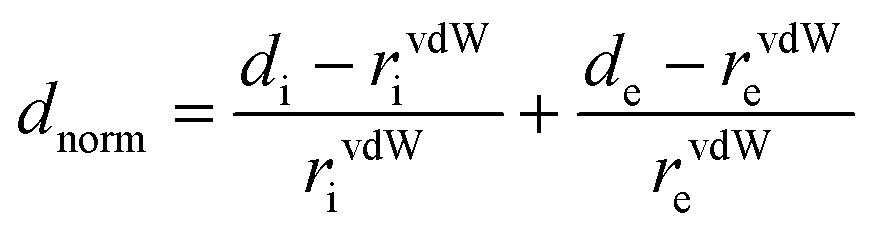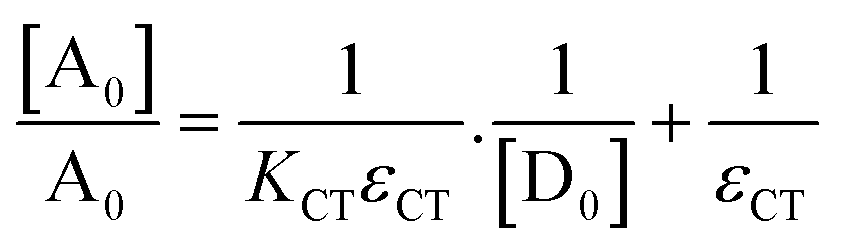DOI:
10.1039/D2TB02362H
(Paper)
J. Mater. Chem. B, 2023,
11, 1262-1278
Protective effect of the newly synthesized and characterized charge transfer (CT) complex against arecoline induced toxicity in third-instar larvae of transgenic Drosophila melanogaster (hsp70-lacZ)Bg9: experimental and theoretical mechanistic insights†
Received
30th October 2022
, Accepted 25th December 2022
First published on 27th December 2022
Abstract
Agents that suppress the toxic effect of arecoline (a chemical present in the Areca nut fruit) have become a need of the hour owing to its several harmful effects on human beings. Although some drug molecules have been developed for this purpose, yet, simple, easy to prepare, and economical molecules with remarkable potency are still a challenge to design. The present work thus becomes important as it involves the synthesis of a new charge transfer complex (CTC) material, which has, for the first time, been screened to investigate its effect on the toxic effects of arecoline. The newly designed material (CL), which is generated from the reaction between 2,4,6-trinitrophenol (TNP) and pyrazole (PYZ), has been crystallized by a slow evaporation method and characterized by employing spectral studies including single crystal X-ray crystallography. Spectrophotometry studies with the inclusion of the Benesi–Hildebrand equation reveal 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry and physical parameters of CL. Assays were used for determining the protective effect of CL against arecoline. CL was found to (dose-dependently) decrease β-galactosidase activity, damage in tissue and DNA damage caused by arecoline (80 μM) in the third-instar larvae of the transgenic Drosophila melanogaster (hsp70-lacZ)Bg9. The possible mechanism of this effect was explored through fluorescence and UV-vis spectroscopy. The possibility of suppression of arecoline action on the muscarinic acetylcholine receptor 1-G11 protein complex (found in the cell membrane) in the presence of CL was studied theoretically by molecular docking. Density functional theory (DFT) also theoretically supported various aspects of the designed material concerning the energy profile of the orbitals (HOMO–LUMO) as well as the energy minimized structure. Furthermore, time dependent (TD) DFT corroborated the electronic properties of the designed material.
:1 stoichiometry and physical parameters of CL. Assays were used for determining the protective effect of CL against arecoline. CL was found to (dose-dependently) decrease β-galactosidase activity, damage in tissue and DNA damage caused by arecoline (80 μM) in the third-instar larvae of the transgenic Drosophila melanogaster (hsp70-lacZ)Bg9. The possible mechanism of this effect was explored through fluorescence and UV-vis spectroscopy. The possibility of suppression of arecoline action on the muscarinic acetylcholine receptor 1-G11 protein complex (found in the cell membrane) in the presence of CL was studied theoretically by molecular docking. Density functional theory (DFT) also theoretically supported various aspects of the designed material concerning the energy profile of the orbitals (HOMO–LUMO) as well as the energy minimized structure. Furthermore, time dependent (TD) DFT corroborated the electronic properties of the designed material.
1. Introduction
Arecoline (methyl-1,2,5,6 tetrahydro-1-methyl-nicotinamide) is an alkaloid, which is isolated from Areca catechu.1 Though an important pharmacological component for various ailments the toxicity of this component is an important concern for pharmacologists.2 The long-term use of arecoline could lead to oral cancers.1 Therefore, it is necessary to find an effective and safe strategy to minimize its harmful effects. Arecanut chewing in the form of betel quid (with lime and with or without tobacco) is very common in South East Asian countries and has been associated with the development of oral cancers or other pre-cancerous symptoms.3 The study on cultured human keratin type cells showed that the exposure of arecoline induced the generation of ROS and arrested cell cycle at the G1/G0 phase.3 The toxicity of arecoline has been studied in several models, which have been extensively reviewed by Oliviera et al. (2021).4 Arecoline has also been reported to be genotoxic in various experimental models.5–9 We have studied the toxicity of arecoline and the protective effect of natural plant products (Geraniol) in the third instar larvae as well as adult flies of the transgenic Drosophila melanogaster (hsp70-lacZ)Bg9.10–12 The toxic effects of arecoline could be reduced by using natural plant products and the supplementation of various vitamins.13
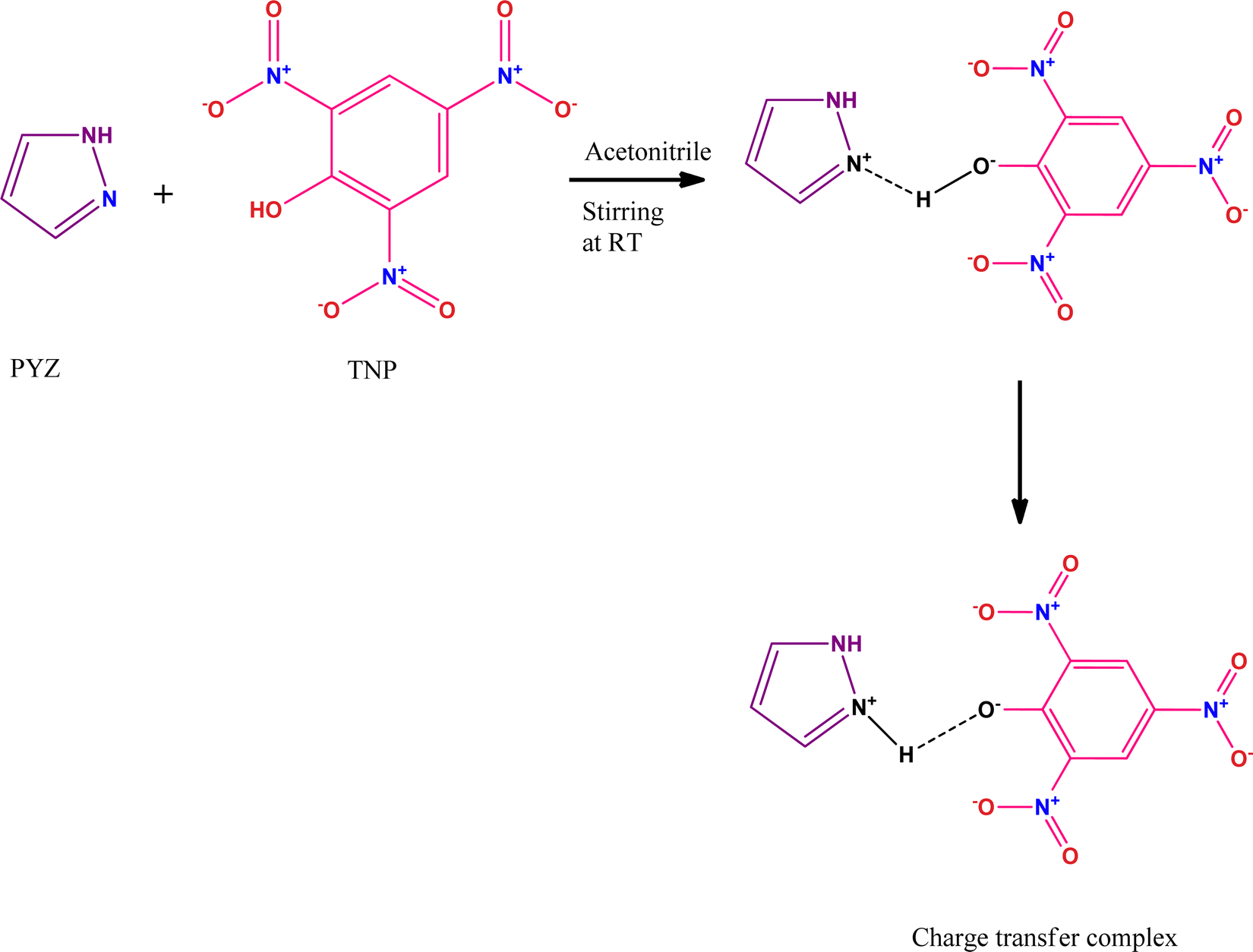
The charge transfer complexes (CTCs) are self-assembled organic materials formed from the interaction between donor and π-acceptor moieties through N+–H⋯O− bonding, and this concept was first introduced by Mulliken and Foster.14,15 CTCs have widespread applications in drug actions, dendrimers’ second-order non-linear optical activity, redox processes, organic semiconductors, and micro-emulsion.16–22 Nowadays, CTCs have emerged as promising and potential chemosensors for detecting hazardous materials.23–25 CTCs of 2,4,6-trinitrophenol (TNP) with donors like 4-(2-thiazolylazo)resorcinol (TAR),26 heterocyclic 4-methylpyridine-2-azomethine-p-benzene derivatives,27 1-naphthylamine,28p-toluidine,29p-phenylenediamine,30 norfloxacin and ciprofloxacin drugs,31 2-hydroxypyridine,32o-tolidine,33 and 2,2′-bipyridine34 have been studied before. The formation of a CTC of TNP and pyrazole (PYZ) for exploring its protective effect against arecoline induced carcinogenicity has not been achieved. The reactant PYZ has effective therapeutic and biologically active scaffolds. PYZ has been reported to possess an extensive range of biological activities like anti-cancer, anti-convulsant, anti-microbial, anti-viral, anti-tubercular, anti-inflammatory, anti-fungal, etc. Chemical modification like adding suitable functional groups on the PYZ moiety revealed useful biological activities and many PYZ derivatives like-1,3,4-trisubstituted pyrazole derivatives, ethyl-5-amino-3-methylthio-1H-pyrazole-4-carboxylates, 4-thiazolyl pyrazolyl derivatives, etc.,35 have already found their application as nonsteroidal anti-inflammatory drugs clinically. Recent observations suggest that substituted pyrazole heterocycles, which are the structural isosteres of nucleotides having fused heterocyclic nuclei in their structures that allow them to interact easily with biopolymers, possess potential activity with lower toxicity in the chemotherapeutic approach in humans.36
In this study, we have designed a CTC material (CL) having N+–H⋯O− bonding between TNP and PYZ, which is evident in single crystal XRD data. This CTC was characterized and studied through FTIR, 1H NMR, TGA-DTA, powder-XRD, and SEM. Hirshfeld surface analysis of the designed material (CL) was used to show dnorm, curvedness and shape index surfaces, along with the fingerprint (2D) plot showing the percentage of close contacts in the crystal. Electronic spectroscopy was employed to calculate the molar extinction coefficient and formation constant to obtain other important physical parameters of the designed material.
Drosophila melanogaster (fruit fly) has been used as an in vivo model organism not only for the study of genetics and development but also for toxicological studies.37Drosophila melanogaster shows various similarities with the human genome38 and also has a functional homology of approximately 75% of the known human disease genes.39 In this context, the heat shock proteins are often associated with the cellular response to harmful environmental agents.40 The stress proteins are highly conserved from bacteria to humans and the most highly conserved family of stress proteins is hsp70.41 Therefore, the present study was carried out on the third instar larvae of transgenic Drosophila melanogaster (hsp70-lacZ)Bg.9 The synthesized compound (CL) was studied for its protective effect against arecoline induced toxicity in the third instar larvae of transgenic Drosophila melanogaster (hsp 70-lac Z)Bg9. We have performed and explored the in situ histochemical β-galactosidase assay, trypan blue exclusion test and comet assay (DNA damage) to study the protective effect of CL in the arecoline exposed third-instar larvae of the transgenic Drosophila melanogaster (hsp70-lacZ)Bg9.
The possible mechanism was explored through UV-visible and fluorescence titration. Molecular docking was used to explore the pharmaceutical action of CL by targeting the muscarinic acetylcholine receptor 1-G11 protein complex as arecoline is known to be a partial agonist of muscarinic acetylcholine M1. Experimental data were supported with the help of DFT theoretical calculations (B-3LYP/6-31G**), and chemical, structural, spectroscopic, thermodynamic and vibrational phenomena of the synthesized material were obtained.
2. Experimental
2.1. Materials, synthesis and crystallization of CL
All reagents of analytical grade, including pyrazole (Aldrich) and 2,4,6-trinitrophenol (Merck), were purchased commercially and used without any further purification. Arecoline and stains (acridine orange and ethidium bromide) were procured from Sigma-Aldrich.
The CTC (CL) of pyrazole (PYZ) and 2,4,6-trinitrophenol (TNP) was synthesized from individually prepared saturated solutions of TNP (1.145 g, 5 mmol) and PYZ (0.34 g, 5 mmol) in acetonitrile. The saturated solutions of TNP and PYZ were mixed slowly with continuous stirring for 2 hours at room temperature. A yellow precipitate started to appear in the solution, which was filtered and washed using acetonitrile. The filtrate was allowed to dry in a desiccator (over anhydrous CaCl2); this dried filtrate is our desired CTC material (CL). The crystallization of CL was achieved by preparing a saturated solution of CL in acetonitrile:ethanol. This saturated solution was stirred for about 3 hours at 35 °C and then filtered to eliminate impurities. The filtered solution was then left undisturbed for 4 days, after which yellow crystals could be seen at the bottom of the beaker.
2.2. Preparation of different solutions
Stock solutions of TNP and PYZ in ethanol at 10−2 M concentration were obtained by mixing 0.228 g of TNP in 100 mL of ethanol and 0.068 g of PYZ in 50 mL of ethanol. The stock solution of PYZ was then diluted to prepare different concentrations (1 × 10−4, 1.5 × 10−4, 2.0 × 10−4, 2.5 × 10−4, and 3.0 × 10−4 M) of the solution, while TNP was diluted to obtain only 1 × 10−4 M solution. Similar concentration solutions were also prepared with other polar solvents (acetonitrile and methanol).
2.3. Characterization of CL
The FTIR (Fourier transform infrared spectroscopy) spectra of CL, PYZ, and TNP were obtained separately by using a PerkinElmer FTIR spectrometer with a KBr disc. The UV-visible spectra of CL, PYZ, and TNP were observed in three polar solvents separately (acetonitrile, methanol, and ethanol). The spectra were recorded in the region of 200–550 nm using a PerkinElmer UV Lambda 45-visible spectrophotometer. 1H NMR of CL was obtained through a Bruker Avance II-400 MHz spectrometer. The P-XRD pattern of CL was recorded through a Bruker Advance D8-diffractometer, at 40 kV in the range of 4–80 °C using Cu K radiation having λ = 0.1542 nm. The TG/DTA (thermogravimetric and differential thermal) study of CL, PYZ, and TNP was done with an EXSTAR TG/DTA 6300 model using a nitrogen (N) atmosphere and 20 °C min−1 heating rate. An aqueous solution of CL (10−4 M) was prepared and fluorescence was measured following excitation at 390 nm using a F-2700 FL-Spectrophotometer. The slit widths of 5.0 and 5.0 nm for the detector and source, respectively, were set. For understanding the microstructure and morphology of CL, scanning electron microscopy (SEM, Quanta FEG-250 instrument) was used. A voltage of 20 kV was supplied to the instrument.
2.4. Single crystal X-ray study
For obtaining crystallographic data of CL, an appropriate crystal was chosen on a Bruker APEX-II CCD diffractometer. The selected crystal of CL was kept at 100 (2) K at the time of collecting data. The structure was solved Using Olex2,42 with the olex2.solve43 structure solution program using Charge Flipping and the data were refined using Gauss-Newton minimization with the olex2.refine43 refinement package.
2.5. Design and assays for determining the protective effect of CL against arecoline
A transgenic fly strain of the Drosophila melanogaster line was used, which expresses bacterial β-galactosidase as a response to stress.44 The transformation vector in this fly strain is inserted with a P-element, i.e., the line contains the wild type hsp70 sequence up to the lac Z fusion point. The larvae and flies were cultured on standard Drosophila food containing agar, maize powder, sugar, and yeast at 24 °C ± 1 °C.45
Larvae were individually exposed to 0.0010, 0.0020, 0.0030, 0.0040, 0.0050, 0.0060, 0.0070, 0.0080, and 0.0090 M CL for 24 hours, and 50% mortality was observed at 0.0070 M CL. This was undertaken as a pilot study to determine LC50. The highest tested dosage was therefore 1/4th of LC50.46 Third instar larvae were permitted to consume CL at final concentrations of 0.2187 × 10−3 M, 0.4375 × 10−3 M, 0.875 × 10−3 M and 1.75 × 10−3 M mixed in food alone and combined with 80 μM arecoline for 24 hours.
2.5.1. Trypan blue exclusion test.
A dye exclusion test was employed to study the tissue damage extent in the larvae.47,48 The explantation of internal tissues of larvae (5 larvae per treatment and 5 replicates per group) was done in the Poles Salt Solution (PSS) drop and the tissues were washed thoroughly in phosphate buffer saline (PBS). The washed explanted tissues were then stained for 30 min in 0.2 mg mL−1 trypan blue in PBS and washed again in PBS, and were immediately scored for dark blue staining. The scoring for the trypan blue staining was done on an average composite index per larva: complete staining of most cells in the tissue = 4; large patches of darkly stained cells = 3; darkly stained = 2; any blue = 1; or no color = 0.47
2.5.2. Soluble O-nitrophenyl-β-D-galactopyranoside (ONPG) assay.
The larvae were washed in PBS and kept in an Eppendorf tube (20 larvae per tube; 5 replicates per group), permeabilized for 10 minutes by acetone, and incubated overnight at 37 °C in 600 μL of ONPG buffer. The reaction was stopped after incubation by adding 300 μL of Na2CO3. The absorbance at 420 nm was used to quantify the extent of the reaction. The ONPG test measures the cytotoxicity of hsp70 expression by measuring the activity of β-galactosidase.49
2.5.3.
In situ histochemical β-galactosidase activity.
The larvae (10 larvae/treatment; 5 replicates/group) were dissected out in PSS and X-gal staining was performed using the method described by Chowdhuri et al. (1999).50 The larval explants were fixed in glutaraldehyde (2.5%), washed in 50 μM sodium phosphate buffer (pH 8.0) and stained overnight at 37 °C in the dark with X-gal staining solution.
2.5.4. Comet assay (DNA damage).
The comet assay experiment was done according to Mukhopadhyay et al. (2004).51 The larval midgut was explanted in PSS (20 larvae/treatment and 4 replicates per group); 300 mL (0.5 mg mL−1 in PBS) of collagenase (pH ∼ 7.4) was used to replace PSS in the microcentrifuge tube which was left at 25 °C for about 15 min. For preparing the suspension of cells, the pellet was washed using phosphate buffer saline and cells were suspended in 80 μL of PBS. 1.5% of 80 μL low melting agarose was mixed with 75 μL of cell suspension, which was then layered on the slides precoated with 1% agarose having a regular melting point. These prepared slides were engrossed in chilled lysing solution (100 mM EDTA, 2.5 M NaCl, 1% Triton-X-100 pH ∼ 10.0 and 10 mM Tris pH ∼ 10.0) at 4 °C for 2 h. Further, they were transferred to the chilled solution of electrophoresis (300 mM NaOH, 1 mM Na2 EDTA, pH > 13). The unwinding of DNA was allowed by keeping it for 10 min in this solution. Electrophoresis was conducted at 4 °C (300 mA) for 15 min and 0.7 V cm−1. After electrophoresis, all the slides were washed using 0.4 M Tris buffer (neutralizing buffer). Slides were then stained for 10 min in the dark using EB (75 μL per slide and 20 μg per mL) and washed with chilled distilled water. At constant gel depth, 20 cells per slide and 4 replicates per group were captured randomly. To calculate the DNA damage, the mean tail length (a.u.) was measured through software Comet ScoreTM v1.5, Su-merduck, TriTek Corporation.52
2.5.5. Statistical analysis.
Using the GraphPad Prism software [version 5.0], the data were statistically analyzed using one-way analysis of variance (ANOVA) and the post hoc Tukey test. The significance threshold was held at p < 0.05. Mean ± SEM was used to express the findings.
2.6. Computational studies
2.6.1. Molecular docking.
The muscarinic acetylcholine receptor 1-G11 protein complex found in the cell membrane, which is responsible for the end-receptor stimulation by releasing acetylcholine, was used as a target molecule as arecoline is known to be a partial agonist of muscarinic acetylcholine M1. Muscarinic acetylcholine M1 (PDB IB = 6OIJ) was downloaded online from the Protein Data Bank (https://www.rcsb.org/) to study the possible interaction of arecoline with muscarinic acetylcholine M1 in the presence and absence of CL. Hex 6.153 software was used for this study using shape + DARS + electrostatic correlation and Grid: 118 × 118 × 118. Visualization of the docking pose was obtained using Chimera software (https://www.cgl.ucsf.edu/chimera). The overall docking experiment was run on a processor (Intel(R) Core(TM) i5-4200U CPU @ 1.60 GHz 2.10 GHz 2.30 GHz, 64 bit).
2.6.2. DFT calculations.
The Gaussian 09 software package54 was used for DFT studies to characterize the molecular geometry and electronic transitions and to better understand the spectral assignments in the synthesized material CL. Optimized structures and Mulliken charges of CL, TNP and PYZ were obtained through the Pople basis set (B3LYP/6-31G**). DFT in combination with the gradient-corrected correlation functional and Becke's three-parameter hybrid exchange function having the Pople basis set were used for full geometrical optimization.55 Moreover, structure-based molecular properties like atomic charges, total energy, electronic properties, frontier molecular orbital energy eigenvalues with their gap and molecular electrostatic potential were also calculated by theory in the gas phase. For visualization of the obtained DFT results, ChemCraft 1.5 software was used.
2.6.3. Hirshfeld surface analyses of the CL crystal.
The partitioning space of the molecule in the crystal into regions where the procrystal is being dominated by the promolecule is the Hirshfeld surface of the crystal.56 CrystalExplorer 3.157 was used to obtain Hirshfeld surfaces (dnorm, curvedness, shape index, etc.) and fingerprint plots (2D). The color scheme (blue-red-white) in the Hirshfeld surfaces represents different types of contacts. The areas of white color represent van der Waals contact separation, bright red areas are for shorter contacts and areas of blue lack close contacts. de and di are the two distances, where di represents the distance from the Hirshfeld surface to the nearest nucleus internal to the surface and de is for the outer surface. The normalized contact distance (dnorm) of the Hirshfeld surfaces, based on de and di, is represented as
where rvdWe and rvdWi are the van der Waals radii of the atoms.58
3. Results and discussion
3.1. Instrumental characterization
3.1.1.
1H NMR study for CL.
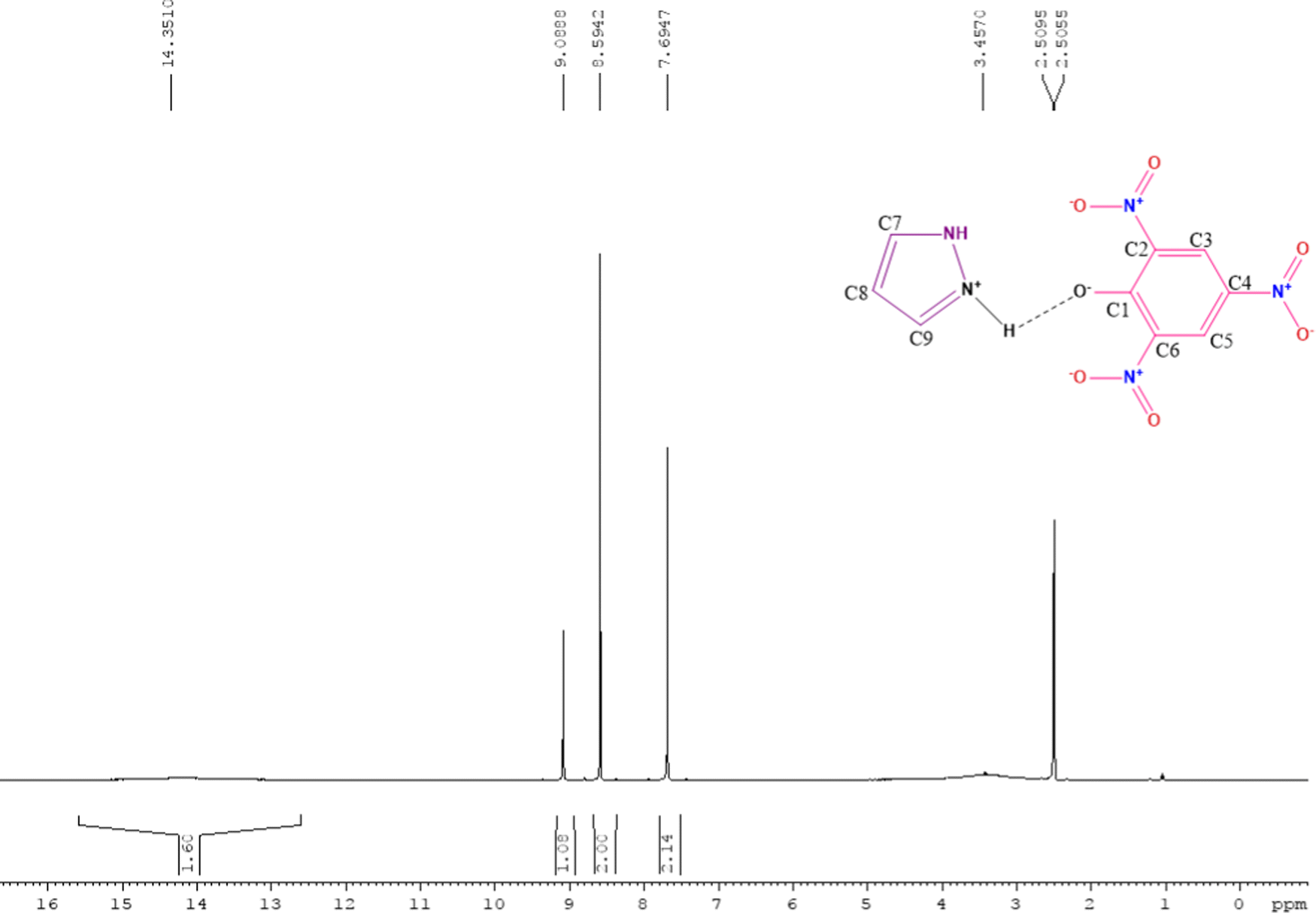
The 1H NMR spectra of free reactants (TNP and PYZ) were compared with the 1H NMR spectrum of CL obtained using DMSO-d6 as a solvent and tetramethylsilane (TMS) as a standard reference to study the proton shifts (Fig. 1). A signal for the N–H proton can be observed at δ = 13.7 ppm in the spectra of PYZ, while it is observed at δ = 14.35 ppm in the case of CL. A signal for the O–H proton at δ = 5.35 ppm, which was observed in the spectrum of free TNP, cannot be seen in the 1H NMR spectra of CL [(PYZH)+(TNP)−].59–61 On the other hand, a new broad signal appeared at δ = 3.45 ppm, which can be designated as a new center (+N–H) due to the electron transfer to the pyrazole N atom from TNP. A triplet at δ = 7.69 ppm for the C8 proton and a doublet at δ = 8.59 ppm for C7 and C9 protons can be seen in the spectrum of CL, which were observed at δ = 7.55 and δ = 6.25 ppm respectively in the PYZ spectrum. The singlet at δ = 9.14 ppm for the C5 and C3 protons of TNP can be seen at δ = 9.08 ppm in CL. The peaks at 2.51 and 2.50 ppm appeared in the spectrum of CL due to the DMSO-d6 solvent. The shift in the frequency is caused by the transfer of hydrogen to TNP and increasing electron density on PYZ.
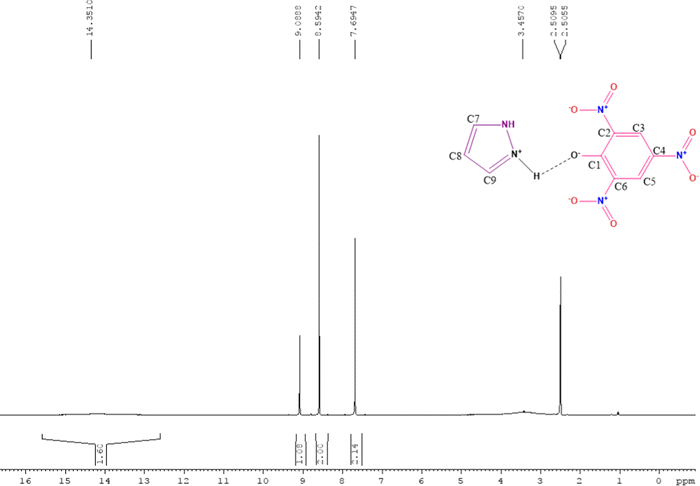 |
| | Fig. 1
1H NMR of the synthesized charge transfer complex (CL) of PYZ and TNP. | |
3.1.2. Size and morphology of CL (SEM images).
Scanning electron microscopy (SEM) analysis was also performed to elucidate the morphology of CL. SEM provides plentiful information regarding the morphology and texture of CL. From Fig. 2 it is clear that the morphological analysis of CL shows a bunch of rectangular plate-shaped crystals with a partial size of 90 × 40 × 20 micrometers approximately. The detailed analysis on zooming out (×600) indicates that the size of the material is larger and the study rules out the presence of any nanoparticle.
 |
| | Fig. 2 SEM micrographs showing the morphology of the synthesized charge transfer complex (CL). | |
3.2. Electronic spectra
The UV-visible spectra were recorded for the different stock solution (PYZ and TNP) mixtures (1 × 10−4 M) in acetonitrile, methanol and ethanol in the range of 200–550 nm. It was observed that new absorption peaks appeared with λmax 374, 361 and 358 nm in acetonitrile, methanol and ethanol, respectively, due to the n → n* transition. These new peaks were not present in the respective acceptor and donor spectra, which indicates the transfer of an electron between reactants, resulting in CL formation (Scheme 1). This transfer of an electron results in the appearance of a hydrogen bond between TNP and PYZ. The UV-visible spectra of PYZ and TNP and the resulting CTC CL [(PYZH)+(TNP)−] in acetonitrile, methanol and ethanol are shown in Fig. S3 (ESI†). The absorption intensity was recorded at λmax in the respective solvents for different solutions prepared by mixing different concentrations of PYZ with a fixed concentration of TNP (Table S1, ESI†). The Gaussian function fitting was applied to analyze the charge transfer band,
| y = y0 + [A/(w √(π/2))] exp [−2(x − xc)2/w2] |
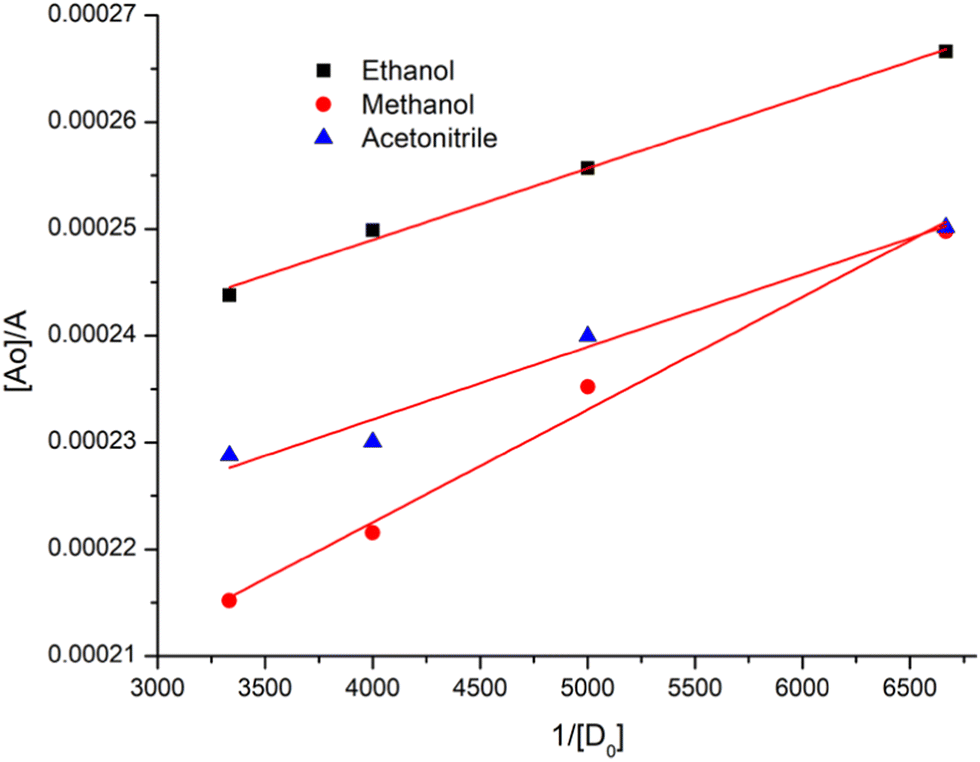
Here, y and x are for absorbance and wavelength, respectively. Table S2 (ESI†) represents the Gaussian fitting results. The Benesi–Hildebrand equation62 was applied to obtain the [A0]/A0vs. 1/[D0] Benesi–Hildebrand plot,
Here, [A0] denotes the initial concentration of TNP, [D0] denotes the initial concentration of PYZ, A denotes absorbance at λCT, KCT denotes the formation constant and εCT denotes the molar extinction coefficient. A straight line plot was obtained for all solvents suggesting 1:1 stoichiometry of CL (Fig. 3). For calculation of the molar extinction coefficient (εCT) and formation constant (KCT) of CL, the intercept and the slope were obtained from the Benesi–Hildebrand plot (straight line method) (Table S1, ESI†).25,63 It can be noted that KCT decreases with the increase in the dielectric constant of the solvent, while εCT significantly increases. This is because of the formation of a strong and stabilized CTC in a less polar solvent. Taken as a measure of solvent polarity, the smaller value of εCT means lower polarity and smaller ability to stabilize charges and hence greater formation constant of the CT complex.64 The data in Table S1 (ESI†) show that KCT is strongly dependent on the solvent used with the order ethanol > methanol > acetonitrile. The least polar aprotic solvent, acetonitrile, exhibits the smallest value of the formation constant relative to both polar protic solvents, methanol and ethanol, due to electric permittivity of the investigated solvents.65
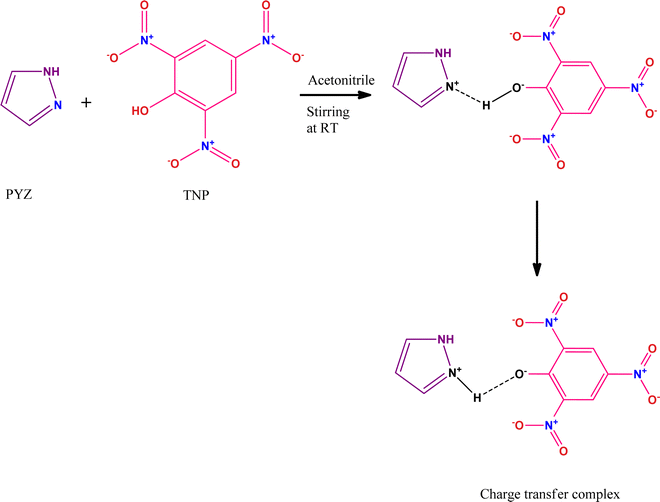 |
| | Scheme 1 Mechanism of the reaction between donor (PYZ) and acceptor (TNP) reactants. | |
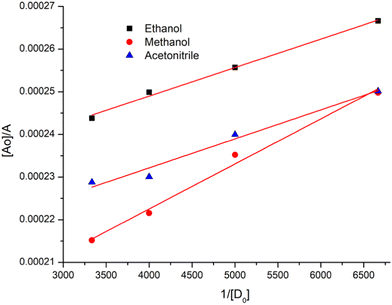 |
| | Fig. 3 Benesi–Hildebrand plots of CL, [A0]/A vs. 1/[D]0, in ethanol, methanol and acetonitrile at room temperature. | |
With the help of calculated εCT and KCT values, other important physical parameters like ID (ionization potential) by Aloisi and Piganatro,66ECT (energy of interaction), RN (resonance energy) by Brieglab and Czekalla,67 ΔG° (free energy),68f (oscillator strength),69 and μEN (dipole moment) by Tsubumora and Lang70 were also calculated using the following equations:
| ID (eV) = 5.76 + 1.53 × 10−4νCT, |
| ECT = 1243.667/λCTnm, |
| εCT = 7.7 × 104/[hνCT/|RN| − 3.5], |
ΔG° = −2.303 RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logKCT, logKCT, |
| F = 4.32 × 10−9εCTΔν1/2, and |
| μEN = 0.0952 [εCTΔν1/2/Δν]1/2 |
The above calculations reveal that with the increase in the polarity of the solvent, the value of ionization potential and energy of interaction decreases.
RN is the resonance energy of the complex in the ground state, which serves as a contributing factor to the stability of
CL. The transition probability of the CT-band is expressed by oscillator strength (
f) which is a dimensionless quantity, which is lowest in acetonitrile. This indicated that the stability of
CL would be more in less polar solvents (observed on the basis of calculated
εCT and
KCT values)
64 as the dative structure D
+–A
− should be stabilized in less polar solvents. The calculated data are shown in
Table 1.
Table 1 Wavelength (λCT), ionization potential (ID), energy of interaction (ECT), resonance energy (RN), oscillator strength (f), dipole moment (μEN), free energy (ΔG) and correlation coefficient (r) of CL
| Solvent |
Wavelength λCT (nm) |
Ionization potential ID (eV) |
Energy of interaction ECT (eV) |
Resonance energy RN |
Free energy −ΔG (kJ mol−1) |
Oscillator strength (f × 10−3) |
Dipole moment μEN (D) |
Correlation coefficient (r) |
| Ethanol |
358 |
10.03 |
3.47 |
0.168 |
25.79 |
9.74 |
23.26 |
0.9984 |
| Methanol |
361 |
9.99 |
3.44 |
0.178 |
25.55 |
6.91 |
22.89 |
0.9989 |
| Acetonitrile |
374 |
9.85 |
3.32 |
0.191 |
24.15 |
3.55 |
19.54 |
0.9971 |
3.3. Assays for determining the protective effect of CL against arecoline
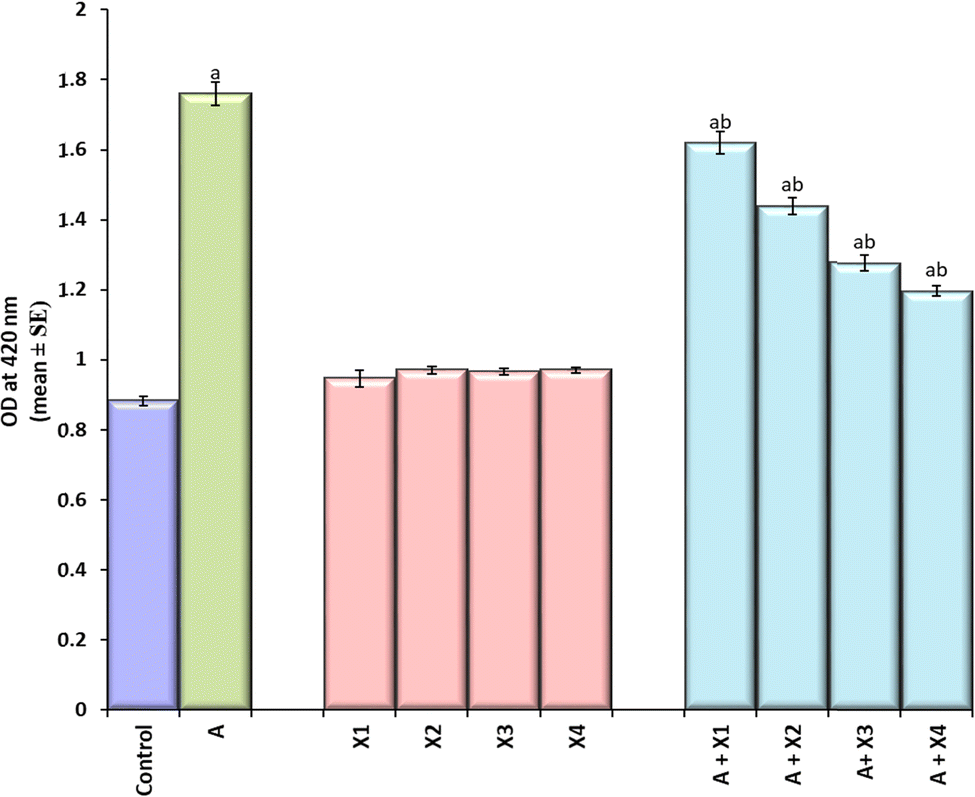
The larvae exposed to 80 μM arecoline showed a significant 2.01 fold increase in the expression of hsp70 compared to the control (Fig. 4; p < 0.05). The larvae exposed to 80 μM arecoline along with 0.2187 × 10−3, 0.4375 × 10−3, 0.875 × 10−3 and 1.75 × 10−3 M CL showed a significant dose dependent decrease of 1.09, 1.22, 1.38, and 1.47 fold, respectively, in the expression of hsp70 compared to the larvae exposed to 80 μM arecoline alone (Fig. 4; p < 0.05).
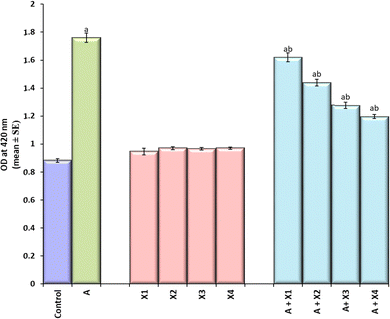 |
| | Fig. 4 Quantification of β-galactosidase activity in the third instar larvae of transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 after exposure to various doses of CL alone and along with 80 μM arecoline for 24 h. [A = 80 μM arecoline; X = CL; X1 = 0.2187 × 10−3 M, X2 = 0.4375 × 10−3 M, X3 = 0.875 × 10−3 M, X4 = 1.75 × 10−3 M; asignificant at p < 0.05 compared to control; bsignificant at p < 0.05 compared to control]. | |
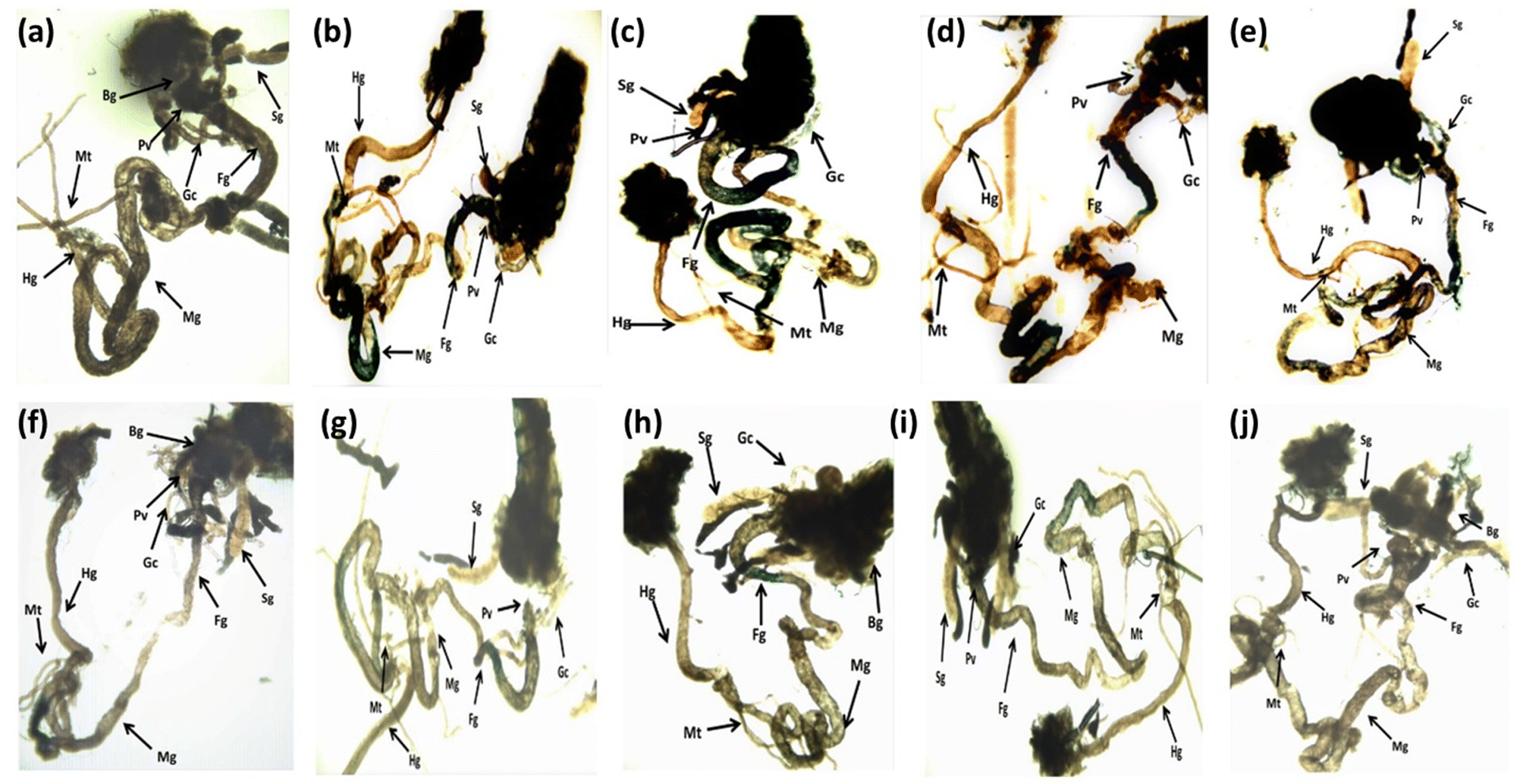
The results obtained for X-gal staining are presented in Fig. 5(a–j). The larvae exposed to 80 μM arecoline showed the highest activity of β-galactosidase in the midgut region compared to control (Fig. 5a and b). The larvae exposed to 0.2187 × 10−3, 0.4375 × 10−3, 0.875 × 10−3, and 1.75 × 10−3 M CL showed a dose dependent decrease in X-gal staining (Fig. 5c–f) compared to larvae exposed to 80 μM arecoline.
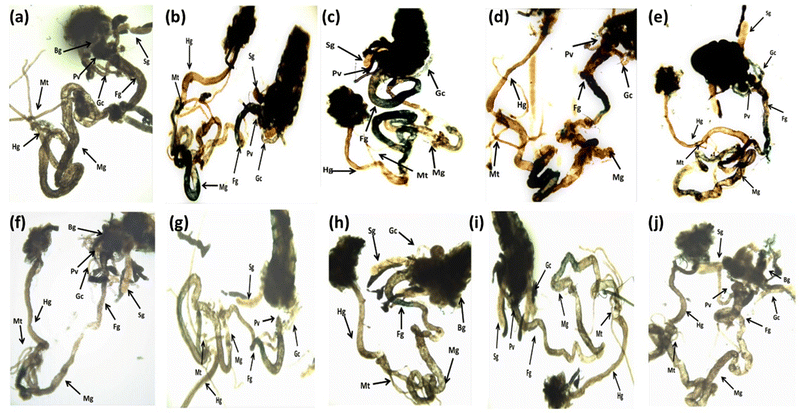 |
| | Fig. 5 X-gal staining performed on the third instar larvae of transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 after exposure to various doses of CL alone and along with 80 μM arecoline for 24 h. [a – control; b – A; c – A + X1; d – A + X2; e – A + X3; f – A + X4; g – X1; h – X2; i – X3; j – X4; A = 80 μM arecoline; X1 = 0.2187 × 10−3 M, X2 = 0.4375 × 10−3 M, X3 = 0.875 × 10−3 M, X4 = 1.75 × 10−3 M; X = CL; asignificant at p < 0.05 compared to control; bsignificant at p < 0.05 compared to control; BG – brain ganglia, SG – salivary gland, PV – proventriculus, FG – foregut, MG – midgut, HG – hindgut, MT – Malpighian tubule, GC – gastric caeca]. | |
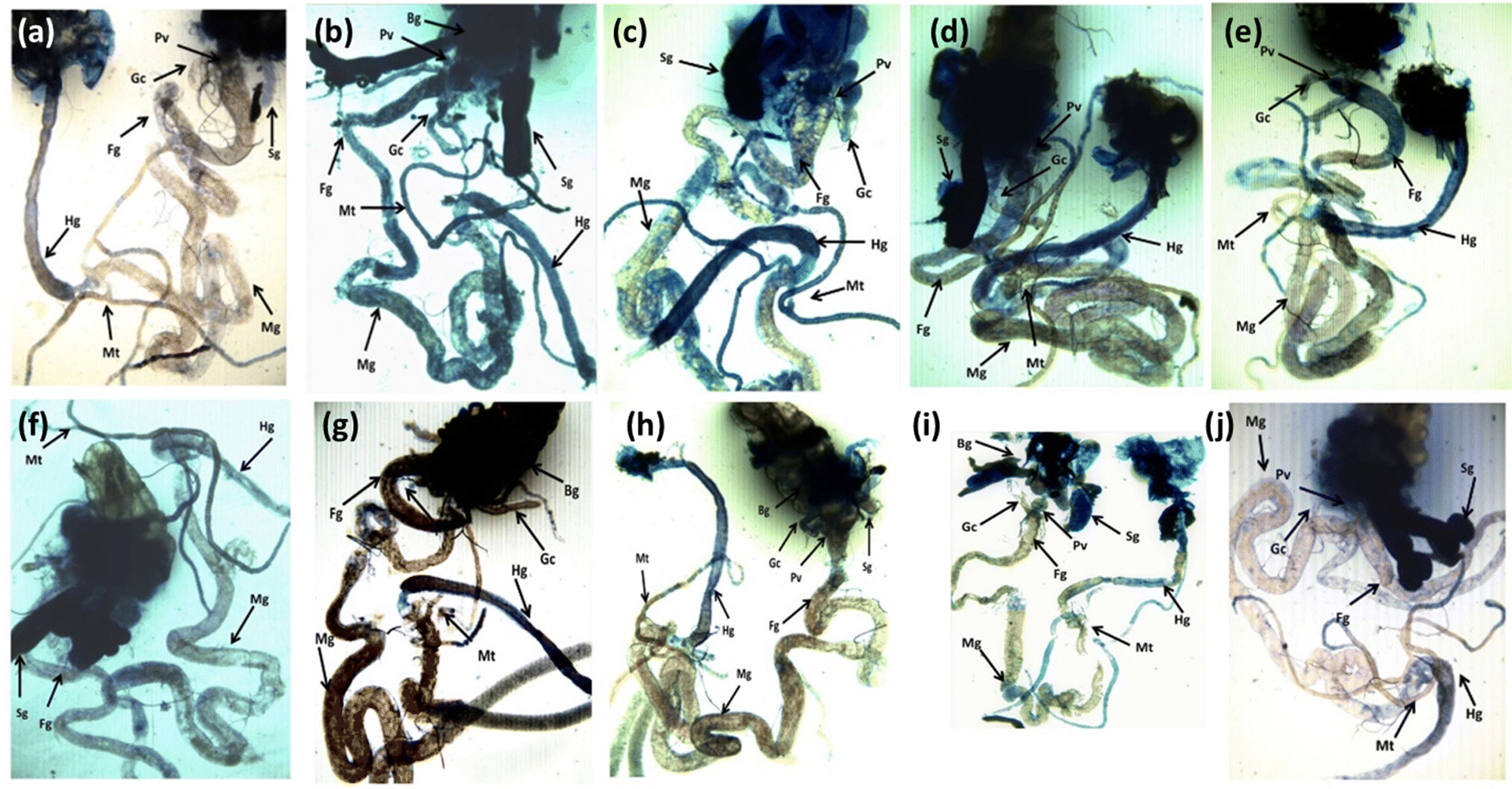
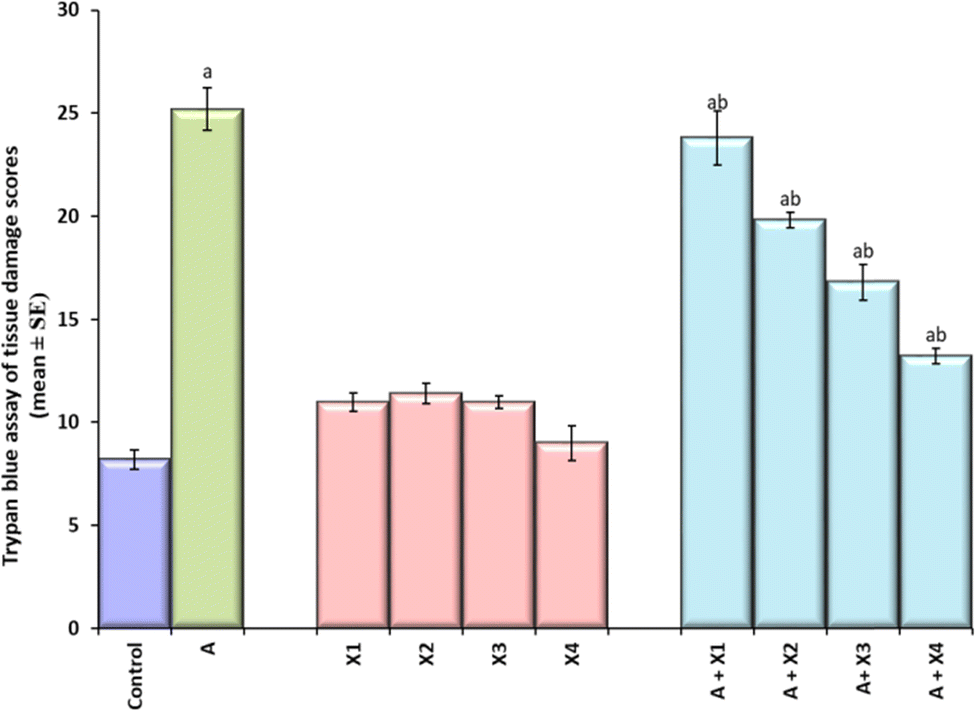
The result obtained for trypan blue staining is shown in Fig. 6(a–j) and the same has been quantified in Fig. 7. The highest tissue damage was observed in the larvae exposed to 80 μM arecoline (Fig. 6a). A dose dependent decrease in the tissue damage was observed in the larvae exposed to 80 μM arecoline along with 0.2187 × 10−3, 0.4375 × 10−3, 0.875 × 10−3, and 1.75 × 10−3 M CL, respectively (Fig. 6a). The larvae exposed to 80 μM arecoline showed a significant increase of 3.07 fold in the tissue damage compared to control (Fig. 5; p < 0.05). The larvae exposed to 80 μM arecoline along with 0.2187 × 10−3, 0.4375 × 10−3, 0.875 × 10−3, and 1.75 × 10−3 M CL showed a significant decrease of 1.10, 1.27, 1.5 and 1.90 fold, respectively, in the tissue damage compared to arecoline alone (Fig. 7; p < 0.05).
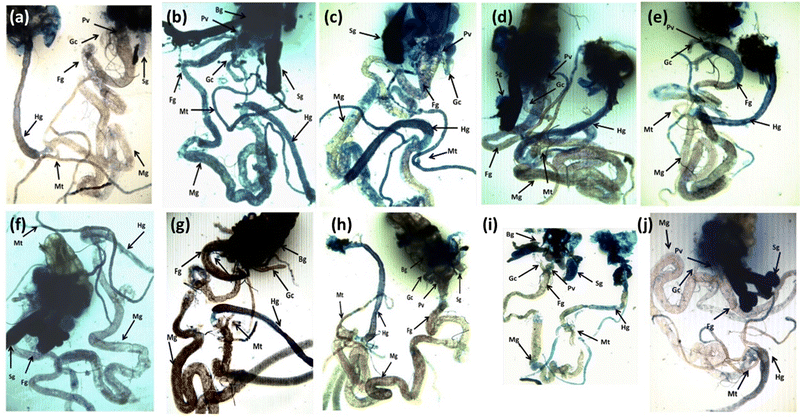 |
| | Fig. 6 Trypan blue staining performed on the third instar larvae of transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 after exposure to various doses of CL alone and along with 80 μM arecoline for 24 h. [a – control; b – A; c – A + X1; d – A + X2; e – A + X3; f – A + X4; g – X1; h – X2; i – X3; j – X4; A = 80 μM arecoline; X1 = 0.2187 × 10−3 M, X2 = 0.4375 × 10−3 M, X3 = 0.875 × 10−3 M, X4 = 1.75 × 10−3 M; X = CL; asignificant at p < 0.05 compared to control; bsignificant at p < 0.05 compared to control; BG – brain ganglia, SG – salivary gland, PV – proventriculus, FG – foregut, MG – midgut, HG – hindgut, MT – Malpighian tubule, GC – gastric caeca]. | |
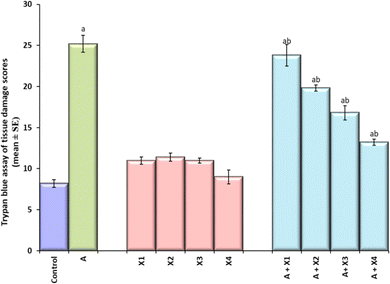 |
| | Fig. 7 Quantification of tissue damage after performing trypan blue staining on the third instar larvae of transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 exposed to various doses of CL alone and along with 80 μM arecoline for 24 h. [A = 80 μM arecoline; X1 = 0.2187 × 10−3 M, X2 = 0.4375 × 10−3 M, X3 = 0.875 × 10−3 M, X4 = 1.75 × 10−3 M; X = CL; asignificant at p < 0.05 compared to control; bsignificant at p < 0.05 compared to control]. | |
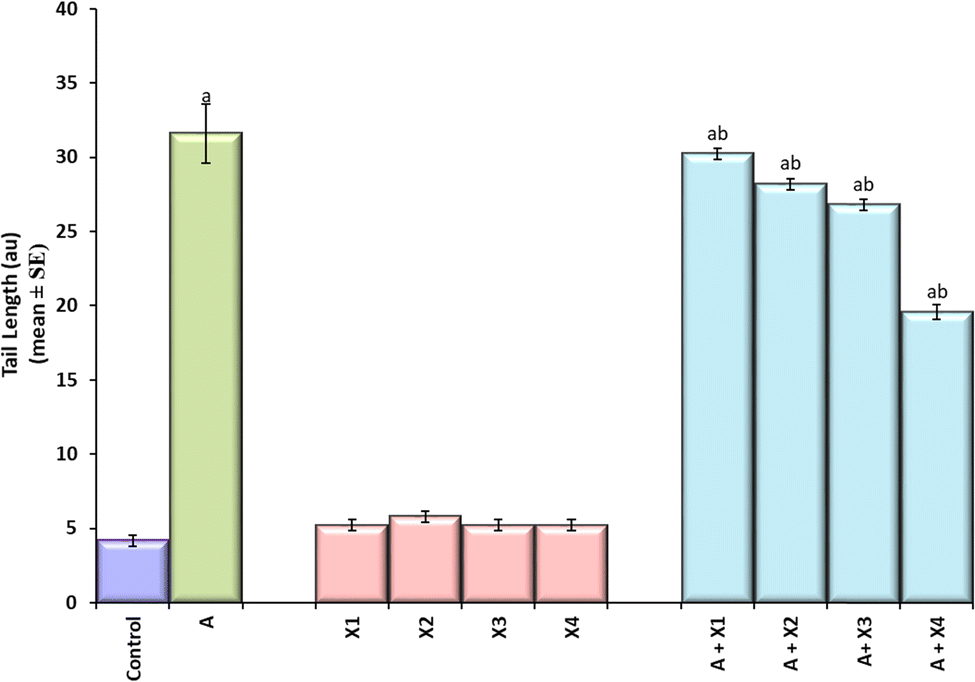
The result obtained for the comet assay is shown in Fig. 8(a–f) and the same has been quantified in Fig. 8. The larvae exposed to 80 μM arecoline showed an increase of 7.52 fold in the DNA damage compared to control (Fig. 9; p < 0.05). The larvae exposed to 80 μM arecoline along with 0.2187 × 10−3, 0.4375 × 10−3, 0.875 × 10−3, and 1.75 × 10−3 M CL showed a decrease of 1.04, 1.12, 1.17, and 1.61 fold, respectively, in the DNA damage compared to 80 μM arecoline treatment (Fig. 9; p < 0.05).
 |
| | Fig. 8 Comet assay performed on the midgut cells of the third instar larvae of transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 exposed to various doses of CL alone and along with 80 μM arecoline for 24 h. [a – control; f – arecoline treated; b – A + X1; c – A + X2; d – A + X3; e – A + X4; X1 = 0.2187 × 10−3 M, X2 = 0.4375 × 10−3 M, X3 = 0.875 × 10−3 M, X4 = 1.75 × 10−3 M; X = CL; A = 80 μM arecoline; asignificant at p < 0.05 compared to control; bsignificant at p < 0.05 compared to control]. | |
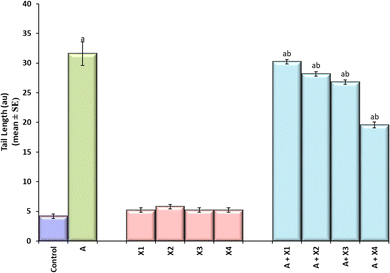 |
| | Fig. 9 Quantification of DNA damage after performing comet assay on the midgut cells of the third instar larvae of transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 exposed to various doses of CL alone and along with 80 μM arecoline for 24 h. [A = 80 μM arecoline; X1 = 0.2187 × 10−3 M, X2 = 0.4375 × 10−3 M, X3 = 0.875 × 10−3 M, X4 = 1.75 × 10−3 M; X = CL; asignificant at p < 0.05 compared to control; bsignificant at p < 0.05 compared to control]. | |
The results obtained in our present study suggested that CL is potent enough to counter the toxic effects of arecoline. The exposure of arecoline, induced the hsp70 expression as evidenced by the increase in the activity of β-galactosidase (estimated by performing ONPG assay). The results were corroborated by the results obtained for X-gal staining. The larvae exposed to varying concentrations/doses of CL showed a dose dependent decrease in the activity of β galactosidase. Similarly, the tissue damage was also reduced in a dose dependent manner as evidenced by the reduction in staining of the tissues. Arecoline is metabolized in the liver and the two major metabolically active products are arecaidine and arecoline N-oxide (AO).4 Arecaidine is mainly considered for oxidation of proteins or other adducts.71 It was also evident in our earlier study performed on the third instar larvae of transgenic Drosophila melanogaster (hsp 70-lacZ)Bg9.11
Cytochrome p-450 is a large and diverse family of heme containing proteins capable of catalyzing a diverse range of chemical reactions required for the developmental processes and also the detoxification of foreign compounds.72 In insects, most of the cytochrome p-450 enzymes are expressed in the larval midgut, Malpighian tubules, and fat body, suggesting the potential roles in detoxification processes and protection from the harmful compounds.72 The study on Drosophila by using the cytochrome inhibitors showed that the impact of mutagenicity of some nitrosamines, triazenes, hydrazines, and seneciphylline was reduced at a significant level suggesting a promising role of cytochromes p450 in the metabolic activation of these compounds.73 Metabolic activation property of Drosophila for other compounds (i.e., aryldialkyltriazenes, cyclophosphamides, nitrosamines, azon, hydrazo and azozyalkanes, aflatoxins and polycyclic hydrocarbons) and polychlorinated biphenyl has already been validated (Vogel,1975; Idda et al., 2020). In our present study, the ingested arecoline by the larvae may be metabolized by the cytochrome P450s present in the gut of the larvae and exhibit toxicity.11 The supplementation of arecoline along with the newly synthesized compound CL showed a marked reduction in cytotoxicity as well as genotoxicity. It may be due to the inhibition of cytochrome P450 by CL that may prevent/reduce the metabolic activation of arecoline by cytochrome P450. Therefore, it may also reduce the probability of oral cancers or the pre-cancerous situations. However, it requires a separate study to be conducted on cell lines to establish its property as an anti-cancerous agent; but at present, we can conclude that CL has potential to reduce the toxicity induced by arecoline.
3.4. Possible mechanism
3.4.1. UV absorption changes.
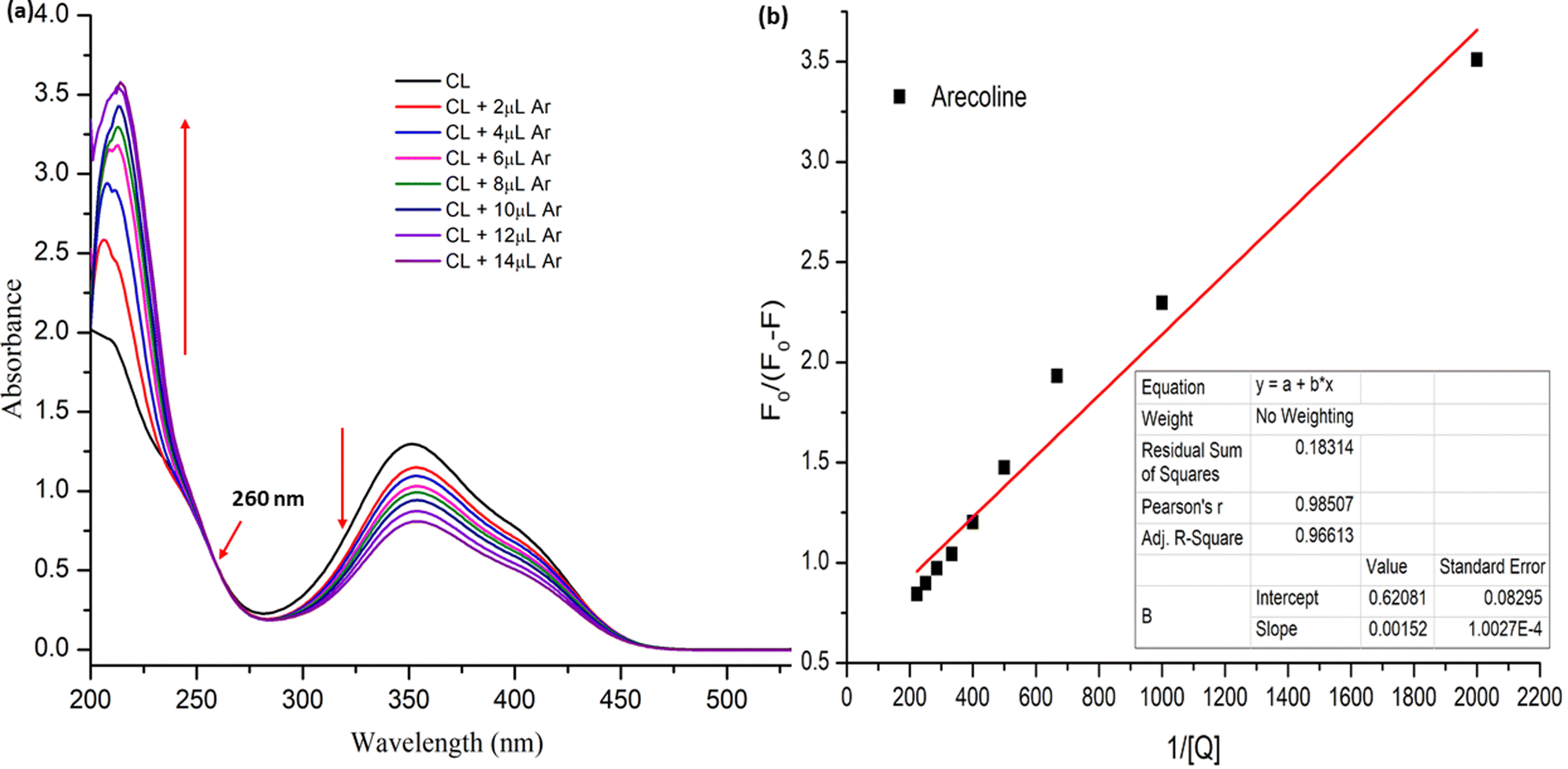
The UV spectra of CL in aqueous solution (10−4 M) are represented in Fig. 9. The characteristic absorption appearing in the range of 286–460 nm with a λmax of 352 nm can be observed. With the incremental addition of arecoline to CL aqueous solution, a decrease in the absorbance with red shift of λmax by 2 nm is observed and a new peak appears at 214 nm. An isosbestic point can also be observed at 260 nm. This indicates the smooth non-emissive ground state interaction between arecoline and CL.
The binding constant was calculated to be 4.084 × 102 M−1 (R2 = 0.985) with the help of the modified Stern Volmer equation (MSV),74
where the intensity before the addition of arecoline and after the addition of arecoline is denoted by
F0 and
F, respectively, [Q] denotes the arecoline concentration and
Ka is the binding constant (
Fig. 10).
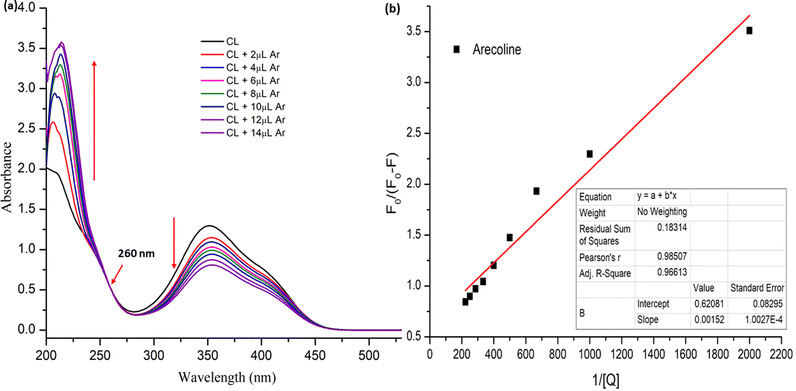 |
| | Fig. 10 (a) UV-vis spectra of CL upon gradual addition of arecoline showing spectral change with the appearance of a new band at 214 nm and the isosbestic point at 260 nm; and (b) modified Stern-Volmer plot for calculating the binding constant. | |
3.4.2. Fluorescence response of CL to arecoline.
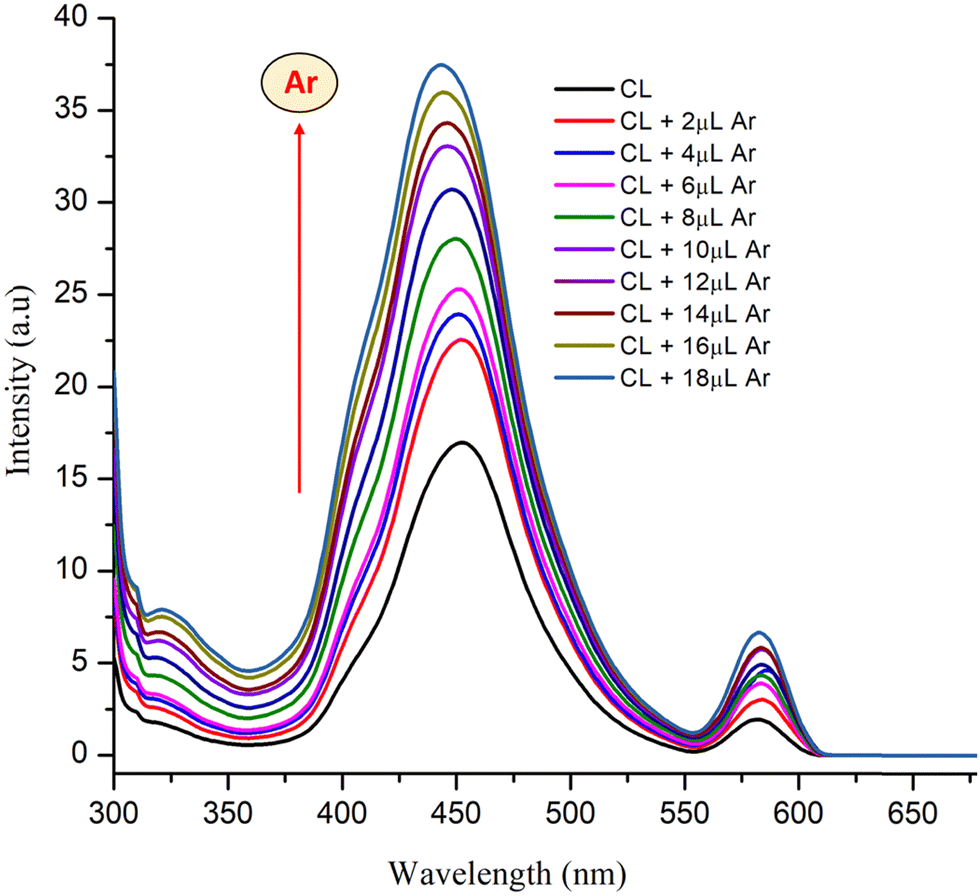
The synthesized material CL showed average fluorescence with λem = 453 nm upon excitation at 390 nm in aqueous solution. On incremental addition of arecoline (1 mM), the fluorescence intensity of CL increased correspondingly (Fig. 11). The results obtained from fluorescence are essentially in agreement with the UV-vis titrations performed in the previous section. No major shift in the position of λex and λem was observed. On the other hand, PYZ and TNP (free reactants) solutions show a slight and negligible change in fluorescence intensity (Fig. S4, ESI†). Structures with rigid conjugation are known to produce strong fluorescence,75 and H2O, H+, and OH− molecules in solution have the ability to form effective hydrogen bonds in the excited and ground states with the chromophore donor group.76 Therefore, CL can form H-bonds with H2O, H+ and OH− in solution. The rigidity of CL will be enhanced by such H-bonds and thus contributes to the moderate fluorescence of CL. A cooperative host–guest complexation may enhance the host conformation rigidity and thus increase its fluorescence.77
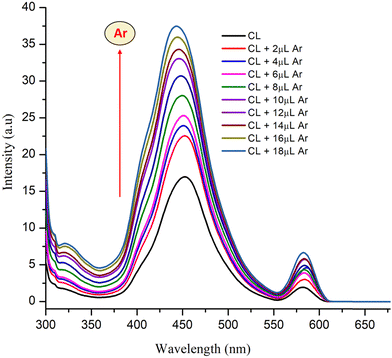 |
| | Fig. 11 Fluorescence enhancement of CL with the increasing amount of arecoline (1 mM) in aqueous solution. | |
3.4.3. Interaction study between CL and arecoline by molecular docking.
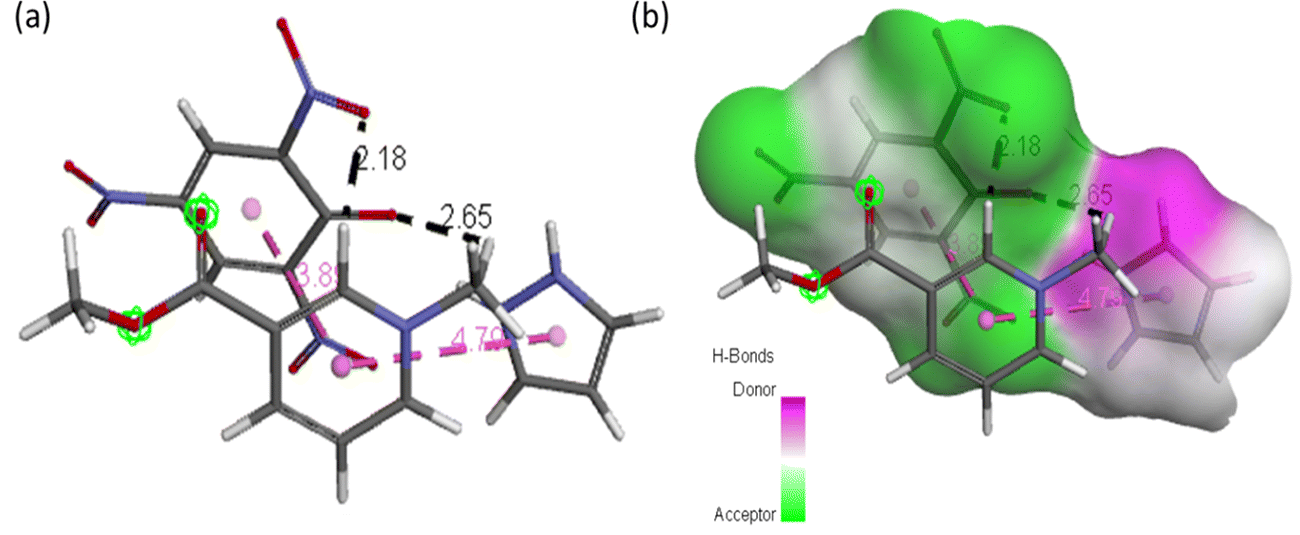
The best possible interaction between CL and arecoline was investigated and optimized at the M06-2X/6-311**G (d,p) level of theory and represented in Fig. 12 and named ACL. In ACL, arecoline and CL interact through π–π stacking (3.8 and 4.7 Å) and carbon hydrogen bonding (2.1 and 2.6 Å).
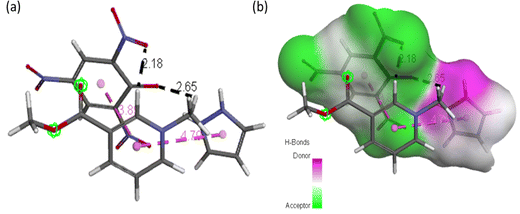 |
| | Fig. 12 (a) ACL model showing possible interaction types between CL and arecoline and (b) representation of hydrogen bond surface around CL of ACL. | |
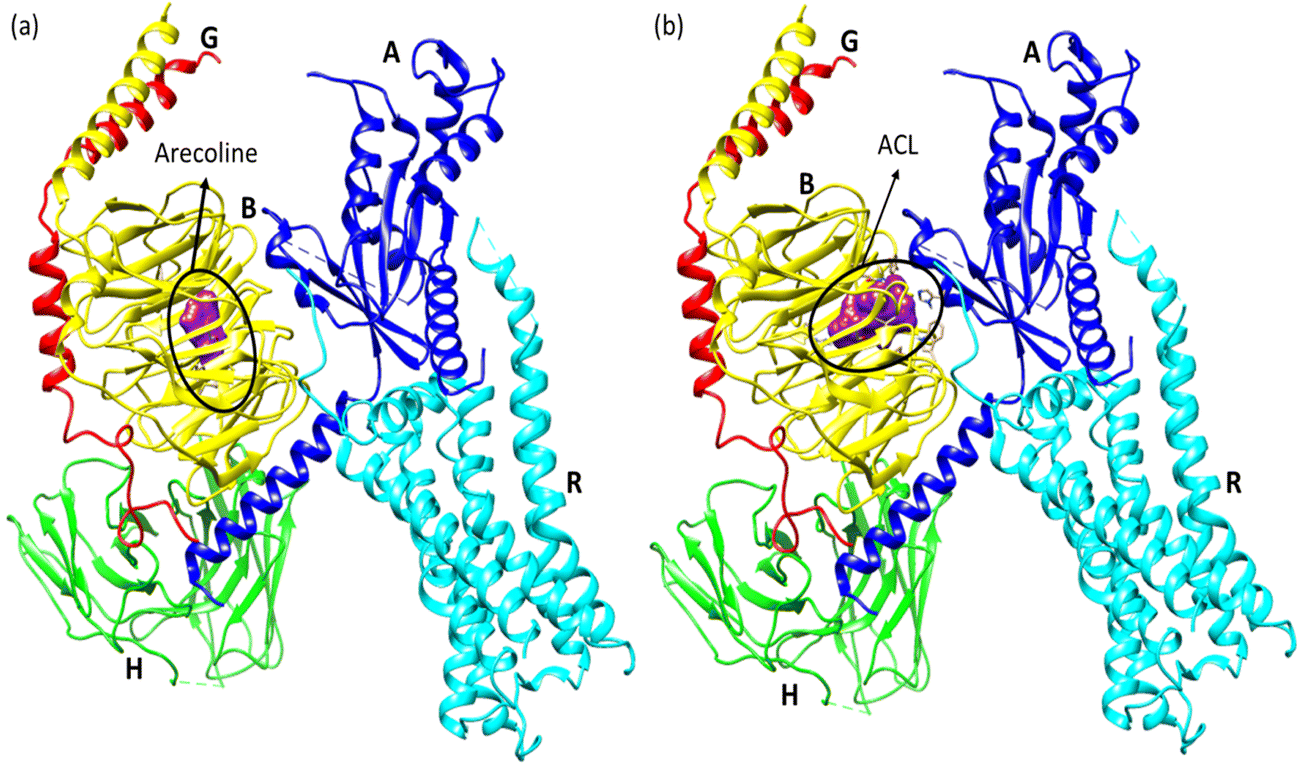
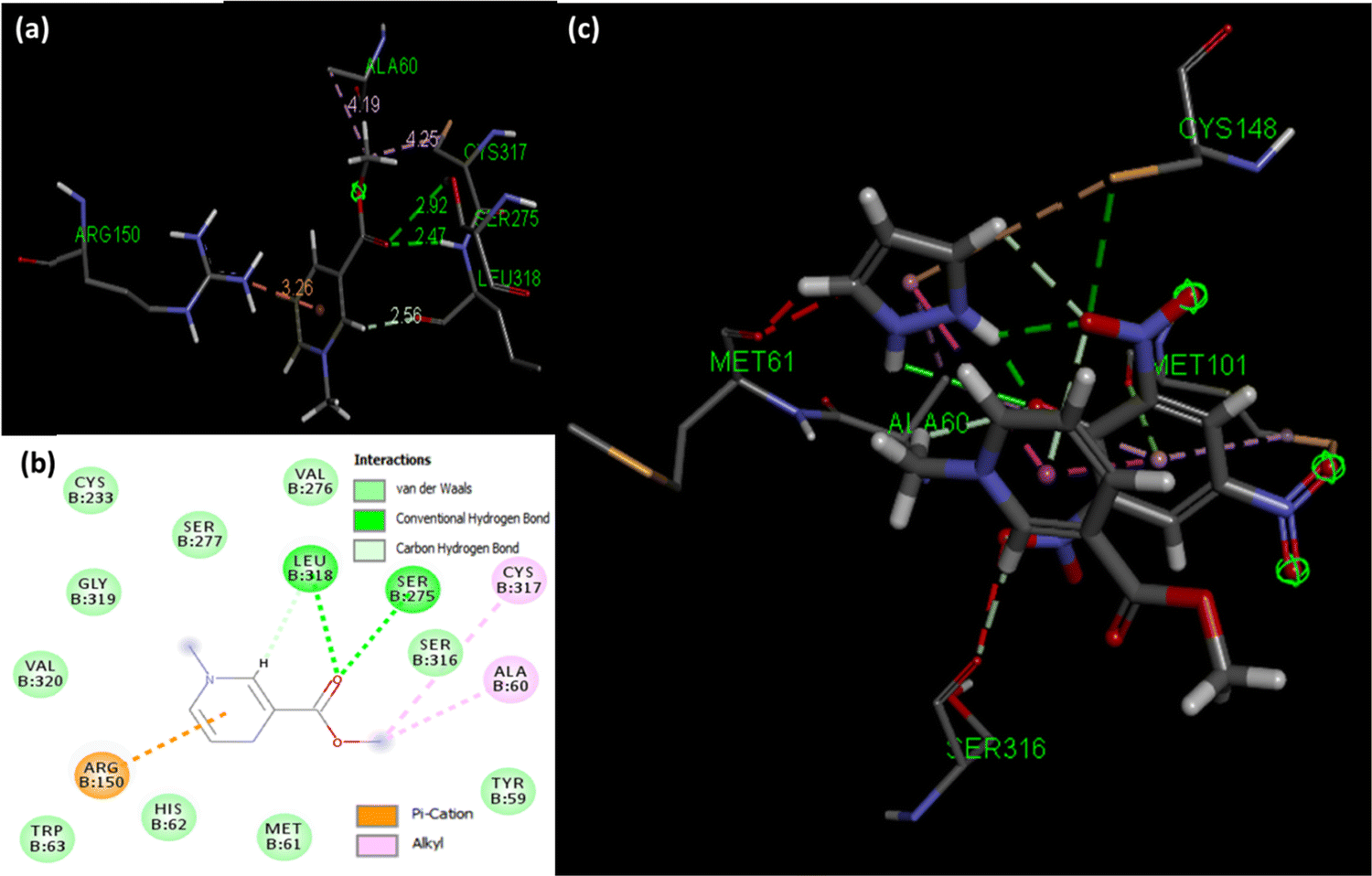
Molecular docking poses for arecoline and muscarinic acetylcholine M1, and ACL and muscarinic acetylcholine M1 were obtained and it was concluded that they bind to chain B of muscarinic acetylcholine M1 (Fig. 13). Arecoline individually forms conventional hydrogen bonds with LEU 318 and SER 275; carbon hydrogen bonds with HIS 62 and LEU 318; van der Waals, π-cation and alkyl interactions are also present (Fig. 14). The interactions/hydrogen bonds between arecoline and the target protein result in the stimulation of parasympathetic effects. On the other hand, the docking pose of ACL and muscarinic acetylcholine M1 reveals that the CL portion of ACL blocks the active binding sites and thus acts as a hindrance between muscarinic acetylcholine M1 and arecoline (Fig. 13). This may result in the reduction of the arecoline effect on muscarinic acetylcholine M1.78
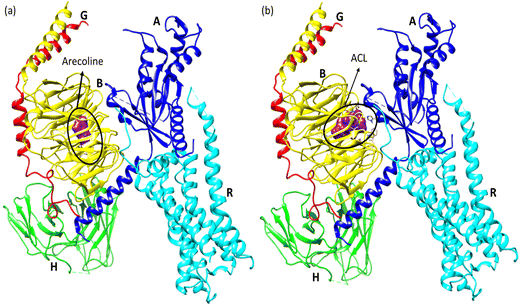 |
| | Fig. 13 (a) Molecular docking pose between arecoline and muscarinic acetylcholine M1 and (b) molecular docking pose between ACL and muscarinic acetylcholine M1 (A, B, G, H, and R are the chains of the respective protein). | |
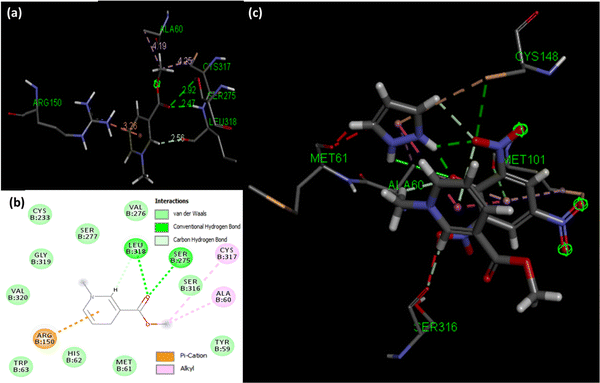 |
| | Fig. 14 (a) Binding sites between arecoline and muscarinic acetylcholine M1, (b) 2D diagram showing binding modes between arecoline and muscarinic acetylcholine M1, and (c) binding sites between ACL and muscarinic acetylcholine M1. | |
3.5. Single crystal XRD studies of CL
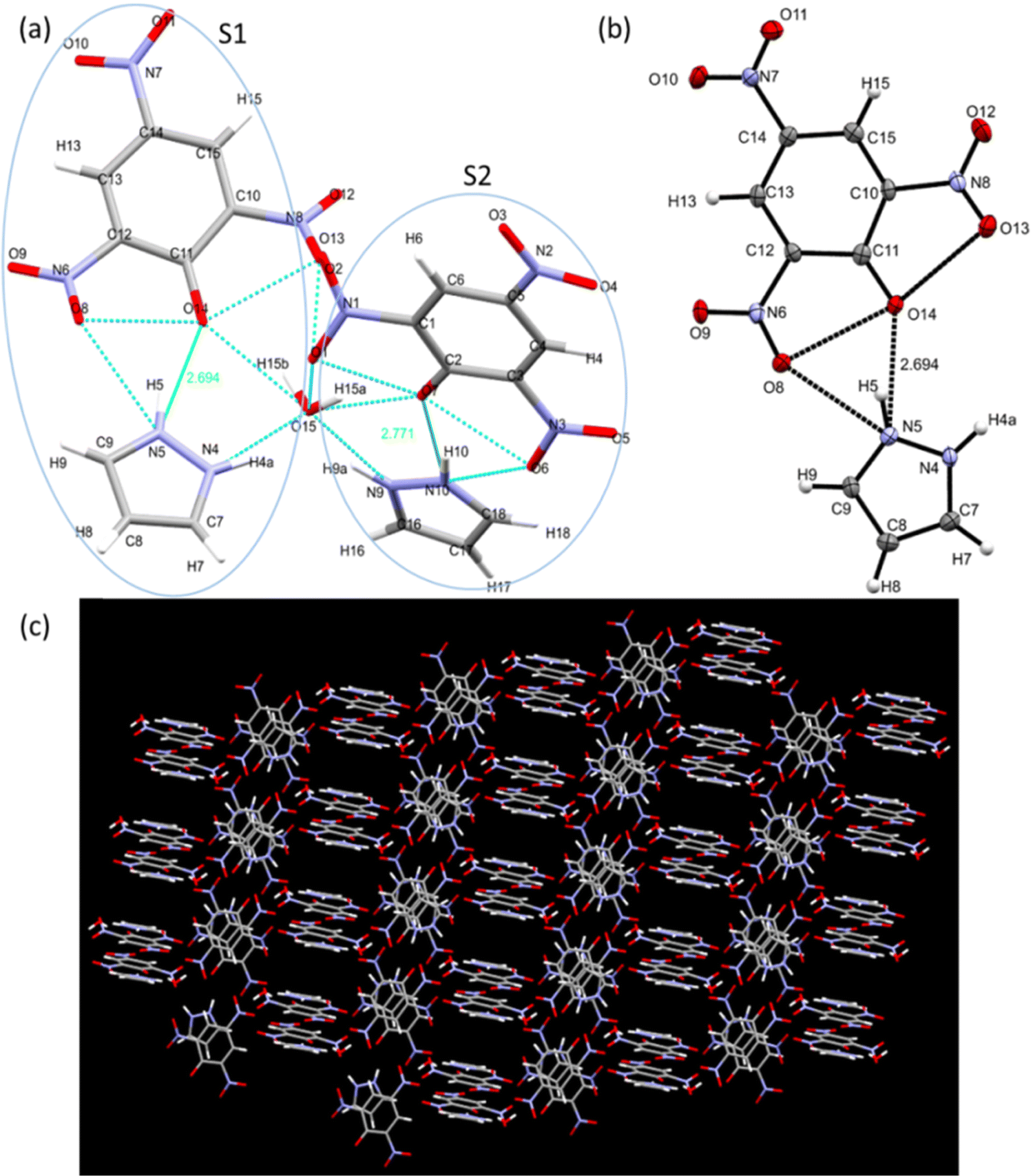
Single crystal X-ray diffraction data help us to study the bonding and structural features of the synthesized crystal CL. The data obtained from SC-XRD reveal the empirical formula of CL to be “2[(C9H7N5O7).0.5H2]” and show H-bonding between TNP and PYZ in a stable crystal lattice.79 Twin sisters S1 and S2 can be seen connected with the water molecule (Fig. 15). PYZ and TNP form a bond through the transfer of the hydrogen atom from O14 of TNP to the N5 atom of PYZ, with a bond length of 2.69 Å (S1). A similar structure (S2) with a hydrogen bond length of 2.79 Å between O7 of TNP and N10 of PYZ was found to be connected through H2O with S1. The water molecule forms a hydrogen bond between N4 of PYZ of S1 with 2.68 Å and N9 of PYZ of S2 with 2.77 Å. Table 2 and Table S3 (ESI†) provide the refinement parameters and bond lengths for the synthesized crystal (CL), respectively. Hydrogen atom coordinates (Å × 104) and isotropic displacement parameters (Å2 × 103) for CL are shown in Table S4 (ESI†). The ortep view and hydrogen bonding network in the crystal are represented in Fig. 14. The CCDC no. of the crystal is 1973751.†
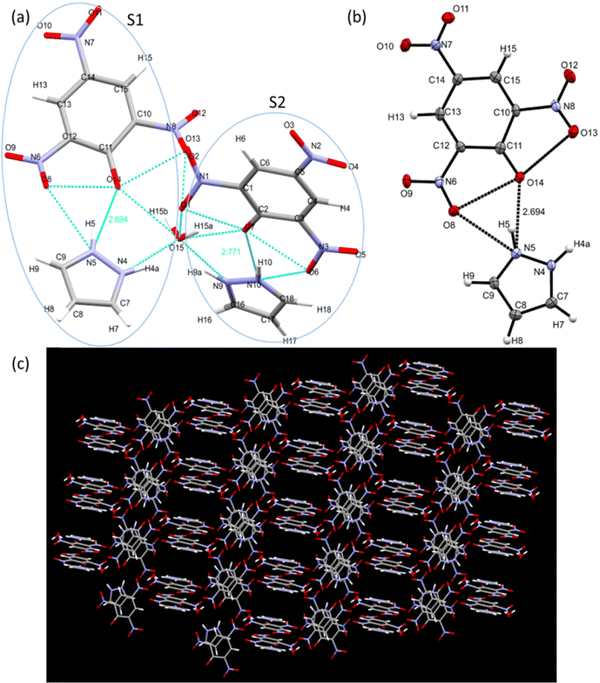 |
| | Fig. 15 (a) Structure showing twin sisters S1 and S2 bonded with a water molecule, (b) an ortep view showing interaction between the donor and acceptor moieties, and (c) crystal packing showing the hydrogen bonding network. | |
Table 2 Crystal data and structure refinement for CL
| Crystal |
CL
|
| Empirical formula |
C18H16N10O15 |
| Formula weight |
612.39 |
| Temperature/K |
100(2) |
| Crystal system |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
a, b, c/Å |
7.6872(6), 11.8217(8), 13.5376(10) |
|
α, β, γ/° |
98.528(2), 101.825(2), 91.802(2) |
| Volume/Å3 |
1188.39(15) |
|
Z
|
2 |
|
ρ
calc/g cm−3 |
1.7112 |
|
μ/mm−1 |
0.152 |
|
F(000) |
628.4 |
| Crystal size/mm3 |
0.38 × 0.26 × 0.16 |
| Radiation |
Mo Kα (λ = 0.71073) |
| 2θ range for data collection/° |
5.66 to 50.1 |
| Index ranges |
−10 ≤ h ≤ 10, −15 ≤ k ≤ 15, −18 ≤ l ≤ 18 |
| Reflections collected |
16890 |
| Independent reflections |
4139 [Rint = 0.0444, RSigma = 0.0562] |
| Data/restraints/parameters |
4139/0/412 |
| Goodness-of-fit on F2 |
1.099 |
| Final R indexes [I ≥ 2σ(I)] |
R
1 = 0.0496, wR2 = 0.1001 |
| Final R indexes [all data] |
R
1 = 0.0607, wR2 = 0.1065 |
| Largest diff. peak/hole/e Å−3 |
0.39/−0.34 |
3.6. DFT/TD-DFT studies, Mulliken charge, and electrostatic potential map
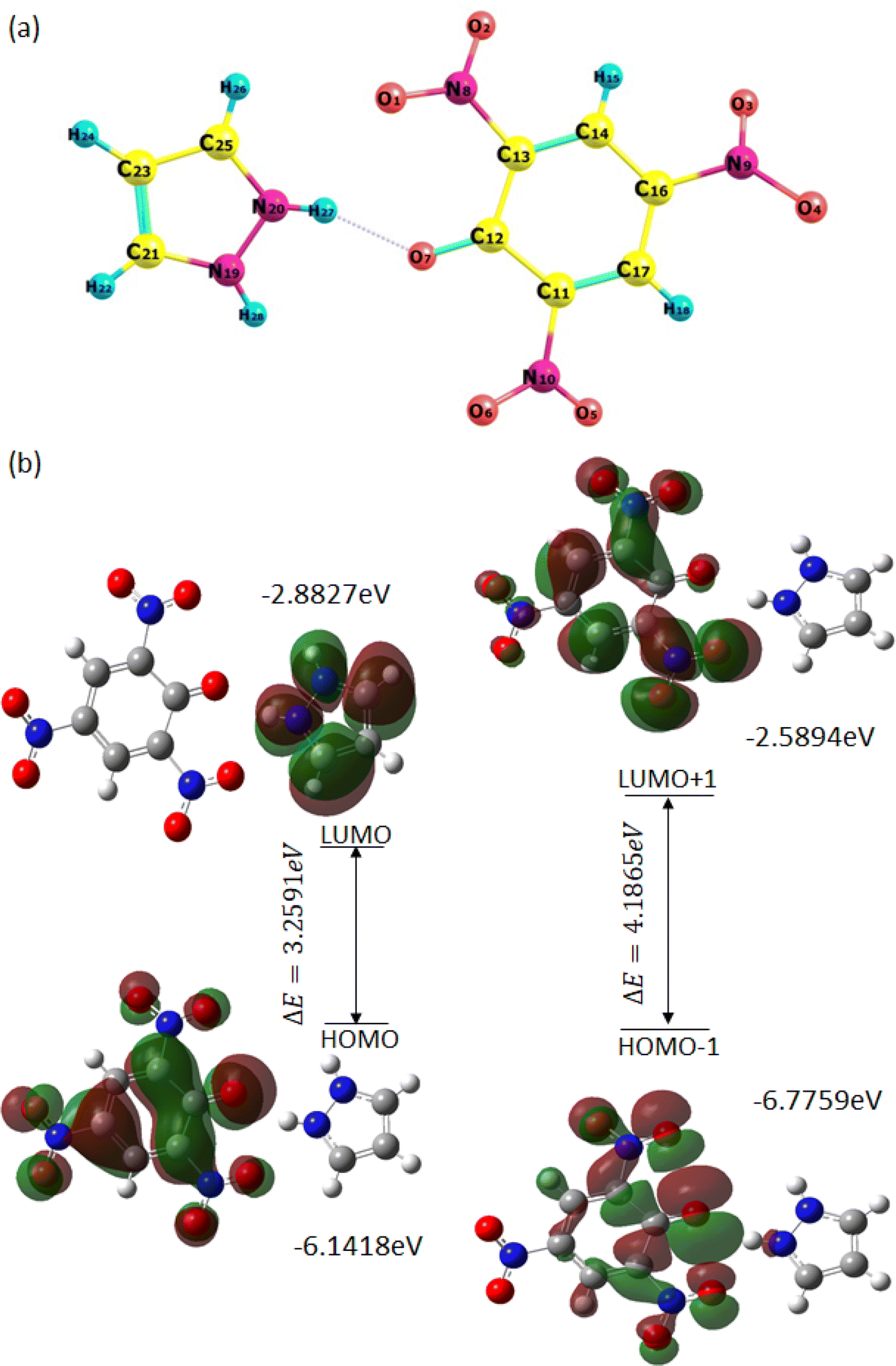
The minimization energy for TNP, PYZ, and CL was obtained and it was observed that the total energy of TPN is −920.57 a.u., PYZ is −226.11 a.u., and CL is −1146.53 a.u., and the stabilization energy is calculated to be −77.18 kcal mol−1 for CL. The optimized structure of CL with atomic numbers is represented in Fig. 16. Mulliken charges for PYZ, TNP, and CL were also calculated and a shift in the Mulliken charges of the atom of CL to the respective PYZ and TNP atom was observed (Table 3). The increase in the negative Mulliken charge on O7 was observed to be 0.54444, which was 0.52425 in free TNP. This shift in the charge on the O7 atom of CL is due to the dissociation of the O7–H27 bond in CL. Similarly, the decrease in the negative Mulliken charge on N20 was observed to be 0.34883, which was 0.39889 in free PYZ, which is caused by the transfer of the proton to N20. H27 of CL also showed a decrease in the positive Mulliken charge to 0.35857 from 0.40197. The decrease in Mulliken charges of CL atoms with respect to PYZ and increase with respect to TNP suggest the transfer of proton between PYZ and TNP. All these shifts in Mulliken charges suggest the transfer of electrons and N+–H⋯O− interaction, as observed in single crystal XRD results. In order to know the preferred binding sites of the nucleophilic and electrophilic attack, the molecular electrostatic potential (MEP) map of PYZ, TNP, and CL was generated (Fig. S5, ESI†). The electrostatic potentials are shown by different colors on the surface of the MEP map. The blue region represents positive electrostatic potential, red represents negative electrostatic potential and zero electrostatic potential is represented by green regions. Negative electrostatic potential and the region with red color are located over the oxygen atom, while positive electrostatic potential and the blue color region are located over the hydrogen atom. The regions with positive electrostatic potential (blue color) and negative electrostatic potential (red color) may be the preferable sites of nucleophilic and electrophilic attacks.61
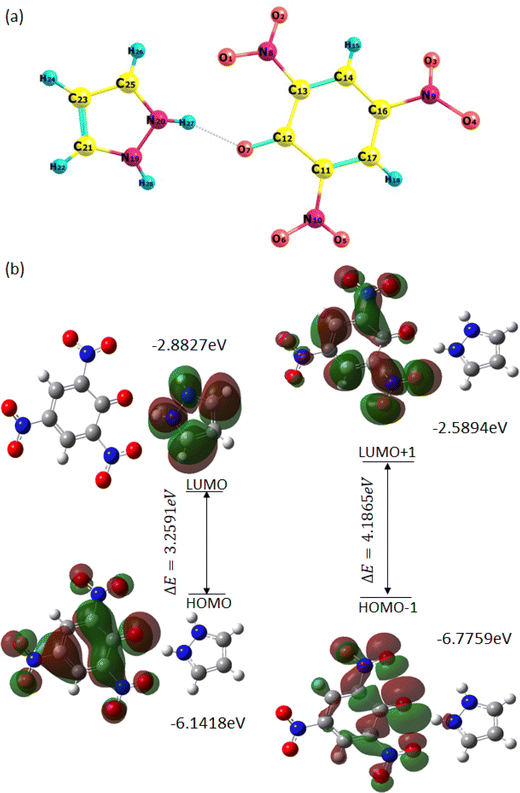 |
| | Fig. 16 (a) Optimized structure of CL with the atom numbering scheme and (b) the pictorial representation of frontier molecular orbitals of CL showing the HOMO–LUMO energy gap. | |
Table 3 Mulliken atomic charges of CL atoms
| Atom |
CT complex |
TNP |
Atom |
CT complex |
PYZ |
| O1 |
−0.35997 |
−0.29579 |
H27 |
0.35857 |
0.40197 |
| O2 |
−0.29808 |
−0.22189 |
N19 |
−0.37390 |
−0.53453 |
| O3 |
−0.29906 |
−0.27743 |
N20 |
−0.34883 |
−0.39889 |
| O4 |
−0.29878 |
−0.23741 |
C21 |
0.25568 |
0.12516 |
| O5 |
−0.30442 |
−0.29248 |
H22 |
0.19358 |
0.16723 |
| O6 |
−0.29324 |
−0.29248 |
C23 |
−0.21810 |
−0.21452 |
| O7 |
−0.54444 |
−0.52425 |
H24 |
0.15519 |
0.13588 |
| N8 |
0.07219 |
0.04076 |
C25 |
0.28996 |
0.02788 |
| N9 |
0.04469 |
0.03065 |
H26 |
0.21101 |
0.15672 |
| N10 |
0.06547 |
0.01169 |
H28 |
0.348887 |
0.33596 |
| C11 |
0.23190 |
0.34143 |
|
|
|
| C12 |
0.31506 |
0.23572 |
|
|
|
| C13 |
0.24807 |
0.36277 |
|
|
|
| C14 |
−0.03060 |
−0.10351 |
|
|
|
| H15 |
0.16008 |
0.24117 |
|
|
|
| C16 |
0.26521 |
0.33504 |
|
|
|
| C17 |
−0.00470 |
−0.08943 |
|
|
|
| H18 |
0.15856 |
0.24304 |
|
|
|
The FMOs (frontier molecular orbitals), i.e., the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO), of CL were also obtained to explain chemical reactivity, kinetic stability, and the charge delocalization within the molecule. The UV spectra and FTIR spectra of CL were obtained through TD-DFT and DFT using the 6-31G**/B3LYP basis set to support experimental data (Fig. S6, ESI†). The HOMO to LUMO energy gap (ΔE) is 3.2591 eV and the HOMO−1 to LUMO+1 energy gap (ΔE) is 4.1865 eV, obtained from FMOs (Fig. 16). The molecular orbital (MO) diagram obtained through theoretical calculations is represented in Fig. S8 (ESI†) and MO energies are given in Table S5 (ESI†). The coordinates of the molecules (TNP, PYZ, and CL) were obtained from DFT and represented in Tables S6–S8 (ESI†). Based on the optimized structure and HOMO–LUMO, some molecular parameters related to chemical reactivity (SCF minimum energies, polarizability, thermodynamic parameters, dipole moments) for the complexes are presented in Table 4 in the gas phase.61,80
Table 4 Various other theoretical molecular parameters of CL and their constituents
| Parameters |
CL (B3LYP/6-31G**) |
| Minimum SCF energy (a.u.) |
−1146.63765124 |
| Stabilization energy (kcal mol−1) |
−77.18 kcal mol−1 |
| Polarizability (a.u.) |
242.629 |
| Zero point vibrational energy (kcal mol−1) |
116.01423 |
| Total thermal energy (kcal mol−1) |
127.139 |
| Field independent dipole moment (Debye) μtotal |
11.7943 |
| Electronic spatial extent (a.u.) |
7534.5215 |
| Frontier MO energies (eV) |
|
| LUMO |
−2.8827 |
| HOMO |
−6.1418 |
| LUMO+1 |
−2.5894 |
| HOMO−1 |
−6.7759 |
| Gap [LUMO–HOMO] |
3.2591 |
| Gap [(LUMO+1)–(HOMO−1)] |
4.1865 |
3.7. Hirshfeld surface analyses of the synthesized crystal (CL)
CrystalExplorer 03.157 software was used to map the Hirshfeld surface in the dnorm range of 0.5 to 2.5 Å. The partitioning space of the molecule in the crystal into regions where the procrystal is being dominated by the promolecule is the Hirshfeld surface of the crystal. Different surfaces like dnorm, di, de, shape index, and curvedness are represented in Fig. S9 (ESI†). The shape index represents the two complementary shapes with blue (bumps) and red (hollow) shapes touching each other. The root-mean-square curvature is represented by the curvedness function, which shows green regions that are separated by deep blue color edges. The depressions with red color on the dnorm surface represent contacts having hydrogen bonds. di represents the distance from the Hirshfeld surface to the nearest nucleus internal to the surface and de is for the outer surface. The 2-D fingerprint plot between these di and de provides more information about the different types of close contacts (Fig. S10, ESI†).81 The two equal sharp peaks in the 2-D fingerprint plot represent the O–H bonding of N–O⋯H interaction. Other important close interactions like C–H⋯π, C–O⋯C, N–O⋯H and C–H⋯H for CL were also shown with C–H = 4.5%, C–O = 43.3%, O–H = 52.2%, N–H–O = 2.7% and N–O = 5.7% of occurrence in the molecule.
3.8. Thermograms study
TGA-DTA analysis was done for free reactants (PYZ and TNP) and the resulting complex CL [(PYZH)+(TNP)−] to study the thermal stability and confirming the interaction between PYZ and TNP (Fig. S11, ESI†). Table 5 shows the useful data obtained from the thermograms study. The DTA of [(PYZH)+(TNP)−] reveals three endothermic peaks which appeared at 122.4 °C (ΔH = −148.1 mJ), 167.2 °C (ΔH = −125.9 mJ) and 285.8 °C (ΔH = −119.4 mJ), respectively, while three endothermic peaks at 121.5 °C (ΔH = −276.1 mJ), 240.9 °C (ΔH = −386.5 mJ) and 281.4 °C (ΔH = −430.6 mJ) were observed in the DTA of TNP (acceptor) and only two endothermic peaks at 75.4 °C (ΔH = −91.2 mJ) and 147.2 °C (ΔH = −202.8 mJ) in the case of PYZ. The TGA of [(PYZH)+(TNP)−] shows decomposition with the weight loss of 74.3 and 99.9% at 265.7 and 292.3 °C (mid points), respectively. PYZ was decomposed with the weight loss of almost 99.6% at 130.9 °C (mid points) and TNP was decomposed with the weight loss of 99.3% at 274.4 °C (mid points). From these above results, it can be concluded that CL [(PYZH)+(TNP)−] is thermodynamically more stable as compared to free PYZ and TNP due to the formation of H-bonding interaction (N+–H⋯O−) between TNP and PYZ to form the CT complex.82 The TGA is not consistent with the acceptor and donor molecules due to strong H-bonding between these moieties providing extra robustness to the final product (CL). In the TGA curve from 190 to 290 °C there is weight loss of both these moieties as we did not get a sharp peak to distinguish them. This may be due to the strong interaction between these two moieties. Thus we can suggest that after 300 °C both donor and acceptor decompose subsequently giving only the residue after 300 °C.83 Moreover, deviation in the thermograms of [(PYZH)+(TNP)−] in comparison with PYZ and TNP confirms the formation of [(PYZH)+(TNP)−].84
Table 5 Weight loss, enthalpy (ΔH), and degradation temperature (T) for pyrazole, 2,4,6-trinitrophenol and CL
| Step |
Pyrazole |
2,4,6-Trinitrophenol |
CT complex [(PYZH)+(TNP)−] |
| Weight loss (%) |
T (°C) |
ΔH (mJ) |
Weight loss (%) |
T (°C) |
ΔH (mJ) |
Weight loss (%) |
T (°C) |
ΔH (mJ) |
| 1 |
99.6 |
130.9 |
−91.2 |
99.3 |
274.4 |
−276.1 |
74.3 |
265.7 |
−148.1 |
| 2 |
|
|
−202.8 |
|
|
−386.5 |
99.9 |
292.3 |
−125.9 |
| 3 |
|
|
|
|
|
−430.6 |
|
|
−119.4 |
4. Conclusion
In summary, the biological activity of an organic material CL is maneuvered by various techniques like in situ histochemical β-galactosidase activity and comet assay. Herein, the synthesized charge transfer complex CL was characterized by various spectroscopic and crystallographic techniques and was observed to be formed through N+–H⋯O− bonding between TNP and PYZ; it was found to cause a remarkable dose-dependent decrease in the toxic effect of arecoline. β-Galactosidase activity, apoptosis and DNA damage in the midgut cells of third-instar larvae of the transgenic Drosophila melanogaster (hsp70-lac Z)Bg9 exposed to 80 μM arecoline were found to be decreased on increasing the dose of the synthesized material (up to 20 μM). A slight and negligible decrease in the toxic effects caused by arecoline was observed on using the highest dose (20 μM) of free reactants (PYZ and TNP) suggesting that CL as a whole is responsible for the reduction of these toxic effects. The decrease in the arecoline toxic effects occurs because of the ground state interaction between arecoline and CL. Molecular docking analysis for interaction of arecoline with muscarinic acetylcholine M1 shows perfect binding at the active sites of muscarinic acetylcholine M1, triggering its end-receptor stimulation. Interestingly, after complexation of CL and arecoline (ACL), it is observed that CL moieties of ACL inhibit the interaction between arecoline and muscarinic acetylcholine M1 by blocking the active sites available for arecoline and it itself interact with arecoline. In this way, a route to design and develop new simple organic molecules with desired efficacy in lowering the toxic effect of arecoline could be achieved in future endeavors.
Author contributions
SS, IMK and YHS conceived and designed research. SS, BS and HV conducted the experiment. SS, SJ, IMK and YHS analyzed the data. SS and YHS wrote the first draft of the manuscript. SS and IMK investigated the results. SS performed the software analysis. SS and BS revised and edited the manuscript. All authors read and approved the manuscript and all data were generated in-house and that no paper mill was used.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgements
The authors thank the Chairman, Department of Chemistry, AMU, Aligarh, for providing research facilities at Physical Chemistry lab. The authors also thank UGC for the fellowship.
References
- Y. J. Liu, W. Peng, M. B. Hu, M. Xu and C. J. Wu, Pharm. Biol., 2016, 54, 2753–2760 CrossRef CAS PubMed.
- Y. T. Shih, P. S. Chen, C. H. Wu, Y. T. Tseng, Y. C. Wu and Y. C. Lo, Free RadicBiol Med., 2010, 49, 1471–1479 CrossRef CAS PubMed.
- G. S. Thangjam and P. Kondaiah, J. Periodontal Res., 2009, 44, 673–682 CrossRef CAS PubMed.
- N. G. Oliveira, D. L. Ramos and R. J. Dinis-Oliveira, Arch. Toxicol., 2021, 95, 375–393 CrossRef CAS PubMed.
- K. Sundqvist, Y. Liu, J. Nair, H. Bartsch, K. Arvidson and R. C. Grafström, Cancer Res., 1989, 49, 5294–5298 CAS.
- G. B. Panigrahi and A. R. Rao, Mutat. Res. Lett., 1982, 103, 197–204 CrossRef CAS PubMed.
- Y. C. Chang, K. W. Tai, M. H. Cheng, L. S. S. Chou and M. Y. Chou, J. Oral Pathol. Med., 1998, 27, 68–71 CrossRef CAS PubMed.
- A. Chatterjee and S. Deb, Cancer Lett., 1999, 139, 23–31 CrossRef CAS PubMed.
- A. H. Trivedi, B. J. Dave and S. C. Adhvaryu, Cancer Lett., 1993, 74, 105–110 CrossRef CAS PubMed.
- B. Shakya, S. Shakya and Y. H. Siddique, Toxicol. Mech. Methods, 2019, 29, 187–202 CrossRef CAS PubMed.
- B. Shakya and Y. H. Siddique, Toxicol. Res., 2018, 7, 432–443 CrossRef CAS PubMed.
- B. Shakya and Y. H. Siddique, J. Basic Appl. Zool., 2018, 79, 47 CrossRef.
- J. Zhou, Q. Sun, Z. Yang and J. Zhang, Korean J. Physiol. Pharmacol., 2014, 18, 143 CrossRef CAS PubMed.
-
(a) R. S. Mulliken, J. Am. Chem. Soc., 1952, 2, 811–824 CrossRef;
(b) D. K. Roy, A. Saha and A. K. Mukherjee, Spectrochem. Acta A, 2005, 9, 2017–2022 CrossRef PubMed;
(c) M. S. Refat, B. Albogami, A. M. A. Adam, H. A. Saad, A. M. Alsuhaibani, L. Miyan and M. S. Hegab, J. Mol. Liq., 2022, 363, 119878 CrossRef CAS PubMed.
-
(a)
R. Foster, Organic Charge Transfer Complexes, Academic Press, London, 1969, p. 15 Search PubMed;
(b) G. H. Al-Hazmi, A. M. Hassanien, A. A. Atta, M. S. Refat, H. A. Saad, S. Shakya and A. M. A. Adam, J. Mol. Liq., 2022, 362, 119757 CrossRef CAS.
- M. Kidwai, S. Saxena, S. Rastogi and R. Venkataramanan, Curr. Med. Chem., 2004, 2, 269–286 Search PubMed.
- R. Jakubiak, Z. Bao and L. Rothberg, Synth. Metals, 2000, 114, 61–64 CrossRef CAS.
- A. Eychmüller and A. L. Rogach, Pure Appl. Chem., 2000, 72, 179–188 CrossRef.
- K. Sathya, P. Dhamodharan and M. Dhandapani, Opt. Laser Technol., 2018, 101, 328–340 CrossRef CAS.
- I. M. Khan, S. Shakya, M. Islam, S. Khan and H. Najnin, Phy. Chem. Liq., 2021, 59, 753–769 CrossRef CAS.
- I. M. Khan, M. Islam, S. Shakya, K. Alam, N. Alam and M. Shahid, Bioorg. Chem., 2020, 99, 103779 CrossRef CAS PubMed.
- I. M. Khan, S. Shakya, R. Akhtar, K. Alam, M. Islam and N. Alam, Bioorg. Chem., 2020, 100, 103872 CrossRef CAS PubMed.
- S. Shakya and I. M. Khan, J. Hazard. Mater., 2021, 403, 123537 CrossRef CAS PubMed.
- I. M. Khan and S. Shakya, ACS Omega, 2019, 4, 9983–9995 CrossRef CAS PubMed.
- S. Shakya, I. M. Khan and M. Ahmad, J. Photochem. Photobio. A, 2020, 392, 112402 CrossRef CAS.
- A. Karmakar, P. Bandyopadhyay, S. Banerjee, N. C. Manda and B. Singh, J. Mol. Liq., 2020, 299, 112217 CrossRef CAS.
- L. M. Al-Harbi, E. H. El-Mossalamy, A. Y. Obaid and A. H. Al-Jedaani, Spectrochim. Acta, Part A, 2014, 120, 25–31 CrossRef CAS PubMed.
- N. Singh and A. Ahmad, J. Mol. Struct., 2010, 977, 197–202 CrossRef CAS.
- N. Singh, I. M. Khan and A. Ahmad, Spectrochim. Acta, Part A, 2010, 75, 1347–1353 CrossRef PubMed.
- I. M. Khan, A. Ahmad and M. Oves, Spectrochim. Acta, Part A, 2010, 77, 1059–1064 CrossRef PubMed.
-
(a) M. S. Refat, A. Elfalaky and E. Elesh, J. Mol. Struct., 2011, 990, 217–226 CrossRef CAS;
(b) A. M. A. Adam, H. A. Saad, A. A. Atta, M. Alsawat, M. S. Hegab, M. S. Refat, T. A. Altalhi, E. H. Alosaimi and A. A. Younes, J. Mol. Liq., 2022, 349, 118188 CrossRef CAS;
(c) A. M. A. Adam and M. S. Refat, J. Mol. Liq., 2022, 348, 118466 CrossRef CAS;
(d) A. M. A. Adam, H. A. Saad, M. S. Refat and M. S. Hegab, J. Mol. Liq., 2022, 353, 118819 CrossRef CAS.
- A. S. Gaballa and A. S. Amin, Preparation, Spectrochim. Acta, Part A, 2015, 145, 302–312 CrossRef CAS PubMed.
-
(a) M. S. Refat, H. A. Saad and A. M. A. Adam, Spectrochim. Acta, Part A, 2011, 79, 672–679 CrossRef CAS PubMed;
(b) A. M. A. Adam, M. S. Hegab, M. S. Refat and H. H. Eldaroti, J. Mol. Struct., 2021, 1231, 129687 CrossRef CAS;
(c) A. M. Adam, H. A. Saad, A. A. Atta, M. Alsawat, M. S. Hegab, T. A. Altalhi and M. S. Refat, Crystals, 2021, 11, 882 CrossRef CAS;
(d) A. M. Adam, H. A. Saad, A. A. Atta, M. Alsawat, M. S. Hegab, T. A. Altalhi and M. S. Refat, Crystals, 2021, 1, 882 CrossRef.
- S. M. Teleb and A. S. Gaballa, Spectrochim. Acta, Part A, 2005, 62, 140–145 CrossRef PubMed.
-
(a) S. G. Alegaon, K. R. Alagawadi, M. K. Garg, K. Dushyant and D. Vinod, Bioorg. Chem., 2014, 54, 51–59 CrossRef CAS PubMed;
(b) A. A. Bekhit, H. T. Fahmy, S. A. Rostom, A. El-Din and A. Bekhit, Eur. J. Med. Chem., 2010, 45, 6027–6038 CrossRef CAS PubMed.
-
(a) M. J. Naim, O. Alam, F. Nawaz, Md. J. Alam and P. Alam, J. Pharm. BioAllied Sci., 2016, 8, 2 CrossRef CAS PubMed;
(b) J. V. Faria, P. F. Vegi, A. G. C. Miguita, M. S. D. Santos, N. Boechat and A. M. R. Bernardino, Bioorg. Med. Chem., 2017, 25, 5891–5903 CrossRef CAS PubMed.
- C. Ong, L. Y. L. Yung, Y. Cai, B. H. Bay and G. H. Baeg, Nanotoxicology, 2015, 9, 396–403 CrossRef CAS PubMed.
- D. J. Benford, A. B. Hanley, K. Bottrill, S. Oehlschlager, M. Balls, F. Brance, J. J. Castegnara, J. Descotes, K. Hemminiky, D. Lindsay and B. Schilter, Alt. Lab. Anim., 2000, 28, 119–131 CrossRef.
- M. D. Rand, Neurotoxicol. Teratol., 2010, 32, 74–83 CrossRef CAS PubMed.
- I. Mukhopadhyay, A. Nazir, D. K. Saxena and D. K. Chowdhuri, Biomarkers, 2002, 7, 501–510 Search PubMed.
- S. Lindquist and E. A. Craig, Annu. Rev. Genet., 1988, 22, 631–677 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Cryst. A, 2015, 71, 59–75 CrossRef CAS PubMed.
- J. T. Lis, J. A. Simon and C. A. Sutton, Cell, 1983, 35, 403–410 CrossRef CAS PubMed.
- Y. H. Siddique, G. Ara, S. Jyoti, F. Naz and M. Afzal, Pharmacologyonline, 2011, 3, 667–684 Search PubMed.
- A. Fatima, S. Khanam, S. Jyoti, F. Naz, Rahul, F. Ali, T. Beg and Y. H. Siddique, J. Diet Suppl., 2017, 15, 893–909 CrossRef.
- R. A. Krebs and M. E. Feder, J. Exper. Bio., 1997, 200, 2007–2015 CrossRef CAS PubMed.
- A. Nazir, I. Mukhopadhyay, D. K. Saxena, M. S. Siddiqui and D. K. Chowdhuri, J. Biochem. Mol. Toxicol., 2003, 17, 98–107 CrossRef CAS PubMed.
- A. Nazir, D. K. Saxena and D. K. Chowdhuri, Biochim. Biophys. Acta, 2003, 1621, 218–225 CrossRef CAS PubMed.
- D. K. Chowdhuri, D. K. Saxena and P. N. Vishwanathan, Pest Biochem. Physiol., 1999, 63, 15–25 CrossRef CAS.
-
(a) I. Mukhopadhyay, D. K. Chowdhuri, M. Bajpayee and A. Dhawan, Mutagenesis, 2004, 19, 85–90 CrossRef CAS PubMed;
(b) H. Nosrati, Y. Baghdadchi, R. Abbasi, M. Barsbay, M. Ghaffarlou, F. Abhari, A. Mohammadi, T. Kavetskyy, S. Bochani, H. Rezaeejam and S. Davaran, J. Mater. Chem. B, 2021, 9, 4510–4522 RSC.
- H. Zhang, J. Chen, X. Zhu, Y. Ren, F. Cao, L. Zhu, L. Hou, H. Zhang and Z. Zhang, J. Mater. Chem. B, 2018, 6, 6108–6121 RSC.
- D. W. Ritche and V. Venkataraman, Bioinformatics, 2010, 26, 2398 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef PubMed;
(c) P. C. Hariharan and J. A. Pople, Chem. Phys. Lett., 1972, 16, 217–219 CrossRef CAS.
- M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef CAS.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer. Version 3.0, University of Western Australia, 2012 Search PubMed.
-
(a) T. Dey, P. Chatterjee, A. Bhattacharya, S. Pal and A. K. Mukherjee, Cryst. Growth Des., 2016, 16, 1442–1452 CrossRef CAS;
(b) I. M. Khan, S. Shakya and N. Singh, J. Mole. Liq., 2018, 250, 150–161 CrossRef CAS.
- I. M. Khan and A. Ahmad, Spectrochim. Acta, Part A, 2010, 76, 315–321 CrossRef PubMed.
- A. M. A. Adam, H. A. Saad, A. A. Atta, M. Alsawat, M. S. Hegab, M. S. Refat, T. A. Altalhi, E. H. Alosaimi and A. A. Younes, Crystals, 2021, 11, 960 CrossRef CAS.
- A. M. A. Adam, H. A. Saad, A. A. Atta, M. Alsawat, M. S. Hegab, M. S. Refat, T. A. Altalhi, E. H. Alosaimi and A. A. Younes, Crystals, 2021, 11, 960 CrossRef CAS.
-
(a) A. M. Hindawey, A. M. G. Nassar, R. M. Issa and Y. M. Issa, Indian J. Chem., 1980, 19, 615–619 Search PubMed;
(b) Y. M. Issa, A. M. Hindawey, A. E. El-Kholya and R. M. Issa, Chim. Ital., 1981, 111, 27–33 CAS.
-
(a) M. El-Sayed, M. El-Sayed and S. Agrawl, Talanta, 1982, 29, 535–537 CrossRef CAS PubMed;
(b) M. D. Ansari, H. Sagir, V. B. Yadav, N. Yadav and I. R. Siddiqui, ChemistrySelect, 2018, 3, 5326–5329 CrossRef CAS.
- L. Miyan and A. Ahmad, J. Mol. Liq., 2016, 219, 614–623 CrossRef CAS.
- A. El-Dissouky, T. E. Khalil, H. A. Elbadawy, D. S. El-Sayed, A. A. Attia and S. Foro, J. Mol. Struct., 2020, 1200, 127066 CrossRef.
- G. Aloisi and S. Pignatoro, J. Chem. Soc., Faraday Trans., 1973, 69, 534–539 RSC.
- G. Briegleb and Z. Angew, Angew. Chem., 1960, 72, 401–413 CrossRef CAS.
-
A. N. Martin, J. Swarbrick and A. Cammarata, Physical Pharmacy, 3rd edn, 1969 Search PubMed.
-
A. B. P. Lever, Inorganic Electronic Spectroscopy, 2nd edn, 1985 Search PubMed.
- H. Tsubumora and R. Lang, J. Am. Chem. Soc., 1961, 83, 2085–2092 CrossRef.
- W. W. Chou, J. Y. Guh and J. F. Tsai,
et al.
, Toxicology, 2008, 243, 1–10 CrossRef CAS PubMed.
-
(a) H. Chung, T. Sztal, S. Pasricha, M. Sridhar, P. Batterham and P. J. Daborn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5731–5736 CrossRef CAS PubMed;
(b) T. Yang, Y. Liang, J. Hou, Y. Dou and W. Zhang, J. Mater. Chem. B, 2017, 5, 5137–5144 RSC.
- J. A. Zijlstra and E. W. Vogel, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1988, 202, 251–267 CrossRef CAS PubMed.
- M. Shukla, N. Srivastava and S. Saha, J. Mol. Struct., 2012, 1021, 153–157 CrossRef CAS.
-
(a) N. I. Nijegorodov and W. S. Downey, J. Phys. Chem., 1994, 98, 5639–5643 CrossRef CAS;
(b) L. Gao, Y. Wang, J. Wang, L. Huang, L. Shi, X. Fan, Z. Zou, T. Yu, M. Zhu and Z. Li, Inorg. Chem., 2006, 45, 6844–6850 CrossRef CAS PubMed;
(c) M. S. Khan, M. Khalid, M. S. Ahmad, M. Shahid and M. Ahmad, Dalton Trans., 2019, 48, 12918–12932 RSC.
- H. Y. Woo, B. Liu, B. Kohler, D. Korystov, A. Mikhailovsky and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 14721–14729 CrossRef CAS PubMed.
-
(a) H. Nakashima and N. Yoshida, Org. Lett., 2006, 8, 4997–5000 CrossRef CAS PubMed;
(b) M. Akram, H. Lal, S. Shakya and Kabir-ud-Din, ACS Omega, 2020, 5, 3624–3637 CrossRef CAS PubMed;
(c) M. Alhomrani, W. F. Alsanie, A. S. Alamri, H. Alyami, H. Habeeballah, H. A. Alkhatabi, R. I. Felimban, J. M. Haynes, S. Shakya, B. M. Raafat and M. S. Refat, Pharmaceuticals, 2022, 15, 285 CrossRef CAS PubMed.
- A. H. Hasan, S. Murugesan, S. I. Amran, S. Chander, M. M. Alanazi, T. B. Hadda, S. Shakya, M. R. F. Pratama, B. Das, S. Biswas and J. Jamalis, Bioorg. Chem., 2022, 119, 105572 CrossRef CAS PubMed.
- I. M. Khan, K. Alam, M. Afshan, S. Shakya and M. Islam, J. Mol. Struct., 2020, 1206, 127758 CrossRef.
-
(a) M. D. Khan, S. Shakya, H. H. T. Vu, L. Habte and J. W. Ahn, J. Environ. Manage., 2021, 280, 111652 CrossRef CAS PubMed;
(b) M. S. Refat, H. A. Saad, A. A. Gobouri, M. Alsawat, A. M. A. Adam, S. Shakya, A. Gaber, A. M. Alsuhaibani and S. M. El-Megharbel, J. Mol. Liq., 2022, 349, 118121 CrossRef CAS;
(c) A. Ahmed, A. Fatima, S. Shakya, Q. I. Rahman, M. Ahmad, S. Javed, H. S. AlSalem and A. Ahmad, Molecules, 2022, 27, 1724 CrossRef CAS PubMed.
-
(a) F. L. Hirshfeld and H. Hope, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 406–415 CrossRef;
(b) G. Moss and P. Coppens, Chem. Phys. Lett., 1980, 75, 298–302 CrossRef CAS.
- L. Miyan, S. Qamar and A. Ahmad, J. Mol. Liq., 2017, 225, 713–722 CrossRef CAS.
- N. Singh, I. M. Khan, A. Ahmad and S. Javed, J. Mol. Liq., 2016, 221, 1111–1120 CrossRef CAS.
- S. M. Sondhi, M. Johar, S. Rajvanshi, S. G. Datidar, R. Shukla, R. Raghubir and J. W. Lown, Aust. J. Chem., 2001, 54, 169–174 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |

 a,
Ishaat M.
Khan
a,
Ishaat M.
Khan