
Spin-dependent active centers in Fe–N–C oxygen reduction catalysts revealed by constant-potential density functional theory†
Tao
Zheng
 a,
Jincheng
Wang
a,
Zhenhai
Xia
b,
Guofeng
Wang
*c and
Zhiyao
Duan
*a
a,
Jincheng
Wang
a,
Zhenhai
Xia
b,
Guofeng
Wang
*c and
Zhiyao
Duan
*a
aState Key Laboratory of Solidification Processing, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an, Shaanxi Province 710072, P. R. China. E-mail: zhiyao.duan@nwpu.edu.cn
bSchool of Chemical Engineering, University of New South Wales, Sydney, NSW, 2052 Australia
cDepartment of Mechanical Engineering and Materials Science, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States. E-mail: guw8@pitt.edu
First published on 27th July 2023
Abstract
Iron and nitrogen co-doped carbon (Fe–N–C) catalysts have shown great promise in promoting the oxygen reduction reaction (ORR) in proton exchange membrane fuel cells. Experimental characterization studies, including Mössbauer and X-ray emission spectroscopy, have revealed the crucial role of spin states in Fe–N–C catalysts in ORR catalysis, but comprehensive theoretical understanding in this aspect is still lacking. Herein, using the grand-canonical density functional theory, we systematically investigate the interplay of the oxidation state, spin state, and applied potentials on the catalytic activity of an FeN4C10 moiety. We have identified two stable spin states of Fe(II)N4C10 at ORR-relevant potentials, namely, a high-spin state with out-of-plane Fe displacement and an in-plane intermediate-spin state. Our results show that the FeN4C10 moiety at the two different spin states exhibits distinct abilities to bind ORR intermediates and ORR activities. Our study provides valuable insights into the spin-correlated catalytic performances of Fe–N–C catalysts.
1 Introduction
The oxygen reduction reaction (ORR) is essential for clean and sustainable energy conversion technologies, such as proton exchange membrane fuel cells (PEMFCs). The sluggish ORR kinetics and high cost of platinum-based catalysts are hindering the commercialization of PEMFCs.1 Iron and nitrogen co-doped carbon materials (denoted as Fe–N–C) are by far the most promising Pt-free ORR electrocatalysts with activity approaching that of state-of-the-art Pt/C.2,3 It is of importance to understand the origin of high-performance Fe–N–C catalysts for their further improvement. Now, it is widely accepted that FeNxCy moieties embedded in carbon layer are responsible for the observed ORR activity. However, the exact chemical nature of active sites in Fe–N–C catalysts is still under debate after intensive research efforts over the past few years.4–8Experimental investigations have been conducted to elucidate the spin states, oxidation states, and local structures of different FeNxCy centers in Fe–N–C catalysts and to identify the intrinsic ORR activity and durability of different FeNxCy centers.9 Mössbauer spectroscopy has been used to identify different FeNxCy moieties in pyrolyzed Fe–N–C catalysts.10–19 Mössbauer parameters obtained at room temperature for various iron species in Fe–N–C catalysts have been summarized in recent publications.11,15 Two distinct quadrupole doublets (D1 and D2) associated with two different iron nitrogen sites have been identified ubiquitously. Doublet D1 typically has a quadrupole splitting energy (ΔEQS) between 0.9 and 1.25 mm s−1 and is assigned to a bare ferrous (Fe2+) low spin (S = 0) or ferric (Fe3+) high spin (S = 5/2) FeN4 moiety at which either O2 or OH is adsorbed.17 Doublet D2 having a ΔEQS of 2.0–2.8 mm s−1 is assigned to a bare ferrous intermediate spin (S = 1) or ferrous low spin FeN4 (S = 0) site.17
The respective activity and durability of D1 and D2 sites have been pursued in recent studies using operando57Fe Mössbauer spectroscopy.14,15 Li et al. demonstrated that the D1 site, assigned to a high spin ferric FeN4 moiety embedded in a pyrrolic nitrogen environment (FeN4C12), initially contributed to the ORR activity but was not durable in operating PEMFCs, quickly transforming into ferric oxide.14 In contrast, a ferrous low- or medium-spin D2 site embedded in a pyridinic nitrogen environment (FeN4C10) was found to be more durable, with no measurable decrease in the number of active sites after 50 h of operation. Ni et al.12,15 also assigned the D1 site to the same high-spin Fe(III)N4C12 with the same pyrrolic FeN4C12 structure. However, they claimed that the changes in D1 sites during the ORR are fully reversible and attributed the observed irreversibility in ref. 14 to the lack of acid leaching during Fe–N–C preparation. Nevertheless, the assignment of the D1 site to pyrrolic FeN4C12 has not yet reached consensus. Scanning transmission electron microscopy (STEM) only confirmed the presence of pyridinic FeN4C10 in Fe–N–C catalysts.20,21
X-ray absorption spectroscopy (XAS) is also widely employed to identify the oxidation states and structural fingerprints of FeNxCy moieties.13,22–26 Using in situ XAS, the D1 FeN4 moiety has been demonstrated to reversibly switch between an out-of-plane ferrous state and an in-plane ferric state when the applied potential crosses the Fe2+/3+ redox potential, which accounts for the weakened Fe–O binding energy, higher Fe2+/3+ redox potential, and superior ORR activity compared to that of the D2 sites.13 The Fe K-edge X-ray absorption near-edge spectra (XANES) were used to identify the potential-dependent redox of Fe ions in an O2-free environment.13,25 The potential-dependent shift of XANES starts at applied potentials as low as 0.2 V, indicating easy oxidation of Fe2+ to Fe3+.13,25 The square-wave voltammogram (SWV), on the other hand, only possesses a single redox peak at approximately 0.75 V.25 The discrepancy between XANES and SWV leads to proposals attributing the shift of XANES at low potentials to the potential-induced spin state transition (spin crossover).25 Recently, potential-dependent spin changes have been identified by in situ X-ray emission spectroscopy (XES).13 Nevertheless, since only 0.2 V and 0.9 V were examined in the XES study, it is still not clear whether the spin changes stem from spin crossover within the same oxidation state or from changes in the oxidation state.
First-principles calculations have been employed to facilitate the assignment of the active FeNxCy centers for the ORR.27 Various FeNxCy moieties have been proposed, including pyridinic FeN4C10,28,29 pyrrolic FeN4C12,30 FeN4C10 with an axial ligand,16,31–36 and FeN4 motif at the defected carbon matrix,37etc. Most theoretical studies focused on the local geometry and oxidation state of FeNxCy moieties; however, few theoretical studies have systematically explored the complete interplay of spin states, oxidation states, and applied potentials in determining the catalytic properties of Fe–N–C catalysts. Recent experimental38–41 and theoretical42–46 studies have evidently demonstrated that spin states are essential for understanding and tuning the activity of Fe–N–C catalysts. More importantly, the theoretical exploration of the spin–valency–potential interactions is required to carry out direct comparisons with experimental characterization since all these interactions are involved in real Fe–N–C catalysts under operando conditions. The insights gained by in-depth theory-experiment collaboration could potentially lead to a reconciled picture of the fundamental understanding of the active FeNxCy moiety for the ORR and thus facilitate the rational design of Fe–N–C catalysts with enhanced activity and durability.
To this end, we employed the grand-canonical density functional theory (GC-DFT) to systematically investigate the potential-dependent spin and oxidation states of the FeN4C10 moiety. It is found that FeN4C10 is bistable under ORR-relevant conditions. The bi-stability consists of a high-spin state with an out-of-plane Fe atom and an intermediate-spin state with an in-plane Fe atom. Intriguingly, the two spin states exhibit dramatically dissimilar behavior for ORR intermediate adsorption, and hence very different ORR catalytic activity. Our work provides new insights into the spin-dependent catalytic performance of Fe–N–C catalysts.
2 Computational methods
Spin-polarized DFT calculations were performed by employing the Vienna ab initio simulation package (VASP).47,48 Electronic exchange and correlation were described using the Perdew–Burke–Ernzerhof (PBE) functional.49 Core electrons were treated within the projector augmented wave (PAW) framework.50 The energy cutoff for the plane-wave basis was set to 400 eV in all the calculations. The atomic structures were fully relaxed until the magnitude of residual force on each atom was less than 0.05 eV Å−1. The DFT + U method was employed to alleviate the self-interaction error in the 3d orbital of Fe ions.51 In this work, the value of Ueff = U – J for the 3d orbital of Fe was set to 3.29 eV.52A slab model consisting of three graphene layers in a 4 × 4 supercell was employed to simulate the surface of the graphite substrate. An FeN4C10 center is embedded in the topmost graphene layer. A vacuum layer of at least 25 Å in the z direction was used to eliminate the interaction between the periodic images. The employed atomic model can be viewed in Fig. S1.† The Brillouin zone integration in k space was performed on a 6 × 6 × 1 gamma-centered k-point mesh.
Constant-potential calculations were performed by optimizing the coordinates and the number of electrons of the system simultaneously according to the target applied potential on the standard hydrogen electrode (SHE) scale, i.e., USHE. The methodology for the constant-potential calculations has been described in detail in our previous publication.42,53,54 The constant-potential calculations utilized implicit solvent at the level of the linear polarizable continuum model (PCM) for electrolyte simulation as implemented in the VASPsol code.55,56 The relative permittivity of the solvent was set to 80 to model the aqueous electrolyte. The surface tension parameter was set to zero to neglect the cavitation energy contribution. For charged systems, a counter-ion charge with generalized Poisson–Boltzmann distribution is included in the implicit solvent region. We used a Debye length of 3 Å to mimic the experimental conditions where 1 M of a strong acid is used. The calculated potential-dependent energies were used to construct a free energy diagram of the electrochemical ORR using the computational hydrogen electrode method assuming the four-step associative mechanism.57
The occupation matrix control plugin for the VASP was employed to generate initial structures according to different metastable spin states.58 This was achieved by constraining the FeN4 spin state by setting the occupation matrix of d-electron localization and then relaxing the geometry. The constraint on the spin state was subsequently removed, and the system was allowed to relax toward the targeted metastable state.
3 Results and discussion
3.1 Validation of the DFT + U method
To examine the predictive performance of DFT + U (Ueff = 3.29 eV for the 3d orbitals of Fe) for spin-related properties, we calculated the stability order of different spin states for intact and oxidized Fe phthalocyanine (FePc) and single-layer FeN4C10 using PBE, PBE + U, and various hybrid functionals (HSE06, B3LYP, and PBE0). An FePc molecule and single-layer FeN4C10 are chosen for the validation to reduce the formidable computational overheads of hybrid functional calculations. And the k-point mesh is reduced to 2 × 2 × 1 for single-layer FeN4C10. The obtained spin state orders are shown in Fig. 1a and b, respectively for FePc and single-layer FeN4C10. Hybrid functionals generally predict that ELS > EHS > EIS for FePc and single-layer FeN4C10, where LS, HS, and IS stand for low-spin, high-spin, and intermediate-spin, respectively. DFT + U calculations predict the same stability order. The DFT + U calculated ELS − EHS is smaller than those obtained using HSE06 and PBE0, but very close to the result of the B3LYP functional. For *OH/FePc and *OH/FeN4C10, hybrid functionals predict a small positive value for EIS − EHS, which can also be reproduced by DFT + U calculations, whereas the plain PBE erroneously predicts EIS to be more stable for *OH/FePc. Hence, DFT + U with Ueff = 3.29 eV for the 3d orbitals of Fe generally reproduces the results of hybrid functionals for the stability order of spin states.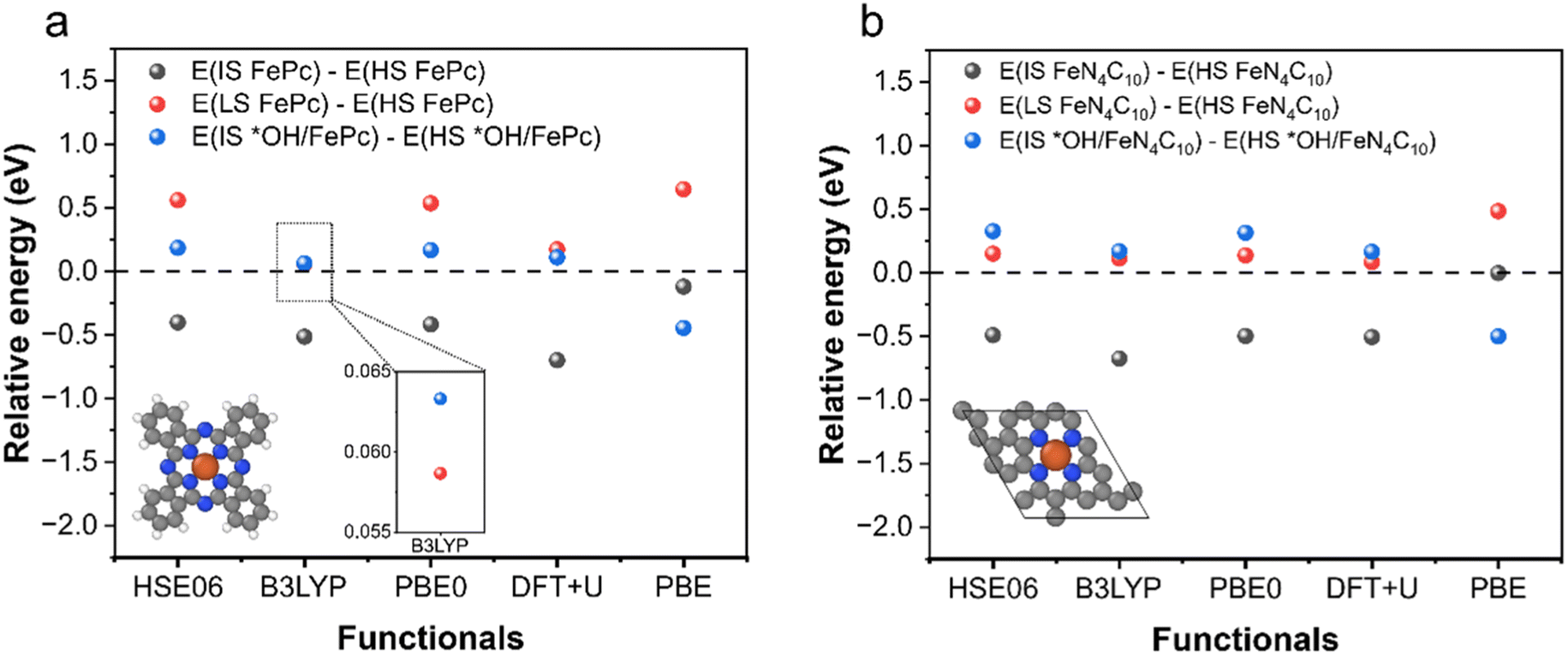 | ||
| Fig. 1 Relative energy of different spin states of intact and oxidized FePc and single-layer FeN4C10 calculated using different functionals. (a) FePc and (b) single-layer FeN4C10. | ||
3.2 FeN4C10
We first performed an extensive search for the metastable spin states of the FeN4C10 moiety. The ferrous Fe ion in tetra-coordinated Fe(II)N4C10 has a formal d6 electron configuration. The six electrons can adopt different occupation configurations over the five d-orbitals (dxy, dxz, dyz, dz2, and dx2−y2) leading to different spin states. The interaction of Fe d-orbitals with the orbitals in pyridinic N (Np) in a square-planar ligand field is schematically plotted in Fig. 2a. The dxy orbital is a non-bonding orbital. The dxz and dyz orbitals are anti-bonding dπ–pπ orbitals due to the interaction between the Fe dxz/dyz orbital and the pz orbitals in Np. The dz2 orbital is also a non-bonding orbital due to the absence of axial ligand. The dx2−y2 orbital is a σ anti-bonding orbital resulting from the hybridization between the dx2−y2 orbital and the sp2 hybridized orbitals in the Np ligands. We exhausted all possible spin states by using the occupation matrix control method to obtain the desired spin states. In total, we have searched 45 different spin states. Seven spin states are found to be local minima, including three high spin (HS) states, three intermediate spin (IS) states, and one low spin (LS) state. The atomic structures and projected density of states (pDOS) plots of bare FeN4C10 in various spin states are shown in Fig. 2b. The spin density plots of various spin states are shown in Fig. S2.† Detailed information about the obtained spin states, including their spin multiplicity, relative stability, structural information, Fe Bader charges and potential of zero charges (PZCs) is listed in Table 1. The detailed Fe 3d orbital occupations of different spin states are summarized in Table S1.†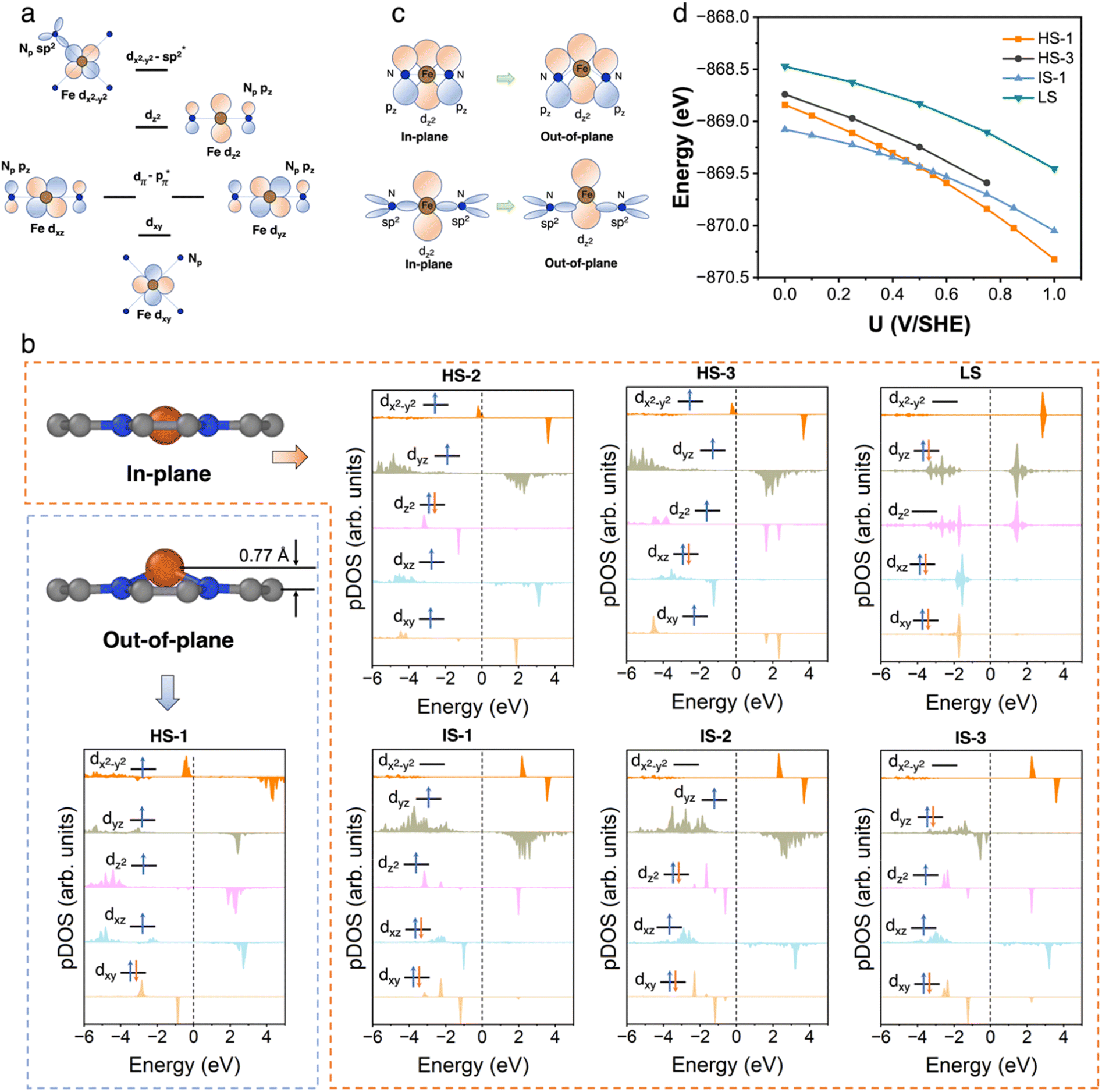 | ||
| Fig. 2 FeN4C10. (a) Schematics of the interactions of Fe d orbitals with the orbitals in pyridinic N (Np) in a square-planar ligand field. (b) Atomic structures and pDOS plots of seven stable spin states of bare FeN4C10. (c) Schematic plots of additional bonding interactions between the Fe dz2 orbital and N pz or sp2 orbitals when Fe transforms from an in-plane structure to an out-of-plane structure in FeN4C10. (d) Potential-dependent energies of FeN4C10 with different spin states under constant-potential conditions. | ||
| Fe spin state | Spin multiplicity | Relative energy (eV) | Fe–N bond length (Å) | Fe–O bond length (Å) | Out of plane displacement of Fe (Å) | Bader charge (e) | PZC (V/SHE) |
|---|---|---|---|---|---|---|---|
| Bare FeN 4 C 10 | |||||||
| HS-1 | 2 | 0 | 2.04 | — | 0.77 | +1.33 | −1.02 |
| HS-2 | 2 | −0.07 | 1.96 | — | 0 | +1.23 | −0.63 |
| HS-3 | 2 | −0.13 | 1.96 | — | 0 | +1.31 | −0.73 |
| IS-1 | 1 | −0.57 | 1.92 | — | 0 | +1.09 | −0.61 |
| IS-2 | 1 | −0.44 | 1.92 | — | 0 | +0.98 | −0.54 |
| IS-3 | 1 | −0.35 | 1.93 | — | 0 | +1.06 | −0.60 |
| LS | 0 | 0.02 | 1.91 | — | 0 | +1.06 | −0.60 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| *OOH/FeN 4 C 10 | |||||||
| HS | 5/2 | 0 | 2.04 | 1.89 | 0.74 | +1.53 | −0.49 |
| IS | 3/2 | 0 | 1.96 | 1.93 | 0.46 | +1.33 | −0.40 |
| LS | 1/2 | +0.02 | 1.94 | 1.85 | 0.34 | +1.18 | −0.39 |
|
|||||||
| *O/FeN 4 C 10 | |||||||
| HS | 2 | 0 | 2.07 | 1.71 | 0.86 | +1.39 | −0.01 |
| IS | 1 | +0.11 | 1.95 | 1.67 | 0.42 | +1.35 | −0.24 |
| LS | 0 | +0.55 | 195 | 1.66 | 0.47 | +1.22 | −0.29 |
|
|||||||
| *OH/FeN 4 C 10 | |||||||
| HS | 5/2 | 0 | 2.06 | 1.86 | 0.81 | +1.52 | −0.50 |
| IS | 3/2 | +0.14 | 1.97 | 1.90 | 0.50 | +1.36 | −0.40 |
| LS | 1/2 | +0.48 | 1.93 | 1.88 | 0.35 | +1.19 | −0.35 |
|
|||||||
| *O 2 /FeN 4 C 10 | |||||||
| HS | 5/2 | 0 | 2.09 | 1.97 | 0.89 | +1.52 | −0.09 |
| IS (end-on) | 3/2 | −0.43 | 1.94 | 2.12 | 0.31 | +1.27 | −0.65 |
| IS (side-on) | 3/2 | −0.25 | 1.96 | 2.18 | 0.49 | +1.32 | −0.32 |
The ground spin state of bare FeN4C10 is recognized as an IS triplet state (S = 1) named IS-1, in which the Fe ion has a formal (dxy)2(dxz)2(dyz)1(dz2)1(dx2−y2)0 d-electron configuration. The other two triplet states, named IS-2 and IS-3 (refer to Fig. 2b for their formal d-electron configurations), are 0.13 and 0.22 eV higher in energy, respectively, compared to the IS-1 state. The three HS quintet states (S = 2) of bare FeN4C10 are energetically less stable than IS states. The Fe ion in HS-1 features a d-orbital occupation of (dxy)1(dxz)2(dyz)1(dz2)1(dx2−y2)1 and is 0.57 eV higher in energy than that in the IS-1 ground state. The HS-2 and HS-3 states are slightly lower in energy than the HS-1 state by −0.07 and −0.13 eV, respectively. The Fe ion in the singlet LS state (S = 0), (dxy)2 (dxz)2(dyz)2(dz2)0(dx2−y2)0, is 0.59 eV higher in energy than that in the ground state and is the highest energy state among all spin states.
All the obtained spin states have a high-symmetry planar-square FeN4 structure with the Fe ion staying in the graphene plane with varying Fe–N bond lengths, except for the HS-1 state. The Fe ion in the HS-1 state has an out-of-plane displacement of 0.77 Å as shown in Fig. 2b, which is quite unexpected since the out-of-plane displacement normally results from axial ligand attachment. Inspecting the orbital-wise occupations of different spin states shown in Table S1,† we attribute the out-of-plane displacement to the simultaneous occupation of the in-plane dxy (doubly occupation) and dx2−y2 orbitals in the HS-1 state. The high-energy dx2−y2 states are dx2−y2-sp2 antibonding states lying above the Fermi level at 2–4 eV, as schematically displayed in Fig. 2a. In all HS states, the anti-bonding dx2−y2 states are partially occupied resulting in dx2−y2 states just below the Fermi level, as seen in the pDOS plots of HS states shown in Fig. 2b. The occupation of anti-bonding dx2−y2 states results in elongation and weakening of Fe–N bonds in HS states. Nevertheless, the occupation of anti-bonding dx2−y2 states does not necessarily lead to out-of-plane movement as in the cases of HS-2 and HS-3 states. Specifically in the HS-1 state, the other in-plane dxy orbital is doubly occupied simultaneously with the dx2−y2 orbital. The double occupation of the dxy orbital causes strong in-plane on-site Coulomb repulsion between dxy and dx2−y2. Because of this destabilizing effect, the Fe ion moves out of the plane to stabilize itself by forming new bonding interactions. The out-of-plane displacement results in non-zero dz2-pz and dz2-sp2 overlaps that are not present in the in-plane structure, thus producing a covalence contribution to the bonding, as schematically illustrated in Fig. 2c. Above all, the out-of-plane displacement of the Fe atom in the HS-1 state could be categorized as a pseudo Jahn-Teller effect (PJTE),59 in which the occupation of dx2−y2 and the double occupation of dxy destabilize the HS-1 state of the in-plane structure. In turn, the out-of-plane displacement stabilizes the HS-1 state by forming dz2-pz and dz2-sp2 bonding interactions.
The Bader charge analysis in Table 1 shows that the charge states of Fe ions in the three HS states (∼+1.3 e) tend to be more positive, i.e., more “oxidized” than the Fe ions in the IS and LS states (∼+1.1 e). The more electron-rich Fe ions in IS and LS states originate from the stronger hybridization between the dx2−y2 orbital and the sp2 hybridized orbital in Np ligands, which can be manifested by the significant occupation of the bonding part of the dx2−y2 orbitals in IS and LS states as shown in Table S1.† In HS states, the hybridization is weaker due to the occupation of the anti-bonding dx2−y2 states resulting in Fe–N bond elongation, so the occupation of the dx2−y2 orbital due to the hybridization is reduced. In the HS-1 state, the out-of-plane displacement further reduces the electron population in the dx2−y2 bonding states as evidenced by its lowest minority spin population in dx2−y2 as shown in Table S1.† The total electron population of Fe 3d orbitals in all three HS states is about 6.2 e by summing the population in each dm orbital. The electron population of Fe 3d orbitals in IS and LS states is slightly more electron-enriched by 0.2 e due to stronger Fe dx2−y2–Np sp2 hybridization, which is also consistent with the Bader analysis.
We further carried out constant-potential calculations to examine the stability of all seven spin states of bare FeN4C10 at ORR-relevant voltage. The calculated potential-dependent energies of bare FeN4C10 with different spin states are shown in Fig. 2d. We noticed that the HS-2, IS-2, and IS-3 states spontaneously transformed into other states (HS-2 to HS-3; IS-2 and IS-3 to IS-1) under constant-potential conditions, so these electrochemically unstable states are not presented in Fig. 2d. The calculation results show that the IS-1 and HS-1 states are the most stable states depending on the range of the applied potential. The IS-1 state is the most thermodynamically stable at U < 0.5 V vs. SHE, while the HS-1 state becomes the ground state at U > 0.5 V. Electrostatic spin crossover occurs because the potential-dependent energy of the HS-1 state drops more quickly than that of the IS-1 state as the applied potential increases. Assuming a simple parallel plate capacitor model for the electric double layer (EDL), the potential-dependent energy can be expressed as 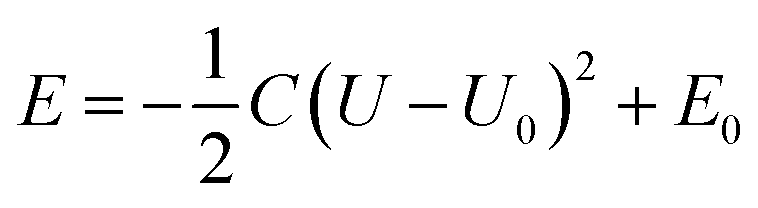 , where C is the capacitance of the EDL, U is the applied potential, U0 is the potential of zero charge (PZC), and E0 is the energy corresponding to the PZC. Provided that the HS-1 state has a more negative PZC of −1.02 V (see Table 1) compared to that of the IS-1 state (−0.61 V), the HS-1 state experiences a higher potential bias, i.e., (U − U0), in the interested potential range and thus has a faster potential-dependent energy decrease. Consequently, the initial 0.57 eV energy gap between the IS-1 and HS-1 states at their zero-charge states is closed at U = 0.5 V vs. SHE. The more negative PZC of the HS-1 state could be traced to the special out-of-plane displacement of the Fe ion. The out-of-plane displacement of the Fe ion causes separation of the positive charge center (Fe ion) and the negative charge center (N atoms), resulting in a surface electric dipole pointing in the +z direction, which leads to a work function decrease and hence lower PZC. Despite the spin crossover, the relative stability of the HS-1 and IS-1 states is within an absolute energy difference of 0.2 eV in the potential range of 0 to 1 V. Hence, the two spin states could coexist at ORR-relevant voltages.
, where C is the capacitance of the EDL, U is the applied potential, U0 is the potential of zero charge (PZC), and E0 is the energy corresponding to the PZC. Provided that the HS-1 state has a more negative PZC of −1.02 V (see Table 1) compared to that of the IS-1 state (−0.61 V), the HS-1 state experiences a higher potential bias, i.e., (U − U0), in the interested potential range and thus has a faster potential-dependent energy decrease. Consequently, the initial 0.57 eV energy gap between the IS-1 and HS-1 states at their zero-charge states is closed at U = 0.5 V vs. SHE. The more negative PZC of the HS-1 state could be traced to the special out-of-plane displacement of the Fe ion. The out-of-plane displacement of the Fe ion causes separation of the positive charge center (Fe ion) and the negative charge center (N atoms), resulting in a surface electric dipole pointing in the +z direction, which leads to a work function decrease and hence lower PZC. Despite the spin crossover, the relative stability of the HS-1 and IS-1 states is within an absolute energy difference of 0.2 eV in the potential range of 0 to 1 V. Hence, the two spin states could coexist at ORR-relevant voltages.
3.3 *OOH/FeN4C10
We further explored different spin states of the OOH-adsorbed FeN4C10 (*OOH/FeN4C10) moiety. Three different spin states, i.e., HS, IS, and LS, are found to be stable for *OOH/FeN4C10. The atomic structures and pDOS plots of the three spin states can be visualized in Fig. 3b–d. The Fe ion in *OOH/FeN4C10 has a d5 electron configuration since one electron transfers from Fe to the adsorbed OOH. The HS state is a sextet state (S = 5/2) with a 3d occupation of (dxy)1(dxz)1(dyz)1(dz2)1(dx2−y2)1 on the Fe ion. The IS state of the Fe ion has a 3d electron occupation of (dxz)1(dyz)1(dz2)1(dxy)2(dx2−y2)0 with a total spin of S = 3/2. The LS state, (dxz)2(dyz)1(dz2)0(dxy)2(dx2−y2)0, has a total spin of S = 1/2. The spin density plots of various spin states are shown in Fig. S3.† For precise d-orbital occupation, please refer to Table S1.† In all the different spin states, the OOH binds on the Fe ion with an end-on configuration, which leads to a strong σ bonding due to the overlap between dz2 and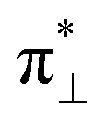 (
(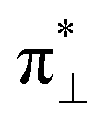 denoting an OOH π* orbital oriented perpendicular to the FeN4C10 plane) as shown in Fig. 3a. The dz2 states in Fig. 3b–d manifest this
denoting an OOH π* orbital oriented perpendicular to the FeN4C10 plane) as shown in Fig. 3a. The dz2 states in Fig. 3b–d manifest this 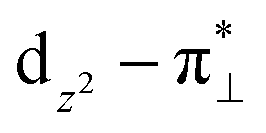 interaction since the dz2 states are further split compared to those in bare FeN4C10. Accordingly, the electron occupation of dz2 increases significantly in the minority spin channel as revealed in Table S1.† There also exists a weaker π bond between dxz and
interaction since the dz2 states are further split compared to those in bare FeN4C10. Accordingly, the electron occupation of dz2 increases significantly in the minority spin channel as revealed in Table S1.† There also exists a weaker π bond between dxz and 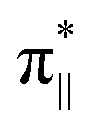 (
(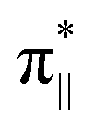 denoting an OOH π* orbital oriented parallel to the FeN4C10 plane) as shown in Fig. 3a. The other two dxy and dx2−y2 orbitals are non-bonding with OOH orbitals. The energies of different spin states of *OOH/FeN4C10 are nearly energetically degenerate. The Bader charge analysis reveals that the Fe ion further loses electrons and becomes Fe(III) upon *OOH adsorption compared to bare FeN4C10 with respective spin states.
denoting an OOH π* orbital oriented parallel to the FeN4C10 plane) as shown in Fig. 3a. The other two dxy and dx2−y2 orbitals are non-bonding with OOH orbitals. The energies of different spin states of *OOH/FeN4C10 are nearly energetically degenerate. The Bader charge analysis reveals that the Fe ion further loses electrons and becomes Fe(III) upon *OOH adsorption compared to bare FeN4C10 with respective spin states.
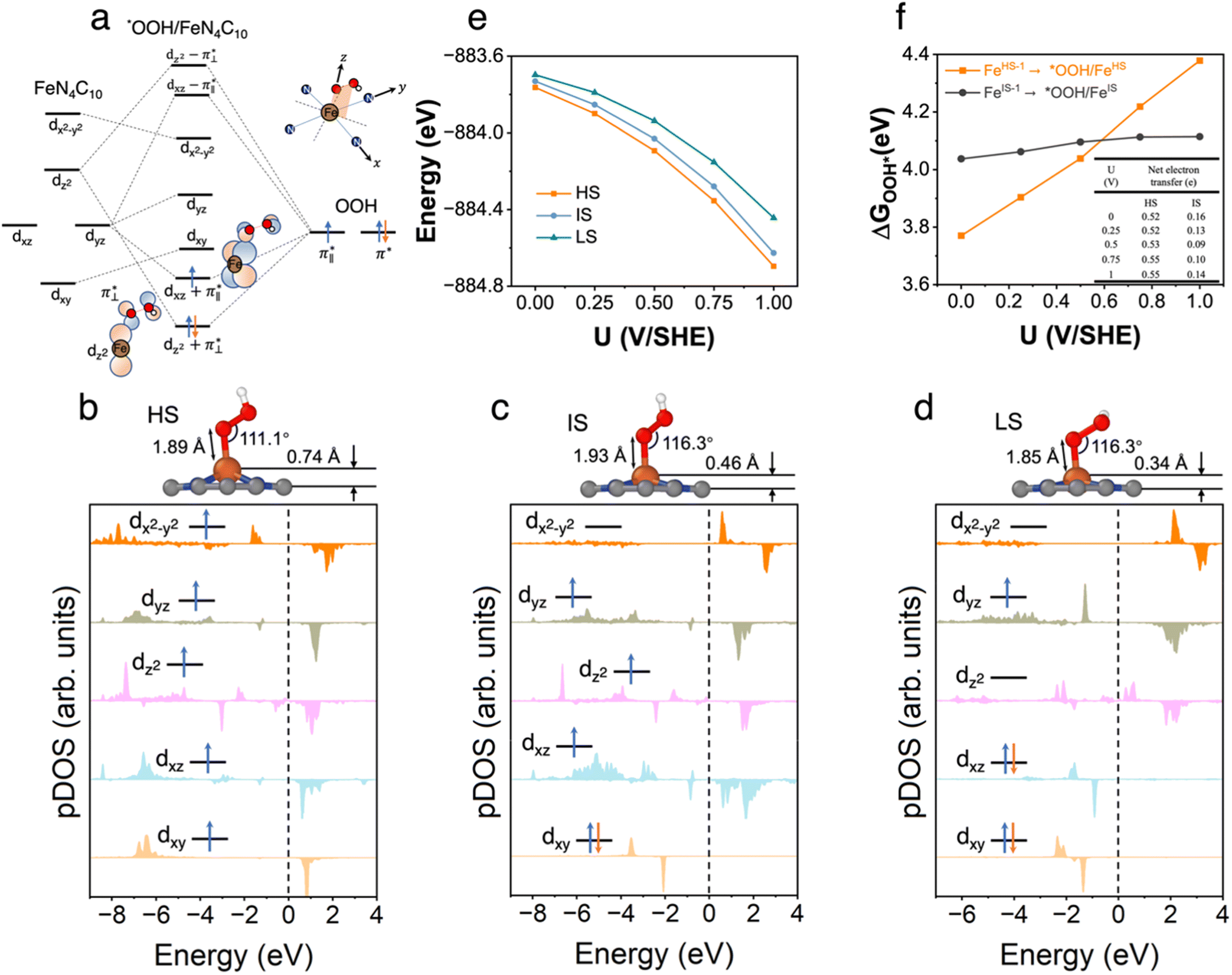 | ||
| Fig. 3 *OOH/FeN4C10. (a) Schematics of orbital hybridization of Fe 3d orbitals in a square-planar ligand field with the molecular orbitals in the OOH adsorbate. (b)–(d) Atomic structures and pDOS plots of the HS, IS, and LS spin states of *OOH/FeN4C10. (e) Spin- and potential-dependent energies of *OOH/FeN4C10 under constant-potential conditions. (f) Spin and potential-dependent *OOH adsorption energies. The inset table shows the net electron transfer in the adsorption process. | ||
The potential-dependent energies of the three spin states of *OOH/FeN4C10 are shown in Fig. 3e. The HS state of *OOH/FeN4C10 has the lowest energies among the three spin states in the interested potential range 0–1 V vs. SHE. The IS state is slightly destabilized by less than 0.1 eV compared to the HS state within the potential range. The LS state is approximately 0.1–0.25 eV higher than the energy of the ground HS state depending on the applied potential. The spin- and potential-dependent *OOH adsorption energies are calculated and shown in Fig. 3f. Due to the bistability of bare FeN4C10 (HS-1 and IS-1 states), we considered two different *OOH adsorption energies corresponding to two spin-dependent adsorption processes, namely, FeIS-1N4C10 ⇒ *OOH/FeISN4C10 and FeHS-1N4C10 ⇒ *OOH/FeHSN4C10. Adsorption of *OOH on FeLSN4C10 is not considered since the LS state is unstable compared to the IS-1 and HS-1 states. Adsorption of *OOH on FeLSN4C10 is not considered since the LS state is highly unstable compared to the IS-1 and HS-1 states.
The potential-dependent *OOH adsorption energies exhibit spin-dependent behavior, as shown in Fig. 3e. The FeIS-1N4C10 ⇒ *OOH/FeISN4C10 adsorption, abbreviated as *OOH/FeIS adsorption hereafter, results in an *OOH adsorption energy of about 4.1 eV with a weak dependence on the voltage. In contrast, the FeHS-1N4C10 ⇒ *OOH/FeHSN4C10 adsorption process (*OOH/FeHS adsorption) has a strong potential dependence. The *OOH/FeHS adsorption energy increases from 3.8 eV at U = 0 V to 4.4 eV at U = 1.0 V. The different potential-dependent *OOH adsorption energy trends of *OOH/FeISvs. *OOH/FeHS adsorption rely on different surface charging behaviors upon *OOH adsorption. In *OOH/FeHS adsorption, the PZC of the system shifts from −1.02 V to −0.49 V without and with *OOH adsorption, which would lead to significant surface electron charging. Our constant-potential calculations show that the system obtains an additional ∼0.5 e negative charge upon *OOH/FeHS adsorption as shown in the table inside Fig. 3f. The *OOH/FeIS adsorption process, on the other hand, results in a smaller surface charge of approximately −0.1 e, leading to a much milder potential dependence due to the smaller PZC shift upon *OOH adsorption.
3.4 *O/FeN4C10
The spin-dependent atomic structures and pDOS of *O/FeN4C10 are shown in Fig. 4b–d. The optimized HS, IS, and LS states of *O/FeN4C10 are quintet (S = 2), triplet (S = 1), and singlet (S = 0), respectively. The spin density plots of various spin states are shown in Fig. S4.† The detailed Fe 3d orbital occupations of different spin states are presented in Table S1.† It could be expected that strong σ bonding formed due to the overlap between Fe dz2 and O 2pz and π bonding formed through Fe dxz–O 2px and Fe dyz–O 2py interactions. These orbital interactions are depicted schematically in Fig. 4a. The significant occupations in the dxz, dyz, and dz2 orbitals presented in Table S1† support the existence of Fe 3d–O 2p hybridizations. The pDOS plots in Fig. 4b–d also demonstrate the splitting of the three interacting dxz, dyz, and dz2 orbitals. In the HS and IS states, the adsorbed *O possesses a singly occupied electron in the O 2p orbital (see Fig. S2† for spin density plots). Hence, the adsorbed *O atom in the HS and IS states is in the form of an oxyl radical (O˙−). Moreover, the Bader analysis shows that the Fe ion in HS *O/FeN4C10 has a Bader charge of 1.39 e, which is less than 1.53 e in HS *OOH/FeN4C10 and close to 1.33 e in HS bare FeN4C10. Hence, we tend to assign the HS to a ferrous Fe(II)⋯O˙− species, which indicates that the electron transfer occurs between the catalyst and *O rather than transferring from the Fe center into the O adduct. In contrast, the Bader charge of the Fe ion of the IS state is 1.35 e, which is close to that in IS *OOH/FeN4C10 (1.33 e) indicating one electron transfer from Fe to the adsorbed *O, resulting in a ferric Fe(III)–O˙− species. The LS state is spin unpolarized suggesting the formation of Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxo species.
O oxo species.
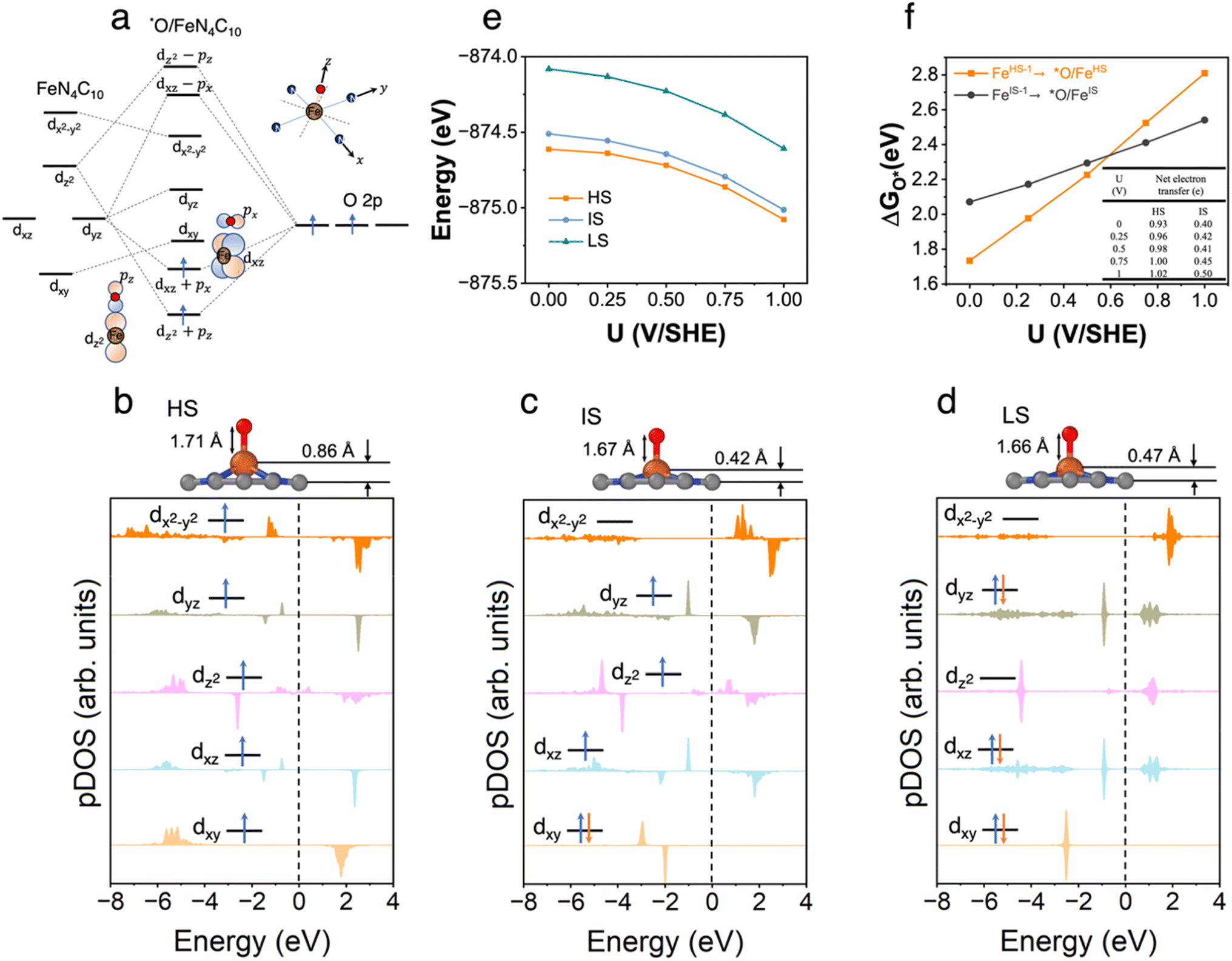 | ||
| Fig. 4 *O/FeN4C10. (a) Schematics of orbital hybridizations of Fe 3d orbitals in a square-planar ligand field with the 2p orbitals in the O adsorbate. (b)–(d) Atomic structures and pDOS plots of the HS, IS, and LS states of *O/FeN4C10. (e) Spin- and potential-dependent energies of *O/FeN4C10 under constant-potential conditions. (f) Spin and potential-dependent *O adsorption energies. The inset table shows the net electron transfer in the adsorption process. | ||
The potential-dependent energies of HS, IS, and LS *O/FeN4C10 are shown in Fig. 4e. The HS state is the energetically favorable state in the potential range of interest. The energy of the IS state is only slightly higher by ∼0.1 eV. The LS is significantly higher (by ∼0.5 eV) in energy than the HS and IS states. Fig. 4f describes the potential-dependent adsorption energies for the FeHS–1N4C10 ⇒ *O/FeHSN4C10 and FeIS–1N4C10 ⇒ *O/FeISN4C10 adsorption processes, which are referred to as *O/FeHS and *O/FeIS adsorption, respectively. The LS state is found to be unstable. Both *O/FeHS and *O/FeIS adsorption energies increase with increasing applied potential. The slope of the potential-dependent *O/FeHS adsorption energy is higher than that of the *O/FeIS adsorption energy due to stronger electron transfer, as shown in the inset table in Fig. 4f. The electron transfer upon *O/FeIS adsorption is composed of two parts: one is the above-discussed electron transfer from the catalyst to the adsorbed O˙−; the other is the surface charging due to the diminished surface dipole resulting from the anionic adduct. The sum of the two electron transfer processes results in a dramatic change in the PZC before and after *O/FeHS adsorption (−1.02 vs. −0.01 V). In *O/FeIS adsorption, the adsorbed O˙− obtains an electron from the Fe ion, so the PZC changes moderately from −0.61 V to −0.24 V, thus resulting in a moderate potential dependence of *O/FeIS adsorption.
3.5 *OH/FeN4C10
The pz and px non-bonding orbitals in the OH adsorbate could interact with Fe dz and dxz orbitals respectively as shown in Fig. 5a. The atomic structures and pDOS plots of *OH/FeN4C10 with different spin states are shown in Fig. 5b–d. The Fe ion has the d5 electron configuration in *OH/FeN4C10. The HS, IS, and LS states of *OH/FeN4C10 have similar electron configurations to those of *OOH/FeN4C10. The Fe ion in the HS state has an electron configuration of (dxz)1(dyz)1(dz2)1(dxy)1(dx2−y2)1 with a total spin of S = 5/2. The electron occupation of the Fe ion in the IS state is (dxz)1(dyz)1(dz2)1(dxy)2(dx2−y2)0 with a total spin of S = 3/2. The LS state, (dxz)2(dyz)1(dz2)0(dxy)2(dx2−y2)0, has a total spin of S = 1/2. The spin density plots of various spin states are shown in Fig. S5.† The potential-dependent energies of the three spin states of *OH/FeN4C10 are shown in Fig. 5e. The HS state of *OH/FeN4C10 has the lowest energy among the three spin states in the interested potential range 0–1 V vs. SHE. The IS state is slightly destabilized by about 0.2 eV compared to the HS state. The LS has the highest energy in the potential range, which is about 0.6 eV higher than that of the HS state. The potential-dependent adsorption energies of *OH/FeHS and *OH/FeIS are shown in Fig. 5f. Similar to the *OOH adsorption, the *OH/FeHS adsorption energy exhibits a stronger potential-dependence than the *OH/FeIS adsorption energy due to a larger PZC shift upon adsorption.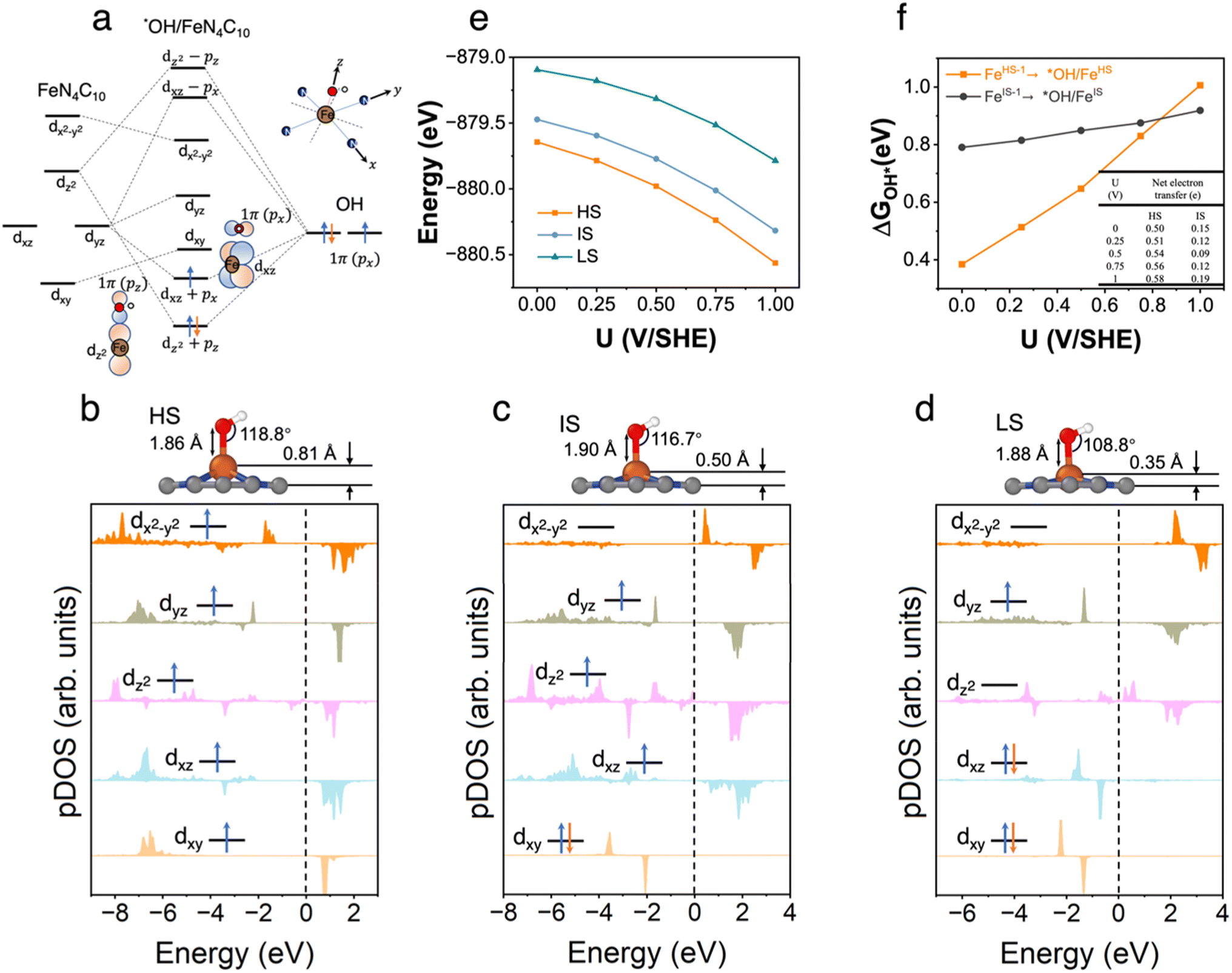 | ||
| Fig. 5 *OH/FeN4C10. (a) Schematics of orbital hybridizations of Fe 3d orbitals in a square-planar ligand field with the 2p orbitals in the OH adsorbate. (b)–(d) Atomic structures and pDOS plots of the HS, IS, and LS states of *OH/FeN4C10. (e) Spin- and potential-dependent energies of *OH/FeN4C10 under constant-potential conditions. (f) Spin and potential-dependent *OH adsorption energies. The inset table shows the net electron transfer in the adsorption process. | ||
3.6 Spin-dependent ORR activity
Our calculation results have shown that the spin states of the FeN4C10 moiety have significant influences on the adsorption behavior of ORR intermediates. This would lead to the spin-dependent ORR activity on FeN4C10. For bare and adsorbed FeN4C10, the HS and IS versions of each intermediate state have nearly degenerate energies in the interested potential range. Hence, we propose two spin-dependent ORR pathways, whose reaction cycles are depicted in Fig. 6a and b, respectively. In the ORR cycle starting from the HS-1 state (called the HS pathway hereafter), the spin states of the ORR intermediates, i.e., *OOH/FeN4C10, *O/FeN4C10, and *OH/FeN4C10, stay in their corresponding HS states along the reaction coordinates. Specifically, the spin multiplicity of the system evolves from S = 2 (FeN4C10) to S = 5/2 (OOH*/FeN4C10), then to S = 2 (O*/FeN4C10) and to S = 2 (OH*/FeN4C10), and finally returns to S = 2 (FeN4C10). We note that the initial spin multiplicity S = 2 only accounts for the spin multiplicity of the Fe center without considering the triplet O2 molecule. The highest possible spin for the combined bare FeN4C10 (HS) + O2 system is S = 3 (ferromagnetic) and the lowest spin is S = 1 (antiferromagnetic). Hence, the ORR reaction coordinate on HS-1 FeN4C10 is composed of several spin transitions (or spin crossovers). Thus, the ORR on HS-1 FeN4C10 is intrinsically a spin-forbidden reaction. Based on the potential-dependent adsorption energies of ORR intermediates in HS states, the free energy diagrams are constructed at various applied potentials as shown in Fig. 6c. The predicted ORR onset potential of this HS pathway is 0.77 V. The potential limiting step is *OOH formation due to the weak *OOH binding strength on HS-1 FeN4C10 at relatively high applied potential.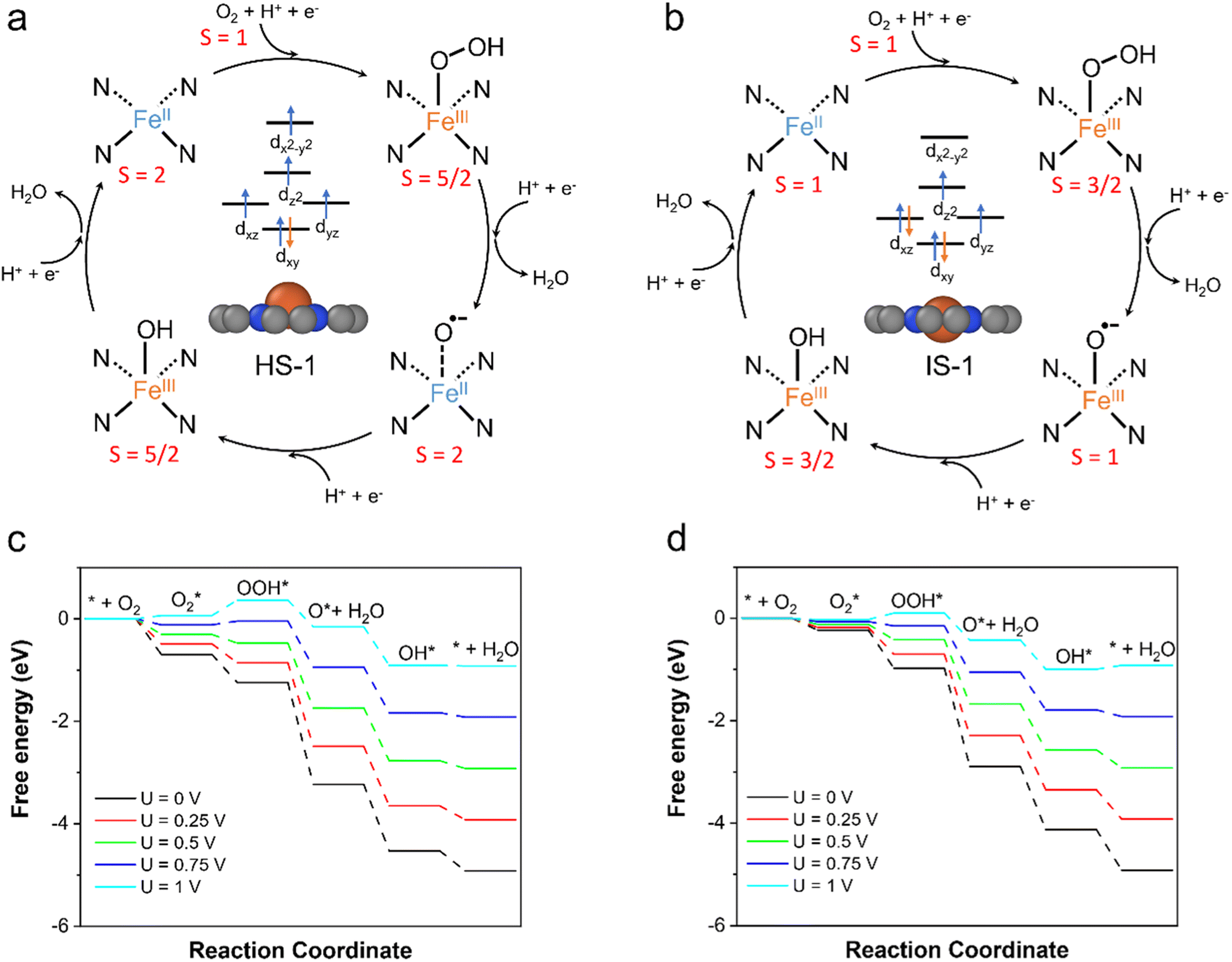 | ||
| Fig. 6 Proposed spin-dependent ORR mechanism on FeN4C10. (a) High-spin ORR cycle. (b) Intermediate-spin ORR cycle. (c) High-spin ORR free energy diagram. (d) Intermediate-spin ORR free energy diagram. | ||
The ORR following the IS pathway will start with IS-1 FeN4C10 as shown in Fig. 6b. The spin multiplicity of the system evolves from S = 2 or 0 (bare FeN4C10 + O2) to S = 3/2 (OOH*/FeN4C10), to S = 1 (OH*/FeN4C10), and to S = 3/2 (OH*/FeN4C10). Consequently, the IS pathway of the ORR is also a spin-forbidden reaction. The constructed free energy diagrams of the ORR via the IS pathway at various applied potentials are shown in Fig. 6d. The predicted onset potential for the ORR along the IS pathway is 0.89 V, which is significantly higher than that of the HS pathway. The potential limiting step is the reduction of *OH to H2O, while the *OOH formation step also has very similar reaction free energy. The IS-1 FeN4C10 exhibits better ORR activity than that of the HS-1 state mainly because of the stronger *OOH adsorption at high applied potentials.
3.7 Adsorption of O2 on FeN4C10
O2 adsorption is an important process for the ORR, but it is somewhat overlooked in studies because O2 adsorption is usually regarded as a non-electrochemical process with no net electron transfer and hence the energetics of this step are not considered for limiting-potential assignment. However, in recent studies, electron transfer and spin selection occurred during O2 activation on magnetic single-atom catalyst sites.60,61 These studies inspire us to further examine the spin- and potential-dependent adsorption of O2.We explore the spin-dependent atomic structures of *O2/FeN4C10 in HS and IS states as shown in Fig. 7a–c. The detailed structural and electronic information about *O2/FeN4C10 is included in Table 1. The spin density plots are shown in Fig. S6.† The detailed Fe 3d orbital occupations of different spin states are presented in Table S1.† In the HS *O2/FeN4C10, the adsorbed O2 molecule adopts a side-on configuration with the O–O bond length elongating to 1.42 Å, which indicates the formation of a peroxide (O22−) species. In the IS *O2/FeN4C10, the adsorbed O2 can adopt two different structures, namely side-on and end-on configurations. They both have O–O bond lengths near 1.3 Å, which is the fingerprint of a superoxide (O2−) species. The Bader charge analysis also supports these assignments since HS *O2/FeN4C10 has more electrons (1.52 e) in the adsorbed O2 than those of the IS states (1.27 e and 1.30 e for end-on and side-on IS *O2/FeN4C10, respectively) as shown in Table 1. The HS *O2/FeN4C10 is more stable than the two IS *O2/FeN4C10 as shown in Fig. 7d. The HS *O2/FeN4C10 also exhibits distinct potential-dependent adsorption behavior compared to the two IS states as presented in Fig. 7e. The adsorption energy of the HS state strongly depends on the applied potential, whereas the adsorption energies of the two IS states only weakly change with respect to the applied potential. The strong potential dependence of HS *O2/FeN4C10 could be attributed to the pronounced electron transfer from the electrode to the adsorbed O2 as evidenced in the chart of electron transfer in Fig. 7e. We also plug the O2 adsorption energy into the free energy diagrams as shown in Fig. 6c and d. The potential limiting steps of the ORR on both HS and IS FeN4C10 are now shifted to the formation of *OOH from adsorbed *O2.
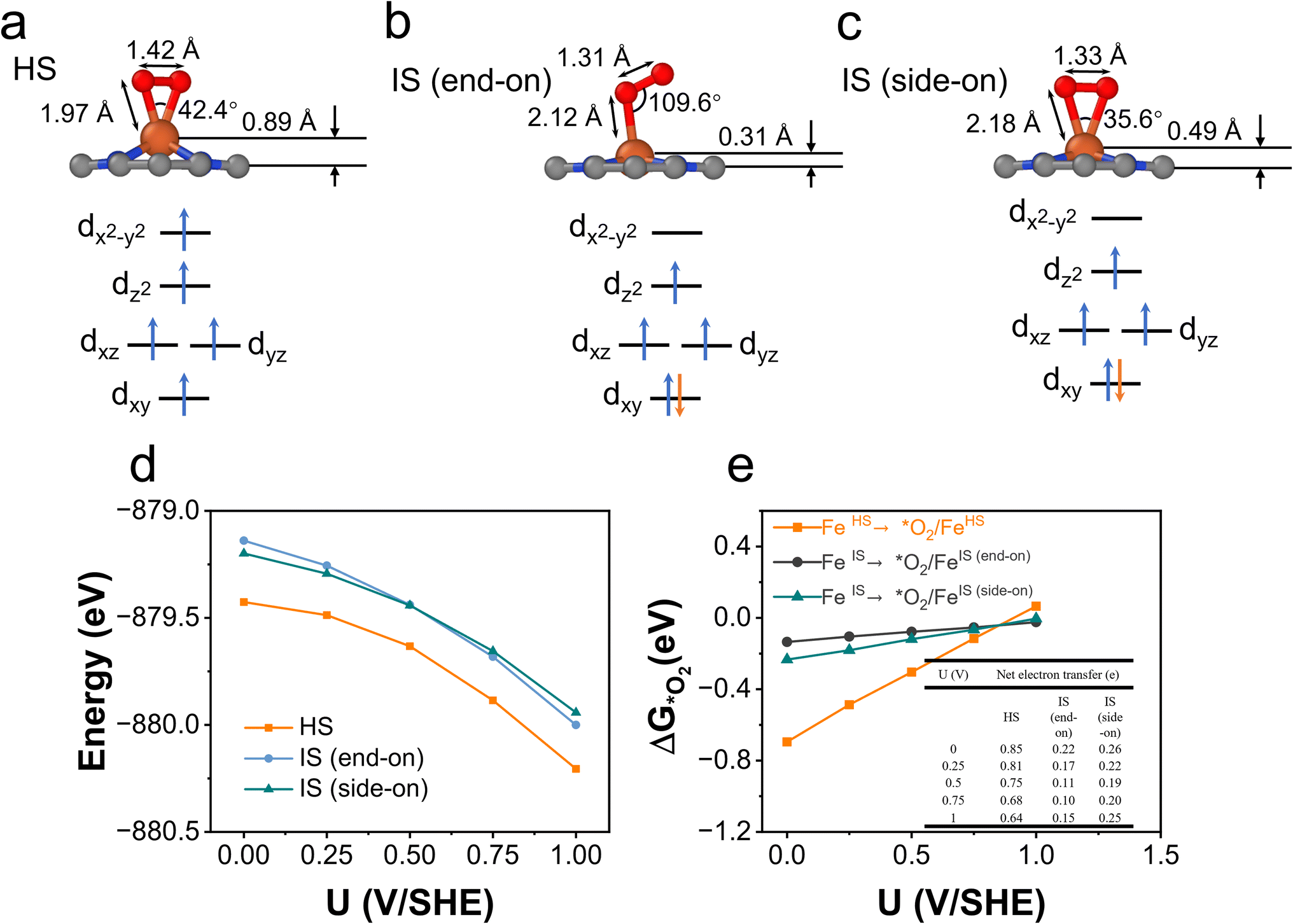 | ||
| Fig. 7 *O2/FeN4C10. (a)–(c) Atomic structures and Fe ion spin configuration in the HS, IS (end-on), and IS (side-on) states of *O2/FeN4C10. (e) Spin- and potential-dependent energies of *OH/FeN4C10 under constant-potential conditions. (f) Spin- and potential-dependent O2 adsorption energies. The inset table shows the net electron transfer in the adsorption process. | ||
3.8 Comparison with experiments
We have identified a HS structure with an out-of-plane structure for bare FeN4C10, which is similar to the non-planar ferrous FeN4 moiety with out-of-plane Fe displacement proposed experimentally based on XAS characterization.13,23 In addition, the shift of XANES spectra associated with Fe(II)/Fe(III) transition starts at applied potential as low as 0.2 V in an O2-free environment,13,25 which is inconsistent with the SWV result showing an Fe(II)/Fe(III) redox peak at 0.75 V for Fe–N–C catalysts.25 Our results have shown that the HS state of FeN4C10 adsorbed oxygenated species much more strongly at lower electrode potential because of strong cathodic electron transfer upon adsorption. Hence, we tend to attribute the early Fe(II)/Fe(III) redox observed in the in situ XANES spectra to the oxidation of the HS FeN4C10, whereas the redox peak at 0.75 V observed in the SWV corresponds to the oxidation of the IS state. Finally, the strong adsorption of oxygenated species on the HS FeN4C10 identified using our calculations could also explain the non-durable sites observed in a recent experimental study14 since the strong adsorption of oxygenated intermediates could transform FeN4C10 into ferric iron oxide. And, more interestingly, the experiment also assigns the non-durable site to a HS state. The above comparisons are only tentative and qualitative; we plan to carry out more quantitative comparisons by simulating spectroscopic spectra.4 Conclusion
In summary, based on the GC-DFT calculations, we have identified that FeN4C10 moieties with two different spin states, i.e., HS and IS states, could coexist under ORR-relevant conditions because their energies are within a 0.2 eV energy window under constant-potential conditions. The HS FeN4C10 has a special out-of-plane structure, whereas the IS state FeN4C10 has an in-plane structure. The out-of-plane displacement in the HS state is caused by the pseudo Jahn–Teller effect. This distortion induces a positive surface dipole moment and could be stabilized in a more positive applied potential relative to the PZC of the HS state. Consequently, the applied potential induces a spin crossover between the HS and IS states of FeN4C10.The two spin states perform quite differently in binding various ORR intermediates. In general, the HS state binds ORR intermediates much more strongly than the IS state at relatively low potentials at U < 0.7 V. The origin of the stronger adsorption on the HS state at lower potentials is attributed to the significant cathodic electron transfer upon adsorption. Because of the adsorption-induced cathodic electron transfer, the adsorption energy of ORR intermediates on the HS FeN4C10 can be strongly modulated by the applied potential. As a result, the adsorption energy of ORR intermediates on HS FeN4C10 increases pronouncedly (weaker adsorption) as the applied potential increases. In comparison, the adsorption energy of ORR intermediates on IS FeN4C10 exhibits a much weaker potential dependence due to slight adsorption induced electron transfer. The different adsorption capabilities of the HS and IS states lead to different ORR activities. Specifically, we predicted that the HS and IS states have ORR onset potentials of 0.77 V and 0.89 V, respectively, suggesting that the IS state would outperform the HS state in ORR activity.
Our findings have demonstrated the spin-dependent activity of Fe–N–C catalysts in the ORR. The obtained insights highlight that a comprehensive understanding of the interplay between spin, valency, and potential is required to fully comprehend the catalytic properties of magnetic single-atom catalysts. This research is expected to inspire further investigations into the role of spin states in spin-mediated catalysis and encourage the deliberate manipulation of the spin states of active centers to enhance catalytic performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (Grant No. 52272238) and Natural Science Foundation of Shaanxi Province (Grant No. 2022-JM-097).References
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Carbon-Supported Pt-Based Alloy Electrocatalysts for the Oxygen Reduction Reaction in Polymer Electrolyte Membrane Fuel Cells: Particle Size, Shape, and Composition Manipulation and Their Impact to Activity, Chem. Rev., 2015, 115(9), 3433–3467 CrossRef CAS PubMed.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Atomically dispersed iron sites with a nitrogen–carbon coating as highly active and durable oxygen reduction catalysts for fuel cells, Nat. Energy, 2022, 7(7), 652–663 CrossRef CAS.
- X. Zhang, L. Truong-Phuoc, T. Asset, S. Pronkin and C. Pham-Huu, Are Fe–N–C Electrocatalysts an Alternative to Pt-Based Electrocatalysts for the Next Generation of Proton Exchange Membrane Fuel Cells?, ACS Catal., 2022, 12(22), 13853–13875 CrossRef CAS.
- Y. Wang, D. Wang and Y. Li, Atom-level interfacial synergy of single-atom site catalysts for electrocatalysis, J. Energy Chem., 2022, 65, 103–115 CrossRef CAS.
- J. Li, Y. Li and T. Zhang, Recent progresses in the research of single-atom catalysts, Sci. China Mater., 2020, 63(6), 889–891 CrossRef.
- H. Shen, T. Thomas, S. A. Rasaki, A. Saad, C. Hu, J. Wang and M. Yang, Oxygen Reduction Reactions of Fe-N-C Catalysts: Current Status and the Way Forward, Electrochem. Energy Rev., 2019, 2(2), 252–276 CrossRef CAS.
- M. Sun, C. Chen, M. Wu, D. Zhou, Z. Sun, J. Fan, W. Chen and Y. Li, Rational design of Fe-N-C electrocatalysts for oxygen reduction reaction: from nanoparticles to single atoms, Nano Res., 2022, 15(3), 1753–1778 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications, Joule, 2018, 2(7), 1242–1264 CrossRef CAS.
- J. Li and F. Jaouen, Structure and activity of metal-centered coordination sites in pyrolyzed metal–nitrogen–carbon catalysts for the electrochemical reduction of O2, Curr. Opin. Electrochem., 2018, 9, 198–206 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials, Nat. Mater., 2015, 14(9), 937–942 CrossRef CAS PubMed.
- L. Ni, C. Gallenkamp, S. Paul, M. Kübler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, B. Albert, V. Krewald and U. I. Kramm, Active Site Identification in FeNC Catalysts and Their Assignment to the Oxygen Reduction Reaction Pathway by In Situ 57Fe Mössbauer Spectroscopy, Adv. Energy Sustain. Res., 2021, 2(2), 2000064 CrossRef CAS.
- L. Ni, P. Theis, S. Paul, R. W. Stark and U. I. Kramm, In situ 57Fe mössbauer study of a porphyrin based FeNC catalyst for ORR, Electrochim. Acta, 2021, 395, 139200 CrossRef CAS.
- J. Li, S. Ghoshal, W. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Jia, Structural and mechanistic basis for the high activity of Fe–N–C catalysts toward oxygen reduction, Energy Environ. Sci., 2016, 9(7), 2418–2432 RSC.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Identification of durable and non-durable FeNx sites in Fe–N–C materials for proton exchange membrane fuel cells, Nat. Catal., 2021, 4(1), 10–19 CrossRef CAS.
- L. Ni, C. Gallenkamp, S. Wagner, E. Bill, V. Krewald and U. I. Kramm, Identification of the Catalytically Dominant Iron Environment in Iron- and Nitrogen-Doped Carbon Catalysts for the Oxygen Reduction Reaction, J. Am. Chem. Soc., 2022, 144(37), 16827–16840 CrossRef CAS PubMed.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Identification of the Electronic and Structural Dynamics of Catalytic Centers in Single-Fe-Atom Material, Chem, 2020, 6(12), 3440–3454 CAS.
- T. Mineva, I. Matanovic, P. Atanassov, M.-T. Sougrati, L. Stievano, M. Clémancey, A. Kochem, J.-M. Latour and F. Jaouen, Understanding Active Sites in Pyrolyzed Fe–N–C Catalysts for Fuel Cell Cathodes by Bridging Density Functional Theory Calculations and 57Fe Mössbauer Spectroscopy, ACS Catal., 2019, 9(10), 9359–9371 CrossRef CAS.
- Y. Zeng, X. Li, J. Wang, M. T. Sougrati, Y. Huang, T. Zhang and B. Liu, In situ/operando Mössbauer spectroscopy for probing heterogeneous catalysis, Chem. Catal., 2021, 1(6), 1215–1233 CrossRef CAS.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J.-P. Dodelet, Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells, Phys. Chem. Chem. Phys., 2012, 14(33), 11673–11688 RSC.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang and H. Sun, General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities, Nat. Catal., 2018, 1(1), 63–72 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst, Science, 2017, 357(6350), 479–484 CrossRef CAS PubMed.
- Q. Jia, N. Ramaswamy, U. Tylus, K. Strickland, J. Li, A. Serov, K. Artyushkova, P. Atanassov, J. Anibal, C. Gumeci, S. C. Barton, M.-T. Sougrati, F. Jaouen, B. Halevi and S. Mukerjee, Spectroscopic insights into the nature of active sites in iron–nitrogen–carbon electrocatalysts for oxygen reduction in acid, Nano Energy, 2016, 29, 65–82 CrossRef CAS.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity, ACS Nano, 2015, 9(12), 12496–12505 CrossRef CAS PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, Activity Descriptor Identification for Oxygen Reduction on Nonprecious Electrocatalysts: Linking Surface Science to Coordination Chemistry, J. Am. Chem. Soc., 2013, 135(41), 15443–15449 CrossRef CAS PubMed.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Identification of catalytic sites in cobalt-nitrogen-carbon materials for the oxygen reduction reaction, Nat. Commun., 2017, 8(1), 957 CrossRef PubMed.
- U. Tylus, Q. Jia, K. Strickland, N. Ramaswamy, A. Serov, P. Atanassov and S. Mukerjee, Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal–Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems, J. Phys. Chem. C, 2014, 118(17), 8999–9008 CrossRef CAS PubMed.
- B. Li, E. F. Holby and G. Wang, Mechanistic insights into metal, nitrogen doped carbon catalysts for oxygen reduction: progress in computational modeling, J. Mater. Chem. A, 2022, 10(45), 23959–23972 RSC.
- S. Kattel and G. Wang, A density functional theory study of oxygen reduction reaction on Me–N4 (Me = Fe, Co, or Ni) clusters between graphitic pores, J. Mater. Chem. A, 2013, 1(36), 10790–10797 RSC.
- S. Kattel and G. Wang, Reaction Pathway for Oxygen Reduction on FeN4 Embedded Graphene, J. Phys. Chem. Lett., 2014, 5(3), 452–456 CrossRef CAS PubMed.
- X. Hu, S. Chen, L. Chen, Y. Tian, S. Yao, Z. Lu, X. Zhang and Z. Zhou, What is the Real Origin of the Activity of Fe–N–C Electrocatalysts in the O2 Reduction Reaction? Critical Roles of Coordinating Pyrrolic N and Axially Adsorbing Species, J. Am. Chem. Soc., 2022, 144(39), 18144–18152 CrossRef CAS PubMed.
- N. Yang, L. Peng, L. Li, J. Li, Q. Liao, M. Shao and Z. Wei, Theoretically probing the possible degradation mechanisms of an FeNC catalyst during the oxygen reduction reaction, Chem. Sci., 2021, 12(37), 12476–12484 RSC.
- P. Hutchison, P. S. Rice, R. E. Warburton, S. Raugei and S. Hammes-Schiffer, Multilevel Computational Studies Reveal the Importance of Axial Ligand for Oxygen Reduction Reaction on Fe–N–C Materials, J. Am. Chem. Soc., 2022, 144(36), 16524–16534 CrossRef CAS PubMed.
- K. Liu, J. Fu, T. Luo, G. Ni, H. Li, L. Zhu, Y. Wang, Z. Lin, Y. Sun, E. Cortés and M. Liu, Potential-Dependent Active Moiety of Fe–N–C Catalysts for the Oxygen Reduction Reaction, J. Phys. Chem. Lett., 2023, 3749–3756 CrossRef CAS PubMed.
- P. Nematollahi, B. Barbiellini, A. Bansil, D. Lamoen, J. Qingying, S. Mukerjee and E. C. Neyts, Identification of a Robust and Durable FeN4Cx Catalyst for ORR in PEM Fuel Cells and the Role of the Fifth Ligand, ACS Catal., 2022, 12(13), 7541–7549 CrossRef CAS.
- X. Yang, D. Xia, Y. Kang, H. Du, F. Kang, L. Gan and J. Li, Unveiling the Axial Hydroxyl Ligand on Fe-N4-C Electrocatalysts and Its Impact on the pH-Dependent Oxygen Reduction Activities and Poisoning Kinetics, Adv. Sci., 2020, 7(12), 2000176 CrossRef CAS PubMed.
- Y. Wang, Y.-J. Tang and K. Zhou, Self-Adjusting Activity Induced by Intrinsic Reaction Intermediate in Fe–N–C Single-Atom Catalysts, J. Am. Chem. Soc., 2019, 141(36), 14115–14119 CrossRef CAS PubMed.
- K. Liu, G. Wu and G. Wang, Role of Local Carbon Structure Surrounding FeN4 Sites in Boosting the Catalytic Activity for Oxygen Reduction, J. Phys. Chem. C, 2017, 121(21), 11319–11324 CrossRef CAS.
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Thermally Driven Structure and Performance Evolution of Atomically Dispersed FeN4 Sites for Oxygen Reduction, Angew. Chem., Int. Ed., 2019, 58(52), 18971–18980 CrossRef CAS PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity, Nat. Commun., 2021, 12(1), 1734 CrossRef CAS PubMed.
- Z. Chen, H. Niu, J. Ding, H. Liu, P. H. Chen, Y. H. Lu, Y. R. Lu, W. Zuo, L. Han and Y. Guo, Unraveling the Origin of Sulfur-Doped Fe-N-C Single-Atom Catalyst for Enhanced Oxygen Reduction Activity: Effect of Iron Spin-State Tuning, Angew. Chem., Int. Ed., 2021, 133(48), 25608–25614 CrossRef.
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Direct Growth of Carbon Nanotubes Doped with Single Atomic Fe–N4 Active Sites and Neighboring Graphitic Nitrogen for Efficient and Stable Oxygen Reduction Electrocatalysis, Adv. Funct. Mater., 2019, 29(49), 1906174 CrossRef CAS.
- Z. Duan and G. Henkelman, Surface Charge and Electrostatic Spin Crossover Effects in CoN4 Electrocatalysts, ACS Catal., 2020, 10(20), 12148–12155 CrossRef CAS.
- F. Sun, F. Li and Q. Tang, Spin State as a Participator for Demetalation Durability and Activity of Fe–N–C Electrocatalysts, J. Phys. Chem. C, 2022, 126(31), 13168–13181 CrossRef CAS.
- W. Orellana, Catalytic Properties of Transition Metal–N4 Moieties in Graphene for the Oxygen Reduction Reaction: Evidence of Spin-Dependent Mechanisms, J. Phys. Chem. C, 2013, 117(19), 9812–9818 CrossRef CAS.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Spin-Related Electron Transfer and Orbital Interactions in Oxygen Electrocatalysis, Adv. Mater., 2020, 32(39), 2003297 CrossRef CAS PubMed.
- D. Wu, Z. Zhuo, Y. Song, P. Rao, J. Luo, J. Li, P. Deng, J. Yang, X. Wu and X. Tian, Synergistic spin–valence catalysis mechanism in oxygen reduction reactions on Fe–N–C single-atom catalysts, J. Mater. Chem. A, 2023, 11(25), 13502–13509 RSC.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Self-interaction correction to density functional approximation for many electron systems, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57(3), 1505–1509 CrossRef CAS.
- I. V. Solovyev, P. H. Dederichs and V. I. Anisimov, Corrected atomic limit in the local-density approximation and the electronic structure of d impurities in Rb, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(23), 16861–16871 CrossRef CAS PubMed.
- Z. Duan and G. Henkelman, Identification of Active Sites of Pure and Nitrogen-Doped Carbon Materials for Oxygen Reduction Reaction Using Constant-Potential Calculations, J. Phys. Chem. C, 2020, 124(22), 12016–12023 CrossRef CAS.
- Z. Duan and P. Xiao, Simulation of Potential-Dependent Activation Energies in Electrocatalysis: Mechanism of O–O Bond Formation on RuO2, J. Phys. Chem. C, 2021, 125(28), 15243–15250 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140(8), 084106 CrossRef PubMed.
- M. Fishman, H. L. Zhuang, K. Mathew, W. Dirschka and R. G. Hennig, Accuracy of exchange-correlation functionals and effect of solvation on the surface energy of copper, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(24), 245402 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
- J. P. Allen and G. W. Watson, Occupation matrix control of d- and f-electron localisations using DFT + U, Phys. Chem. Chem. Phys., 2014, 16(39), 21016–21031 RSC.
- I. B. Bersuker, Jahn–Teller and Pseudo-Jahn–Teller Effects: From Particular Features to General Tools in Exploring Molecular and Solid State Properties, Chem. Rev., 2021, 121(3), 1463–1512 CrossRef CAS PubMed.
- L. Zhang, X. Ren, X. Zhao, Y. Zhu, R. Pang, P. Cui, Y. Jia, S. Li and Z. Zhang, Synergetic Charge Transfer and Spin Selection in CO Oxidation at Neighboring Magnetic Single-Atom Catalyst Sites, Nano Lett., 2022, 22(9), 3744–3750 CrossRef CAS PubMed.
- Y. Wang, X. Ren, B. Jiang, M. Deng, X. Zhao, R. Pang and S. F. Li, Synergetic Catalysis of Magnetic Single-Atom Catalysts Confined in Graphitic-C3N4/CeO2(111) Heterojunction for CO Oxidization, J. Phys. Chem. Lett., 2022, 13(27), 6367–6375 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural model, table of occupation numbers for Fe 3d orbitals, and spin density plots. See DOI: https://doi.org/10.1039/d3ta03271j |
| This journal is © The Royal Society of Chemistry 2023 |