
Macrocyclization-induced phosphorescence enhancement of pyridinium-based macrocycles†
Shuo
Li‡
ab,
Zhi-Yuan
Zhang‡
 a,
Jing-Fang
Lv
a,
Ling
Li
a,
Jian
Li
c and
Chunju
Li
*a
a,
Jing-Fang
Lv
a,
Ling
Li
a,
Jian
Li
c and
Chunju
Li
*a
aTianjin Key Laboratory of Structure and Performance for Functional Molecules, College of Chemistry, Tianjin Normal University, Tianjin 300387, P. R. China. E-mail: cjli@shu.edu.cn
bSchool of Advanced Study, Taizhou University, Taizhou 318000, Zhejiang, P. R. China
cSchool of Chemistry and Chemical Engineering, Henan Normal University, P. R. China
First published on 19th January 2023
Abstract
Organic room temperature phosphorescence (RTP) has exhibited various applications in optoelectronics and photobiology. Reported here is an effective phosphorescence enhancement strategy through the macrocyclization of phosphorescent pyridine/pyridinium units by methylene linkers. A pyridine macrocycle was synthesized by the condensation of the 3,5-bis(2,4-dimethoxyphenyl)pyridine monomer with paraformaldehyde under the catalysis of a Lewis acid. The methylation reaction of the pyridine macrocycle with methyl iodide and subsequent ion exchange with ammonium hexafluorophosphate and tetrabutyl ammonium halide gave pyridinium macrocycles. Compared with pyridinium monomers, the macrocycles exhibited enhanced phosphorescence with up to 59-fold prolonging of lifetime and 3.7-fold increase of quantum yields. A mechanism study revealed that macrocyclization restrained the rotation/vibration of luminescent units and therefore suppressed nonradiative decay. Moreover, the macrocyclization slowed down the nonradiative and radiative decay processes of the triplet state. Such macrocyclization-induced phosphorescence enhancement (MIPE) would provide a novel and general strategy for enhancing organic room temperature phosphorescence (RTP) and find wide applications beyond phosphorescence.
Introduction
Organic room-temperature phosphorescence (RTP) has shown great promise in the applications of bioimaging,1–5 optoelectronics,6–8 data encryption,9–14etc. It also has great advantages of high molecular diversity, good biocompatibility and low cost compared with inorganics. Therefore, much effort has been devoted to organic RTP. In general, three key factors should be considered for realizing organic RTP with long lifetime, high efficiency and robustness: boosting intersystem crossing (ISC) to produce more triplet state population, stabilizing triplet states and lowering the radiative/nonradiative decays, and shielding luminescent units against quenchers such as oxygen and impurities. Hitherto, notable methods have been reported for pursuing high performance organic RTP. The common strategies to boost the ISC process are to incorporate n-electron groups (e.g., halogens and heteroatoms) or charge transfer states into the luminescent motif,15–20 which help to realize efficient RTP, especially when these phosphors are restrained by rigid environments such as those resulting from polymerization,21–26 crystallization,27–33 matrix rigidification,34–43 host–guest complexation and supramolecular assembly.44–46 However, these n-electron groups accelerate radiative decay of triplet excitons; most of the RTP molecules featuring charge transfer tend to undergo a distinct structure transformation in the excited state transition process, thereby resulting in limited suppression of the non-radiative decay of triplet excitons. Thus, it is significant to develop a new strategy for efficient RTP enhancement in the presence of n-electron groups or charge transfer, which will provide new guidelines to design smart luminescent materials.Our recent studies have demonstrated a good phosphorescent precursor, phenylpyridinium.47,48 We anticipated that the macrocyclization of the phenylpyridinium units through methylenes would enhance the phosphorescence. Such phosphorescence enhancement is reasonable since the methylenes can restrict vibration and rotation of phosphorescent units by locking them into the skeleton of the macrocycle and therefore suppress nonradiative decay. Moreover, the unconjugated structure of methylene will not disturb the emission of phosphorescent units. Herein, we designed and synthesized pyridine/pyridinium-based macrocycles PC and PC2+, using our synthetic method for functional biphen[n]arenes.49,50 All these macrocycles exhibited prolonged phosphorescence lifetime and improved quantum efficiencies in comparison with their monomers, thus exhibiting macrocyclization-induced phosphorescence enhancement (MIPE). Single crystal X-ray diffraction analysis and photophysical characterization revealed that this enormous enhancement is because of the restriction of the macrocyclic skeleton for the phosphorescent units which efficiently suppressed the radiative decay of the singlet state and triplet state, and nonradiative decay of the triplet state.
Results and discussion
First, we examined the synthesis of a pyridine-based macrocycle PC (Scheme 1). As the most commonly used reaction conditions for synthesizing functional biphen[n]arenes,51 a BF3·Et2O catalyst and dichloromethane (DCM) solvent were chosen to achieve the condensation of the monomer PM and paraformaldehyde, giving the cyclic product PC in 15% yield. PC was confirmed by 1H, 13C NMR, and HRMS (Fig. S1–S6†). The fairly low yield spurred us to screen out good reaction conditions by evaluating solvents, catalysts and the reaction time. As illustrated in Table S1,† chloroform (CHCl3) gave highest yield of 37% for BF3·Et2O and 1,2-dichloroethane (DCE) gave the highest yield of 65% for the trifluoromethanesulfonic acid (TfOH) catalyst after screening DCM, CHCl3, DCE and CH3CN solvents (entries 1–8). Then FeCl3, AlCl3, BF3·Et2O, and TfOH catalysts were studied in DCE and TfOH was proved to be the best (entries 3, 7, 9, and 10). Therefore, the optimized conditions (DCE as the solvent, TfOH as the catalyst, and at 25 °C) were utilized for large-scale preparation (Schemes S1 and S2†). Cationic macrocycle PC·2I was synthesized by methylation of PC with excess CH3I and recrystallization from EtOH. After the anion exchange of NH4PF6, tetrabutylammonium bromide (TBAB) and tetrabutylammonium chloride (TBAC), another two pyridinium macrocycles with counterions of Br− (PC·2Br) and Cl− (PC·2Cl) were successfully obtained (Scheme S3 and Fig. S7–S12†). Moreover, the monomers PM·X (PM·Cl and PM·Br) were synthesized using similar procedures (Scheme S4 and Fig. S13–S18†). Single crystal X-ray diffraction analysis revealed the box-like cavity structure of PC with a size of 6.2 Å × 7.0 Å (Fig. 1a, b and Table S2†). Dimethoxyphenyl paralleled with the opposite one and was perpendicular to the adjacent one. Surface electrostatic potential maps showed that this macrocycle possessed a relatively electronegative cavity (Fig. 1e). PC·2Cl has a similar box-like shape and size (6.3 Å × 7.0 Å) compared with PC (Fig. 1c, d and Table S3†). The 0.1 Å increase in size probably arises from the difference of torsion angles. As illustrated in Fig. 3b and S19b,† the torsion angles between central pyridine/pyridinium units and dimethoxyphenyl units are 47°, 49°, 51°, and 54°for PC·2Cl and 65°, 42°, 42°, and 65° for PC. The entire molecule exhibited an obviously more positive surface electrostatic potential than neutral PC (Fig. 1f).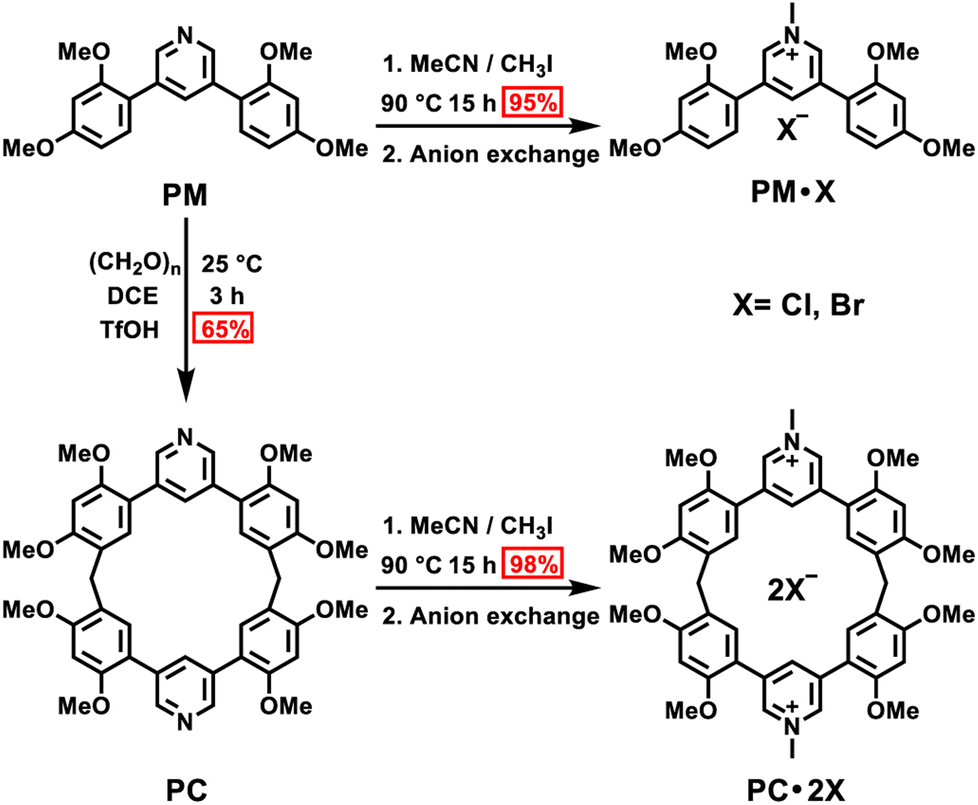 | ||
| Scheme 1 The synthetic route of monomer PM·X and macrocycles PC·2X. | ||
 | ||
| Fig. 1 Single-crystal structures of (a) and (b) PC and (c) and (d) PC·2Cl (top view and side view) and electrostatic potential maps of (e) PC and (f) PC·2Cl (top view and side view). Note: red region represents a low potential area with the characterization of an abundance of electrons. Blue region represents a high potential area with the characterization of a relative absence of electrons. C, marine blue or green; O, red; N, blue; H, white. Counter anions of Cl− are omitted for clarity. | ||
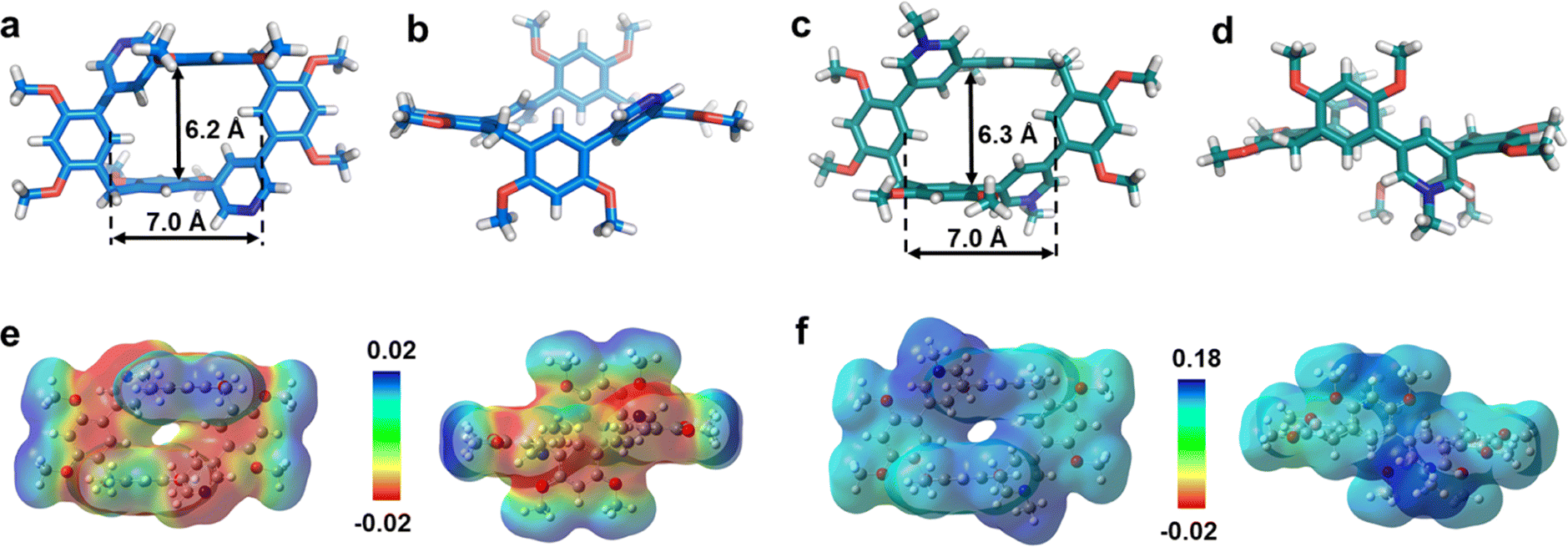
The photoluminescence (PL) spectra, phosphorescence spectra and time-resolved spectra were measured to illustrate the effect of macrocyclization on photophysical properties at room temperature. Both monomer PM·Cl and macrocycle PC·2Cl exhibited a cyan emission peak at 470 nm with nanosecond-scale lifetimes in the solid state (Fig. 2a, b and S20, S21†). The phosphorescence spectra revealed that PM·Cl displayed a maximal RTP peak around 500 nm with a lifetime of 0.29 ms, along with a shoulder peak at 565 nm. For the macrocycle PC·2Cl, it showed RTP bands at 518 nm with a lifetime of 16.7 ms (Fig. 2b–d). Significantly, compared with the monomer, macrocycle PC·2Cl showed a 58-fold improvement in the phosphorescence lifetime. Moreover, an enhanced phosphorescence intensity (3.7 fold) was observed in the phosphorescence spectra (Fig. 2b). Actually, PC·2Cl also exhibited higher RTP efficiency than PM·Cl (Fig. S22 and S23†). The above results indicated that the macrocyclization strategy is highly effective in promoting RTP.
 | ||
| Fig. 2 Photophysical properties of PC, and PC·2X in the solid state. (a) Normalized photoluminescence (solid black line) and phosphorescence spectra (dashed red line) of PC·2Cl under 350 nm excitation (inset: photograph of PC·2Cl under a 365 nm UV lamp). (b) Phosphorescence spectra of PM·Cl (deep red) and PC·2Cl (red) under 350 nm excitation. (c) Lifetimes of macrocycles (PC, PC·2Cl, and PC·2Br) and monomers (PM, PM·Cl, and PM·Br) in the solid state at room temperature. (d) Luminescence photographs of PC·2Cl and PC·2Br under 365 nm light at 77 K and at different time intervals after ceasing irradiation. | ||
Next, PC2+ with a bromide counterion and the neutral macrocycle PC were evaluated. PC·2Br (λmax = 481 nm and τF = 8.0 ns) displayed red-shifted emission and prolonged lifetimes compared to PM·Br (λmax = 470 nm and τF = 5.6 ns) (Fig. S24 and S25†). Compared with PM·Br, both phosphorescence lifetime and quantum efficiency of PC·2Br exhibited a prominent enhancement. The lifetime prolonged from 0.15 ms of PM·Br to 7.0 ms of PC·2Br, a 47-fold improvement (Fig. 2c). The phosphorescence intensity also showed 8.3-fold improvement (Fig. S24c†). Similarly, the phosphorescence efficiency improved from 5.9% of the monomer to 7.2% of the macrocycle (Fig. S26b and S27b†). Moreover, the neutral macrocycle PC also showed such MIPE. The monomer PM emitted blue fluorescence (λmax = 338 nm) with a lifetime of 0.85 ns (Fig. S28†). PC showed a red-shifted emission band at 355 nm with a lifetime of 0.91 ns, accompanied by a shoulder peak at 458 nm (Fig. S29†). The phosphorescence spectra gave a 2.6-fold improvement in intensity (Fig. S29c†). Time-resolved PL decay indicated a prolonged lifetime (10.9 ms for PC and 2.10 ms for PM) (Fig. 2c). This phosphorescence enhancement was also confirmed by improved quantum efficiencies of PC (3.3%) compared with those of PM (1.8%) (Fig. S30 and S31†). Furthermore, a green afterglow could be observed for both PC·2Cl and PC·2Br after ceasing the UV irradiation at 77 K (Fig. 2d). Phosphorescence spectra also showed enormous enhancements as the temperature decreased from 298 K to 77 K (Fig. S32a and b†). Moreover, their lifetimes prolonged from 16.7 ms of PC·2Cl and 7.00 ms of PC·2Br to 241 ms and 51.2 ms, respectively, proving that lowering temperature is beneficial for the phosphorescence intensity and lifetime, which is a typical characteristic of phosphorescent compounds (Fig. S32c and d†). These remarkable enhancements of phosphorescence for all macrocycles (PC, PC·2Cl, and PC·2Br) proved the universality of the MIPE strategy.
On the basis of the obtained photophysical properties, the radiative and nonradiative decay rate constants were calculated following the standard methods (Table 1).52,53 According to Kasha's rule, an excited molecule usually deactivates to the lowest excited singlet S1. Then S1 undergoes three competing decay processes: decay to the ground state S0via a nonradiative process with a rate constant kFnr = (1 − ΦF − ΦP)/τF; radiative decay to the S0 by emitting fluorescence with a rate constant kFr = ΦF/τF; conversion to the triplet state (usually is Tn and then fast decay to the lowest triplet state T1) via intersystem crossing with a rate constant kisc = ΦP/τF. T1 can decay to S0via a radiative process by emitting phosphorescence with a rate constant kPr = ΦP/τP or a nonradiative process with a rate constant kPnr = (1 − ΦP)/τP.
| Entry | Compound | λ F (nm) | λ P (nm) | τ F (ns) | τ P (ms) | Φ F (%) | Φ P (%) | k Fr (s−1) | k Fnr (s−1) | k isc (s−1) | k Pr (s−1) | k Pnr (s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The radiative decay rate constant of fluorescence kFr = ΦF/τF. b The singlet state nonradiative decay rate constant of fluorescence kFnr = (1 − ΦF − ΦP)/τF. c The intersystem crossing rate constant kisc = ΦP/τF. d The radiative decay rate constant of phosphorescence kPr = ΦP/τP. e The triplet state nonradiative decay rate constant kPnr = (1 − ΦP)/τP. | ||||||||||||
| 1 | PM | 338 | 495 | 0.85 | 2.10 | 6.1 | 1.8 | 7.2 × 107 | 1.1 × 109 | 2.1 × 107 | 8.6 | 468 |
| 2 | PC | 355 | 495 | 0.91 | 10.9 | 3.5 | 3.3 | 3.8 × 107 | 1.0 × 109 | 3.6 × 107 | 3.0 | 89 |
| 3 | PM·Cl | 470 | 500 | 14.1 | 0.29 | 10.4 | 17.3 | 7.4 × 106 | 5.1 × 107 | 1.2 × 107 | 597 | 2.8 × 103 |
| 4 | PC·2Cl | 470 | 518 | 9.45 | 16.7 | 14.6 | 18.6 | 1.5 × 107 | 7.1 × 107 | 2.0 × 107 | 11 | 49 |
| 5 | PM·Br | 470 | 503 | 5.6 | 0.16 | 3.9 | 5.9 | 7.0 × 106 | 1.6 × 108 | 1.0 × 107 | 369 | 5.9 × 103 |
| 6 | PC·2Br | 481 | 522 | 8.0 | 7.00 | 4.2 | 7.2 | 5.2 × 106 | 1.1 × 108 | 9.0 × 106 | 10 | 133 |
The intersystem crossing rate constant is kisc = (0.9–3.6) × 107 s−1, which guaranteed the production of sufficient triplet state population. The macrocycles stabilized triplet states and lowered the decay rates of both radiative and nonradiative processes. They jointly provided two indispensable conditions for phosphorescence. The radiative decay rate constant of the triplet state was reduced to kPr = 11 s−1 for PC·2Cl, about 54-fold lower than that of monomer PM·Cl (kPr = 597 s−1). Such reduction was also found for PC·2Br (kPr = 10 s−1) and PC (kPr = 3.0 s−1). Moreover, the nonradiative decay rate constants of the macrocycles (kPnr = 49 s−1 for PC·2Cl kPnr = 133 s−1 for PC·2Br, and kPnr = 89 s−1 for PC) were also much smaller than that of the monomers (kPnr = 2.8 × 103 s−1 for PM·Cl, kPnr = 5.9 × 103 s−1 for PM·Br, and kPnr = 468 s−1 for PM). According to eqn (1) in Fig. 3g, smaller (kPr + kPnr) should result in a longer lifetime. These results confirmed that the macrocyclization slowed down the decay of triplet states and favored long phosphorescence lifetime. Moreover, the radiative decay rate constants (kFr) of the singlet state were also slightly slowed down after macrocyclization which was beneficial for the ISC and hence probably promoted the phosphorescence enhancement. Eqn (2) indicated that quantum efficiency of phosphorescence ΦP was dependent on Φisc and kPnr/kPr. Therefore, the decreased kPnr/kPr (from 4.7 of PM·Cl to 4.5 of PC·2Cl, 16 of PM·Br to 13 pf PC·2Br, and 54 of PM to 30 of PC) means increased (kPr)/(kPr + kPnr). The improved ΦP makes it impossible to confirm the change (increase or decrease) of the Φisc (Table 1).
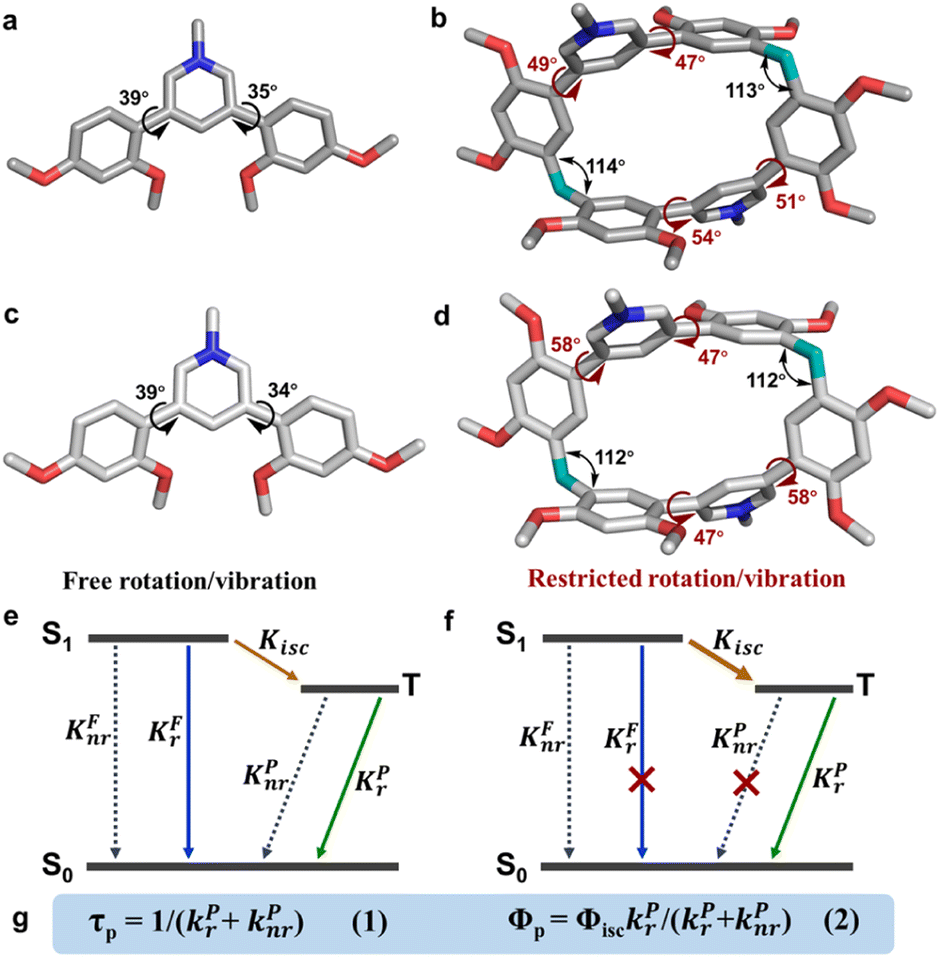 | ||
| Fig. 3 Mechanism of the macrocyclization strategy for phosphorescence enhancement. The single crystal structures of the monomers (a) PM·Cl and (c) PM·Br and the macrocycles (b) PC·2Cl and (d) PC·2Br. All chloride and bromide ions and hydrogen atoms are omitted for clarity. (e) and (f) Simplified Jablonski diagram of monomers and macrocycles. (g) The equation of τP and ΦP. | ||
Single-crystal X-ray diffraction analysis revealed atomic-level structures for further understanding of such phosphorescence enhancement (Tables S2–S7 and Fig. S33†). As shown in Fig. 3a and b, the torsion angles between pyridinium and dimethoxyphenyls in PM·Cl are 39° and 35°, respectively. Macrocycle PC·2Cl, made up of two PM·Cl and two methylene linkers, has larger torsion angles of 47°, 49°, 51° and 54°. For PC·2Br, the torsion angles are 47° and 58°, larger than those of monomer PM·Br (39° and 34°) (Fig. 3c and d). The increased torsion angles were also found in PC (41° and 52°) and PM (42° and 65°) (Fig. S19†). Because of the definite orientation of methylene, this more twisted structure is the result of structural adjustment after macrocyclization. Rotation of dimethoxyphenyls was prohibited and the vibration was also suppressed because of the restriction of the macrocycle skeleton. As a result, the nonradiative decay processes were greatly restrained and the radiative decay process was promoted. According to the Jablonski diagram and eqn (1) and (2) in Fig. 3e–g, phosphorescence lifetime τP = 1/(kPr + kPnr) and quantum yield ΦP = ΦisckPr/(kPr + kPnr), the promoted radiative process and decreased (kPr + kPnr) boosted the prolonging of τP and improvement of ΦP and hence phosphorescence enhancement was realized. Furthermore, to thoroughly eliminate the influence of noncovalent interactions (π⋯π, C–H⋯π interaction, hydrogen bonding, etc.) in crystals, the photoluminescence spectra of macrocycle PC·2Cl and monomer PM·Cl in aqueous solution were measured. These noncovalent interactions in the solid state would be completely destroyed by solvation in water and hence reflect the conformation in a free state. Moreover, macrocycle PC·2Cl possessed two monomers of PM·Cl and the photoluminescent spectra of PC·2Cl in a 1 × 10−6 mol L−1 solution should be the same as those of PM·Cl in a 2 × 10−6 mol L−1 solution if the nonconjugated methylene had no influence on the luminescent units. However, a 4-fold enhancement was observed and proved that the macrocyclization promoted the emission even in solution and the restriction is ever present (Fig. S34†).
To evaluate the intermolecular aggregation, concentration-dependent UV-vis absorption spectra and photoluminescence spectra in an aqueous solution were measured (Fig. S35†). Both the macrocycle and monomer have identical normalized UV-vis absorption spectra, indicating that there is no aggregation which was also confirmed by the identical normalized photoluminescence spectra of PC·2Cl with the concentration changed from 1 × 10−6 to 5 × 10−5 mol L−1. We further evaluated their packing mode in the solid state. Monomers PM·Cl and PM·Br mutually stacked with a typical π–π interaction of 3.56 Å, hinted the H aggregates (Fig. S36 and S37†). However, macrocycles PC·2Cl and PC·2Br stacked to form the tubular superstructures without π–π interactions (Fig. S38 and S39†). Therefore, there are no apparent aggregate modes.54–57
Experimental
All reagents and solvents were commercially available and used without further purification, unless otherwise noted. 1H NMR and 13C NMR spectra were recorded using a Bruker Avance 500 MHz spectrometer. High-resolution mass spectra (HRMS) was recorded on an SCIEX, X-500R QTOF instrument. Photoluminescence spectra and lifetime were obtained on an FLS900 and FLS1000. Fluorescence and phosphorescence quantum efficiencies were measured on a HAMAMATSU C9920-02. Melting points were obtained on an X-4 digital melting point apparatus. Single crystal X-ray diffraction data were collected on a Bruker Smart Apex 2 and Bruker D8 Venture. The electrostatic potential maps of PC and PC·2Cl were obtained by using the Gaussian 09 program with B3LYP-D3(BJ)/6-31G+(d,p) and B3LYP-D3(BJ)/6-31G++(d,p) levels, respectively.Conclusions
In conclusion, we present an effective phosphorescence enhancement strategy of macrocyclization based on pyridine/pyridinium macrocycles. A dimeric pyridine macrocycle was synthesized in 65% yield by the condensation of the 3,5-bis(2,4-dimethoxyphenyl)pyridine monomer and paraformaldehyde under the catalysis of TfOH in DCE. Two pyridinium macrocycles with different counterions were prepared by the methylation of the pyridine macrocycle and subsequent ion exchange in high yields. Significantly, all of them show enhanced phosphorescence compared with monomers. The phosphorescence lifetime increased 59-fold and quantum efficiency showed a 3.7-fold increase after macrocyclization. The mechanism study revealed that the macrocyclic skeleton locked the luminescent units by prohibited rotation and suppressed vibration and therefore suppressed nonradiative decay. Furthermore, the radiative process of the triplet state was also slowed down which favored the improvement of lifetime. This result provided a new strategy and typical examples for realizing organic room temperature phosphorescence with high performance and may promote the development of phosphorescence materials. Considering the universality of this phosphorescence enhancement strategy, more macrocycle-based phosphorescent materials with longer lifetime and higher efficiency will be prepared in the near future.Author contributions
Chunju Li and Zhi-Yuan Zhang provided facilities and funding and supervised the overall research work. Shuo Li and Zhi-Yuan Zhang designed and conducted experiments, analysis and characterization. Jing-Fang Lv and Ling Li performed mechanism analysis and discussion. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (21971192, 21772118, and 22201211), the Natural Science Foundation of Tianjin City (20JCZDJC00200), and Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University.Notes and references
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed.
- X. Zhen, C. Xie and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3938 CrossRef CAS PubMed.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- W.-L. Zhou, Y. Chen, Q. Yu, H. Zhang, Z.-X. Liu, X.-Y. Dai, J.-J. Li and Y. Liu, Nat. Commun., 2020, 11, 4655 CrossRef CAS PubMed.
- M. Cui, P. Dai, J. Ding, M. Li, R. Sun, X. Jiang, M. Wu, X. Pang, M. Liu, Q. Zhao, B. Song and Y. He, Angew. Chem., Int. Ed., 2022, 61, e202200172 CAS.
- S. Hirata, K. Totani, T. Yamashita, C. Adachi and M. Vacha, Nat. Mater., 2014, 13, 938 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655 CrossRef CAS PubMed.
- X. Wu, C.-Y. Huang, D.-G. Chen, D. Liu, C. Wu, K.-J. Chou, B. Zhang, Y. Wang, Y. Liu, E. Y. Li, W. Zhu and P.-T. Chou, Nat. Commun., 2020, 11, 2145 CrossRef CAS PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Chem. Mater., 2017, 29, 4866 CrossRef CAS.
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2018, 57, 6216 CrossRef CAS PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406 CrossRef CAS.
- A. Abdollahi, H. Roghani-Mamaqani, B. Razavi and M. Salami-Kalajahi, ACS Nano, 2020, 14, 14417 CrossRef CAS PubMed.
- J. Tan, Q. Li, S. Meng, Y. Li, J. Yang, Y. Ye, Z. Tang, S. Qu and X. Ren, Adv. Mater., 2021, 33, 2006781 CrossRef CAS PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384 CrossRef CAS PubMed.
- P. Alam, N. L. C. Leung, J. Liu, T. S. Cheung, X. Zhang, Z. He, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, C. C. S. Chan, K. S. Wong, Q. Peng and B. Z. Tang, Adv. Mater., 2020, 32, 2001026 CrossRef CAS PubMed.
- P. Alam, T. S. Cheung, N. L. C. Leung, J. Zhang, J. Guo, L. Du, R. T. K. Kwok, J. W. Y. Lam and Z. Zeng, J. Am. Chem. Soc., 2022, 144, 3050 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451 CrossRef CAS PubMed.
- G. Farias, C. A. M. Salla, M. Aydemir, L. Sturm, P. Dechambenoit, F. Durola, B. d. Souza, H. Bock, A. P. Monkman and I. H. Bechtold, Chem. Sci., 2021, 12, 15116 RSC.
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CAS.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 10854 CrossRef CAS PubMed.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed.
- X. Lin, J. Wang, B. Ding, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 3459 CrossRef CAS PubMed.
- S. Cai, Z. Sun, H. Wang, X. Yao, H. Ma, W. Jia, S. Wang, Z. Li, H. Shi, Z. An, Y. Ishida, T. Aida and W. Huang, J. Am. Chem. Soc., 2021, 143, 16256 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090 CrossRef CAS.
- Y. Gong, Y. Tan, H. Li, Y. Zhang, W. Yuan, Y. Zhang, J. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1183 CrossRef CAS.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 CrossRef CAS PubMed.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728 CrossRef CAS PubMed.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962 CrossRef CAS PubMed.
- J.-X. Wang, Y.-G. Fang, C.-X. Li, L.-Y. Niu, W.-H. Fang, G. Cui and Q.-Z. Yang, Angew. Chem., Int. Ed., 2020, 59, 10032 CrossRef CAS PubMed.
- B. Roy, I. Maisuls, J. Zhang, F. C. Niemeyer, F. Rizzo, C. Wölper, C. G. Daniliuc, B. Z. Tang, C. A. Strassert and J. Voskuhl, Angew. Chem., Int. Ed., 2022, 61, 202111805 Search PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081 RSC.
- R. Gao and D. Yan, Chem. Sci., 2017, 8, 590 RSC.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393 CrossRef CAS PubMed.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054 CrossRef CAS PubMed.
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970 CrossRef CAS PubMed.
- Z.-A. Yan, X. Lin, S. Sun, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 19735 CrossRef CAS PubMed.
- Z. Xie, X. Zhang, H. Wang, C. Huang, H. Sun, M. Dong, L. Ji, Z. An, T. Yu and W. Huang, Nat. Commun., 2021, 12, 3522 CrossRef CAS PubMed.
- Y. Zhang, L. Gao, X. Zheng, Z. Wang, C. Yang, H. Tang, L. Qu, Y. Li and Y. Zhao, Nat. Commun., 2021, 12, 2297 CrossRef CAS PubMed.
- X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Small, 2022, 18, 2104073 CrossRef CAS PubMed.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738 CrossRef CAS PubMed.
- X.-K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed.
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720 CrossRef CAS PubMed.
- Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028 CrossRef CAS PubMed.
- Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773 RSC.
- Z.-Y. Zhang and C. Li, Acc. Chem. Res., 2022, 55, 916 CrossRef CAS PubMed.
- S. Li, K. Liu, X.-C. Feng, Z.-X. Li, Z.-Y. Zhang, B. Wang, M. Li, Y.-L. Bai, L. Cui and C. Li, Nat. Commun., 2022, 13, 2850 CrossRef CAS PubMed.
- K. Xu, Z.-Y. Zhang, C. Yu, B. Wang, M. Dong, X. Zeng, R. Gou, L. Cui and C. Li, Angew. Chem., Int. Ed., 2020, 59, 7214 CrossRef CAS PubMed.
- C. C. Byeon, M. M. McKerns, W. Sun, T. M. Nordlund, C. M. Lawson and G. M. Gray, Appl. Phys. Lett., 2004, 84, 5174 CrossRef CAS.
- P. C. Y. Chow, S. Albert-Seifried, S. Gélinas and R. H. Friend, Adv. Mater., 2014, 26, 4851 CrossRef CAS PubMed.
- J. Vollbrecht, New J. Chem., 2018, 42, 11249–11254 RSC.
- S. Ma, S. Du, G. Pan, S. Dai, B. Xu and W. Tian, Aggregate, 2021, 2, e96 CAS.
- A. Eisfeld and J. S. Briggs, Chem. Phys., 2006, 324, 376–384 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2169350–2169354 and 2065557. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ta09993d |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |