
Dual metal site-mediated efficient C–N coupling toward electrochemical urea synthesis†
Sourav
Paul
a,
Sougata
Sarkar
a,
Ashadul
Adalder
 a,
Amitava
Banerjee
b and
Uttam Kumar
Ghorai
*a
a,
Amitava
Banerjee
b and
Uttam Kumar
Ghorai
*a
aDepartment of Industrial Chemistry and Applied Chemistry, Swami Vivekananda Research Center, Ramakrishna Mission Vidyamandira, Belur Math, Howrah 711202, West Bengal, India. E-mail: uttam.indchem@vidyamandira.ac.in
bDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology Jodhpur, Jodhpur, Rajasthan 342030, India
First published on 17th May 2023
Abstract
Electrochemical urea synthesis is a promising technology for carbon utilization. Herein, we report a CoPc–MoS2 system for promoting urea synthesis by a C–N coupling reaction. Dual metal sites mediate N2 activation and CO2 adsorption insertion to produce a urea yield of 175.6 μg h−1 mgcat−1 at −0.7 V vs. RHE.
Urea is a vital compound for societal development, and the high presence of 46% nitrogen (by weight) makes it a major player of the fertilizer industry.1,2 The growth of the urea based industry is crucial to satisfy the demand of the growing population. Till now, the commercial urea production has depended on the energy intensive reaction between ammonia and carbon dioxide, which utilizes high temperature and elevated pressure conditions (150–200 °C and 150–250 bar).1 The industrial process requires almost 80% of the globally produced ammonia, produced from artificial nitrogen fixation.3 However the challenge lies in nitrogen fixation, due to the inert nature of the N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond. Therefore industrial ammonia production depends on the century old Haber–Bosch (H–B) process, which demands high temperature and high pressure to break the NN triple bond, having a high bond energy of 941 kJ mol−1.4,5 The H–B process consumes 2–3% of the global energy annually and results in significant carbon dioxide emissions.6 To achieve the goal of a carbon neutral society, a green and clean technology to produce urea under mild conditions is an absolute necessity. An electrocatalytic approach using renewable energy to unify the carbon and nitrogen cycles is a technologically viable and a net zero-carbon emission avenue.7–10 It is a promising step for the decarbonization of the global economy and a positive move towards energy sustainability. In recent times, researchers have successfully produced urea using electrocatalysis by a C–N coupling reaction in aqueous media under ambient conditions. Chen et al. have coupled N2 and CO2 using PdCu–TiO2 as an electrocatalyst, and the metal alloy system aided in enhancing electronic interaction, chemical adsorption, and active site exposure.8 Recently, we reported about copper phthalocyanine nanotubes as an efficient electrocatalyst to produce urea.9 Yuan et al. designed a Mott–Schottky heterostructural interface of Bi–BiVO4, BiFeO3/BiVO4, which aids in specific adsorption and activation of CO2 and N2 to form urea.11,12 Apart from experimental reports, theoretical studies were done, which are important with respect to catalyst selectivity and fundamental understanding of reaction mechanisms. Roy et al. have carried out the theoretical calculation of a novel non-hazardous metal free catalyst for electrochemical urea synthesis.13 Zhu et al. have systematically studied with DFT about the two-dimensional MBenes (Mo2B2, Ti2B2 and Cr2B2) and their capability of coupling of N2 and CO2 to form urea.14
N triple bond. Therefore industrial ammonia production depends on the century old Haber–Bosch (H–B) process, which demands high temperature and high pressure to break the NN triple bond, having a high bond energy of 941 kJ mol−1.4,5 The H–B process consumes 2–3% of the global energy annually and results in significant carbon dioxide emissions.6 To achieve the goal of a carbon neutral society, a green and clean technology to produce urea under mild conditions is an absolute necessity. An electrocatalytic approach using renewable energy to unify the carbon and nitrogen cycles is a technologically viable and a net zero-carbon emission avenue.7–10 It is a promising step for the decarbonization of the global economy and a positive move towards energy sustainability. In recent times, researchers have successfully produced urea using electrocatalysis by a C–N coupling reaction in aqueous media under ambient conditions. Chen et al. have coupled N2 and CO2 using PdCu–TiO2 as an electrocatalyst, and the metal alloy system aided in enhancing electronic interaction, chemical adsorption, and active site exposure.8 Recently, we reported about copper phthalocyanine nanotubes as an efficient electrocatalyst to produce urea.9 Yuan et al. designed a Mott–Schottky heterostructural interface of Bi–BiVO4, BiFeO3/BiVO4, which aids in specific adsorption and activation of CO2 and N2 to form urea.11,12 Apart from experimental reports, theoretical studies were done, which are important with respect to catalyst selectivity and fundamental understanding of reaction mechanisms. Roy et al. have carried out the theoretical calculation of a novel non-hazardous metal free catalyst for electrochemical urea synthesis.13 Zhu et al. have systematically studied with DFT about the two-dimensional MBenes (Mo2B2, Ti2B2 and Cr2B2) and their capability of coupling of N2 and CO2 to form urea.14
In spite of the aforementioned catalysts, the progress of the electrochemical process of urea synthesis is hindered significantly due to poor selectivity and low catalytic activity. The reports suggest that the electrocatalyst are synthesized to address the issue of adsorption and activation of reactant molecules, but the C–N coupling process, which is important for urea production has received microscopic attention.15,16 The challenge exists in byproduct formation, difficulty of separation of urea, low yield and poor faradaic efficiency (FE). In order to overcome the challenge, it is vital to rationally design an electrocatalyst with multiple active sites that enhances the adsorption of gaseous reactants and facilitates the C–N coupling reaction to produce urea. The selection of the catalyst should be made on the following aspects: (i) facile adsorption of the gaseous reactants (N2 and CO2), (ii) facilitation of the carbon dioxide reduction reaction (CO2RR) to CO*, (iii) activity towards the nitrogen reduction reaction (NRR) to NH2*, (iv) the catalyst must have a similar reduction potential range for the NRR and CO2RR, (v) suppression of the hydrogen evolution reaction (HER), and (vi) enhancement of the C–N bond formation.
Prof. Robert and co-workers have shown cobalt phthalocyanine (CoPc) as a highly active, selective and stable electrocatalyst for the electroreduction of CO2 to CO in a wide pH range (4 to 14).17 Zhu et al. have studied CoPc supported on pyridine functionalized carbon nanotubes (CoPc-py-CNT), which showed superior activity (TOFCO: 34.5 s−1 at −0.63 V versus RHE) and high selectivity (FECO > 98%).18 Furthermore, research reports suggest that transition metal phthalocyanine (MPc) can also play the role of an effective NRR catalyst.19–23 However, MPc requires a substrate for support, and the substrates are generally carbon based.19,20 Generally, the substrates are not electrocatalytically active. However, if we select a substrate which possess active sites demonstrates electrochemical activity and provides a framework to support the MPc, it would be highly beneficial to promote urea production via C–N bond formation. It is well known that 2D materials (graphene, MoS2, TiO2, etc.)8,24 can effectively act as a support and participate in electrocatalysis. Prof. Sun and co-workers have shown MoS2 as an effective NRR electrocatalyst, which showed a FE of 1.17% and ammonia yield of 8.08 × 10−11 mol s−1 cm−1 at −0.5 V.24 Chou et al. have shown that MoS2 nanosheets can be utilized to tune the NRR as well as HER25via defect engineering. MoS2 can act as an effective catalyst for the CO2RR.26 Inspired by the research, which shows that CoPc and MoS2 are highly active and selective towards the CO2RR and NRR in a similar potential range, we designed an electrocatalyst by combining CoPc with MoS2, and formed a CoPc–MoS2 composite system, where MoS2 nanosheets act as a catalytically active substrate to which CoPc is anchored. The CoPc–MoS2 electrocatalyst can simultaneously activate and reduce N2 and CO2 due to the presence of various active sites and electrochemically produce urea under ambient conditions, which is unexplored by the research fraternity.
In this work, CoPc–MoS2 composite was prepared by embedding CoPc on MoS2nanosheets, which plays the role of efficient electrocatalyst for urea synthesis (Fig. 1a). Even though MoS2 or CoPc alone showed inferior electrochemical activity, CoPc–MoS2 showed exuberant electrochemical performance, with a urea yield rate and FE of 175.6 μg h−1 mgcat−1 and 15.12% respectively at −0.7 V vs. RHE and it remains similar for five consecutive cycles. The “acceptor–donor” mechanism21 suggests that the dual metal active sites of ‘Co’ and ‘Mo’ of the CoPc–MoS2 system have orbitals with appropriate symmetry. So, the vacant d orbitals can accept lone pair electrons of N2, and the partially filled d electrons feed electrons back to the N2 antibonding orbitals. Hence, the NN triple bonds are polarized and activated. In the case of single catalytic sites, the empty d orbital of a metal site can accommodate one lone pair electron of the N2 molecule. To enhance the activation process, the empty d-orbitals of the dual metal sites can pull two lone pair electrons at two ends of the N2 molecule (Fig. 1b). The polarization of the N2 molecule facilitates the insertion of the intermediate CO species, produced by the CO2 reduction at the Mo or Co sites. The Mo sites are preferred active sites for CO2 reduction due to the localized induced negatively charged surface on the MoS2 nanosheets. The C–N coupling reaction occurs via the CO insertion into the activated N2 molecule, and the reaction propagates via the formation of the triangular shape intermediate state of CONN* which induces angular strain, and it aids the breaking of the N–N bond and thereafter protonation occurs to form urea as a product.9,15 The N2 activation process is further supported by DFT based electronic structure and Bader charge analysis (shown in the ESI†). It has been observed that the dual metal mediated activation process transferred almost 12% more charge to N2 as compared to single metal center activation. The exchange of charge causes a change in the bond length of N2 to 1.196 Å for the dual metal center and 1.155 Å for the single metal center compared to the bond length of 1.130 Å for inactive N2 gas molecules and eventually activate the N2 molecule. The charge density difference image (Fig. 1c) depicts this charge donation/back-donation to and from N2 by cyan and magenta isosurfaces.
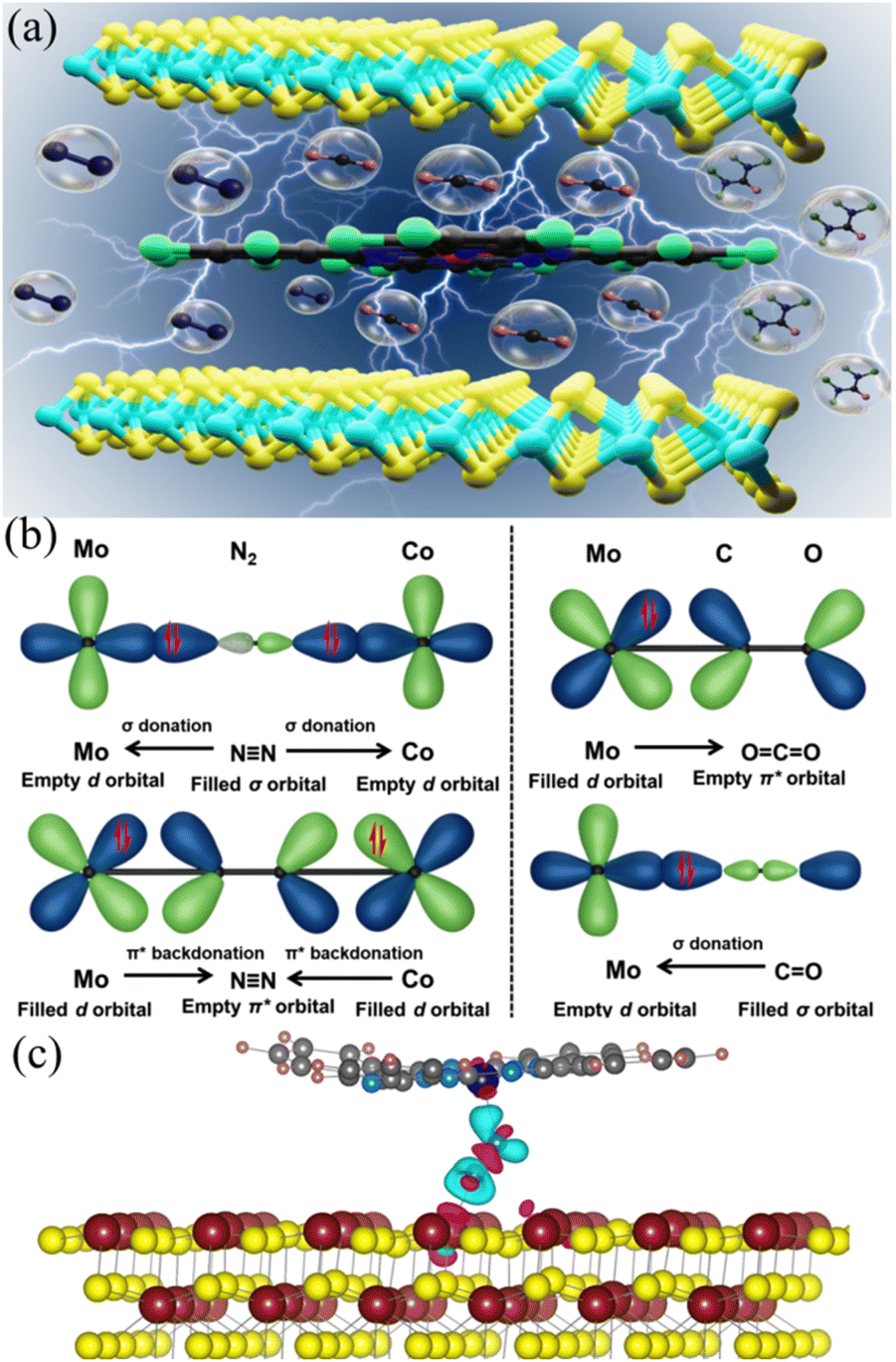 | ||
| Fig. 1 (a) Pictorial representation of urea synthesis. (b) Schematic diagram showing N2 bonding to the dual metal site in CoPc–MoS2. (c) Charge density difference during N2 activation by the double metal center in CoPc–MoS2. Mo, Co, S, N, C, and H atoms are in maroon, blue, yellow, sky-blue, gray, and pink, respectively. Cyan and magenta isosurfaces (0.01e) show charge depletion and accumulation, respectively. | ||
The isotopic tracing experiment by 1H-NMR was carried out to unveil the origin of urea formation. The catalyst showed stability for 12 hours without any decrease in electrochemical performance. The catalyst showed brilliant electrochemical performance due to the following factors: CoPc–MoS2 interfacial coupling interaction which played an essential role in synergistic stimulation of the Mo-edge sites of MoS2 and the active site of CoPc toward the urea synthesis, while concurrently protecting the exposed sites from the competing HER.17,18 The work provides a detailed understanding of the catalytic activity and showed a route to strategically develop an electrocatalyst that can couple N2 and CO2 to produce urea. An efficient hydrothermal route was utilized to prepare MoS2 nanosheets, which played the role of the matrix of the CoPc–MoS2 composite system in which cobalt phthalocyanine molecules were embedded via mechano-chemical blend formation (ESI†); the process can be utilized to produce the electrocatalyst on a large scale.
The crystallinity patterns of CoPc, MoS2 and CoPc–MoS2 were analyzed by the X-ray diffraction technique as shown in Fig. 2a. In the composite, CoPc was uniformly distributed over the MoS2 nanosheets, as illustrated by the presence of the characteristics diffraction peaks at (100), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 02), (002), (
02), (002), (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 02), (04), (112), and (
02), (04), (112), and (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 11), which are well matched with CoPc (JCPDS no. 14-0948),27 together in unison with the presence of broad diffraction peaks of MoS2 corresponding to (100), (103), (105) and (110) planes (JCPDS no. 37-1492).28 Next, high resolution XPS spectra analyses were carried out to probe the electronic properties and chemical composition of CoPc–MoS2. In Fig. 2b, the binding energies at 780.2 and 795.6 eV correspond to the Co 2p3/2 and 2p1/2 regions, respectively.21 Meanwhile the peaks detected at 228.7 and 231.9 eV (shown in Fig. 2c) are assigned to the Mo 3d5/2 and 3d3/2 electronic states, indicating the presence of the tetravalent state of Mo in CoPc–MoS2.28,29 Similarly, the peaks observed at 161.8 and 162.7 eV (represented in Fig. S2†) are assigned to the electronic states of S 2p3/2 and 2p1/2 respectively, which represents the divalent state of S in the catalytic system.28,30Fig. 2d represents the high-resolution N 1s XPS spectra, and binding energies at 398.7 and 399.6 eV are shown due to the presence of pyridinic nitrogen and pyrrolic nitrogen respectively in CoPc–MoS2.21 In CoPc, the pyridinic nitrogen is bonded to the carbon atom of the phthalocyanine ring while the pyrrolic nitrogen atoms are attached to the central Co atom.17 The different functional groups of the CoPc–MoS2 catalyst were identified by using the FTIR spectra (Fig. S3†). The TEM imaging reveals that the catalytic surfaces are rough in nature with distinct microporous domains (Fig. 2e). The morphology containing porous regions are expected to provide a greater surface area leading to increase in the exposure of catalytically active sites, thereby enhancing the mass transport.31 Thus, it leads to efficient N2 and CO2 adsorption and electrocatalytic co-reduction to facilitate urea synthesis. The TEM-EDS elemental mapping confirmed that cobalt, molybdenum, sulfur and nitrogen were uniformly distributed in the CoPc–MoS2 composite (Fig. 2f).
11), which are well matched with CoPc (JCPDS no. 14-0948),27 together in unison with the presence of broad diffraction peaks of MoS2 corresponding to (100), (103), (105) and (110) planes (JCPDS no. 37-1492).28 Next, high resolution XPS spectra analyses were carried out to probe the electronic properties and chemical composition of CoPc–MoS2. In Fig. 2b, the binding energies at 780.2 and 795.6 eV correspond to the Co 2p3/2 and 2p1/2 regions, respectively.21 Meanwhile the peaks detected at 228.7 and 231.9 eV (shown in Fig. 2c) are assigned to the Mo 3d5/2 and 3d3/2 electronic states, indicating the presence of the tetravalent state of Mo in CoPc–MoS2.28,29 Similarly, the peaks observed at 161.8 and 162.7 eV (represented in Fig. S2†) are assigned to the electronic states of S 2p3/2 and 2p1/2 respectively, which represents the divalent state of S in the catalytic system.28,30Fig. 2d represents the high-resolution N 1s XPS spectra, and binding energies at 398.7 and 399.6 eV are shown due to the presence of pyridinic nitrogen and pyrrolic nitrogen respectively in CoPc–MoS2.21 In CoPc, the pyridinic nitrogen is bonded to the carbon atom of the phthalocyanine ring while the pyrrolic nitrogen atoms are attached to the central Co atom.17 The different functional groups of the CoPc–MoS2 catalyst were identified by using the FTIR spectra (Fig. S3†). The TEM imaging reveals that the catalytic surfaces are rough in nature with distinct microporous domains (Fig. 2e). The morphology containing porous regions are expected to provide a greater surface area leading to increase in the exposure of catalytically active sites, thereby enhancing the mass transport.31 Thus, it leads to efficient N2 and CO2 adsorption and electrocatalytic co-reduction to facilitate urea synthesis. The TEM-EDS elemental mapping confirmed that cobalt, molybdenum, sulfur and nitrogen were uniformly distributed in the CoPc–MoS2 composite (Fig. 2f).
 | ||
| Fig. 2 (a) XRD image, (b–d) high resolution XPS scan of Co, Mo, and N, and (e) TEM image of CoPc–MoS2; (f) elemental distribution of Co, Mo, N and S in the CoPc–MoS2 system. | ||
To study the electrocatalytic activity of CoPc–MoS2, electrochemical measurements were performed in Ar and (N2 + CO2) purged saturated 0.1 M KHCO3 electrolyte solution in a H-type cell. Fig. 3a shows the LSV curve of CoPc–MoS2 recorded at 10 mV s−1 when compared to the Ar saturated electrolyte, there is an increase in current densities in the potential range of −0.5 to −0.8 V (vs. RHE) for N2 + CO2 purged electrolyte.8,9,11 Time dependent chronoamperometry measurements were executed at various potentials in N2 and CO2 saturated electrolytes (shown in Fig. 3b).The UV-vis absorption spectrum of the different electrolytes with characteristic absorption of 525 nm at different potentials is shown in Fig. S6† and the maximum absorbance was found at −0.7 V versus RHE. The produced urea was estimated by the diacetylmonoxime (DAMO) method (ESI Experimental section†). The UV-vis absorbance and the related calibration curve for urea quantification are shown in Fig. S4 and S5.† The urea yield rate and corresponding FE in a potential window are displayed in Fig. 3c. CoPc–MoS2 delivers a maximum urea yield rate and FE of 175.6 μg h−1 mgcat−1 and 15.12% at −0.7 V versus RHE, respectively. It is observed that the urea formation rate rises with increase in negative potential until −0.7 V versus RHE and the rate drops down due to the competitive reaction of H2 and N2 on the surface of the electrode. Furthermore, when MoS2 and CoPc were individually tested, they demonstrated decent electrocatalytic performance, but excellent performance was observed for the CoPc–MoS2 system (Fig. S16†). The high urea yield rate is attributed to the C–N bond formation through co-reduction of N2 and CO2 in the Co metal center, and the Mo edge sites of CoPc–MoS2. The vital existence of chemical bonds after the electrochemical synthesis of urea is characterized using the ATR-FTIR technique. Stretching vibrations present at ∼1690 and 1165–1334 cm−1 correspond to the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O respectively. The result clearly indicates that CO2 gas is reduced during the electro-reduction process. The stretching vibration at ≈1431 cm−1 corresponds to the important C–N bond and confirms the C–N coupling leading to the formation of urea during the electrocatalytic process (Fig. 3f).8,32
O and C–O respectively. The result clearly indicates that CO2 gas is reduced during the electro-reduction process. The stretching vibration at ≈1431 cm−1 corresponds to the important C–N bond and confirms the C–N coupling leading to the formation of urea during the electrocatalytic process (Fig. 3f).8,32
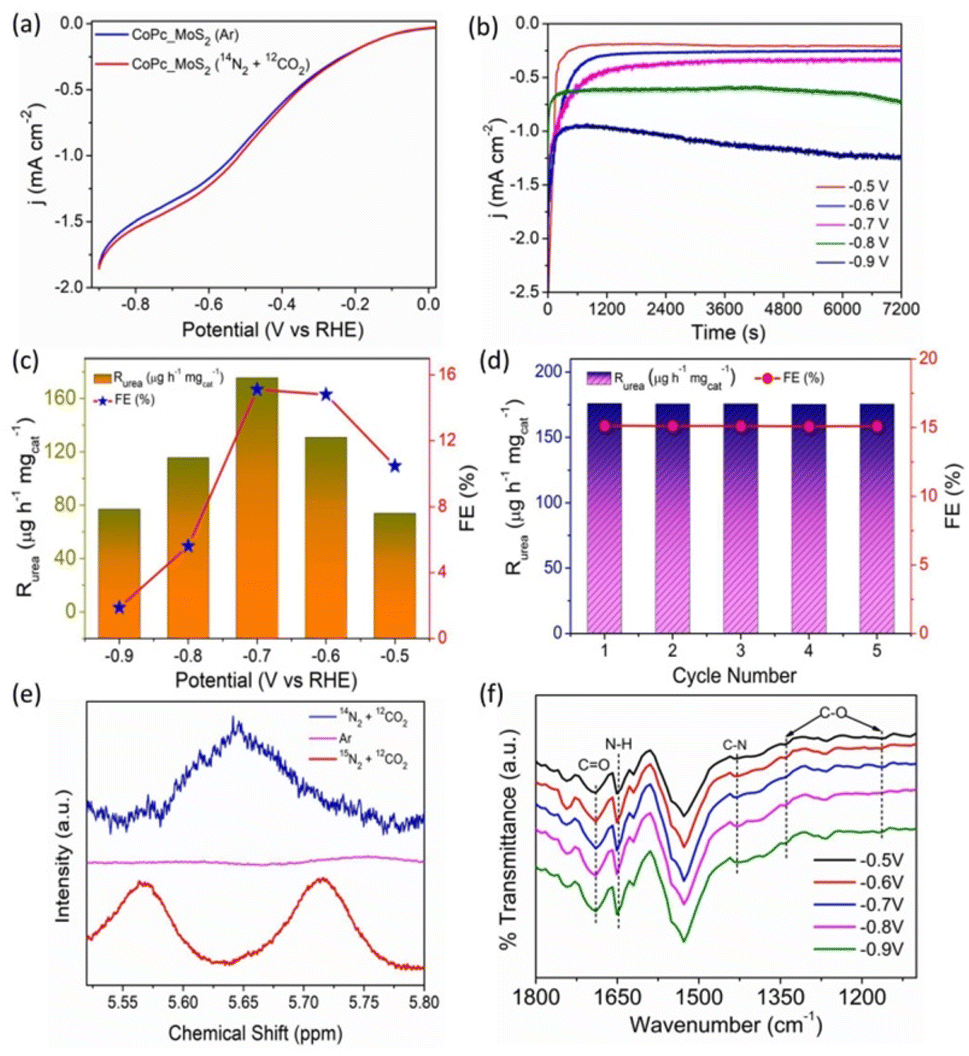 | ||
| Fig. 3 (a) LSV profile of CoPc–MoS2 in Ar and N2 + CO2 saturated 0.1 M KHCO3 electrolytes; (b) time dependent current density (j) curves for CoPc–MoS2 at different potentials; (c) urea yield rate and FE with N2 and CO2 as the feeding gas at different potentials for CoPc–MoS2; (d) the urea yield rate and FE of CoPc–MoS2 at −0.7 V vs. RHE during recycling tests for five cycles. (e) 1H NMR spectra of electrolytes saturated with 14N2 + 12CO2, Ar and 15N2 + 12CO2 after 20 h of electrolysis. (f) ATR-FTIR spectroscopy experiments of the electrolytes at different potentials during electrocatalytic co-reduction of N2 and CO2 using CoPc–MoS2. | ||
Recyclability and stability are the two crucial parameters of an electrocatalyst for practical utility. The recycling tests were conducted in a N2 and CO2 saturated 0.1 M KHCO3 solution at −0.7 V potential for five cycles, and the catalyst maintains almost the same urea yield rate in the entire process (Fig. 3d), indicating the stable electroreduction performance of the catalyst. The chemical composition of CoPc–MoS2 after five consecutive electrocatalytic cycles was investigated by XPS. The high resolution spectra of Co 2p, Mo 3d, and N 1s demonstrate indistinguishable nature with the electrocatalyst before the electrochemical test (shown in Fig. S7–S9†). To further check the stability of the electrocatalyst, a long duration stability test was done at −0.7 V. The catalyst showed an almost stable electroreduction performance without much fluctuation in current density when the electrocatalysis was executed for 12 h (Fig. S10†). Furthermore, to assess the stability of the electrocatalyst, XRD analysis was done post electrolysis, and the result clearly indicates that the structural integrity of the catalyst is maintained (Fig. S11†). In order to find the origin of urea formation, control experiments were done in an Ar (N2 and CO2) saturated electrolyte, and carbon paper without the catalyst, at −0.7 V versus RHE for 2 h. An insignificant amount of urea was detected for all the cases as shown in Fig. S23,† except (N2 and CO2) purged conditions. The selectivity of the catalyst is good, as a negligible amount of hydrazine and a very low amount of ammonia are produced as a side-product (Fig. S19 and S22†). Therefore, the result suggests that CoPc–MoS2 shows high activity and selectivity towards urea synthesis. The characteristic singlet peak of the formation of urea in 1H NMR appeared at ≈5.65 ppm when the electrolysis was carried out by purging of 14N2 and 12CO2 gases and a doublet peak was observed when isotopic 15N2 and 12CO2 gases were purged during the electrocatalytic reduction process (Fig. 3e). However, no such singlet and doublet were detected when only Ar gas was purged during the electrolysis process, which clearly shows that the urea produced is solely due to the electrocatalytic co-reduction of N2 and CO2 gases and not by any contaminants.8
The catalytic activity can be estimated with the free energy diagram, which provides a visual representation of the reaction mechanism. The free energy profile (Fig. 4) has identified various possible intermediates and their contribution to the reaction mechanism. In the entire urea formation mechanism, only two uphill reactions have been identified, one is the N2 activation process and the other is the fourth protonation process, i.e. *NHNH2CO to *NH2NH2CO formation step. The first uphill step is the rate determining step, where proton/electron transfer does not occur, whereas the second uphill reaction is the potential determining step with an over potential of ∼2 V to make all electrochemical steps exothermic. The catalytic origin of the CoPc_MoS2 system derives from the spatial distribution of the charge in between the dual metal center and N2 as well as facile formation of the *NNCO intermediate. The free energy profile depicts that *NNCO is form as a result of a favourable downhill reaction. We have also found that, during the formation of *NNCO, the NN triple bond breaks immediately as soon as CO comes close to N2. As *NNCO forms, the fourth protonation step takes place to form urea.
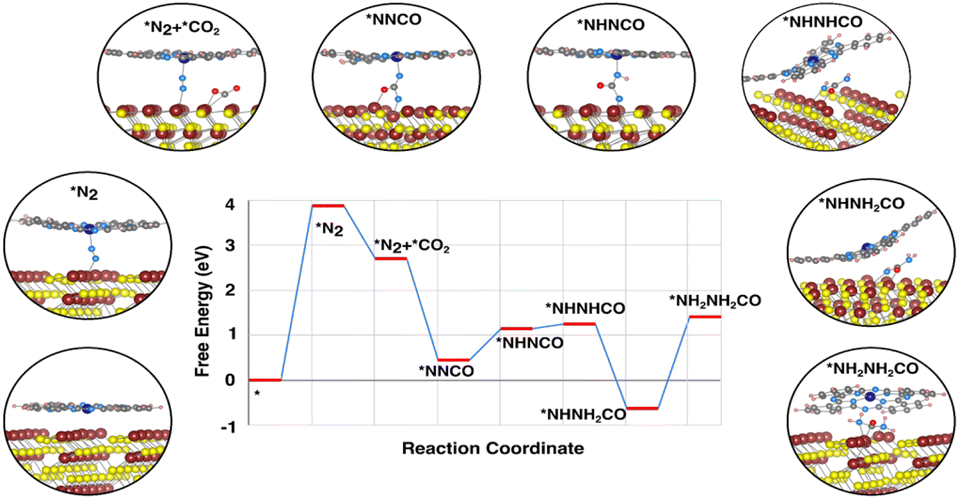 | ||
| Fig. 4 Free energy profile of the urea synthesis mechanism. Insets show the possible intermediates in the plausible mechanism. Mo, Co, S, N, C, H and O atoms are in maroon, blue, yellow, sky-blue, gray, pink and red, respectively. | ||
Conclusions
In summary, CoPc_MoS2 is theoretically predicted and experimentally confirmed as an active and robust electrocatalyst for the co-reduction of N2 and CO2 gases under ambient conditions. When tested in 0.1 M KHCO3, the urea yield rate and FE can reach 175.6 μg h−1 mgcat−1 and 15.12% at −0.7 V versus RHE, respectively. Impressively, CoPc–MoS2 exhibits good electroreduction activity and effectively suppresses the hydrogen evolution reaction. This study not only offers us an efficient cost-effective electrocatalyst for high performance N2 and CO2 co-reduction under ambient conditions but would open up an exciting new route to explore transition metal dichalcogenide/transition metal phthalocyanine as an efficient catalyst for applications toward urea electrosynthesis.Author contributions
UKG conceived the idea and supervised the entire project. SP synthesized the materials and characterised them. SP, SS, and AA performed the electrochemical experiments and analysed the results. SP wrote the original manuscript. SP and AA did the XPS analysis. AB performed the theoretical (DFT) calculations. AB and SP analysed the DFT results. All authors approved the final version of the manuscript.Conflicts of interest
There is no conflict of interest.Acknowledgements
AA thanks SERB-CRG for the fellowship. SS acknowledges the UGC, Govt. of India for the JRF Fellowship (Scholarship ID: 191620014156). The authors would like to thank IACS Kolkata for providing the instrumentation facilities. The authors acknowledge the financial support from the SERB-Core Research Grant (CRG/2022/009427), WBDSTBT research grant (199 (Sanc.)/ST/P/S&T/6G-12/2018), SERB-Start-up Research Grant (SRG/2022/001377), IIT Jodhpur Seed grant (I/SEED/PRJ//DSN/AB/20220044) and supercomputing facility. AB also acknowledges Dr. Appala Naidu and Department of Metallurgical and Materials Engineering of IIT Jodhpur for software support.Notes and references
- F. Barzagli, F. Mani and M. Peruzzini, Green Chem., 2011, 13, 1267–1274 RSC
.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS
- A. Adalder, S. Paul and U. K. Ghorai, J. Mater. Chem. A, 2023, 11, 10125–10148 RSC
- A. Banerjee, Catal. Today, 2023, 418, 114079 CrossRef CAS
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H. L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed
- A. Biswas, S. Kapse, R. Thapa and R. S. Dey, Nano-Micro Lett., 2022, 14, 1–17 CrossRef PubMed
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 128(12), 717–724 CrossRef PubMed
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS
- A. Biswas, S. Kapse, B. Ghosh, R. Thapa and R. S. Dey, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204638119 CrossRef CAS PubMed
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC
- P. Roy, A. Pramanik and P. Sarkar, J. Phys. Chem. Lett., 2021, 12, 10837–10844 CrossRef CAS PubMed
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 121(12), 1–9 Search PubMed
- D. Jiao, Y. Dong, X. Cui, Q. Cai, C. R. Cabrera, J. Zhao and Z. Chen, J. Mater. Chem. A, 2022, 11, 232–240 RSC
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 131(13), 1–9 Search PubMed
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y. F. Han, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS
- S. Murmu, S. Paul, A. Santra, M. Robert and U. K. Ghorai, Catal. Today DOI:10.1016/J.CATTOD.2022.10.020
- C. He, Z. Y. Wu, L. Zhao, M. Ming, Y. Zhang, Y. Yi and J. S. Hu, ACS Catal., 2019, 9, 7311–7317 CrossRef CAS
- U. K. Ghorai, S. Paul, B. Ghorai, A. Adalder, S. Kapse, R. Thapa, A. Nagendra and A. Gain, ACS Nano, 2021, 15, 5230–5239 CrossRef CAS PubMed
- S. Murmu, S. Paul, S. Kapse, R. Thapa, S. Chattopadhyay, A. Nagendra, S. N. Jha, D. Bhattacharyya and U. K. Ghorai, J. Mater. Chem. A, 2021, 9, 14477–14484 RSC
- J. Mukherjee, A. Adalder, N. Mukherjee and U. K. Ghorai, Catal. Today DOI:10.1016/J.CATTOD.2022.09.011
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed
- I. Matanovic, K. Leung, S. J. Percival, J. E. Park, P. Lu, P. Atanassov and S. S. Chou, Appl. Mater. Today, 2020, 21, 100812 CrossRef
- K. Lv, C. Teng, M. Shi, Y. Yuan, Y. Zhu, J. Wang, Z. Kong, X. Lu and Y. Zhu, Adv. Funct. Mater., 2018, 28, 1802339 CrossRef
- S. Paul, S. Sarkar, A. Adalder, S. Kapse, R. Thapa and U. K. Ghorai, ACS Sustainable Chem. Eng., 2023, 11, 6191–6200 CrossRef CAS
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed
- X. Wang, H. Feng, Y. Wu and L. Jiao, J. Am. Chem. Soc., 2013, 135, 5304–5307 CrossRef CAS PubMed
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS
- Y. Li, Z.-S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang, X. Bao, Y. G. Li, Z. Wu, P. F. Lu, X. Wang, W. Liu, X. H. Bao, Z. B. Liu, W. C. Ren, J. Y. Ma and Z. Jiang, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustain., 2021, 410(4), 868–876 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S26, and Tables S1 and S2. See DOI: https://doi.org/10.1039/d3ta01011b |
| This journal is © The Royal Society of Chemistry 2023 |