
Photocatalytic hydrogen production: an overview of new advances in structural tuning strategies
Fenghua
Xu
and
Baicheng
Weng
 *
*
Department of Chemistry and Chemical Engineering, Central South University, Changsha, Hunan Province 410083, China. E-mail: bcweng@mail.csu.edu.cn
First published on 30th January 2023
Abstract
Hydrogen production from photocatalytic water decomposition provides a promising approach to harvest solar energy for addressing the challenges of energy shortage and environmental pollution. To meet the commercialization target set by the US Department of Energy, three great challenges of photocatalytic hydrogen production should be well addressed: the capture of solar light, the separation/transfer of photo-excited charge pairs and the surface redox reactions to produce useable fuels. The component, morphological and structural designs are highly promising solutions for solving these challenges. Among them, structural designs are more effective and easier; in particular, the incorporation of single atoms into the structure, defect control and formation of S-scheme heterojunctions has attracted considerable attention. The recent advancements in these topics are highlighted and discussed in this review.
 Fenghua Xu | Fenghua Xu joined Professor Baicheng Weng's group at Central South University as a lecturer after completing her postdoctoral work at the University of Texas-RGV, USA. She received her PhD from Shanghai Institute of Applied Physics, Chinese Academy of Sciences. Her scientific interest includes design and synthesis of nanocomposites and their application in catalysis and biosensing. |
 Baicheng Weng | Baicheng Weng is currently a professor at Central South University, China. He received his PhD from Shanghai Institute of Microsystem and information technology, Chinese Academy of Sciences, in 2011. His research interest concentrates on discovering and designing new materials for photocatalytic and electrocatalytic water splitting. |
Introduction
Energy shortage and environmental pollution are two major issues hindering the sustainability of modern civilization.1–3 The search for sustainable and green energy sources is extremely urgent and highly demandable. Among all the intensively studied alternative energy sources, such as wind and tidal power, solar energy is an energy source that can meet our long-term needs because of its advantages of zero cost and being abundant and inexhaustible.4–8 Recently, conversion of solar energy to useable fuels (e.g., hydrogen, ethane, and propanol) using semiconductor photocatalysts has attracted considerable attention,9–12 especially in the fields of solar-driven overall water splitting, photocatalytic CO2 reduction and photocatalytic degradation of pollutants.13–17 Among the useable fuels, hydrogen is a highly efficient energy source because of its ultra-high energy density and zero emission.18,19 Photocatalytic water decomposition provides a promising approach to harvest solar energy for generating environment-friendly hydrogen energy for our long-term needs.20–22 The hydrogen production cost from solar-driven water splitting should be limited below $2.0 kg−1 on the terawatt scale to compete with the traditional hydrogen production methods, such as coal gasification, in the global market. To achieve this goal, analysis suggests a solar-to-hydrogen efficiency (STH) of 10% as a threshold for energy return on investment.23Photocatalytic processes involve three essential steps: the capture of solar light, the separation/transfer of photo-excited charge pairs, and the surface redox reactions to produce useable fuels.23–26 To reach such STH, challenges in the photocatalytic processes should be addressed. Additionally, a photocatalyst should also have decent stability for practical usage. Unfortunately, none of the existing single photocatalysts show decent efficiency in all requirements. For instance, TiO2 has a band gap of 3.2 eV, which means that it can only absorb parts of ultra-violet (UV) light. UV light only accounts for less than 5% of the total solar energy, while visible light and infrared light contribute 43% and 52% of solar energy, respectively.27–29 A good photocatalyst, therefore, should show decent light absorption in the visible light and near infrared light regions. Another example is Fe2O3 that has a bandgap of 2.1 eV and can absorb visible light up to 590 nm. Unfortunately, it suffers from the fast recombination of photoexcited charge pairs and the average charge carrier migration length is less 10 nm.30
To improve the solar to hydrogen efficiency, many strategies have been proposed. Among them, the search for new materials is the most effective way to improve the STH. Solar-driven hydrogen production began with the observation of water photooxidation on semiconducting TiO2; its limited STH efficiency, however, is only 1.3% due to a wide bandgap of 3.2 eV.31 The investigations on semiconducting transition metal oxides with empty d bands (i.e., V5+, Nb5+ and W6+) show that their valence-band energy levels are dictated strongly by the O 2p levels, making their valence band maximum (VBM) higher than that of TiO2 but still thermodynamically capable for water oxidation. In addition, incorporating s2 cations (e.g., Sn2+ and Bi3+) can narrow the band gap through a coupling between s and O 2p bands that pushes the VBM upward and another coupling between the empty d bands and the p bands of the s2 cations that pushes the conduction band minimum (CBM) downward.32 For instance, our previous work demonstrated that the composition tunning of Bi element, which has both d0 band (Bi5+) and 6s2 cations(Bi3+), in a Ba2Bi1+xNb1−xO6-type double perovskite oxide can greatly shift down the CBM and shift up the VBM and therefore improve the light absorption.9,10 Novel photocatalysts, such as BiVO4, WO3 and Fe2O3, which possess much higher theoretical STH, have been proposed. For instance, the theoretical STH of BiVO4 approaches 9.2% with a photocurrent of 7.5 mA cm−2 under the standard AM1.5G solar light illumination.33 Accordingly, these materials have garnered increasing attention for decades. Unfortunately, high ionic character of these oxides induces a large separation between the band edges, limiting efficient solar energy harvesting especially in visible and near infrared light regions. Until now, no single semiconductor can achieve the targeted STH because of tremendous difficulties in searching for new materials that can fulfill all three requirements. The most promising materials, therefore, are still the semiconductors that are intensively studied for decades.
Another approach is the structural tuning strategy, which includes the modification (e.g., heteroatom doping and defect control) of existing semiconductors and the fabrication of tandem devices for taking full advantage of synergistic effects among varied semiconductors. Also, the latter has already shown high potential for practical applications with high STH. For instance, in 2017, a photoelectrochemical (PEC) STH of 16.2% was achieved using a Si/GaInAs/GaInP tandem device with a newly found semiconductor, 1.8 eV-bandgap p-type GaInP.23 In 2018, the efficiency was improved to 19% using a tandem cell comprising TiO2, AlInPOx, AlInP, GaInP, GaInAs and GaAs, using the same AM1.5 light source.34 Most recently, the Mi group reported a STH over 9.2% for photocatalytic hydrogen production from pure water and concentrated solar light using InGaN/GaN nanowires as the photocatalysts.35 Besides the three requirements for high conversion efficiency mentioned above, the device must be composed of widely available and low-cost component atoms, be inexpensive to be fabricated and be robust enough for long-term operation. Although these devices manifest high STH, their dramatically high fabrication cost is the greater challenge for hydrogen production cost control below $2.0 kg−1.
Moreover, the structural tuning strategy is much easier than developing new materials and is more effective than other strategies such as morphology control. For instance, heteroatom doping demonstrates the most effective way to improve the performance of a semiconductor. Doping Mo6+ or W6+ into the V5+ sites of BiVO4 increases the electronic conductivity and thus carrier collection efficiencies.36–39 Incorporation of Cu into WO3 induces strong Cu 3d character in the valence band, which raises the VBM and narrows the bandgap.40,41 The most widely used doping elements are “N” and “S/Se” because the substitution of O by N and S/Se will decrease the bandgap by introducing higher-lying N 2p states and S/Se p-band energy levels, respectively.42–46 Our recent work shows that the incorporation of S into the perovskite oxide can push the CBM down by S 2p orbital coupling, which pushes the light absorption edge to the near infrared light region. The bandgap tuning of perovskite oxides increases the light absorption but is not at the expense of losing stability due to the natural chemical stability of oxides.47 Moreover, the fabrication of heterojunction or tandem structures is also one of the effective structural tuning methods. For PEC water splitting, tandem structures already demonstrate the capability for achieving high STH as mentioned above,23,34,35 while for photocatalytic hydrogen production, a heterojunction formed by the direct contact of two semiconductors is widely used to improve the efficiency of the separation of charge pairs. Three traditional type heterojunctions, i.e., straddling gap, staggered gap, and broken gap, have been intensively studied for decades.48–50
It is well-known that the small size of metal catalysts is beneficial to the improvement of metal utilization efficiency, which is particularly important for noble metal components. Classic metal nanoparticles (or nanocrystals) have low metal utilization efficiency due to the existence of abundant bulk atoms. Contrastively, single-atom catalysts (SACs) with a theoretical metal utilization rate of 100% and subnanometer nanoclusters with nearly 100% atom efficiency for specified reactions have received increasing attention in recent years.51 SACs contain isolated metal atoms or mononuclear metal complexes on appropriate supports, while a nanocluster with well-defined structures in the size regime of a few sub-nanometers to several nanometers consists of tens to hundreds of metal atoms.52,53 Both SACs and nanoclusters all have the following advantages over their nanoparticle counterparts: (1) low-coordination environment; (2) quantum size effects; (3) metal–support interactions, originating from the chemical bonding effect and the charge transfer between metal species and supports.51 Remarkably, compared to nanoclusters, SACs have exclusively higher activity and selectivity brought by their unsaturated coordination sites and unique electronic structures. In addition, embedding SACs into transition metal oxides has been proved to be an effective strategy not only to minimize the loading amount of precious metals but also to improve the intrinsic performance of catalysts.54 For example, Sun et al. dispersed Ir atoms into the WO3 matrix lattice via a two-step method, which exhibited improved activity and durability for acidic overall water electrolysis.55 In this case, the valence of an isolated metal single atom should not be necessarily zero to boost the metal–support synergistic effects. More importantly, the appropriate supports can be in the form of clusters, trimers, dimers, monomers, and even nanoclusters. SACs, therefore, could be the ultimate size limit for nanoclusters and broaden the scope of nanoclusters.
To address the irreconcilable conflict between a wide light absorption range and high redox ability in single semiconductors, people have never stopped the studies on heterojunctions since 1984.56 With the continuous deep understanding on the light absorption and charge transfer in heterojunctions, fine control of the heterojunction structure became more necessary. Recent calculations on PEC tandem devices indicate that theoretical STH can reach 21%, 24% and 27% for a junction with top/bottom bandgaps of 1.7/1.2, 1.8/1.2 and 1.7/1.05 eV, respectively.23 For a photocatalytic hydrogen production heterojunction, the interpretation of the charge-transfer route is controversial. Recently, the Yu group raised a different charge transfer route (S-scheme heterojunction) compared to the widely acknowledged type-II and Z-scheme heterojunctions.57
Given that traditional three type heterojunctions and heteroatom doping have been intensively studied and reviewed previously, in this review, we will brief the fundamentals of structural tuning strategies of the newly emerged approaches, i.e., singe atom photocatalysts, defect control and S-scheme heterojunctions. We present an analysis on their advantages for photocatalytic hydrogen production and mainly focus on how these strategies address the three challenges mentioned above in photocatalytic processes. Since no single attempt can solve all challenges, in the final section, we discussed the limitations of each approach, and the future research direction was also discussed.
Structural tunning approaches
Single atom metal photocatalysts
To address the issues of rapid recombination of photo-generated electron–hole pairs and insufficient surface active sites on the photocatalyst, SACs have been employed and become a hot topic in water-splitting photocatalysts because of their unique advantages, such as the highest atom utilization rate and excellent catalytic activity.58–66 Single atoms (SAs) can be incorporated into the crystal lattice of the photocatalyst substrate or in the form of isolated metal atoms or mononuclear metal complexes on varied substrates. Since Zhang et al. firstly reported single Pt atoms on the FeOx carrier with a Pt–O–Fe structure in 2011,67 most atoms in the periodic table can be synthesized as SA materials. Therefore, the versatile nature of SAs and the diversity of photocatalyst substrates can enable the flexible engineering of both electronic structures and crystal structures, thereby facilitating the charge separation, narrowing the bandgap, and increasing the surface reaction kinetics. The material obtained by this method not only has the advantages of a homogeneous catalyst, including the uniformity of active sites and the adjustable interaction with ligands, but also inherits the high durability and excellent recoverability of a heterogeneous catalyst.68–75 Yang's group prepared single metal atoms (Pt, Pd, Rh, and Ru) on TiO2 for the first time,68 and the catalyst showed higher photocatalytic activity than the metal clusters supported on TiO2 for hydrogen production. This work opened a door for the application of SACs in photocatalysis. The introduction of SACs can not only effectively improve the light absorption capacity, broaden the light collection range, and improve the efficiency of charge separation/transfer, but also maximize the utilization rate of atoms and reduce the use of catalytic metals due to the unique exposure mode. They exhibit remarkable stability and long service life, which has been verified in many photocatalytic reactions.76–81 Since then, SACs have attracted wide attention and developed rapidly in the field of photocatalysis.Noble metal SAs
SAs can not only accept photo-generated electrons and promote the separation and transfer of interface charges, but also provide proton reduction sites.82 Besides that, noble metal single atoms or nanoparticles are also active photo-absorbers, such as Au. The hot electron flow between noble metal single atoms and the substrate greatly promotes the charge transfer and surface reactions. There are eight kinds of noble metals, which are used in the field of water decomposition photocatalysis. Herein, we will discuss the widely studied four kinds of them, i.e., Pt,83–86 Rh,87 Ru,88 and Ag.89,90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 423 h−1, under UV-visible light illumination (300 W Xe lamp). Hu et al. (Fig. 1) also investigated Pt SA loaded defective TiO2 using TiO2 nanosheets as the substrate.97 Their computational analysis treated Pt SAs as homogeneous catalysts rather than heterogenous catalysts in Chen's work. They found that defective TiO2 nanosheets could effectively stabilize the single-atomic Pt sites through constructing a three-center Ti–Pt–Ti structure. More importantly, the Ti–Pt–Ti structure can hold the stability of isolated single-atomic Pt sites and facilitate the separation and transfer of photoinduced charge carriers, resulting in improvements in photocatalytic H2 evolution. The photocatalyst delivers a hydrogen production rate of 13460.7l mol h−1 g−1 under UV-visible light illumination (300 W xenon lamp).
423 h−1, under UV-visible light illumination (300 W Xe lamp). Hu et al. (Fig. 1) also investigated Pt SA loaded defective TiO2 using TiO2 nanosheets as the substrate.97 Their computational analysis treated Pt SAs as homogeneous catalysts rather than heterogenous catalysts in Chen's work. They found that defective TiO2 nanosheets could effectively stabilize the single-atomic Pt sites through constructing a three-center Ti–Pt–Ti structure. More importantly, the Ti–Pt–Ti structure can hold the stability of isolated single-atomic Pt sites and facilitate the separation and transfer of photoinduced charge carriers, resulting in improvements in photocatalytic H2 evolution. The photocatalyst delivers a hydrogen production rate of 13460.7l mol h−1 g−1 under UV-visible light illumination (300 W xenon lamp).
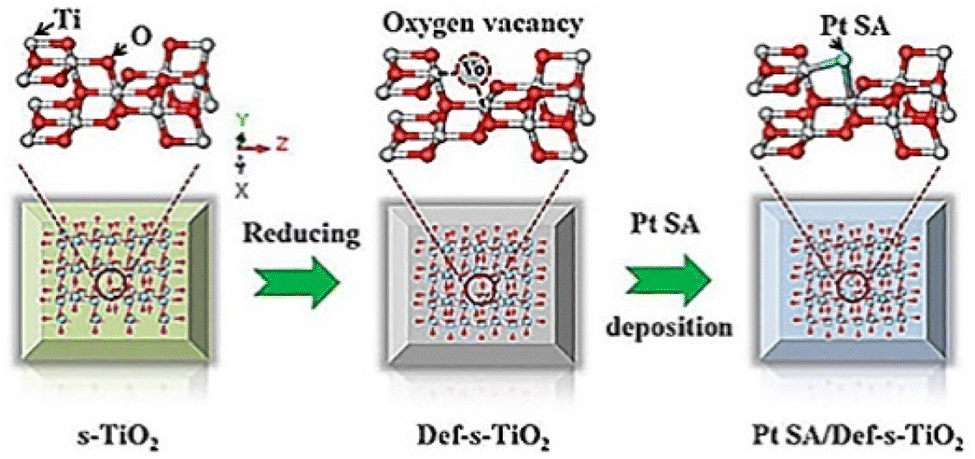 | ||
| Fig. 1 The synthesis strategy for the Pt SA/Def-s-TiO2 photocatalyst. Copyright 2021, Elsevier. | ||
Polymeric graphitic carbon nitride (g-C3N4) has attracted extensive attention in recent years because of its high chemical stability and attractive electronic structure. Li et al. took Pt SAs and g-C3N4 as an example to demonstrate that metal–ligand charge transfer could adjust the light absorption and improve the carrier utilization rate,98 resulting from strong interaction between the d0 orbitals of Pt and N-forming π-conjugated graphitic planes (Fig. 2). Li et al. synthesized a new single Pt atom co-catalyst by anchoring Pt SAs on g-C3N4 with high dispersion and stability.99 When the Pt loading amount reached 0.38%, Pt atoms dispersed and formed sub-nano clusters. The isolated Pt SAs induced the intrinsic change of the surface trap state, which prolonged the lifetime of photo-generated electrons.
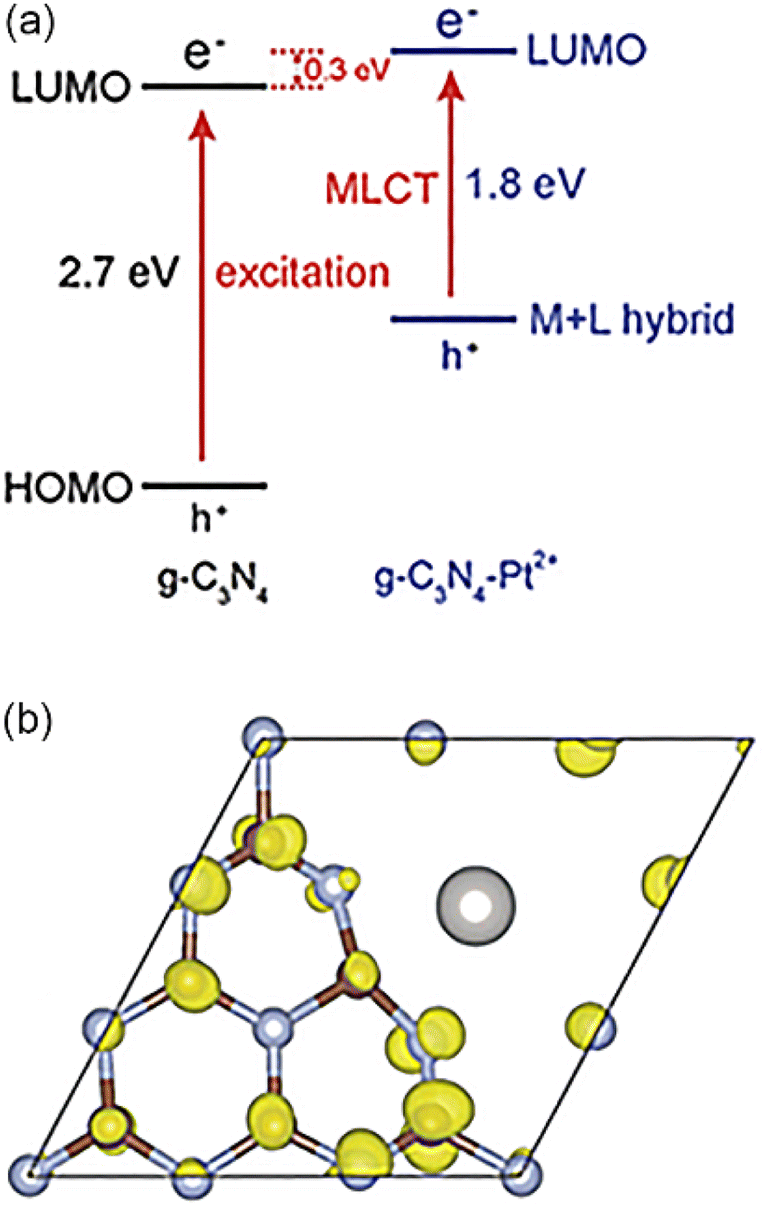 | ||
| Fig. 2 (a) Energy-band alignment diagrams for g-C3N4 and g-C3N4-Pt2+. (b) Photoexcited charge density transition from the Pt2+-induced hybrid highest occupied molecular orbital (HOMO) states to the lowest unoccupied molecular orbital (LUMO) of g-C3N4-Pt2+. Copyright 2016, Wiley-VCH. | ||
Instability of SAs is a great challenge preventing their massive applications, especially the aggregation issue, since unanchored SAs are too fluid.51 MOFs or covalent organic frameworks (COFs) are an important class of porous crystals with various customizable structures as well as functions and large surface area. They can provide the most effective coordination to anchor metal SAs, which mitigates the aggregation during the photocatalytic processes. In addition, the interaction between SAs and the MOF/COF substrate produces active sites and further regulates the light absorption capability. For instance, Zuo et al. prepared the first SACs based on 2-dimensional (2D) MOFs,100 as shown in Fig. 3. 12.0 wt% 2D monoatomic catalyst showed a photocatalytic hydrogen production rate of 11320 mol h−1 g−1 under visible light irradiation (λ > 420 nm).
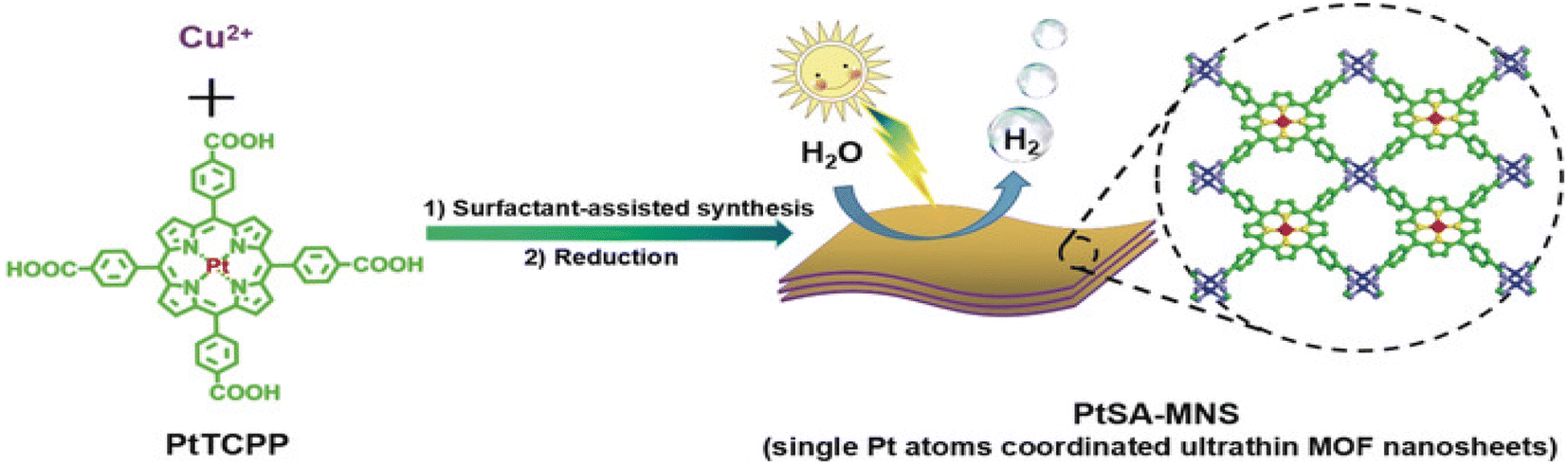 | ||
| Fig. 3 Illustration of the synthetic route towards Pt single-atom-coordinated ultrathin MOF nanosheets (PtSA-MNSs) for photocatalytic hydrogen production. Copyright 2019, Wiley-VCH. | ||
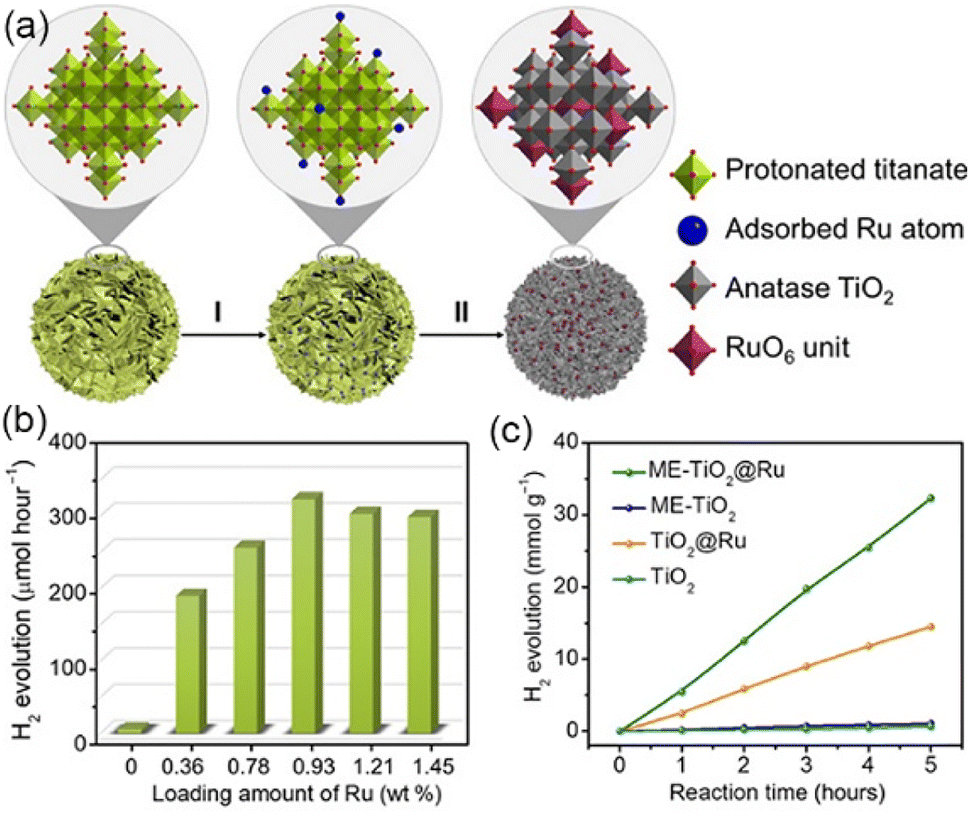 | ||
| Fig. 4 (a) Illustration of the synthetic route to Ru SAs in TiO2. (b) Photocatalytic H2 generation rates of ME-TiO2@Ru with different Ru loading amounts. (c) Time-dependent hydrogen evolution activities. Copyright 2020, the author, and American Association for the Advancement of Science. | ||
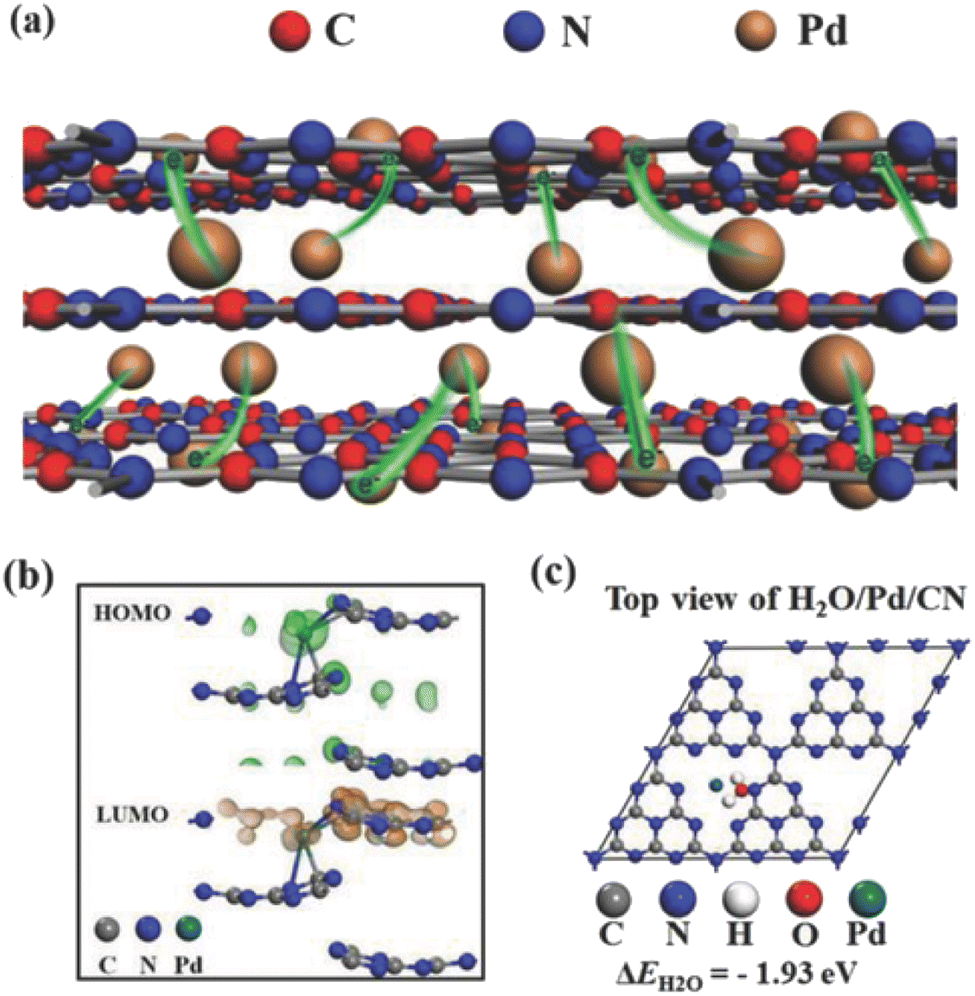 | ||
| Fig. 5 (a) Conceptual illustration of the Pd/g-CN hybrid with the interlayer intercalation and surface anchoring of Pd atoms. (b) The HOMO/LUMO calculation of Pd/g-CN. (c) The calculation of adsorption energy of a H2O molecule on the Pd/g-CN. Copyright 2018, Wiley-VCH. | ||
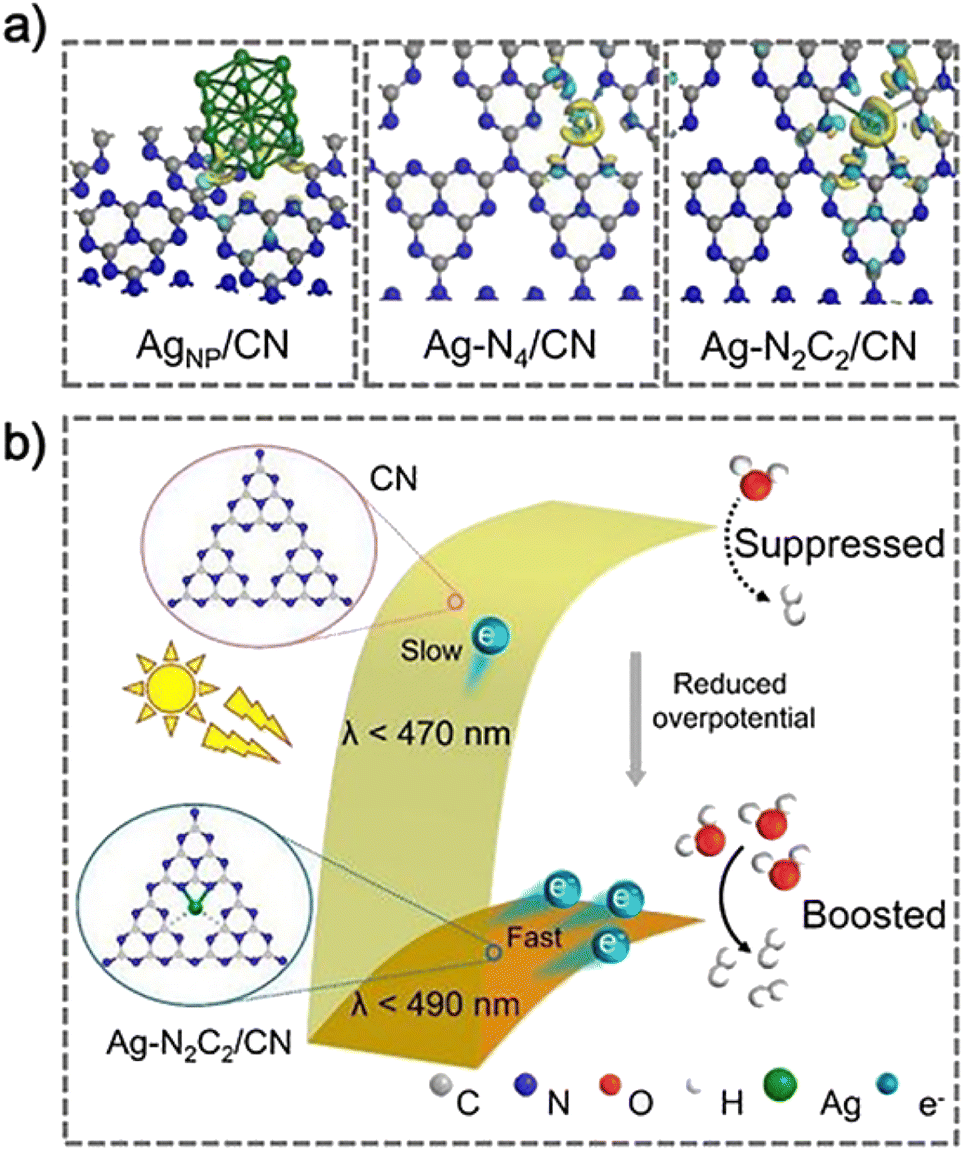 | ||
| Fig. 6 (a) Charge density difference maps, and (b) proposed mechanism for photocatalytic H2 evolution over CN and Ag–N2C2/CN. Copyright 2020, Wiley-VCH. | ||
Non-noble metal SAs
Although noble metals are widely used in the field of photocatalytic water decomposition, their scarcity and high price limit the large-scale application, so it is highly demanded to develop efficient and low-cost non-noble metal photocatalysts.105,106 The introduction of non-noble metal atoms to synthesize photocatalysts not only reduces the utilization rate of noble metals but also breaks the limit of activity noble metal atom sites.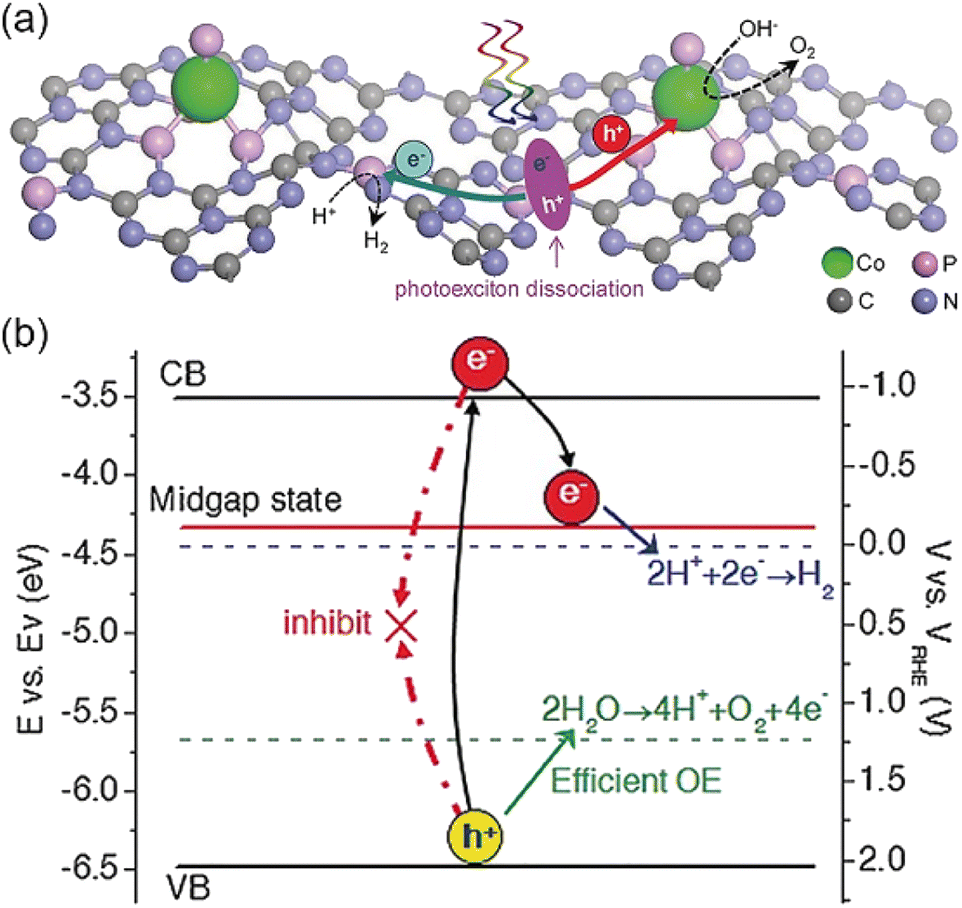 | ||
| Fig. 7 (a) Schematic illustration of the solar-driven overall water splitting on the Co1-phosphide/PCN photocatalyst, in which the photogenerated electrons and holes (h+) are spatially separated. (b) Electronic band structure diagram for the Co1-phosphide/PCN photocatalyst. Copyright 2017, Wiley-VCH. | ||
Aiming at mitigating the aggregation or sintering of SACs during the synthetic procedures and to achieve high loading amounts of SACs, Shi et al. developed a new in situ preparation strategy on carbon nitride,109 which could synthesize separated cobalt SAs with a high metal loading amount up to 5.9 wt%. Furthermore, due to the rapid electron transfer from carbon nitride to isolated Co SAs, the photocatalytic H2 production rate reached 1180 μmol g−1 h−1 using a light-emitting diode light source (12 W, λ = 420 ± 10 nm).
712 μmol h−1 g−1, which is 14.23-fold higher than that of pristine CdS. Ni can also be used as the cocatalyst to improve the hydrogen evolution performance. Su et al. atomically dispersed Ni in zinc sphalerite cadmium-zinc sulfide quantum dots (ZCS QDs).111 The photocatalyst delivered an efficient and durable photocatalytic performance for water splitting under visible light irradiation (λ > 420 nm) (Fig. 8). The finely tuned Ni SAs dispersed in ZCS QDs exhibit a photocatalytic H2 production activity of 18.87 mmol h−1 g−1. The improvements could be attributed to the favorable surface engineering to achieve highly active sites of monovalent Ni(I) and the surface heterojunctions to reinforce the carrier separation owing to the suitable energy band structures, built-in electric field, and optimized surface H2 adsorption thermodynamics.
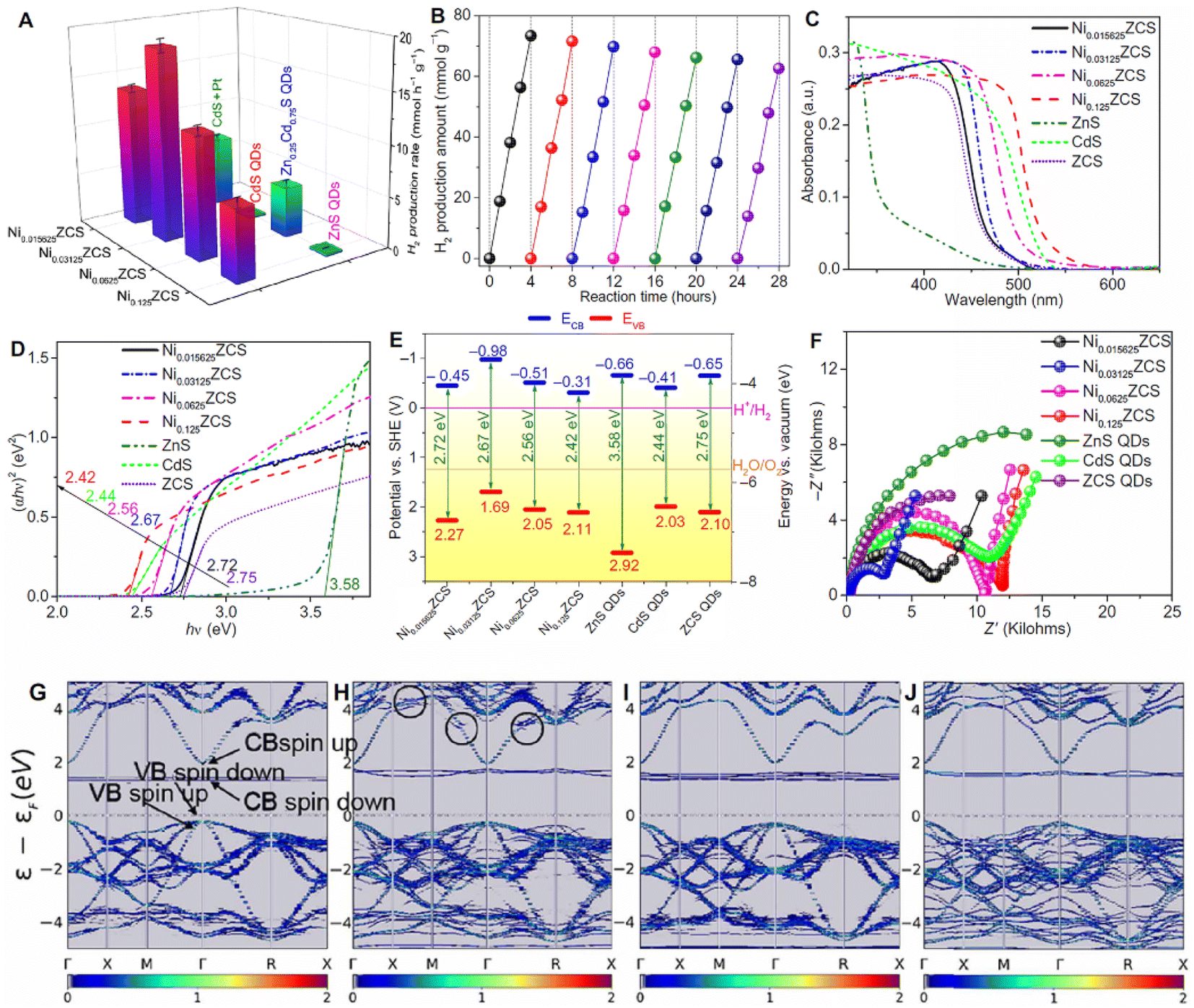 | ||
| Fig. 8 Photocatalytic performance and spectroscopy/(photo)electrochemical characterization. (A) A comparison of the photocatalytic H2 production activities of NixZCS (x = 0.015625, 0.03125, 0.0625, and 0.125) QDs, Pt–CdS QDs, CdS QDs, ZCS QDs, and ZnS QDs. The error bars denote SD. (B) Time course of photocatalytic H2 production over Ni0.03125ZCS; every 4 hours, the reaction system was purged with Ar for 30 min to remove H2. (C) Ultraviolet-visible (UV-vis) diffuse reflectance spectra of NixZCS (x = 0.015625, 0.03125, 0.0625, and 0.125) QDs, CdS QDs, ZCS QDs, and ZnS QDs. (D) The plots of transformed Kubelka–Munk function of the NixZCS (x = 0.015625, 0.03125, 0.0625, and 0.125) QDs, CdS QDs, ZCS QDs, and ZnS QDs. (E) Electronic band structures of NixZCS (x = 0.015625, 0.03125, 0.0625, and 0.125) QDs, Pt–CdS QDs, CdS QDs, ZCS QDs, and ZnS QDs. (F) EIS Nyquist plots of NixZCS (x = 0.015625, 0.03125, 0.0625, and 0.125) QD, CdS QD, ZCS QD, and ZnS QD electrodes measured under the open-circuit potential of electrodes with visible-light irradiation. (G to J) Unfolded band structure of the supercell for bulk NixZCS (x = 0.015625, 0.03125, 0.0625, and 0.125) crystals, demonstrating the direct bandgap. The Fermi level moves to 0, as indicated by the dashed lines. Copyright 2020, American Association for the Advancement of Science. | ||
Defect control
Engineering defects is an effective method to improve the activity of the photocatalyst by adjusting the coordination number and electronic structure of atoms, improving the mobility as well as conductivity of carriers, and adjusting the surface properties such as the interaction with reactants or intermediates.112–114 In the photocatalytic water decomposition reaction, due to the lack of efficient catalytic active sites on the semiconductor surface, it is not only difficult to achieve effective adsorption and activation of reaction molecules, but also it seriously hinders the transfer of photo-generated charges to reaction molecules, which greatly limits the catalytic reaction efficiency. Therefore, defect construction on the surface of materials can not only provide active sites with coordination unsaturation for adsorption and activation of reactive small molecules, but also regulate the semiconductor energy band structure, accumulate photo-generated charges, and eliminate oxidation holes. At the same time, defects can be formed as part of the catalytic cycle and interact with the electrolyte.115–119Wang et al. synthesized three dimensionally ordered macroporous (3DOM) WO3 with large holes by defect engineering using H2 treatment.120 The oxygen evolution rate of 3DOM WO3 (W270-400) was 40.1 μmol h−1, under visible light irradiation (300 W Xe lamp, cutoff at λ = 400 nm). It is much higher than that of untreated 3DOM WO3 and defect engineered WO3 nanoparticles, which was caused by good light collection and excellent charge transport, resulting from the formation of an amorphous layer with abundant surface oxygen vacancies on the catalyst surface. Ma et al. synthesized MOFs with controlled structural defects, UiO-66-NH2-X (X is the molar equivalent of the regulator acetic acid relative to the linking agent during synthesis, Fig. 9).121 With the increase of structural defects, the photocatalytic H2 production rate showed a volcanic trend. Among them, Pt@UiO-66-NH2-100 displayed the highest activity. Ultra-fine transient absorption spectrum analysis indicated that UiO-66-NH2-100 with moderate structural defects showed the fastest relaxation kinetics and the highest charge separation efficiency, while excessive defects delayed the relaxation and reduced the charge separation efficiency. Therefore, the generation of moderate structural defects was crucial to promote the efficient separation of electrons–holes.
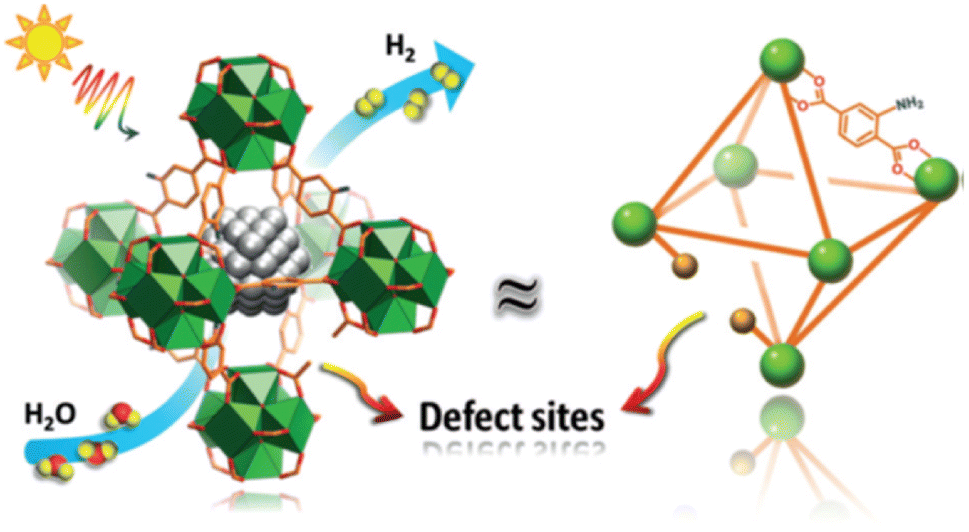 | ||
| Fig. 9 Photocatalytic hydrogen production over Pt@UiO-66-NH2-X with structural defects. Copyright 2019, Wiley-VCH. | ||
Vacancies are the main surface defects; however, the reports on vacancies in the crystal lattice are relatively few, especially for photocatalysis. Lin et al. prepared one-dimensional Cd1−xZnxS@O–MoS2/NiOx hybrid nanostructures by adjusting zinc doping content, defect-rich O–MoS2 layer and the growth of NiOx nanoparticles,122 and optimized the energy band arrangement, morphology, surface active sites and interface charge separation to enhance the photocatalytic performance of hydrogen production. With the increase of Zn doping amount, HER activity of Cd1−xZnxS (x = 0, 0.05, 0.10, and 0.15) increased, due to the increase of negative conduction potential. At the same time, after crossing Cd1−xZnxS with defect-rich O–MoS2 and NiOx, the H2 formation rate of Cd1−xZnxS @ O–MoS2/NiOx containing 15% Zn and 0.2 wt% O–MoS2 (CZ0.15S-0.2 M -NiOx) reached up to 66.08 mmol h−1 g−1, under visible light irradiation (a 300 W Xe lamp equipped with a 420 nm cutoff filter), because of the introduction of a large amount of active sites and the promotion of carrier separation, as shown in Fig. 10. Notably many different defects can exist simultaneously. A ZnIn2S4 monolayer with double defects (Ag dopant and nanopore) was obtained by Pan et al. through cation exchange.123 These two defects act as active sites for water oxidation (Ag dopant) and reduction (nanopore), which makes the ZnIn2S4 monolayer have optimized light absorption and carrier dynamics, resulting in stable photocatalytic overall water splitting performance without an auxiliary catalyst, under visible light irradiation (λ > 420 nm).
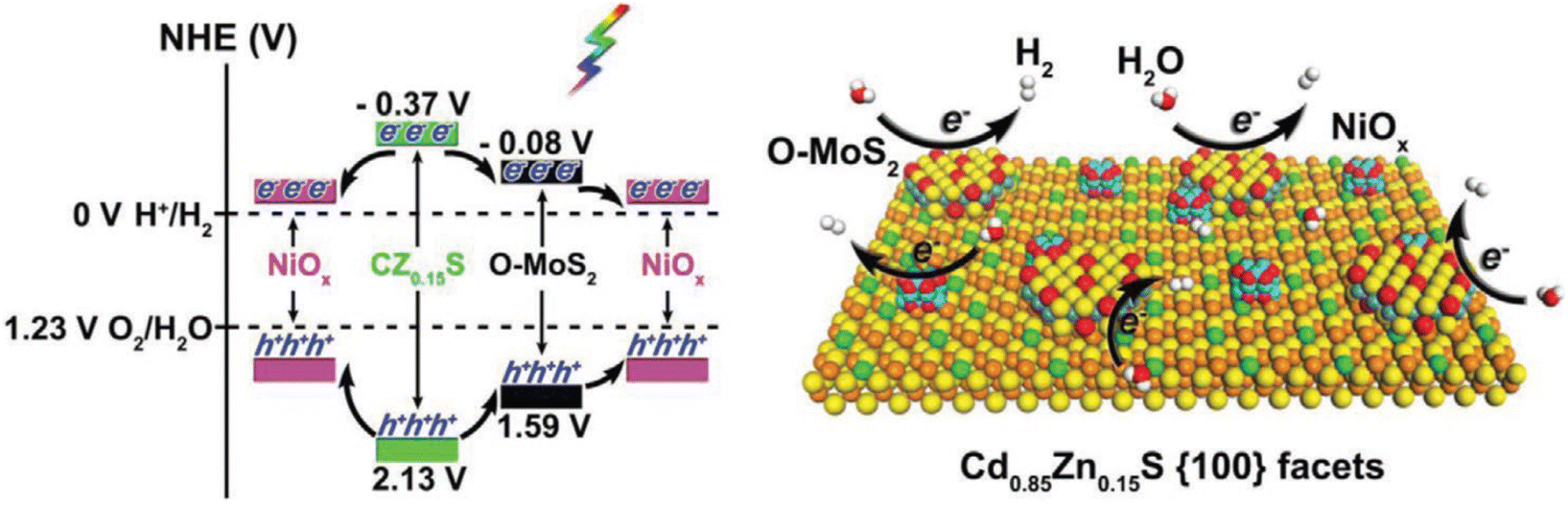 | ||
| Fig. 10 Energy-band alignments of CZ0.15S, O–MoS2, and NiOx (left) and a schematic showing the photocatalytic hydrogen evolution over CZ0.15S@O–MoS2/NiOx (right image). Copyright 2019, Wiley-VCH. | ||
S-scheme heterojunction
A photocatalyst can combine several semiconductors to form a heterojunction, which can broaden light collection characteristics and improve chemical stability, enhancing the separation of photo-generated carriers, reducing the recombination probability of photo-generated electrons and holes, and promoting photocatalytic water decomposition.124–131 Straddling, staggered, and broken gaps have been intensively studied, and the Z-type heterojunction offers another solution for facilitating charge transfer in the junction. Notably, the Yu Group proposed a new type of heterojunction, S-scheme (or step-scheme) in 2019,132 which could effectively realize the separation of electron–hole pairs with strong redox. Photocatalysts can be categorized into reduction photocatalysts (RPs) and oxidation photocatalysts (OPs), based on their CB and VB positions. RPs have high CB, while OPs have low VB. Therefore, photogenerated electrons are effective in RPs, while photogenerated holes are the contributive factors in OPs. An S-scheme heterojunction is composed of a RP and OP with staggered band structures, as indicated in Fig. 11. In the junction, the powerful photogenerated electrons and holes are reserved in the CB of the RP and VB of the OP, respectively, while the pointless photogenerated charge carriers are recombined, introducing a strong redox potential (Fig. 11). Although the structure is similar to type-II heterojunction, the charge-transfer route is totally different. If a charge-transfer route in a heterojunction goes with the S-scheme mechanism, the useless photogenerated charge carriers with weak redox ability are recombined with each other, while the useful photogenerated charge carriers with strong redox power are spatially separated and reserved, endowing it with strong redox ability and high charge-separation efficiency.57 As a result, the S-scheme system possesses strong redox ability, which is endowed with a thermodynamic advantage in overall water splitting reaction. Since then, S-scheme heterojunctions have attracted more and more attention.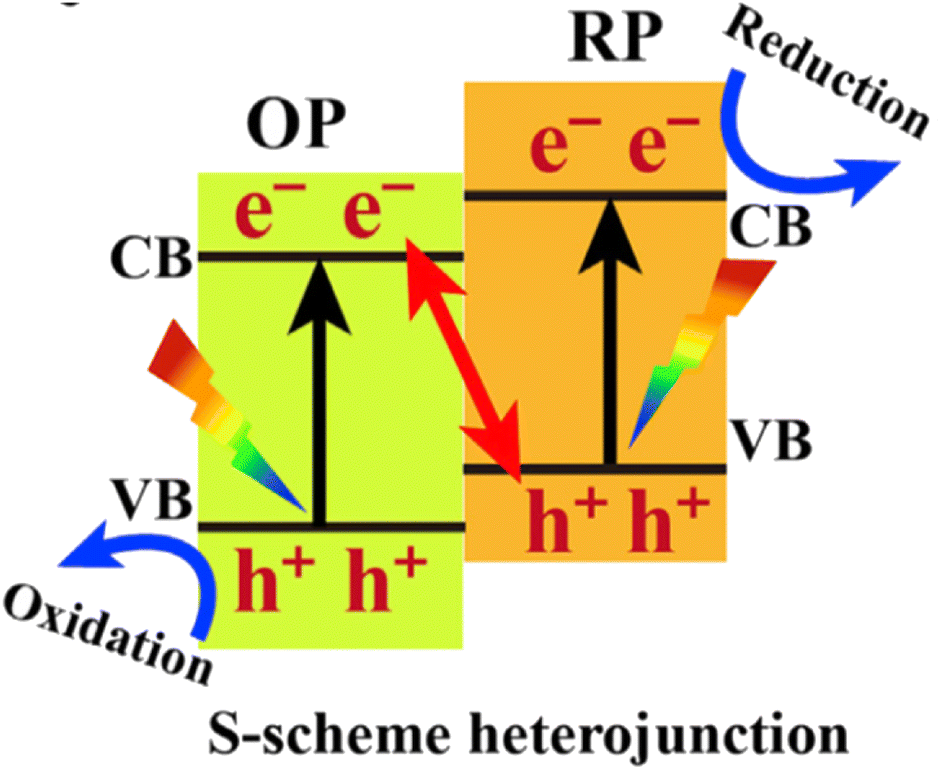 | ||
| Fig. 11 Band structure configuration and charge-transfer route of the S-scheme heterojunction. Copyright 2020, Elsevier Inc. | ||
The investigation on the junction of ultrathin WO3/g-C3N4 nanosheets generates the first report on S-scheme heterojunctions, whose photocatalytic hydrogen production rate is ∼1.7 times higher than that of pure g-C3N4.132 With the help of X-ray photoelectron spectroscopy, work function and electron paramagnetic resonance characterization, the enhanced photocatalytic performance was ascribed to the formation of the S-scheme heterojunction, which inhibited the recombination of useful electrons and holes, but expedited the recombination of relatively useless electrons and holes. Then many S-scheme heterojunctions are constructed, such as oxide/oxide (e.g., NiFe2O4/TiO2–Ag+),133 oxide/sulfide (e.g., ZnWO4–ZnIn2S4, W18O49/Mn0.2Cd0.8S, O–ZnIn2S4/TiO2−x, MoS2@MoO3, and Ti3C2 MXene@TiO2/CuInS2),134–138 MOF/oxide (e.g., Ni-MOF-74/BiVO4/P and Ni-MOF-74/In2O3)139,140 and layered double hydroxides (e.g., CoSx/NiAl-LDH and ZIF-67@NiAl LDH).141,142 For instance, the junction of NiFe2O4/TiO2–Ag+ had enhanced light absorption with an absorption edge of 457 nm, and its maximum hydrogen production rate was 27.4 μmol g−1 h−1, under simulated sunlight illumination (HAL-320 Compact Xenon Light Source Solar Simulator, 1000 W m−2), owing to enhanced light absorption and mitigated e−/h+ recombination, originating from the S-scheme heterojunction.133 Zhang et al. reported in situ partial sulfidation of MoO3 powder to obtain a MoS2@MoO3 S-scheme heterojunction photocatalyst.136 The introduction of sulfur atoms could replace oxygen atoms to build a sulfur-rich surface and generate molybdenum sulfide. The hydrogen production rate was 12416.8 μmol h−1 g−1, illuminated with 300 W xenon light. Dai et al. successfully synthesized an S-scheme ZnWO4–ZnIn2S4 heterojunction with a two-dimensional coupling interface by a simple solvothermal method.134 The H2 evolution activity of the ZnWO4–ZnIn2S4 composite with good stability reached 4925.3 μmol h−1 g−1, illuminated under 500 W xenon light. Chen et al. constructed S-doped g-C3N4 (SCN) and N-doped MoS2 (NMS) ladder heterojunctions by one-step thermal polycondensation.143 Within 4 hours of illumination under a 300 W xenon lamp, the hydrogen production rate of the material reached 658.5 μmol−1 h−1, which was about 23 times and 38 times higher than that of pure SCN (28.8 μmol−1 h−1) and NMS (17.4 μmol−1 h−1), respectively.
It is worth noting that the combination of junctions with other structural tuning approaches has attracted intensive studies. For example, Ruan et al. proposed a double S-scheme heterojunction, composed of g-C3N4 nanosheets with hydrogen-doped rutile TiO2 nanorods and anatase TiO2 nanoparticles (Fig. 12).144 The photocatalytic hydrogen evolution rate of the catalyst at 365 nm reached 62.37 mmol−1 h−1, irradiated with a 300 W Xe lamp (200–1000 nm), and the apparent quantum efficiency was up to 45.9%. The remarkable improvement of photocatalytic performance was ascribed to the unique double S-shaped structure, which induced efficient charge separation and transfer. Another example is the combination of the Schottky junction and S-scheme heterojunction. Ti3C2 MXene@TiO2/CuInS2, integrating Schottky/S-scheme heterojunction, was synthesized by a two-step hydrothermal process.138 It exhibited synergistic effects for enhanced visible-light harvesting, improved charge transfer and increased active sites for photocatalytic H2 evolution. As a result, it delivered a H2 evolution rate of 356.27 μmol g−1 h−1, illuminated with a 300 W xenon lamp. Wang et al. constructed an O–ZnIn2S4/TiO2−x photocatalyst by combining the S-scheme heterojunction with oxygen vacancies.136 Owing to the synergistic effect of oxygen defects and the S-scheme heterojunction, a tailored energy band structure alignment with higher redox potentials and a larger Fermi level potential difference was achieved, resulting in more efficient S-scheme interface charge transfer and separation efficiency. As a result, the photocatalyst showed a H2 production rate of 2584.9 μmol g−1 h−1 under AM1.5G illumination.
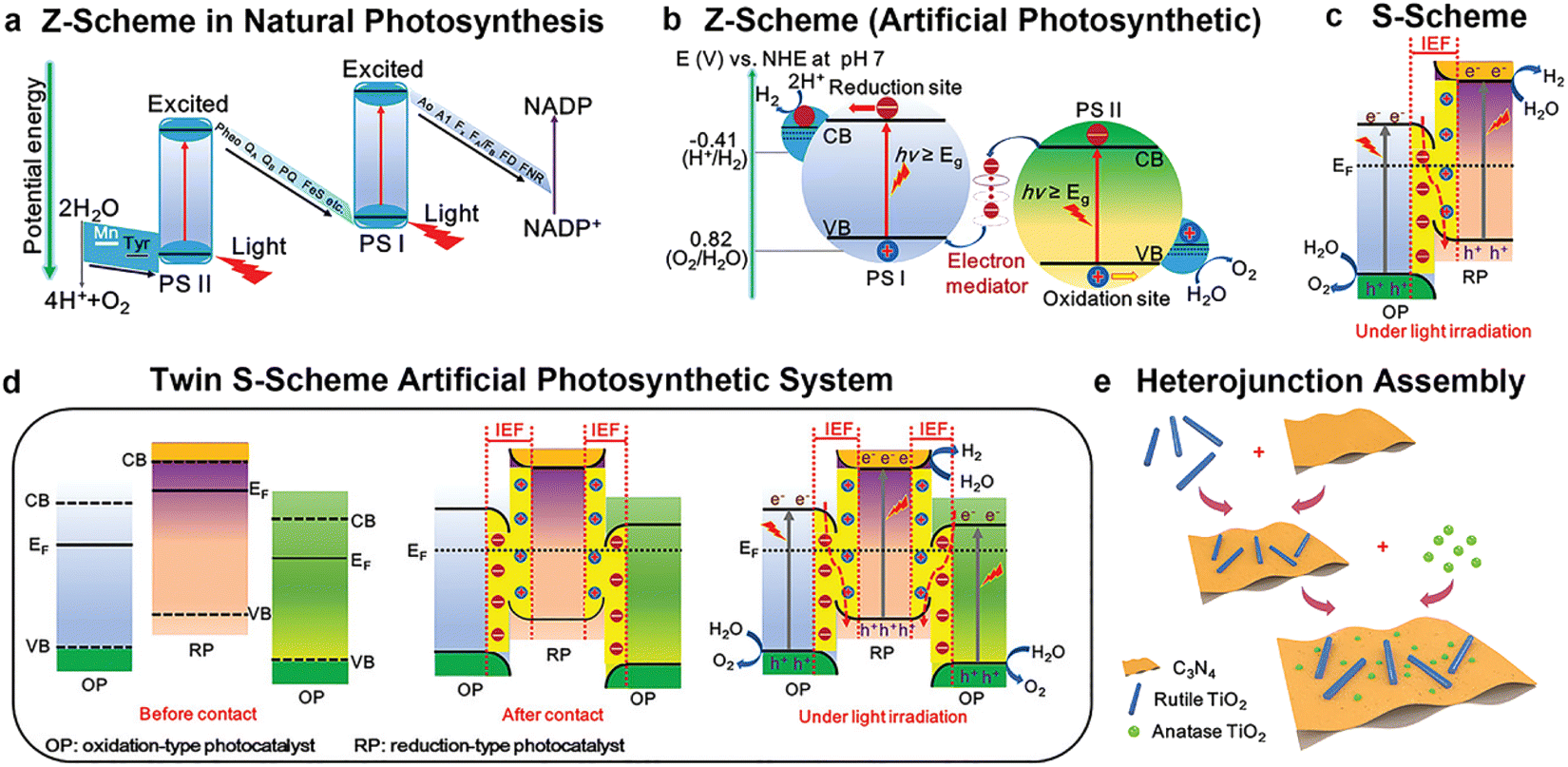 | ||
| Fig. 12 Schematic illustration of (a) the Z-scheme in natural photosynthesis, (b) Z-scheme in an artificial photosynthetic system, (c) S-scheme, and (d) twin S-scheme artificial photosynthetic system before and after contact, and under illumination. (e) Schematic diagram of electrostatic self-assembly of material. Copyright 2022, Wiley-VCH. | ||
Limitations of the structural tuning strategy
Currently SAs, defect control and S-scheme heterojunctions are the hot topics in related research areas including solar-driven hydrogen production. Great advancements have been achieved in the comprehension of the physicochemical properties of structurally tuned semiconductors or heterojunctions. Limitations, however, also exist for each tuning approach, and they are strongly time-dependent, because the shortcomings will be resolved or changed with deep investigations on the structure–activity relationship and the development of synthesis methods. These shortcomings are also important further research directions.First complexity that all structural tuning approaches are facing is the potential structural reconstruction, which will induce defects such as strain and grain boundaries. It is well-known that strain and grain boundaries as well as reconstructed structures are all highly active centers. Therefore, the understanding on the mechanism for the enhancement of photocatalytic performance and the clarification of how SACs, defects or heterojunctions contribute in a catalyst are extremely crucial in future research. Another challenge is that three essential steps in photocatalytic processes are not isolated. For example, the enhancement of light absorption by vacancy control may induce the increase of charge recombination.145 Nonetheless, the overall performance could still be improved if the extent of light absorption enhancement is much higher than that of charge recombination. This problem is general in all photocatalytic systems.
The metals in SACs are mostly rare and expensive; the production cost to the final form will be a challenge. When the SACs can be achieved in a cost-effective and mass producible way, it can be expected that they can play an important role in practical photocatalytic applications. Most recently, researchers have done some advancements in this topic, which shows high potential for resolving this issue. For instance, our recent work146 demonstrated that Ir SACs could be easily synthesized on Co3O4 nanowires using a simple dip coating and sol-flame annealing method. The total process lasts no more than 100 seconds without any instruments. This method could also be applicable to other metals (Fig. 13).
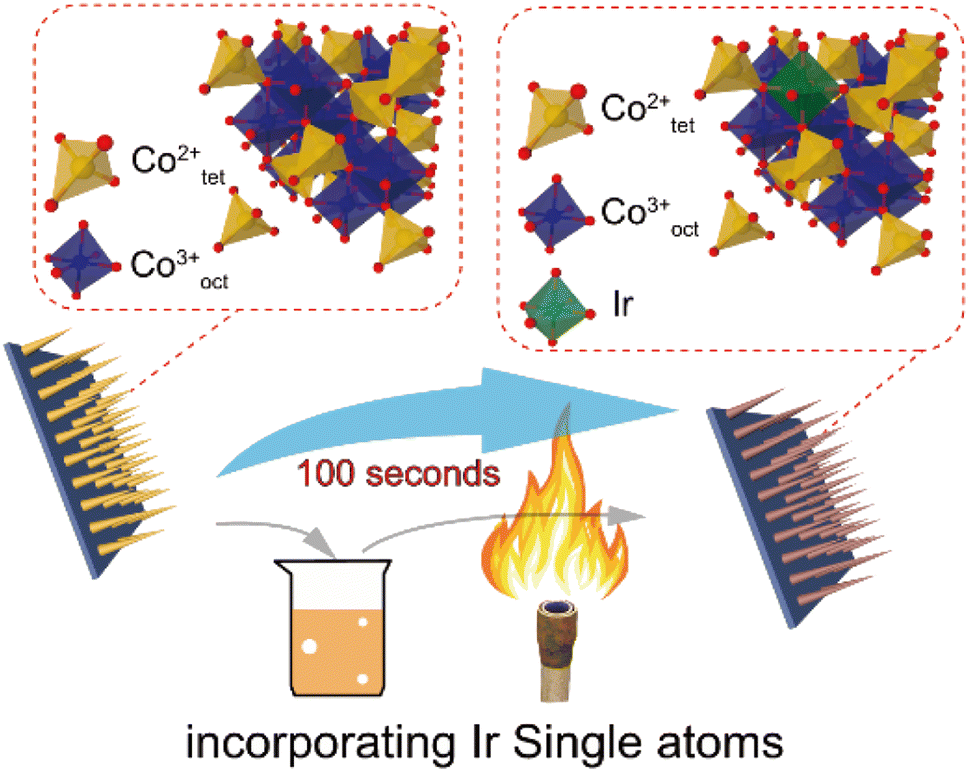 | ||
| Fig. 13 Schematic illustration of synthesizing Ir SA incorporated Co3O4 NWs. Copyright 2022, Wiley-VCH. | ||
Moreover, the increase of the SAC content (>10 wt%) will undoubtedly enhance the photocatalytic activities, but the stability will be compromised, because the high surface energy of SACs will induce the rapid aggregation or the reaction with the surrounding reactant to form other undesired species.
SACs with single metals cannot provide the sites of multiple metal atoms or metal ensemble required by some catalytic reactions, which limits their applications. In contrast, classic metal nanoparticles can offer the sites of multiple metal atoms or metal ensemble, but they have low metal utilization efficiency compared with SACs. In this case, many research studies focus on the subnanometer nanoclusters rather than SACs.
A significant gap remains between the experimental and computational studies on SAC involved semiconductors or devices. Density functional theory (DFT) is generally an extremely helpful, powerful, and reliable tool to provide fundamental understanding of electrocatalysis. Unfortunately, SACs are a real challenge for simulations, due to their elusive nature at the boundary between homogeneous and heterogeneous catalysis. When treated as homogeneous catalysts, SACs are individual atoms, whose chemical reactivity directly relates to their electron configuration and orbital diagram. In contrast, when treated as heterogeneous catalysts, SACs are supposed to stay put through the reaction's cycle, due to strong interaction with the underlying support, which in turn influences their charge and spin state, as well as their chemical reactivity. Authentically modeling SAC-support bonds is highly demanded for the accurate prediction of electronic and band structures. However, this modeling is also affected by the real operating conditions, which often differ from the low-temperature or high-vacuum conditions for DFT calculations. The complexity makes the DFT calculation highly correlated with the selected spin polarization and adopted DFT functional.
The limitation of computational studies on SACs is also applicable to defect control and heterojunctions because inaccurate structural modeling will provide meaningless or even false understanding on the structure–activity relationship. Defects such as vacancies that could be anywhere in the crystal lattice and in multiple forms make the DFT calculations extremely difficult, owing to the challenges in structural characterization on defects as well as the limitation in computer processors for processing an extremely large super cell. Existing characterization techniques can only provide indirect information about the atomic structure of defects at low resolutions, which hinders the establishment of the structure–activity relationship in photocatalytic reactions.
To boost the photocatalytic activities, bulk, surface, and interfacial defects should be finely controlled. Unfortunately, the existing synthetic methods are incapable of achieving defect control at such levels and can only tune one parameter of defects. Only when the defects are precisely controlled in photocatalytic materials can the rational defect design come into reality. More importantly, the defects such as vacancies could evolve during the photocatalytic process. For instance, bulk defects could evolve to surface defects and the latter could participate in the photocatalytic reactions. Therefore, how to measure the evolution of defects also remains a challenge. Present in situ and operando Raman, X-ray powder diffraction and infrared spectroscopy still have limitations at spatial, spectral, and temporal resolutions.
The S-scheme heterojunction is a milestone that researchers attempt to understand the charge transfer in the heterojunctions. It is a proposed new interpretation, but its scope is mainly limited to powder photocatalysts. It cannot be applied for PEC hydrogen production and solar cells with an external circuit until now. In addition, the S-scheme route is mainly used for the junction composed of two n-type semiconductors, which should have suitable band structures and obvious Fermi level differences.
Conclusion and outlook
A promising photocatalytic hydrogen production system should have the following features: (1) high light absorption ability for visible and near infrared light. High light absorption coefficiency is the same important as narrow bandgap. (2) Low recombination rate of photo-excited charge carriers. The charge pair transportation length should be higher than light absorption depth. (3) Fast surface redox reaction kinetics as well as decent stability. To achieve the goal, photocatalytic system designs especially on the composition and structure are highly demanded, and new composition and novel structures should be proposed. Given that it is a dramatic challenge for single semiconductors to possess all the features, a multi junction with engineered architectures is necessary to achieve high solar to hydrogen conversion efficiency. In addition, owing to the highly active nature of SACs, the application of SACs in photocatalysis will be more intensively studied. Herein a short overview on the development of SAs, defect control and S-heterojunctions is given, and tries to inspire more novel ideas in the research on structural engineering approaches. In addition, from our point of view, further work will put more efforts in the following aspects. The clarification of the charge transfer mechanism, which is closely related to the catalytic performance. The careful and precise control of the band structure and electronic structures among the multiple junctions will be developed based on the clarification. The massive production of SAs with multiple elemental components and their applications for photocatalytic hydrogen production will be an important direction. And the synergetic effects between SAs with semiconductor substrates will be deeply studied using advanced experimental and theoretical methods, since the related topics are barely investigated. Active centers should be identified, which is crucial for interpreting the photocatalytic mechanism and developing more active and stable photocatalytic systems. Since the water splitting to produce hydrogen involves multiple electron transfer, the surface structure will definitely experience structural transformation. How to control the defects to make it both active and stable will be a hot topic. SACs with multiple elemental components should be developed to bridge SACs and subnanometer nanoclusters, and they could be ideal catalyst candidates because they can not only provide multiple metal atom sites, but also bear higher surface atom ratios than nanocluster counterparts. The structure of S-scheme photocatalysts needs to be rationally designed. The interface, junction morphology and band structures of each semiconductor should be preciously controlled. The interaction among various engineering strategies should be identified so as to effectively enhance the photocatalytic hydrogen evolution performance. To deepen the understanding on the structure–activity relationship and pinpoint the contribution of each structural tuning strategy, advanced in situ and operando observations at higher spatial, spectral, and temporal resolutions or multiple characterizations should be developed. Overall, the rational structural design strategy promotes quick advancements in photocatalytic water splitting and still needs further intensive studies.Author contributions
F. Xu, and B. Weng devised and outlined the draft of the manuscript.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was financially supported by Hunan Key Technology R&D Program (2022GK2045) and the foundation of Hunan Environmental Protection Department.References
- Y. Jiang and Y. Lu, Nanoscale, 2020, 12, 9327–9351 RSC.
- W. Qi, Y. Zhou, S. Liu, H. Liu, L. S. Hui, A. Turak, J. Wang and M. Yang, Appl. Mater. Today, 2020, 18, 100476 CrossRef.
- B. Weng, X. Wang, C. R. Grice, F. Xu and Y. Yan, J. Mater. Chem. A, 2019, 7, 7168–7178 RSC.
- L. Gibson, E. N. Wilman and W. F. Laurance, Trends Ecol. Evol., 2017, 32, 922–935 CrossRef PubMed.
- B. Weng, C. R. Grice, W. Meng, L. Guan, F. Xu, Y. Yu, C. Wang, D. Zhao and Y. Yan, ACS Energy Lett., 2018, 3, 1434–1442 CrossRef CAS.
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- C. An, Z. Wang, W. Xi, K. Wang, X. Liu and Y. Ding, J. Mater. Chem. A, 2019, 7, 15691–15697 RSC.
- B. Weng, Z. Song, R. Zhu, Q. Yan, Q. Sun, C. G. Grice, Y. Yan and W. J. Yin, Nat. Commun., 2020, 11, 3513 CrossRef CAS.
- B. Weng, C. R. Grice, J. Ge, T. Poudel, X. Deng and Y. Yan, Adv. Energy Mater., 2018, 8, 1701655 CrossRef.
- B. Weng, Z. Xiao, W. Meng, C. R. Grice, T. Poudel, X. Deng and Y. Yan, Adv. Energy Mater., 2017, 7, 1602260 CrossRef.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- Y. Zhang, B. Zhou, H. Chen and R. Yuan, Sci. Total Environ., 2023, 856, 159048 CrossRef CAS PubMed.
- M. Saeed, M. Muneer, A. Haq and N. Akram, Environ. Sci. Pollut. Res., 2022, 29, 293–311 CrossRef CAS PubMed.
- Q. Wang and Z. Pan, Nano Res., 2022, 15, 10090–10109 CrossRef CAS.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- W. Sun, J. Zhu, M. Zhang, X. Meng, M. Chen, Y. Feng, X. Chen and Y. Ding, Chin. J. Catal., 2022, 43, 2273–2300 CrossRef CAS.
- M. Aniruddha and V. Alberto, Adv. Funct. Mater., 2022, 2208994 Search PubMed.
- T. Iqbal, A. Riaz and M. Zafar, Int. J. Environ. Anal. Chem., 2021, 1–11 CrossRef.
- M. Tayyab, Y. Liu, Z. Liu, Z. Xu, W. Yue, L. Zhou, J. Lei and J. Zhang, Chem. Eng. J., 2022, 140601 Search PubMed.
- A. Gupta, B. Likozar, R. Jana, W. C. Chanu and M. K. Singh, Int. J. Hydrogen Energy, 2022, 47, 33282–33307 CrossRef CAS.
- A. M. Mehta, S. Basu, N. P. Shetti, K. R. Reddy, T. A. Saleh and T. M. Aminabhavi, J. Environ. Manage., 2019, 250, 109486 CrossRef CAS.
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Nat. Energy, 2017, 17028 CrossRef CAS.
- W. Lei, Y. Yu, H. Zhang, Q. Jia and S. Zhang, Mater. Today, 2022, 52, 133–160 CrossRef CAS.
- C. Kranz and M. Wächtler, Chem. Soc. Rev., 2020, 50, 1407–1437 RSC.
- Y. Fang, Y. Hou, X. Fu and X. Wang, Chem. Soc. Rev., 2022, 122, 4204–4256 CrossRef CAS PubMed.
- S. J. Babu, V. N. Rao, D. H. K. Murthy, M. Shastri, M. M. Murthy, M. Shetty, K. S. A. Raju, P. D. Shivaramu, C. S. A. Kumar, M. V. Shankar and D. Rangappa, Ceram. Int., 2021, 47, 14821–14828 CrossRef CAS.
- X. Zhang, C. Bo, S. Cao, Z. Cheng, Z. Xiao, X. Liu, T. Tan and L. Piao, J. Mater. Chem. A, 2022, 10, 24381–24387 RSC.
- K. Priyalakshmi Devi, P. Goswami and H. Chaturvedi, Appl. Surf. Sci., 2022, 591, 153226 CrossRef CAS.
- J. Li, X. Wu, W. Pan, G. Zhang and H. Chen, Angew. Chem., Int. Ed., 2017, 57, 491–495 CrossRef.
- L. Guo, J. Luo, T. He, S. Wei and S. Li, Phys. Rev. Appl., 2018, 10, 064059 CrossRef CAS.
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S. H. Wei, Chem. Mater., 2009, 21, 547–551 CrossRef CAS.
- S. Wang, P. Chen, Y. Bai, J. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef.
- W. Cheng, M. H. Richter, M. M. May, J. Ohlmann, D. Lackner, F. Dimroth, T. Hannappel, H. A. Atwater and H. J. Lewerenz, ACS Energy Lett., 2018, 3(8), 1795–1800 CrossRef CAS.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS.
- Y. Liang, T. Tsubota, L. P. A. Mooij and R. van de Krol, J. Phys. Chem. C, 2011, 115, 16955–16972 Search PubMed.
- M. Li, L. Zhao and L. Guo, Int. J. Hydrogen Energy, 2010, 35, 7127–7133 CrossRef CAS.
- H. Ye, J. Lee, J. S. Jang and A. J. Bard, J. Phys. Chem. C, 2010, 114, 13322–13328 CrossRef CAS.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS.
- J. E. Yourey and B. M. Bartlett, J. Mater. Chem., 2011, 21, 7651–7660 RSC.
- M. V. LaliĆ, Z. S. PopoviĆ and F. R. VukajloviĆ, Comput. Mater. Sci., 2011, 50, 1179–1186 CrossRef.
- A. Dabirian and R. van de Krol, Chem. Mater., 2015, 27, 708–715 CrossRef CAS.
- A. Suzuki, Y. Hirose, D. Oka, S. Nakao, T. Fukumura, S. Ishii, K. Sasa, H. Matsuzaki and T. Hasegawa, Chem. Mater., 2014, 26, 976–981 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS.
- T. Minegishi, H. Kumagai, M. Katayama, J. Kubota and K. Domen, J. Am. Chem. Soc., 2013, 135, 3733–3735 CrossRef.
- L. Djellal, S. Omeiri, A. Bouguelia and M. Trari, J. Alloys Compd., 2009, 476, 584–589 CrossRef CAS.
- F. Xu, Z. Li, R. Zhu, Y. Chu, Z. Pan, S. Xia, J. Fu, Z. Xiao, X. Ji, M. Liu and B. Weng, Appl. Catal., B, 2022, 316, 121615 CrossRef CAS.
- K. Xu, Y. Xu, H. Zhang, B. Peng, H. Shao, G. Ni, J. Li, M. Yao, H. Lu, H. Zhu and C. M. Soukoulis, Phys. Chem. Chem. Phys., 2018, 20, 30351–30364 RSC.
- C. E. Pachón, L. F. Mulcué-Nieto and E. Restrepo, Mater. Today Energy, 2020, 17, 100457 CrossRef.
- Y. Ding and Y. Wang, Appl. Surf. Sci., 2022, 593, 153317 CrossRef CAS.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- T. He, A. R. Puente-Santiago, S. Xia, M. A. Ahsan, G. Xu and R. Luque, Adv. Energy Mater., 2022, 12, 2200493 CrossRef CAS.
- J. Li, Y. Chen, Y. Zhou, L. Li, B. Qiao, A. Wang, J. Liu, X. Wang and T. Zhang, AIChE J., 2017, 63, 4003–4012 CrossRef.
- W. Sun, L.-M. Cao and J. Yang, J. Mater. Chem. A, 2016, 4, 12561–12570 RSC.
- P. Li, X. Duan, Y. Kuang and X. Sun, Small, 2021, 17, 2102078 CrossRef CAS.
- N. Serpone, E. Borgarello and M. Gratzel, J. Chem. Soc., Chem. Commun., 1984, 342–344 RSC.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- J. Guo, J. Liang, X. Yuan, L. Jiang, G. Zeng, H. Yu and J. Zhang, Chem. Eng. J., 2018, 352, 782–802 CrossRef CAS.
- S. Masudy-Panah, R. Katal, N. D. Khiavi, E. Shekarian, J. Hu and X. Gong, J. Mater. Chem. A, 2019, 7, 22332–22345 RSC.
- R. K. Chava, J. Y. Do and M. Kang, J. Mater. Chem. A, 2019, 7, 13614–13628 RSC.
- H. Ren, J.-L. Yang, W.-M. Yang, H.-L. Zhong, J.-S. Lin, P. M. Radjenovic, L. Sun, H. Zhang, J. Xu, Z.-Q. Tian and J.-F. Li, ACS Mater. Lett., 2021, 3, 69–76 CrossRef CAS.
- X. Lv, X. Li, C. Yang, X. Ding, Y. Zhang, Y. Z. Zheng, S. Li, X. Sun and X. Tao, Adv. Funct. Mater., 2020, 30, 1910830 CrossRef CAS.
- H. Sun, Y. Ma, Q. Zhang and C. Su, Trans. Tianjin Univ., 2021, 27, 313–330 CrossRef CAS.
- J. He, P. Liu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 6835–6871 RSC.
- G. Wang, C.-T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem. - Eur. J., 2014, 20, 2138–2144 CrossRef CAS PubMed.
- Z. H. Xue, D. Luan, H. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- P. Chen, B. Lei, X. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef CAS PubMed.
- H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. Lou, J. Am. Chem. Soc., 2021, 143, 2173–2177 CrossRef CAS PubMed.
- Y. Cong, J. Yi, S. Liang, F. Zhang, Y. Zhang and S. Lv, Environ. Res., 2022, 208, 112739 CrossRef CAS PubMed.
- X. Lin, S. Ng and W. Ong, Coord. Chem. Rev., 2022, 471, 214743 CrossRef CAS.
- S. Zhang, B. Bai, J. Liu and J. Zhang, Catalysts, 2021, 11, 1168 CrossRef CAS.
- X. Niu, Q. Zhu, S. Jiang and Q. Zhang, J. Phys. Chem. Lett., 2020, 11, 9579–9586 CrossRef CAS PubMed.
- S. Xu, Z. Zhang, D. Wang, J. Yang, J. Lu, Y. Guo, S. Kang and X. Chang, Chem. - Eur. J., 2022, 10, 972496 CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- Y. Sui, S. Liu, T. Li, Q. Liu, T. Jiang, Y. Guo and J. Luo, J. Catal., 2017, 353, 250–255 CrossRef CAS.
- L. Yi, F. K. Lan, J. Li and C. Zhao, ACS Sustainable Chem. Eng., 2018, 6, 12766–12775 CrossRef CAS.
- Y. Pang, W. Zang, Z. Kou, L. Zhang, G. Xu, J. Lv, X. Gao, Z. Pan, J. Wang and Y. Wu, Nanoscale, 2020, 12, 4302–4308 RSC.
- L. Sun, L. Han, J. Huang, X. Luo and X. Li, Int. J. Hydrogen Energy, 2022, 47, 17583–17599 CrossRef CAS.
- M. Xiao, L. Zhang, B. Luo, M. Lyu, Z. Wang, H. Huang, S. Wang, A. Du and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 7230–7234 CrossRef CAS PubMed.
- Y. Xue, Y. Y. Lei, X. Liu, Y. Li, W. Deng, F. Wang and S. Min, New J. Chem., 2018, 42, 14083–14086 RSC.
- Z. Zeng, Y. Sun, X. Quan, W. Choi, G. Zhang, N. Liu, B. Kim, S. Chen, H. Yu and S. Zhang, Nano Energy, 2020, 69, 104409 CrossRef CAS.
- Y. Hu, Y. Qu, Y. Zhou, Z. Wang, H. Wang, B. Yang, Z. Yu and Y. Wu, Chem. Eng. J., 2021, 412, 128749 CrossRef CAS.
- L. Wang, R. Tang, A. Kheradmand, Y. Jiang, H. Wang, W. Yang, L. Jiang, L. Zhang, C. Cheng, Z. Sun and L. Hu, Appl. Catal., B, 2021, 284, 119758 CrossRef.
- P. Zhu, N. Li, Y. Chao, W. Zhang, F. Lv, L. Wang, W. Yang, P. Gao and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 14184–14188 CrossRef PubMed.
- X. Guan, Q. Wu, H. Li, S. Zeng, Q. Yao, R. Li, H. Chen, Y. Zheng and K. Qu, Appl. Catal., B, 2023, 323, 122145 CrossRef CAS.
- X. H. Jiang, L. S. Zhang, H. Y. Liu, D. S. Wu, F. Y. Wu, L. Tian, L. Liu, J. Zou, S. Luo and B. Chen, Angew. Chem., Int. Ed., 2020, 59, 23112–23116 CrossRef CAS PubMed.
- X. Li, S. Zhao, X. Duan, H. Zhang, S. Yang, P. Zhang, S. Jiang, S. Liu, H. Sun and S. Wang, Appl. Catal., B, 2021, 283, 119660 CrossRef CAS.
- H. Chen, F. Zhang, X. Sun, W. Zhang and G. Li, Int. J. Hydrogen Energy, 2018, 43, 5331–5336 CrossRef CAS.
- J. Wang, Q. Zhou, Y. Shen, X. Chen, S. Liu and Y. Zhang, Langmuir, 2019, 35, 12366–12373 CrossRef CAS PubMed.
- H. Liu, M. Cheng, Y. Liu, J. Wang, G. Zhang, L. Li, L. Du, G. Wang, S. Yang and X. Wang, Energy Environ. Sci., 2022, 15, 3722–3749 RSC.
- H. Su, W. Che, F. Tang, W. Cheng, X. Zhao, H. Zhang and Q. Liu, J. Phys. Chem. C, 2018, 122, 21108–21114 CrossRef CAS.
- L. Zhang, R. Long, Y. Zhang, D. Duan, Y. Xiong, Y. Zhang and Y. Bi, Angew. Chem., Int. Ed., 2020, 59, 6224–6229 CrossRef CAS PubMed.
- Y. Chen, S. J. W. Sun, Y. Lei, Q. Wang, A. Li, W. Chen, G. Zhou, Z. Zhang, Y. Wang, L. Zheng, Q. Zhang, L. Gu, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 1295–1301 CrossRef CAS PubMed.
- X. Hu, J. Song, J. Luo, H. Zhang, Z. Sun, C. Li, S. Zheng and Q. Liu, J. Energy Chem., 2021, 62, 1–10 CrossRef CAS.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long, Q. Xu, C. Wang, L. Song, J. Zhu, J. Jiang and Y. Xiong, Adv. Mater., 2016, 28, 6959–6965 CrossRef CAS PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 CrossRef CAS PubMed.
- H. Zhang, S. ZUo, M. Qiu, S. Wang, Y. Zhang, J. Zhang and X. Lou, Sci. Adv., 2020, 6, eabb9823 CrossRef CAS PubMed.
- B. Yan, D. Liu, X. Feng, M. Shao and Y. Zhang, Adv. Funct. Mater., 2020, 30, 2003007 CrossRef CAS.
- S. Cao, H. Li, T. Tong, H. C. Chen, A. Yu, J. Yu and H. M. Chen, Adv. Funct. Mater., 2018, 28, 1802169 CrossRef.
- X. H. Jiang, L. S. Zhang, H. Y. Liu, D. S. Wu, F. Y. Wu, L. Tian, L. L. Liu, J. P. Zou, S. L. Luo and B. B. Chen, Angew. Chem., Int. Ed., 2020, 132, 23312–23316 CrossRef.
- L. Li, Z. Deng, L. Yu. Z. Lin, W. Wang and G. Yang, Nano Energy, 2016, 27, 103–113 CrossRef CAS.
- H. Zhao, J. Wang, Y. Dong and P. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 8053–8060 CrossRef CAS.
- Y. Cao, S. Chen, Q. Luo, H. Yan, Y. Lin, W. Liu, L. Cao, J. Lu, J. Yang, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 12191–12196 CrossRef CAS PubMed.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 CrossRef CAS PubMed.
- R. Shi, C. Tian, X. Zhu, C. Peng, B. Mei and L. He, Chem. Sci., 2019, 10, 2585–2591 RSC.
- B. Xia, Y. Yang, Y. Zhang, Y. Xia, M. Jaroniec, J. Yu, J. Ran and S. Qiao, Chem. Eng. J., 2022, 431, 133944 CrossRef CAS.
- D. W. Su, J. Ran, Z. W. Zhuang, C. Chen, S. Z. Qiao, Y. D. Li and G. X. Wang, Sci. Adv., 2020, 6, eaaz8447 CrossRef CAS PubMed.
- Y. C. Zhang, N. Afzal, L. Pan, X. Zhang and J. Zou, Adv. Sci., 2019, 6, 1900053 CrossRef PubMed.
- N. Zhang, C. Gao and Y. Xiong, J. Energy Chem., 2019, 37, 43–57 CrossRef.
- X. Wang, H. Huang, J. Wang, Z. Li and Z. Zou, J. Phys. Chem. Lett., 2020, 11, 1708–1713 CrossRef PubMed.
- F. Amano and S. Nakayama, J. Environ. Chem. Eng., 2022, 10, 108089 CrossRef CAS.
- T. Wei, Y. Zhu, X. An, L. Liu, X. Cao, H. Liu and J. Qu, ACS Catal., 2019, 9, 8346–8354 CrossRef CAS.
- J. Bown and A. J. Page, J. Mater. Chem. A, 2019, 7, 13029–13035 RSC.
- Y. Xu, Y. Yao, W. Yin, J. Cao, M. Chen and X. Wei, RSC Adv., 2020, 10, 10816–10825 RSC.
- C. Zhu, M. Zhu, Y. Sun, Y. Zhou, H. Huang, Y. Lifshitz, S. Lee, J. Zhong, Y. Liu and Z. Kang, Appl. Catal., B, 2018, 237, 166–174 CrossRef CAS.
- Y. Wang, C. Peng, T. Jiang, J. Zhang, Z. Jiang and X. Li, J. Mater. Chem. A, 2021, 9, 3036–3043 RSC.
- X. Ma, L. Wang, Q. Zhang and H. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12175–12179 CrossRef CAS PubMed.
- H. Lin, B. Sun, H. Wang, Q. Ruan, Y. Geng, Y. Li, J. Wu, W. Wang, J. Liu and X. Wang, Small, 2019, 15, 1804115 CrossRef PubMed.
- R. Pan, M. Hu, J. Liu, D. Li, X. Wan, H. Wang, Y. Li, X. Zhang, X. Wang, J. Jiang and J. Zhang, Nano Lett., 2021, 21, 6228–6236 CrossRef CAS PubMed.
- X. Chen, C. Zhao, H. Wu, Y. Shi, C. Chen and X. Zhou, Materials, 2022, 15, 3786 CrossRef CAS PubMed.
- R. Liu, S. Y. Fang, C. D. Dong, K. C. Tsai and W. D. Yang, Int. J. Hydrogen Energy, 2021, 46, 28462–28473 CrossRef CAS.
- P. Dong, A. Zhang, T. Cheng, J. Pan, J. Song and L. Zhang, Chin. J. Catal., 2022, 43, 2592–2605 CrossRef CAS.
- V. Hasija, A. Kumar, A. Sudhaik, P. Raizada, P. Singh, Q. Van Le, T. Le and V. Nguyen, Environ. Chem. Lett., 2021, 19, 2941–2966 CrossRef CAS.
- P. Shan, C. Zhang, M. Zhou, C. He, T. Ouyang, J. Li, C. Tang and J. Zhong, J. Mater. Chem. C, 2021, 9, 7734–7744 RSC.
- Z. Zhang, L. Huang, J. Zhang, F. Wang, Y. Xie, X. Shang, Y. Guo, H. Zhao and X. Wang, Appl. Catal., B, 2018, 233, 112–119 CrossRef CAS.
- M. Dai, Z. He, P. Zhang, X. Li and S. Wang, J. Mater. Sci. Technol., 2022, 122, 231–242 CrossRef.
- Y. Chen, F. Su, H. Xie, R. Wang, C. Ding, J. Huang, Y. Xu and L. Ye, Chem. Eng. J., 2021, 404, 126498 CrossRef CAS.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- I. Firtina-Ertis and O. Kerkez-Kuyumcu, J. Photochem. Photobiol., A, 2022, 432, 114106 CrossRef CAS.
- M. Dai, Z. He, P. Zhang, X. Li and S. Wang, J. Mater. Sci. Technol., 2022, 122, 231–242 CrossRef.
- X. Ma, D. Li, P. Su, Z. Jiang and Z. Jin, ChemCatChem, 2021, 13, 2179–2190 CrossRef CAS.
- Y. Wang, X. Meng, Q. Hu, M. Zhang, X. Cao, C. Xu and Y. Ding, Int. J. Hydrogen Energy, 2021, 46, 6262–6271 CrossRef CAS.
- L. Zhang, Z. Jin and N. Tsubaki, Chem. Eng. J., 2022, 438, 135238 CrossRef CAS.
- W. Yang, G. Ma, Y. Fu, K. Peng, H. Yang, X. Zhan, W. Yang, L. Wang and H. Hou, Chem. Eng. J., 2022, 429, 132381 CrossRef CAS.
- H. Li, H. Gong and Z. Jin, Appl. Catal., B, 2022, 307, 121166 CrossRef CAS.
- X. Jiang, M. Li, H. Li and Z. Jin, New J. Chem., 2021, 45, 16155–16167 RSC.
- Y. Liu, X. Ma and Z. Jin, J. Colloid Interface Sci., 2022, 609, 686–697 CrossRef CAS PubMed.
- K. Wang, S. Liu, Y. Li, G. Wang, M. Yang and Z. Jin, Appl. Surf. Sci., 2022, 601, 154174 CrossRef CAS.
- Y. Chen, F. Su, H. Xie, R. Wang, C. Ding, J. Huang, Y. Xu and L. Ye, Chem. Eng. J., 2021, 404, 126498 CrossRef CAS.
- X. Ruan, C. Huang, H. Cheng, Z. Zhang, Y. Cui, Z. Li, T. Xie, K. Ba, H. Zhang, L. Zhang, X. Zhao, J. Leng, S. Jin, W. Zhang, W. Zheng, S. Ravi, Z. Jiang, X. Cui and J. Yu, Adv. Mater., 2023, 35, 2209141 CrossRef CAS PubMed.
- F. Xu, W. Zhao, X. Hu and B. Weng, J. Colloid Interface Sci., 2023, 636, 470–479 CrossRef CAS PubMed.
- W. Zhao, F. Xu, Z. Wang, Z. Pan, Y. Ye, S. Hu, B. Weng and R. Zhu, Small, 2022, 18, 2270275 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |