
How carbon contamination on the photocatalysts interferes with the performance analysis of CO2 reduction†
Jiakang
You
a,
Mu
Xiao
a,
Siqi
Liu
 a,
Haijiao
Lu
a,
Peng
Chen
a,
Zhi
Jiang
b,
Wenfeng
Shangguan
b,
Zhiliang
Wang
*a and
Lianzhou
Wang
*a
a,
Haijiao
Lu
a,
Peng
Chen
a,
Zhi
Jiang
b,
Wenfeng
Shangguan
b,
Zhiliang
Wang
*a and
Lianzhou
Wang
*a
aNanomaterials Centre, School of Chemical Engineering and Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Queensland 4072, Australia
bResearch Center for Combustion and Environment Technology, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: zhiliang.wang@uq.edu.au; l.wang@uq.edu.au
First published on 24th April 2023
Abstract
Photocatalytic carbon dioxide (CO2) reduction reaction (CO2RR) for the production of valuable chemicals is a promising solar-driven strategy to mitigate CO2 emissions. However, carbon contamination on the photocatalysts interferes with the investigation of CO2RR performance. This work quantitatively investigates the significant impact of carbon contamination on performance analysis of photocatalytic CO2RR, which can lead to false-positive results of photocatalysts with different types of band structure (i.e., TiO2, CuO, and BiVO4) due to photoinduced oxidation process. Moreover, the commonly used organic solvent in a laboratory environment (e.g., ethanol) was proved to have a profound impact on photocatalytic CO2RR behaviour wherein 1 microliter of ethanol could boost the apparent methane generation by 17 times. To solve this issue, oxygen plasma treatment is demonstrated to be effective in removing surface carbon contamination. To minimise the impact of surface carbon contamination and eliminate false-positive results, it is expected to further enhance the photocatalytic performance and store catalysts in a carbon-free atmosphere.
The rapid consumption of fossil fuels in human activities, such as transport, industry, and household sectors, causes a striking increase in CO2 concentration in the atmosphere, leading to serious environmental issues, such as global warming and ocean acidification.1,2 Thus, these problems must urgently be addressed by reducing CO2 emissions and making full utilisation of the existing CO2. Photocatalytic CO2 reduction reaction (CO2RR) has attracted global research interest over the past decades.3 In an ideal process, it is expected that the photogenerated electrons in the photocatalysts are applied to reduce CO2, meanwhile, photogenerated holes are consumed for water oxidation. Considering the stable structure of CO2 with a high bond energy of 750 kJ mol−1,4 there is a high energy barrier for the activation of CO2, which requires sophisticated photocatalyst designs.
Despite tremendous research efforts, the production rates of carbon monoxide (CO) and methane (CH4), which are the most common products, stay at low levels (e.g., <17.33 μmol g−1 h−1 for CO and <2000 μmol g−1 h−1 for CH4).5–12 However, external factors, such as organic vapours in a lab, surface carbon contamination, etc., are more likely to produce these carbon products via oxidation reaction, other than CO2 reduction, and therefore they might result in false-positive signals. For example, methanol (CH3OH), a common hole scavenger in photocatalytic reactions, can produce significant amounts of CO through a photocatalytic oxidation process.13–16 Although isotope analysis is regarded as an effective approach to verify the carbon source by tracing the 13C transfer from CO2 molecules to the products, the possible isotopic substitution makes this method less reliable.17–20 To provide reproducible and convincing data for CO2RR analysis, some recent perspectives invoked the elimination of contamination sources as completely as possible.21,22 Yet, there is still no quantitative investigation on how carbon contamination interferes with the performance analysis of photocatalytic CO2RR.
In this work, we have provided a quantitative analysis of the apparently over-estimated product amount due to hole-induced contamination oxidation, other than the CO2RR process. Photocatalysts with different valence band (VB) positions have been applied to verify that the contamination oxidation reaction exaggerates the apparent CO2RR activity. Using a prototypical Au/TiO2 photocatalyst for CO2RR, introducing a trace amount (i.e., 1 μL) of ethanol (EtOH) caused over 17 times higher CH4 production rate and higher stability for CO production. Moreover, facile oxygen plasma pre-treatment was confirmed to be an effective protocol to minimise the influence of carbon contamination during gas-phase photocatalytic CO2 conversion. These findings provide new insights into CO2RR research and will enable the acquisition of more consistent and reliable quantitative results across the research community.
The Au/TiO2 has been intensively investigated to be an effective photocatalyst for CO2RR, which makes it a good benchmark for investigating the carbon contamination issue. The prototypical Au/TiO2 photocatalysts have been widely reported to be active towards gas-phase CO2 conversion to CO and CH4, with a production rate ranging from 3 to 210 μmol g−1 h−1 depending on reaction conditions.5,23 Considering the ultralow dosage of photocatalyst (c.a. 10 mg) in the reported research, the absolute yields of CO2RR products are negligible. In our research, we adopted a similar procedure to coat the as-prepared Au/TiO2 photocatalyst (ESI†) on a glass substrate with a rough surface. Herein, Au was deposited via chemical reduction by sodium borohydride to avoid introducing organic sources on the TiO2 surface.24–26 The X-ray diffraction (XRD) (Fig. S1†) and transmission electronic microscopy (TEM) (Fig. S2†) of the as-produced Au/TiO2 indicate that the Au nanoparticles were deposited on the TiO2 surface.
Photocatalytic CO2 conversion was carried out in a batch reactor, wherein the system was filled with pure CO2 (>99.9%) before turning on the lighting (ESI†). Fig. 1a and b show the CO and CH4 with a mass-specific production (MSP) of 30 μmol g−1 and 67.5 μmol g−1, respectively, in 90 minutes. This photocatalytic activity is comparable to literature results based on similar Au/TiO2 systems.27–31 Unexpectedly, in the controlled experiment, where the photocatalytic system was filled with argon gas (Ar), obvious CH4 and CO production was also detected. Especially, the absolute yields of CH4 were very close to the case in the presence of CO2, indicating that the apparent CO2RR activity has been overestimated as shown in Fig. 1b. In addition, profound H2 production (Fig. S3†) was observed in both cases (while no oxygen was detected), which may lead to a noticeable CO decrease as shown in Fig. 1avia hydrogenation. With the above results, there is a question on the origin of CO and CH4 under an Ar atmosphere.
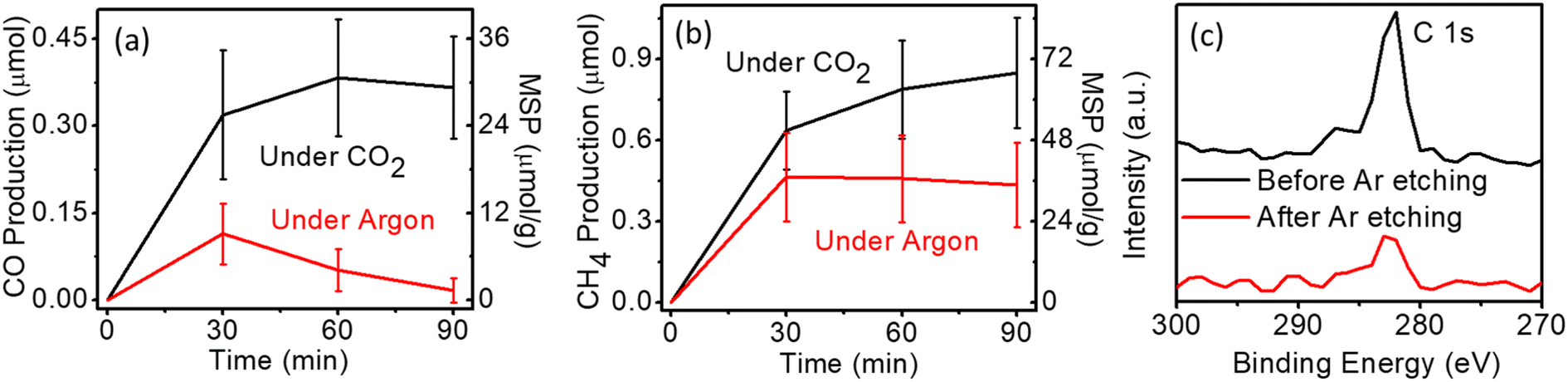 | ||
| Fig. 1 (a) CO production and (b) CH4 production from photocatalytic reactions on Au/TiO2 in CO2 (black line) or Ar (red line). (c) Comparison of C 1s XPS spectrum of the same TiO2 sample before and after Ar etching. MSP: mass-specific production. | ||
X-ray photoelectron spectroscopy (XPS) was used to determine the surface chemical environment of TiO2 (Fig. 1c). Although we avoided the organic sources during photocatalyst preparation, the carbon peak could always be observed even on the pure inorganic metallic samples, which is indexed to ubiquitous carbon contamination from air exposure.32 This peak is commonly used as a reference value to calibrate XPS data, representing C–C or C–H bond.32–34 These carbon species can produce extra CO or CH4 with the interaction with the photogenerated charges.
The applied TiO2 holds a large bandgap, where it is capable of producing CO and CH4 either by CO2 reduction reaction or via carbon contamination oxidation. To get more insight into whether photogenerated electrons or holes contribute more to the CO and CH4 generation, the other two semiconductors, BiVO4 and CuO, were selected due to their band structure features (Fig. 2a, see ESI† for details on preparation). Ultraviolet photoelectron spectroscopy (UPS) and ultraviolet-visible (UV-vis) spectroscopy were used to further determine the band positions as shown in Fig. S4 and S5.† The properties of the valence band (VB) and conduction band (CB) of BVO4, CuO, and TiO2 are summarised in Fig. 2a, which shows the CB of CuO and VB of BiVO4 to be close to the CB and VB of TiO2, respectively, in accordance with the literature.35–37 From the relative position of CB (−3.73 eV) to the redox potential of CO2/CO (−4.38 eV) and CO2/CH4 (−4.67 eV), it can be concluded that only CuO can facilitate the CO2RR. While the relatively shallow VB (−5.05 eV) of CuO makes it unlikely to process contamination oxidation. The BiVO4 has the opposite situation in that CO2RR is unlikely to happen due to the thermodynamic limit of CB (−5.03 eV), but the contamination oxidation process is relatively easy due to the deep VB (−7.43 eV). Therefore, the photocatalytic performance of CuO and BiVO4 can help distinguish whether the CO2RR or oxidation of carbon contaminations contributes to the apparent CO and CH4 generation. Surface carbon content on the TiO2, CuO, and BiVO4 was determined to be 14.96, 26.02, and 8.44 wt%, respectively, as analysed by XPS (Table S1†).
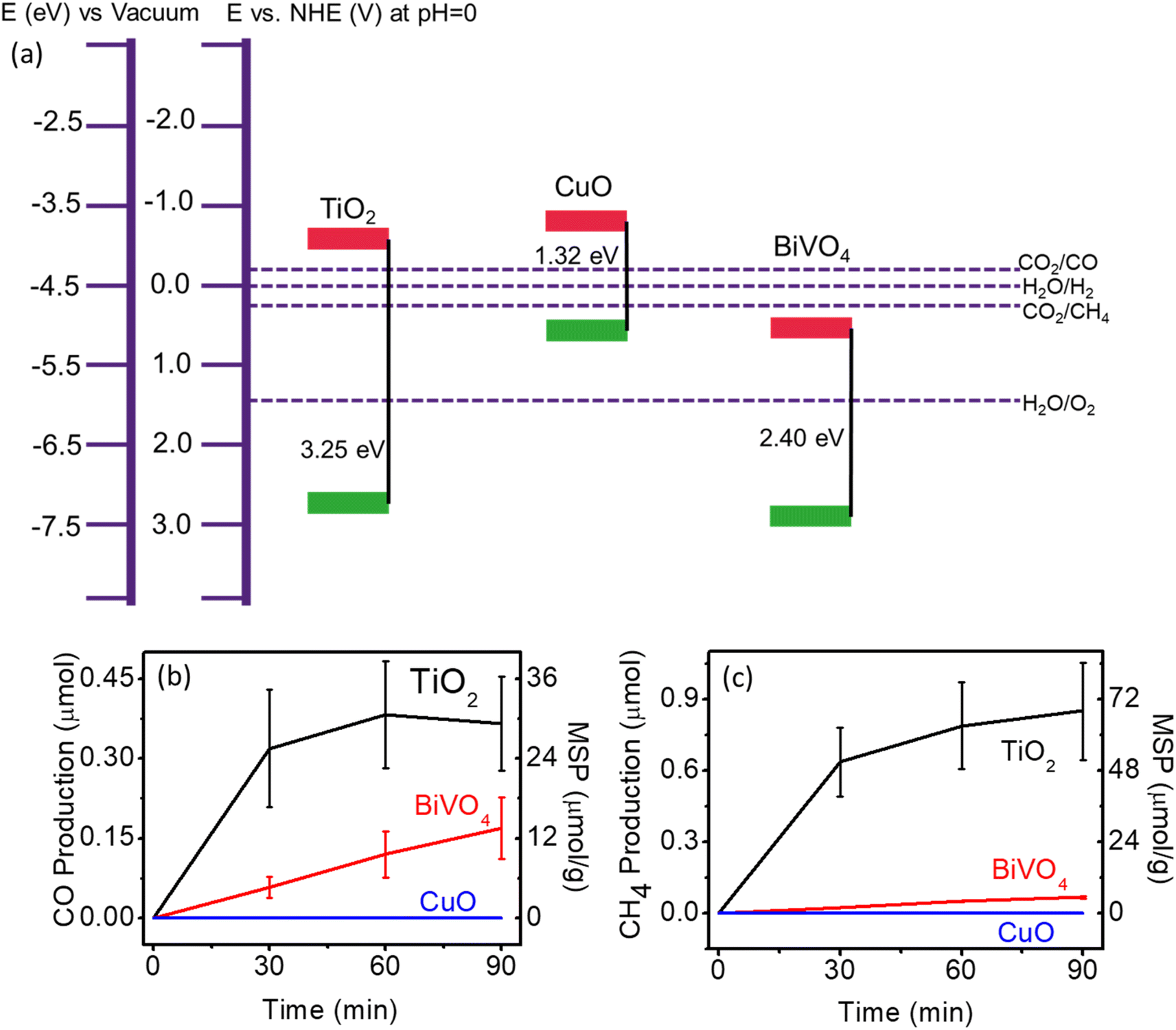 | ||
| Fig. 2 (a) Band position of TiO2, CuO, and BiVO4 in line with CO2RR, redox potentials of hydrogen and oxygen evolution reactions. Photocatalytic CO2RR performance over TiO2, CuO, and BiVO4 (b) CO production; (c) CH4 production. MSP: mass-specific production. | ||
Their photocatalytic performance is shown in Fig. 2b and c and S7.† Interestingly, BiVO4 produced significant amounts of CH4 (5.4 μmol g−1) and CO (13.5 μmol g−1), while CuO produced nearly null. As mentioned above, products from BiVO4 indicate that carbon contamination oxidation occurs, which can lead to false-positive CO2RR results, despite the consensus that BiVO4 is not capable of this process. Another photocatalyst, SnO2, also possesses a low CB which is challenged for CO2RR.38 However, a considerable amount of CH4 (3.8 μmol g−1) and CO (13.1 μmol g−1) is observed upon light irradiation (Fig. S8†). We suppose this is also a false-positive result for CO2RR caused by carbon contamination oxidation. The absence of a product from CuO further indicates the significance of the oxidation process for the apparent CO and CH4 generation.
Carbon-containing photocatalysts have been extensively researched for CO2RR because of their attractive physicochemical properties. Graphitic carbon nitride (g-C3N4) is one of the most investigated metal-free organic photocatalysts due to its low cost, visible light harvesting, and suitable band position for CO2RR.39 However, the carbon in g-C3N4 will interfere with the CO2RR performance analysis. Fig. 3 shows the photocatalytic performance on g-C3N4 under Ar and CO2 atmospheres. The production rates of CO (47.1 μmol g−1) and CH4 (3.5 μmol g−1) under the Ar atmosphere have very small and even negligible difference, as compared to the rates under the CO2 atmosphere. These results are consistent with the report of light-induced self-decomposition of g-C3N4, rather than CO2RR.40 Theoretical calculations have indicated that self-decomposition reaction is thermodynamically more favourable than CO2RR.40
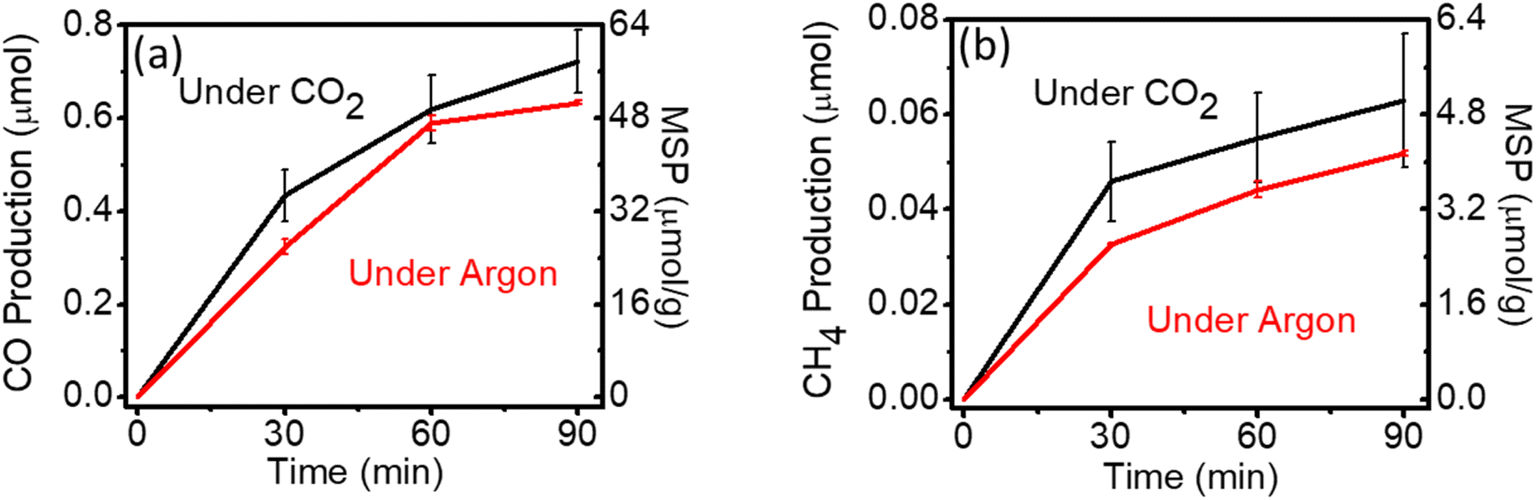 | ||
| Fig. 3 (a) CO production and (b) CH4 production from photocatalytic reactions on g-C3N4 in CO2 (black line) or Ar (red line). MSP: mass-specific production. | ||
The impact of organic pollution on photocatalytic CO2RR is even more pronounced in the presence of ethanol (EtOH), which is a widely used organic solvent for synthesis and a sacrificial agent for photocatalysis. Take the Au/TiO2 system as an example, when 1 μL of EtOH is deliberately added to the reaction system, the CH4 production rate was boosted by 17 times from 68 μmol g−1 to 1244.8 μmol g−1 as shown in Fig. 4a because of the alcohol and carboxyl acid decomposition under the light.41–45 Meanwhile, a large amount of hydrogen was produced (Fig. S7†), which could inhibit the generation of CO as shown in Fig. 4b. The photocatalytic performance of pure TiO2 was also evaluated as a reference (Fig. S9†), which showed overall lower activity in the absence of Au cocatalyst. A plausible pathway for EtOH oxidation under light is shown in Fig. 4c. Upon light irradiation, ethanol was oxidised to ethanal with hydrogen generation. Ethanal can be decomposed via three different routes to mislead the photocatalytic CO2RR test. Ethanal can be directly converted to CO2 and hydrogen via the photocatalytic process. Under ambient conditions, some of the ethanal decomposes to produce CH4 and CO spontaneously. While, in some cases, the above products would be further oxidised to acetic acid and H2 with acetic acid being further decomposed to CH4 and CO2 upon light illumination. Thus, the introduction of EtOH into the reaction system not only affects the activity analysis, but also influences the selectivity analysis (Table S2†) with considerable hydrogen production (Fig. S7†). Therefore, possible false-positive results can be obtained due to the organic residuals in the system, which must be eliminated when conducting photocatalytic CO2RR experiments. Due to the dramatic performance boost by the trace amount of EtOH, researchers in this field should be extremely careful about organic solvent vapours generated in the laboratory environment.
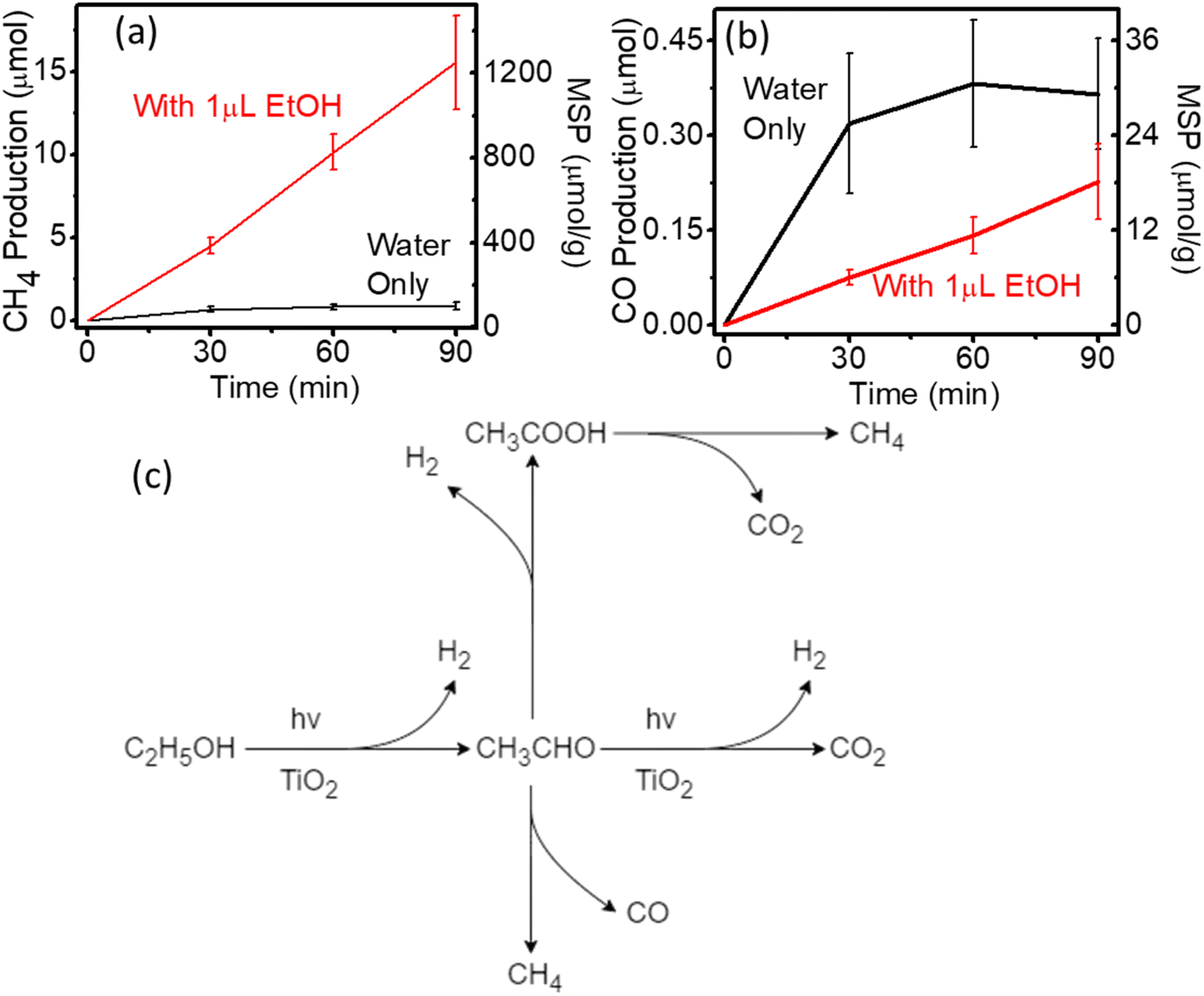 | ||
| Fig. 4 Photocatalytic performance of Au/TiO2 with different solvents (a) CO production; (b) CH4 production. (c) Schematic illustration of photocatalytic ethanol oxidation. MSP: mass-specific production. | ||
Oxygen plasma treatment is commonly used to eliminate residual organic ligands from the material surface. To avoid the interference of surface contamination, oxygen plasma cleaning was performed to etch the surface of the photocatalyst (i.e., Au/TiO2). Fig. 5 and S10† show the photocatalytic performance of treated Au/TiO2 photocatalysts. In the Ar environment, the CO and CH4 production rates decreased dramatically to a negligible level, especially for CH4; the production rate for CH4 dropped from 34 to 4.69 μmol g−1, indicating the effectiveness of the oxygen plasma treatment. Therefore, oxygen plasma treatment can be used to effectively clean the surface of the materials before conducting photocatalytic experiments as shown in Fig. 5c.
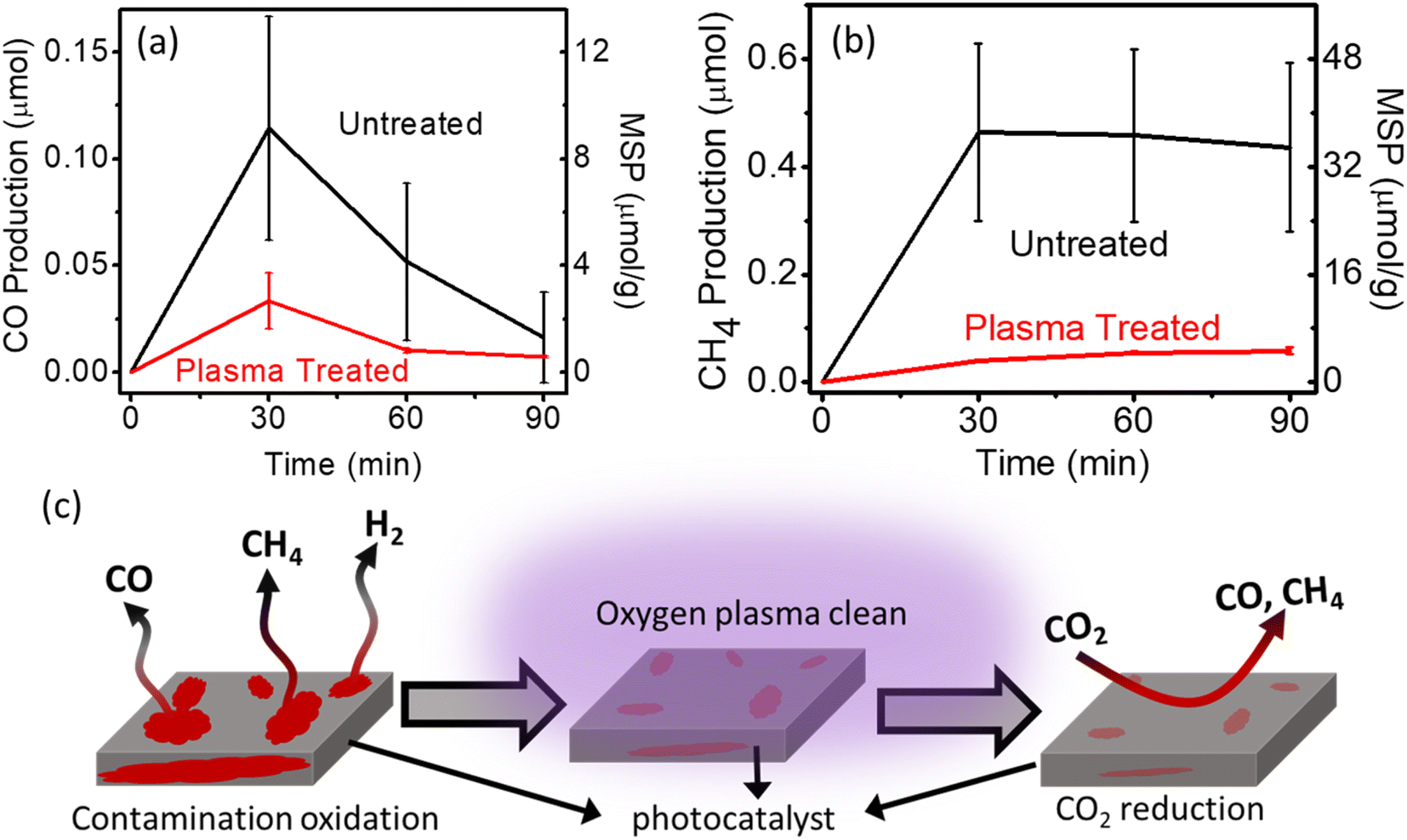 | ||
| Fig. 5 Controlled experiments before and after plasma treatment under the Ar atmosphere (a) CO production; (b) CH4 production. MSP: mass-specific production rate. (c) schematic representation of oxygen plasma cleaning. | ||
In summary, we have quantitatively shown the influence of carbon contamination on photocatalysts during photocatalytic CO2RR activities. When the photocatalytic CO2RR activity is low, the impact of the carbon contamination oxidation is therefore significant. The commonly used organic solvent in laboratories (e.g., ethanol) was demonstrated to have a serious impact on photocatalytic CO2RR behaviour, leading to false-positive results. To address this issue, oxygen plasma treatment is effective in removing carbon contamination by cleaning the surface of the materials before conducting photocatalytic experiments. The reason for such a significant impact of carbon contamination is the extremely low production rate and carbon conversion rates of photocatalytic CO2RR.46–49 If the production and carbon conversion rates are high enough (e.g., >10 mmol g−1 h−1), the carbon contamination issue would be negligible. Some strategies can be utilised for higher CO2RR performance, such as defect engineering, nanostructure design, cocatalysts design, heterostructure design, and Z-scheme construction. Furthermore, photocatalysts should be stored in a carbon-free environment (e.g., an N2/Ar-filled glove box), if possible, to minimise carbon contamination. Long-term stability tests could decrease the effect of carbon contaminations. Future research should focus on improving the production rate and selectivity and developing highly efficient photocatalysts for CO2RR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of the Australian Research Council through its DECRA (DE210100930), Discovery (DP200101900), and Laureate Fellowship (FL190100139) schemes. This work was performed in part at the Queensland Node of the Australian National Fabrication Facility – a company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers. The authors acknowledge the facilities and the scientific and technical assistance provided by the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. J. Y. acknowledges scholarship support from the UQ Graduate School.References
- Global Monitoring Laboratory, Trends in Atmospheric Carbon Dioxide, https://gml.noaa.gov/ccgg/trends/mlo.html, accessed November 1, 2021 Search PubMed.
- R. Heinberg, in The Community Resilience Reader: Essential Resources for an Era of Upheaval, ed. D. Lerch, Island Press/Center for Resource Economics, Washington, DC, 2017, pp. 65–78, DOI:10.5822/978-1-61091-861-9_4.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- Ş. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed.
- V. A. de la Peña O'Shea, D. P. Serrano and J. M. Coronado, in From Molecules to Materials: Pathways to Artificial Photosynthesis, ed. E. A. Rozhkova and K. Ariga, Springer International Publishing, Cham, 2015, DOI:10.1007/978-3-319-13800-8_7, pp. 171–191.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Sol. RRL, 2020, 4, 1900546 CrossRef CAS.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 3169 CrossRef PubMed.
- Z. Jiang, H. Sun, T. Wang, B. Wang, W. Wei, H. Li, S. Yuan, T. An, H. Zhao, J. Yu and P. K. Wong, Energy Environ. Sci., 2018, 11, 2382–2389 RSC.
- W. Shangguan, Q. Liu, Y. Wang, N. Sun, Y. Liu, R. Zhao, Y. Li, C. Wang and J. Zhao, Nat. Commun., 2022, 13, 3894 CrossRef CAS PubMed.
- W. Wang, C. Deng, S. Xie, Y. Li, W. Zhang, H. Sheng, C. Chen and J. Zhao, J. Am. Chem. Soc., 2021, 143, 2984–2993 CrossRef CAS PubMed.
- F. Chen, Z. Ma, L. Ye, T. Ma, T. Zhang, Y. Zhang and H. Huang, Adv. Mater., 2020, 32, 1908350 CrossRef CAS PubMed.
- M. Xiao, L. Zhang, B. Luo, M. Lyu, Z. Wang, H. Huang, S. Wang, A. Du and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 7230–7234 CrossRef CAS PubMed.
- J. Zhao, R. Shi, Z. Li, C. Zhou and T. Zhang, Nano Select, 2020, 1, 12–29 CrossRef.
- M. Whitbeck, Atmos. Environ., 1983, 17, 121–126 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- N. N. Lavrentieva, A. S. Dudaryonok and J. V. Buldyreva, Atmos. Oceanic Opt., 2012, 25, 311–316 CrossRef CAS.
- O. A. Schaeffer and H. R. Owen, J. Chem. Phys., 1955, 23, 1305–1309 CrossRef CAS.
- K. Zhang, Q. Gao, C. Xu, D. Zhao, Q. Zhu, Z. Zhu, J. Wang, C. Liu, H. Yu, C. Sun, X. Liu and Y. Xuan, Carbon Neutrality, 2022, 1, 10 CrossRef.
- M. Heuillet, F. Bellvert, E. Cahoreau, F. Letisse, P. Millard and J.-C. Portais, Anal. Chem., 2018, 90, 1852–1860 CrossRef CAS PubMed.
- Y. Zhang, D. Yao, B. Xia, M. Jaroniec, J. Ran and S.-Z. Qiao, ACS Energy Lett., 2022, 1611–1617, DOI:10.1021/acsenergylett.2c00427.
- R. Das, S. Chakraborty and S. C. Peter, ACS Energy Lett., 2021, 6, 3270–3274 CrossRef CAS.
- H. Zhao, X. Zheng, X. Feng and Y. Li, J. Phys. Chem. C, 2018, 122, 18949–18956 CrossRef CAS.
- M. V. Dozzi, L. Prati, P. Canton and E. Selli, Phys. Chem. Chem. Phys., 2009, 11, 7171–7180 RSC.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- J. Polte, R. Erler, A. F. Thünemann, S. Sokolov, T. T. Ahner, K. Rademann, F. Emmerling and R. Kraehnert, ACS Nano, 2010, 4, 1076–1082 CrossRef CAS PubMed.
- N. Shehzad, M. Tahir, K. Johari, T. Murugesan and M. Hussain, J. CO2 Util., 2018, 26, 98–122 CrossRef CAS.
- L. Collado, A. Reynal, J. M. Coronado, D. P. Serrano, J. R. Durrant and V. A. de la Peña O'Shea, Appl. Catal., B, 2015, 178, 177–185 CrossRef CAS.
- W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS.
- W. Tu, Y. Zhou, H. Li, P. Li and Z. Zou, Nanoscale, 2015, 7, 14232–14236 RSC.
- M. Compagnoni, G. Ramis, F. S. Freyria, M. Armandi, B. Bonelli and I. Rossetti, Rend. Lincei Sci. Fis. Nat., 2017, 28, 151–158 CrossRef.
- G. Greczynski and L. Hultman, ChemPhysChem, 2017, 18, 1507–1512 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, Sci. Rep., 2021, 11, 11195 CrossRef CAS PubMed.
- Y. Yamada, J. Kim, S. Matsuo and S. Sato, Carbon, 2014, 70, 59–74 CrossRef CAS.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- S. J. A. Moniz and J. Tang, ChemCatChem, 2015, 7, 1659–1667 CrossRef CAS.
- K. Ding, B. Chen, Y. Li, Y. Zhang and Z. Chen, J. Mater. Chem. A, 2014, 2, 8294–8303 RSC.
- J. A. Torres, G. T. S. T. Da Silva, F. Barbosa de Freitas Silva and C. Ribeiro, ChemPhysChem, 2020, 21, 2392–2396 CrossRef CAS PubMed.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- P. Chen, X. a. Dong, M. Huang, K. Li, L. Xiao, J. Sheng, S. Chen, Y. Zhou and F. Dong, ACS Catal., 2022, 12, 4560–4570 CrossRef CAS.
- M. A. Nadeem, M. Murdoch, G. I. N. Waterhouse, J. B. Metson, M. A. Keane, J. Llorca and H. Idriss, J. Photochem. Photobiol., A, 2010, 216, 250–255 CrossRef CAS.
- T. Sakata and T. Kawai, Chem. Phys. Lett., 1981, 80, 341–344 CrossRef CAS.
- X. Fu, D. Y. C. Leung, X. Wang, W. Xue and X. Fu, Int. J. Hydrog. Energy, 2011, 36, 1524–1530 CrossRef CAS.
- S. Jia, X. Shu, H. Song, Z. An, X. Xiang, J. Zhang, Y. Zhu and J. He, Ind. Eng. Chem. Res., 2021, 60, 12282–12291 CrossRef CAS.
- C. A. Walenta, S. L. Kollmannsberger, J. Kiermaier, A. Winbauer, M. Tschurl and U. Heiz, Phys. Chem. Chem. Phys., 2015, 17, 22809–22814 RSC.
- M. Marx, A. Mele, A. Spannenberg, C. Steinlechner, H. Junge, P. Schollhammer and M. Beller, ChemCatChem, 2020, 12, 1603–1608 CrossRef CAS.
- M. Qureshi and K. Takanabe, Chem. Mater., 2017, 29, 158–167 CrossRef CAS.
- K. Takanabe, J. Catal., 2019, 370, 480–484 CrossRef CAS.
- C.-H. Lim, S. Ilic, A. Alherz, B. T. Worrell, S. S. Bacon, J. T. Hynes, K. D. Glusac and C. B. Musgrave, J. Am. Chem. Soc., 2019, 141, 272–280 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00834g |
| This journal is © The Royal Society of Chemistry 2023 |