltration of inflammatory cells in PBS-treated mice, while PANP-treated mice showed pathological tissue structures similar to those in control mice (Fig. 5e). Especially, 100-PANPs significantly reduced colonic damage in colitis models (Fig. 5f). When orally delivered PANPs are localized in inflamed intestinal colon mucosa, they may effectively scavenge excessive ROS and suppress inflammatory responses via NF-κB nuclear translocation in macrophages.15 This anti-inflammatory process may prevent the disruption of epithelial barriers and immune cell infiltration, promoting tissue regeneration. These results suggest that orally administered anti-inflammatory PANPs can be used to treat ulcerative colitis by suppressing the inflammatory responses in the colon.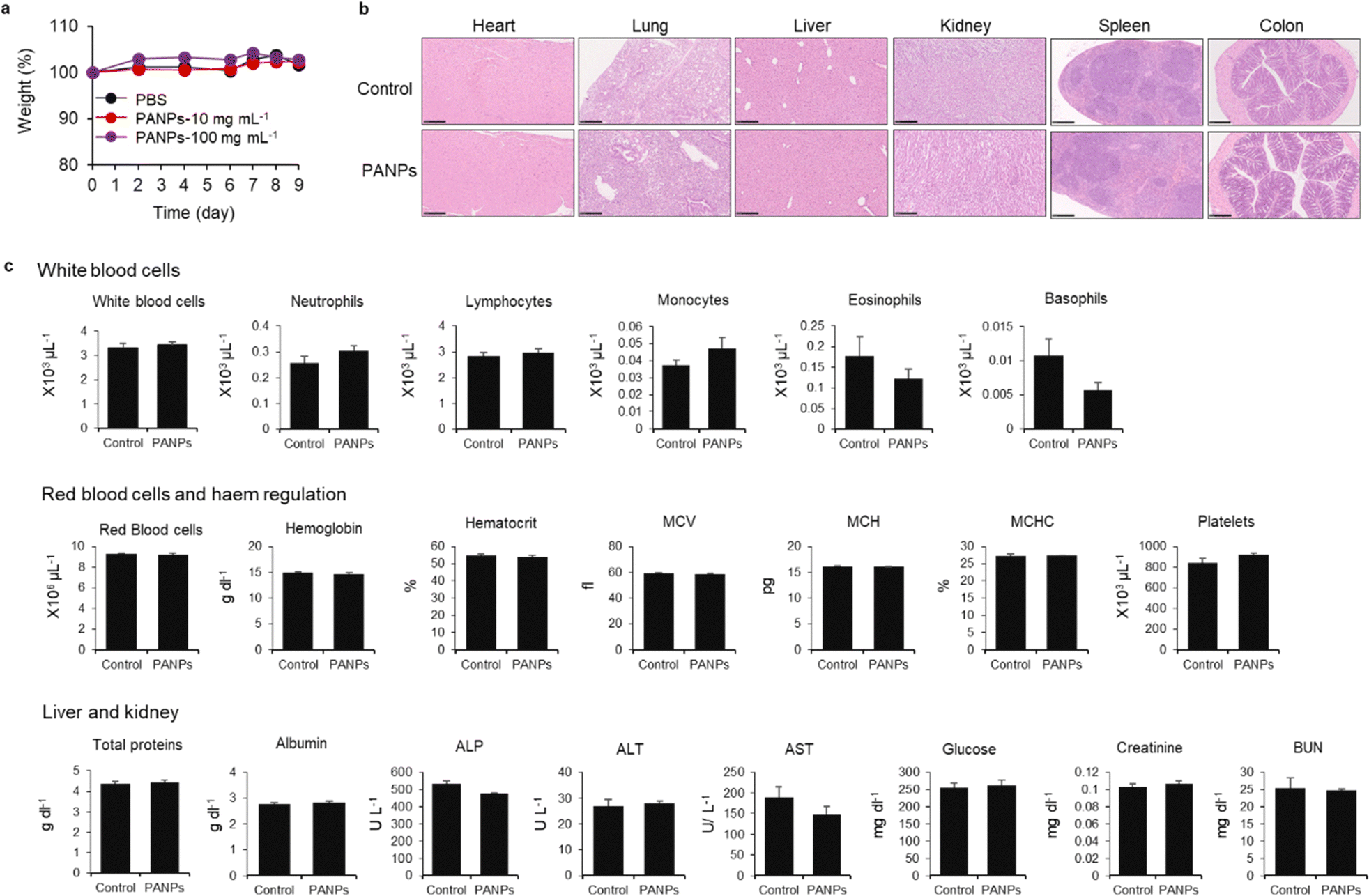 | ||
| Fig. 4 (a) Mice (C57BL/6J) were orally administered phosphate-buffered saline (PBS) and 100-PANPs (10 and 100 mg kg−1) on days 0, 2, 4, and 6. Body weight changes were observed for 9 d. (b) Hematoxylin and eosin (HE)-stained histological images of heart, lungs, liver, kidneys, spleen, and colon tissues. (c) Blood was analyzed via blood chemistry for systemic toxicity evaluation. Data are presented as the mean ± SEM (a) and SD (c) from one representative experiment (n = 3; biologically independent samples). | ||
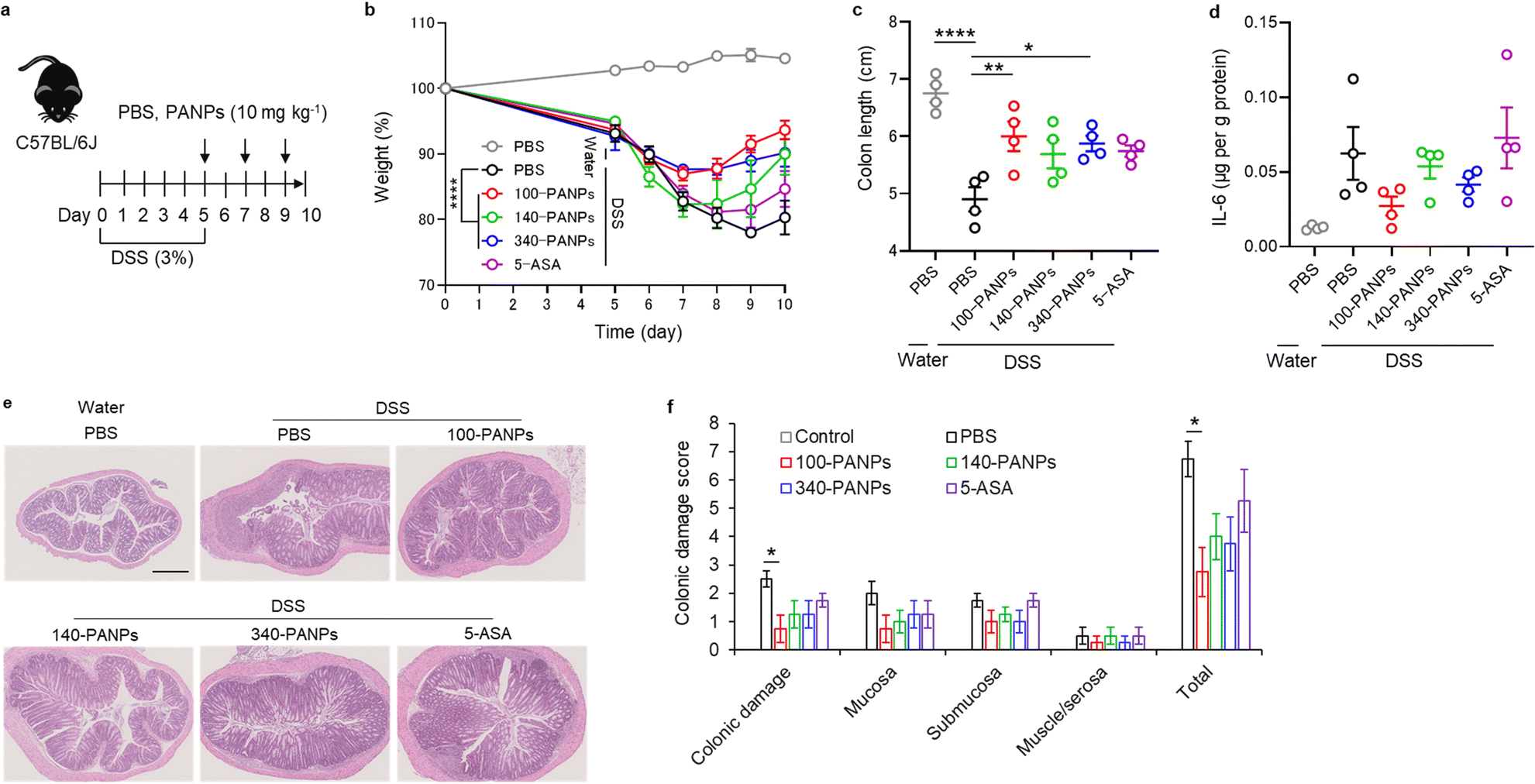 | ||
| Fig. 5 (a) Mice (C57BL/6J) were provided water or 3% DSS-containing water for 5 d and orally administered PBS, 100-PANPs (10 mg kg−1), and 5-aminosalicylic acid (5-ASA; 10 mg kg−1) on days 5, 7, and 9. (b) Changes in daily body weight for 10 d (n = 4). (c) On day 10, mice were euthanized, and colon length was measured (n = 4). (d) ELISA measurements of interleukin (IL)-6 levels in the colon collected on day 10. (e) HE-stained images and (f) damage scores of colon on day 10 (n = 4). Scale bar, 500 μm. Data are presented as the mean ± SEM from one representative experiment (biologically independent samples. *P < 0.05, **P < 0.01, ****P < 0.0001 analyzed via one-way (c and f) or two-way (b) ANOVA with Tukey's multiple comparison post-hoc test. | ||
4. Conclusions
In this study, we developed oral ROS-scavenging PANPs for the treatment of ulcerative colitis in mice. PANPs were prepared via self-assembly of HA in organic solvents and crosslinking with PA. PANPs were found to suppress excessive inflammatory responses in macrophages via the potent ROS-scavenging functions of bOEI against hydroxyl radicals. Nanoparticulation of bOEI and HA enhanced their stability against enzymatic degradation and revealed targeting ability to inflamed intestinal tissues. Moreover, PANPs recovered inflammatory responses and pathological disorders in mice with DSS-induced UC. Therefore, PA-based nanomedicines with high biocompatibility and ROS-scavenging activity may serve as anti-inflammatory drugs for the treatment of inflammatory diseases in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Japan Society for the Promotion of Science KAKENHI (grant numbers 22K19947, 22H03962, and 20H02470), Project for Translational Research program, Strategic PRomotion for practical application of INnovative medical Technology from the Japan Agency of Medical Research and Development (grant number JP20lm0203010), “Innovation inspired by Nature” Research Support Program, Uehara Memorial Foundation, and NIMS Molecule & Material Synthesis Platform in “Nanotechnology Platform Project” by the Ministry of Education, Culture, Sports, Science and Technology, Japan.References
- E. Cario, Inflammatory Bowel Dis., 2010, 16, 1583–1597 CrossRef PubMed.
- M. Saleh and G. Trinchieri, Nat. Rev. Immunol., 2011, 11, 9–20 CrossRef CAS PubMed.
- K. Papamichael, A. Gils, P. Rutgeerts, B. G. Levesque, S. Vermeire, W. J. Sandborn and N. Vande, Casteele, Inflammatory Bowel Dis., 2015, 21, 182–197 CrossRef PubMed.
- P. D. Lu and Y. H. Zhao, Chin. Med., 2020, 15, 15 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- L. A. Sena and N. S. Chandel, Mol. Cell, 2012, 48, 158–167 CrossRef CAS PubMed.
- M. B. Grisham, Lancet, 1994, 344, 859–861 CrossRef CAS PubMed.
- H. A. Almenier, H. H. Al Menshawy, M. M. Maher and S. Al Gamal, Front. Biosci., 2012, 4E, 1335–1344 CrossRef PubMed.
- G. Bjelakovic, D. Nikolova, L. L. Gluud, R. G. Simonetti and C. Gluud, JAMA, 2007, 297, 842–857 CrossRef CAS PubMed.
- Z. Cheng, A. A. Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- T. Yoshitomi, D. Miyamoto and Y. Nagasaki, Biomacromolecules, 2009, 10, 596–601 CrossRef CAS PubMed.
- Y. Lee, K. Sugihara, M. G. Gillilland 3rd, S. Jon, N. Kamada and J. J. Moon, Nat. Mater., 2019, 19, 118–126 CrossRef PubMed.
- Q. Zhang, H. Tao, Y. Lin, Y. Hu, H. An, D. Zhang, S. Feng, H. Hu, R. Wang, X. Li and J. Zhang, Biomaterials, 2016, 105, 206–221 CrossRef CAS PubMed.
- J. F. Watchko and C. Tiribelli, N. Engl. J. Med., 2013, 369, 2021–2030 CrossRef CAS PubMed.
- H. C. Ha, N. S. Sirisoma, P. Kuppusamy, J. L. Zweier, P. M. Woster and J. R. A. Casero, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11140–11145 CrossRef CAS PubMed.
- A. Nishiguchi and T. Taguchi, Adv. Funct. Mater., 2021, 31, 2100548 CrossRef CAS.
- S. Misra, V. C. Hascall, R. R. Markwald and S. Ghatak, Front. Immunol., 2015, 6, 201 Search PubMed.
- H. Zhao and N. D. Heindel, Pharm. Res., 1991, 8, 400–402 CrossRef CAS PubMed.
- J. L. Reissig, J. L. Storminger and L. F. Leloir, J. Biol. Chem., 1955, 217, 959–966 CrossRef CAS.
- R. C. S. Bicudo and M. H. A. Santana, J. Nanosci. Nanotechnol., 2012, 12, 2849–2857 CrossRef CAS PubMed.
- L. B. Vong, T. Tomita, T. Yoshitomi, H. Matsui and Y. Nagasaki, Gastroenterology, 2012, 143, 1027–1036 CrossRef CAS PubMed.
- G. Wang, X. Cao, H. Dong, L. Zeng, C. Yu and X. Chen, Polymers, 2018, 10, 949 CrossRef PubMed.
- L. F. Chen and W. C. Greene, Nat. Rev. Mol. Cell Biol., 2004, 5, 392–401 CrossRef CAS PubMed.
- S. Akira and K. Takeda, Nat. Rev. Immunol., 2004, 4, 499–511 CrossRef CAS PubMed.
- Q. Li and I. M. Verma, Nat. Rev. Immunol., 2002, 2, 725–734 CrossRef CAS PubMed.
- I. Atreya, R. Atreya and M. F. Neurath, J. Intern. Med., 2008, 263, 591–596 CrossRef CAS PubMed.
- I. Okayasu, S. Hatakeyama, M. Yamada, T. Oohusa, Y. Inagaki and R. Nakaya, Gastroenterology, 1990, 98, 694–702 CrossRef CAS PubMed.
- A. Lamprecht, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 195–204 CrossRef CAS PubMed.
- A. Lamprecht, U. Schäfer and C. M. Lehr, Pharm. Res., 2001, 18, 788–793 CrossRef CAS PubMed.
- B. Tirosh, N. Khatib, Y. Barenholz, A. Nissan and A. Rubinstein, Mol. Pharm., 2009, 6, 1083–1091 CrossRef CAS PubMed.
- S. Zhang, J. Ermann, M. D. Succi, A. Zhou, M. J. Hamilton, B. Cao, J. R. Korzenik, J. N. Glickman, P. K. Vemula, L. H. Glimcher, G. Traverso, R. Langer and J. M. Karp, Sci. Transl. Med., 2015, 7, 300ra128 Search PubMed.
- A. E. Pegg, Chem. Res. Toxicol., 2013, 26, 1782–1800 Search PubMed.
- J. Kao, K. Kwok and K. M. Das, J. Clin. Gastroenterol., 2010, 44, 531–535 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |