
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversibility limitations of metal exsolution reactions in niobium and nickel co-doped strontium titanate†
Moritz L.
Weber‡
 *abc,
Yoo Jung
Sohn
c,
Regina
Dittmann
b,
Rainer
Waser
bd,
Norbert H.
Menzler
ce,
Olivier
Guillon
ce,
Christian
Lenser
c,
Slavomír
Nem
*abc,
Yoo Jung
Sohn
c,
Regina
Dittmann
b,
Rainer
Waser
bd,
Norbert H.
Menzler
ce,
Olivier
Guillon
ce,
Christian
Lenser
c,
Slavomír
Nem![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) ák
af and
Felix
Gunkel
b
ák
af and
Felix
Gunkel
b
aAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: mlweber@lbl.gov
bPeter Gruenberg Institute (PGI-7) and JARA-FIT, Forschungszentrum Juelich GmbH, Wilhelm-Johnen-Straße, 52425 Juelich, Germany
cInstitute of Energy and Climate Research (IEK-1), Forschungszentrum Juelich GmbH, Wilhelm-Johnen-Straße, 52425 Juelich, Germany
dInstitute for Electronic Materials (IWE 2), RWTH Aachen University, Sommerfeldstraße 18/24, 52074 Aachen, Germany
eInstitute of Mineral Engineering (GHI), RWTH Aachen University, Forckenbeckstraße 33, 52074, Aachen, Germany
fDepartment of Physics, University of California, Davis, California 95616, USA
First published on 12th July 2023
Abstract
Metal exsolution reactions enable the preparation of metal-oxide nano-composites from oxide parent materials in a single thermal reduction step. In this process, reducible metals are released from the doped oxide and nucleate in the form of finely dispersed, supported nanoparticles. A reversible exsolution and re-dissolution reaction could provide an effective way to regenerate catalysts, where the surface structure and functionality dynamically adapt to the ambient gas environment. However, the reversibility of exsolution reactions is often limited. We investigate reversibility limitations in the niobium and nickel co-doped perovskite SrTi0.95−xNb0.05NixO3−δ with varying Ni doping concentrations between x = 0.005–0.1. Combined morphological, structural and chemical analyses of the material response upon consecutive thermal treatments in reducing and oxidizing environments reveal a non-correlated bulk and surface response of the material upon redox treatment. While the bulk structural changes are mostly reversible, no re-dissolution of the exsolved surface nanoparticles is detected for the investigated time–temperature window (T = 800 °C, t = 5 h for reduction and reoxidation, respectively). Instead, a modification in the nanoparticle distribution and an increased surface wetting of the support by the exsolved metal species are observed upon reoxidation of the nanoparticles.
 Moritz L. Weber | Dr Moritz L. Weber is a materials scientist and analytical chemist who specializes in catalyst design principles for green hydrogen technologies. His research focuses on controlling the defect structure and surface properties of oxides and oxide-metal composites at the nanoscale using oxide epitaxy, in order to gain mechanistic insights into catalyst self-assembly and performance. He earned his PhD with honors from RWTH Aachen University & Forschungszentrum Jülich and joined Lawrence Berkeley National Laboratory as a postdoctoral fellow in 2022. He has been recognized with several awards and fellowships for his proficiency in science communication, scientific reviewing, and independent research projects conducted within the framework of international collaborations. |
1 Introduction
Nanostructuring of electrode materials is an effective strategy to tailor their electrochemical functionality and to improve the performance of energy conversion devices such as solid oxide cells.1–3 However, nanostructured electrodes are challenging to manufacture due to the high processing temperatures of solid oxide cells, which leads to severe particle coarsening and growth. The idea of “self-regenerating” catalysts on the basis of reducible metals that reversibly occupy either the B-sites of perovskite host oxides or nucleate in the form of dispersed nanoparticles at the oxide surface was introduced by Nishihata and co-workers as an innovative strategy to counteract nanoparticle coarsening and poisoning.4 Later, particularly Neagu et al. refined and popularized the synthesis of homogeneously dispersed, supported nanoparticles by thermal reduction of doped perovskites coining the term metal exsolution.5 The concept of metal exsolution enables the generation of metallic nanoparticles under the reducing operation conditions of the fuel electrode, which may serve as catalytic centers for the electrochemical conversion reaction.4–7 For this purpose, oxide parent materials – oftentimes donor-type perovskite host materials – are co-doped with catalytically active and reducible metals. Under a reducing thermal treatment, a fraction of surface-near metal dopants is released to the oxide surface where they nucleate in the form of metal nanoparticles, while others remain buried in the oxide matrix.8–10 Besides the simplicity of the synthesis concept, the extraordinarily homogeneous distribution and controllable density of the generated exsolution nanoparticles are beneficial for the design of high-performance fuel electrode materials.Of particular interest is that the exsolution process was reported to be reversible4,11–15 or partially reversible16,17 in some cases, which may offer the possibility to reactivate the catalysts in the case of deactivation by coking, sulfur poisoning or coarsening of the nanoparticles by re-dissolution of the metallic phase within the oxide host under oxidizing thermal treatment and repeated exsolution under reducing operation conditions. Reversible metal exsolution reactions would hence promise considerable technological advances, extending the lifetimes of catalysts by regeneration upon redox cycling to prevent a loss in electrochemically active triple phase boundaries. However, not all exsolution-active perovskites exhibit a reversible exsolution response.18–20 Here, several reports point out that the exsolved nanoparticles may remain pinned to the perovskite surface during the reoxidation of the material.
A large structural and chemical variety of exsolution-active materials and a broad range of processing conditions are presented in the literature.21,22 The reasons for reversibility limitations of metal exsolution reactions, hence, can be manifold and may be specific to the respective material system. A brief overview of exemplary literature references that address reversibility and reversibility limitations of exsolution reactions, summarizing the respective material compositions, sample properties, annealing conditions and the applied methodology is given in Table S1 in the ESI.† In the present study, we aim to discuss the reversibility limitations observed for doped strontium titanate, which was demonstrated as a candidate for the development of alternative electrode materials for solid oxide fuel cells.23–28 To date, most studies on this matter have been conducted using ceramic oxides with complex micro- and nano-structures (cf. Table S1†). Ceramic oxides typically exhibit comparably rough surfaces, a high degree of chemical and structural anisotropy as well as a high density of extended defects such as grain boundaries. In contrast, we employ epitaxial thin films with single-crystal character and atomically flat surfaces enabling the high-precision analysis of nanoparticles during nucleation and redox treatment. Our study hence focuses on the redox response of exsolution catalysts at the “single-grain” level, whereas grain boundaries that may serve as fast diffusion pathways are not present in our samples.
For this purpose, ceramic oxide pellets are synthesized, which are applied as target materials for the pulsed laser deposition (PLD) of high-quality epitaxial thin films of SrTi0.95−xNb0.05NixO3−δ with varying Ni doping concentrations between x = 0.0–0.1 (STNNix). The thin films are employed as model systems for annealing studies. The material response is investigated upon consecutive reducing and oxidizing thermal treatments with respect to the morphological, structural and chemical evolution of the perovskite and the nanostructured metal-oxide composite, respectively.
2 Results & discussion
2.1 Structural evaluation of ceramic STNNix pellets
Fig. 1 shows X-ray diffraction (XRD) patterns obtained from sintered ceramic STNNix pellets with a Ni doping concentration in the range between x = 0.0–0.1 (air, T = 1200 °C, t = 12 h). The Nb doping level was kept constant for all STNNix compositions. The material system crystallizes in the perovskite structure, where Rietveld refinement of the diffraction patterns (cf. Fig. S1 and S2 in the ESI†) reveals lattice parameters of a = 3.910–3.913 Å, i.e. increased values with reference to undoped SrTiO3−δ (STO) with a nominal lattice parameter of a = 3.905 Å.29 Independent of the Ni doping level x, however, no systematic shifts in the position of the diffraction peaks can be observed in comparison to the purely Nb-doped material (x = 0.0). The observed lattice expansion of the STO crystal lattice hence appears to be mainly induced by Nb doping30,31 and is in good agreement with the larger ionic radius of Nb(V) cations (r = 0.64 Å) relative to the Ti(IV) cations (r = 0.605 Å). As a multivalent cation, Ni dopants may be present in different oxidation states with the corresponding ionic radii of Ni(II) (r = 0.69 Å), Ni(III) (r = 0.60 Å) and Ni(IV) (r = 0.48 Å). Since no systematic expansion or decrease in the lattice parameters is detected across the Ni doping series (x = 0.0–0.1), it is likely that Ni dopants occupying the B-sites of the STNNi perovskite to a large extent are present in an oxidation state of Ni(III) with similar ionic radius to the Ti(IV) host cations.32 Therefore, lattice expansion due to steric effects may be negligible, albeit electrostatic modifications of the crystal lattice involved in chemical doping may be expected to induce a systematic variation in the lattice parameter of the perovskite oxide depending on the Ni substitution level. The exact influence of chemical doping on the structural properties of the perovskite lattice, however, depends on the complex defect structure of multi-cation perovskites. For instance, charge compensation of Nb dopants can be accomplished electronically or ionically, where the formation of Sr vacancies may compete with the compensation by lower valent Ni(III) dopants under the applied oxidizing synthesis conditions.31,33–37 In addition, Nb-substitution may result in the formation of Ti(III) valence states (r = 0.67 Å)32 which contributes to the expansion of the crystal lattice. This effect might, however, be partially compensated for by Ni doping and depends on the extent of Ni incorporation into the lattice, whereas the charge of the Ni acceptors can, in principle, be compensated for by the formation of oxygen vacancies in perovskite oxides, which can induce considerable changes in the structural properties of the material.38,39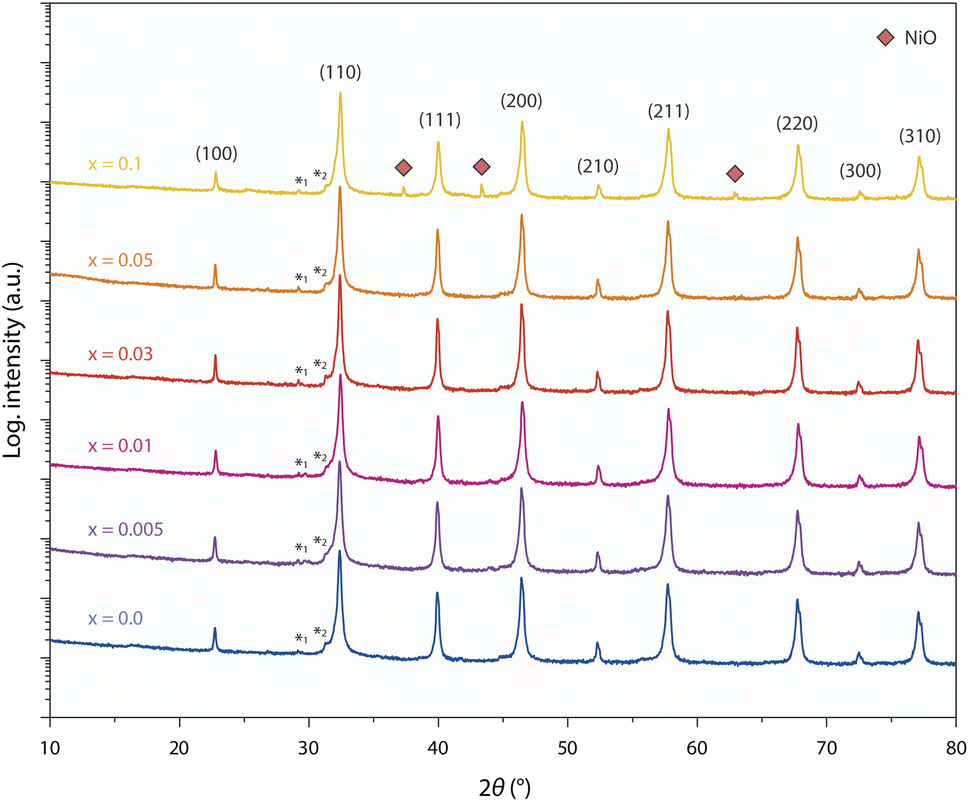 | ||
| Fig. 1 X-ray diffraction analysis of sintered ceramic pellets of SrTi0.95−xNb0.05NixO3−δ with systematically varying Ni doping concentrations between x = 0.0–0.1. No secondary phases are detected between x = 0.0–0.05, while a minor NiO signature becomes visible for x = 0.1 (red rhombi). Miller-indices assigned to the respective diffraction peaks of the perovskite oxide are given in the figure. Figure was modified from the ESI of ref. 8. A small contribution of Cu-Kβ radiation is denoted by asterisk 1. Shoulders visible in close proximity to diffraction peaks originate from spectral features of the primary radiation that result from passing the Ni-Kβ filter on the primary beam side, due to its characteristic absorption edge. The artefacts are exemplarily denoted by asterisk 2 for the peak of maximum intensity. | ||
The finding of a non-measurable systematic change in the structural properties of the STNNix doping series may hence point towards finite dissolution of the Ni dopants within the crystal lattice as revealed in our previous work on SrTi0.9Nb0.05Ni0.05O3−δ (i.e. x = 0.05),8 where the dopants might either segregate to grain boundaries of the ceramic oxide or form separated nano-phase inclusions. Notably, the low dopant concentration and nanometre size of the separated phases result in small X-ray coherence lengths, which are likely to prevent detection by XRD.40 Here, significant broadening of the XRD peaks will render the nano-phases non-detectable as the signals may disappear in the background signal of the main phase. A minor NiO secondary phase becomes apparent on the logarithmic intensity scale solely for the highest doping concentration of x = 0.1. Here, quantitative phase analysis of the diffractogram by Rietveld refinement reveals a NiO secondary phase of ∼0.4 wt%.
It is likely that the Ni dopants partly dissolve within the perovskite lattice and partly remain in the form of a secondary phase across the STNNix series, while the Nb dopants fully dissolve within the perovskite lattice. Consequently, the present material system is most probably not an ideal solid solution parent perovskite, but the dopant distribution varies with respect to the crystallographic sites of the perovskite host lattice and at the grain boundaries of the ceramic oxides or in the form of other extended defects such as nano-inclusions. In terms of design strategies for exsolution catalysts it is important to note that similar inhomogeneities are likely to occur to a certain degree for many material systems (for example shown elsewhere18), which can have a significant impact on the defect engineering approaches that need to be applied to control exsolution reactions.5,8
2.2 Epitaxial growth and characterization of STNNix thin film model systems
Epitaxial thin film model systems with well-defined surface morphology and a controlled thickness of 50 nm were synthesized for detailed studies of the materials redox response. The thin films of varying Ni concentrations were deposited by employing the ceramic STNNix pellets as targets for reflection high-energy electron diffraction (RHEED)-controlled PLD. Here, equal deposition conditions were applied for all Ni doping levels x, resulting in a layer-by-layer growth mode and comparable deposition rates (Fig. 2a). Investigations of the surface morphology by atomic force microscopy (AFM) reveal a defined step terrace structure for the as-deposited thin films, representatively shown for x = 0.1 in Fig. 2b and for all thin films with x = 0.0–0.1 in the ESI (Fig. S3 and S4).† A low root mean square (RMS) surface roughness with values in the range between RMS = 171–334 pm was detected for the epitaxial model systems.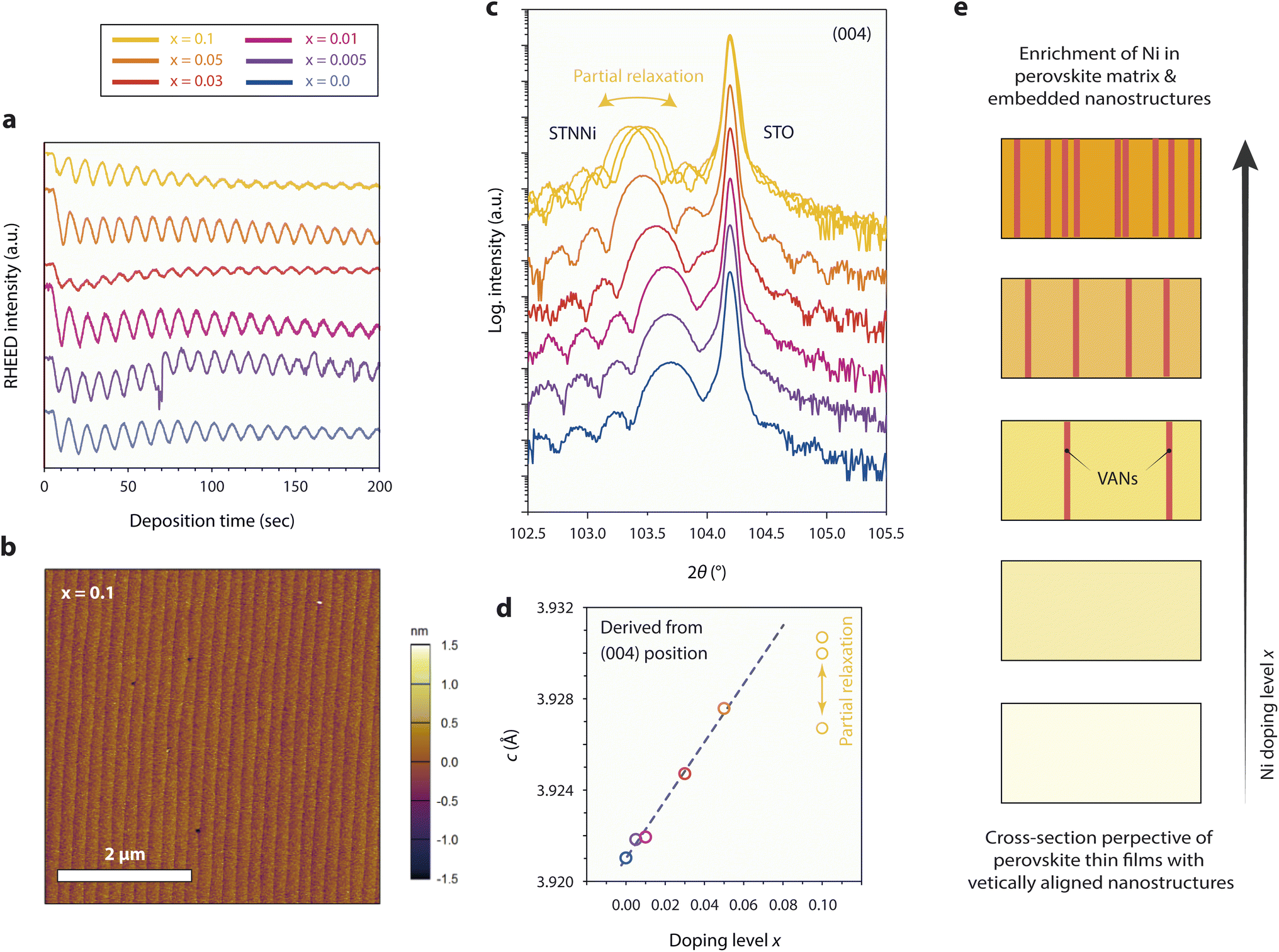 | ||
| Fig. 2 Pulsed laser deposition and characterization of 50 nm thick, [001]-oriented epitaxial STNNix thin films with different Ni doping levels x = 0.0–0.1 deposited on (100) STO single crystal substrates. (a) RHEED intensity evolution during the initial PLD of STNNix. (b) Representative surface morphology of STNNix thin films, exemplarily shown for x = 0.1. (c) X-ray diffraction analysis in the vicinity of the (004) diffraction signals of the STO substrate and respective thin films. The structural properties of three samples that were deposited under equal conditions are compared for x = 0.1, where a partial relaxation of the STNNi crystal lattice is apparent. (d) Dependence of the c-lattice parameter on the Ni-doping level x derived from the (004) diffraction peak positions shown in (c). (e) Schematic illustration of the expected structural evolution of the epitaxial STNNix thin films. Ni doping of the perovskite oxide is expected to result in an enrichment of Ni in the perovskite structure and for larger doping levels x in the form of phase-separated, embedded nanostructures. | ||
High-resolution X-ray diffraction analysis (HR-XRD) was performed to compare the structural properties of the thin films. Fig. 2c shows the diffraction patterns obtained from the as-prepared thin films, where a broader thin film diffraction signal is visible in close vicinity of the sharp (004) diffraction signal of the STO substrate, respectively. In addition, distinct Laue oscillations are visible, indicating high crystallinity and coherence of the epitaxial layers and the top and bottom interfaces. Remarkably, and in contrast to the ceramic oxides, a systematic shift of the position of the thin film peak towards smaller diffraction angles is apparent for the epitaxial STNNix thin films (Fig. 2c), corresponding to an expansion of the out-of-plane c-lattice parameter of the crystal lattice with increasing Ni doping level x (Fig. 2d). It is worth pointing out that the in-plane a-lattice parameter is constrained with respect to the lattice parameter of the substrate due to the epitaxial character of the STNNix-to-STO interface. As can be seen, the STNNix c-lattice parameter increases monotonically with an increasing Ni concentration, while a considerable deviation from the trend is apparent for high doping concentrations of x = 0.1 (Fig. 2d).
As we have depicted in our earlier publication,8 the structural evolution of the STNNi thin films is not only related to the substitution of the Ti B-site cations by Ni dopants, but is additionally influenced by the spontaneous formation of vertically aligned nanostructures (VANs).41,42 While the presence of minor amounts of separated Ni-rich nano-phases will not influence the structural properties of ceramic oxides, the vertical alignment and related interfacial superstructure of the embedded nanostructures with the perovskite host lattice will considerably influence the structural properties of the epitaxial perovskite layers. This is due to the specific structural relationship between the minority phase (VANs) and the majority phase (perovskite host lattice), where a domain matching mechanism results in the formation of a semi-coherent vertical interface as we have demonstrated at the atomic scale for a Ni doping concentration of x = 0.05.8 Consequently, a large portion of the observed lattice expansion is related to elastic deformation of the host lattice in order to compensate for the residual misfit of the semi-coherent interface between the host and the embedded phase. The total interfacial area and therefore the volume fraction of the embedded phase, which is presumably somewhat proportional to the doping level x, are of particular importance for the structural properties of the material system.41 As discussed above, point defects such as cation vacancies may cause an additional expansion of the perovskite lattice.43–45 For large dopant concentrations, misfit dislocations were reported to form at the vertical interface between the host phase and the embedded VANs resulting in partial relaxation of the crystal lattice.41 For STNNi with x = 0.1 most probably partial relaxation of the host lattice becomes apparent by a deviation from the linear relationship between the doping level x and the c-lattice parameter, as depicted by comparing the measurements of the three samples that were deposited under equal conditions.
Fig. 2e illustrates schematic cross sections of epitaxial STNNix thin films, depicting the expected structural evolution of the samples with increasing Ni doping level x. At low doping concentrations, Ni dopants may be preferentially accommodated by dissolution within the perovskite crystal lattice, whereas larger Ni concentrations are likely to result in progressive phase separation and assembly of Ni-rich VANs, which are embedded within the perovskite host lattice. In consequence, the out-of-plane lattice parameter in epitaxial STNNix thin films is determined using a variety of entangled parameters, namely the epitaxial strain at the horizontal thin film-to-substrate interface, the effective concentration of extrinsic Ni and Nb dopants and the interfacial area of the vertical interface between the perovskite host and the embedded nanostructures. In addition, cation and oxygen point defects as well as extended defects such as misfit dislocations will impact the average lattice parameter derived from XRD investigations.
2.3 Redox behavior of exsolution-active STNNix thin films
In order to investigate the exsolution behavior and reversibility of the exsolution reaction in STNNix, consecutive reducing and oxidizing annealing experiments were performed at T = 800 °C, comparable to typical operating temperatures of solid oxide fuel cells, to study the material response. Here, the smooth morphology of the (001) epitaxial thin film surfaces allows to study the nanoparticle exsolution behavior with high precision. Redox studies on the basis of the Nb:STO (x = 0.0) reference sample are not further discussed as the material shows degradation under oxidizing conditions (cf. Fig. S3†). After thermal reduction of the samples (4% H2/Ar gas flow, T = 800 °C, t = 5 h), the nucleation of finely dispersed nanoparticles is detected by AFM (cf. Fig. S4†), where a monotonic increase in the nanoparticle density is visible for an increasing Ni doping level x (Fig. 3a, green line). It is worth noting that a considerable deviation from the monotonic trend in the nanoparticle density is visible for a maximum Ni concentration of x = 0.1, which coincides with the partial relaxation of the as-prepared perovskite host lattice linked to the formation of misfit dislocations (cf.Fig. 2c and d). Accordingly, the nanoparticle density is approximately proportional to the Ni doping level x in the range between x = 0.005–0.05 but drops for x = 0.1 relative to the monotonic trend. This observation indicates that the nanoparticle yield, achievable by successive doping of the parent perovskite is limited, presumably due to increasing inhomogeneity of the dopant distribution within the host lattice and the associated incorporation of extended defects that are expected to moderate the embedment of large volumes of phase-separated VANs.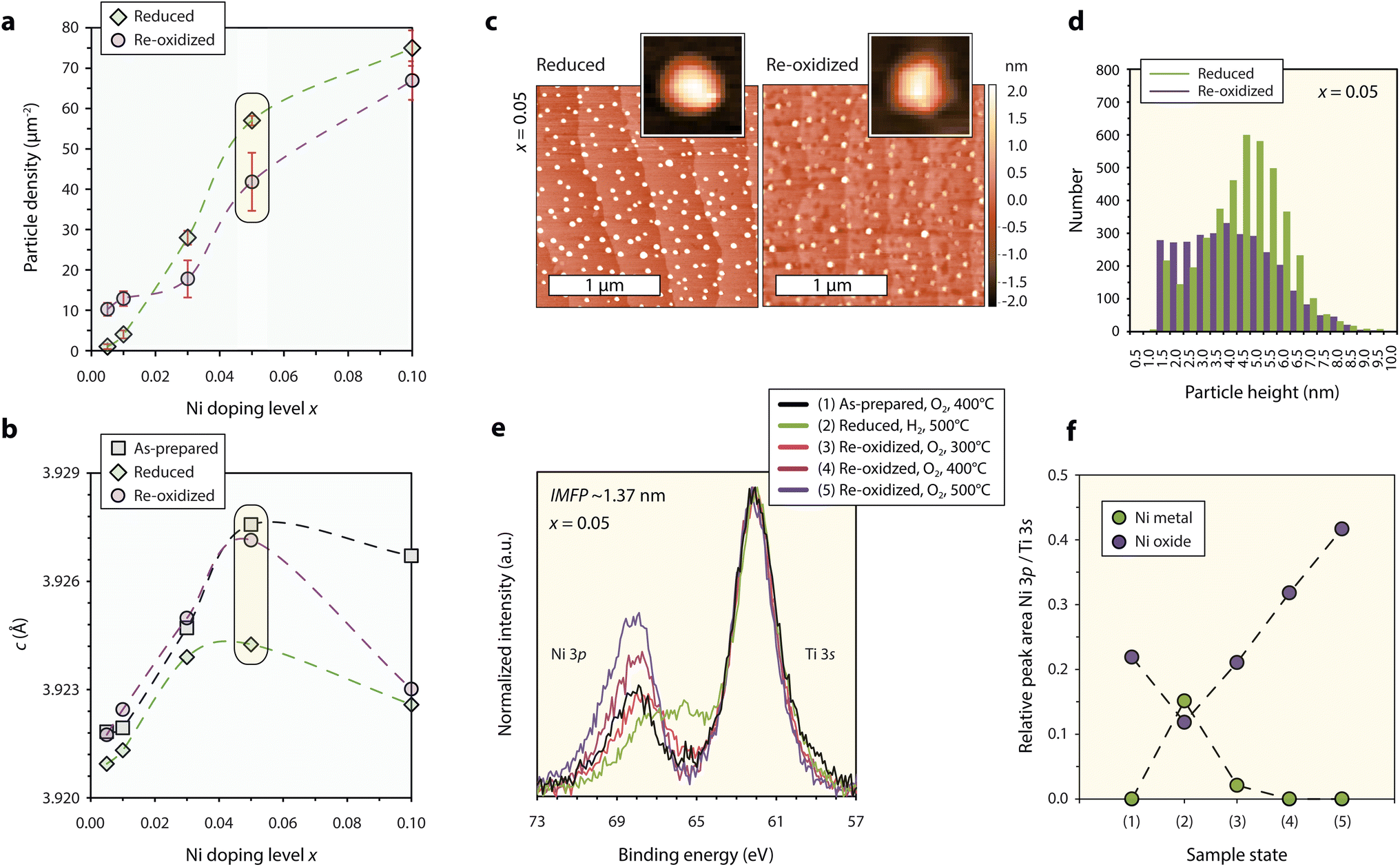 | ||
| Fig. 3 Characterization of the redox response of epitaxial STNNix thin films. (a) Nanoparticle density detected for STNNix (x = 0.005–0.05) by atomic force microscopy using a lower threshold of 1 nm. The error bars denote the standard deviation of average values obtained by the measurement of three different 5 × 5 μm2 scans. (b) Out-of-plane c-lattice parameter of the as-prepared, reduced and reoxidized thin films determined from the position of the (004) diffraction peak obtained from STNNix thin films (x = 0.005–0.05). (c) Atomic force microscopy images of STNNix (x = 0.05) in the reduced and in the reoxidized states. A single particle image cut from the larger scans is shown as the inset image, respectively. (d) Frequency distribution of the maximum height of exsolved nanoparticles after reduction and reoxidation determined on the basis of three 5 x 5 μm2 scans. For (a–d) the reduction step was performed ex situ in continuous flow of a 4% H2/Ar gas mixture at T = 800 °C while the reoxidation step was performed in air at T = 800 °C. (e) Ambient-pressure X-ray photoelectron spectroscopy of the Ni 3p-Ti 3s core-level region obtained from STNNix (x = 0.05). The core-level signature is compared in situ after consecutive oxidation, reduction and reoxidation steps at different temperatures. The hydrogen and oxygen partial pressures were p(H2) = 0.5 mbar and p(O2) = 0.5 mbar. The binding energies of all spectra were aligned to Ti 2p = 458.4 eV and a Shirley-type background was subtracted. (f) Relative Ni 3p/Ti 3s peak area ratio derived from (e) based on deconvolution of the spectra by peak fitting as specified in the ESI.† Yellow boxes in (a and b) highlight data points obtained from STNNix (x = 0.05). Dashed lines serve as a guide for the eye. | ||
As can be seen in Fig. 3b, the thermal reduction step is accompanied by a decrease in the c-lattice parameter of the host lattice, as derived from a shift of the (004) thin film diffraction peaks towards larger diffraction angles (cf. Fig. S5†). The structural changes may be interpreted as a consequence of the release of Ni dopants onto the perovskite surface upon thermal reduction; however, the exsolution response is strongly limited to the surface region.9,10,18,46 Furthermore, the degree of the decrease in the c-lattice parameter appears not to be directly correlated to the doping level x. In fact, large changes in the lattice parameters are only visible for larger doping levels of x = 0.05 and x = 0.1, while only a minor lattice shrinkage is apparent for thin films with x ≤ 0.03. Therefore, changes in the c-lattice parameter after the thermal reduction treatment are likely to be related to secondary processes. Most importantly, changes in the superstructure of the semi-coherent interface lattice may result in modifications in the average perovskite strain state due to the transition of the embedded phase between NiOx and Ni metal.8 A strong decrease in the c-lattice at larger doping levels x hence might be related to an expected greater share of Ni dopants present in the form of VANs (cf.Fig. 2e). Consequently, the lattice parameter is not a suitable indicator for the interpretation of exsolution reactions in epitaxial thin films as it can be strongly affected by secondary processes that impact the bulk structure. The exsolution of surface nanoparticles, relevant for catalysis, hence should be regarded as largely independent from the bulk structural changes.
The thin film diffraction peaks are detected close to their initial position after thermal reoxidation of the STNNix samples (air, T = 800 °C, t = 5 h), again except for STNNix with x = 0,1 (Fig. 3b). While the bulk structural modifications appear to be mostly reversible upon reoxidation for x ≤ 0.05, the exsolved nanoparticles for the greater part remain stable at the surface (Fig. 3a, purple line). Here, only minor changes in the nanoparticle density are detected with respect to the applied lower threshold of 1 nm with respect to the nanoparticle height. This finding implies that, although the bulk properties may reversibly adapt to the reactive gas environment, the exsolved nanoparticles do not or only to a limited extent re-dissolve within the host lattice during the applied time–temperature window of thermal oxidation. This is also the case for very low Ni doping concentrations x, where the formation of phase-separated VANs is likely to be negligible and Ni dopants are expected to be homogeneously dissolved within the parent perovskite.
In the following, the redox response of STNNix will be discussed in more detail on the basis of SrTi0.9Nb0.05Ni0.05O3−δ (x = 0.05). Notably, a reversible lattice decrease and expansion were detected after reduction and reoxidation for this specific stoichiometry. The nanoparticle density, however, only slightly decreased after reoxidation (cf. data points highlighted using yellow boxes in Fig. 3a and b). Detailed AFM images of the surface morphology comparing the reduced and the reoxidized states are shown in Fig. 3c. Although only minor changes in the nanoparticle density were detected on the basis of 5 × 5 μm2 scans (Fig. 3a), distinct morphological changes of the nanostructured surface became apparent on smaller length scales. While initially spherical nanoparticles are exsolved under reducing conditions, the particles have a faceted appearance after reoxidation, which can be attributed to the oxidation of Ni to rock-salt NiO.19,47 Furthermore, a large share of the nanoparticles appear to be decreasing in height, which is also reflected by the frequency distribution of the nanoparticle height shown in Fig. 3d. As can be seen, the nanoparticle population consists of a larger percentage of smaller nanoparticles below z = 3 nm while the share of nanoparticles in the range between z = 3–7 nm has decreased. Here, we expect several competing processes to influence the nanoparticle height. Partial re-dissolution (if any) may result in a loss of metal cations and hence a decrease in the average nanoparticle size. Furthermore, the wetting behaviour and correlated contact angle between the metal nanoparticle and oxide support are likely to considerably differ from the properties of the oxidized nanoparticles and the oxide support. Increased wetting i.e. decreased contact angles at the oxide–oxide interface will presumably result in a decrease in the average nanoparticle height. In contrast, oxidation of Ni to NiO is related to a volume expansion causing an increase in the nanoparticle size. If these entangled processes result in a net increase or decrease in nanoparticle height may be material specific. In our system it is apparent that an average decrease is resulting from the reoxidation step.
The morphological changes imply a dynamic response of the Ni chemistry during reoxidation, which was further characterized by ambient-pressure X-ray photoelectron spectroscopy (AP-XPS). To study the chemical evolution of the near-surface region, the Ni 3p–Ti 3s core-level region was recorded upon reduction and subsequent reoxidation (cf. Fig. S8 and S9† for O 1s, Ti 2p, Sr 3d and Nb 3d states). In order to vary the surface sensitivity, a photon energy of Ehv = 680 eV corresponding to an inelastic mean free path (IMFP) of IMFP ∼1.37 nm and Ehv = 900 eV corresponding to an IMFP ∼1.72 nm was applied for the analysis. The evolution of the core-level signature, obtained at room temperature after five consecutive annealing steps in reactive O2 and H2 gas environments, is shown in Fig. 3e. Here, the binding energies of all spectra were aligned with respect to the Ti 2p position B.E. = 458.4 eV in order to compare relative peak shifts between Ti and Ni and to correct for space charge induced peak shifts upon change in ambient atmosphere.10,48–50 First, a mild annealing step in oxygen was performed to desorb carbon species from the surface. The Ni 3p doublet gives rise to a slightly asymmetric peak at higher binding energies relative to the Ti 3s signal. Subsequent reduction of the sample results in a relative shift of the Ni 3p signal towards the Ti 3s peak indicating the formation of metallic surface species, which is correlated to the exsolution of Ni nanoparticles. Notably, the enrichment of Ni at the surface is less pronounced than that demonstrated in our previous study,51 which can be attributed to the oxidizing pre-treatment10 and the lower hydrogen partial pressure applied during reduction of the sample.
Reoxidation of the sample was performed by annealing in oxygen gas in three consecutive steps with increasing temperatures of T = 300 °C, T = 400 °C and T = 500 °C. As can be seen, the first oxidation step results in a reverse shift of the Ni 3p peak to higher binding energies, indicating partial oxidation of the metal nanoparticles, while a low binding energy shoulder remains present. The following oxidation steps, however, result not only in a shift to the initial binding energy, but also in a significant increase in the relative intensity between the Ni 3p and Ti 3s signals. While a similar trend can be seen for the two different information depths, the detected chemical changes are much less pronounced for larger information depths (cf.Fig. 3eversus S6†). The corresponding evolution of the Ni oxide and Ni metal components, derived from the deconvolution of the Ni 3p–Ti 3s core-level region by peak fitting, is shown in Fig. 3f (cf. Fig. S7† for details on the peak fitting model). As can be seen, a Ni-rich surface is formed under oxidizing conditions, which is in line with previous reports describing the formation of extensive surface phases by exsolved metal species upon reoxidation11,17 and observations of the metal enrichment of the near-surface region of the perovskite upon redox cycling.52
The oxidation of metallic Ni nanoparticles to NiO is expected to be accompanied by both a change in the particle volume and a decrease in the relative number of Ni atoms probed by XPS due to the dilution with oxygen anions. Due to the complex geometry of the nanostructured surface, it is challenging to predict the net change in the Ni coverage. Since the relative Ni concentration, however, increases by a factor of almost three, we assume that the fragmentation of the nanoparticles and surface diffusion of ionic Ni species result in increased surface wetting under oxidizing conditions which is frequently observed for metal particles on oxide supports.53–55 Nanoparticle fragmentation and surface diffusion of Ni ions could be a crucial step in reversible exsolution catalysts given the fact that ionic species need to be incorporated into the perovskite structure.
It is likely that reversibility limitations, i.e. irreversibility or significant differences in the timescales of exsolution and re-dissolution of the metal species are a direct result of thermodynamics and kinetic limitations. The driving force for metal exsolution is the change in Gibbs free energy ΔG and is associated with a decrease in the entropy of the system due to demixing of the multi-cation perovskite oxide. Upon reoxidation, slow metal re-dissolution competes with the rapid oxidation of the metallic nanoparticles. In consequence, the oxidized nanoparticles and the perovskite oxide support are in quasi-equilibrium, while both are stable at high oxygen partial pressure. In contrast to the exsolution step, the rapid reoxidation of metal nanoparticles at the perovskite support renders predictions of the thermodynamic driving force for metal re-dissolution on the basis of Gibbs free energy ΔG values difficult. Nanoparticle re-dissolution is associated with an increase in entropy. Hence, it could conceivably be hypothesized that the reversibility of exsolution reactions towards a full recovery of the original parent oxide is typically limited by thermodynamic and kinetic boundary conditions causing a considerable offset between the required exsolution and re-dissolution time–temperature window. It remains unclear to what extent the thermodynamic and kinetic boundary conditions can be fulfilled under operation conditions of a working solid oxide cell. High temperatures that are close to the synthesis conditions of the parent oxide or long reoxidation times, typically not feasible to be applied in operational devices, may be required to achieve a full regeneration of exsolution catalysts.
Most importantly, the incorporation of the exsolution-active cations must be energetically favourable in comparison to the co-existence of the oxidized nanoparticle phase (e.g. binary oxide) at the perovskite parent surface. Moreover, a sufficient physical or chemical gradient is required to provide for sufficient reaction kinetics promoting indiffusion of cations into the perovskite host lattice. If a sufficient physico-chemical gradient promoting the incorporation of cations back into the perovskite host oxide is present, a high cation mobility within the perovskite lattice may be beneficial for the reaction kinetics. Notably, the orientation-specific properties of exsolved nanoparticle populations (e.g. size and special distribution), which have been shown to be related to anisotropy in the interfacial energy between metal species and different crystallographic planes56,57 as well as surface reconstructions,58 may further influence the re-dissolution kinetics. Here, small and finely dispersed nanoparticles may be favourable for metal re-dissolution within the perovskite lattice, if a sufficient physical or chemical gradient is present to promote incorporation.
Furthermore, defect structures and their associated charge and strain fields play a significant role in moderating metal exsolution reactions.5,8,10,59–64 For instance, the mass transport of exsolution-active dopants to the surface by ionic diffusion results from the concentration gradient that forms between the surface and the sub-surface region, where the exsolution kinetics of surface nanoparticles is strongly entangled with the defect chemistry of the perovskite host lattice.10 The dynamic formation of space charge regions results in a p(O2)-dependant redistribution of the point defect distribution under oxidizing and reducing conditions, which moderates the exsolution reaction.
For materials of low redox stability, increasing point defect concentrations result in an increased probability of interactions between the charged defects (electrostatic and strain interactions), which may finally result in the formation of defect structures of higher dimensions.65 Sample annealing at elevated temperatures, hence, can be associated with significant modifications of the defect structure and surface chemistry9,60,66 and may result in irreversible material changes that may hinder the indiffusion of exsolved species upon reoxidation. Therefore, fully reversible changes of the underlying defect structure of the perovskite parent upon reduction and reoxidation may be required to realize reversibility in exsolution catalysts.
Observations of a limited exsolution reversibility cast doubt on the feasibility of smart catalysts that passively adapt with respect to the redox environment by dynamically switching between a single perovskite phase and a nanostructured composite. Interestingly, indications for (partial) reversibility of metal exsolution reactions have so far only been reported for ceramic oxides with large densities of grain boundaries (cf. Table S1†) or, to a very small extent, for defect-rich thin films.18,46 However, it remains unclear if reversible exsolution and re-dissolution upon redox cycling are possible across a coherent crystal surface with a low concentration of defects. We therefore recommend complementing studies of real-world but structurally complex ceramic catalysts with studies on well-defined epitaxial thin film model systems to further the understanding of reversibility limitations in exsolution catalysts. Moreover, it is necessary to use multimodal analysis approaches that provide complementary surface and bulk information (chemistry, structure, and morphology) in order to reduce the risk of delusive interpretation of the findings. For instance, it was reported that despite morphological changes being detected, i.e., the absence of spherical nanoparticles after reoxidation, hard-to-detect metal-rich phases may remain at the surface.17 Furthermore, a reversible change in the spectroscopic signature may be observed, but it may not be directly correlated to the exsolution and re-dissolution of catalytically relevant surface nanoparticles.4,18,46 Alternatively to a reversible exsolution and re-dissolution process, catalyst regeneration may be possible if extraordinarily stable nanostructured catalysts can be synthesized via metal exsolution. Here, supported (pinned) nanoparticles may be repeatedly reduced and oxidized during redox cycles, while the nanoparticle density is preserved.
3 Conclusions
We have investigated reversibility limitations in the metal exsolution response of nickel in STNNix perovskites. The bulk structures of ceramic oxide pellets and epitaxial thin films of SrTi0.95−xNb0.05NixO3−δ with systematically varying Ni doping levels x = 0.005–0.1 exhibit distinct differences with respect to the structural characteristics of the host lattice pointing towards an increasing dopant inhomogeneity with increasing doping levels. The bulk structure of the epitaxial thin films responds dynamically to thermal reduction and reoxidation (for x ≤ 0.05); however, the magnitude of the structural changes upon reduction appears not to be proportional to the doping level x. While the nanoparticle density that exsolves at the perovskite surface monotonically correlates with the doping concentration, the bulk and the surface responses appear to be mostly independent. Here, reoxidation results in fragmentation of the nanoparticles and increased surface wetting of the perovskite surface by the exsolved species, which goes along with a particle reconstruction towards a faceted shape and reduced average particle height. We presume that the nanoparticle re-dissolution is limited by the thermodynamic and kinetic boundary conditions, where high temperatures and long reoxidation times relative to the energetically favorable exsolution of metal dopants may be required.4 Experimental
The ceramic PLD targets with different Ni-doping levels were synthesized by a solid–state reaction of homogenized stoichiometric amounts of TiO nanopowder (99.7%), SrCO3 (≥98%), Nb2O5 (99.99%) and NiO nanopowder (99.99%). After calcination at 1100 °C for 8 h, a slurry in isopropanol was obtained from the product and ball milled for 24 h. The powder was dried using a rotary evaporator and ceramic pellets were prepared by uniaxial and subsequent cold-isostatic pressing. The pellets were sintered at 1200 °C for 12 h. X-ray diffraction of the ceramic pellets was performed in 2θ − ω geometry using a Bruker D4 Endeavor (Massachusetts, USA).PLD of 50 nm thick epitaxial thin films was performed using a Demcon TSST system (Enschede, The Netherlands) equipped with RHEED-monitoring to control the growth kinetics. The STNNix thin films were deposited on TiO2-terminated (001) SrTiO3 single crystal substrates. An excimer laser was used for ablation, where the repetition rate was f = 5 Hz and the laser fluence was J = 1.14 J cm−2. The substrate temperature was T = 650 °C and the oxygen partial pressure was p(O2) = 0.108 mbar. The distance between the ceramic target and the substrate is d = 60 mm. After the deposition, the samples were cooled down to room temperature with a maximum cooling rate of 50 °C min−1, which decreases during the cooling period owing to the resistive heating equipment.
The crystal structure of the epitaxial thin films was investigated by high-resolution XRD (D8 Discover, Bruker, Karlsruhe, Germany). A Goebel mirror, a Cu Kα monochromator, a centric Eulerian cradle and a Lynxeye XE detector were used for the analysis. A divergence aperture of 0.2 mm and a pinhole adapter of 2 mm diameter were applied.
The surface morphology of the epitaxial thin films was studied by AFM (Cypher, Oxford Instruments Asylum Research Inc., Santa Barbara). A probing tip with a curvature of ∼8 nm was used for the analysis. The particle density was determined by AFM imaging of three different locations of the thin films using a scan size of 5 × 5 μm2, respectively and Gwyddion 2.52 for data evaluation. A lower threshold of 1 nm was applied to determine the nanoparticle characteristics. The error bars denote the standard deviation of the averaged values.
Ex situ thermal reduction of the samples was performed under continuous 4% H2/Ar gas flow at T = 800 °C for 5 h using a quench oven. The reoxidation of the samples was performed in air at T = 800 °C in a box oven.
AP-XPS was performed at beamline 9.3.2 of the Advanced Light Source using soft X-rays with a photon energy of Ehv = 680 eV and Ehv = 900 eV. A hemispherical analyzer (Scienta R4000 HiPP) was differentially pumped to maintain ultra-high vacuum conditions at the detector. The inelastic mean free path was calculated with the QUASES-IMFP software using the TPP-2M equation67 on the basis of the material properties of as-prepared STNNi (x = 0.05). The binding energies of all spectra were aligned with respect to the Ti 2p position B.E. = 458.4 eV in order to compare relative peak shifts between Ti and Ni and to correct for space charge-induced, rigid peak shifts upon change in an ambient atmosphere. A Shirley-type background was subtracted. An initial annealing step in oxygen (p(O2) = 0.1 mbar, T = 400 °C, 25 min) was performed to clean the surface of adsorbed carbon species, and the first spectra were recorded under the same conditions. All other data sets shown were recorded subsequent to the respective annealing procedures, after cooling the sample to room temperature and evacuating the chamber. The reduction step was performed in hydrogen (p(H2) = 0.5 mbar, T = 500 °C, 127 min). Reoxidation was performed in oxygen by a three-step procedure ((1) p(O2) = 0.5 mbar, T = 300 °C, 80 min, (2) p(O2) = 0.5 mbar, T = 400 °C, 82 min, and (3) p(O2) = 0.5 mbar, T = 500 °C, 34 min). The STNNix (x = 0.05) sample employed for AP-XPS was deposited using a ceramic target that was synthesized by the Pechini method.
Author contributions
M. L. W. synthesized the ceramic oxide pellets and epitaxial thin films and performed the XRD, AFM and AP-XPS analyses and data evaluation (data curation, formal analysis, investigation, methodology, project administration, validation, and visualization) where C. L., S. N. and F. G. contributed to the evaluation of the data. Y. J. S. performed the Rietveld refinement of the X-ray diffractograms obtained from the ceramic oxide pellets. M. L. W., C. L., S. N. and F. G. conceptualized the experiments. R. W., O. G., R. D., N. H. M., C. L., S. N. and F. G. supervised the research. M. L. W. wrote the original draft and edited the manuscript with contributions from all authors. All authors reviewed the manuscript and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research used beamline 9.3.2 of the Advanced Light Source, a U.S. DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231. M. L. W. was supported in part by an ALS Collaborative Postdoctoral Fellowship. M. L. W. sincerely thanks Terry Mcafee and Grigory Potemkin for their experimental support.Notes and references
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- T. Z. Sholklapper, H. Kurokawa, C. P. Jacobson, S. J. Visco and L. C. De Jonghe, Nano Lett., 2007, 7, 2136–2141 CrossRef CAS.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164–167 CrossRef CAS PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- J. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- B. Hua, M. Li, Y.-F. Sun, J.-H. Li and J.-L. Luo, ChemSusChem, 2017, 10, 3333–3341 CrossRef CAS PubMed.
- M. L. Weber, M. Wilhelm, L. Jin, U. Breuer, R. Dittmann, R. Waser, O. Guillon, C. Lenser and F. Gunkel, ACS Nano, 2021, 15, 4546–4560 CrossRef CAS PubMed.
- J. Wang, A. Kumar, J. L. Wardini, Z. Zhang, H. Zhou, E. J. Crumlin, J. T. Sadowski, K. B. Woller, W. J. Bowman, J. M. LeBeau and B. Yildiz, Nano Lett., 2022, 22, 5401–5408 CrossRef CAS PubMed.
- M. L. Weber, B. Šmíd, U. Breuer, M.-A. Rose, N. H. Menzler, R. Dittmann, R. Waser, O. Guillon, F. Gunkel and C. Lenser, Space charge governs the kinetics of metal exsolution, 2023, DOI:10.26434/chemrxiv-2022-v9z6n.
- H. Lv, L. Lin, X. Zhang, Y. Song, H. Matsumoto, C. Zeng, N. Ta, W. Liu, D. Gao and G. Wang, et al. , Adv. Mater., 2020, 32, 1906193 CrossRef CAS PubMed.
- Z. Shang, J. Zhang, L. Ye and K. Xie, J. Mater. Chem. A, 2022, 10, 12458–12463 RSC.
- D. Hosseini, F. Donat, P. M. Abdala, S. M. Kim, A. M. Kierzkowska and C. R. Müller, ACS Appl. Mater. Interfaces, 2019, 11, 18276–18284 CrossRef CAS PubMed.
- D. Burnat, R. Kontic, L. Holzer, P. Steiger, D. Ferri and A. Heel, J. Mater. Chem. A, 2016, 4, 11939–11948 RSC.
- H. Tanaka, M. Taniguchi, M. Uenishi, N. Kajita, I. Tan, Y. Nishihata, J. Mizuki, K. Narita, M. Kimura and K. Kaneko, Angew. Chem., Int. Ed., 2006, 45, 5998–6002 CrossRef CAS PubMed.
- M. Santaya, C. E. Jiménez, H. E. Troiani, E. A. Carbonio, M. D. Arce, L. M. Toscani, R. Garcia-Diez, R. G. Wilks, A. Knop-Gericke, M. Bär and L. V. Mogni, J. Mater. Chem. A, 2022, 10, 15554–15568 RSC.
- K.-Y. Lai and A. Manthiram, Chem. Mater., 2018, 30, 2838–2847 CrossRef CAS.
- M. B. Katz, S. Zhang, Y. Duan, H. Wang, M. Fang, K. Zhang, B. Li, G. W. Graham and X. Pan, J. Catal., 2012, 293, 145–148 CrossRef CAS.
- D. Neagu, E. I. Papaioannou, W. K. Ramli, D. N. Miller, B. J. Murdoch, H. Ménard, A. Umar, A. J. Barlow, P. J. Cumpson and J. T. Irvine, et al. , Nat. Commun., 2017, 8, 1855 CrossRef PubMed.
- A. K. Opitz, A. Nenning, V. Vonk, S. Volkov, F. Bertram, H. Summerer, S. Schwarz, A. Steiger-Thirsfeld, J. Bernardi and A. Stierle, et al. , Nat. Commun., 2020, 11, 4801 CrossRef CAS PubMed.
- J. H. Kim, J. K. Kim, J. Liu, A. Curcio, J.-S. Jang, I.-D. Kim, F. Ciucci and W. Jung, ACS Nano, 2021, 15, 81–110 CrossRef CAS PubMed.
- O. Kwon, S. Joo, S. Choi, S. Sengodan and G. Kim, J. Phys.: Energy, 2020, 2, 032001 CAS.
- R. Martínez-Coronado, J. Alonso, A. Aguadero, D. Perez-Coll and M. Fernández-Díaz, J. Appl. Phys., 2013, 113, 123708 CrossRef.
- Q. Ma, B. Iwanschitz, E. Dashjav, S. Baumann, D. Sebold, I. Arul Raj, A. Mai and F. Tietz, J. Power Sources, 2015, 279, 678–685 CrossRef CAS.
- F. Horikiri, L. Han, N. Iizawa, K. Sato, K. Yashiro, T. Kawada and J. Mizusaki, J. Electrochem. Soc., 2007, 155, B16 CrossRef.
- O. A. Marina, N. L. Canfield and J. W. Stevenson, Solid State Ion., 2002, 149, 21–28 CrossRef CAS.
- Q. Fu, F. Tietz, D. Sebold, S. Tao and J. T. Irvine, J. Power Sources, 2007, 171, 663–669 CrossRef CAS.
- M. R. Pillai, I. Kim, D. M. Bierschenk and S. A. Barnett, J. Power Sources, 2008, 185, 1086–1093 CrossRef CAS.
- M. Schmidbauer, A. Kwasniewski and J. Schwarzkopf, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 8–14 CrossRef CAS PubMed.
- C. Rodenbücher, M. Luysberg, A. Schwedt, V. Havel, F. Gunkel, J. Mayer and R. Waser, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- P. Blennow, A. Hagen, K. K. Hansen, L. R. Wallenberg and M. Mogensen, Solid State Ionics, 2008, 179, 2047–2058 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Moos and K. H. Hardtl, J. Am. Ceram. Soc., 1997, 80, 2549–2562 CrossRef CAS.
- R. Meyer, A. F. Zurhelle, R. A. De Souza, R. Waser and F. Gunkel, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 94, 115408 CrossRef.
- F. Gunkel, R. Waser, A. H. H. Ramadan, R. A. De Souza, S. Hoffmann-Eifert and R. Dittmann, Phys. Rev. B, 2016, 93, 245431 CrossRef.
- P. Blennow, K. K. Hansen, L. R. Wallenberg and M. Mogensen, J. Eur. Ceram. Soc., 2007, 27, 3609–3612 CrossRef CAS.
- U. Balachandran and N. G. Eror, J. Electrochem. Soc., 1982, 129, 1021 CrossRef CAS.
- E. Enriquez, A. Chen, Z. Harrell, P. Dowden, N. Koskelo, J. Roback, M. Janoschek, C. Chen and Q. Jia, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- F. Gunkel, D. V. Christensen, Y. Chen and N. Pryds, Appl. Phys. Lett., 2020, 116, 120505 CrossRef CAS.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- A. Chen, J.-M. Hu, P. Lu, T. Yang, W. Zhang, L. Li, T. Ahmed, E. Enriquez, M. Weigand and Q. Su, et al. , Sci. Adv., 2016, 2, e1600245 CrossRef PubMed.
- J. L. MacManus-Driscoll, P. Zerrer, H. Wang, H. Yang, J. Yoon, A. Fouchet, R. Yu, M. G. Blamire and Q. Jia, Nat. Mater., 2008, 7, 314–320 CrossRef CAS PubMed.
- D. J. Keeble, S. Wicklein, R. Dittmann, L. Ravelli, R. A. Mackie and W. Egger, Phys. Rev. Lett., 2010, 105, 226102 CrossRef CAS PubMed.
- E. Breckenfeld, R. Wilson, J. Karthik, A. R. Damodaran, D. G. Cahill and L. W. Martin, Chem. Mater., 2012, 24, 331–337 CrossRef CAS.
- F. Gunkel, S. Wicklein, S. Hoffmann-Eifert, P. Meuffels, P. Brinks, M. Huijben, G. Rijnders, R. Waser and R. Dittmann, Nanoscale, 2015, 7, 1013–1022 RSC.
- M. B. Katz, G. W. Graham, Y. Duan, H. Liu, C. Adamo, D. G. Schlom and X. Pan, J. Am. Chem. Soc., 2011, 133, 18090–18093 CrossRef CAS PubMed.
- A. P. LaGrow, D. C. Lloyd, P. L. Gai and E. D. Boyes, Chem. Mater., 2018, 30, 197–203 CrossRef CAS.
- M. Andrä, F. Dvořák, M. Vorokhta, S. Nemšák, V. Matolín, C. M. Schneider, R. Dittmann, F. Gunkel, D. N. Mueller and R. Waser, APL Mater., 2017, 5, 056106 CrossRef.
- M. Andrä, H. Bluhm, R. Dittmann, C. Schneider, R. Waser, D. Mueller and F. Gunkel, Phys. Rev. Mater., 2019, 3, 044604 CrossRef.
- M. Andrä, C. Funck, N. Raab, M.-A. Rose, M. Vorokhta, F. Dvorák, B. Šmíd, V. Matolín, D. N. Mueller and R. Dittmann, et al. , Adv. Electron. Mater., 2020, 6, 1900808 CrossRef.
- H. Kersell, M. Weber, L. Falling, Q. Lu, C. Baeumer, N. Shirato, V. Rose, C. Lenser, F. Gunkel and S. Nemšák, Faraday Discuss., 2022, 236, 141–156 RSC.
- H. Lv, L. Lin, X. Zhang, R. Li, Y. Song, H. Matsumoto, N. Ta, C. Zeng, Q. Fu and G. Wang, et al. , Nat. Commun., 2021, 12, 5665 CrossRef CAS PubMed.
- E. Ruckenstein and S. H. Lee, J. Catal., 1987, 104, 259–278 CrossRef CAS.
- E. Ruckenstein and J. Chen, J. Colloid Interface Sci., 1982, 86, 1–11 CrossRef CAS.
- I. Sushumna and E. Ruckenstein, J. Catal., 1985, 94, 239–288 CrossRef CAS.
- K. J. Kim, H. Han, T. Defferriere, D. Yoon, S. Na, S. J. Kim, A. M. Dayaghi, J. Son, T.-S. Oh and H. M. Jang, et al. , J. Am. Chem. Soc., 2019, 141, 7509–7517 CrossRef CAS PubMed.
- D. Jennings, S. Ricote, J. Santiso, J. Caicedo and I. Reimanis, Acta Mater., 2022, 228, 117752 CrossRef CAS.
- M. L. Weber, M. Kindelmann, E. Wessel, A. Sarantopoulos, N. H. Menzler, R. Dittmann, R. Waser, O. Guillon, C. Lenser and F. Gunkel, J. Phys.: Energy, 2022, 5, 014002 Search PubMed.
- Y. Gao, Z. Lu, T. L. You, J. Wang, L. Xie, J. He and F. Ciucci, J. Phys. Chem. Lett., 2018, 9, 3772–3778 CrossRef CAS PubMed.
- E. Calí, M. P. Thomas, R. Vasudevan, J. Wu, O. Gavalda-Diaz, K. Marquardt, E. Saiz, D. Neagu, R. R. Unocic, S. C. Parker, B. S. Guiton and D. J. Payne, Nat. Commun., 2023, 14, 1754 CrossRef PubMed.
- J. Wang, J. Yang, A. K. Opitz, W. Bowman, R. Bliem, G. Dimitrakopoulos, A. Nenning, I. Waluyo, A. Hunt, J.-J. Gallet and B. Yildiz, Chem. Mater., 2021, 33, 5021–5034 CrossRef CAS.
- H. Han, Y. Xing, B. Park, D. I. Bazhanov, Y. Jin, J. T. Irvine, J. Lee and S. H. Oh, Nat. Commun., 2022, 13, 6682 CrossRef CAS PubMed.
- S. Singh, E. Prestat, L.-F. Huang, J. M. Rondinelli, S. J. Haigh and B. A. Rosen, Sci. Rep., 2017, 7, 10080 CrossRef PubMed.
- T. Heisig, J. Kler, H. Du, C. Baeumer, F. Hensling, M. Glöβ, M. Moors, A. Locatelli, T. O. Menteş and F. Genuzio, et al. , Adv. Funct. Mater., 2020, 30, 2004118 CrossRef CAS.
- P. A. Cox, Transition metal oxides: an introduction to their electronic structure and properties, Oxford University Press, 2010, vol. 27.
- K. Szot, W. Speier, U. Breuer, R. Meyer, J. Szade and R. Waser, Surf. Sci., 2000, 460, 112–128 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02927a |
‡ Present address: Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 720, United States & Peter Gruenberg Institute (PGI-7) & JARA-FIT, Forschungszentrum Juelich GmbH, Wilhelm-Johnen-Straße, 52425 Juelich, Germany. 720, United States & Peter Gruenberg Institute (PGI-7) & JARA-FIT, Forschungszentrum Juelich GmbH, Wilhelm-Johnen-Straße, 52425 Juelich, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |