
Cationic vacancies accelerate the generation of CoOOH in perovskite hydroxides for the electrooxidation of biomass†
Rixin
Peng‡
a,
Yimin
Jiang‡
a,
Chung-Li
Dong‡
 b,
Ta Thi
Thuy Nga‡
b,
Yuxuan
Lu
a,
Shengkai
Li
a,
Yun
Fan
a,
Chao
Xie
ac,
Shuangyin
Wang
a and
Yuqin
Zou
*a
b,
Ta Thi
Thuy Nga‡
b,
Yuxuan
Lu
a,
Shengkai
Li
a,
Yun
Fan
a,
Chao
Xie
ac,
Shuangyin
Wang
a and
Yuqin
Zou
*a
aState Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Advanced Catalytic Engineering Research Center of the Ministry of Education, Hunan University, Changsha 410082, China. E-mail: yuqin_zou@hnu.edu.cn
bDepartment of Physics, Tamkang University, New Taipei City 25137, Taiwan
cShenzhen Institute of Hunan University, Shenzhen 518057, China
First published on 28th June 2023
Abstract
The electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid is an environmentally friendly strategy for enhancing biomass energy. Defect engineering has been demonstrated as a promising strategy for increasing the catalytic activity of the HMF oxidation reaction (HMFOR). However, the exact function of cationic vacancies has been rarely studied due to the higher formation energy. Herein, Co-based perovskite hydroxide [SnCo(OH)6] was used as a mode material to investigate the critical role of cationic vacancies in HMFOR. A simple plasma-engraving strategy was applied to prepare Co-based perovskite hydroxides with abundant Sn cation vacancies [SnCo(OH)6-VSn]. Compared to SnCo(OH)6, the advanced potential of SnCo(OH)6-VSn is 200 mV, indicating that cationic vacancies are a promising strategy for enhancing the performance of the HMFOR. Operando characterization and theoretical calculations revealed that the short-range ordered amorphous structure broke the periodicity of the crystalline lattice to produce a large number of cation vacancies. Subsequently, these vacancies facilitated the transfer of electrons, the oxidation of transition metal Co at the B site to a higher valence state, and the formation of deprotonated Co(OH)2 active species, thereby promoting the adsorption of HMF on the catalyst surface. Thus, the current study provided significant insights into the critical role of cationic vacancies in enhancing the electrocatalytic performance of biomass upgrading via Co-based catalysts.
 Yuqin Zou | Yuqin Zou received her PhD in 2017 from the University of Manchester, UK. She is currently a professor of the College of Chemistry and Chemical Engineering, Hunan University. Her research interests are the organic electrocatalytic conversion. |
Introduction
With the rapid development of modern society, the global consumption of non-renewable energy sources such as coal, oil, and natural gas has increased, resulting in a number of environmental issues.1 Biomass, an abundant and renewable resource, can be used as a substitute for fossil fuels to achieve sustainable development.2,3 5-Hydroxymethylfurfural (HMF) is a vital biomass platform molecule.4,5 2,5-furandicarboxylic acid (FDCA), the typical oxidation product of HMF, can be used as an environmentally friendly alternative to the petroleum-based monomer terephthalic acid for the preparation of renewable bio-based polyethylene terephthalate (PET).6,7 In this regard, the HMF electrocatalytic oxidation reaction (HMFOR) is regarded as a green, efficient, and promising method for producing high-value chemicals, which can replace the kinetically slow oxygen evolution reaction (OER) at the anode, while concurrently achieving high-efficiency hydrogen production at the cathode.8–10Perovskite hydroxide [AB(OH)6] is a type of hydroxide with a typical perovskite structure. Typically, a rare earth metal occupies the A site, while a transition metal occupies the B site.11 By substituting A and B cations, the electronic structure, oxidation state, and catalytic performance of perovskite structures can be altered. It has been reported that both perovskite hydroxides and oxides can accommodate multiple A- and B-site cations, allowing for the fine-tuning of their electronic and catalytic properties and making them suitable for examining the relationship between structure and performance in electrocatalysis.12 Compared to perovskite oxides, perovskite hydroxides have a distorted ReO3-type structure that is more susceptible to defect formation.13,14 This unique property endows perovskite hydroxides with enormous potential as a mode electrode for the design and development of high-efficiency electrocatalysts. Prior to now, the application of perovskite hydroxide in electrocatalysis has been limited, and the lack of intrinsic active sites is regarded as one of the greatest obstacles limiting their applications.15–20
Creating vacancies has been demonstrated to be an effective strategy for regulating the electronic structure and surface/interface microenvironment of electrocatalysts. This strategy has been extensively utilized for HMFOR.21–24 For instance, our group demonstrated that the nucleophilic OH− was easily incorporated into the oxygen-deficient lattice of the Co3O4 electrocatalyst to facilitate the 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) dehydrogenation, resulting in improved HMFOR performance.25 Wang et al., demonstrated that oxygen vacancies (Vo) at low oxidation potential promoted the pre-oxidation of low-valent Ni in NiO, inducing catalyst surface reconstruction and synergistically promoting HMFOR.26 In addition to the aforementioned efforts, numerous others have been made to create Vo in order to improve the HMFOR performance. However, studies on the use of cationic defects to accomplish this goal have been very limited, probably because the formation energy of cationic vacancies is significantly higher than that of Vo.
Herein, we used Co-based perovskite hydroxide [SnCo(OH)6] as a mode material to reveal the critical role of cation vacancies in HMFOR by employing a simple plasma-engraving strategy to create cationic vacancies in SnCo(OH)6. Due to the lower chemical bonding energy and lattice energy of Sn(OH)4, plasma treatment favored the separation of A-site Sn4+ from the perovskite host to form amorphous surface structures with abundant Sn cation vacancies. This short-range ordered amorphous structure effectively disrupted the periodicity of the crystalline material. In addition, the amorphous structure provided abundant vacancies, facilitated electron transfer, the oxidation of transition metal Co at the B site to a higher valence state, and the production of deprotonated Co(OH)2 active species, thereby enhancing the adsorption of HMF on the catalyst surface. This study, therefore, provided new insights into the creation of cationic vacancies in Co-based catalysts to enhance the electrocatalytic upgrading of biomass.
Results and discussion
The SnCo(OH)6 was prepared using a simple co-precipitation method, and the plasma-treated SnCo(OH)6 was designated as SnCo(OH)6-VSn (the fabrication procedures are detailed in the ESI†). First, X-ray diffraction (XRD) analysis was utilized to confirm the crystal structures of SnCo(OH)6 and SnCo(OH)6-VSn. As shown in Fig. S1,† all the diffraction peaks of SnCo(OH)6 can be attributed to its perovskite hydroxide phase (JCPDS card No. 13-0356). A broad peak at around 33° was observed in the XRD pattern of SnCo(OH)6-VSn, demonstrating its amorphous structure.27 Morphological changes of the material before and after the plasma treatment were further confirmed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SnCo(OH)6 exhibited a nanocube structure with an average particle size of 80 nm (Fig. S2a–c†). After plasma treatment, the morphology changed to echinus-like nanospheres with a significantly smaller average particle size of ∼68 nm (Fig. S2d–f†). Moreover, the high-resolution TEM (HR-TEM) image of SnCo(OH)6 revealed that it had a smooth surface and excellent crystallization (Fig. 1a). The 0.44 nm lattice fringe spacing in HR-TEM corresponds to the (111) crystal plane of ReO3-type perovskite hydroxides SnCo(OH)6.28 The SAED pattern further demonstrated the high crystallinity of SnCo(OH)6 (inset of Fig. 1a). In contrast, no prominent lattice fringe was observed in the HR-TEM of SnCo(OH)6-VSn. No diffraction spot was observed in the SAED pattern, indicating that SnCo(OH)6-VSn had a distinct amorphous structure (Fig. 1b).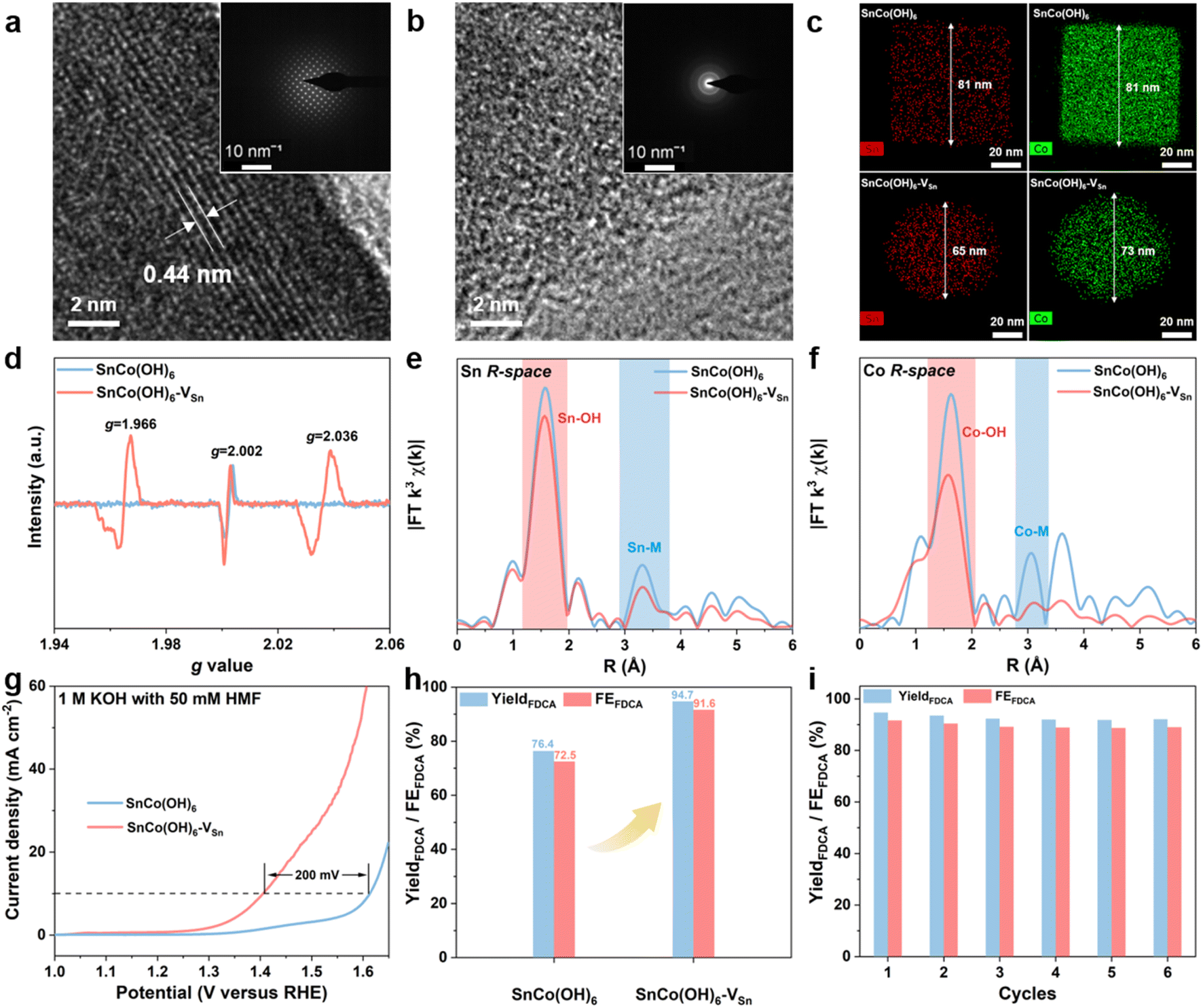 | ||
| Fig. 1 HR-TEM images and SAED patterns of (a) SnCo(OH)6 and (b) SnCo(OH)6-VSn; (c) HAADF-mapping images of SnCo(OH)6 and SnCo(OH)6-VSn; (d) EPR spectra of SnCo(OH)6 and SnCo(OH)6-VSn; R-space of (e) Sn and (f) Co in SnCo(OH)6 and SnCo(OH)6-VSn; (g) LSV curves for SnCo(OH)6 and SnCo(OH)6-VSn in 1 M KOH with 50 mM HMF; (h) yield and FE of FDCA at 1.47 VRHE for SnCo(OH)6 and SnCo(OH)6-VSn; (i) yield and FE of FDCA on SnCo(OH)6-VSn for six successive electrolysis cycles. | ||
The distribution of Sn and Co elements in SnCo(OH)6 and SnCo(OH)6-VSn was also examined using high-angle annular dark-field (HAADF)-mapping imaging (Fig. 1c). In both SnCo(OH)6 and SnCo(OH)6-VSn, Sn and Co signals were detected. In addition, Co had a smaller element domain in SnCo(OH)6-VSn (73 nm) than in SnCo(OH)6 (81 nm). Moreover, the element domain of Sn (65 nm) was smaller than that of Co (73 nm) in SnCo(OH)6-VSn, indicating that the Ar plasma reduced the particle size of the hydroxides and selectively etched Sn in SnCo(OH)6-VSn.
In order to confirm the presence of vacancies in SnCo(OH)6 and SnCo(OH)6-VSn, electron paramagnetic resonance (EPR) analyses were performed (Fig. 1d). Due to the intrinsic dislocation of the structure, a characteristic peak of oxygen vacancy at g = 2.002 was observed in both samples.29,30 After plasma etching, two additional vacancy characteristic peaks appeared at g = 1.966 and g = 2.036 for SnCo(OH)6-VSn, indicating the formation of Sn cationic vacancies.31 It was reasonable to conclude that the lack of Sn elements on the SnCo(OH)6-VSn surface led to the collapse of the crystal structure, resulting in the formation of vacancies and the amorphous surface.
To further investigate the coordination environment and valence state of SnCo(OH)6-VSn, X-ray photoelectron spectroscopy (XPS) and X-ray absorption near-edge structure (XANES) experiments were performed (Fig. S3 and S4†). The binding energies of Co 2p in the XPS spectra exhibited a positive shift in Fig. S3b,† indicating that Co sites gained electrons following plasma treatment.32 In addition, the Co K-edge XANES in Fig. S4a† revealed that the spectral oscillation of SnCo(OH)6-VSn was weaker than that of SnCo(OH)6. Moreover, the intensity of the prominent absorption peak at 7725 eV was reduced due to the fact that plasma treatment disrupted the crystallinity of the catalyst, resulting in modifications to the electronic structure and coordination environment. Compared to SnCo(OH)6, the absorption edge of SnCo(OH)6-VSn shifted to lower energy (Fig. S4a inset†).33 This was because Sn vacancies increased the local electron density of Co, indicating a slight decrease in the valence state of Co. The prominent absorption peaks and spectral distribution of Co and Sn were comparable to those of CoO and SnO2, respectively (Fig. S5†), confirming the XPS results (Fig. S3†). In addition, extended X-ray absorption near-edge structure (EXAFS) analyses were utilized to investigate the coordination environment of Co and Sn. As shown in Fig. 1e and f, the peaks at approximately 1.6 Å and 3.1 Å were attributed to the M–OH and M–M bonds (M = Co or Sn), respectively.34 The coordination number of M–M in SnCo(OH)6-VSn was significantly lower than that in SnCo(OH)6, indicating the presence of metal vacancies, and the reduced M–OH coordination number at 1.6 Å was due to deprotonation caused by the cation vacancies.27,35
Furthermore, we evaluated the effect of vacancies by measuring the electrocatalytic HMFOR performance of the resulting materials. As depicted in Fig. 1g, the current density of SnCo(OH)6 for HMFOR reached 10 mA cm−2 at 1.61 VRHE. In order to achieve the same current density with SnCo(OH)6-VSn, the oxidation potential of HMFOR was shifted by 200 mV. In addition, the electrochemically active surface area (ECSA) of the catalysts was calculated based on the current density in the non-faradaic reaction region at various scan rates (Fig. S6†). The ECSA of SnCo(OH)6-VSn (51.82 mF cm−2) was significantly higher than that of SnCo(OH)6 (13.83 mF cm−2), indicating that the amorphous structure exposed more active sites on the surface. In addition, based on the N2 adsorption and desorption experiment and the Brunauer–Emmett–Teller (BET) equation, the specific surface area of SnCo(OH)6-VSn was 62.19 m2 g−1, which was significantly greater than that of SnCo(OH)6 (27.19 m2 g−1) (Fig. S7†). The HMFOR current density was normalized by both BET and ECSA (Fig. S8†). Additionally, the normalized current density of SnCo(OH)6-VSn was greater than that of SnCo(OH)6, indicating that SnCo(OH)6-VSn possessed greater intrinsic activity. In addition, potentiostatic electrolysis was applied to the electrooxidation of HMF in a divided cell at 1.47 VRHE. Using high-performance liquid chromatography (HPLC) (Fig. S9†), the oxidation products were identified and quantified. As shown in Fig. 1h, a high performance of 94.7% yield and 91.6% faradaic efficiency (FE) was achieved for FDCA production at the SnCo(OH)6-VSn electrode, which was considerably superior to the SnCo(OH)6 electrode (76.4% yield and 72.5% FE). Finally, six consecutive potentiostatic cyclic electrolysis were performed on the same SnCo(OH)6-VSn electrode. All of these compounds maintained a high yield and FE, indicating that SnCo(OH)6-VSn exhibited excellent stability for HMFOR (Fig. 1i).
Initially, a number of electrochemical techniques were employed to determine the cause of the enhanced electrocatalytic performance of SnCo(OH)6-VSn. Using electrochemical impedance spectroscopy (EIS) measurements, the reaction kinetics and electron transfer between electrode and electrolyte were further investigated. By fitting the Nyquist plots (Fig. S11 and 12†), the equivalent circuit (Fig. S10†) and its parameters (Table S1–S4†) were determined. Fig. 2a shows the Nyquist plots of SnCo(OH)6 and SnCo(OH)6-VSn at 1.35 VRHE in 1 M KOH with 50 mM HMF. The lower charge transfer resistance of SnCo(OH)6-VSn compared to SnCo(OH)6 indicates that SnCo(OH)6-VSn has superior electronic conductivity and a quicker electron transfer rate. Moreover, the Tafel slope of SnCo(OH)6-VSn (133.6 mV dec−1) was lower than that of SnCo(OH)6 (340.3 mV dec−1), indicating a faster electron-transfer rate and improved reaction kinetics (Fig. 2b). In addition, the redox behavior of the two catalysts were evaluated using cyclic voltammetry (CV) tests in 1 M KOH (Fig. S13†). SnCo(OH)6-VSn exhibited a more pronounced oxidation peak of Co2+ to Co3+, indicating easier generation of high-valence Co3+ species in SnCo(OH)6-VSn. Typically, the change in valence of a metal site is closely related to HMFOR activity. As a result, it is necessary to provide more direct evidence to prove the conversion between Co2+ and Co3+ in SnCo(OH)6-VSn during the HMFOR process through quasi-in situ and in situ characterization.
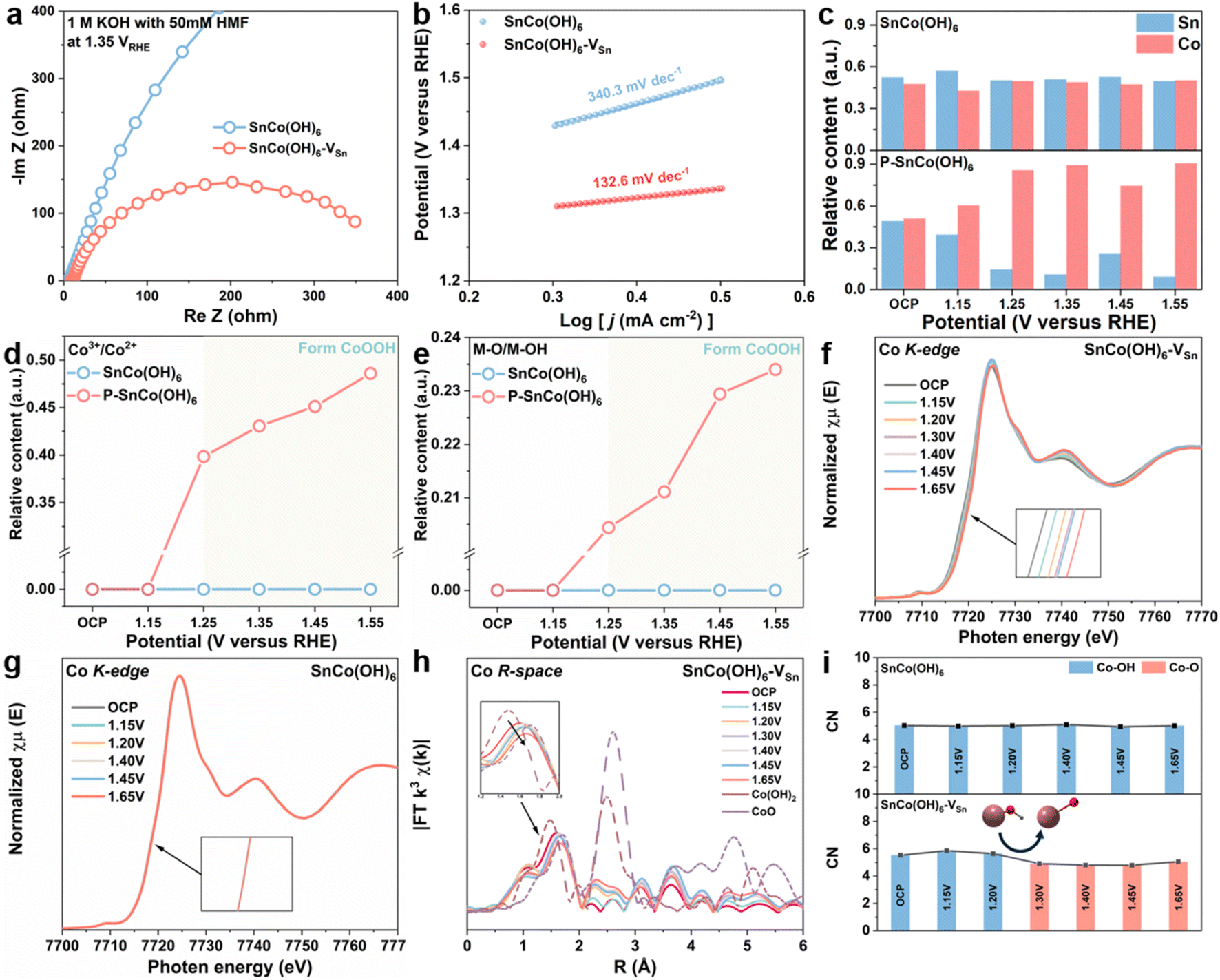 | ||
| Fig. 2 (a) Nyquist plots for SnCo(OH)6 and SnCo(OH)6-VSn in 1 M KOH with 50 mM HMF at 1.35 VRHE. (b) Tafel slope of SnCo(OH)6 and SnCo(OH)6-VSn. (c) The relative content of Sn and Co at different potentials for SnCo(OH)6 and SnCo(OH)6-VSn. The relative ratio of (d) Co3+/Co2+ and (e) M–O/M–OH for SnCo(OH)6 and SnCo(OH)6-VSn at different potential. The Co K-edge of (f) SnCo(OH)6-VSn and (g) SnCo(OH)6 from operando XANES spectra. (h) The Co R-space of CoO, Co(OH)2 and SnCo(OH)6-VSn from operando EXAFS spectra. (i) The CNs at various potentials for SnCo(OH)6 and SnCo(OH)6-VSn obtained from the fitting of EXAFS spectra. | ||
To determine the relationship between valence state and electrocatalytic reactivity, quasi-in situ XPS was used to examine the electronic structure and surface composition of SnCo(OH)6 and SnCo(OH)6-VSn during the HMFOR process (Fig. S14–S16†). As depicted in Fig. 2c, the relative content of Sn in SnCo(OH)6-VSn was less than that in SnCo(OH)6 at open circuit potential, further validating the selective etching of Sn by plasma treatment. At various reaction potentials, the ratio of Sn to Co in SnCo(OH)6 did not change significantly. However, when the potential reached 1.25 V, the Sn relative content of SnCo(OH)6-VSn decreased dramatically. In addition, the microporous structure and the amorphous surface of SnCo(OH)6-VSn likely promoted alkaline etching and contributed to the further dissolution of Sn, which led to the aforementioned results.15,17 Moreover, the dissolution process increased the number of Sn vacancies in SnCo(OH)6-VSn during the reaction process. Notably, Co3+ (780.0 eV) and metal-O (529.8 eV) were detected in SnCo(OH)6-VSn beginning at 1.25 VRHE,36 and the content of Co3+ and metal-O increased as the potential increased. However, neither Co3+ nor metal-O for SnCo(OH)6 was detected until the potential reached 1.55 VRHE (Fig. 2d and e). Thus, these results illustrate that the valence state of Co increased to maintain charge equilibrium after the dissolution of high-valent Sn, and Co(OH)2 was deprotonated to form the novel species of CoOOH. As a result, CoOOH was considered to be the active species of Co-based catalysts for HMFOR.37,38
The changes in the coordination environment of the catalyst during the HMFOR process were further investigated using in situ XANES and EXAFS analyses. As depicted in Fig. 2f, as the reaction potential increased, the Co K-edge XANES of SnCo(OH)6-VSn shifted to higher photon energies at 7720 eV, indicating the valence state of Co.33 However, no such trend was observed for SnCo(OH)6 in Fig. 2g, indicating that the valence state of Co was maintained throughout the reaction. Fig. 2h depicted the Co R-space of SnCo(OH)6-VSn at various reaction potentials, which revealed a gradual transition from Co–OH to Co–O as the reaction potentials increased. As compared, no discernible transition shift was observed for SnCo(OH)6 in Fig. S17.† The coordination number (CN) derived from the least-squares curve fitting of EXAFS was utilized to reveal the evolution of the coordination environment of SnCo(OH)6 and SnCo(OH)6-VSn at various potentials (Fig. 2i, Tables S5 and S6†). For SnCo(OH)6, only Co–OH was detected at OCP and various reaction potentials. In contrast, for SnCo(OH)6-VSn, Co–OH was observed to transform Co–O at 1.30 VRHE. Moreover, after the transformation, there was a slight decrease in CN of Co–O, which could be attributed to the dehydrogenation of Co(OH)2 to CoOOH. This finding further illustrated that the amorphous surface with abundant cation vacancies promoted the oxidation of Co, deprotonation of Co(OH)2, and formation of CoOOH species.
In addition, electrochemical experiments and theoretical calculations were used to reveal the distinct adsorption behavior caused by changes in the surface electronic structure of the catalyst. First, density functional theory (DFT) calculations were performed to investigate the adsorption behavior of SnCo(OH)6 and SnCo(OH)6-VSn. The energetically favorable SnCo(OH)6 (100) crystal plane was selected, and the B-site Co was used as the active catalytic site.13,19 The carbon in the furan ring of the HMF molecule was attached to the oxygen on the surface of SnCo(OH)6 and SnCo(OH)6-VSn, which was the optimal adsorption model determined by the calculated results. To simulate the Sn vacancies and structural distortions caused by plasma treatment, Sn atoms were then selectively removed from the metal layer (Fig. 3a inset). The adsorption of HMF on SnCo(OH)6 was a complex process with a barrier of 1.01 eV. Comparatively, SnCo(OH)6-VSn underwent a spontaneous thermodynamic process with a barrier of −0.50 eV (Fig. 3a). The density of states (DOS) of free HMF molecules and adsorbed HMF on SnCo(OH)6 and SnCo(OH)6-VSn were also calculated (Fig. 3b). The DOS of free HMF molecules and adsorbed HMF on SnCo(OH)6 coincided, indicating a lack of interaction between HMF and SnCo(OH)6 molecule. Nevertheless, the DOS distribution of the adsorbed HMF for SnCo(OH)6-VSn was significantly different from that of the free HMF molecule, indicating a strong interaction between SnCo(OH)6-VSn and the HMF molecule. These results demonstrated that the adsorption of HMF molecules on SnCo(OH)6-VSn was more facile than on SnCo(OH)6, which was attributable to the decreased OH− coverage on the catalyst surface. The H atoms of the M–OH on the surface of the catalyst would repel the H atoms of the HMF molecule. Meanwhile, the generation of vacancies decreased M–OH. Thus, the repulsive force between the catalyst surface and the HMF molecules was decreased, facilitating adsorption even more (Fig. 3c).
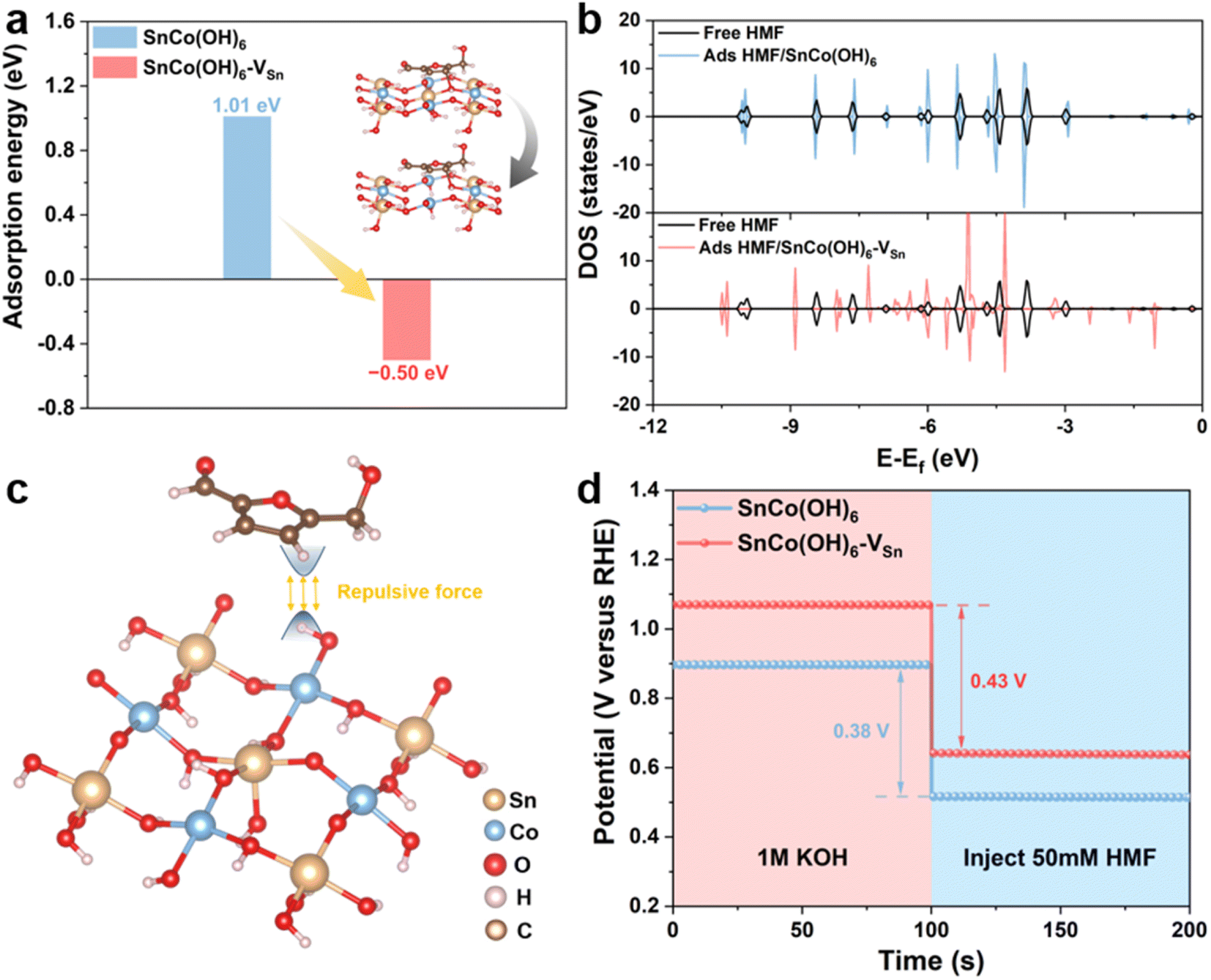 | ||
| Fig. 3 (a) The adsorption energy of HMF on SnCo(OH)6 and SnCo(OH)6-VSn (inset: the adsorption model of HMF on two electrocatalysts); (b) DOS for HMF molecules in SnCo(OH)6 and SnCo(OH)6-VSn; (c) the repulsive force between the catalyst surface and the HMF molecules; (d) OCP curves of SnCo(OH)6 and SnCo(OH)6-VSn when 50 mM HMF was injected subsequently. | ||
Meanwhile, open circuit potential (OCP) was carried out to assess the adsorption behavior of HMF on the catalyst surface. As shown in Fig. 3d, the OCP of SnCo(OH)6-VSn decreased by 0.43 V after the addition of 50 mM HMF, which was greater than that of SnCo(OH)6 (0.38 V), indicating that SnCo(OH)6-VSn could adsorb more HMF molecules.39
Real-time monitoring of complex reaction processes and intermediates could reveal the crucial role of HMFOR vacancies. Since HMF has two functional groups, hydroxyl and aldehyde, there are two pathways for the oxidation of HMF to FDCA. As shown in Fig. 4a, HMF first adsorbs on the surface of the catalyst. In pathway 1, the aldehyde group is oxidized to the carboxyl group first, resulting in the formation of HMFCA; in pathway 2, the hydroxyl group is oxidized to the aldehyde group first, resulting in the formation of DFF. Then, both HMFCA and DFF are oxidized to FFCA and desorb from the surface of the catalyst to produce FDCA.40–42 To determine the reaction pathway, intermediates were identified by HPLC at various electrolysis coulombs (Fig. S18†). Fig. 4b and S19† illustrate the HMFOR products for SnCo(OH)6-VSn and SnCo(OH)6 with varying electrolysis coulombs. Pathway 1 was the primary oxidation route for HMFOR on both SnCo(OH)6 and SnCo(OH)6-VSn, as evidenced by the presence of HMFCA throughout the entire electrolysis process. Meanwhile, it indicated the complexity of the transition from HMFCA to FFCA. Thus, we amplified the HMFCA concentration Y-axis in Fig. 4c. HMFCA reached a maximum (2.42 mM) for SnCo(OH)6-VSn during electrolysis at 40 C, then converted to FDCA and decreased to a minimum value at 60 C (0.87 mM). In contrast, for SnCo(OH)6, HMFCA maintained a high concentration (2.16 mM) during electrolysis at 60 C, indicating that the presence of vacancies accelerated the conversion of HMFCA to FFCA.
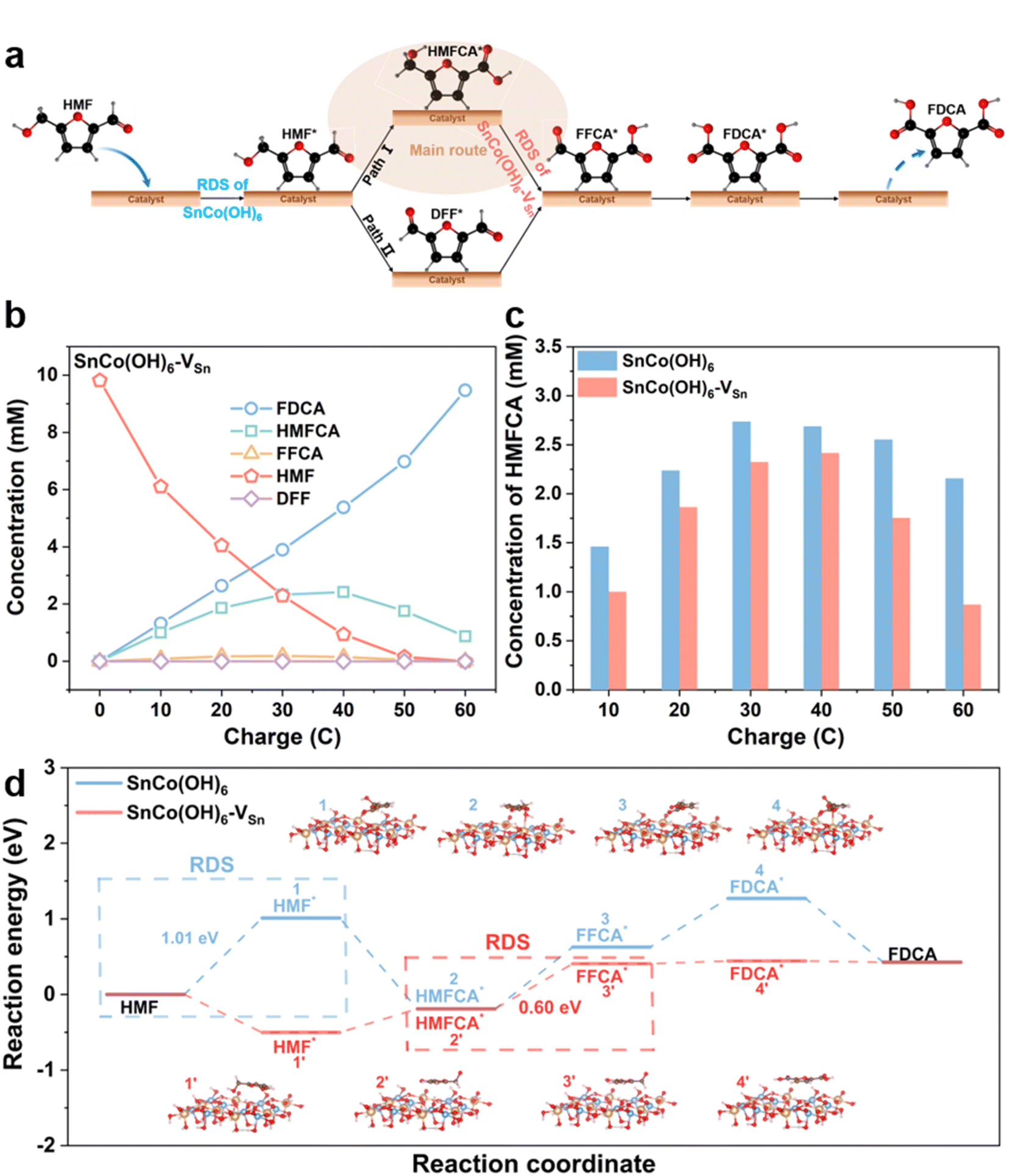 | ||
| Fig. 4 (a) Two possible reaction pathways for HMF oxidation. (b) Concentration changes of HMF and its oxidation products during the electrochemical oxidation of HMF at 1.47 VRHE in 1.0 M KOH with 10 mM HMF for SnCo(OH)6-VSn. (c) The concentration of HMFCA during HMFOR for SnCo(OH)6 and SnCo(OH)6-VSn. (d) The calculated free energy diagram for HMFOR on SnCo(OH)6 and SnCo(OH)6-VSn. | ||
Based on literature and experimental evidence, we calculated the free energy diagram of SnCo(OH)6 and SnCo(OH)6-VSn during HMFOR in Fig. 4d. The rate-determining step (RDS) for SnCo(OH)6 in the HMFOR process was the adsorption of HMF molecules onto the catalyst surface, which exhibited the highest free energy barrier (1.01 eV). For SnCo(OH)6-VSn, the RDS for the dehydrogenation of HMFCA to FFCA (0.60 eV) decreased from that of SnCo(OH)6. Notably, the free energy barrier of SnCo(OH)6 in the dehydrogenation of HMFCA to FFCA (0.82 eV) was higher than that of SnCo(OH)6-VSn. These findings suggested that vacancies accelerated the formation of CoOOH, lowered the free energy barrier of RDS, facilitated the dehydrogenation step of the HMFCA intermediate, and increased the reaction activity.
Conclusions
Herein, Co-based perovskite hydroxide (SnCo(OH)6) was utilized as a mode material to elucidate the significance of cationic vacancies in HMFOR. A series of characterizations revealed that vacancies increased the specific surface area of the catalyst to form an amorphous surface layer and expose more active sites, modulating the electrical conductivity and decreasing the charge transfer resistance. Significantly, the absence of Sn at the A site promoted the oxidation of Co at the B site, producing CoOOH active species and resulting in a catalyst with higher intrinsic activity. The introduction of cationic vacancies promoted the adsorption of HMF on the catalyst surface and accelerated the dehydrogenation step of the HMFCA intermediate, according to DFT calculations. Therefore, SnCo(OH)6-VSn catalyst demonstrated excellent catalytic performance for the electrochemical oxidation of HMF to FDCA, resulting in a low onset potential of 1.41 VRHE, a high FDCA yield of 94.7%, and adequate stability. This research determined that introducing cationic vacancies into Co-based electrocatalysts could improve the electrocatalytic performance of HMF, which could inform the design of efficient and advanced HMFOR electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Program of China (No. 2020YFA0710000), the National Natural Science Foundation of China (No. 22122901), the Provincial Natural Science Foundation of Hunan (No. 2021JJ20024, and 2021RC3054), and the Shenzhen Science and Technology Program (No. JCYJ20210324140610028).References
- S. Pacala and R. Socolow, Sci, 2004, 305, 968–972 CrossRef CAS PubMed.
- P. Prabhu, Y. Wan and J.-M. Lee, Matter, 2020, 3, 1162–1177 CrossRef.
- H. Zhao, D. Lu, J. Wang, W. Tu, D. Wu, S. W. Koh, P. Gao, Z. J. Xu, S. Deng, Y. Zhou, B. You and H. Li, Nat. Commun., 2021, 12, 2008 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- S. Rajendran, R. Raghunathan, I. Hevus, R. Krishnan, A. Ugrinov, M. P. Sibi, D. C. Webster and J. Sivaguru, Angew. Chem., Int. Ed., 2015, 54, 1159–1163 CrossRef CAS PubMed.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- C. Tang, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- M. Yang, Z. Yuan, R. Peng, S. Wang and Y. Zou, Energy Environ. Mater., 2022, 5, 1117–1138 CrossRef CAS.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- J. R. Neilson, J. A. Kurzman, R. Seshadri and D. E. Morse, Inorg. Chem., 2011, 50, 3003–3009 CrossRef CAS PubMed.
- D. Liu, P. Zhou, H. Bai, H. Ai, X. Du, M. Chen, D. Liu, W. F. Ip, K. H. Lo, C. T. Kwok, S. Chen, S. Wang, G. Xing, X. Wang and H. Pan, Small, 2021, 17, 2101605 CrossRef CAS PubMed.
- D. Chen, M. Qiao, Y.-R. Lu, L. Hao, D. Liu, C.-L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691–8696 CrossRef CAS PubMed.
- N. H. Bashian, S. Zhou, M. Zuba, A. M. Ganose, J. W. Stiles, A. Ee, D. S. Ashby, D. O. Scanlon, L. F. J. Piper, B. Dunn and B. C. Melot, ACS Energy Lett., 2018, 3, 2513–2519 CrossRef CAS.
- F. Song, K. Schenk and X. Hu, Energy Environ. Sci., 2016, 9, 473–477 RSC.
- S. R. Ede, T. K. Bijoy, S. S. Sankar, P. Murugan and S. Kundu, Inorg. Chem., 2020, 59, 4816–4824 CrossRef CAS PubMed.
- B.-Q. Li, Z.-J. Xia, B. Zhang, C. Tang, H.-F. Wang and Q. Zhang, Nat. Commun., 2017, 8, 934 CrossRef PubMed.
- S. Liu, Y. Zhang, X. Mao, L. Li, Y. Zhang, L. Li, Y. Pan, X. Li, L. Wang, Q. Shao, Y. Xu and X. Huang, Energy Environ. Sci., 2022, 15, 1672–1681 RSC.
- Y. Fang, Y. Fang, R. Zong, Z. Yu, Y. Tao and J. Shao, J. Mater. Chem. A, 2022, 10, 1369–1379 RSC.
- S. Li, R. Ma, Y. Lu, Y. Pei, M. Yang, J. Wang and D. Liu, J. Mater. Chem. A, 2020, 8, 5919–5926 RSC.
- B. Zhu, Y. Qin, J. Du, F. Zhang and X. Lei, ACS Sustainable Chem. Eng., 2021, 9, 11790–11797 CrossRef CAS.
- Y.-F. Qi, K.-Y. Wang, Y. Sun, J. Wang and C. Wang, ACS Sustainable Chem. Eng., 2022, 10, 645–654 CrossRef CAS.
- B. Liu, S. Xu, M. Zhang, X. Li, D. Decarolis, Y. Liu, Y. Wang, E. K. Gibson, C. R. A. Catlow and K. Yan, Green Chem., 2021, 23, 4034–4043 RSC.
- R. Zhong, Q. Wang, L. Du, Y. Pu, S. Ye, M. Gu, Z. Conrad Zhang and L. Huang, Appl. Surf. Sci., 2022, 584, 152553 CrossRef CAS.
- Y. Lu, T. Liu, C.-L. Dong, C. Yang, L. Zhou, Y.-C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2022, 34, 2107185 CrossRef CAS PubMed.
- H. Wang, J. Zhang and S. Tao, Chem. Eng. J., 2022, 444, 136693 CrossRef CAS.
- S. Liu, S. Geng, L. Li, Y. Zhang, G. Ren, B. Huang, Z. Hu, J.-F. Lee, Y.-H. Lai, Y.-H. Chu, Y. Xu, Q. Shao and X. Huang, Nat. Commun., 2022, 13, 1187 CrossRef CAS PubMed.
- X. Lin, Y. Gao, M. Jiang, Y. Zhang, Y. Hou, W. Dai, S. Wang and Z. Ding, Appl. Catal., B, 2018, 224, 1009–1016 CrossRef CAS.
- K. Zhang, G. Zhang, J. Qu and H. Liu, Small, 2018, 14, 1802760 CrossRef PubMed.
- J. Zheng, Y. Lyu, C. Xie, R. Wang, L. Tao, H. Wu, H. Zhou, S. Jiang and S. Wang, Adv. Mater., 2018, 30, 1801773 CrossRef PubMed.
- E. M. Golden, S. A. Basun, D. R. Evans, A. A. Grabar, I. M. Stoika, N. C. Giles and L. E. Halliburton, J. Appl. Phys., 2016, 120, 133101 CrossRef.
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- F. Lin, Y. Liu, X. Yu, L. Cheng, A. Singer, O. G. Shpyrko, H. L. Xin, N. Tamura, C. Tian, T.-C. Weng, X.-Q. Yang, Y. S. Meng, D. Nordlund, W. Yang and M. M. Doeff, Chem. Rev., 2017, 117, 13123–13186 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- Y. Fu, H.-Y. Yu, C. Jiang, T.-H. Zhang, R. Zhan, X. Li, J.-F. Li, J.-H. Tian and R. Yang, Adv. Funct. Mater., 2018, 28, 1705094 CrossRef.
- X. Deng, G.-Y. Xu, Y.-J. Zhang, L. Wang, J. Zhang, J.-F. Li, X.-Z. Fu and J.-L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed.
- Y. Lu, C.-L. Dong, Y.-C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- Y. Lu, C.-L. Dong, Y.-C. Huang, Y. Zou, Y. Liu, Y. Li, N. Zhang, W. Chen, L. Zhou, H. Lin and S. Wang, Sci. China: Chem., 2020, 63, 980–986 CrossRef CAS.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02813e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |