
Protein-induced modifications in crystal morphology of a hydrogen-bonded organic framework†
Kate L.
Flint
 a,
Jack D.
Evans
a,
Francesco
Carraro
b,
Simon
Renner
b,
Oliver M.
Linder-Patton
a,
Heinz
Amenitsch
c,
Robert J.
Falconer
d,
Nicholas G.
White
e,
Christopher J.
Sumby
*a,
Paolo
Falcaro
*b and
Christian J.
Doonan
*a
a,
Jack D.
Evans
a,
Francesco
Carraro
b,
Simon
Renner
b,
Oliver M.
Linder-Patton
a,
Heinz
Amenitsch
c,
Robert J.
Falconer
d,
Nicholas G.
White
e,
Christopher J.
Sumby
*a,
Paolo
Falcaro
*b and
Christian J.
Doonan
*a
aDepartment of Chemistry, School of Physics, Chemistry and Earth Sciences, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: christopher.sumby@adelaide.edu.au; christian.doonan@adelaide.edu.au
bInstitute of Physical and Theoretical Chemistry, Graz University of Technology, Stremayrgasse 9, Graz 8010, Austria. E-mail: paolo.falcaro@tugraz.at
cInstitute for Inorganic Chemistry, Graz University of Technology, Stremayrgasse 9, Graz 8010, Austria
dSchool of Chemical Engineering, The University of Adelaide, Adelaide, SA 5005, Australia
eResearch School of Chemistry, The Australian National University, Canberra, ACT 2601, Australia
First published on 5th October 2023
Abstract
In this work, we studied the encapsulation of a range of proteins in a hydrogen-bonded organic framework (HOF) comprised of a tetraamidinium cation and diazobenzene-based dicarboxylate anion. We explore the use of external stimuli: light and temperature to modulate HOF crystal growth and size. In particular, we found photo-isomerisation can be used to control the concentration of the trans-azobenzene building block that contributes to HOF formation. When HOF growth was slowed sufficiently, deformation of the crystals and ultimately multicrystal aggregates were observed in the presence of some proteins. We propose that the extent of crystal deformation, consistent with better protein association, may be governed by differences in the type and strength of interactions between proteins and the surface of the growing HOF crystals.
Introduction
Enzymes have been widely explored as catalysts for chemical synthesis, particularly due to their excellent chemo-, regio-, and enantioselectivity.1 However, a number of challenges need to be overcome to broaden their commercial application including their lack of stability in non-native environments.2,3 A common approach to overcome this limitation involves immobilisation of enzymes within porous supports. This has been shown to extend their use in conditions that would otherwise cause denaturation, such as exposure to organic solvents, elevated temperatures and pH extremes.4–6Metal–organic frameworks (MOFs) are considered promising solid supports for enzymes due to their high accessible porosity and molecular-level control of functionality.7–10 Indeed an emerging body of research has shown that enzymes, and other biomacromolecules, can be encapsulated within MOF particles via a one-pot synthetic approach. In such cases, the MOF forms a protective shell around the enzyme that imparts enhanced durability,11–13 presumably by confining the enzyme within a rigid cavity that prevents denaturation.14 The majority of enzyme@MOF studies have focused on zeolitic imidazolate framework-8 (ZIF-8), and its structural polymorphs, due to its facile synthesis under biologically compatible conditions.15,16 However, ZIF-8 has limitations as a platform material for biocomposites; it is unstable to pH < 6,17 certain buffers,18,19 and metal-chelating agents,20 and has relatively narrow pore apertures which can restrict substrate diffusion and lead to reduced catalytic activity.21–23
To overcome some of these challenges there is increasing interest in employing hydrogen-bonded organic frameworks (HOFs) as alternative enzyme support materials for one-pot encapsulation.24–29 HOFs are porous solids formed via intermolecular hydrogen bonding of discrete organic components30–37 and analogous to MOFs, are assembled via a modular building block approach.38 Furthermore, some HOFs can be readily synthesized in biocompatible conditions, avoiding the use of organic solvents, and elevated temperature or pH extremes which typically lead to enzyme denaturation.38,39 In contrast to ZIF-8, HOFs are metal free systems, and many have shown good stability to buffers and chelating agents over a biologically relevant pH range.24
While the aforementioned properties have been capitalised on to synthesize HOF-based biocomposites that afford protection to a range of encapsulated enzymes, the mechanism of encapsulation is not understood as well as their ZIF counterparts, which limits the potential to design systems with tailored properties. Previously we assessed the kinetics of formation of a HOF (Bio-HOF 1) composed of water soluble tetraamidinium and tetracarboxylate building blocks in both the presence and absence of protein using time-resolved small-angle scattering experiments.24 These data revealed that nuclei formed within 100 ms in both instances suggesting that, unlike ZIF-8 biomimetic mineralization, the protein does not seed HOF crystallisation but rather the enzyme is incorporated into the crystal during the growth stage. If enzyme@HOF biocomposites are formed via this mechanism then the rate of HOF crystal growth and the affinity of the protein to the crystal surface are two parameters that could influence properties such as the morphology of the biocomposite and enzyme occlusion within the lattice.40,41
Herein we report the synthesis and characterisation of HOF-based biocomposites synthesised from tetraamidinium (1·Cl4) and diazobenzene dicarboxylate (K2·2) building blocks, that have previously been studied for intracellular biocatalysis,29 and explore how proteins can influence HOF crystal morphology under controlled crystal growth kinetics achieved by exploiting the photoswitching ability of 2, temperature, or through simple building block dilution (Fig. 1). In addition, we examine the protein–HOF interactions by modifying the surface chemistry of the protein via amination or acetylation. These data provide a better understanding of the primary interactions that influence the formation of enzyme@HOF biocomposites and thus, will inform the design of new materials with tailored properties.
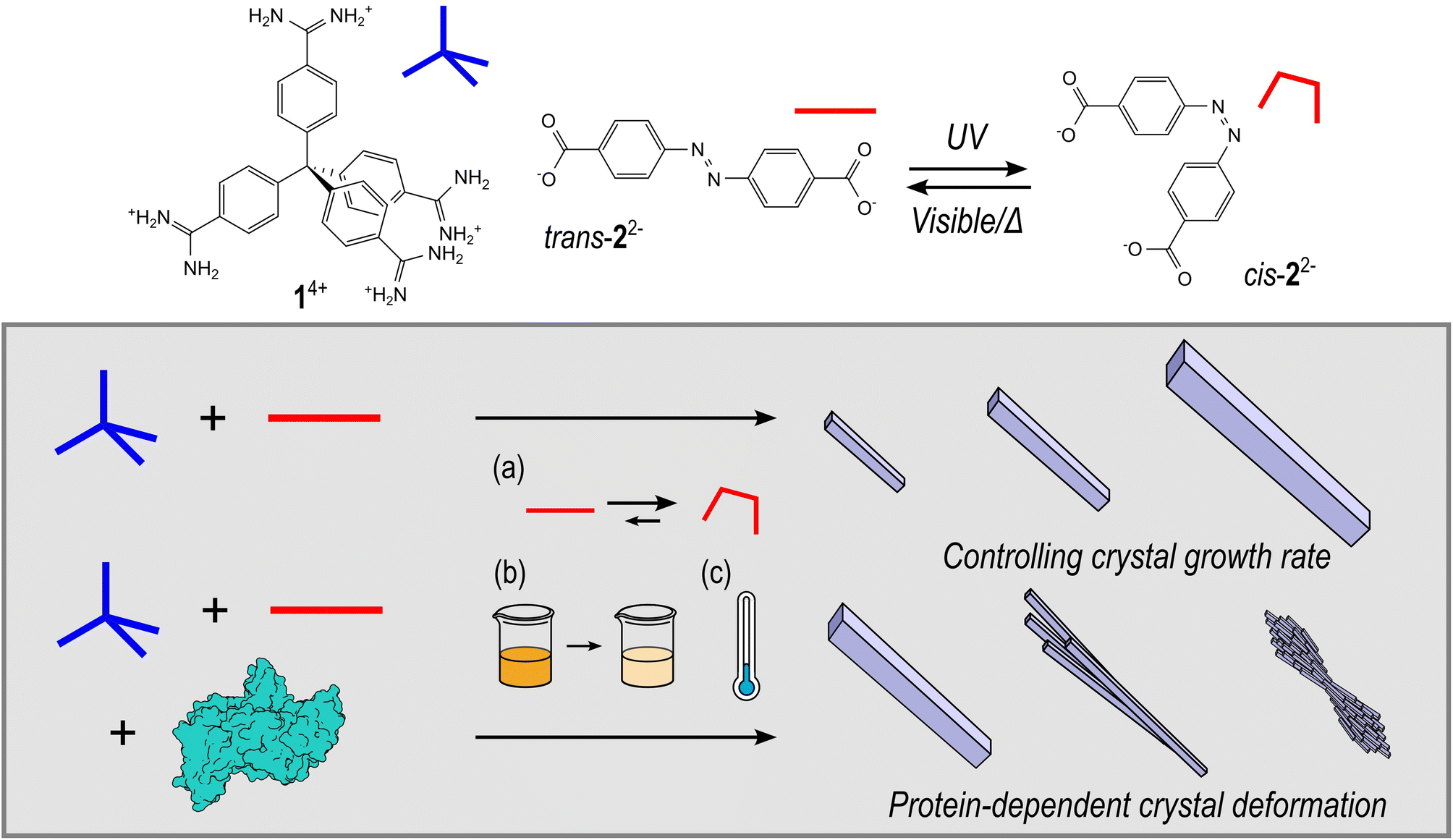 | ||
| Fig. 1 The chemical structures of HOF components: tetraamidinium (1·Cl4) and dicarboxylate (K2·2), and a schematic representation of the synthesis HOF crystals, where the rate of crystallisation can be controlled by (a) use of the inbuilt photoswitch, (b) total precursor concentration, or (c) lowering the reaction temperature. Furthermore, the HOF crystal morphology can be perturbed by addition of a protein to form protein@HOF biocomposites. | ||
Results and discussion
HOF and HOF-biocomposite synthesis
The assembly and encapsulation of proteins in this 1·2 HOF system has been previously described by Tang et al.; however, the photoswitching properties of the anionic building block were not explored. Indeed, the authors noted that the self-assembly process was not affected in the presence of protein under their synthesis conditions.29 Here, we prepared HOF samples by mixing aqueous solutions of 1·Cl4 (5 mL, 1 mM) and K2·2 (5 mL, 2 mM). The mixture was left undisturbed for 24 hours, and the resulting precipitate recovered by centrifugation (2000 g, 10 minutes), and washed twice with water. For samples containing protein the appropriate molar equivalents were pre-mixed with a solution of 1·Cl4 before addition of K2·2. Furthermore, the rate of crystal growth was controlled by addition of K2·2 as the cis isomer (after irradiation with UV light), by decreasing the overall concentrations of precursors (50%, 15%), and by reducing the reaction temperature with protein concentration and volume remaining constant (see ESI† for details).Effect of protein addition on HOF morphology
Initial screening of HOF crystal growth was undertaken by employing the same precursor concentrations as reported by White and coworkers,42 utilising both the photo- and non-photoswitched forms of 2. The growth rate was qualitatively assessed in the absence and presence of a model protein, namely Bovine Serum Albumin (BSA). The crystal size and morphology of the final products were analysed by scanning electron microscopy (SEM). During the screening process it was observed that the rate of crystal formation was largely dependent on the isomer used. For example, where K2·2 was added in the trans form precipitation of particles was immediate, while addition of the cis form resulted in slower crystal growth (i.e. a noticeable precipitate formed only after several hours) and significantly larger crystals (Fig. S3†). The crystallinity of both samples was analysed by performing powder X-ray diffraction (PXRD) experiments (Fig. S4†). Upon drying the materials became amorphous but, upon careful addition of water, crystallinity could be restored, giving PXRD patterns consistent with those calculated from the single crystal X-ray diffraction (SCXRD) structure reported previously.42 In the presence of BSA, crystals of the HOF formed using trans-K2·2 closely resembled those obtained in the absence of protein, with needle-like morphology (Fig. 2a). In contrast, crystals formed using cis-K2·2 in the presence of BSA were comprised of small intergrown aggregates, forming larger feather-like structures (Fig. 2b). The observed modification to crystal morphology suggests that slowing crystal growth facilitates greater surface coverage of the protein at step edges.43 Indeed, the ability of macromolecular additives to impact crystallisation processes is well known,44 of particular note being the variety of protein-specific and concentration-dependent morphologies able to be obtained in the crystallisation of salts.45,46 Furthermore, amino acids and proteins have been shown to direct the crystal morphology of ZIF-8 materials.12,47 | ||
| Fig. 2 SEM images of BSA@HOF composites showing crystals grown upon addition of the (a) trans- and (b) cis-K2·2. | ||
Given the observed effect of trans-K2·2 and cis-K2·2 on the morphology of the HOF crystals we further investigated their role in the nucleation and growth of HOF particles via time-resolved Small Angle X-ray Scattering (SAXS) at a synchrotron facility. Using a stopped-flow setup, we rapidly mixed the K2·2 and the 1·Cl4/BSA solutions and collected SAXS patterns of the growing HOF particles in solution with a time resolution of 100 ms (see ESI†). While the use of the photoswitch to slow crystal growth had been effective in the experiments described above, we additionally sought to ascertain whether lowering the overall building block concentration (component) could achieve similar outcomes. To that end two ratios of HOF component![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :protein were selected for SAXS measurements; 500:1 and 75:1 (mol mol−1), annotated as high and low, respectively. We note that the high ratio (500:1) was used in the initial screening using photoswitched cis-K2·2 as described above. Fig. 3 reports representative SAXS patterns of the HOF (110) reflection collected at different reaction times (0.1–180 s range). Fig. S31a–d† show the time evolution of the SAXS pattern in the q range from 0.16 nm−1 to 3 nm−1. In this q range, we did not detect any significant difference in the SAXS patterns over time. The Porod Invariant calculated from these patterns does not change over time (Fig. S30†). We ascribed this to rapid particle growth (>100 ms) that engenders crystallite dimensions larger than the resolution limit of the used set-up (>40 nm, qmin = 0.16 nm−1 thus no Guinier regime were detected).48 We analysed the diffraction patterns in the same time range (Fig. 3a–d) and detected the appearance of the most intense HOF diffraction peak ((110) at 3.4 nm−1)42 for the samples prepared at highest total ligand concentration (500:1, protein/ligand) after ca. 120 s. Notably, at 180 s for the sample prepared using trans-K2·2 the (110) is more intense than the peak detected for the sample prepared using cis-K2·2. For the samples prepared at the lowest total ligand concentration (75:1, protein:ligand), we hypothesize that the concentration of crystalline particles in the investigated volume was not high enough to detect the HOF diffraction peaks with the collection time used for these in situ experiments. Overall, these data demonstrate that the trans form of K2·2 determines the kinetics of nucleation of the HOF, independently of the presence of a fraction of cis-K2·2 in the solution and its concentration (within the investigated concentration range, Table S1†). Furthermore, the presence of cis-K2·2 slows the crystal growth process, leading to a lower yield of crystalline material within 180 s of reaction compared to trans-K2·2.
:protein were selected for SAXS measurements; 500:1 and 75:1 (mol mol−1), annotated as high and low, respectively. We note that the high ratio (500:1) was used in the initial screening using photoswitched cis-K2·2 as described above. Fig. 3 reports representative SAXS patterns of the HOF (110) reflection collected at different reaction times (0.1–180 s range). Fig. S31a–d† show the time evolution of the SAXS pattern in the q range from 0.16 nm−1 to 3 nm−1. In this q range, we did not detect any significant difference in the SAXS patterns over time. The Porod Invariant calculated from these patterns does not change over time (Fig. S30†). We ascribed this to rapid particle growth (>100 ms) that engenders crystallite dimensions larger than the resolution limit of the used set-up (>40 nm, qmin = 0.16 nm−1 thus no Guinier regime were detected).48 We analysed the diffraction patterns in the same time range (Fig. 3a–d) and detected the appearance of the most intense HOF diffraction peak ((110) at 3.4 nm−1)42 for the samples prepared at highest total ligand concentration (500:1, protein/ligand) after ca. 120 s. Notably, at 180 s for the sample prepared using trans-K2·2 the (110) is more intense than the peak detected for the sample prepared using cis-K2·2. For the samples prepared at the lowest total ligand concentration (75:1, protein:ligand), we hypothesize that the concentration of crystalline particles in the investigated volume was not high enough to detect the HOF diffraction peaks with the collection time used for these in situ experiments. Overall, these data demonstrate that the trans form of K2·2 determines the kinetics of nucleation of the HOF, independently of the presence of a fraction of cis-K2·2 in the solution and its concentration (within the investigated concentration range, Table S1†). Furthermore, the presence of cis-K2·2 slows the crystal growth process, leading to a lower yield of crystalline material within 180 s of reaction compared to trans-K2·2.
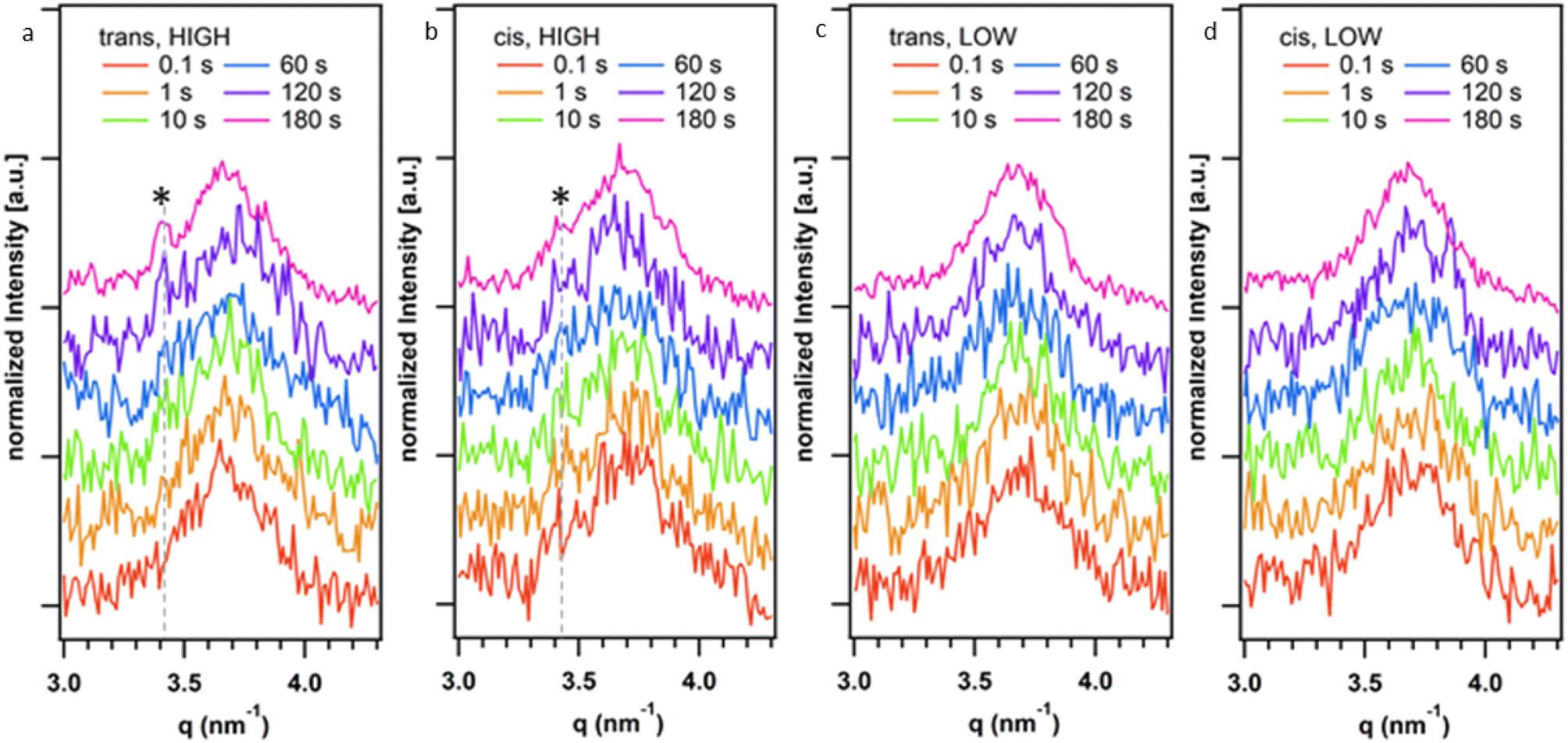 | ||
| Fig. 3 Time evolution of the (110) HOF diffraction peak (3.4 nm−1) from time-resolved SAXS measurements during the synthesis of the HOF crystals using trans-K2·2 with (a) 500:1 (HIGH) and (c) 75:1 (LOW); and cis-K2·2 at (b) 500:1 (HIGH) and (d) 75:1 (LOW) total ligand concentration. The broad peak centred at 3.7 nm−1 is associated to the Kapton windows of the experimental setup. The SAXS patterns were shifted along the Y-axis for the sake of clarity. | ||
From the SAXS measurements it was clear that utilisation of the azobenzene photoswitch and reduction of overall building block concentration significantly impacted crystallisation of the HOF, and we were interested to see whether this led to changes in crystal morphology. For these syntheses an intermediate ratio of HOF component:protein was added; and 500:1, 250:1, and 75:1 (mol mol−1) used, annotated as high, medium, and low, respectively. The samples were again amorphous upon drying (Fig. S5†), but PXRD data obtained for solvated samples indicated HOF formation (Fig. S6†). Unfortunately, PXRD characterisation was not possible for all samples, due to low yields and difficulties in achieving optimal solvation to restore crystallinity of the material.
In the absence of protein, the SEM micrographs showed pristine, needle-like HOF crystals for all concentrations (Fig. S12†), with the lower dilutions resulting in the formation of larger crystals. In the presence of BSA the high ratio of trans-K2·2 yielded crystals with a needle-like morphology analogous to those formed in the absence of protein. However, when the building block concentration was decreased, intergrown clusters like those produced using cis-K2·2 were observed (Fig. 4b). Lowering the ligand concentration further resulted in spherical bud-like clusters of directionally intergrown crystals (Fig. 4c). We then examined the spatial distribution of the fluorescently tagged protein, FITC-BSA, in the HOF-based biocomposites via confocal laser scanning microscopy (CLSM). For high ratio samples CLSM imaging indicated that the protein was likely surface bound, where protein can be seen in a ring coating the exterior of the crystals (Fig. S22 and S23†). In contrast at the medium and low concentrations the protein appeared to be internalised, with fluorescently-tagged protein evident throughout the microcrystalline composite (Fig. S25–S28†). However, we note that the resolution of CLSM does not allow us to determine if the protein is completely encapsulated within the framework, or if it is partially or completely coating the surface of many intergrown crystals within the crystalline aggregate.
 | ||
| Fig. 4 SEM images of BSA@HOF composites showing crystals grown upon addition of K2·2 at three different protein/total ligand ratios. Upon addition of trans-K2·2 at (a) 500:1; (b) 250:1; (c) 75:1; and cis-K2·2 at (d) 500:1; (e) 250:1; (f) 75:1. | ||
For samples synthesised using cis-K2·2, SEM micrographs show that similar crystal morphologies were observed for both the high and medium ligand concentrations (Fig. 4d and e). In these cases, clusters of small crystals were seen to grow directionally to form long seaweed-like structures. When the ligand concentration was lowered further intergrown structures comprised of many bud-like spheroids clumped together were observed (Fig. 4f). Analysis of these samples using CLSM (and FITC-BSA) showed similar localisation of the protein to that in the trans samples of similar morphology (Fig. S25–S28†).
The same syntheses were repeated at reduced temperature (4 °C) to investigate if it was possible to further slow the crystal growth and study the effect on HOF morphology across the range of ligand concentrations. In the presence of BSA crystal deformation was observed by SEM at all three ligand concentrations, upon addition of either trans- or cis-K2·2 (Fig. S14†). Most interestingly, a change in crystal morphology could be seen for samples synthesised at the highest total ligand concentration utilising the trans form of K2·2. This combination had produced HOF crystals with a pristine needle form when grown at room temperature; however, at reduced temperature a combination of isolated and intergrown strands of needle-like crystals could be seen by SEM imaging (Fig. S14a†). These small crystals became more intergrown for samples utilising the photoswitch or having greater ligand dilution, with the slowest growing crystals forming bud-like chains and clusters.
Given that pristine HOF crystals were observed in the absence of BSA, it is apparent that BSA has a significant impact on crystal growth, resulting in a change in HOF crystal morphology. This influence becomes more pronounced in samples where growth is intentionally slowed down through methods such as utilising the photoswitch of 22−, diluting the total ligand concentration, reducing the reaction temperature, or a combination of these factors. An increase in crystal deformation with decreasing crystallisation rate, resulting in clusters of intergrown crystals for the slowest growing samples, suggests that this phenomenon may arise from the binding of proteins at advancing step edges on the HOF crystal surface.
Effect of protein surface charge and chemistry
Previous studies have shown that protein surface chemistry plays a key role in the formation of ZIF-8-based biocomposites,49 and surface residue modification can be a useful tool to improve protein incorporation in other HOF materials.26,27 Thus, we were motivated to understand if modifying protein functionality had a similar effect in the 1·2 HOF system. We prepared two samples of BSA where the acidic and basic surface residues were chemically modified by amination or acetylation to decrease or increase the negative surface charge, respectively (see ESI† for details). Following modification, the proteins were reacted with the HOF precursors under identical conditions to those employed for screening the native protein, including varying ligand form (cis/trans), and ligand concentration. To limit the number of repeats, the effect of reduced temperature was not further investigated, and all samples were crystallised at room temperature.Despite significant differences in surface charge and zeta potential of the functionalised BSA, SEM analysis indicated that all samples followed similar trends in terms of crystal morphology (Fig. S15 and S16†). At the highest ratio (500:1 mol mol−1 protein:ligand) using trans-K2·2 all samples gave crystals with a needle-like morphology, and as ligand concentration decreased (250:1, 75:1) intergrown clusters and bud-like structures were observed. For cis K2·2 (where growth was further slowed) the crystal morphology was essentially identical to samples containing the unmodified protein.
Simulations were undertaken to better understand the surface electronic structure of the 1·2 HOF and the limited effect of surface charge on protein–HOF interaction. Surface models of 1·2 and, for comparison ZIF-8, were generated. To examine the electrostatic forces experienced by a protein on these surfaces the average electrostatic potential was computed normal to surface (Fig. 5). ZIF-8 has greater overall electrostatic potential near the surface owing to a high density of Zn sites compared to the surface of the 1·2 HOF which only has localised areas of large electrostatic potential at greater distances (i.e. lower density) originating from the carboxylate and amidinium groups. These simulations demonstrate that, as opposed to ZIF-8, electrostatics are not expected to play an important role in surface–protein interactions. This finding is in agreement with those of Tang et al., who showed that protein encapsulation in the same HOF was not meaningfully affected by protein surface chemistry, using green fluorescent protein variants bearing a range of surface charges.29
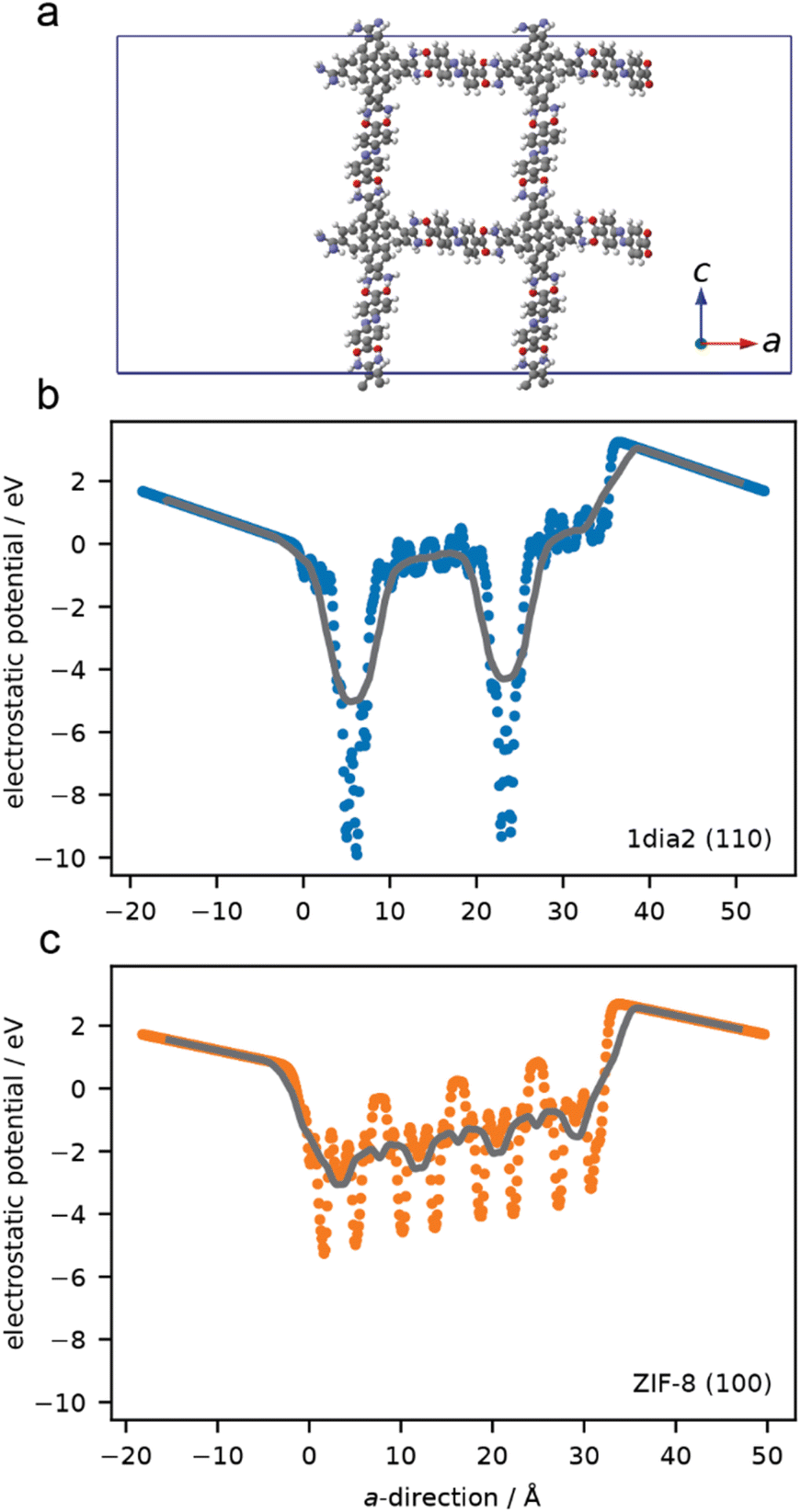 | ||
| Fig. 5 Atomistic model of the (110) surface for the HOF (a) average one-dimensional electrostatic potential computed in the direction normal to the surface. Points are from 0.01 Å slices and lines correspond to smoothing this data over 0.5 Å (b and c). | ||
To better understand how proteins influence HOF crystal morphology, a selection of proteins and enzymes with different native surface chemistry were encapsulated to assess the impact on crystal growth. The size and zeta potentials of the various proteins are recorded in Table S2.† Analysis of the SEM micrographs show that Horseradish Peroxidase (HRP) and urease afford intergrown crystals assembled in chains or clumps, similar to BSA, when HOF growth was slowed, either by utilising the photoswitch of 22− or lowering the total ligand concentration. In contrast, for Lysozyme, Lipase B from Candida Antarctica (CALB), and myoglobin the SEM shows only slight changes in crystal morphology, even at the lowest concentrations of ligand. PXRD patterns of the solvated samples were consistent with that calculated from the SCXRD structure, although we note significant differences in crystallinity depending on the protein present. Samples containing HRP, urease, and lysozyme showed only broad peaks, and achieving optimal resolvation to recover crystallinity was not possible for all samples, particularly due to low yields at the lowest ligand concentrations (Fig. S7–S9†). By contrast, CALB and myoglobin, which showed the least crystal deformation, generally exhibited higher crystallinity after rehydration, with CALB in particular giving well resolved diffraction peaks (Fig. S10 and S11†).
Studying protein–HOF interactions
Given that modification of the protein surface had not greatly impacted crystal morphology and computational studies had suggested electrostatics did not play a key role in surface–protein interactions we sought to understand what type of interactions were present in this system.To examine the type and strength of interactions between the surface of the HOF crystals and proteins we carried out isothermal titration calorimetry (ITC) studies. ITC is a powerful technique to measure the thermodynamics of molecular interactions, allowing characterisation of protein–protein, and protein-small molecule binding events in solution.50,51 Increasingly, ITC has been applied in the fields of polymer and supramolecular chemistry,52,53 including MOFs,54 to study small molecule adsorption55–57 and gain mechanistic insight into the formation of materials.58,59 ITC has also been used to investigate the binding of various proteins with ZIF-8, with electrostatic interactions being dominant.60 Crucially, the authors found that smaller anionic proteins exhibited the strongest adsorption onto the cationic ZIF nanoparticles.
To gain insight into the protein–HOF interactions present we selected two proteins for ITC studies that had shown opposing effects on crystal growth; BSA and lysozyme. Each protein was titrated into a solution of HOF particles and the heat of adsorption recorded. Control titrations of protein into buffer (without the HOF particles present) to assess the heat of dilution were also carried out (see ESI†).
Initial experiments with BSA at 25 °C showed a poorly defined isotherm where the heat of dilution was comparable to the reaction heat. However, when the temperature was increased to 35 °C exothermic binding was observed which is indicative of the formation of non-covalent bonds, such as van der Waals forces, coulombic interactions or hydrogen-bonding (Fig. S32†).61 Further experiments at increased temperature (40 °C, and 45 °C) confirmed the presence of exothermic binding, ruling out the possibility of hydrophobic interactions between BSA and the HOF crystal surface (Fig. S33 and S34†).
Given that BSA showed only modest interactions with the HOF crystals by ITC, it was anticipated that very weak interactions would be detected in the case of lysozyme, which had showed limited crystal deformation during protein@HOF synthesis. This proved to be the case, with minimal reaction heat change beyond the heat of dilution recorded for lysozyme titrated into a suspension of HOF crystals at 35 °C (Fig. S35†).
When taken in conjunction with the protein modification and computational experiments described above, the binding detected by ITC is due to hydrogen-bonding rather than electrostatic interactions. Given that the HOF itself is assembled through hydrogen-bonding interactions, it is likely that residues on the protein surface could interact with available amidinium or carboxylate groups on the crystal surfaces and at crystal steps through similar non-covalent interactions. The same phenomenon could also occur during the HOF crystallisation process, terminating crystal growth and leading to the deformation observed. The importance of surface functionality has previously been demonstrated for the related tetraamidinium/tetracarboxylate-based BioHOF-1, where it was found that enzyme loading and activity could be boosted by incorporation of an arginine-rich moiety to enhance surface-binding interactions; however the underlying mechanism for enhanced loading was not studied.27
Overall, the ITC data for BSA titration into HOF showed higher signal-to-noise ratio compared to the Tris–HCl buffer control due to the presence of HOF particles in the sample cell. The binding isotherm did not fit a single site binding model which is consistent with non-specific binding across multiple sites. In other studies, proteins have been shown to adsorb to neutral hydrophilic surfaces in random orientations, allowing the tertiary and quaternary protein structure to remain intact.62 Furthermore, our findings are broadly in agreement with those seen in studies with ZIF-8, where large, electrostatically-favourable proteins (such as BSA) bind with no preferred orientation.60
Overall, the ITC results revealed exothermic binding for BSA to the HOF particle surface, which, taken in conjunction with protein modification and computational studies, are likely hydrogen-bonding interactions. These findings illustrate the complexity of protein–HOF interactions and emphasise the need for careful consideration of the strength and types of interactions present to improve the immobilisation capacity of these materials.
Conclusions
Herein, we have thoroughly investigated the immobilisation of proteins in an amidinium/carboxylate-based HOF that has previously been investigated for biomacromolecule encapsulation.29 We identified that crystal size and morphology could be modulated by controlling ligand concentration via an intrinsic photoswitching component, and/or reducing reaction temperature. A range of proteins that possess distinct surface charge and chemistry were immobilised and these data showed that protein charge did not have a significant influence on the level of HOF crystal deformation. This observation was consistent with ITC studies and computational analysis that revealed weak exothermic binding, with hydrogen-bonding interactions rather than electrostatics likely driving non-specific binding of the protein to the HOF surface. Furthermore, our results provide an explanation as to why increasing the number of surface arginine residues on a protein led to an increased loading of immobilised protein for an analogous amidinium-carboxylate system.27 In summary, our results indicate that studies focused on optimising the secondary interactions between proteins and amidinium-carboxylate HOF materials, rather than surface charge, will aid the synthesis of HOF-based biocomposites with tailored properties and functionality.Author contributions
C. J. D., C. J. S. and N. G. W. conceived the project. K. L. F. and O. M. L.-P. performed all the synthesis and characterisation of the samples. F. C., S. R. and H. A. carried out and analysed the data from the SAXS measurements. J. D. E. performed the computational calculations. R. J. F. analysed the ITC data and provided all the equipment for the measurements. K. L. F. and C. J. D. prepared the final manuscript and ESI. C. J. D., P. F. and C. J. S. supervised this work and acquired the financial support for the project. All authors collectively discussed the results, provided insightful inputs, and reviewed the manuscript prior to submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council Discovery Project (DP200102411). The authors acknowledge the instruments and expertise of Microscopy Australia at Adelaide Microscopy, The University of Adelaide, enabled by NCRIS, university, and state government support. J. D. E. is the recipient of an Australian Research Council Discovery Early Career Award (project number DE220100163) funded by the Australian Government. Phoenix HPC service at the University of Adelaide are thanked for providing high-performance computing resources. This research was supported by the Australian Government's National Collaborative Research Infrastructure Strategy (NCRIS), with access to computational resources provided by Pawsey Supercomputing Research Centre through the National Computational Merit Allocation Scheme. We acknowledge the CERIC-ERIC consortium for access to the SAXS beamline at ELETTRA – Synchrotron Trieste and financial support. F. C. acknowledges the TU Graz Initial Funding Programme.References
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef PubMed.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- C. Scharnagl, M. Reif and J. Friedrich, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1749, 187–213 CrossRef PubMed.
- I. Gill and A. Ballesteros, J. Am. Chem. Soc., 1998, 120, 8587–8598 CrossRef.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef PubMed.
- C. Ispas, I. Sokolov and S. Andreescu, Anal. Bioanal. Chem., 2009, 393, 543–554 CrossRef PubMed.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- R. J. Drout, L. Robison and O. K. Farha, Coord. Chem. Rev., 2019, 381, 151–160 CrossRef.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef PubMed.
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef PubMed.
- R. Murty, M. K. Bera, I. M. Walton, C. Whetzel, M. R. Prausnitz and K. S. Walton, J. Am. Chem. Soc., 2023, 145, 7323–7330 CrossRef PubMed.
- W. Liang, R. Ricco, N. K. Maddigan, R. P. Dickinson, H. Xu, Q. Li, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Mater., 2018, 30, 1069–1077 CrossRef.
- K. Liang, C. J. Coghlan, S. G. Bell, C. Doonan and P. Falcaro, Chem. Commun., 2015, 52, 473–476 RSC.
- C.-Y. Sun, C. Qin, X.-L. Wang, G.-S. Yang, K.-Z. Shao, Y.-Q. Lan, Z.-M. Su, P. Huang, C.-G. Wang and E.-B. Wang, Dalton Trans., 2012, 41, 6906–6909 RSC.
- M. de J. Velásquez-Hernández, R. Ricco, F. Carraro, F. T. Limpoco, M. Linares-Moreau, E. Leitner, H. Wiltsche, J. Rattenberger, H. Schröttner, P. Frühwirt, E. M. Stadler, G. Gescheidt, H. Amenitsch, C. J. Doonan and P. Falcaro, CrystEngComm, 2019, 21, 4538–4544 RSC.
- M. A. Luzuriaga, C. E. Benjamin, M. W. Gaertner, H. Lee, F. C. Herbert, S. Mallick and J. J. Gassensmith, Supramol. Chem., 2019, 31, 485–490 CrossRef PubMed.
- Y. Gao, C. M. Doherty and X. Mulet, ChemistrySelect, 2020, 5, 13766–13774 CrossRef.
- K. Zhang, R. P. Lively, C. Zhang, R. R. Chance, W. J. Koros, D. S. Sholl and S. Nair, J. Phys. Chem. Lett., 2013, 4, 3618–3622 CrossRef.
- C. Zhang, R. P. Lively, K. Zhang, J. R. Johnson, O. Karvan and W. J. Koros, J. Phys. Chem. Lett., 2012, 3, 2130–2134 CrossRef PubMed.
- X. Wu, H. Yue, Y. Zhang, X. Gao, X. Li, L. Wang, Y. Cao, M. Hou, H. An, L. Zhang, S. Li, J. Ma, H. Lin, Y. Fu, H. Gu, W. Lou, W. Wei, R. N. Zare and J. Ge, Nat. Commun., 2019, 10, 5165 CrossRef PubMed.
- W. Liang, F. Carraro, M. B. Solomon, S. G. Bell, H. Amenitsch, C. J. Sumby, N. G. White, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 14298–14305 CrossRef PubMed.
- G. Chen, L. Tong, S. Huang, S. Huang, F. Zhu and G. Ouyang, Nat. Commun., 2022, 13, 4816 CrossRef PubMed.
- G. Chen, S. Huang, Y. Shen, X. Kou, X. Ma, S. Huang, Q. Tong, K. Ma, W. Chen, P. Wang, J. Shen, F. Zhu and G. Ouyang, Chem, 2021, 7, 2722–2742 Search PubMed.
- P. Wied, F. Carraro, J. M. Bolivar, C. J. Doonan, P. Falcaro and B. Nidetzky, Angew. Chem., Int. Ed., 2022, 61, e202117345 CrossRef PubMed.
- Z. Tang, X. Li, L. Tong, H. Yang, J. Wu, X. Zhang, T. Song, S. Huang, F. Zhu, G. Chen and G. Ouyang, Angew. Chem., Int. Ed., 2021, 60, 23608–23613 CrossRef PubMed.
- J. Tang, J. Liu, Q. Zheng, W. Li, J. Sheng, L. Mao and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 22315–22321 CrossRef PubMed.
- M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696–4698 CrossRef.
- V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef PubMed.
- J. Luo, J.-W. Wang, J.-H. Zhang, S. Lai and D.-C. Zhong, CrystEngComm, 2018, 20, 5884–5898 RSC.
- N. G. White, Chem. Commun., 2021, 57, 10998–11008 RSC.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef PubMed.
- P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef PubMed.
- S. A. Boer, M. Morshedi, A. Tarzia, C. J. Doonan and N. G. White, Chem.–Eur. J., 2019, 25, 10006–10012 CrossRef PubMed.
- M. Morshedi, M. Thomas, A. Tarzia, C. J. Doonan and N. G. White, Chem. Sci., 2017, 8, 3019–3025 RSC.
- K. Rae Cho, Y.-Y. Kim, P. Yang, W. Cai, H. Pan, A. N. Kulak, J. L. Lau, P. Kulshreshtha, S. P. Armes, F. C. Meldrum and J. J. De Yoreo, Nat. Commun., 2016, 7, 10187 CrossRef PubMed.
- Y. Ning and S. P. Armes, Acc. Chem. Res., 2020, 53, 1176–1186 CrossRef PubMed.
- P. Muang-Non, H. D. Toop, C. J. Doonan and N. G. White, Chem. Commun., 2022, 58, 306–309 RSC.
- Y.-Y. Kim, K. Ganesan, P. Yang, A. N. Kulak, S. Borukhin, S. Pechook, L. Ribeiro, R. Kröger, S. J. Eichhorn, S. P. Armes, B. Pokroy and F. C. Meldrum, Nat. Mater., 2011, 10, 890–896 CrossRef PubMed.
- A. G. Shtukenberg, M. D. Ward and B. Kahr, Chem. Rev., 2017, 117, 14042–14090 CrossRef PubMed.
- B. Szabó and T. Vicsek, Phys. Rev. E, 2003, 67, 011908 CrossRef PubMed.
- B. Pathak, J. Christy, K. Sefiane and D. Gozuacik, Langmuir, 2020, 36, 9728–9737 CrossRef PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles and P. Falcaro, CrystEngComm, 2016, 18, 4264–4267 RSC.
- F. Carraro, J. D. Williams, M. Linares-Moreau, C. Parise, W. Liang, H. Amenitsch, C. Doonan, C. O. Kappe and P. Falcaro, Angew. Chem., Int. Ed., 2020, 59, 8123–8127 CrossRef PubMed.
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217–4223 RSC.
- M. W. Freyer and E. A. Lewis, in Methods in Cell Biology, Academic Press, 2008, vol. 84, pp. 79–113 Search PubMed.
- R. J. Falconer, B. Schuur and A. K. Mittermaier, J. Mol. Recognit., 2021, 34, e2901 CrossRef PubMed.
- W. R. Archer and M. D. Schulz, Soft Matter, 2020, 16, 8760–8774 RSC.
- A. Arnaud and L. Bouteiller, Langmuir, 2004, 20, 6858–6863 CrossRef PubMed.
- F. Sha, T.-Y. Tai, M. A. Gaidimas, F. A. Son and O. K. Farha, Langmuir, 2022, 38, 6771–6779 CrossRef PubMed.
- Y.-B. Wei, M.-J. Wang, D. Luo, Y.-L. Huang, M. Xie, W. Lu, X. Shu and D. Li, Mater. Chem. Front., 2021, 5, 2416–2424 RSC.
- S. Kato, K. Otake, H. Chen, I. Akpinar, C. T. Buru, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2568–2576 CrossRef PubMed.
- R. J. Drout, S. Kato, H. Chen, F. A. Son, K. Otake, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 12357–12366 CrossRef PubMed.
- Y.-C. Zhang, Z.-Y. Xu, Z.-K. Wang, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, ChemPlusChem, 2020, 85, 1498–1503 CrossRef.
- S. Li, M. Dharmarwardana, R. P. Welch, C. E. Benjamin, A. M. Shamir, S. O. Nielsen and J. J. Gassensmith, ACS Appl. Mater. Interfaces, 2018, 10, 18161–18169 CrossRef PubMed.
- J. G. Turner and C. J. Murphy, Langmuir, 2021, 37, 9910–9919 CrossRef PubMed.
- D. Prozeller, S. Morsbach and K. Landfester, Nanoscale, 2019, 11, 19265–19273 RSC.
- R. A. Latour, Colloids Surf., B, 2020, 191, 110992 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04707e |
| This journal is © The Royal Society of Chemistry 2023 |