
Regulating the electronic structure of single-atom catalysts for electrochemical energy conversion
Wei
Ma
 *a,
Zhe
Deng
a,
Xinjie
Zhang
a,
Zhang
Zhang
ab and
Zhen
Zhou
*a
*a,
Zhe
Deng
a,
Xinjie
Zhang
a,
Zhang
Zhang
ab and
Zhen
Zhou
*a
aInterdisciplinary Research Center for Sustainable Energy Science and Engineering (IRC4SE2), School of Chemical Engineering, Zhengzhou University, Henan 450001, China. E-mail: mawei@zzu.edu.cn; zhenzhou@zzu.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China
First published on 28th February 2023
Abstract
Single-atom catalysts (SACs) are highly effective in electrochemical energy conversion due to their abundant active sites. Their exceptional electrocatalytic efficiency can be achieved by regulating the electronic structure to optimize the binding energy of intermediates and reduce the energy barrier of electrocatalytic reactions. This is achieved through the metal–support interaction at the interface of SACs. Controlling the electronic structure at the atomic level is a promising strategy for promoting activity, selectivity, and stability. To further understand the influence of electronic structure and accelerate the development of SACs, we summarize various fabrication strategies to construct SACs and further adjust the electronic structure to achieve excellent hydrogen/oxygen evolution and O2/N2/CO2 reduction reactions. Finally, we discuss the challenges and prospects of SACs for controllable electrocatalytic processes and provide guidance for the future exploration of electrochemical energy conversion.
 Wei Ma | Wei Ma received his Bachelor's degree in Inorganic Nonmetal Materials Engineering from Central South University, China in 2009, followed by his PhD in Mineral Materials Science from the same university in 2015. During his doctoral studies, he spent two years (2013 to 2015) at the National Institute for Materials Science (NIMS), Japan, as a joint PhD student. Since 2016, he has been a faculty member at Zhengzhou University, China, where he is now an associate professor at the School of Chemical Engineering. His research focuses on the development of advanced nanomaterials for electrocatalysis. |
 Zhe Deng | Zhe Deng is a graduate student at the School of Chemical Engineering, Zhengzhou University, China, having earned his Bachelor's degree in Materials Science and Engineering from Central South University, China in 2021. His current research focuses on the development of efficient catalysts for electrochemical energy conversion. |
 Zhen Zhou | Zhen Zhou earned both his B.S. (in Applied Chemistry in 1994) and PhD (in Inorganic Chemistry in 1999) degrees from Nankai University, China. He joined the faculty at Nankai University as a lecturer in 1999. Two years later, he began to work as a postdoctoral fellow at Nagoya University, Japan. In 2005, he returned to Nankai University as an associate professor and became a full professor in 2011. In 2021, he moved to Zhengzhou University, China, as a Distinguished Professor and he is now Dean of the School of Chemical Engineering. His main research interest lies in the integration of high-throughput computations, experiments and machine learning for energy storage and conversion. |
10th Anniversary StatementThe Journal of Materials Chemistry A is an exceptional platform for sharing findings in the field of materials for energy and sustainability. We have published approximately 50 reviews and research papers since the journal's inception in 2012, including Zhen Zhou, one of the corresponding authors, who serves as an Associate Editor for the journal. Our research focuses on exploring novel materials to address environmental challenges and energy crises. Recently, single-atom catalysts have garnered significant attention due to their remarkable performance and potential in advancing green and sustainable energy. The unique low-coordination environment, quantum size effect, and metal–support interactions of single-atom catalysts make them an exciting avenue of research. In this review, we summarize the recent developments of single-atom catalysts in electrochemical energy conversion. We believe that the integration of advanced preparation methods, in situ characterizations, and artificial intelligence will accelerate the development of novel materials, including single-atom catalysts, for energy and environmental applications. We are honored to celebrate the 10th anniversary of the Journal of Materials Chemistry and are excited to continue contributing to its growth through the exploration and development of novel energy materials. |
1 Introduction
The excessive consumption of fossil energy has led to severe environmental issues and energy crises.1–4 The development and application of clean energy via electrocatalytic reactions is a promising strategy for overcoming these hurdles, i.e., splitting water into hydrogen, reducing N2 into NH3, and converting CO2 into high-value chemicals and fuel cells.5–10 Single-atom catalysts (SACs) anchored on substrates have been attracting significant attention for increasing the electrocatalytic efficiency resulting from the separated energy levels between the highest occupied and lowest unoccupied molecular orbitals.11–15 The super-high utilization of active atoms in SACs (almost 100%) reduces the loading of metal elements, especially noble metals, and maximizes active centers.15–18 SACs optimize the adsorption/desorption of reaction intermediates to promote the selectivity of products.19–21 Overall, improved catalytic activity can be achieved by increasing the loading of metal single atoms (SAs) or controlling the carrier to expose a larger surface area to increase the number of active sites, or by modulating the electronic structure and geometry of active sites to increase the intrinsic activity of each active site.14,22–24 Therefore, SACs can generate higher catalytic activity, selectivity, and stability, and achieve more efficient chemical reactions and energy conversion.25–29Since SACs were first reported in 2011, numerous precious metal SACs, such as Pt, Ir, Ru, and Au, have been obtained through different synthetic strategies, i.e., co-precipitation, impregnation, atomic layer deposition, high-temperature pyrolysis, and low-temperature chemical reduction.30–49 To further lower the cost of precious metal SACs, 3d transition metal SACs, such as Fe, Co, and Ni, were also fabricated via bonding with O, N, S, and P atoms or anchoring at the defects to increase the durability.50–52 Metal SAs anchored to the substrates interact with each other to generate many active sites.53,54 The number of active sites and the intrinsic activity of SACs play significant roles in the electrocatalytic processes.55,56 However, the low loading and the instability of metal SACs depress the electrocatalytic efficiency due to the aggregation of isolated metal atoms because of high surface energy and weak interactions between metal atoms and substrates.24,57,58 Therefore, the regulation of the interaction between the isolated metal atoms and the support provides a promising strategy for stabilizing metal SACs and strengthening the intrinsic activity to advance the electrocatalytic efficiency.59–62
Various central metal atoms, coordination atoms, and electronic metal–support interaction (EMSI) of SACs have a great influence on the electronic properties and charge distribution of active sites in electrocatalysis, optimizing the binding energy of intermediates, reducing the energy barrier of the rate-determining step (RDS), and thus accelerating the electrocatalytic reaction. For example, M–N–C (M, metal)-based SACs with metals of small atomic radii (such as Cr, Mn, Fe, Co, and Ni) prefer to adsorb the C atom of CO2 at the metal site, while those with large radii (such as Sc) coadsorb C and O for the CO2 reduction reaction (CO2RRR). Ga SACs with different coordination atoms such as P, S, and N exhibit varied selectivity and stability in the CO2RR. In particular, the Co3O4 carrier modulates the unoccupied 5d state of Pt SAs by EMSI and weakens the hydrogen adsorption at Pt SAs, thus greatly improving hydrogen production.63–70 Therefore, some strategies, e.g., adjusting the central atoms or structure of substrates, doping heteroatoms, and changing the strain, have been achieved to regulate the coordination environment of SACs.9,71–74 The uniform and well-defined structural characteristics of SACs are beneficial for investigating the electrocatalytic mechanism.75–77 The rapid development of in situ/operando characterizations and high-performance computations can disclose the relationship between the electronic structure and electrocatalytic efficiency.78–80 Although many strategies have been implemented to fabricate/design various SACs and numerous reviews have focused on SACs, few emphasize the influence of the electronic structure on the adsorption/desorption of the intermediates and the electrocatalytic activity.
In this review, first, we briefly summarize the common preparation and characterization methods to fabricate and analyze SACs with different structures (Fig. 1). Then, the influences of the optimized bonding energy, electronic structure, and charge transfer of active sites on the improvement in electrocatalytic activity are discussed. Additionally, we elaborate on recent applications of SACs in electrocatalytic processes including the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), nitrogen reduction reaction (NRR), and CO2 reduction reaction (CO2RR), providing a brief conclusion on the impact of metal SACs in electrocatalytic processes. Finally, we discuss the challenges and prospects of SACs in future electrocatalytic applications.
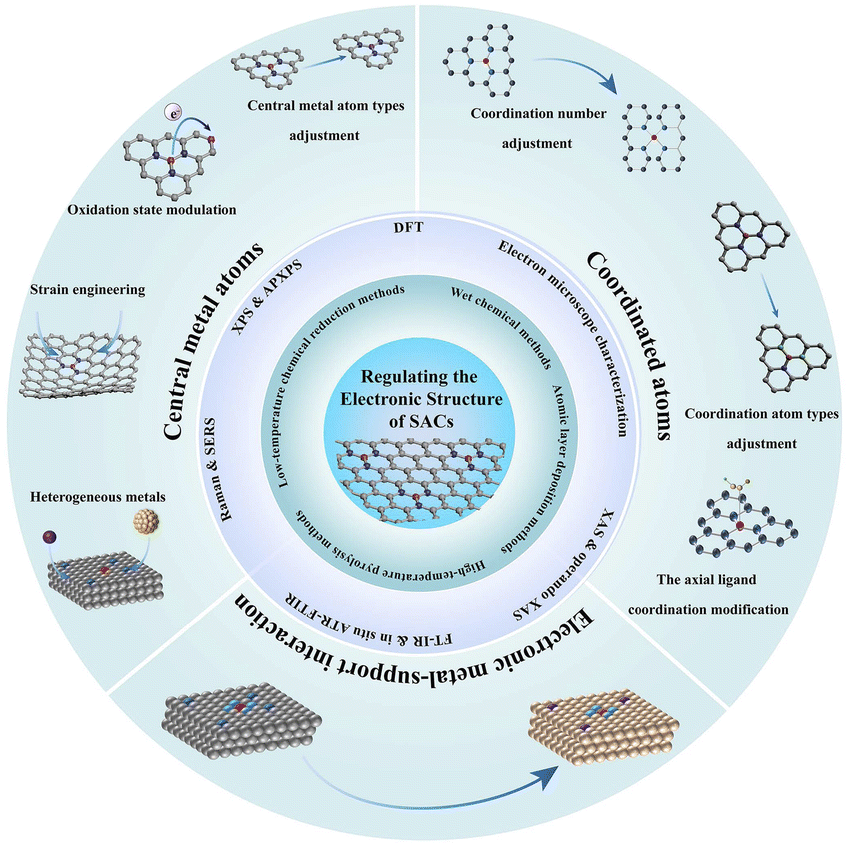 | ||
| Fig. 1 An overview of preparation methods, characterization techniques and design strategies of SACs. | ||
2 Preparation of SACs
SACs are proposed to avoid the agglomeration of isolated metal atoms in the surface of the substrates via the confinement or coordination of isolated metal atoms in substrates.81–83 To date, several fabrication strategies have been employed to prepare SACs by trapping metal SAs at defects or heteroatoms in the substrates to enhance the stability of SACs.84–862.1 Wet chemical methods
The conventional wet chemical method is optimal for fabricating SACs as commercial products due to its easy operation, low cost, and massive production.87–89 The SACs are fabricated by loading isolated metal atoms on anchored sites.90–93 The co-precipitation method is performed by adding precipitating agents to aqueous solutions containing various metal cations and adjusting the pH to obtain uniformly dispersed SACs, and the SACs are approached by lowering the loading of metal atoms to prevent aggregation, e.g., Ru1/FeOx SACs with the low loading of 0.18 wt% was prepared through the co-precipitation method by dissolving ferric nitrate and ruthenium chloride in a NaOH aqueous solution with the pH value of 7.0–8.0.94 The impregnation method is a traditional route to prepare catalysts by adsorbing metal cations in the solution to the pore channels of the supports.30,31 The chelation and the viscosity of the solution leach the support and remove the metal element to strengthen the electrostatic adsorption, further dispersing the metal atoms. Therefore, the number of defects greatly influences the loading of metal atoms, and the precise control for SACs is inaccessible.2.2 Atomic layer deposition
Atomic layer deposition (ALD) is a method for depositing the catalysts on the surface of supports at a certain temperature.95 The deposition process can be precisely controlled by adjusting the number of cycles.43–45,96 Therefore, the SACs can be approached by controlling the ALD cycle. Ramesh et al. prepared Pt SACs on a porous nitrogen–carbon-powder-coated carbon substrate (NC-CC) by ALD, and the controllable loading of Pt SAs can be approached by adjusting the number of ALD cycles from 2 to 60.97 However, the high cost and the high-temperature experimental environment would cause the impossibility of large-scale commercial applications.2.3 High-temperature pyrolysis
High-temperature pyrolysis is approached by calcinating the organic precursors, including metal-containing complexes, irregular metal-containing complexes, metal-containing polymers, and a mixture of metal and carbon sources, at 600–1000 °C.46–48 The isolated metal atoms usually coordinate with the N, S, and P atoms or are trapped by the defects in carbon materials to increase the metal loading and prevent the agglomeration of metal atoms.49 Therefore, the substrates with numerous defects and heteroatoms provide a promising pathway to fabricating SACs.98 Al-Zoubi et al. used the high-temperature pyrolysis method to prepare non-platinum group metal (non-PGM) electrocatalysts by pyrolyzing a metal–organic framework (MOF) precursor at 750 °C in an ammonia atmosphere.992.4 Low-temperature chemical reduction
Low-temperature chemical reduction is an accessible method for fabricating SACs in a low-temperature environment.41,42 The precursors containing metal cations frozen in ice are reduced by ultraviolet irradiation or reducing agents (such as NaBH4).100 The size of the metal catalysts can be controlled by adjusting the growth and nucleation process.101 The Pt SACs were fabricated by irradiating the frozen low-concentration Pt precursor solution with a xenon lamp.42 Therefore, it was necessary to lower the reaction temperature and the concentration of the precursor solution to fabricate SACs.2.5 Other classical preparation methods
Some classical preparation methods have been adopted to fabricate SACs, i.e., the milling method, microwave-assisted method, and electrochemical method. The preparation of SACs with the milling method relies mainly on mechanochemical forces to create defects in the substrate and capture the atomized metal, in which no other by-products or wastes are generated and the metal loading can be easily tuned by controlling the milling rate. Jeong et al. used the milling method to directly atomize the bulk metals onto different supports, such as carbon frameworks, oxides, and nitrides.102 The microwave-assisted method is considered to be efficient for fabricating SACs within a reduced reaction time. Fei et al. rapidly loaded a variety of metal SAs (Co, Ni, or Cu) onto graphene by the microwave-assisted method. The high temperature generated by the microwave heating process allows the N doping and the simultaneous embedding of metal into graphene.103 Electrochemical deposition is a universal method for the preparation of various SACs, and the SA metal loading can be approached by adjusting the precursor concentration, deposition rate, etc. Zeng et al. loaded Ir on a Co(OH)2 substrate by the oxidation–reduction reaction in the electrolyte and the loading of metal SAs can be precisely adjusted by the concentration of the Ir precursors and the number of scanning cycles.104Although substantial fabrication methods have been implemented to prepare SACs, further fabrication strategies should be developed to generate SACs with high loading, excellent efficiency, and selectivity. The high activity and selectivity of SACs are attributed to the interaction between active atoms and supports, resulting in changes in the electronic structure. Therefore, it is of great significance to study more straightforward and more environmentally-friendly technologies for industrial application.
3 Characterizations of SACs
To investigate the morphology and structure of SACs, some characterization techniques have been adopted, including, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), in situ-scanning transmission electron microscopy (in situ-STEM), scanning tunneling microscopy (STM), X-ray absorption spectroscopy (XAS), Fourier transform-infrared spectroscopy (FT-IR), Raman spectroscopy, and X-ray photoelectron microscopy (XPS). These characterization techniques not only provide local structural information at the atomic scale but also allow further analysis of catalytic mechanisms and structural evolution of isolated atoms and supports, making great significance for studying the reaction mechanism in the electrocatalytic process of SACs.105,1063.1 Electron microscopes
HAADF-STEM was employed to directly observe the dispersed sites in the substrates, which was first applied by Treacy et al. in Cavendish Laboratory in 1996.112 The SACs were detected from the Z-contrast atomic resolution imaging by focusing the electron probe on the specimen and detecting the scattering electrons on an annular dark-field detector.113,114 HAADF-STEM is considered an efficient technology for observing isolated metal atom bonding in the supports. The uniform contrast of the HAADF-STEM image and energy dispersive X-ray spectroscopy (EDX) in Fig. 2a indicate that single Pt atoms are uniformly distributed on the edge of Ru nanoparticles.107,115 In recent years, the development of in situ-STEM techniques has allowed the observation of dynamic structural changes in SACs. For example, the in situ-STEM proved the mobility of Pt SAs dispersed on anatase TiO2 under different conditions. Pt SAs are located at the original sites under low-temperature oxidation or reduction conditions. However, the reduction conditions (5% H2) at 450 °C for 30 min led to the shifting of Pt single atoms and the changing of the lattice fringe of TiO2.108 Additionally, STM was used to image the surface of SACs at the atomic level by the quantum tunneling effect of electrons, and investigate the differences in the electronic properties of the samples caused by the tip current under the applied voltage.116 The STM image of Fe atoms deposited on GaAs(110) is shown in Fig. 2e, and the Fe atom-induced feature is limited to an area of ∼13 Å in diameter.109 Several different peaks were observed in the dI/dV spectra of Fe atoms, indicating that the substitution of Ga by Fe atoms on the GaAs surface resulted in hybridization between the d orbitals of the Fe atoms and the p-like orbitals of the neighboring As atoms (Fig. 2f).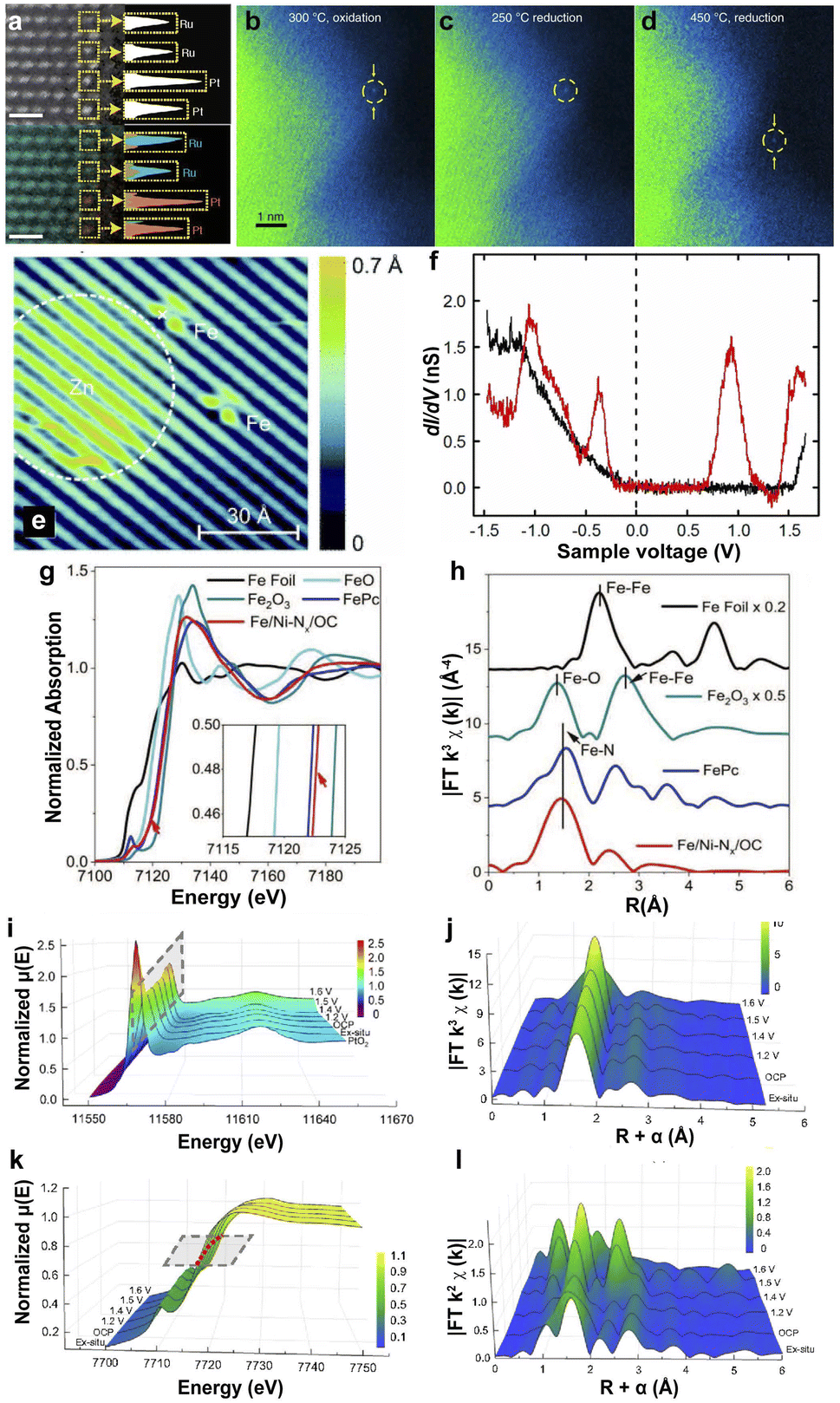 | ||
| Fig. 2 (a) HAADF-STEM image (top) and EDX map (bottom) of single Pt atoms on a Ru nanoparticle; single Pt atoms (orange), and Ru atoms (blue) are identified in yellow boxes.107 Copyright 2022, Nature Publishing Group. In situ-STEM images of PtSA/TiO2 under different annealing conditions: 300 °C, O2 for 30 min (b); 250 °C, 5% H2 for 30 min (c); 450 °C, 5% H2 for 30 min (d).108 Copyright 2019, Nature Publishing Group. (e) STM image of Fe- and Zn-induced structures on GaAs(110). (f) Spectra of dI/dV as a function of the sample voltage on the clean GaAs(110) surface (black curve) and on the Fe atom-induced structure (red curve).109 Copyright 2013, American Physical Society. (g) Fe K-edge XANES. (h) EXAFS at the Fe K-edge.110 Copyright 2020, Wiley. Operando potential-dependent XAFS of the XANES (i and k) and EXAFS (j and l) at the Pt L3-edge (i and j) and Co K-edge (k and l) of Pt1/CoHPO4.111 Copyright 2022, Nature Publishing Group. | ||
3.2 XAS
XAS includes X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine spectroscopy (EXAFS). XANES is sensitive to the oxidation state and the coordination environment of SACs, and EXAFS is employed to detect the coordination number, atomic distances, and the species of the neighboring SAs.117,118 For example, Zhu et al. loaded single-atom Fe and Ni sites on hierarchically ordered porous carbon to prepare Fe/Ni–Nx/OC SACs.110 The absorption edge position of Fe in Fe/Ni–Nx/OC is close to that of iron phthalocyanine (FePc), indicating the +2 valence state (Fig. 2g). The similar pre-edge profiles between Fe in Fe/Ni–Nx/OC and FePc, and the close Fe peak centered in Fe/Ni–Nx/OC (1.48 Å) to that of FePc (1.50 Å) suggest the formation of Fe–Nx coordination (Fig. 2h). Furthermore, the SAs with Fe–N4 coordination were proven by the least-squares EXAFS fitting for Fe/Ni–Nx/OC SACs. Operando XAS is more accurate in identifying active sites and electron transfer under working conditions. The operando XAS spectra in Fig. 2i–l suggest the reduced d-band occupancy state of the Co atoms in the CoHPO4 substrate because of the low oxidation state of Pt and an apparent shift in the absorption threshold of the Co K-edge to higher energies.111 The electron transfer from the neighboring Co to Pt and shrinking of the Co–O bond prevent the dissolution of Pt. Similarly, Jiang et al. investigated Ni SACs in the CO2RR with in situ XAS and found no significant changes in Ni XANES under different applied potentials, demonstrating the high stability of Ni SAs in graphene nanosheets.119 Therefore, XAS provides a promising method for investigating the interfacial electronic structure and the interaction between metal SAs and supports.3.3 FT-IR
FT-IR is a structural analysis method based on the selective absorption of electromagnetic radiation in the infrared light region, which is employed to probe the local structure, oxidation state, and coordination environment of SACs.44,120 In particular, in situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy was used to monitor the evolution of COO− and COOH intermediates during electrocatalysis (Fig. 3a), proving that the support of In2O3 accelerated the CO2RR on the surface of PdIn@In2O3 and CO was the major product along with minor HCOOH (Fig. 3b and c).120,121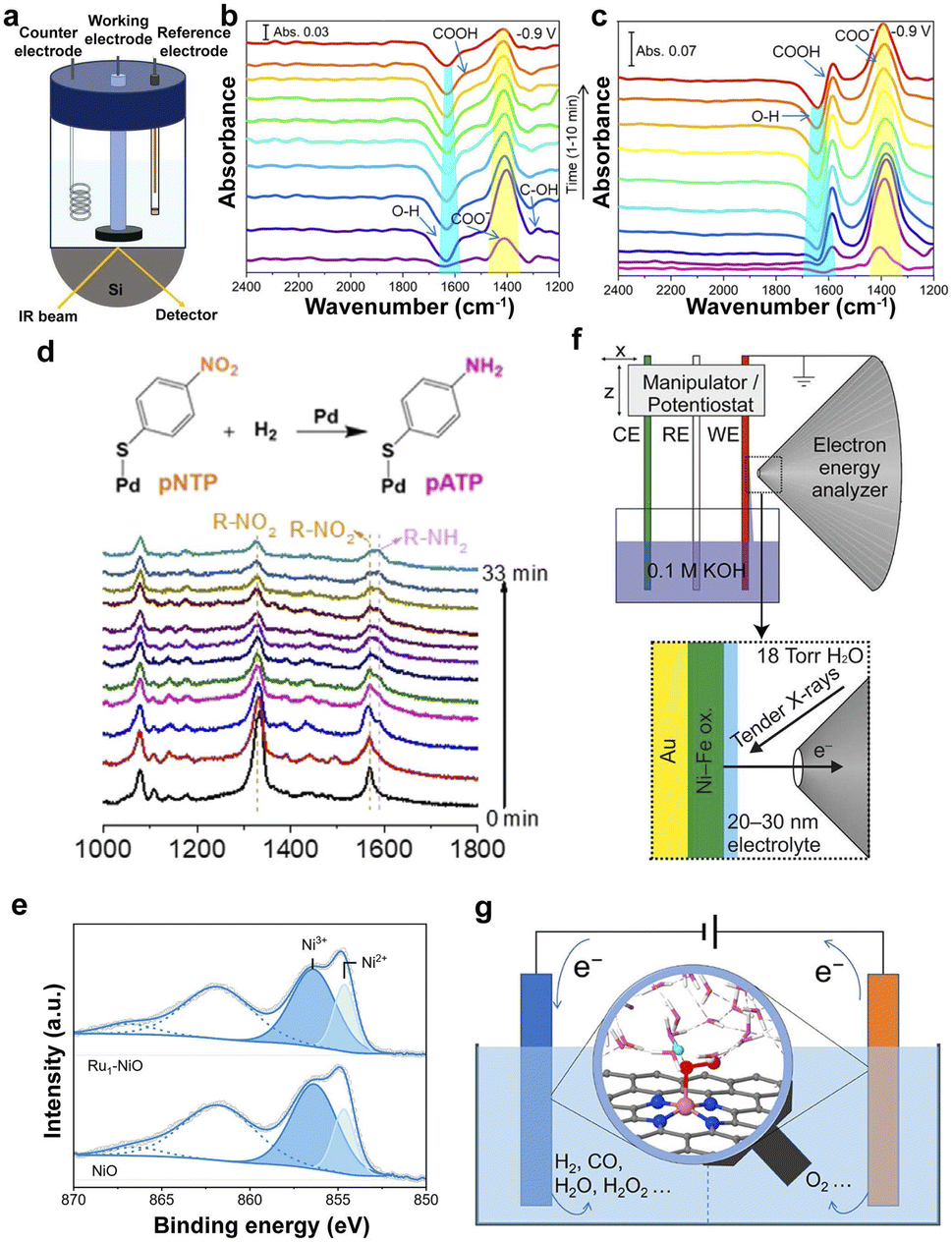 | ||
| Fig. 3 (a) Schematic illustration of the in situ FT-IR configuration for the CO2RR. Time-dependent in situ IR spectra taken at −0.9 V (vs. RHE) in the CO2RR conditions on PdIn@In2O3 (b) and PdIn alloy (c) during the second chronoamperometric test at −0.9 V.121 Copyright 2022, American Chemical Society. (d) Diagram and in situ SERS of the hydrogenation of pNTP over Pd SACs under H2 at 80 °C.127 Copyright 2021, Wiley. (e) XPS in the Ni 2p region for NiO and Ru1–NiO.129 Copyright 2022, Wiley. (f) Schematic illustration of an electrochemical cell.130 Copyright 2016, American Chemical Society. (g) Understanding electrocatalysis at SACs from simulations under realistic electrochemical environments.135 Copyright 2022, American Chemical Society. | ||
3.4 Raman scattering spectroscopy
Raman scattering spectroscopy is very sensitive to molecular bonding and structure. Therefore, it was employed to characterize the active sites of SACs via the fingerprint information of the supports and the change of the species in the solid–liquid interfaces in the low-wavenumber regions, such as oxygen species and hydroxy groups.122 The surfaces of nanostructured Au, Ag, and Cu can provide enhanced Raman signals, namely, surface-enhanced Raman spectroscopy (SERS), which is more sensitive to trace species on the surface of the catalysts, providing a promising technology for probing the SACs and the reaction mechanisms.123–126 It was employed to probe the hydrogenation process from para-nitrothiophenol (pNTP) to para-aminothiophenol (pATP) according to the decreased Raman peak of R–NO2 and the increased peak of R–NH2 (Fig. 3d).1273.5 XPS
XPS, as a typical surface analysis tool, is used to analyze the elemental composition of the catalyst surface (except H and He elements), the valence information, and the electronic state.128The XPS spectra in Fig. 3e indicate that the loading of single Ru atoms leads to a decreased oxidation state of the Ni in Ru1–NiO.129 The ambient-pressure XPS (APXPS) overcomes the limitation of the vacuum conditions to analyze the changes in the microstructure under the electrochemical reaction conditions by configuring the electrochemical cell (Fig. 3f), proving the initial stages of the oxidation of Ni(OH)2 to NiOOH.130–132 Therefore, APXPS is a promising technique for investigating the evolution of the catalysts in the electrocatalytic process.
3.6 Computation and simulations
Density functional theory (DFT) computation provides an available method for investigating the adsorption energy changes in the intermediates on the catalysts and possible reaction processes and helps to screen and optimize SACs.133 However, the complicated electrocatalysis processes on the solid–liquid interfaces make it difficult to mimic the realistic electrochemical reaction environment because of the sophisticated dynamic electronic charge on the surface and solvation effects. Simulations under realistic electrochemical environments, including solvent and constant-potential effects, can determine the true active site and understand the catalytic mechanism (Fig. 3g).134–137 Therefore, the computations and simulations provide indispensable guidelines for the rational design of SACs.The development of various advanced characterization techniques has greatly aided in identifying the structural and electronic properties of active sites on electrocatalysts. However, the breaking of the original chemical bonds and the formation of new chemical bonds on active sites during catalysis will lead to changes in the local coordination environment. Conventional characterization techniques fail to identify the dynamic evolution of active sites, leading to critical process information being missed. Therefore, the development of in situ characterization techniques is of great significance for a deeper recognition of active sites and a clearer understanding of catalytic principles.
4 Strategies for regulating the electronic structures of SACs
The change in the electronic structure of SACs has a great influence on the adsorption/desorption of the intermediates, thus regulating the electrocatalytic processes and efficiency. Therefore, a suitable electronic structure of SACs is of great significance for accelerating electrocatalysis. To date, there have been some successful strategies for adjusting the electronic structure of SACs via central metal atoms, coordination atoms, and metal–support interaction, to improve the electrocatalytic efficiency in different electrocatalytic processes.4.1 Central metal atoms
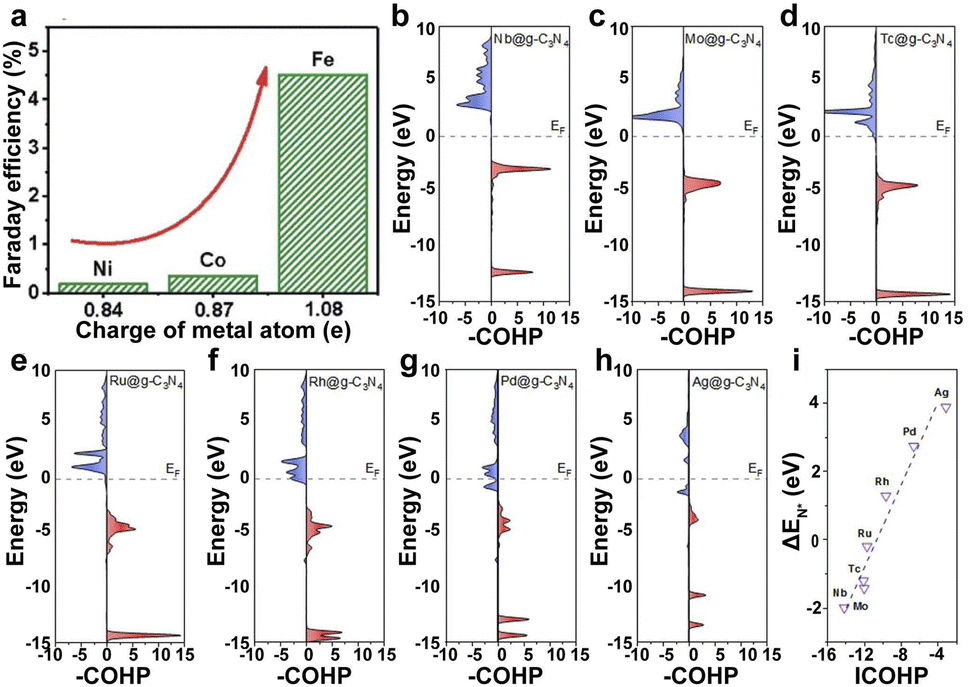 | ||
| Fig. 4 (a) FE of Fe1–N–C, Co1–N–C, and Ni1–N–C.138 Copyright 2019, The Royal Society of Chemistry. (b–h) pCOHP between the metal centers from Nb to Ag and the nitrogen atom. (i) Illustration of the correlation between ICOHP and ΔEN*. Red represents bonding contributions, and blue represents antibonding contributions.139 Copyright 2019, American Chemical Society. | ||
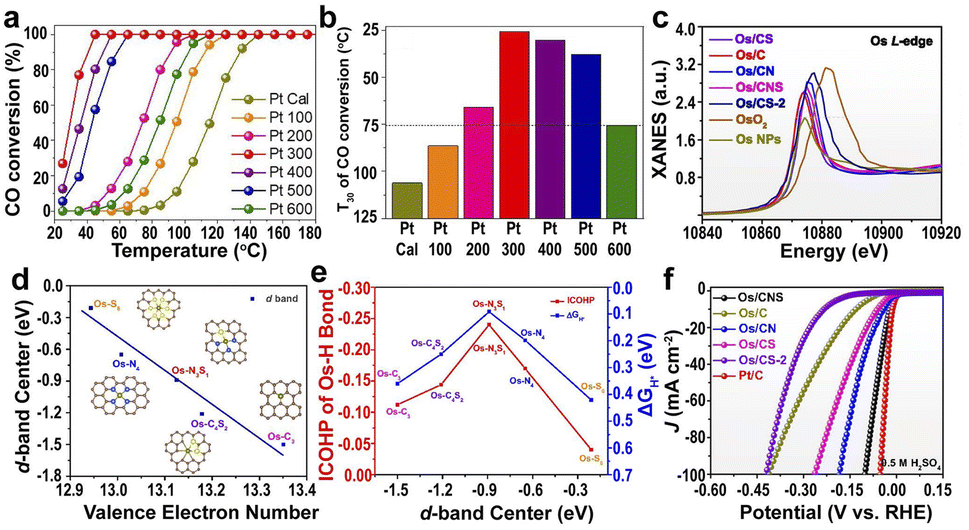 | ||
| Fig. 5 (a and b) Catalytic activity of Pt SACs (from Pt 100 to Pt 500) and Pt NPs (Pt 600).141 Copyright 2020, Wiley. (c) XANES of the Os L-edge for different samples. (d) d-band center of the Os single-atom as a function of the valence electron number. (e) ΔGH* and ICOHP of the Os–H bond for H* on Os SACs as a function of the d-band center of the Os single-atom. (f) HER polarization curves in acidic media.142 Copyright 2022, Nature Publishing Group. | ||
The supports with different heteroatoms accordingly have a great influence on the oxidation state of SAs and electrocatalytic activity.142 Five kinds of Os SAs with different oxidation states, by modifying the coordination environment of the support, follow the order Os/C < Os/CS < Os/CNS < Os/CN < Os/CS-2 (Fig. 5c), suggesting that the heteroatoms in the supports regulate the oxidation state. The increased oxidation state leads to the decreased occupation of the anti-bonding states of the Os–H bond and further enhances the adsorption of H atoms to Os (Fig. 5d). The low occupancy of the antibonding state around 0 and the increased oxidation state lead to the decreased occupancy of the Os–H bonding states near the Fermi level, decreasing the H* adsorption intensity (Fig. 5e). Therefore, the HER activities of different Os SACs follow the order Os/CNS > Os/CN > Os/CS > Os/C > Os/CS-2 (Fig. 5f), indicating that the doping of N and S heteroatoms provided an available pathway to regulate the HER activity.
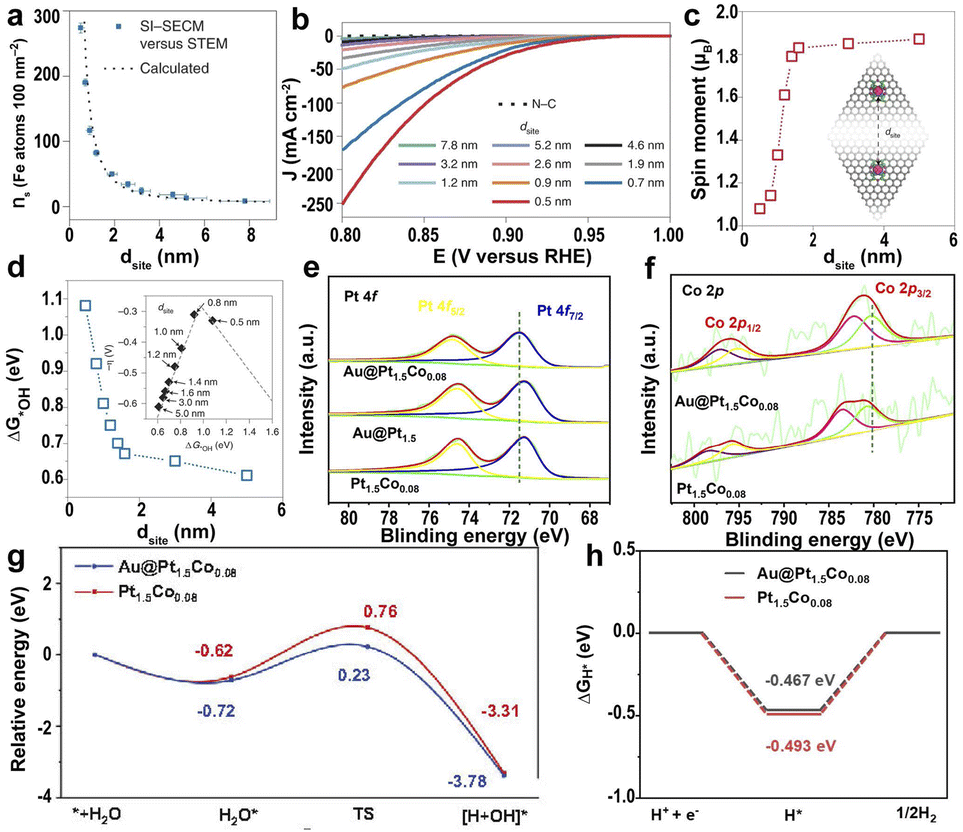 | ||
| Fig. 6 (a) A plot of Fe atom density (ns, Fe atoms per 100 nm2) measured with surface interrogation scanning electrochemical microscopy (SI-SECM) vs. the dsite. (b) Linear sweep voltammetry (LSV) of the PPy-derived N–C and Fe SACs with different dsite values. (c) DFT-calculated on-site magnetic moments of Fe atoms interacting with neighboring sites with different dsite values. (d) The dsite-dependent ΔG*OH was obtained by DFT calculations. Inset: volcano plot of calculated overpotentials for the ORR against ΔG*OH.145 Copyright 2021, Nature Publishing Group. High-resolution XPS of (e) Pt 4f and (f) Co 2p for Au@Pt1.5Co0.08, Au@Pt1.5, and Pt1.5Co0.08. (g) The relative energy diagram along with the water adsorption and dissociation processes on Pt1.5Co0.08 and Au@Pt1.5Co0.08 surfaces. (h) ΔGH* calculated at the equilibrium potential of Au@Pt1.5Co0.08 and Pt1.5Co0.08.149 Copyright 2022, Elsevier. | ||
To further improve the catalytic activity of SACs, the strain effect was approached to mediate the catalytic efficiency by constructing 3D structure modulation on the metal sites. The helical carbon with abundant high curvature surfaces was prepared via the carbonization of helical polypyrrole, and the compressive strain was subjected to the Fe–N4 sites in right-handed helical single atomic iron catalysts (D-Fe SAC).147 The bending structure and the compressive strain caused the downward shift of the d-band center of the curved Fe–N4, weakening the adsorption of intermediates and accelerating the charge transfer between Fe–N4 sites. Therefore, the D-Fe SAC has a higher ORR activity.
In addition to the SACs, single-atom alloy (SAA) catalysts can enhance the catalytic efficiency because of the tailoring of the electronic structure and synergy effect.148 The tensile strain induced by Au nuclei in core–shell Au@(PtCo0.05)x affects the electronic structure of the Co atoms in the Pt lattice.149 The strong surface tensile strain of Au@Pt1.5Co0.08 caused the positive shift of Pt atoms and the negative shift of Co atoms, resulting in the charge transfer from Co to Pt (Fig. 6e and f), promoting the breakage of O–H bonds and the desorption of OH groups (Fig. 6g). In addition, Au nuclei induced stronger Co–O bonding and promoted H* desorption, leading to excellent HER activity (Fig. 6h).
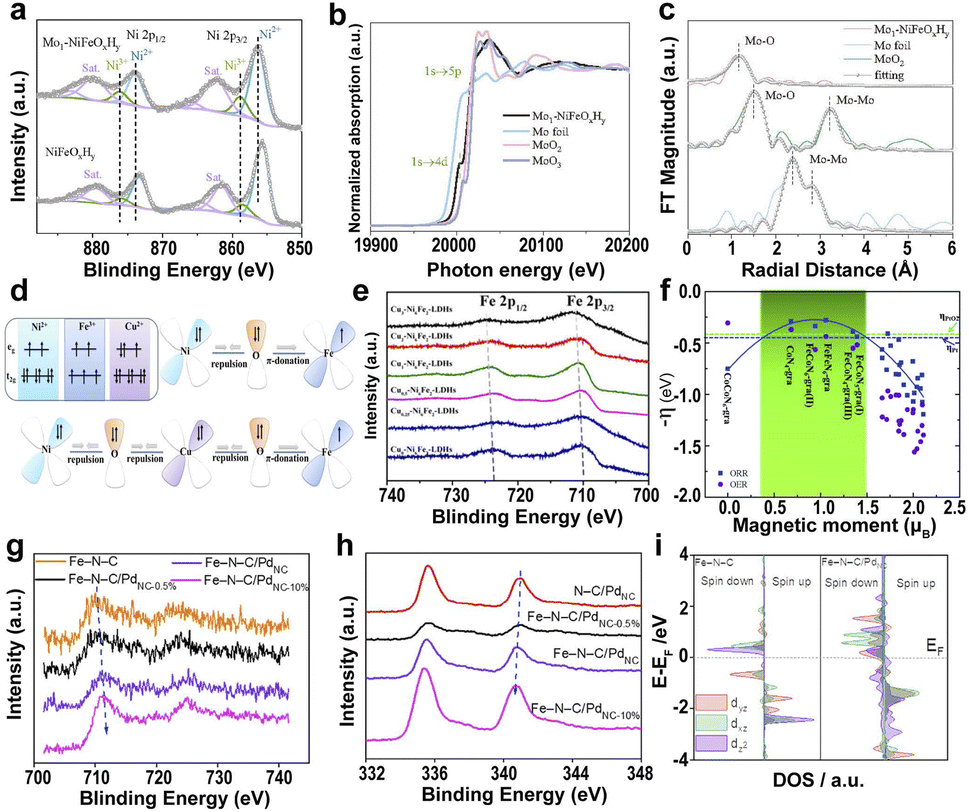 | ||
| Fig. 7 (a) XPS of Ni 2p for Mo1–NiFeOxHy and NiFeOxHy. (b) XANES of Mo K-edge. (c) EXAFS of Mo K-edge.152 Copyright 2022, Wiley. (d) Schematics of electronic interactions in the dxy orbitals of NiFe-LDHs and Cu–NiFe-LDHs. (e) High-resolution XPS scans of Fe 2p.154 Copyright 2022, American Chemical Society. (f) The negative ORR/OER overpotential as a function of the spin moment on the reaction center atoms. The blue and green dotted lines are the ORR overpotential for commercial Pt and the OER overpotential for PtO2.155 Copyright 2019, Royal Society of Chemistry. Deconvoluted XPS of Fe 2p (g) and Pd 3d (h) of different catalysts. (i) Projected DOS diagrams of Fe–N–C and Fe–N–C/PdNC.156 Copyright 2022, Elsevier. | ||
Introducing heterogeneous elements to mediate the spin state of the active center atoms also plays a significant role in achieving superior electrocatalytic activity.157–159 The hybrid electrocatalyst assembled by ultrafine Pd nanoclusters (PdNC) and Fe SAs (Fe–N–C/PdNC) leads to electron transfer from Fe SAs to PdNC.156 The PdNC induce the redistribution of Fe 3d-orbital electrons and increase the spin state of Fe dz2 orbitals because of the high electronegativity of Pd (Fig. 7g–i). The high spin state in Fe–N–C/PdNC creates a wider spin-related channel and promotes the Fe 3d electron spin configuration from low spin to intermediate spin, resulting in high catalytic activity for Fe–N–C/PdNC. Metal-based SACs loaded on carbon substrates exhibit desirable electrocatalytic activity and good electrochemical stability due to their high electrical conductivity and ability to bind metals.160 Particularly, two different transition metals Fe and Ni anchored on carbon supports take advantage of the coexistence and the interaction of two different transition metal SAs to modify the coordination environment of the neighboring atoms, thus improving the ORR performance resulting from the synergetic effects of adjacent Ni–N4 and Fe–N4 through the conjugated π bond of graphene.75,110,161.
4.2 Coordination atoms
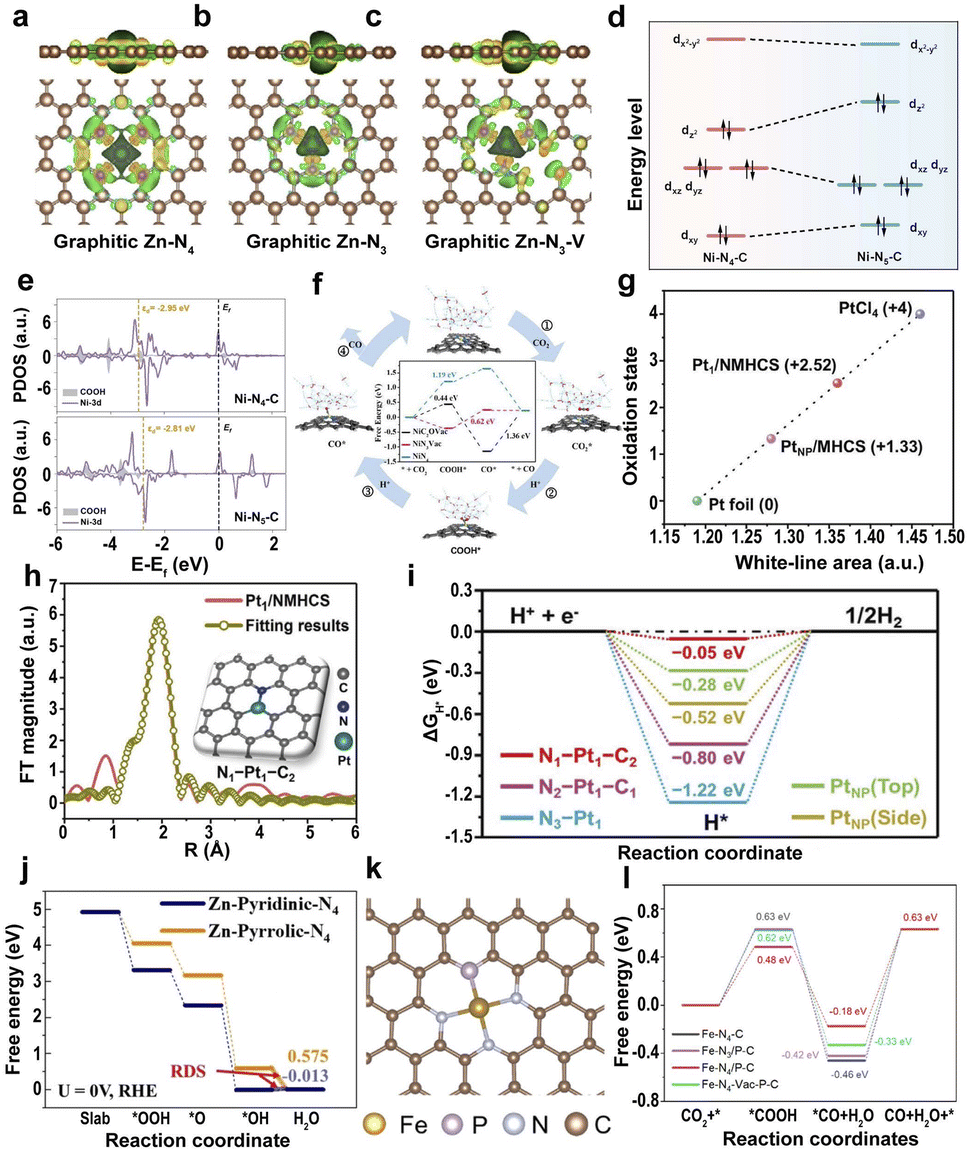 | ||
| Fig. 8 (a–c) Calculated spatial distribution of charge density for graphitic Zn–N4, Zn–N3, and Zn–N3–V (yellow and green iso-surfaces denote the depletion and gain of electron density, respectively).166 Copyright 2021, Wiley. (d) Ni 3d orbital splitting of Ni centers on Ni–N4–C and Ni–N5–C. (e) The d-band center of adsorption structures of *COOH on Ni–N4–C and Ni–N5–C.169 Copyright 2022, Wiley. (f) The proposed reaction pathways and free energy diagrams for the electrochemical reduction of CO2 to CO.170 Copyright 2022, Elsevier. (g) Fitting curve correlating the average oxidation state of Pt of Pt1/NMHCS, PtNP/MHCS, Pt foil, and PtCl4 at the Pt L3-edge. (h) EXAFS and the corresponding fitting curve of the Pt1/NMHCS. (i) Calculated ΔGH* values for the HER at the equilibrium potential.171 Copyright 2021, Wiley. (j) The oxygen-free energy diagrams for Zn-pyridinic-N4 and Zn-pyrrolic-N4.172 Copyright 2022, Wiley. (k) Atomic structure model of the Fe–N/P–C catalyst. (l) Gibbs free energy profiles of the single-Fe-atom structures with different coordination environments for the electroreduction of CO2 to CO.178 Copyright 2022, American Chemical Society. | ||
The introduction of axially coordinated N atoms into the Ni–N4 site to form square-pyramidal Ni–N5 leads to a quick increase in the dz2 orbital energy level of Ni, which facilitates the transfer of electrons from the dz2 orbital of Ni to the lowest unoccupied molecular orbital of CO2, promoting the adsorption and activation of CO2 (Fig. 8d).169 The d-band center of Ni-3d on Ni–N5–C with adsorbed *COOH is closer to the Fermi level than that of centrosymmetric Ni–N4–C (Fig. 8e), leading to the higher adsorption energies of *COOH and lower adsorption energies of *CO on Ni–N5–C, facilitating the formation and desorption of *COOH. The axial coordination of a nitrogen atom increases the dz2 orbital energy level and decreases the dxz/yz orbital energy level on the Ni–N5 active site, increasing the selectivity of the CO2RR and the limiting step of *CO2 to *COOH and CO desorption. Unsaturated Ni–N species also have a great influence on the efficiency of CO2RR.170 The defective Ni–N3 structure (NiN3Vac) is considered the active site of Ni-SACs@NC because of the lowest energy barrier for RDS, facilitating the generation of COOH* and increasing the activity of the catalyst (Fig. 8f).
Different types of coordinated N atoms, such as pyridine N and pyrrole N, have a significant influence on the catalytic performance. For atomically dispersed Zn–N–C SACs, the Zn-pyrrolic-N4 structure is the origin of high catalytic activity.172 Specifically, the binding strength of Zn to O with the pyrrole N coordination is lower than that of the pyridine N coordination, leading to the weaker adsorption of oxygen species at Zn sites, thus increasing the intrinsic ORR activity (Fig. 8j). Furthermore, heteroatoms directly coordinated with SAs, such as O, P, S, Cl, and B, can modulate the electronic structure.173–177 For example, the coordination environment of the Fe site is regulated by introducing P atoms into the Fe–N–C SACs, where the Fe atom co-locates with three N atoms and one P atom (Fig. 8k).178 The introduction of the P element in Fe–N/P–C SACs reduces the oxidation state of Fe SAs and the formation barrier of *CO intermediates, leading to higher electrocatalytic activity (Fig. 8l). Similarly, Liu et al. anchored S to the metal site of Ni SACs, which led to a decrease in the valence state of Ni and promoted the catalytic activity of CO2RR.179 Therefore, the adjustment of the coordination atoms has a great influence on the microstructure and the catalytic efficiency.
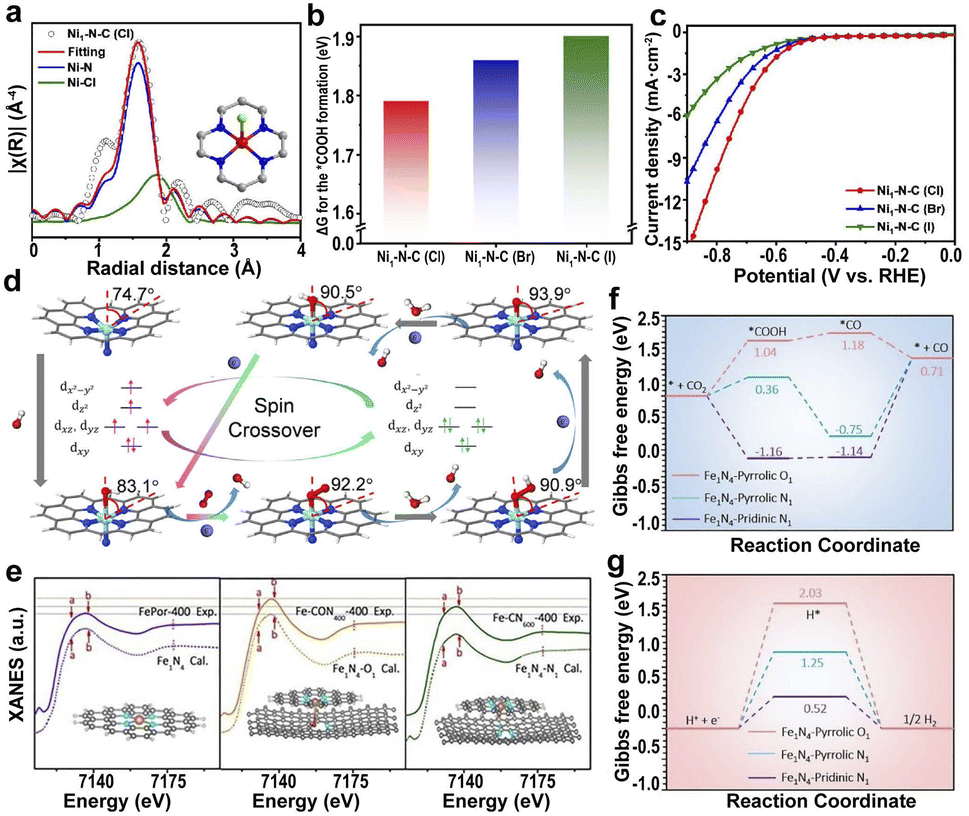 | ||
| Fig. 9 (a) EXAFS fitting of Ni1–N–C(Cl). (b) The ΔG for the *COOH formation in Ni1–N–C(X) catalysts. (c) LSV curves of Ni1–N–C(X).181 Copyright 2022, Springer. (d) Structure and dynamics of the N–FeN4C10 moiety in the ORR with optimized geometries (the angles represent the deviation of iron from the N4-plane).184 Copyright 2020, Elsevier. (e) Experimental XANES curves and the calculated XANES data for Fe1N4 (FePor-400, by calcinating iron porphyrin at 400 °C), Fe1N4–O1 (Fe–CON400-400, by calcinating O, N codoped carbon at 400 °C to form CON400, and loading iron porphyrin on CON400 at 400 °C to fabricate Fe–CON400-400), and Fe1N4–N1 (Fe–CN600-400, by calcinating O, N codoped carbon at 600 °C under 5% H2/Ar to form CN600, and loading iron porphyrin on CN600 at 400 °C to fabricate Fe–CN600-400). Gibbs free energy diagrams for the (f) CO2RR and (g) HER pathways on Fe sites of different configurations.185 Copyright 2021, The Royal Society of Chemistry. | ||
Additionally, axial N atoms have a great influence on the spin state of the center atoms, leading to a greater ORR activity of N–FeN4C10 than FeN4C10 (Fig. 9d).184 The axial Fe–O-coordinated Fe1N4–O1 and the axial Fe–N-coordinated Fe1N4–N1 are the active sites of the respective Fe SACs (Fig. 9e).185 The axial O ligand-induced d-band center of the Fe-3d orbital moved to a lower energy level; therefore, Fe1N4–O1 has the highest catalytic activity because of the increased CO desorption and inhibitory hydrogen precipitation reactions (Fig. 9f and g).
4.3 Electronic metal–support interaction
The strong EMSI between the supports and SAs has a great influence on the electronic structure and catalytic efficiency.70 The Pt SAs loaded on different transition-metal dichalcogenides (TMDs) lead to different average oxidation states of Pt as follows: Pt-SAs/WS2 < Pt-SAs/MoS2 < Pt-SAs/MoSe2 < Pt-SAs/WSe2.186 However, the acidic HER activity follows the order: Pt-SAs/WS2 > Pt-SAs/MoS2 > Pt-SAs/MoSe2 > Pt-SAs/WSe2, suggesting that the high oxidation states of Pt on different supports lead to decreased HER activity (Fig. 10a) because of the narrower d band of Pt SAs resulting from the p–d orbital hybridization between Pt SAs and coordinating atoms (S and Se) (Fig. 10b). The upward-shifted Pd d band leads to more antibonding states of hydrogen above the Fermi level, strengthening the affinity of Pt towards hydrogen (Fig. 10c). Therefore, the high oxidation state leads to the strong hydrogen adsorption of the EMSI and poisons the active site because of slow desorption.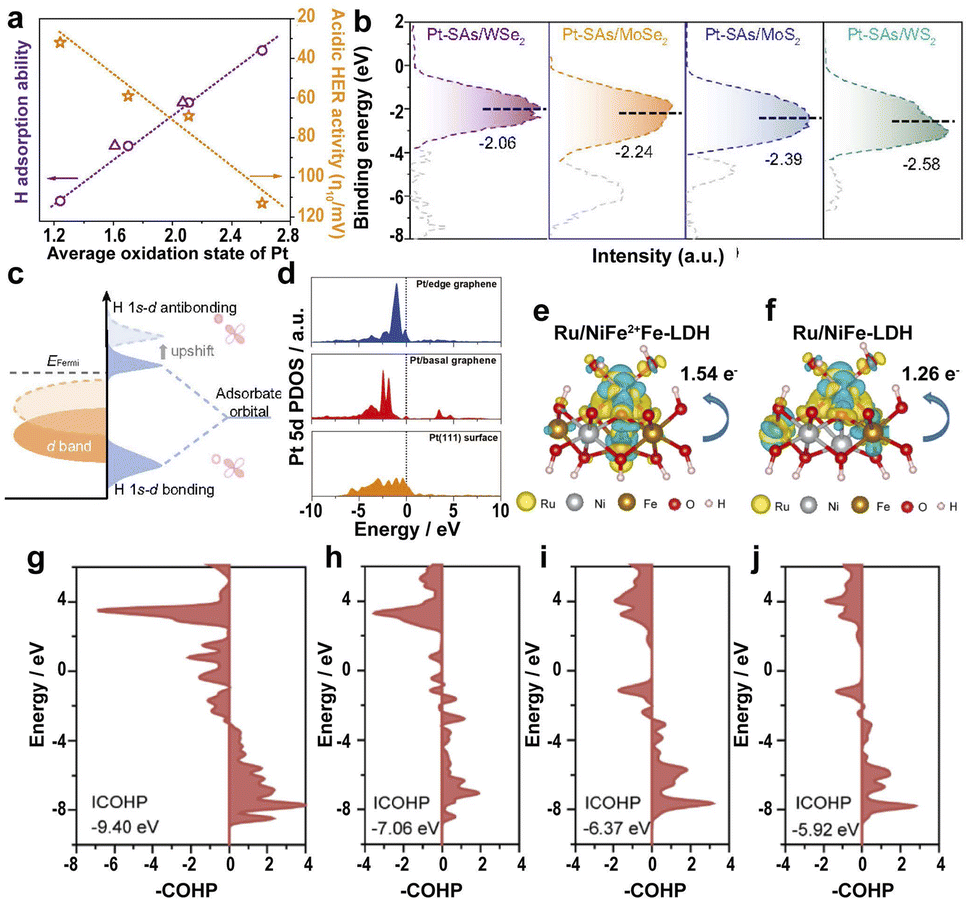 | ||
| Fig. 10 (a) The relationship between the average oxidation state, H adsorption ability, and acidic HER activity of Pt-SAs/TMDs. (b) Ultraviolet photoelectron spectroscopy-valence band spectra (UPS VBS) of Pt SAs relative to the Fermi level. (c) Schematic DOS diagrams illustrating the EMSI effect on the d-band position of Pt SAs, the interaction between Pt, and chemisorbed atomic hydrogen.186 Copyright 2021, Nature Publishing Group. (d) Calculated PDOS of the Pt 5d orbital of the Pt/graphene edge, Pt/basal graphene, and a Pt(111) surface.187 Copyright 2022, Wiley. A comparison of the dissolution of Ru, Ni, and Fe in electrolytes with different charge densities of (e) Ru/NiFe2+Fe-LDH and (f) Ru/NiFe-LDH. Yellow and light blue represent the charge accumulation and depletion regions, respectively. pCOHP between the Fe center at the edge near Ru and around the O atom of (g) Ru/NiFe2+Fe-LDH, (h) Ru/NiFeLDH, (i) NiFe2+Fe-LDH, and (j) NiFe-LDH.191 Copyright 2022, Elsevier. | ||
In addition to the TMDs, the influence of graphene on the electronic states of SACs was also investigated. A high electron population in the 5d orbitals and a low oxidation state for edge-rich vertical graphene (Pt/VG Ar-5) results in low ΔGH* on the Pt/edge graphene.187 The d-band center of Pt/VG Ar-5 is closer to the Fermi level because of the strong coupling between the π electrons of the graphene edge and the Pt SAs (Fig. 10d), resulting in excellent HER efficiency for Pt/VG Ar-5. Therefore, changing the location of metal atoms in the support regulates the electronic structure and controls the adsorption energy of the *H intermediate, accelerating the HER process.
Also, the controllable defects on the transition-metal layered hydroxides with ultra-high surface area and ultra-thin thickness to anchor noble metal SACs allow the fundamental study of the reaction steps and kinetics of the OER.188,189 Loading Ru atoms on the defective NiFe LDH leads to the electronic transfer from Ru to Ni and Fe, lowering ΔGH* on Ru active sites, and resulting in outstanding HER activity.190 The strong interaction of SAs reduces the overpotential of RDS for Ru–O, thus accelerating the OER kinetics. However, the active sites tend to form high valence states and dissolve in the electrolyte, leading to serious electrochemical instability. To solve this problem, anchoring Ru SAs on Fe2+-doped NiFe LDH (Ru/NiFe2+Fe-LDH) leads to the electronic transfer of Fe to high binding energy and Ru to low binding energy in Ru/NiFe2+Fe-LDH, lowering the valence of Ru in Ru/NiFe2+Fe-LDH, thus improving the electrochemical stability in the OER process.191 The strong coupling between Ru and Fe2+ leads to a lower energy barrier of Ru SAs on NiFe2+Fe-LDH and lower overpotential on the Fe sites surrounding Ru atoms in NiFe2+Fe-LDH, benefiting the electron transfer and facilitating the OER kinetics. More electron transfer between Ru and Fe sites in Ru/NiFe2+Fe-LDH than that of Ru/NiFe-LDH resulted in the tight bonding of Ru atoms to NiFe2+Fe-LDH, reducing the dissolution of Ru SAs (Fig. 10e and f). The energy-weighted ICOHP value (Fig. 10g–j) suggests that the more negative the ICOHP value, the greater the bond strength. Therefore, the enhanced Fe–O bond strength in Ru/NiFe2+Fe-LDH significantly improves the stability and electrocatalytic efficiency of the catalyst.
Note that the effects of central metal atoms, the coordination environment, and metal–support interactions are integrated into SACs, and cannot be decoupled definitely and studied separately.
5 Conclusions and outlook
SACs have been extensively employed in various electrocatalytic reactions, including the HER, OER, ORR, NRR, and CO2RR, due to their high electrocatalytic activity and excellent selectivity. The development of electrochemical energy conversion systems is crucial for solving the energy crisis and environmental issues, and SACs hold great promise for advancing these goals. However, the performance of SACs is influenced by several factors, such as electron transfer, metal–support interactions, coordination environment, and the density of the active centers. To further advance the applications of SACs, it is necessary to develop advanced characterization methods that can help us to understand the relationship between the catalyst structure and catalytic activity.The adjustment of the electronic structure of the active site in SACs mainly uses the following aspects. (1) Metal SAs interactions with supports, namely EMSI, leads to electronic hybridization, charge transfer, and energy transformation between the metal SAs and supports as a perturbation of the electronic structure, including the movement of d band centers and changes in charge distribution. Additionally, the electronic hybridization redistributes charge density between the isolated metal and adjacent atoms and allows the modulation of the adsorption and desorption of reactants, intermediates, and products. (2) Interactions of metal SAs with doping elements. Doping elements such as N and S for carbon supports can change the electronic structure of metal atoms and ultimately enhance the intrinsic activity of active sites. In addition, local defects such as vacancies around metal SAs can be used to adjust the type, number, and position of ligand atoms and thus optimize the adsorption reactants/intermediates/products at the active sites. (3) Interactions between active centers. The low loading of active metal SAs results in a relatively far distance and ignorable interactions between two adjacent active sites. However, in industrial applications, high metal loading is often achieved to pursue lower costs and higher activity. When the distance between two active centers is reduced to the atomic scale, the interaction between active centers will greatly affect the catalytic performance (including catalytic selectivity and efficiency) of the catalyst due to synergistic effects. In addition, in the diatomic system, strong electronic interactions between atoms may lead to a change in the spin state of the active metal atom, affecting the electron transfer path. Whether it is a monoatomic system or a diatomic system with the same or different metals, the synergistic effects between the neighboring metal atoms change the adsorption of the catalytic species, activate the chemical bonds, lower the reaction energy barrier and change the reaction path of intermediates, etc.
Accordingly, the hybridization of metal atoms results in more electronic states near the Fermi energy level, improving the electrical conductivity of materials and electron transfer in electrocatalysis, further regulating the charge distribution. The introduction of SAs leads to a dramatic reduction of the energy barrier for the RDS associated with electrochemical reactions.
Despite the rapid development of SACs, there are still many obstacles to overcome in their application. The hindrance of SACs from large-scale industrialization is reflected in high cost, low selectivity, and poor stability. For the practical application of SACs, the development should be accessible in the following aspects.
(1) More advanced characterization techniques. In catalytic processes, the active site often undergoes reversible structural changes due to the interactions with reactants or intermediates, and the structure of active sites often has a decisive influence on the performance. Therefore, advanced characterization techniques are still necessary to precisely identify individual atoms in catalysts, obtain local structural information, and understand the structural evolution of active sites in actual reactions. Additionally, the development and combined use of multiple techniques will play an important role in the development of catalysts. More accurate in situ/operando characterizations and computational simulations allow for a more precise understanding of catalytic mechanisms in realistic reaction systems and the targeted design of SACs.
(2) New materials and preparation methods. In electrocatalysis, all metal atoms, coordination atoms, and supports have a great influence on the activity, selectivity, and stability of the catalysts. Different materials interact with each other to produce distinct active sites, and new active species bring more selectivity and possibility to electrocatalysis. However, huge challenges remain in the low loading, uncontrollable coordination environments, and non-uniform distribution of metal sites, leading to poor catalytic performance. Traditional fabrication methods waste resources and time. Machine learning based on databases from high-throughput experiments and computations provides a promising pathway for screening excellent electrocatalysts. In particular, advanced fabrication methods such as 3D printing would promote SACs for practical application.
(3) The introduction of single-atom alloys. Compared with single active sites, the introduction of a second metal atom would improve metal loading and optimize the electronic structure and spin state of metal atoms, providing adsorption sites for multiple reactants and changing the adsorption sites of key intermediates, thus offering the prospect of breaking the possible linear relationship of adsorption energy of the intermediates on SACs. In addition, SAAs can bridge the model and industrial catalysts, and establish clear structure–performance relationships through characterization, computation, and performance evaluations. New knowledge could also be gained about spin or magnetism in electrocatalysis.
(4) Wider applications of SACs. SACs have not been widely applied due to their unclear catalytic processes. To overcome this challenge and enable their industrial use, advanced characterization techniques such as in situ and operando tools should be developed. Moreover, SACs with high loading capacity must be achieved for large-scale applications in industry. The development of SACs holds significant potential for important industrial conversion processes, but further research and improvements are needed to enable their widespread use.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (U21A20281).Notes and references
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- S. Ding, M. J. Hülsey, J. Pérez-Ramírez and N. Yan, Joule, 2019, 3, 2897–2929 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–801 CrossRef CAS.
- J. Yang, W. H. Li, S. Tan, K. Xu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19085–19091 CrossRef CAS PubMed.
- J. Liu, M. Jiao, B. Mei, Y. Tong, Y. Li, M. Ruan, P. Song, G. Sun, L. Jiang, Y. Wang, Z. Jiang, L. Gu, Z. Zhou and W. Xu, Angew. Chem., Int. Ed., 2019, 58, 1163–1167 CrossRef CAS PubMed.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- J. Ge, J. Y. Zheng, J. Zhang, S. Jiang, L. Zhang, H. Wan, L. Wang, W. Ma, Z. Zhou and R. Ma, J. Mater. Chem. A, 2021, 9, 14432–14443 RSC.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- T. Cui, Y.-P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, 202115219 Search PubMed.
- Y. Guo, S. Yao, Y. Xue, X. Hu, H. Cui and Z. Zhou, Appl. Catal., B, 2022, 304, 120997 CrossRef CAS.
- H. Huang, D. Yu, F. Hu, S.-C. Huang, J. Song, H.-Y. Chen, L. L. Li and S. Peng, Angew. Chem., Int. Ed., 2022, 61, 202116068 Search PubMed.
- W. Ma, H. Wan, L. Zhang, J. Y. Zheng and Z. Zhou, J. Energy Chem., 2021, 63, 170–194 CrossRef CAS.
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569–588 RSC.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. Yang, W. H. Li, K. Xu, S. Tan, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, 202200366 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- Y. Fang, Y. Hou, X. Fu and X. Wang, Chem. Rev., 2022, 122, 4204–4256 CrossRef CAS PubMed.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang, G.-Y. Shi, Y.-Y. Sun, X.-H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Perez-Ramirez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS.
- Y. Liu, B. Wang, Q. Fu, W. Liu, Y. Wang, L. Gu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 22522–22528 CrossRef CAS PubMed.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- L.-S. Zhang, X.-H. Jiang, Z.-A. Zhong, L. Tian, Q. Sun, Y.-T. Cui, X. Lu, J.-P. Zou and S.-L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751–21755 CrossRef CAS PubMed.
- S. Zhang, X. Ao, J. Huang, B. Wei, Y. Zhai, D. Zhai, W. Deng, C. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 9691–9698 CrossRef CAS PubMed.
- D. Cao, J. Wang, H. Xu and D. Cheng, Small, 2021, 17, 2101163 CrossRef CAS PubMed.
- M. Li, S. Zhao, J. Li, X. Chen, Y. Ji, H. Yu, D. Bai, G. Xu, Z. Zhong and F. Su, Nano Res., 2022, 15, 5857–5864 CrossRef CAS.
- C. Mochizuki, Y. Inomata, S. Yasumura, M. Lin, A. Taketoshi, T. Honma, N. Sakaguchi, M. Haruta, K.-i. Shimizu, T. Ishida and T. Murayama, ACS Catal., 2022, 12, 6149–6158 CrossRef CAS.
- S. Si, H. Shou, Y. Mao, X. Bao, G. Zhai, K. Song, Z. Wang, P. Wang, Y. Liu, Z. Zheng, Y. Dai, L. Song, B. Huang and H. Cheng, Angew. Chem., Int. Ed., 2022, 61, 202209446 Search PubMed.
- K. Xiao, R.-T. Lin, J.-X. Wei, N. Li, H. Li, T. Ma and Z.-Q. Liu, Nano Res., 2022, 15, 4980–4985 CrossRef CAS.
- L. Sun, L. R. Cao, Y. Su, C. J. Wang, J. Lin and X. D. Wang, Appl. Catal., B, 2022, 318, 121841 CrossRef CAS.
- G. D. Ding, L. D. Hao, H. P. Xu, L. G. Wang, J. Chen, T. Li, X. M. Tu and Q. Zhang, Chem. Commun., 2020, 3, 43 CrossRef CAS PubMed.
- W. Liu, F. Morfin, K. Provost, M. Bahri, W. Baaziz, O. Ersen, L. Piccolo and C. Zlotea, Nanoscale, 2022, 14, 7641–7649 RSC.
- Q. Shen, H. Jin, P. Li, X. Yu, L. Zheng, W. Song and C. Cao, Nano Res., 2022, 15, 5024–5031 CrossRef CAS.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cheong, X. Tan, H. Lei, H. A. Tahini, A. Henson, B. Subhash, N. M. Bedford, B. K. Miller, P. B. O'Mara, T. M. Benedetti, D. L. Huber, W. Zhang, S. C. Smith, J. J. Gooding, W. Schuhmann and R. D. Tilley, Nat. Catal., 2022, 5, 231–237 CrossRef CAS.
- L. Wang, L. Zhang, W. Ma, H. Wan, X. Zhang, X. Zhang, S. Jiang, J. Y. Zheng and Z. Zhou, Adv. Funct. Mater., 2022, 32, 2203342 CrossRef CAS.
- Z. Li, W. Wei, H. Li, S. Li, L. Leng, M. Zhang, J. H. Horton, D. Wang, W. Sun, C. Guo, W. Wu and J. Wang, ACS Nano, 2021, 15, 10175–10184 CrossRef CAS PubMed.
- Z. Sun, Y. Yang, C. Fang, Y. Yao, F. Qin, H. Gu, Q. Liu, W. Xu, H. Tang, Z. Jiang, B. Ge, W. Chen and Z. Chen, Small, 2022, 18, 2203422 CrossRef CAS.
- S. Jiao, M. Kong, Z. Hu, S. Zhou, X. Xu and L. Liu, Small, 2022, 18, 2105129 CrossRef CAS.
- F. Kong, X. Liu, Y. Song, Z. Qian, J. Li, L. Zhang, G. Yin, J. Wang, D. Su and X. Sun, Angew. Chem., Int. Ed., 2022, 61, 202207524 Search PubMed.
- L. Zhang, Q. Wang, L. Li, M. N. Banis, J. Li, K. Adair, Y. Sun, R. Li, Z.-J. Zhao, M. Gu and X. Sun, Nano Energy, 2022, 93, 106813 CrossRef CAS.
- J. Liu, C. Cao, X. Liu, L. Zheng, X. Yu, Q. Zhang, L. Gu, R. Qi and W. Song, Angew. Chem., Int. Ed., 2021, 60, 15248–15253 CrossRef CAS PubMed.
- J. Yang, J. Zhang, Q. Jiang, Y. Su, Y. Cui, X. Li, S. Zhang, W. Li and B. Qiao, J. Chem. Phys., 2021, 154, 131105 CrossRef CAS PubMed.
- J. Leverett, J. A. Yuwono, P. Kumar, T. Tran-Phu, J. Qu, J. Cairney, X. Wang, A. N. Simonov, R. K. Hocking, B. Johannessen, L. Dai, R. Daiyan and R. Amal, ACS Energy Lett., 2022, 7, 920–928 CrossRef CAS.
- J. Shi, Y. Wei, D. Zhou, L. Zhang, X. Yang, Z. Miao, H. Qi, S. Zhang, A. Li, X. Liu, W. Yan, Z. Jiang, A. Wang and T. Zhang, ACS Catal., 2022, 12, 7760–7772 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- K. Qi, M. Chhowalla and D. Voiry, Mater. Today, 2020, 40, 173–192 CrossRef CAS.
- K. Wu, K. Sun, S. Liu, W.-C. Cheong, Z. Chen, C. Zhang, Y. Pan, Y. Cheng, Z. Zhuang, X. Wei, Y. Wang, L. Zheng, Q. Zhang, D. Wang, Q. Peng, C. Chen and Y. Li, Nano Energy, 2021, 80, 105467 CrossRef CAS.
- K. Jiang, M. Luo, Z. Liu, M. Peng, D. Chen, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2021, 12, 1687 CrossRef CAS PubMed.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887 CrossRef CAS PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- H. Jin, P. Li, P. Cui, J. Shi, W. Zhou, X. Yu, W. Song and C. Cao, Nat. Commun., 2022, 13, 723 CrossRef CAS PubMed.
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569–588 RSC.
- H. Peng, T. Yang, H. Lin, Y. Xu, Z. Wang, Q. Zhang, S. Liu, H. Geng, L. Gu, C. Wang, X. Fan, W. Chen and X. Huang, Adv. Energy Mater., 2022, 12, 2201668 Search PubMed.
- M. Wu, G. Zhang, W. Wang, H. Yang, D. Rawach, M. Chen and S. Sun, Small Methods, 2022, 6, 2100947 CrossRef CAS PubMed.
- M. Gao, F. Tian, Z. Guo, X. Zhang, Z. Li, J. Zhou, X. Zhou, Y. Yu and W. Yang, Chem. Eng. J., 2022, 446, 137127 CrossRef.
- J. Xu, H. Xu, A. Dong, H. Zhang, Y. Zhou, H. Dong, B. Tang, Y. Liu, L. Zhang, X. Liu, J. Luo, L. Bie, S. Dai, Y. Wang, X. Sun and Y. Li, Adv. Mater., 2022, 34, 2206991 CrossRef CAS.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef CAS PubMed.
- G. Chen, H. Wan, W. Ma, N. Zhang, Y. Cao, X. Liu, J. Wang and R. Ma, Adv. Energy Mater., 2020, 10, 1902535 CrossRef CAS.
- H. Wan, X. Xiao, W. Ma, Y. Zhang, X. Liu, Y. Liu, G. Chen, N. Zhang, Y. Cao and R. Ma, Inorg. Chem. Front., 2022, 9, 83–89 RSC.
- J. Zhang, J. Ma, T. S. Choksi, D. Zhou, S. Han, Y. F. Liao, H. B. Yang, D. Liu, Z. Zeng, W. Liu, X. Sun, T. Zhang and B. Liu, J. Am. Chem. Soc., 2022, 144, 2255–2263 CrossRef CAS.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS PubMed.
- J. Yang, W.-H. Li, K. Xu, S. Tan, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, 202200366 Search PubMed.
- L. Gong, D. Zhang, C.-Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, 202215136 Search PubMed.
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- X. Duan, P. Li, D. Zhou, S. Wang, H. Liu, Z. Wang, X. Zhang, G. Yang, Z. Zhang, G. Tan, Y. Li, L. Xu, W. Liu, Z. Xing, Y. Kuang and X. Sun, Chem. Eng. J., 2022, 446, 136962 CrossRef CAS.
- G. Han, X. Zhang, W. Liu, Q. Zhang, Z. Wang, J. Cheng, T. Yao, L. Gu, C. Du, Y. Gao and G. Yin, Nat. Commun., 2021, 12, 6335 CrossRef CAS.
- J. Zhang, G. Zeng, L. Chen, W. Lai, Y. Yuan, Y. Lu, C. Ma, W. Zhang and H. Huang, Nano Res., 2022, 15, 4014–4022 CrossRef CAS.
- S.-N. Zhao, J.-K. Li, R. Wang, J. Cai and S.-Q. Zang, Adv. Mater., 2022, 34, 2107291 CrossRef CAS PubMed.
- H. Cai, G. Zhang, X. Zhang, B. Chen, Z. Lu, H. Xu, R. Gao and C. Shi, Small, 2022, 18, 2200911 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, J. Wu, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Chem. Rev., 2022, 122, 1273–1348 CrossRef CAS PubMed.
- Y. Wang, X. Li, M. Zhang, J. Zhang, Z. Chen, X. Zheng, Z. Tian, N. Zhao, X. Han, K. Zaghib, Y. Wang, Y. Deng and W. Hu, Adv. Mater., 2022, 34, 2107053 CrossRef CAS PubMed.
- L. Cao, X. Liu, X. Shen, D. Wu and T. Yao, Acc. Chem. Res., 2022, 55, 2594–2603 CrossRef CAS PubMed.
- W. Q. Zheng, R. Zhu, H. J. Wu, T. Ma, H. J. Zhou, M. Zhou, C. He, X. K. Liu, S. Li and C. Cheng, Angew. Chem., Int. Ed., 2022, 61, 202208667 Search PubMed.
- C.-X. Zhao, J.-N. Liu, J. Wang, C. Wang, X. Guo, X.-Y. Li, X. Chen, L. Song, B.-Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS.
- X. Liu, Y. Deng, L. Zheng, M. R. Kesama, C. Tang and Y. Zhu, ACS Catal., 2022, 12, 5517–5526 CrossRef CAS.
- Y. Chen, J. Lin, B. Jia, X. Wang, S. Jiang and T. Ma, Adv. Mater., 2022, 34, 2201796 CrossRef CAS PubMed.
- Y. Liu, X. Liu, A. R. Jadhav, T. Yang, Y. Hwang, H. Wang, L. Wang, Y. Luo, A. Kumar, J. Lee, H. T. D. Bui, M. G. Kim and H. Lee, Angew. Chem., Int. Ed., 2022, 61, 202114160 Search PubMed.
- Y. Cheng, H. Guo, X. Li, X. Wu, X. Xu, L. Zheng and R. Song, Chem. Eng. J., 2021, 410, 128359 CrossRef CAS.
- J. Huo, Z. Shen, X. Cao, L. Li, Y. Zhao, H. Liu and G. Wang, Small, 2022, 18, 2202394 CrossRef CAS PubMed.
- Y. Wu, Q. Wu, Q. Zhang, Z. Lou, K. Liu, Y. Ma, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai, B. Huang and P. Wang, Energy Environ. Sci., 2022, 15, 1271–1281 RSC.
- X. I. Pcrcira-Hcrnandcz, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarlk, E. J. M. Hcnscn, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1358 CrossRef.
- R. Santhosh Kumar, S. Ramakrishnan, S. Prabhakaran, A. R. Kim, D. R. Kumar, D. H. Kim and D. J. Yoo, J. Mater. Chem. A, 2022, 10, 1999–2011 RSC.
- N. Logeshwaran, S. Ramakrishnan, S. S. Chandrasekaran, M. Vinothkannan, A. R. Kim, S. Sengodan, D. B. Velusamy, P. Varadhan, J.-H. He and D. J. Yoo, Appl. Catal., B, 2021, 297, 120405 CrossRef CAS.
- W. Liu, H. Zhang, C. Li, X. Wang, J. Liu and X. Zhang, J. Energy Chem., 2020, 47, 333–345 CrossRef.
- S. Tian, B. Wang, W. Gong, Z. He, Q. Xu, W. Chen, Q. Zhang, Y. Zhu, J. Yang, Q. Fu, C. Chen, Y. Bu, L. Gu, X. Sun, H. Zhao, D. Wang and Y. Li, Nat. Commun., 2021, 12, 3181 CrossRef CAS PubMed.
- S. C. Karthikeyan, R. Santhosh Kumar, S. Ramakrishnan, S. Prabhakaran, A. R. Kim, D. H. Kim and D. J. Yoo, ACS Sustainable Chem. Eng., 2022, 10, 15068–15081 CrossRef.
- R. Santhosh Kumar, S. C. Karthikeyan, S. Ramakrishnan, S. Vijayapradeep, A. Rhan Kim, J.-S. Kim and D. Jin Yoo, Chem. Eng. J., 2023, 451, 138471 CrossRef CAS.
- L. Sun, L. Cao, Y. Su, C. Wang, J. Lin and X. Wang, Appl. Catal., B, 2022, 318, 121841 CrossRef CAS.
- J. Fonseca and J. Lu, ACS Catal., 2021, 11, 7018–7059 CrossRef CAS.
- S. Jiao, M. Kong, Z. Hu, S. Zhou, X. Xu and L. Liu, Small, 2022, 18, 2105129 CrossRef CAS PubMed.
- R. Ramesh, S. Han, D. K. Nandi, S. Y. Sawant, D. H. Kim, T. Cheon, M. H. Cho, R. Harada, T. Shigetomi, K. Suzuki and S.-H. Kim, Adv. Mater. Interfaces, 2021, 8, 2001508 CrossRef CAS.
- Y. Li, S. L. Zhang, W. Cheng, Y. Chen, D. Luan, S. Gao and X. W. D. Lou, Adv. Mater., 2022, 34, 2105204 CrossRef CAS PubMed.
- T. Al-Zoubi, Y. Zhou, X. Yin, B. Janicek, C. Sun, C. E. Schulz, X. Zhang, A. A. Gewirth, P. Huang, P. Zelenay and H. Yang, J. Am. Chem. Soc., 2020, 142, 5477–5481 CrossRef CAS PubMed.
- H. Wei, K. Huang, L. Zhang, B. Ge, D. Wang, J. Lang, J. Ma, D. Wang, S. Zhang, Q. Li, R. Zhang, N. Hussain, M. Lei, L.-M. Liu and H. Wu, Angew. Chem., Int. Ed., 2018, 57, 3354–3359 CrossRef CAS PubMed.
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li, M. Lei, C. Zhang, J. Irawan, L. M. Liu and H. Wu, Nat. Commun., 2017, 8, 1490 CrossRef PubMed.
- G.-F. Han, F. Li, A. I. Rykov, Y.-K. Im, S.-Y. Yu, J.-P. Jeon, S.-J. Kim, W. Zhou, R. Ge, Z. Ao, T. J. Shin, J. Wang, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2022, 17, 403–407 CrossRef CAS.
- H. Fei, J. Dong, C. Wan, Z. Zhao, X. Xu, Z. Lin, Y. Wang, H. Liu, K. Zang, J. Luo, S. Zhao, W. Hu, W. Yan, I. Shakir, Y. Huang and X. Duan, Adv. Mater., 2018, 30, 1802146 CrossRef PubMed.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- V. Muravev, G. Spezzati, Y.-Q. Su, A. Parastaev, F.-K. Chiang, A. Longo, C. Escudero, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2021, 4, 469–478 CrossRef CAS.
- X. Wu, H. Zhang, S. Zuo, J. Dong, Y. Li, J. Zhang and Y. Han, Nano-Micro Lett., 2021, 13, 136 CrossRef CAS PubMed.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cheong, X. Tan, H. Lei, H. A. Tahini, A. Henson, B. Subhash, N. M. Bedford, B. K. Miller, P. B. O'Mara, T. M. Benedetti, D. L. Huber, W. Zhang, S. C. Smith, J. J. Gooding, W. Schuhmann and R. D. Tilley, Nat. Catal., 2022, 5, 231–237 CrossRef CAS.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS PubMed.
- S. Mühlenberend, M. Gruyters and R. Berndt, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115301 CrossRef.
- Z. Zhu, H. Yin, Y. Wang, C. H. Chuang, L. Xing, M. Dong, Y. R. Lu, G. Casillas-Garcia, Y. Zheng, S. Chen, Y. Dou, P. Liu, Q. Cheng and H. Zhao, Adv. Mater., 2020, 32, 2004670 CrossRef CAS PubMed.
- L. Zeng, Z. Zhao, F. Lv, Z. Xia, S. Y. Lu, J. Li, K. Sun, K. Wang, Y. Sun, Q. Huang, Y. Chen, Q. Zhang, L. Gu, G. Lu and S. Guo, Nat. Commun., 2022, 13, 3822 CrossRef CAS PubMed.
- M. M. J. Treacy, A. Howie and C. J. Wilson, Philos. Mag., 1978, 32, 569–585 CrossRef.
- S. Hejazi, S. Mohajernia, B. Osuagwu, G. Zoppellaro, P. Andryskova, O. Tomanec, S. Kment, R. Zboril and P. Schmuki, Adv. Mater., 2020, 32, 1908505 CrossRef CAS PubMed.
- S. Liang, Q. Jiang, Q. Wang and Y. Liu, Adv. Energy Mater., 2021, 11, 2101477 CrossRef CAS.
- W. Yang, P. Cheng, Z. Li, Y. Lin, M. Li, J. Zi, H. Shi, G. Li, Z. Lian and H. Li, Adv. Funct. Mater., 2022, 32, 2205920 CrossRef CAS.
- C. J. Chen, Phys. Rev. Lett., 1990, 65, 448–451 CrossRef CAS PubMed.
- T. Zhang, Z. Chen, A. G. Walsh, Y. Li and P. Zhang, Adv. Mater., 2020, 32, 2002910 CrossRef CAS PubMed.
- D. Bagchi, N. Phukan, S. Sarkar, R. Das, B. Ray, P. Bellare, N. Ravishankar and S. C. Peter, J. Mater. Chem. A, 2021, 9, 9319–9326 RSC.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- C. Yang, Z. Miao, F. Zhang, L. Li, Y. Liu, A. Wang and T. Zhang, Green Chem., 2018, 20, 2142–2150 RSC.
- D. Bagchi, S. Sarkar, A. K. Singh, C. P. Vinod and S. C. Peter, ACS Nano, 2022, 16, 6158–6196 CrossRef PubMed.
- W. Zhang, C. Huang, J. Zhu, Q. Zhou, R. Yu, Y. Wang, P. An, J. Zhang, M. Qiu, L. Zhou, L. Mai, Z. Yi and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, 202112116 Search PubMed.
- S. Inoue, T. Nakajima and Y. Kikuchi, Carbon, 2006, 44, 1287–1291 CrossRef CAS.
- J.-H. Zhong, X. Jin, L. Meng, X. Wang, H.-S. Su, Z.-L. Yang, C. T. Williams and B. Ren, Nat. Nanotechnol., 2017, 12, 132–136 CrossRef CAS PubMed.
- J. Chen, G. Liu, Y.-z. Zhu, M. Su, P. Yin, X.-j. Wu, Q. Lu, C. Tan, M. Zhao, Z. Liu, W. Yang, H. Li, G.-H. Nam, L. Zhang, Z. Chen, X. Huang, P. M. Radjenovic, W. Huang, Z.-q. Tian, J.-f. Li and H. Zhang, J. Am. Chem. Soc., 2020, 142, 7161–7167 CrossRef CAS PubMed.
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T.-B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, 202203569 Search PubMed.
- J. Wei, S. N. Qin, J. Yang, H. L. Ya, W. H. Huang, H. Zhang, B. J. Hwang, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2021, 60, 9306–9310 CrossRef CAS PubMed.
- B. Hu, K. Sun, Z. Zhuang, Z. Chen, S. Liu, W.-C. Cheong, C. Chen, M. Hu, X. Cao, J. Ma, R. Tu, X. Zheng, H. Xiao, X. Chen, Y. Cui, Q. Peng, C. Chen and Y. Li, Adv. Mater., 2022, 34, 2107721 CrossRef CAS.
- R. Ge, Y. Wang, Z. Li, M. Xu, S. M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, 202200211 Search PubMed.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 CrossRef.
- L. Wang, X. Li, X. Hu, S. Chen, Z. Qiu, Y. Wang, H. Zhang, Y. Yu, B. Yang, Y. Yang, P. Orgiani, C. Aruta and N. Yang, Nano Res., 2023, 16, 3278–3286 CrossRef CAS.
- L. Nguyen, F. F. Tao, Y. Tang, J. Dou and X. J. Bao, Chem. Rev., 2019, 119, 6822–6905 CrossRef.
- L. Chen, Y. Tian, X. Hu, S. Yao, Z. Lu, S. Chen, X. Zhang and Z. Zhou, Adv. Funct. Mater., 2022, 32, 2208418 CrossRef CAS.
- X. Hu, S. Chen, L. Chen, Y. Tian, S. Yao, Z. Lu, X. Zhang and Z. Zhou, J. Am. Chem. Soc., 2022, 144, 18144–18152 CrossRef CAS PubMed.
- X. Zhao, Z. H. Levell, S. Yu and Y. Liu, Chem. Rev., 2022, 122, 10675–10709 CrossRef CAS PubMed.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS.
- X. Hu, S. Yao, L. Chen, X. Zhang, M. Jiao, Z. Lu and Z. Zhou, J. Mater. Chem. A, 2021, 9, 23515–23521 RSC.
- R. Zhang, L. Jiao, W. Yang, G. Wan and H.-L. Jiang, J. Mater. Chem. A, 2019, 7, 26371–26377 RSC.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS.
- X. Zheng, J. Tang, A. Gallo, J. A. Garrido Torres, X. Yu, C. J. Athanitis, E. M. Been, P. Ercius, H. Mao, S. C. Fakra, C. Song, R. C. Davis, J. A. Reimer, J. Vinson, M. Bajdich and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2101817118 CrossRef.
- H. Jeong, D. Shin, B. S. Kim, J. Bae, S. Shin, C. Choe, J. W. Han and H. Lee, Angew. Chem., Int. Ed., 2020, 59, 20691–20696 CrossRef CAS PubMed.
- D. Cao, H. Xu, H. Li, C. Feng, J. Zeng and D. Cheng, Nat. Commun., 2022, 13, 5843 CrossRef CAS PubMed.
- C. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS.
- X. F. Lu, B. Y. Xia, S.-Q. Zang and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 4634–4650 CrossRef CAS PubMed.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS.
- J. Yang, Z. Wang, C. X. Huang, Y. Zhang, Q. Zhang, C. Chen, J. Du, X. Zhou, Y. Zhang, H. Zhou, L. Wang, X. Zheng, L. Gu, L. M. Yang and Y. Wu, Angew. Chem., Int. Ed., 2021, 60, 22722–22728 CrossRef CAS PubMed.
- Y. Xie, J. Cai, Y. Wu, Y. Zang, X. Zheng, J. Ye, P. Cui, S. Niu, Y. Liu, J. Zhu, X. Liu, G. Wang and Y. Qian, Adv. Mater., 2019, 31, 1807780 CrossRef PubMed.
- R. Wan, M. Luo, J. Wen, S. Liu, X. Kang and Y. Tian, J. Energy Chem., 2022, 69, 44–53 CrossRef CAS.
- M. Li, S. Wang, X. Wang, X. Tian, X. Wu, Y. Zhou, G. Hu and L. Feng, Chem. Eng. J., 2022, 442, 136165 CrossRef CAS.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W.-X. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- Y. Wu, Y. Zhao, P. Zhai, C. Wang, J. Gao, L. Sun and J. Hou, Adv. Mater., 2022, 34, 2202523 CrossRef CAS PubMed.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- Z. Sun, L. Lin, J. He, D. Ding, T. Wang, J. Li, M. Li, Y. Liu, Y. Li, M. Yuan, B. Huang, H. Li and G. Sun, J. Am. Chem. Soc., 2022, 144, 8204–8213 CrossRef CAS PubMed.
- Y. Sun, J. Wang, Q. Liu, M. Xia, Y. Tang, F. Gao, Y. Hou, J. Tse and Y. Zhao, J. Mater. Chem. A, 2019, 7, 27175–27185 RSC.
- X. Wei, S. Song, W. Cai, X. Luo, L. Jiao, Q. Fang, X. Wang, N. Wu, Z. Luo, H. Wang, Z. Zhu, J. Li, L. Zheng, W. Gu, W. Song, S. Guo and C. Zhu, Chem, 2022, 8, 2136–2147 Search PubMed.
- T. Wang, A. Chutia, D. J. L. Brett, P. R. Shearing, G. He, G. Chai and I. P. Parkin, Energy Environ. Sci., 2021, 14, 2639–2669 RSC.
- X. Ao, W. Zhang, B. Zhao, Y. Ding, G. Nam, L. Soule, A. Abdelhafiz, C. Wang and M. Liu, Energy Environ. Sci., 2020, 13, 3032–3040 RSC.
- X. Wei, X. Luo, N. Wu, W. Gu, Y. Lin and C. Zhu, Nano Energy, 2021, 84, 105817 CrossRef CAS.
- C. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14, 2349–2356 RSC.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- M. W. Jia, C. Choi, T. S. Wu, C. Ma, P. Kang, H. C. Tao, Q. Fan, S. Hong, S. Z. Liu, Y. L. Soo, Y. S. Jung, J. S. Qiu and Z. Y. Sun, Chem. Sci., 2018, 9, 8775–8780 RSC.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X. M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS PubMed.
- D. Fang, P. Sun, S. Huang, Y. Shang, X. Li, D. Yan, Y. V. Lim, C.-Y. Su, B.-J. Su, J.-Y. Juang and H. Y. Yang, ACS Mater. Lett., 2022, 4, 1–10 CrossRef CAS.
- X. Liang, D. Wang, Z. Zhao, T. Li, Y. Gao and C. Hu, Adv. Funct. Mater., 2022, 32, 2203001 CrossRef CAS.
- J. R. Huang, X. F. Qiu, Z. H. Zhao, H. L. Zhu, Y. C. Liu, W. Shi, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, 202210985 Search PubMed.
- Y. Guo, S. Yao, Y. Xue, X. Hu, H. Cui and Z. Zhou, Appl. Catal., B, 2022, 304, 120997 CrossRef CAS.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C. W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, 2008599 CrossRef CAS PubMed.
- P. Sun, Z. Qiao, S. Wang, D. Li, X. Liu, Q. Zhang, L. Zheng, Z. Zhuang and D. Cao, Angew. Chem., Int. Ed., 2023, 62, 202216041 Search PubMed.
- X. Long, Z. Li, G. Gao, P. Sun, J. Wang, B. Zhang, J. Zhong, Z. Jiang and F. Li, Nat. Commun., 2020, 11, 4074 CrossRef CAS PubMed.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS PubMed.
- F. Pan, B. Li, E. Sarnello, S. Hwang, Y. Gang, X. Feng, X. Xiang, N. M. Adli, T. Li, D. Su, G. Wu, G. Wang and Y. Li, Nano Energy, 2020, 68, 104384 CrossRef CAS.
- S. Ji, B. Jiang, H. Hao, Y. Chen, J. Dong, Y. Mao, Z. Zhang, R. Gao, W. Chen, R. Zhang, Q. Liang, H. Li, S. Liu, Y. Wang, Q. Zhang, L. Gu, D. Duan, M. Liang, D. Wang, X. Yan and Y. Li, Nat. Catal., 2021, 4, 407–417 CrossRef CAS.
- K. Li, S. Zhang, X. Zhang, S. Liu, H. Jiang, T. Jiang, C. Shen, Y. Yu and W. Chen, Nano Lett., 2022, 22, 1557–1565 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- Y. Wang, Y.-J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS PubMed.
- J.-X. Peng, W. Yang, Z. Jia, L. Jiao and H.-L. Jiang, Nano Res., 2022, 15, 10063–10069 CrossRef CAS.
- Y. Liu, S. Zhang, C. Jiao, H. Chen, G. Wang, W. Wu, Z. Zhuo and J. Mao, Adv. Sci., 2023, 10, 2206107 CrossRef CAS PubMed.
- K.-M. Zhao, S. Liu, Y.-Y. Li, X. Wei, G. Ye, W. Zhu, Y. Su, J. Wang, H. Liu, Z. He, Z.-Y. Zhou and S.-G. Sun, Adv. Energy Mater., 2022, 12, 2103588 CrossRef CAS.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS.
- Z. Chen, A. Huang, K. Yu, T. Cui, Z. Zhuang, S. Liu, J. Li, R. Tu, K. Sun, X. Tan, J. Zhang, D. Liu, Y. Zhang, P. Jiang, Y. Pan, C. Chen, Q. Peng and Y. Li, Energy Environ. Sci., 2021, 14, 3430–3437 RSC.
- Y. Shi, Z. R. Ma, Y. Y. Xiao, Y. C. Yin, W. M. Huang, Z. C. Huang, Y. Z. Zheng, F. Y. Mu, R. Huang, G. Y. Shi, Y. Y. Sun, X. H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef PubMed.
- C. Tsounis, B. Subhash, P. V. Kumar, N. M. Bedford, Y. Zhao, J. Shenoy, Z. Ma, D. Zhang, C. Y. Toe, S. Cheong, R. D. Tilley, X. Lu, L. Dai, Z. Han and R. Amal, Adv. Funct. Mater., 2022, 32, 2203067 CrossRef CAS.
- X. Zheng, J. Tang, A. Gallo, J. A. Garrido Torres, X. Yu, C. J. Athanitis, E. M. Been, P. Ercius, H. Mao, S. C. Fakra, C. Song, R. C. Davis, J. A. Reimer, J. Vinson, M. Bajdich and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2101817118 CrossRef PubMed.
- Z. Li, B. Li, Y. Hu, S. Wang and C. Yu, Materials Advances, 2022, 3, 779–809 RSC.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- X. Duan, P. Li, D. Zhou, S. Wang, H. Liu, Z. Wang, X. Zhang, G. Yang, Z. Zhang, G. Tan, Y. Li, L. Xu, W. Liu, Z. Xing, Y. Kuang and X. Sun, Chem. Eng. J., 2022, 446, 136962 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |