
Structural modulation of low-valent iron in LDH-derived Ni3Se4 nanosheets: a breakthrough electrocatalyst for the overall water splitting reaction†
Arun
Karmakar
 ab,
Abhirami V.
Krishnan
c,
Rahul
Jayan
d,
Ragunath
Madhu
ab,
Md Mahbubul
Islam
*d and
Subrata
Kundu
*ab
ab,
Abhirami V.
Krishnan
c,
Rahul
Jayan
d,
Ragunath
Madhu
ab,
Md Mahbubul
Islam
*d and
Subrata
Kundu
*ab
aAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
bElectrochemical Process Engineering (EPE) Division, CSIR-Central Electrochemical Research Institute (CECRI), Karaikudi-630003, Tamil Nadu, India. E-mail: skundu@cecri.res.in; kundu.subrata@gmail.com; Fax: (+91) 4565-241487; Tel: (+91) 4565-241487
cDepartment of Chemistry, Wayne State University, Detroit-48201, USA
dDepartment of Mechanical Engineering, Wayne State University, Detroit-48201, USA. E-mail: gy5553@wayne.edu
First published on 24th April 2023
Abstract
Transition metal-based layered double hydroxides (LDHs) have attracted significant attention in recent research in electrochemistry and hold promising capacity in a wide range of applications. Owing to their diverse chemical structure, improved mass- and charge-transfer kinetics, and fascinating 2D layered structure, LDHs have emerged to be promising electrocatalysts for water electrolysis. Yet, some limitations, including their low stability and poor conductivity, remain as major obstacles towards the application of LDHs in the field of electrocatalysis. As a result, various LDH-derived nanomaterials, like selenides, sulphides, phosphides, etc., have been employed as effective electrocatalysts towards water splitting. Here in this study, a solvothermal reaction strategy was employed to derive a selenide from NiFe-LDH and the successful selenization resulted in the formation of Fe-doped Ni3Se4 (Fe@Ni3Se4) with a 2D microporous structure. The Fe@Ni3Se4 showed excellent electrocatalytic performance for the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) in alkaline conditions, requiring just 185 and 33 mV overpotentials to drive 10 mA cm−2 current density in the OER and HER, respectively, which outperformed many recently reported benchmark electrocatalysts. Also, a 10-fold increase in turnover frequency (TOF) was noticed in Fe@Ni3Se4 compared to pristine NiFe-LDH, revealing an enormous improvement in specific activity as a result of the selenization. The presence of Fe with a lower valency provided an added advantage over the redox activity and stable structural outcomes, which was confirmed from operando impedance analysis and various other characterizations. First-principles DFT calculations revealed that doping low-valent Fe ions largely increased the 3d-2p repulsion in the Ni–O bonds, thereby facilitating the O2 desorption process as compared to pristine Ni3Se4, as a result of the electronic exchange. Moreover, the downward shifting of the d-band centre with respect to the Fermi level modulated the binding strengths of the Fe-doped substrate towards H* compared to the pristine substrates.
1. Introduction
In view of the increasing globalization and consequently the high energy demand, the utilization of various non-renewable fossil fuels has reached an extreme level. In this situation, finding a proper and effective alternative energy source is a prime importance for the research community as the availability of the currently used fossil fuel stocks are limited.1–4 In this regard, out of various energy sources, hydrogen has emerged as the most effective one owing to its high specific energy density, renewable nature, and zero carbon emission.5 Producing hydrogen from natural hydrogen resources, like from H2O (by electrolysis) or hydrocarbons (stream reforming), is always worthy due to the insufficient availability of free hydrogen in the environment.6 The electrolysis of water to hydrogen and oxygen is considered to be most effective strategy owing to its high efficiency as well as its environmentally friendly nature.7Water splitting by electrochemical means involves two half-cell reactions, namely the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode.8 Since the OER involves a complex four-electron and multi-step process, it can be a high bottleneck towards the overall water splitting reaction compared to the HER.9–13 Therefore, to remove this kinetic barrier, designing earth-abundant and stable electrocatalysts has gained a crucial importance owing to the high cost of the conventional noble metal (Ir, Ru, and Pt)-based electrocatalysts.14 Several, electrocatalysts based on earth-abundant 3d transition metals, like oxides, hydroxides, sulphides, selenides, phosphides, MOFs, etc., have been reported as potential candidates for the water splitting reaction.15–26
Among all the above-mentioned materials, layered double hydroxides (LDHs) with a brucite-like layered structure consisting of bi- and tri-valent metal ions as the metal-ion source along with charge balancing interlayer anions have attracted much attention.27,28 Significant studies on the utilization of LDHs for electrocatalytic water splitting have recently been performed. These studies have revealed that LDH-based materials can be an effective alternative to noble metal-based electrocatalysts. Among various LDHs, NiFe-LDH has recently been considered a benchmark catalyst for the OER. However, NiFe-LDH materials have a few limitations, like poor electrical conductance, restriction towards the number of active sites, and poor stability.29 Apart from these drawbacks, the improper active sites towards the cathodic counterpart of water splitting, limit their practical application. To address these issue, the derivatizations of LDHs to various counterparts, like sulphides, selenides, etc., have been investigated and has proven to be a very effective strategy. The various advantages of this strategy can be summarized as: (1) the transformation to a sulphide, selenide, or phosphide could alter the electronic structure of the parent materials; (2) an increase in conductivity; (3) a retention of a 2D sheet-like morphological structure could be achieved, and so on.30
So, by considering the various advantageous of this derivatization of LDHs, herein we report an effective selenization of the hierarchical flower-like NiFe-LDH material under solvothermal conditions. The selenization of NiFe-LDH led to the formation of an Fe-doped Ni3Se4 hierarchical nanostructure with a modified coordination environment on the active Ni sites. The transformation of the hydroxide moiety to a selenide entity resulted in an increase in the porosity of the material. As a result of such improvement in porosity, the Fe-doped Ni3Se4 possessed a better charge as well as mass transfer at the electrode–electrolyte interfaces. The detailed chemical analysis via X-ray photoelectron spectroscopy (XPS) showed that the doping of iron in the seleno-spinel structure in a reducing environment led to the transformation of Fe3+ ions to active Fe2+ ions. The high crystal-field-stabilization energy (CFSE) guided the Fe2+ ions to replace the Ni3+ in the octahedral (Oh) NiSe6 moiety.31 While to maintain the charge balance, the Fe3+ ions without any CFSE were been placed in the tetrahedral (Td) coordination site to form an FeSe4 unit. The formation of a Ni–Se–Fe linkage largely facilitated the electronic exchange in the electrocatalytic process. The hierarchical Fe-doped Ni3Se4 nanostructure demonstrated excellent OER and HER activity in alkaline conditions by requiring just 185 and 33 mV overpotentials to drive 10 mA cm−2 current density, respectively. First-principles DFT calculations revealed that doping low-valent Fe ions largely stabilized the high valent Ni–O intermediates by transferring their electron density to the active Ni sites. Moreover, the increased 3d-2p repulsion in Ni–O bonds largely facilitated the O2-desorption process compared to with pristine Ni3Se4 as a result of the electronic exchange.
2. Results and discussion
2.1. Characterization
The detailed synthetic procedure of both the NiFe-LDH and Fe-doped Ni3Se4 are given in the ESI† along with various other experimental details. A schematic representation and the overall outcomes of this work are presented in Fig. 1a. The formation of the desired structural outcomes was first confirmed via X-ray diffraction (XRD) analysis and the obtained results are given in Fig. S1.† Fig. S1a† displays the XRD pattern of the synthesized materials (NiFe-LDH and Fe@Ni3Se4) over nickel foam (NF), showing the presence of three strong diffraction peaks corresponding to the (111), (200), and (220) planes of metallic Ni in accordance with the ICDD card no: 65-2865.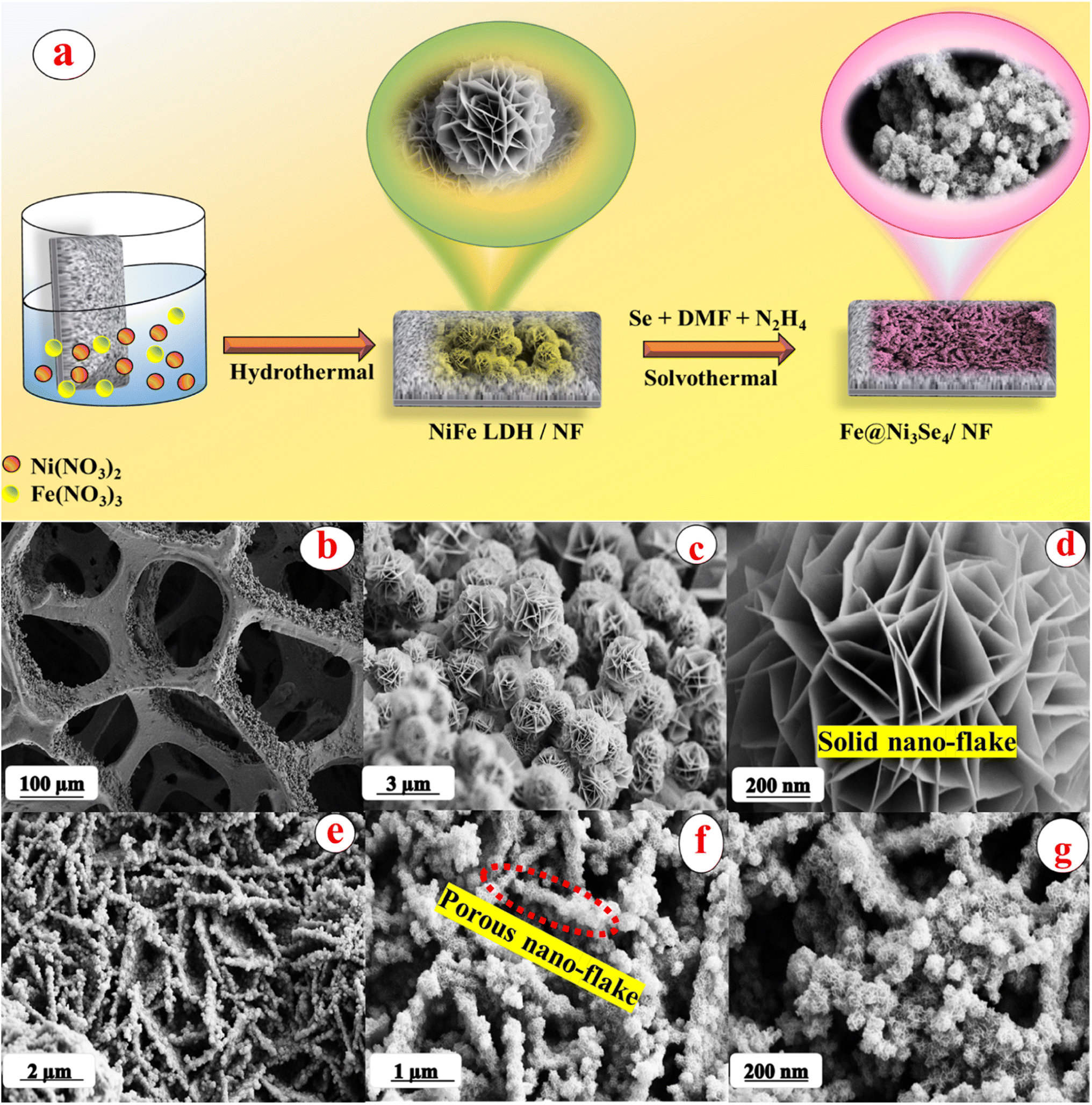 | ||
| Fig. 1 (a) Schematic diagram for the overall design strategy adopted for the synthesis of Fe@Ni3Se4 from NiFe-LDH; (b–d) low- to high-magnification FE-SEM images for pristine NiFe-LDH grown over metallic nickel foam. (e–g) Low to high magnification FE-SEM images of the sheet-like Fe-doped Ni3Se4 with a microporous structure. | ||
Due to the large intensity of the substrate peaks, the other peaks corresponding to our desired materials were diminished. Therefore, XRD analysis of the powder materials obtained during the hydrothermal treatment was carried out and the obtained results are shown in Fig. S1b.† The diffraction pattern corresponding to NiFe-LDH showed different diffraction peaks corresponding to the (003), (006), (012), (110), and (113) planes of NiFe-LDH (ICDD card no: 00-051-0463). The XRD analysis of the selenized NiFe-LDH showed various diffraction peaks corresponding to the (−312), (−313), (−401), (310), and (−714) planes of the selenium counterpart of the spinel Ni, i.e. Ni3Se4.
The morphologies of the NiFe-LDH and Fe-doped Ni3Se4 (Fe@Ni3Se4) materials were analyzed via field emission-scanning electron microscopy (FE-SEM). From the FE-SEM images in Fig. 1b–d, it can be observed that the NiFe-LDH possessed a hierarchical micro-flower structure. The images show the flower-like morphological outcomes were made up by combining numerous uniform nano-flakes. The Fe@Ni3Se4 possessed a similar sheet-like morphology (Fig. 1e–g). It was interesting to note that after the selenization, the solid flakes of the flower-like structure created an internal porous structure. The formation of such an internal porous structure is beneficial for allowing the effective tunnelling of the electrolyte anions to favour the electrochemical process with less applied potential required.32 The highly porous nature of Fe@Ni3Se4 was further verified form the Brunauer–Emmett–Teller (BET) N2 adsorption–desorption isotherm analysis results given in Fig. S2a.† The calculated BET isotherm areas for NiFe-LDH and Fe@Ni3Se4 were 33.46 and 61.65 m2 g−1 respectively. Moreover, the pore diameters were obtained from the adsorption branch by using the Barrett–Joyner–Halenda (BJH) algorithm and the corresponding results are listed in Fig. S2b.† The NiFe-LDH and Fe@Ni3Se4 possessed characteristic BJH diameters of 18.7 and 13.32 nm, respectively. This high specific surface area and low pore diameter with distinct distribution is beneficial for allowing effective mass transfer. The quantitative analysis was carried out on the EDAX spectra given in Fig. S3a and b.† The EDAX spectra showed the presence of the expected elements, like Ni, Fe, C, O, and Se, without any impurity. It was interesting to note that the ratio of Ni to Fe in NiFe-LDH was almost about ‘1’, but after selenization, this ratio decreased substantially, which might be due to an additional selenization of the underlying Ni foam under the selenized conditions and as Fe has been placed as a doped entity in the Ni3Se4 lattice. The ratios of Ni to Fe in both NiFe-LDH and Fe@Ni3Se4 were further confirmed through inductive coupled plasma-mass spectroscopy (ICP-MS) analysis.
The microstructural outcomes of both the materials were analyzed via high-resolution transmission electron microscopy (HR-TEM). Fig. 2a and b show the low to high magnification TEM images of pristine NiFe-LDH, revealing the presence of the particles as a flower-like hierarchical nanostructure, which was in accordance with the FE-SEM results given in Fig. 1d. The selected area electron diffraction (SAED) pattern given in Fig. 2c revealed the polycrystalline nature of the particles and showed diffraction patterns from the (110) and (012) planes of the layered LDH lattice. The porous hierarchical nano-architecture of Fe@Ni3Se4 was further confirmed from the TEM images given in Fig. 2d. Further, the polycrystalline behaviour was confirmed from the SAED pattern given in Fig. 2e.
 | ||
| Fig. 2 (a and b) Low- to high-magnification TEM images for NiFe-LDH showing the micro flower-like structure consisting of distinct nanoflakes; (c) corresponding SAED pattern for NiFe-LDH; (d) microstructural outcomes of the Fe@Ni3Se4 nanosheets; (e) SAED pattern for polycrystalline Fe@Ni3Se4; (f) HAADF area chosen for the colour mapping analysis and (g–i) Colour mapping results for Ni, Fe, and Se K shells, respectively. | ||
The high-resolution TEM image (Fig. S3†) displayed a distinct lattice fringes pattern with a d-spacing value of 0.342 nm corresponding to the (110) plane of the Ni3Se4 structure. Elemental confirmation and their distributions were analyzed from the high angle annular dark field (HAADF) colour mapping analysis. The selected HAADF area is displayed in Fig. 2f. The uniform distribution of all the elements, like Ni, Fe, and Se, was confirmed from the colour mapping results given in Fig. 2g–i, respectively.
The chemical nature of all the elements was analyzed by X-ray photoelectron spectroscopy (XPS). The corresponding survey spectra of NiFe-LDH and Fe@Ni3Se4 are given in Fig. S4 and S5,† respectively, which reveal the presence of all the expected elements, like Ni, Fe, Se, C, and O, without any other impurity. The high-resolution XPS spectrum of the Ni 2p orbital (Fig. 3a) of NiFe-LDH could be well-fitted with two spin–orbit-coupling-originated peaks corresponding to Ni 2p3/2 and Ni 2p1/2 orbitals along with two satellite peaks at 860.8 and 878.7 eV.33 The deconvoluted XPS spectrum showed the presence of Ni in both the +2 and +3 oxidation states. The Fe 2p XPS outcomes of NiFe-LDH are displayed in Fig. 3b, revealing the presence of two well-fitted maxima at 711.1 and 724.52 eV corresponding to Fe 2p3/2 and Fe 2p1/2 orbitals in a +3 oxidation state.
 | ||
| Fig. 3 (a–c) Deconvoluted X-ray photoelectron spectra for Ni 2p, Fe 2p, and O 1s orbitals in pristine NiFe-LDH materials and (d–f) high-resolution XPS spectra for Ni 2p, Fe 2p, and Se 3d orbitals in Fe@Ni3Se4 materials. | ||
The other two peaks at higher binding energies of 715.74 and 733. 03 eV corresponded to Fe 2p3/2 and Fe 2p1/2 satellite peaks, respectively. The maximum for the 2p3/2 peak for Fe@Ni3Se4 was shifted towards a lower binding energy value of 0.62 eV, which indicated the existence or formation of lower valent iron as a result of the selenization under the reducing conditions. Moreover, the higher spin–orbit-coupling constant of Fe@Ni3Se4 (13.63 eV) compared to NiFe-LDH (13.11 eV) indicated the formation of a mixed valency, i.e. the co-existence of Fe2+ and Fe3+ ions. The O 1s XPS results given in Fig. 3c show the presence of three spin–orbit-coupling-originated peaks at binding energy values of 527.87, 529.75, and 530.29 eV, in accordance with the presence of oxygen as metal-hydroxide, metal-oxygen, and intercalated water molecule functionalities, respectively. The XPS spectrum of the Ni 2p orbital for Fe@Ni3Se4 (Fig. 3d) showed a similar XPS pattern as for NiFe-LDH, but maximum corresponding to the Ni 2p3/2 orbital was shifted towards a higher binding energy value by 0.65 eV, which might due to the oxidation of Ni2+ ions to maintain the charge in the seleno-spinel structure. It is interesting to look at the XPS spectrum of the Fe 2p orbital for the selenized counterpart given in Fig. 3e. A substantial amount of Fe3+ ions were reduced to Fe2+ under the reducing conditions, which could be identified from the XPS peaks at 710.9 eV.31 The presence of Fe2+ ions influenced the structural outcomes of the seleno-spinel structure, as confirmed through the DFT analysis discussed later. The Se 3d orbital XPS peak (Fig. 3f) showed two peaks at binding energy values of 53.64 and 54.94 eV, corresponding to the Se 3d5/2 and Se 3d3/2 orbitals and confirming the presence of selenium as Se2− ions. Another two higher energy peaks at 58.9 and 57.6 eV were also observed due to the presence of different surface oxides of Se.
2.2. Structural determination
Usually, Ni3Se4 crystallizes in the orthorhombic crystal system, where the bivalent nickel ions are bonded to four selenides to form NiSe4 tetrahedra that share corners with 12 NiSe6 octahedra. The presence of bi- or trivalent metal ions in the octahedral (Oh) and tetrahedral (Td) holes of a spinel is usually governed by the crystal-field-stabilization energy (CFSE) of the respective metal ions. For the Ni3Se4 structure, Ni3+ ions with the t2g6eg1 electronic configuration having a CFSE value of −1.8 Δo occupied the Oh hole, whereas Ni2+ ions with the eg4t24 electronic configuration with a CFSE value of −0.35 Δo occupied the Td hole, as shown in Scheme 1 (S-1). In the case of Fe-doped Ni3Se4, due to the multiple valences of the Fe ions, the selectivity for occupying the Oh and Td holes significantly varied.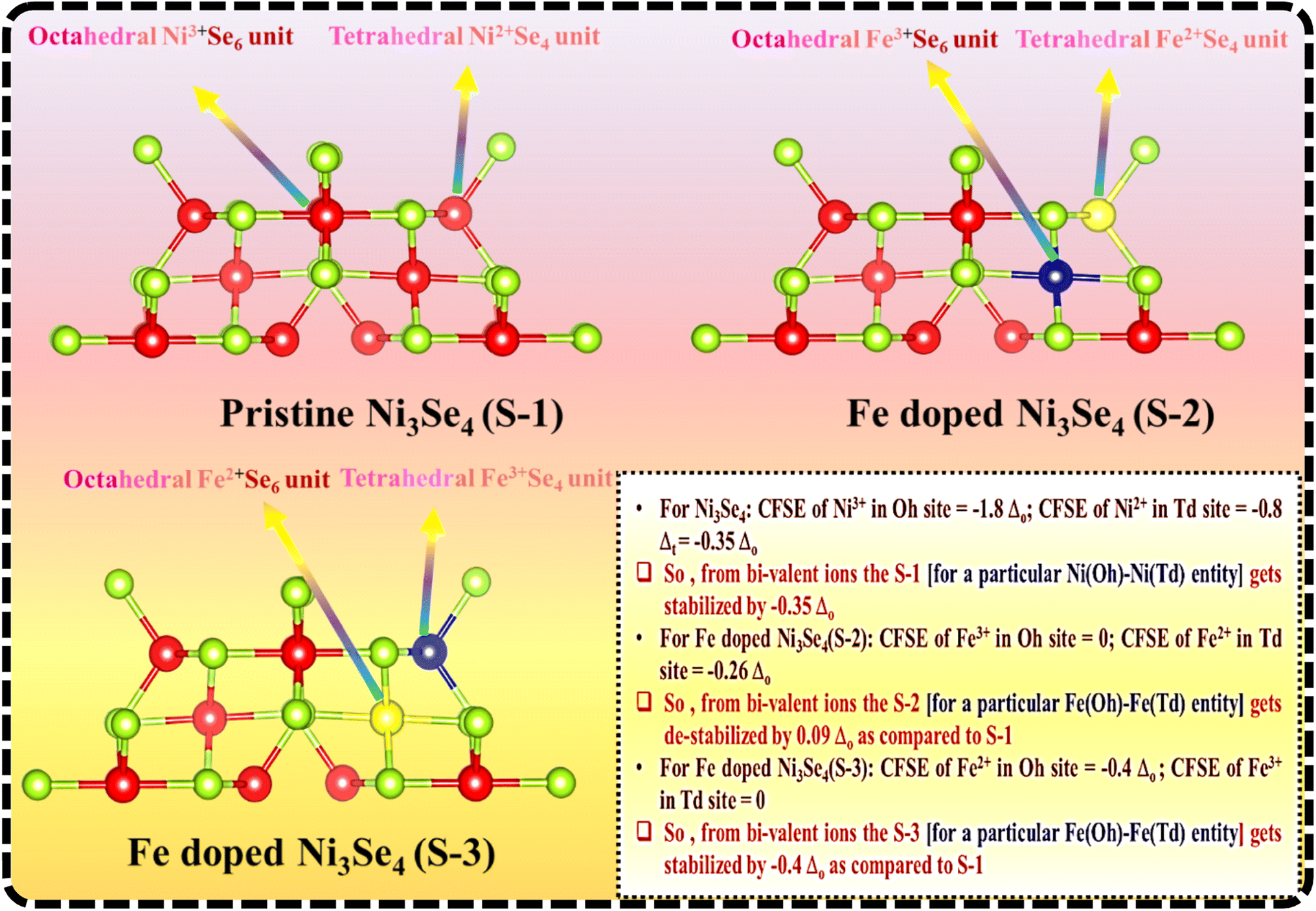 | ||
| Scheme 1 Probable structural analysis of pristine Ni3Se4 and different Fe-doped Ni3Se4 structures. S-1 corresponds to the pristine Ni3Se4; S-2 shows the Fe-doped Ni3Se4 where Fe3+ and Fe2+ ions have been placed over Oh and Td coordination sites, and S-3 displays the Fe-doped Ni3Se4 where Fe3+ and Fe2+ ions have been placed over Td and Oh coordination sites. | ||
According to the chemical nature of the spinel structure, Fe in the +3 and +2 oxidation states should occupy the Oh and Td sites, respectively, and would result in structure S-2. However, the presence of Fe3+ ions with the t2g3eg2 electronic configuration with a zero CFSE value and Fe2+ ions with the eg3t23 electronic configuration with a CFSE value of −0.26 Δo would destabilize the structure by 0.09Δo [CFSCFe2+(Fe@Ni3Se4) − CFSENi2+(Ni3Se4) = 0.09Δo]. Whereas, when considering the structure S-3, the Fe2+ ions with the electronic configuration of t2g4eg2 in Oh could bring additional stabilization to the spinel structure by 0.05Δo [CFSCFe(Oh)2+(Fe@Ni3Se4) − CFSENi(td)2+(Ni3Se4) = −0.05Δo]. The XPS analysis confirmed that Fe2+ and Fe3+ ions were present in equal proportions, and therefore, the even distribution of both metal ions could bring a structural stabilization to the Fe-doped Ni3Se4 structure. The presence of redox-active Fe2+ ions in the Oh coordination sites bought about added advantages over the redox activity of the selenide-based spinel structure with extra stability.
2.3. Electrochemical characterization
The electrochemical OER, HER, and total water splitting (TWS) catalytic activities of all the catalyst were evaluated in 1 M KOH solution. Modified nickel foam (NF) with an effective surface area of 1 cm2 was used as the working electrode. First, all the working electrodes were activated through CV cycling with a scan rate of 50 mV s−1 for 10 cycles in the case of OER in the potential region of 1 to 2 V vs. RHE. The polarization outcome of Fe@Ni3Se4 is given in Fig. 4a, demonstrating its highest OER activity among all the catalysts studied. The Fe@Ni3Se4 required just 201 mV of overpotential value to drive 50 mA cm−2 current density. The other given polarization values inferred that Fe@Ni3Se4 significantly outperformed NiFe-LDH (270 mV), Ni3Se4 (295 mV), and NF (400 mV) at 50 mA cm−2 (Fig. 4b). A predominant and broad oxidation peak could be observed in the potential region of 1.4–1.45 V vs. RHE, which was attributed to the Ni(III)–Ni(IV) redox transformation.34 From the ICP-MS analysis, it was observed that the quantitative ratio of Ni to Fe was about 4.85 in Fe@Ni3Se4, whereas it was about 1 in NiFe-LDH. Therefore, to validate the importance of the selenide structure over the hydroxide entity, the electrocatalytic OER activity was compared with Ni5Fe-LDH where the Ni to Fe ratio was ∼4.85. From the LSV outcomes (Fig. S6†), it was observed that Ni5Fe-LDH required 255 mV overpotential to drive 50 mA cm−2, which was about ∼50 mV higher than that of Fe@Ni3Se4. Moreover, the OER performance of Fe@Ni3Se4 was compared with commercial RuO2 materials, and the acquired LSV results are displayed in Fig. S7.† The commercial RuO2 required 327 mV overpotential to drive 50 mA cm−2 current density, which was 126 mV higher than our Fe@Ni3Se4 materials. To gain further insight into the OER activity, Tafel plots were extracted from the iR-drop free polarization information for all the catalysts, and are presented in Fig. 4c.35 Fe@Ni3Se4 delivered the lowest Tafel slope value of 38 mV dec−1, which indicated faster charge-transfer kinetics at the electrode–electrolyte interface compared to NiFe-LDH (68 mV dec−1), Ni3Se4 (77 mV dec−1), and NF (95 mV dec−1). The double-layered capacitance (Cdl) values of Fe@Ni3Se4 and NiFe-LDH were evaluated by scan-rate-dependent CV measurements (Fig. S8a and b†) and are illustrated in Fig. 4d. The calculated Cdl values were 0.053 and 0.04735 mF cm−2 for Fe@Ni3Se4 and NiFe-LDH, respectively. The electrochemical active surface area (ECSA), which is proportional to the Cdl value, was derived by dividing by Cs (capacitance for a flat surface = 0.04 mF cm−2).2 The results showed that Fe@Ni3Se4 (1.325 cm2) could deliver more exposed active surface area towards the electrolyte ions compared to pristine NiFe-LDH (1.18 cm2).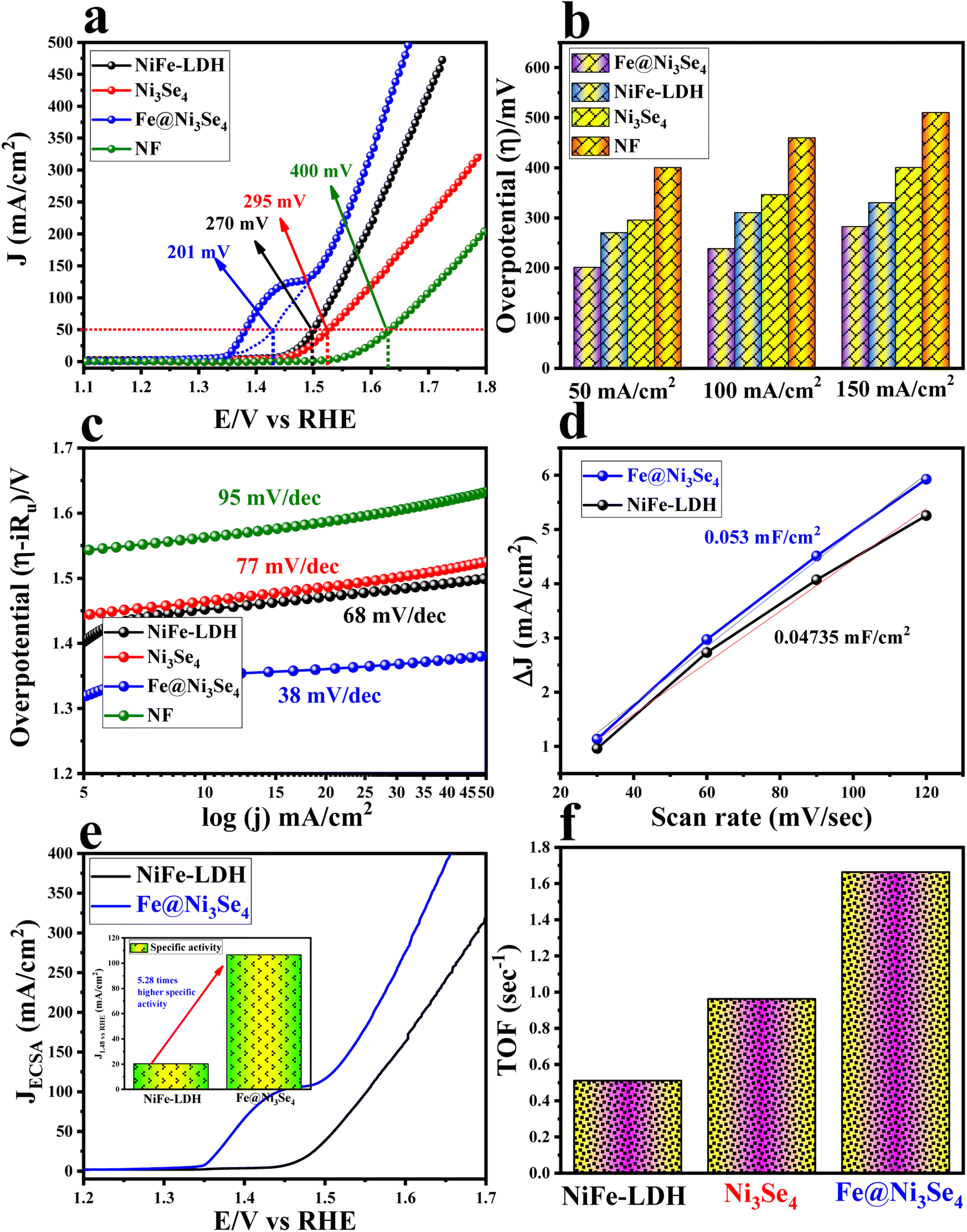 | ||
| Fig. 4 (a) Linear sweep voltammetric outcomes for all the catalyst acquired at a scan rate of 5 mV s−1 and the overpotentials calculated at 50 mA cm−2 current density; (b) bar diagram for the calculated overpotentials at different current densities; (c) Tafel plot acquired from the iR-drop free polarization information; (d) Cdl plot used to calculate the ECSA values of NiFe-LDH and Fe@Ni3Se4 catalyst; (e) ECSA-normalized LSV polarization results acquired to specify the specific activity of the catalysts; inset shows a comparison of the specific activities delivered by NiFe-LDH and Fe@Ni3Se4 at 1.48 V vs. RHE; (f) bar diagram for the TOF values delivered by all the catalysts with an applied potential value of 1.55 V vs. RHE. | ||
The specific activity of Fe@Ni3Se4 was further compared with NiFe-LDH from the ECSA-normalized polarization information given in Fig. 4e. The current density values acquired by Fe@Ni3Se4 and NiFe-LDH was calculated at a thermoneutral potential of 1.48 V vs. RHE. The inset of Fig. 4e displays the calculated current density, and shows that Fe@Ni3Se4 possessed a 5.23 times higher specific activity than the other catalyst. The intrinsic activity of all the catalyst were determined by calculating the turnover frequency (TOF), which is the number of O2 or H2 formed per second per active site.36 The TOF values of all the catalysts were determined from the redox surface area information given in Fig. S9a–c.†Fig. 4f shows the TOF values of Fe@Ni3Se4 (1.66 s−1), NiFe-LDH (0.96 s−1), and Ni3Se4 (0.5 s−1). Thus, by comparing the TOF result, it could be observed that Fe@Ni3Se4 delivered a high specific as well as high intrinsic activity compared to the other catalysts. Further the long-term stability of Fe@Ni3Se4 was verified by CV cycling for 1000 times with a high scan rate of 180 mV s−1. The resulting backwards polarization (to exclude the interference of the oxidation peak current in the forward CV cycles) information was compared with the polarization information before the cycling study (Fig. 5a). Fig. 5a portrays there was minimal degradation of the catalyst under the harsh cycling, inferring the excellent stability of the catalyst. A steady-state long-term stability study was performed via chronoamperometry (CA) with a fixed potential of 1.5 V vs. RHE. The resulting CA curve given in Fig. 5b shows the excellent stability of the catalyst over 50 h. The intrinsic activity of a catalyst demonstrates its site-specific or per-site activity information and thereby is related to the chemical and physical properties of the catalytic sites. The ECSA-normalized TOF, i.e. specific TOF value, is more acceptable than that of geometrically normalized polarization information (Fig. S10a and b†). To gain deeper knowledge about the site-specific intrinsic activity information, the specific TOF values were calculated and are portrayed in Fig. 5c.
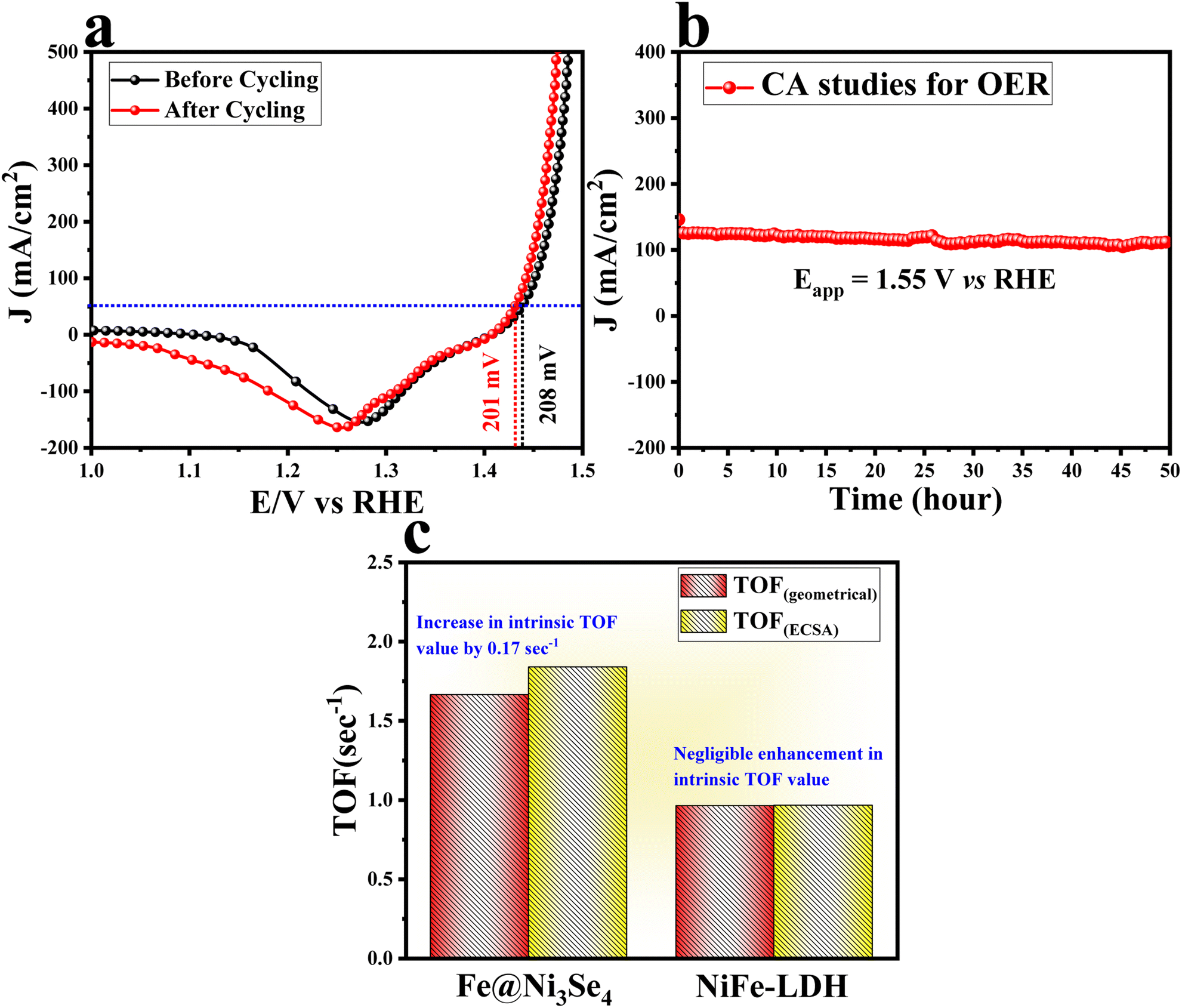 | ||
| Fig. 5 (a) LSV polarization outcomes of Fe@Ni3Se4 catalyst before and after 1000 cycles at a high scan rate of 180 mV s−1; (b) chronoamperometric results of Fe@Ni3Se4 with an applied potential of 1.55 V vs. RHE for 50 h, and (c) comparison of the intrinsic activity (with respect to the TOF value) in the geometrical and ECSA-normalized conditions. | ||
It is interesting to notice that Fe@Ni3Se4 delivered a higher specific TOF value by 0.17 s−1, whereas for the pristine NiFe-LDH, the difference in TOF value was negligible. This is a quite interesting result and the possible reason behind this improvised specific activity is the presence of redox-active Fe2+ ions and the hierarchical porous structure in the selenide counterpart, as was evident from the BET results. The formation of a porous network allows mass transfer quite effectively, which enhances the charge transfer at the electrolyte interfaces. On the other hand, the presence of redox-active low-valent Fe entities could apparently enhance the site-specific active sites for OH* adsorption.31
To visualize the improved adsorption kinetics of OH* over Fe@Ni3Se4, operando electrochemical impedance (EIS) analysis was carried out. Operando EIS is an effective tool to investigate the adsorption and desorption kinetics for OER processes at the electrode–electrolyte interface.37–40 The corresponding Nyquist plots at different applied potentials are displayed in Fig. S11a and b,† which show the electrochemical behaviours of Fe@Ni3Se4 and NiFe-LDH. The Bode plots for both materials are shown in Fig. 6a and b, which could be differentiated into three different regions: the non-faradaic Cdl region, electro-activation or redox region (both at high frequency sites), and the OER region or the region of the concerned faradaic process (at low frequency sites). The localized and higher phase angle shift in Fe@Ni3Se4 in the Bode signal in the high-frequency region (Fig. 6b) revealed the higher charge-transfer kinetics at the electrode compared to pristine NiFe-LDH.
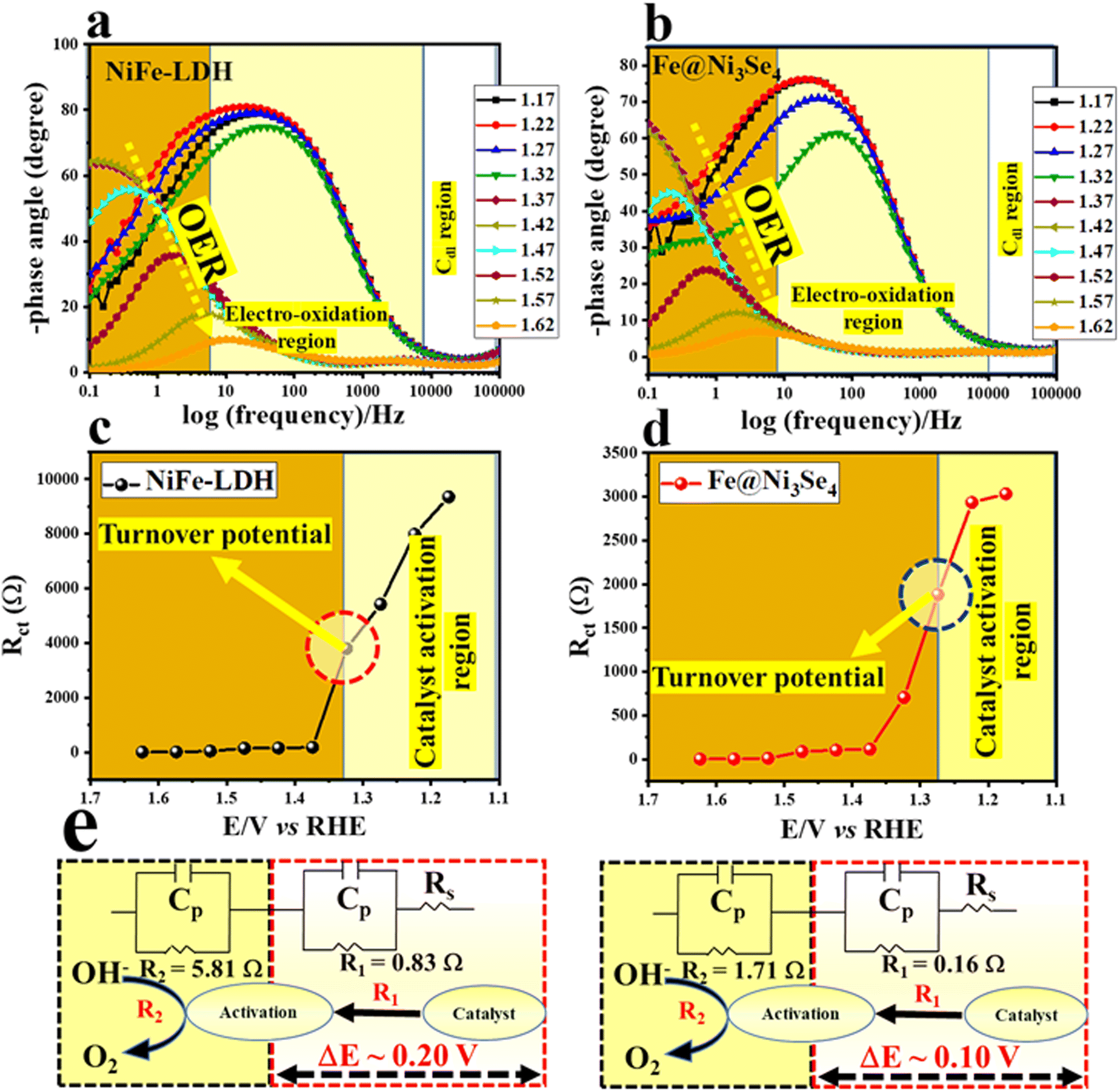 | ||
| Fig. 6 (a and b) Respective Bode plots for NiFe-LDH and Fe@Ni3Se4 with different applied potentials; (c and d) Rctvs. applied potential plots for NiFe-LDH and Fe@Ni3Se4 and (e) the corresponding equivalent circuit diagrams with the respective activation resistance and resistance of the faradaic reaction. | ||
To gain an insight into the improved redox activity by Fe@Ni3Se4, the fitting resistance information (Fig. 6c–e) was analyzed at different applied potentials. An equivalent circuit diagram was noted with two successive redox reactions characterized with two resistance values, i.e. R1 (equivalent to redox transformation to reach the activated state) and R2 (related to the OER process). From the resistance information in Fig. 6c and d, a sharp drop in resistance could be seen at a certain potential (marked as the turnover potential), which indicated the transformation from a non-faradaic reaction to a faradaic process. Fe@Ni3Se4 delivered a turnover potential value of 1.27 V vs. RHE with a potential window of ∼0.1 V, whereas NiFe-LDH demonstrated a turnover potential of 1.32 V vs. RHE with a potential window of ∼0.2 V. Further, the calculated R1 value, i.e. the resistance towards the electrochemical activation steps of Fe@Ni3Se4 (0.16 Ω) and NiFe-LDH (0.83 Ω), indicated the faster redox transformation of the selenide counterpart as compared to NiFe-LDH.41 Therefore, the increase in porosity and redox-active metal ions as a result of the selenization in the sheet-like NiFe-LDH synergistically enhanced the electrocatalytic activity in the resulting Fe-doped Ni3Se4.
Further the potential application of Fe@Ni3Se4 towards the cathodic HER reaction was verified in 1 M KOH solution. As shown Fig. 7a, Fe@Ni3Se4 showed the highest HER activity and it required just 33 and 96 mV of overpotential to achieve 10 and 50 mA cm−2 current densities, respectively; whereas the other pristine materials, like NiFe-LDH and Ni3Se4, showed higher overpotentials of 113 and 143 mV (Fig. S12†) to drive the same 10 mA cm−2 current density. More importantly, from the given polarization information in Fig. 7a, it was interesting to observe that Fe@Ni3Se4 outperformed the commercial Pt/C (20%) catalyst, which required 59 mV overpotential at the benchmark current density.
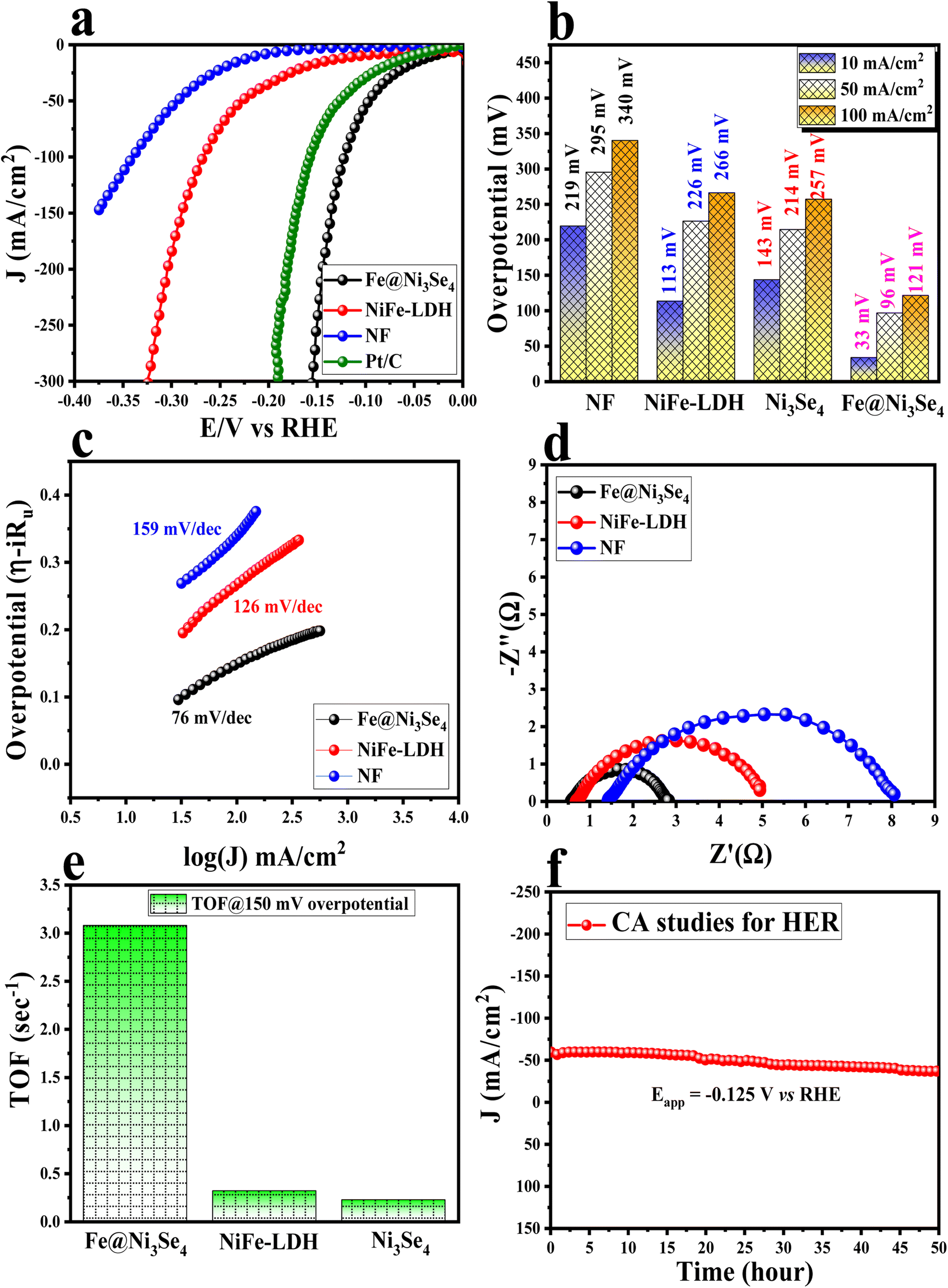 | ||
| Fig. 7 (a) Linear sweep voltammetric outcomes for all the catalysts acquired at a scan rate of 5 mV s−1 and overpotentials calculated at a 50 mA cm−2 current density; (b) bar diagram showing the calculated overpotentials at different current densities; (c) Tafel plots acquired from the iR-drop free polarization information; (d) Nyquist plots for all the catalysts under HER conditions; (e) bar diagram for the TOF values delivered by all the catalysts with an applied potential value of −0.15 V vs. RHE, and (f) chronoamperometric results of Fe@Ni3Se4 with an applied potential of 1.55 V vs. RHE for 50 h. | ||
The excellent electrocatalytic performance of Fe@Ni3Se4 compared to the other materials could further be verified from the bar diagram in Fig. 7b. The charge-transfer kinetics at the interface was verified from the Tafel slope analysis and the obtained results are illustrated in Fig. 7c. Fe@Ni3Se4 possessed a lower Tafel slope value of 76 mV dec−1, whereas pristine NiFe-LDH delivered a higher Tafel slope value of 126 mV dec−1. A lower Tafel slope value indicates faster kinetics, and a value of 76 mV dec−1 indicates that HER has proceeded by following the Volmer–Heyrovsky kinetics process. Faster electron transfer at the interface was further verified from the EIS analysis by fixing a constant over a potential value of −0.175 V vs. RHE (Fig. 7d). The lower Rct value (2.21 Ω) for Fe@Ni3Se4 compared to NiFe-LDH (6.62 Ω) illustrated the low internal resistance for the electron transfer at the interface towards the HER.42
The intrinsic activities of all the catalysts were verified by calculating their TOF values at 150 mV overpotential. Fe@Ni3Se4 delivered the highest TOF value of 3.07 s−1, whereas the TOF values for NiFe-LDH and Ni3Se4 were about 0.31 and 0.22 s−1, respectively. The high TOF value for Fe-doped Ni3Se4 illustrated its improved per-site activity as a result of the selenization and the low-valent Fe ions in the lattice. Moreover, the static stability of Fe@Ni3Se4 was investigated via chronoamperometry (CA) with an applied potential value of −0.125 V vs. RHE. The 50 h CA study revealed the excellent stability of the selenized counterpart.
In light of the exceptional activities and stabilities of Fe@Ni3Se4 towards the OER and HER, it is reasonable to suggest Fe@Ni3Se4 would be an auspicious bifunctional electrocatalyst for the overall water splitting reaction. Therefore, a two electrode set-up was constructed by taking two pieces of Fe@Ni3Se4 with an effective geometrical surface area of 1 × 1 cm2. The LSV polarization curve (Fig. 8a) for the total water splitting showed that the Fe@Ni3Se4‖Fe@Ni3Se4 electrolyser demanded just 1.64 V to drive 50 mA cm−2 current density. Also, the Fe@Ni3Se4‖Fe@Ni3Se4 electrolyser outperformed the state-of-art RuO2‖Pt/C electrolyser. More importantly, the Fe@Ni3Se4‖Fe@Ni3Se4 electrolyser displayed strong stability for total water splitting at a cell voltage of 1.63 V with a current density of 50 mA cm−2 for 33 h (Fig. 8b).
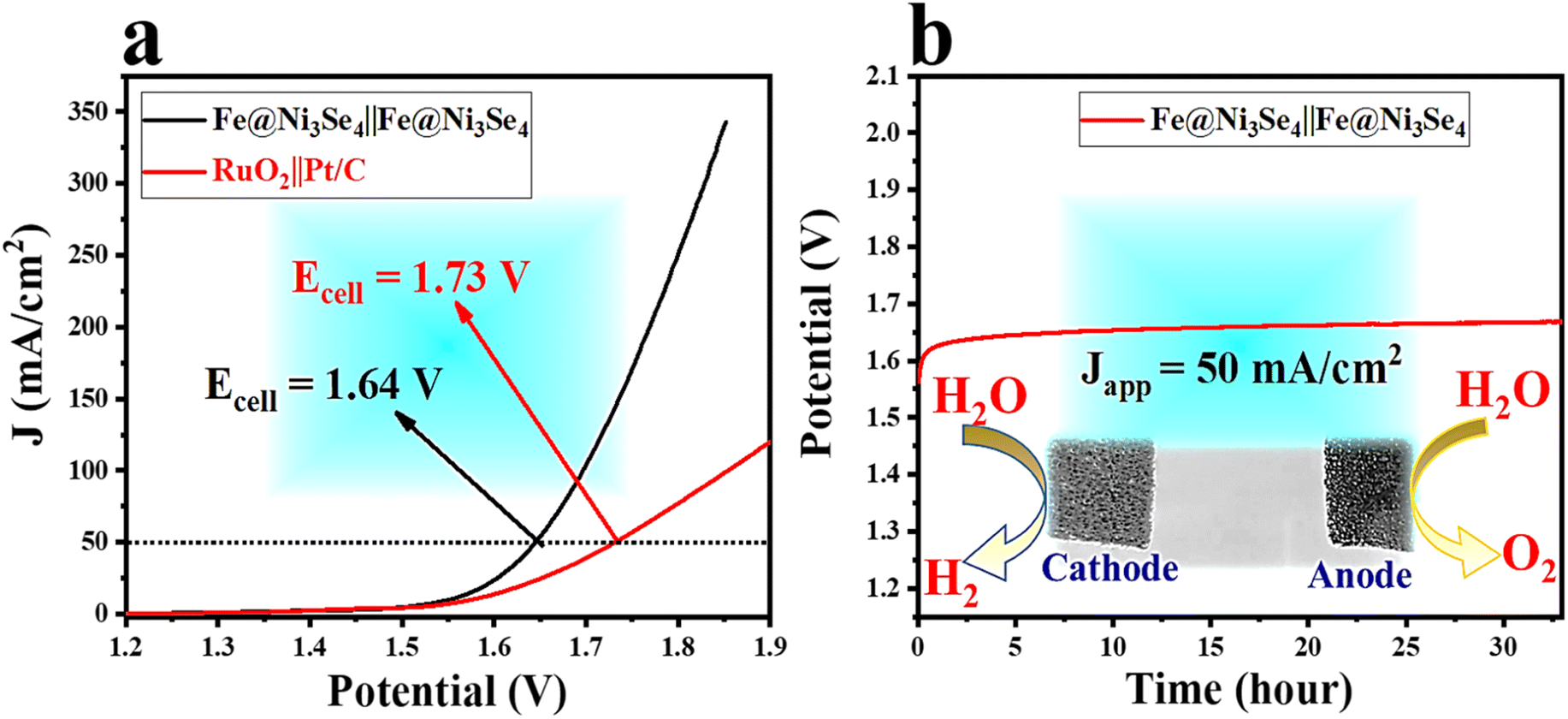 | ||
| Fig. 8 (a) LSV polarization information towards total water splitting activity by using Fe@Ni3Se4 as both anode and cathode materials and (b) chronopotentiometric outcomes for total water splitting for >30 h; inset shows the cathode and anode configuration in the electrochemical cell. | ||
The Fe@Ni3Se4 porous nanostructured electrode exhibited remarkably low OER overpotentials of 201 and 185 mV at 50 and 10 mA cm−2, respectively, which make it among the best NiFe-based OER catalysts (Table S1†), and much more active than the state-of-the-art RuO2 catalyst. Moreover, Fe-doped Ni3Se4 possessed excellent alkaline HER activity too and it demanded just 33 mV overpotential to drive 10 mA cm−2 current density, which is a remarkable result for a non-platinum or non-noble-metal-based electrocatalyst in alkaline conditions, as can be seen from Table S2†.
Further, the structural and morphological stabilities of Fe@Ni3Se4 after long-term electrochemical application were assessed by different characterization techniques, such as XPS, TEM, and Raman analysis. From the XPS plots (Fig. S13a–d†), it was evident that for Ni and Fe, there was definite peak shifting of the XPS peaks towards higher binding energy values compared to the XPS maxima in the pre-OER materials. This peak shifting towards higher binding energy sites revealed the definite oxidation of the metallic elements under the harsh anodic environment. The deconvoluted XPS spectrum of the O 1s orbital displayed three spin–orbit-coupling-originated peaks corresponding to the M-O (oxyhydroxide), M-OH, and H2O molecules. The presence of selenide along with the oxide phase were observed from the Se 3d orbital XPS outcomes. Microstructural analysis of the Fe@Ni3Se4 material after the OER study was performed by TEM analysis, and the corresponding TEM results are portrayed in Fig. S14a–h.† The materials possessed similar 2D sheet-like morphologies after OER analysis, which were also like the catalyst before the OER analysis. The high-resolution TEM image showed different d-spacing patterns corresponding to the basic Ni3Se4 and electrochemically modified oxyhydroxide layer. This electrochemical modification would lead to the formation of a NiOOH@Ni3Se4 heterostructural entity. HADDF colour mapping analysis displayed a finite distribution of all the expected elements. The formation of an –OOH layer was further confirmed from the Raman analysis given in Fig. S15.†43
2.4. DFT analysis
Density functional theory (DFT) computations were used to investigate the interfacial electronic structures and catalytic reactions of the pristine and Fe-doped Ni3Se4 for investigating the underlying OER and HER mechanisms. All the calculation details are provided in the ESI.† According to previous DFT results, the Ni3Se4 (110) faces have the lowest surface energy and hence Ni3Se4 was modelled using the most stable Ni3Se4 facet.44 The most stable structural configurations of both the pristine and Fe-doped Ni3Se4 are illustrated in Fig. S16a and b.† In the crystal structure of Ni3Se4, there are two types of coordinated Ni ions: tetracoordinated Ni4 (a Ni atom coordinated with four Se atoms, Ni2+) and hexacoordinated Ni6 (a Ni atom coordinated with six Se atoms, Ni3+). The Fe doping was studied by replacing one Ni2+ and Ni3+ site in the Ni–Fe–Se spinel, and the calculation results suggested that the energy of the Fe doping in the octahedral site of the Fe–Ni–Se spinel unit cell was 0.192 eV lower compared to that of tetrahedral site, which was predicted before by the CFSE calculations. Furthermore, density of states (DOS) analysis was performed to understand the role of Fe in modulating the electronic structure of pristine Ni3Se4. Compared to the pristine substrate, Fe doping in the Fe–Ni–Se spinel exhibited an improved electron density around the Fermi level, illustrating the improved electrical conductivity (Fig. 9a and b). For both Ni3Se4 and Fe@Ni3Se4, it was noteworthy to see that the selenium 3d states were more dominant in the Fermi level (Ef), and therefore it could be predicted that the addition of Fe ions well retained the electronic interactions among the various metal ions. In the pristine Ni3Se4 system, it was interesting to note that the Ni 3d state was poorly situated near the Fermi level, whereas for Fe@Ni3Se4 a definite Ni 3d state was localized near it. The localization of the Ni 3d states near the Ef could play a crucial role in promoting the OER. Bader charge analysis was performed and illustrated that the Fe atom has a more positive charge (0.593e) compared to its neighbouring Ni atoms 0.3912e). The observed electron accumulation on the Ni site could be anticipated to improve the total water splitting reactions by stabilizing the various intermediates of Ni ions in a higher oxidation state (Fig. 9c). This electronic exchange between higher valent Ni and low-valent Fe ions leads to an increase in the 3d-2p electronic repulsion in the Ni–OOH intermediate and thereby could accelerate the O2 desorption faster than with the pristine Ni3Se4.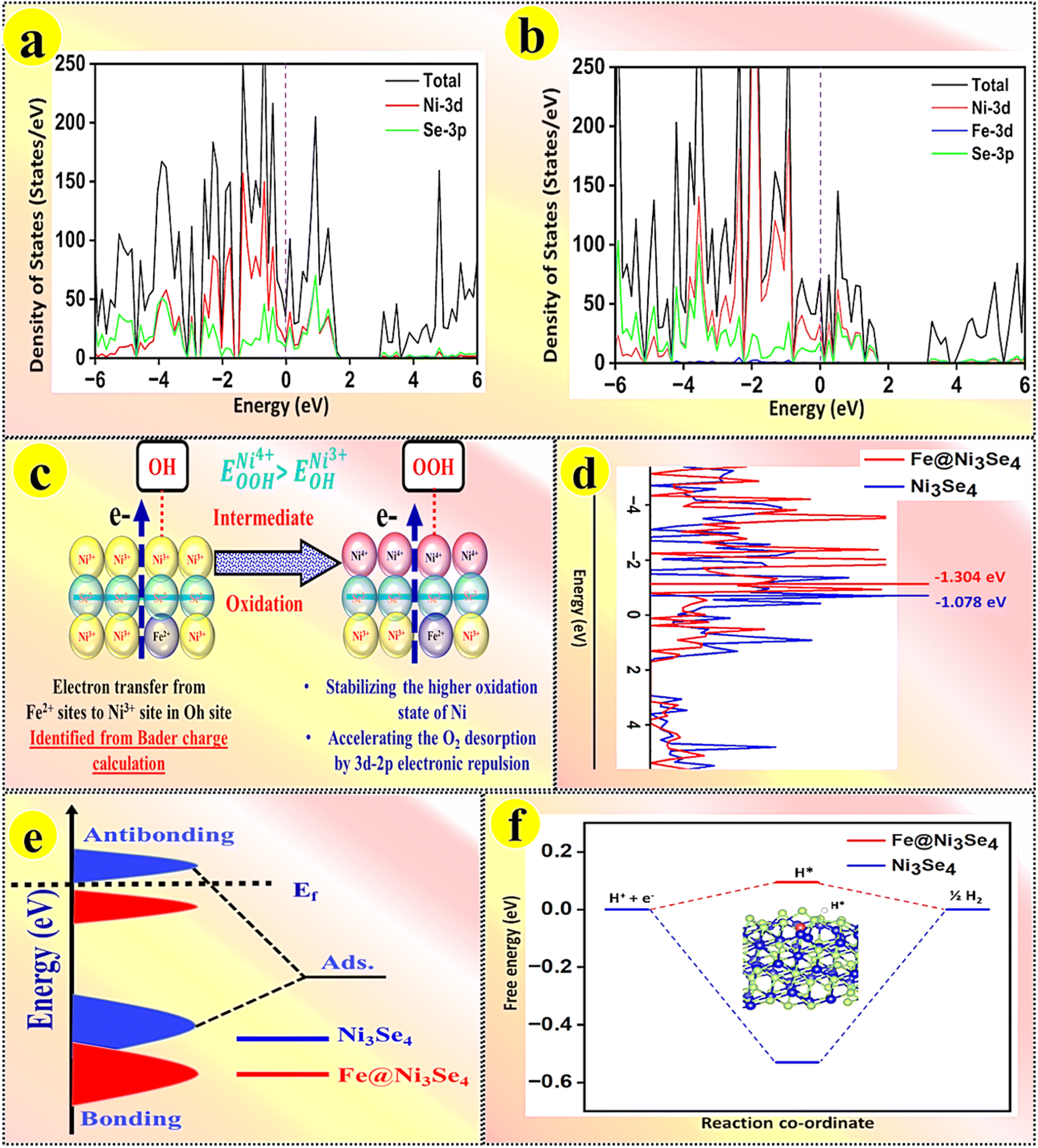 | ||
| Fig. 9 (a and b) Total density of states (TDOS) of both the pristine and Fe@Ni3Se4 materials. The Fermi level is indicated with a purple line; (c) possible electronic exchanges in various Ni–O intermediates and the respective electronic exchange between Ni and Fe via selenide bridging; (d) computed d-band centres of both the pristine and Fe@Ni3Se4 substrates; (e) d-band structure and (f) Gibbs free energy diagram of the HER process on the pristine and Fe@Ni3Se4 substrates. | ||
In addition, we investigated the free energy profile for H* adsorption for both the pristine Ni3Se4 and Fe@Ni3Se4 materials. It was demonstrated that the H* adsorption free energy 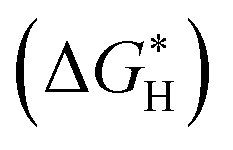 is a critical predictor of the activity of HER catalysts, and that an efficient HER catalyst should have a small H* adsorption energy (Fig. S17a and b†).45,46 From Fig. 9f, the
is a critical predictor of the activity of HER catalysts, and that an efficient HER catalyst should have a small H* adsorption energy (Fig. S17a and b†).45,46 From Fig. 9f, the 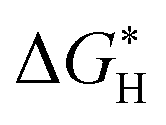 of the studied Fe-doped Ni3Se4 electrocatalyst was 0.094 eV, i.e. closer to the thermoneutral value compared to the pristine substrate (−0.573 eV), and moreover the studied electrocatalyst's HER activity was comparable to that of the state-of-the-art Pt. The d-band centre was determined using the position of the d-orbital, which regulates the adsorption strength of the examined electrocatalysts.47 By examining the energy of the antibonding states located above the Fermi level, the d-band centre can be used as a useful descriptor to understand the adsorption strengths of the investigated electrocatalysts. We noted that the d-band centre of Fe@Ni3Se4 was shifted further away from the Fermi level (−1.304 eV) compared to that of the pristine Ni3Se4 (−1.078 eV), as shown in Fig. 9d. The study of the d-band centres revealed that the binding strength of the Fe-doped substrate towards the HER was weaker than that of the pristine substrate, as evidenced by the shift in the Fermi level to a lower energy level with a greater number of occupied antibonding states (Fig. 9e). As a result, the mild adsorption behaviour of the Fe-doped Ni3Se4 substrate may facilitate the adsorption and desorption of H*, thereby enhancing its HER catalysis. The experimentally observed superior electrocatalytic activity of the Fe-doped Ni3Se4 substrate was in good agreement with the theoretical DFT calculations.
of the studied Fe-doped Ni3Se4 electrocatalyst was 0.094 eV, i.e. closer to the thermoneutral value compared to the pristine substrate (−0.573 eV), and moreover the studied electrocatalyst's HER activity was comparable to that of the state-of-the-art Pt. The d-band centre was determined using the position of the d-orbital, which regulates the adsorption strength of the examined electrocatalysts.47 By examining the energy of the antibonding states located above the Fermi level, the d-band centre can be used as a useful descriptor to understand the adsorption strengths of the investigated electrocatalysts. We noted that the d-band centre of Fe@Ni3Se4 was shifted further away from the Fermi level (−1.304 eV) compared to that of the pristine Ni3Se4 (−1.078 eV), as shown in Fig. 9d. The study of the d-band centres revealed that the binding strength of the Fe-doped substrate towards the HER was weaker than that of the pristine substrate, as evidenced by the shift in the Fermi level to a lower energy level with a greater number of occupied antibonding states (Fig. 9e). As a result, the mild adsorption behaviour of the Fe-doped Ni3Se4 substrate may facilitate the adsorption and desorption of H*, thereby enhancing its HER catalysis. The experimentally observed superior electrocatalytic activity of the Fe-doped Ni3Se4 substrate was in good agreement with the theoretical DFT calculations.
3. Conclusion
In summary, a NiFe-LDH-derived Fe-doped Ni3Se4 nanostructure with a high porosity was designed by a simple and effective selenization under solvothermal conditions. Selenization of the LDH nanostructure resulted in the doping of Fe ions with multiple valences in the Ni3Se4 seleno-spinel structure. In line with the high CFSE values, the Fe ions with bivalency were positioned over the redox-active Oh coordination environment, whereas the trivalent Fe ions were placed over the redox-inactive Td sites. The selective adsorption of low-valent Fe ions over the Oh sites resulted in a high specific activity of the Fe@Ni3Se4 material compared to the pristine NiFe-LDH and Ni3Se4. Operando EIS analysis showed the strong support for the facile redox activity of the active Ni ions for the electrocatalytic performance. The Fe@Ni3Se4 demanded overpotentials of 185 and 33 mV to deliver 10 mA cm−2 current density in the OER and HER, respectively. The calculated TOF value was about 3.07 s−1 in the HER condition, which was almost 10 times higher than that of the pristine materials. Moreover, Fe@Ni3Se4 displayed excellent long-term stability for over 50 h with negligible degradation in both the HER and OER conditions. When Fe@Ni3Se4 was used as both the anode and cathode materials for total water splitting, a constructed cell showed excellent performance. With a limiting current density of about 320 mA cm−2, the Fe@Ni3Se4‖Fe@Ni3Se4 electrolyser demanded just 1.64 V cell voltage to drive 50 mA cm−2 current density. Post-electrocatalytic characterization showed the excellent morphological and structural stabilities of the Fe@Ni3Se4 materials. Our work shows the important role of metal-ion doping with a stabilized coordination environment for the optimization of electrocatalysis. Moreover, the LDH-derived selenide materials outperform the benchmark electrocatalyst in terms of the OER, HER, and total water splitting performances. Therefore, the present work demonstrates a noble approach for catalyst designing. The first-principles DFT calculation revealed that doping low-valent Fe ions largely increased the 3d-2p repulsion in Ni–O bonds and thereby facilitated the O2 desorption process compared to pristine Ni3Se4 as a result of the electronic exchange. Moreover, the downward shift of the d-band centre with respect to the Fermi level thereby weakened the binding strengths of the Fe-doped substrate toward H* with an optimum adsorption energy compared to the pristine substrates and could meet a balance between the adsorption and desorption processes according to Sabatier's principle.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Arun Karmakar wishes to acknowledge CSIR-New Delhi for the award of a Senior Research Fellowship (SRF). S. K. wishes to acknowledge the Department of Science and Technology (DST) for HCF (Hydrogen and Fuel Cell) research funding of number # DST/TMD/HFC/2K18/60 on 7th October 2019 with institute OM number 18-29-03/(31/19)-TTBD-CSIR-CECRI on 24/10/2019 and CRG (Core Research Grant) funding of number # CRG/2021/001089 dated November 20th, 2021. CSIR-CECRI manuscript number: CECRI/PESVC/Pubs/2023-017.References
- N. L. Garland, D. C. Papageorgopoulos and J. M. Stanford, Energy Procedia, 2012, 28, 2–11 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- I. Dincer, Int. J. Hydrogen Energy, 2012, 37, 1954–1971 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- P. P. Edwards, V. L. Kuznetsov, W. I. F. David and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef.
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2002, 27, 731–740 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- D. Liu, Y. Yang, H. Zhu, D. Liu, S. Yan and Z. Zou, J. Phys. Chem. Lett., 2022, 13, 49–57 CrossRef CAS PubMed.
- Y. Jiang and Y. Lu, Nanoscale, 2020, 12, 9327–9351 RSC.
- S. Anantharaj and S. Noda, Small, 2020, 16, 1–24 CrossRef PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- Y. Zhang, X. Chen, Y. Huang, C. Zhang, F. Li and H. Shu, J. Phys. Chem. C, 2017, 121, 1530–1536 CrossRef CAS.
- K. Zhang and R. Zou, Small, 2021, 17, 1–40 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. Kundu, S. Kumaravel, K. Karthick, S. S. Sankar, A. Karmakar, R. Madhu and K. Bera, ChemElectroChem, 2021, 1–49 Search PubMed.
- S. Kumaravel, K. Karthick, S. S. Sankar, A. Karmakar, R. Madhu, K. Bera and S. Kundu, Sustain. Energy Fuels, 2021, 5, 6215–6268 RSC.
- N. Seal, A. Karmakar, S. Kundu and S. Neogi, ACS Sustain. Chem. Eng., 2023, 11, 979–993 CrossRef CAS.
- R. Madhu, R. Jayan, A. Karmakar, S. S. Selvasundarasekar, S. Kumaravel, K. Bera, S. Nagappan, H. N Dhandapani, M. M. Islam and S. Kundu, ACS Sustain. Chem. Eng., 2022, 10, 11299–11309 CrossRef CAS.
- R. Madhu, A. Karmakar, S. Kumaravel, S. S. Sankar, K. Bera, S. Nagappan, H. N. Dhandapani and S. Kundu, ACS Appl. Mater. Interfaces, 2022, 14, 1077–1091 CrossRef CAS PubMed.
- S. Nagappan, A. Karmakar, R. Madhu, H. N Dhandapani, K. Bera, A. De and S. Kundu, ACS Appl. Energy Mater., 2022, 5, 12768–12781 CrossRef CAS.
- K. Ao, Q. Wei and W. A. Daoud, ACS Appl. Mater. Interfaces, 2020, 12, 33595–33602 CrossRef CAS PubMed.
- S. Hu, S. Wang, C. Feng, H. Wu, J. Zhang and H. Mei, ACS Sustain. Chem. Eng., 2020, 8, 7414–7422 CrossRef CAS.
- M. Ali, E. Pervaiz, T. Noor, O. Rabi, R. Zahra and M. Yang, Int. J. Energy Res., 2021, 45, 1190–1226 CrossRef CAS.
- S. Anantharaj and S. Noda, Int. J. Hydrogen Energy, 2020, 45, 15763–15784 CrossRef CAS.
- K. Kannimuthu, K. Sangeetha, S. Sam Sankar, A. Karmakar, R. Madhu and S. Kundu, Inorg. Chem. Front., 2021, 8, 234–272 RSC.
- J. Wei, K. Xiao, Y. Chen, X. P. Guo, B. Huang and Z. Q. Liu, Energy Environ. Sci., 2022, 15, 4592–4600 RSC.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W. F. Lin, X. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- D. Tyndall, J. Craig, L. Gannon, C. Mcguinness, N. Mcevoy, A. Roy, M. Garc, P. Browne and V. Nicolosi, J. Mater. Chem. A, 2023, 11, 4067–4077 RSC.
- A. Hameed, M. Batool, Z. Liu, M. A. Nadeem and R. Jin, ACS Energy Lett., 2022, 7, 3311–3328 CrossRef CAS.
- Z. Cai, D. Zhou, M. Wang, S. M. Bak, Y. Wu, Z. Wu, Y. Tian, X. Xiong, Y. Li, W. Liu, S. Siahrostami, Y. Kuang, X. Q. Yang, H. Duan, Z. Feng, H. Wang and X. Sun, Angew. Chem. Int. Ed., 2018, 57, 9392–9396 CrossRef CAS PubMed.
- W. K. Han, J. X. Wei, K. Xiao, T. Ouyang, X. Peng, S. Zhao and Z. Q. Liu, Angew. Chem. Int. Ed., 2022, 61, e202206050 CAS.
- R. Yang, Y. Zhou, Y. Xing, D. Li, D. Jiang, M. Chen, W. Shi and S. Yuan, Appl. Catal., B, 2019, 253, 131–139 CrossRef CAS.
- Z. K. Goldsmith, A. K. Harshan, J. B. Gerken, M. Vörös, G. Galli, S. S. Stahl and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3050–3055 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem. Int. Ed., 2021, 60, 23051–23067 CrossRef CAS PubMed.
- C. S. Hsu, N. T. Suen, Y. Y. Hsu, H. Y. Lin, C. W. Tung, Y. F. Liao, T. S. Chan, H. S. Sheu, S. Y. Chen and H. M. Chen, Phys. Chem. Chem. Phys., 2017, 19, 8681–8693 RSC.
- G. M. Rupp, M. Kubicek, A. K. Opitz and J. Fleig, ACS Appl. Energy Mater., 2018, 1, 4522–4535 CrossRef CAS PubMed.
- R. Kant and H. Goel, J. Phys. Chem. Lett., 2021, 12, 10025–10033 CrossRef CAS PubMed.
- K. George, M. Van Berkel, X. Zhang, R. Sinha and A. Bieberle-Hütter, J. Phys. Chem. C, 2019, 123, 9981–9992 CrossRef CAS.
- Y. Li, Y. Wu, M. Yuan, H. Hao, Z. Lv, L. Xu and B. Wei, Appl. Catal., B, 2022, 318, 121825 CrossRef CAS.
- J. Chen, X. Luo, H. Zhang, X. Liang, K. Xiao, T. Ouyang, M. Dan and Z. Q. Liu, Electrochim. Acta, 2023, 439, 141711 CrossRef CAS.
- S. Zuo, Z. P. Wu, H. Zhang and X. W. Lou, Adv. Energy Mater., 2022, 12, 2103383 CrossRef CAS.
- K. Wan, J. Luo, C. Zhou, T. Zhang, J. Arbiol, X. Lu, B. W. Mao, X. Zhang and J. Fransaer, Adv. Funct. Mater., 2019, 29, 1900315 CrossRef.
- W. H. Lai, H. Wang, L. Zheng, Q. Jiang, Z. C. Yan, L. Wang, H. Yoshikawa, D. Matsumura, Q. Sun, Y. X. Wang, Q. Gu, J. Z. Wang, H. K. Liu, S. L. Chou and S. X. Dou, Angew. Chem. Int. Ed., 2020, 59, 22171–22178 CrossRef CAS PubMed.
- R. Yan, J. Lin, L. You, K. Guo, X. Yang, J. Zhang, D. Hu, J. Luo and J. T. Zhao, J. Alloys Compd., 2018, 745, 228–233 CrossRef CAS.
- S. Bhattacharjee, U. V. Waghmare and S. C. Lee, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electrodes fabrications, material preparation for different characterizations, data for OER, and comparative electrocatalysis data are given. See DOI: https://doi.org/10.1039/d3ta00868a |
| This journal is © The Royal Society of Chemistry 2023 |