
Advanced oxygen evolution reaction catalysts for solar-driven photoelectrochemical water splitting
Guojun
Dong
 a,
Lianglin
Yan
b and
Yingpu
Bi
*a
a,
Lianglin
Yan
b and
Yingpu
Bi
*a
aState Key Laboratory for Oxo Synthesis & Selective Oxidation, National Engineering Research Center of Fine Petrochemical Intermediates, Lanzhou Institute of Chemical Physics, CAS, Lanzhou 730000, China. E-mail: yingpubi@licp.cas.cn
bGansu Key Laboratory for Environmental Pollution Prediction and Control, College of Earth and Environmental Sciences, Lanzhou University, Lanzhou 730000, China
First published on 18th January 2023
Abstract
Solar-driven photoelectrochemical (PEC) water splitting is a promising technique for converting abundant solar-energy into clean and sustainable hydrogen fuels. However, the sluggish oxygen evolution reaction (OER) kinetics occurring on semiconductor photoanodes greatly hampered its practical application. To address this issue, the decoration of OER catalysts on photoanode surfaces has been proved to be an effective approach to decrease the reaction overpotential and accelerate the surface OER kinetics, thus improving the PEC performances for water splitting. It was generally recognized that OER catalysts could efficiently extract photo-generated holes from bulk photoanodes to their active sites, so that the surface holes are rapidly consumed in the water oxidation process, effectively restraining charge recombination. Herein, the recent developments in OER catalyst decoration on semiconductor photoanodes for promoting PEC water splitting have been described and discussed. Firstly, the basic principles of PEC water splitting and the functions of OER catalysts have been summarized. Secondly, the structure optimization of OER catalysts, including defect construction, interfacial bonding, elemental doping, morphological regulation and multi-component synergy, toward PEC water oxidation have been discussed. Finally, some challenges and opportunities to achieve further developments in highly efficient OER catalysts for PEC water splitting have been summarized.
 Guojun Dong | Guojun Dong obtained his Ph.D. degree from the Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences (CAS) in 2019, and then worked in the group of professor Yingpu Bi. He is an assistant researcher at the Lanzhou Institute of Chemical Physics, CAS. His research interests focus on the development of novel semiconductor materials for photo(electro)catalytic water splitting and N2 reduction. |
 Yingpu Bi | Yingpu Bi obtained his Ph.D. degree in 2008 from the Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences (CAS), and then carried out three years of postdoctoral research at the International Centre for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), Japan. He is currently a professor at the Lanzhou Institute of Chemical Physics, CAS. His research interests include the development of novel nanostructured materials, photocatalytic properties, and novel hydrogen generation reactions. |
1. Introduction
Energy and environmental issues have become the biggest obstacles to the sustainable development of the world due to the over-exploitation of traditional fossil fuels and serious destruction of the ecological environment. Therefore, the exploration and utilization of renewable energy are a promising choice for resolving the above crises to ensure human, social and planetary security.1,2 Owing to its inexhaustible, ubiquitous and clean characteristics, solar energy is widely recognized as the best renewable energy source compared with the current hydroelectric, wind, geothermal, and fossil power sources.3 Up to now, among various technologies for solar-energy conversion, photoelectrochemical (PEC) water splitting could directly decompose water into hydrogen and oxygen by absorbing sunlight with a clean and pollution-free reaction process, which is regarded as the most ideal approach to produce sustainable hydrogen fuels.4–7 Generally, there are two half-reactions in the PEC water splitting process: (1) the oxygen evolution reaction (OER) on the photoanode of an n-type semiconductor and (2) the hydrogen evolution reaction (HER) on the photocathode of a p-type semiconductor. Note that the water oxidation half-reaction requires four-electron transfer for oxygen production, which is more difficult than the two-electron HER due to the higher energy barrier and slower reaction kinetics.8–10 Therefore, during the past few decades, great efforts have been devoted to explore efficient and stable photoanode materials for promoting the oxygen evolution reactivity and enhancing the solar energy conversion efficiency of PEC water splitting.11–15It is well known that there are three crucial steps to determine the PEC conversion efficiency of semiconductor photoanodes: (1) light absorption, (2) charge separation and transfer, and (3) charge utilization. The first two steps are related to the number of generated charge carriers and separated electrons/holes that reached the electrode surfaces. The third step is related to the final yield of oxygen or hydrogen (the faradaic efficiency is often assumed to be 100 percent).16–18 Therefore, ideal photoanodes should have the basic characteristics of broad spectral absorption and low bulk/surface recombination for satisfying the water oxidation reaction. In terms of light absorption, narrow band gap semiconductor materials (BiVO4, Ta3N5, Fe2O3, CdIn2S4, etc.) could efficiently extend the spectral absorption range to the visible light region.19–24 Moreover, the light-capturing capability of some wide band gap semiconductors (TiO2, ZnO, WO3, etc.) has been improved by appropriate methods, including quantum dot sensitization or heterojunction construction.25–28 In the meantime, various strategies, such as morphology regulation, metal/non-metal element doping and surface reconstruction, have been successfully applied to accelerate the photogenerated charge separation and transport.29–32 For the complex surface charge utilization process, the decoration of OER catalysts (also referred to as cocatalysts) is considered a promising method for elevating the efficiency of charge utilization. More specifically, they could efficiently accelerate photogenerated hole transfer, provide more active sites, overcome the reaction overpotential, suppress photo-corrosion etc., which are all beneficial for promoting water oxidation reactivity. Up to now, almost all photoanode studies have adopted one or more OER catalysts to improve the catalytic activity, which plays a crucial role in determining the PEC conversion efficiency for water oxidation.33–35 Thereby, various strategies were explored to optimize OER catalysts for designing high performance photoanode systems in recent years. To date, some techniques, such as oxygen defect construction, selective interfacial bonding, element doping, morphology regulation and multi-component synergy, have been successfully applied to construct highly efficient OER catalysts on photoanode surfaces, which exhibited significantly enhanced PEC water oxidation properties over pristine photoanodes. At present, many important and excellent reviews focusing on the material type and basic functions of OER catalysts in PEC water splitting have been reported.36–39 It should be essential to deeply understand the advanced structure optimization as well as the relevant design principles of OER catalysts for establishing a highly efficient and stable photoanode system with outstanding PEC water oxidation performances.
This review summarizes the recent achievements and progress in structure optimization on OER catalysts for promoting PEC water splitting activity and durability of semiconductor photoanode systems. Firstly, the basic principles of PEC water splitting and the various functions of OER catalysts in the PEC water oxidation reaction have been described. Secondly, the present OER catalyst structure optimization strategies, design principles and the corresponding mechanisms for enhancing PEC performances have been summarized. Finally, some challenges and opportunities for developing highly efficient OER catalysts were briefly discussed.
2. The principles of PEC water splitting
A PEC water splitting system mainly consists of two half-reactions of water oxidation and reduction, which could effectively separate the oxidation and reduction products compared with the conventional photocatalytic process.40 In this part, the single photoanode PEC configuration was adopted to illustrate the basic working principles. As shown in Fig. 1, an n-type semiconductor served as the photoanode for the oxygen evolution reaction (OER), and a metal-based material is applied as the counter electrode (the most common metal is “Pt”) for the hydrogen evolution reaction (HER). Besides, an external bias (≤1.23 V vs. reversible hydrogen potential, RHE) is required to facilitate the electron transfer through an external circuit to the cathode.41 Under dark conditions, electron transfer occurs between the photoanode and electrolyte by the effect of electrostatic balance. The Fermi levels of the semiconductor will align with the redox potential of electrolyte at their interface, inducing an upward energy band bending. Thus, the semiconductor surface will be filled with an excess of positive charge, called the depletion layer, and a Helmholtz layer with negative charge will be generated between the photoanode and the electrolyte.42,43 Under solar light irradiation, when the energy of the incident photon is higher than the bandgap energy (Eg) of semiconductor materials, the electrons will be excited to the conduction band (CB), and the holes will be left in the valence band (VB). The electrons in the CB will be transferred to fluorine doped tin oxide (FTO) or other conductive substrates and subsequently reach the cathode through an external circuit at the applied bias voltage for water reduction. In the meantime, holes in the VB will migrate to the surface of the photoanode for water oxidation.44,45 The water splitting reaction requires a minimum Gibbs free energy of 237.2 kJ mol−1 for converting one water molecule into hydrogen and half oxygen corresponding to a photon energy of 1.23 eV. Therefore, the semiconductor photoanode should provide a bandgap of greater than 1.23 eV for solar light capture. In addition, the redox potentials of O2/H2O and H+/H2 were set to be 1.23 VNHE and 0 VNHE (pH = 0), respectively. However, overpotentials will be generated during the hole travel through the space charge region and the electron transfer through an external circuit. Thereby, the potential of the valence band should be more positive than 1.23 VNHE for water oxidation, and the potential of the conduction band should be more negative than 0 VNHE for water reduction. It should be noted that the bulk phase and surface recombination of photogenerated carriers should be inevitable during the charge separation and transport processes, which mainly affects the number of holes and electrons that arrive at the electrode surface to participate in oxidation and reduction reactions.46,47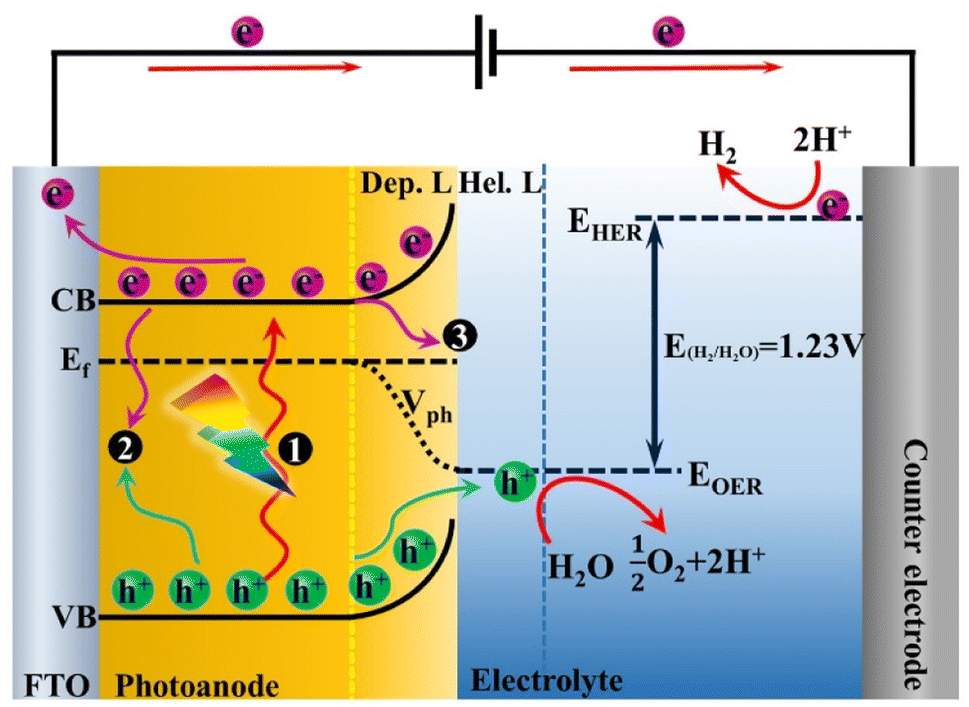 | ||
| Fig. 1 The principles of PEC water splitting for single photoanode configurations. Mark 1 represents the process of charge separation under light irradiation. Marks 2 and 3 represent the bulk and surface recombination, respectively. Dep. L: depletion layer, Hel. L: Helmholtz layer, CB: conduction band, Ef: Fermi energy level, VB: valence band, and Vph: photo-potential. | ||
3. The functions of OER catalysts
Up to now, noble-metal (Ru and Ir etc.) oxides, transition metal (Fe, Co, Ni etc.) oxides/hydroxides/hydroxyl oxides, C-based nonmetal materials, coordination polymers, and molecule catalysts have been widely used as OER catalysts for improving the interface charge transfer and surface water oxidation dynamics in the PEC water splitting process. Specifically, transition metal-based materials were more popular in “OER catalyst/semiconductor” photoanode construction due to the significant advantage in material costs, preparation methods and activity requirements. Recently, molecular catalysts, complexes based on Ru, Ir, Fe, Co, Ni and Cu, have been reported as novel OER catalysts owing to their high activity and tunability, as well as their ability to be integrated into sophisticated molecular assemblies,48,49 which were generally decorated on semiconductor photoanode surfaces by covalent attachment, electrostatic interaction and electro-polymerization.50 Each material, of course, has its advantages and disadvantages. How to choose suitable OER catalysts for constructing the desired “OER catalyst/semiconductor” system? The most basic premise is to understand the intrinsic role and function of OER catalysts. Based on previous relevant studies, some functions of OER catalysts in the PEC water oxidation reaction are summarized in Fig. 2.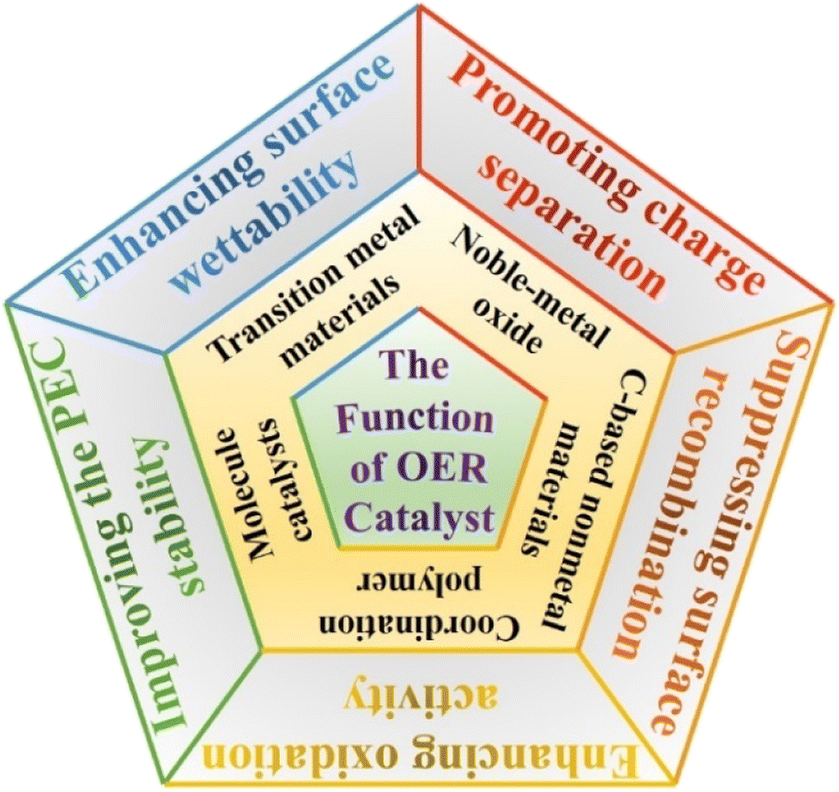 | ||
| Fig. 2 The schematic illustration of the function of OER catalysts in PEC water splitting. | ||
3.1 The promotion of charge separation
Efficient transfer of photogenerated holes from the valence band to photoanode surfaces is crucial to promote the charge separation efficiency as well as PEC water oxidation activity. Except for heterojunction construction and morphological regulation, OER catalyst loading on semiconductor photoanodes should be one of the most feasible strategies.51,52 For example, Co3O4 nanoparticles as OER catalysts could rapidly extract photogenerated holes from the BiVO4 photoanode, and a significantly enhanced photocurrent density was achieved, owing to the improvement of both bulk and surface charge separation efficiencies.53 Cobalt phosphide (CoP) could significantly improve the PEC performances of the Fe2O3 photoanode due to the accelerated water oxidation kinetics and improved hole injection efficiency.54 Moreover, after loading of ultrathin NiFe-layered double hydroxide (LDH), the transfer distance of photogenerated holes at the interface of WO3 nanorods and the OER catalyst has been remarkably shortened, which exhibited an improved charge separation ability and photocurrent density.55 Thereby, OER catalysts not only provide additional driving force for hole transfer from the semiconductor bulk phase to the surface due to the low energy level, but also shorten the interfacial transfer distance by coming into close contact with the semiconductor photoanode for increasing the charge separation capability.3.2 The suppression of surface recombination
Owing to the termination of crystal lattice, the surface of semiconductor material has many surface states, which are often considered to be the recombination centres of charge carriers. Besides, it is easily polluted by external contamination, creating more charge recombination sites. As a result, the photocurrent density of pristine semiconductor photoanodes is much lower than the theoretical values. To address this issue, Wang et al. reported the modification of an IrOx catalyst on the Fe2O3 photoanode for optimising the surface states to reduce surface charge recombination and promote PEC water splitting performances.56 In addition, Krol and co-workers proposed that surface charge recombination is the main reason to limit the photocurrent density of BiVO4 materials by using intensity modulated photocurrent spectroscopy (IMPS). After the decoration of a CoPi OER catalyst, the PEC performances of BiVO4 photoanodes have been obviously improved owing to the efficient surface passivation.57 Therefore, the decoration of OER catalysts should be efficient to suppress surface charge recombination by passivating the semiconductor surfaces for decreasing the surface states.3.3 The enhancement of water oxidation activity
The oxygen evolution reaction is the rate determining step in the PEC water splitting reaction. Because of high interface energy barriers and sluggish water oxidation kinetics, a large number of photogenerated charges would be recombined on photoanode surfaces, resulting in a relatively low photocurrent density and poor OER activity. It should be noted that OER catalysts could also be used to enhance the water oxidation reactivity. For example, Co(OH)x as an OER catalyst with suitable oxidation states could provide more active sites for improving the PEC water oxidation activity of the Bi2WO6 photoanode.58 Similar studies also proved that Co3+/4+ redox pairs could serve as active centres of amorphous CoOOH, which could reduce the interface reaction barrier and provide abundant active sites for promoting the PEC water oxidation activities of the TiO2 photoanode.59 Besides, FeCoOx could promote hole trapping and reduce the OER overpotential, resulting in remarkably enhanced water oxidation activity of the BiVO4 photoanode.60 After coating the NiFeOOH cocatalyst, the Fe2O3 photoanode exhibits significantly enhanced PEC water splitting performance owing to the accelerated OER kinetics and decreased overpotential.61 As a result, the remarkably enhanced PEC activity for water oxidation should be attributed to the low overpotential and abundant active sites of OER catalysts.3.4 The enhancements of water oxidation stability
Stability is another key parameter in determining the practical applications of the PEC water splitting technique. However, owing to the excess accumulation of photogenerated holes on the surfaces, self-oxidation would occur on semiconductor photoanode materials. Additionally, the dissolution of semiconductors also occurs in harsh electrolyte environments under solar irradiation. Both of these will inevitably reduce the PEC stability of photoanodes during the water oxidation process. More specifically, pristine Ta3N5, BiVO4, and S-based semiconductor materials generally exhibit relatively poor PEC water oxidation stability. For the Ta3N5 photoanode, Co3O4, CoPi catalysts could effectively and rapidly extract surface holes to inhibit photo-corrosion reactions and enhance PEC durability.62,63 NiFe-MOFs and FexCo1−xOOH catalysts could prevent V element dissolution for improving the long-term stability of the BiVO4 photoanode.64,65 After the decoration of FeOOH nanolayers, the WO3 photoanode exhibits improved structure stability against photo-corrosion.66 Thereby, the function of OER catalysts in enhancing the PEC water oxidation stability could be attributed to the efficient utilization of surface photogenerated holes and the production of protective layers on the semiconductor photoanode.3.5 The improvement of surface wettability
The surface wettability of semiconductor materials dominates the water adsorption and oxygen desorption processes, affected by the surface texture and the micro-morphology of photoanodes.67,68 Improved hydrophilicity can be achieved through appropriate surface and structural modifications, while the active sites of water oxidation and charge transport properties also need to be taken into account. In this case, hydrophilic OER catalyst decoration becomes a more advantageous strategy. For instance, super-hydrophilic CoAl-LDH has been prepared by plasma treatment, which could accelerate the adsorption of water molecules on surface active sites, resulting in the superior photocurrent density of the BiVO4 photoanode.69 Moreover, owing to the presence of hydrophilic phosphonic acid groups in the phytic acid (PA) molecular structure, CoFe-PA served as an efficient OER catalyst to accelerate interfacial charge transfer by facilitating electrolyte infiltration, inducing a great promotion of water oxidation activity of the BiVO4 photoanode.70 Thus, hydrophilic surface reconstruction or coupling of hydrophilic groups is an effective strategy for achieving excellent wettability of OER catalysts, which should be beneficial for the PEC water oxidation reaction.Although the decoration of OER catalysts has been proved to be a feasible method for promoting the PEC activity of photoanodes, some fundamental principles should be further clarified for guiding the selective construction of OER catalyst materials. Firstly, the energy level of the OER catalyst should match that of semiconductor photoanodes, which was applied to balance the activity and conductivity for achieving optimized PEC water oxidation properties. Secondly, the thickness of the OER catalyst should be regulated, which is related to the light absorption of the photoanode and the proportion of active sites. Thirdly, the stability of OER catalysts should be maintained, which guarantees long-term operation of the photoanode at a high photocurrent density. Finally, the cost of an OER catalyst should also be considered, which determines the economic benefits and practicality of PEC water splitting devices.
4. The structure optimization of OER catalysts in PEC water oxidation
In addition to the exploration of novel and highly efficient OER catalyst materials, the structure optimization of OER catalysts is also considered a feasible strategy for further improving the PEC performances of photoanode systems, which offers the possibility to maximize the activity of the reported OER catalysts. In this section, the optimized nanostructures that could effectively extract photogenerated holes, accelerate hole transfer or provide water oxidation active sites are also recognized to clarify the significance of the design approach of OER catalysts. In contrast to the previous material classification, performance comparison, and function introduction, the main purpose of structure optimization is to further improve the catalytic activity of OER catalysts in PEC water splitting applications. A comprehensive schematic illustration is shown in Fig. 3, the strategies of structure optimization could be broadly categorized into oxygen defect construction, selective interfacial bonding, elemental doping, morphological regulation, and multi-component synergy from previous relevant reports. More importantly, the corresponding design principles, design concepts, and mechanisms of OER catalysts for enhancing PEC water oxidation will be discussed in detail.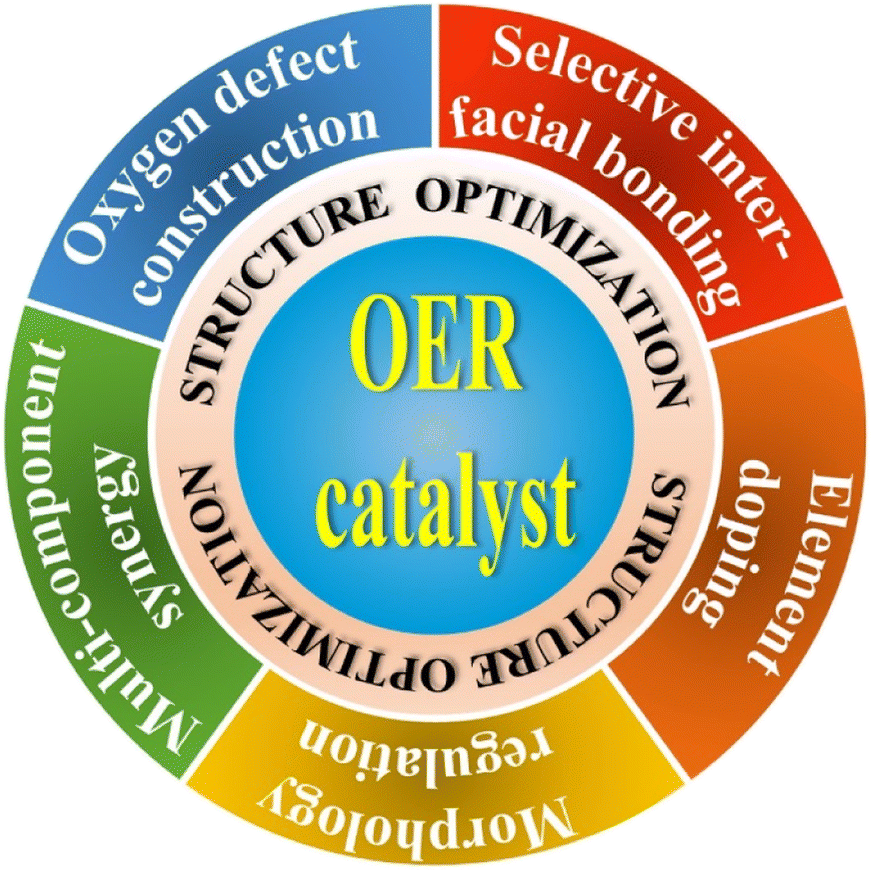 | ||
| Fig. 3 The summary of design strategies for further enhancing the OER catalyst activity. | ||
4.1 Oxygen defect construction
Oxygen vacancies (Ov) are known as one of the most common defects that form in the lattice of metal oxides or other oxygenated compounds due to the deficiency of oxygen atoms or oxygen ions, which play an important role in determining the physical and chemical properties, such as the geometrical structure, the electronic structure, adsorption/desorption characteristics, etc. Therefore, defect engineering is regarded as a promising strategy to construct high performance catalysts for thermocatalysis, electrocatalysis, photocatalysis, and other catalytic applications.71–73 For PEC water splitting, OER catalysts with abundant oxygen vacancies could efficiently improve the water oxidation activity of semiconductor photoanodes. For example, our research group has reported that the OER catalyst of β-FeOOH with abundant oxygen vacancies not only facilitated hole transport and trapping, but also provided more active sites for water oxidation. The as-prepared β-FeOOH/BiVO4 photoanode exhibits a photocurrent of 4.3 mA cm−2 at 1.23 V vs. RHE, which is more than four times that of the pristine BiVO4 photoanode and nearly two times that of the conventional FeOOH decorated BiVO4 photoanode under the same conditions.74 Zhou et al. have demonstrated that a large amount of Ov in carbon quantum dots (CQDs) could induce the outward driving force for hole trapping and migration at the BiVO4 and electrolyte interface; the photocurrent density of Ov-CQDs/BiVO4 is 3.4 times and 2.6 times higher than that of BiVO4 and CQDs/BiVO4, respectively.75Considering the beneficial functions of oxygen defects in OER catalysts, the targeted construction of oxygen defects or rational regulation of the number of oxygen defects is feasible to design high performance OER catalysts for PEC water oxidation. We have adopted Ar-plasma bombardment for constructing oxygen defects on Co3O4 catalysts. As shown in Fig. 4a, Co3O4/TiO2 could achieve a photocurrent density of 2.5 mA cm−2 at 1.23 V vs. RHE under AM 1.5G irradiation after Ar-plasma treatment, up to 2 times higher than that of the untreated Co3O4/TiO2 photoanodes. More importantly, the amount of oxygen defects could be rationally adjusted by changing the treatment time. In addition to the charge transfer resistance and surface charge separation efficiency analyses, density functional theory (DFT) calculations were performed to clarify the role of oxygen defects in improving PEC performances. As shown in Fig. 4b and c, the adsorption energy of Co3O4 with oxygen vacancies (VO-Co3O4: −1.6 eV) is lower than that of pristine Co3O4 (Co3O4: −1.1 eV), indicating reduced thermodynamic overpotential for increasing the reactivity of active sites. Additionally, the new defect states were located in the band gap, resulting in an improvement in the conductivity of Co3O4 for accelerating the hole transfer at the TiO2/electrolyte interface.76 Our group has also successfully constructed a defect-rich CoOOH nanolayer with an ultrathin structure using a similar strategy. With BiVO4 as the model photoanode, the photocurrent density and surface charge separation efficiency have achieved an outstanding enhancement.77 Besides, the high energy Ar-plasma treatment method was adopted to reconstruct the oxygen defects in the bulk of Sn-doped Fe2O3 and the surface OER catalyst of NiOOH by Guo and co-workers. Compared with the original Sn@α-Fe2O3 photoanode, enhanced photocurrent and decreased onset potential could be achieved after reconstruction of oxygen vacancies, owing to the improved charge separation.78
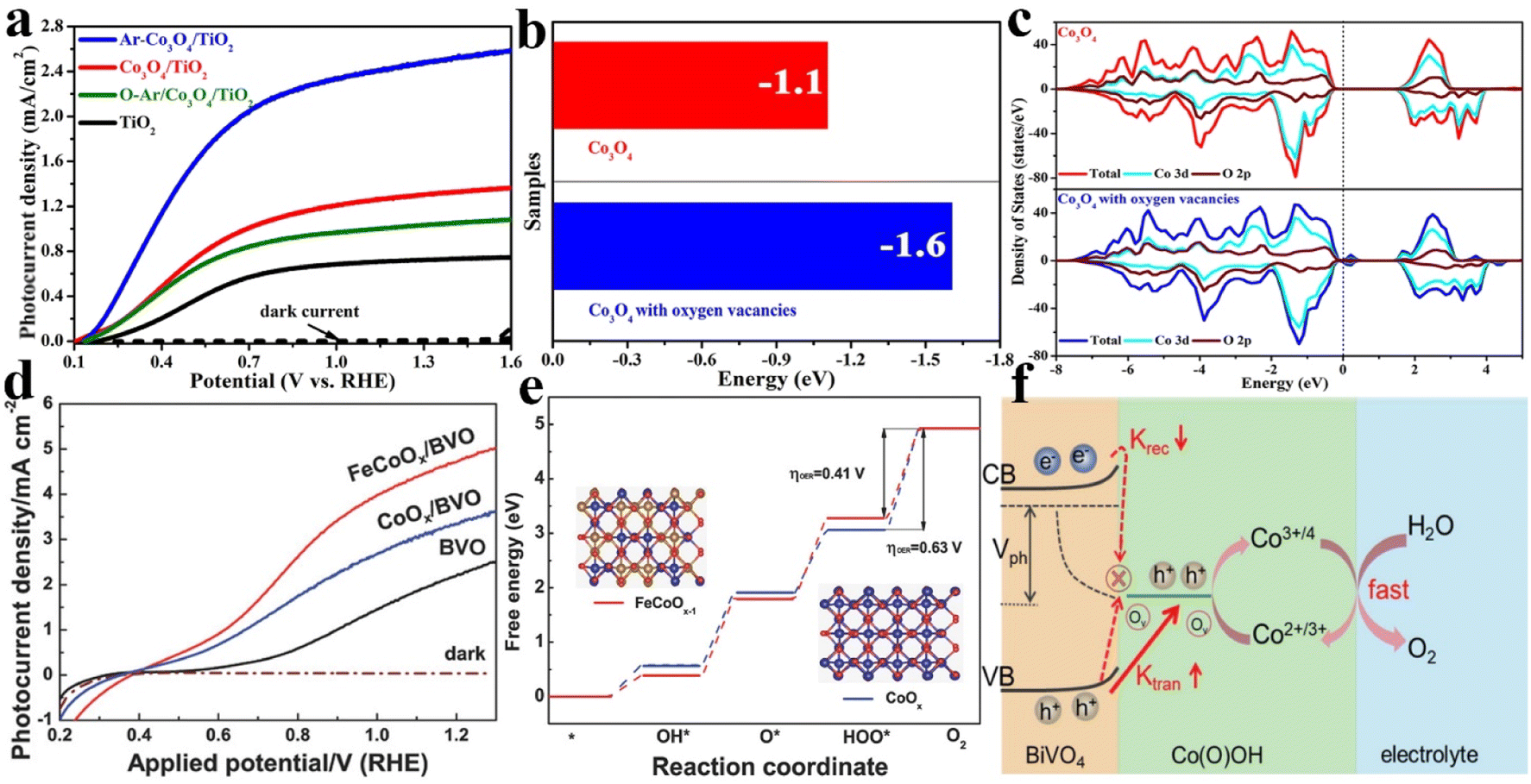 | ||
| Fig. 4 (a) The linear sweep voltammetry curves of the pristine TiO2, Co3O4/TiO2 and Ar–Co3O4/TiO2 photoanodes. The calculated adsorption energy (b) and density of states (c) for Co3O4 and VO-Co3O4. Reproduced with permission.76 Copyright 2018, Royal Society of Chemistry. (d) Photocurrent density versus applied potential curves of the BiVO4, CoOx/BiVO4 and FeCoOx/BiVO4 photoanodes. (e) Mechanism investigation of the free energy diagram of CoOx and FeCoOx−1 (i.e., FeCoOx with oxygen vacancies) for the OER pathway. Insets: the models of FeCoOx−1 (left) and CoOx (right). Reproduced with permission.79 Copyright 2018, Wiley-VCH. (f) The working models of PEC water oxidation in BiVO4/Co(O)OH photoanodes. Reproduced with permission.81 Copyright 2022, Elsevier. | ||
Recently, other strategies such as heteroatom incorporation, in situ activation and precursor regulation have also been demonstrated to construct oxygen defects in OER catalysts for further promoting the PEC water splitting activity. For instance, Wang et al. reported a two-step photo-assisted electrodeposition and subsequent calcination process for incorporating Fe into CoOx to form FeCoOx with abundant oxygen defects, which drastically increased the photocurrent density of the BiVO4 photoanode to 4.82 mA cm−2 at 1.23 VRHE (Fig. 4d). The enhanced PEC activities were ascribed to the promoted hole transport and trapping from BiVO4, and more active site creation for OER catalysts. To further identify the enhanced OER performances of FeCoOx with oxygen defects, the theoretical comprehension of the OER on FeCoOx with abundant oxygen vacancies (FeCoOx−1) and CoOx is shown in Fig. 4e. The overpotential of FeCoOx−1 (0.41 V) is lower than that of CoOx (0.63 V), suggesting a significant enhancement of the OER activities. Moreover, the weakened adsorption of the HOO* intermediate on FeCoOx−1 could facilitate the formation of O2 in the last step.79 Ge et al. displayed the in situ reconstruction of amorphous NiFeMo oxides by cyclic voltammetry (CV) treatment, and enriched surface oxygen vacancies and increased Ni oxidation states were formed owing to the formation of an oxide/hydroxide layer. The photocurrent density of pristine BiVO4 could be improved by up to 5.0 mA cm−2 at 1.23 V vs. RHE with an excellent long-term PEC stability.80 In addition, Wang and co-workers demonstrated that the self-transformation of a precursor metal–organic framework could produce an oxygen defect enriched ultrathin Co(O)OH catalyst for improving the PEC performances of BiVO4 photoanodes. As shown in Fig. 4f, the oxygen-vacancy-rich Co(O)OH nanolayer would accelerate hole transport/extraction and water oxidation kinetics, thus significantly inhibiting the surface charge recombination.81 Regardless of the methods used to construct the oxygen vacancy in OER catalysts, the key mechanism of enhanced OER performances mainly focused on the reduction of coordination numbers in surrounding atoms for generating active sites to promote the water oxidation reaction. As a result, the intrinsic activity of OER catalysts will be promoted once contributing electronic configurations or surface properties are produced. Therefore, the generation of oxygen vacancies in OER catalysts should be one of the reliable strategies for achieving their optimal structure in PEC water splitting.
4.2 Selective interfacial bonding
In general, most OER catalysts are loaded directly onto a specific semiconductor photoanode by random accumulation. Under this circumstance, an empty space is usually formed at the interfaces between the photoanode and OER catalyst, which would act as a depletion layer to prevent the charge transfer; thereby additional driving force will be required to push the charge carriers and overcome this interfacial barrier.82 In order to reduce this hindrance, the principles of the energy band or lattice matching are usually adopted to combine OER catalysts and photoanode materials. For example, Hu et al. reported a BiVO4 photoanode with a stacked structure, in which BiVO4, cadmium sulfide nanosheets, hole transport molecules (carbazole, triphenylamine, and phenothiazine derivatives), and OER catalysts (CoBi) are sequentially assembled by a cascade band alignment for accelerating the hole transfer and water oxidation efficiency.83 Min et al. demonstrated that the formed junction between MnO2 and the (111) plane of TiO2 could provide a stronger interface electric field for boosting the catalytic activity owing to the befitting energy levels.84 Although the above strategies have achieved a favourable effect to some extent, the barrier of charge transfer cannot be completely removed.The interatomic chemical bonding of photoanode materials and OER catalysts may be a practicable route to eliminate charge depletion space for improving PEC activity and stability. Inspired by this principle, as shown in Fig. 5a, we have adopted a facile nitrogen plasma engraving strategy for constructing an oxygen vacancy enriched N-doped surface on TiO2 nanotube arrays. Owing to the strong coordination of O–metal and N–metal bonds, the MoO2 catalyst was deposited on the TiO2 surface through selective bonding between the Ti/O atom in TiO2 and the O/Mn atom in MnO2. The obtained MnO2/N–TiO2 photoanode exhibited a photocurrent density of 1.95 mA cm−2 at 1.23 VRHE as shown in Fig. 5b, which is much higher than that of the pristine TiO2 nanotube arrays decorated with MnO2 (0.72 mA cm−2 at 1.23 VRHE). From the results of binding energy shift of X-ray photoelectron spectroscopy (XPS), as well as the corresponding electrochemical characteristics, the rapid hole transfer from TiO2 to MnO2 could be ascribed to the advantageous bonding configuration.85 In addition, the FeNi oxyhydroxide was in situ coated on the BiVO4 surface by a pH-modulated route. As shown in Fig. 5c, the prepared BiVO4/FexNi1−xOOH exhibits a photocurrent density of 5.8 mA cm−2 with excellent stability at 1.23 VRHE under AM 1.5G solar irradiation. Although the BiVO4/FeOOH photoanode shows an initial photocurrent value close to that of BiVO4/FexNi1−xOOH, it presents a very poor PEC stability, while BiVO4/NiOOH with high stability gives a low photocurrent under the same test conditions. The detailed comparison experiments and angle-resolved XPS results demonstrated that Fe and Ni could selectively tailor the electronic structure and chemical bonds of surface Bi and V sites on the BiVO4 photoanode. The possible formation process is shown in Fig. 5d and e, revealing that the priority formation of Bi–O–Fe bonds is due to the higher bind energy of Fe atoms and Bi sites, and then V–O–Ni bonds are formed at the interface. More specifically, the formation of Bi–O–Fe interfacial bonds could efficiently promote the hole transfer for promoting OER activities, while V–O–Ni bond formation effectively restrains the dissolution of V5+ ions from the BiVO4 lattice and enhances the PEC stability.86
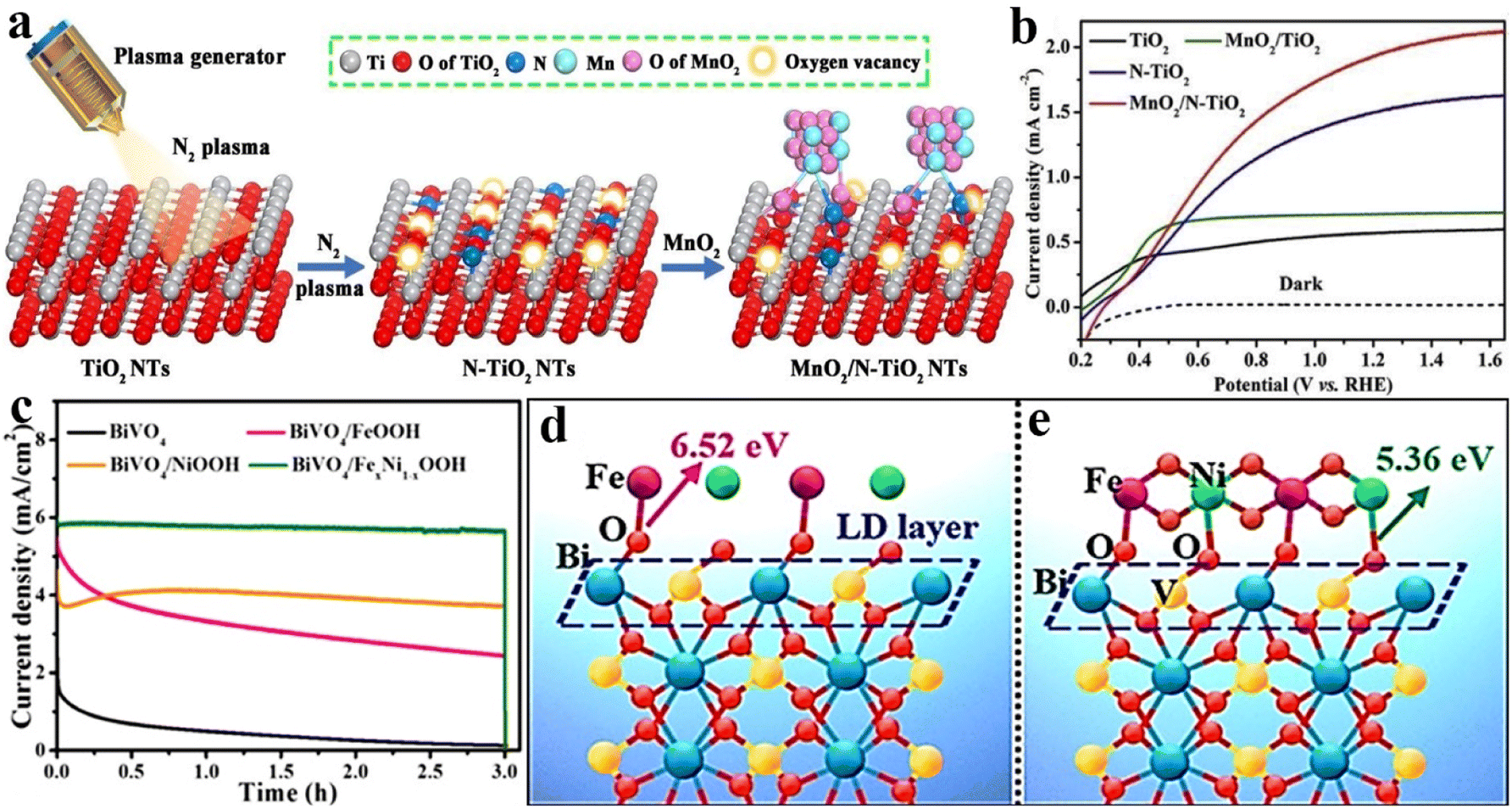 | ||
| Fig. 5 (a) Schematic illustration of the crystal structure for the fabrication process of N–TiO2 and MnO2/N–TiO2 photoanodes. (b) Linear-sweep voltammograms of TiO2, MnO2/TiO2, N–TiO2 and MnO2/N–TiO2 nanotube arrays. Reproduced with permission.85 Copyright 2020, Elsevier. (c) Linear-sweep voltammograms for BiVO4, BiVO4/FeOOH, BiVO4/NiOOH and BiVO4/FexNi1−xOOH photoanodes. (d) and (e) The possible formation process of FexNi1−xOOH on BiVO4 photoanodes. Reproduced with permission.86 Copyright 2020, Wiley-VCH. | ||
To achieve further breakthroughs in the performance of BiVO4 photoanodes for PEC water oxidation, our group has developed a phosphorus–oxygen bonding strategy to rationally regulate the interfacial electronic coupling between FeNi catalysts and BiVO4 photoanodes. The produced FeNiPOx catalyst on BiVO4 could significantly promote the PEC activity for water oxidation, which exhibits an outstanding photocurrent density of 6.73 mA cm−2 at 1.23 vs. RHE, accompanying a lower onset potential and high durability in PEC water splitting. The main role of P–O interfacial bonding is to promote the electron transfer from Bi sites to Fe atoms and the electron injection from Ni atoms to the V sites, resulting in the formation of high-valence Bi(3+x)+ and low-valence V(5−x)+ in the BiVO4 lattice. Under light excitation, owing to the enrichment of electrons on Fe sites, the photogenerated holes could rapidly transfer from BiVO4 to Fe active sites to participate in the water oxidation reaction, which could effectively reduce the charge recombination and promote the interface hole transfer. Additionally, benefiting from the electron injection of Ni atoms, the V(5−x)+ sites with low valence states in the BiVO4 lattice could effectively restrain V dissolution during the PEC water oxidation process for achieving superior photoelectric stability.87 Besides, Hou et al. and co-workers utilized atomically dispersed Ni–N4 sites coordinated with an axial direction oxygen atom to generate Ni–N4–O configuration on the BiVO4 surface for boosting the PEC activity. The constructed FeOOH/Ni–N4–O/BiVO4 photoanode exhibits an excellent photocurrent density of 6.0 mA cm−2 at 1.23 VRHE and long-term photostability under AM 1.5G simulated sunlight, which have been ascribed to the formation of a chemical bonding bridge of Ni–O–Bi or Ni–O–V at the intimate interface between Ni–N4–O and BiVO4.88 Therefore, employing selective interfacial bonding OER catalysts and semiconductor photoanodes is an advanced strategy to further enhance the PEC activity, which can provide a fast channel for hole transport through interatomic bridging at their contact interfaces.
4.3 Element doping
Elemental doping in semiconductors is the incorporation of small amounts of heteroatoms into the lattice for improving electrical, optical, magnetic properties, etc. In general, owing to the formation of impurity states in the forbidden band, the light absorption edge and the electrical conductivity will be regulated by producing the donor/acceptor level or increasing the carrier density after the doping of heteroatoms. Besides, the dopants could induce the formation of shallow potential traps or cause lattice defects to inhibit photogenerated carrier recombination and create oxygen vacancies. Therefore, elemental doping was widely used in photocatalysis, electrocatalysis and photoelectrocatalysis for constructing anticipant catalyst materials.89–93Inspired by previous doping studies, this strategy was also applied to design OER catalysts with superior activity for PEC water splitting applications. For example, Zheng et al. reported that an Ni doped FeOOH catalyst (Ni:FeOOH) could be coated on various photoanodes by a one-step hydrothermal process. As shown in Fig. 6a, the amount of Ni doping affects the PEC performance of the Ni:FeOOH-coated BiVO4 film, which has been ascribed to the adjusted charge-transfer efficiency.94 Ao and co-workers reported the Ru doped SrTiO3 (Ru–SrTiO3) perovskite as the OER catalyst on the BiVO4 photoanode, which could provide oxidation active sites during the PEC water oxidation process by reducing the reaction energy barrier at the solid–electrolyte interface. Furthermore, the corresponding theoretical calculations shown in Fig. 6b and c indicate that the water oxidation reaction is more likely to occur on the (110) crystal plane of Rh–SrTiO3 because the oxygen evolution reaction overpotential on the (110) crystal plane is reduced significantly. Therefore, the obtained BiVO4/Rh5%–SrTiO3 photoanode exhibits an optimized photocurrent density of ∼0.74 mA cm−2 at 1.23 VRHE, which is much higher than that of BiVO4/Rh–SrTiO3 (∼0.27 mA cm−2 at 1.23 VRHE).95 Jang et al. demonstrated that Ti doping could promote the charge extraction properties and conductivity of SiOx, and the stability of the as-prepared photoanode was also enhanced by the high chemical inertness of the Ti–SiOx layers.96 They also reported that Ti doping could reduce the particle size of FeOOH as well as lead to less deposition on the Fe2O3 surface, which could avoid light blocking and facilitate hole transport.97 Mo et al. reported that NiOx could serve as a hole transport layer owing to its improved hole transfer ability after Cu doping.98
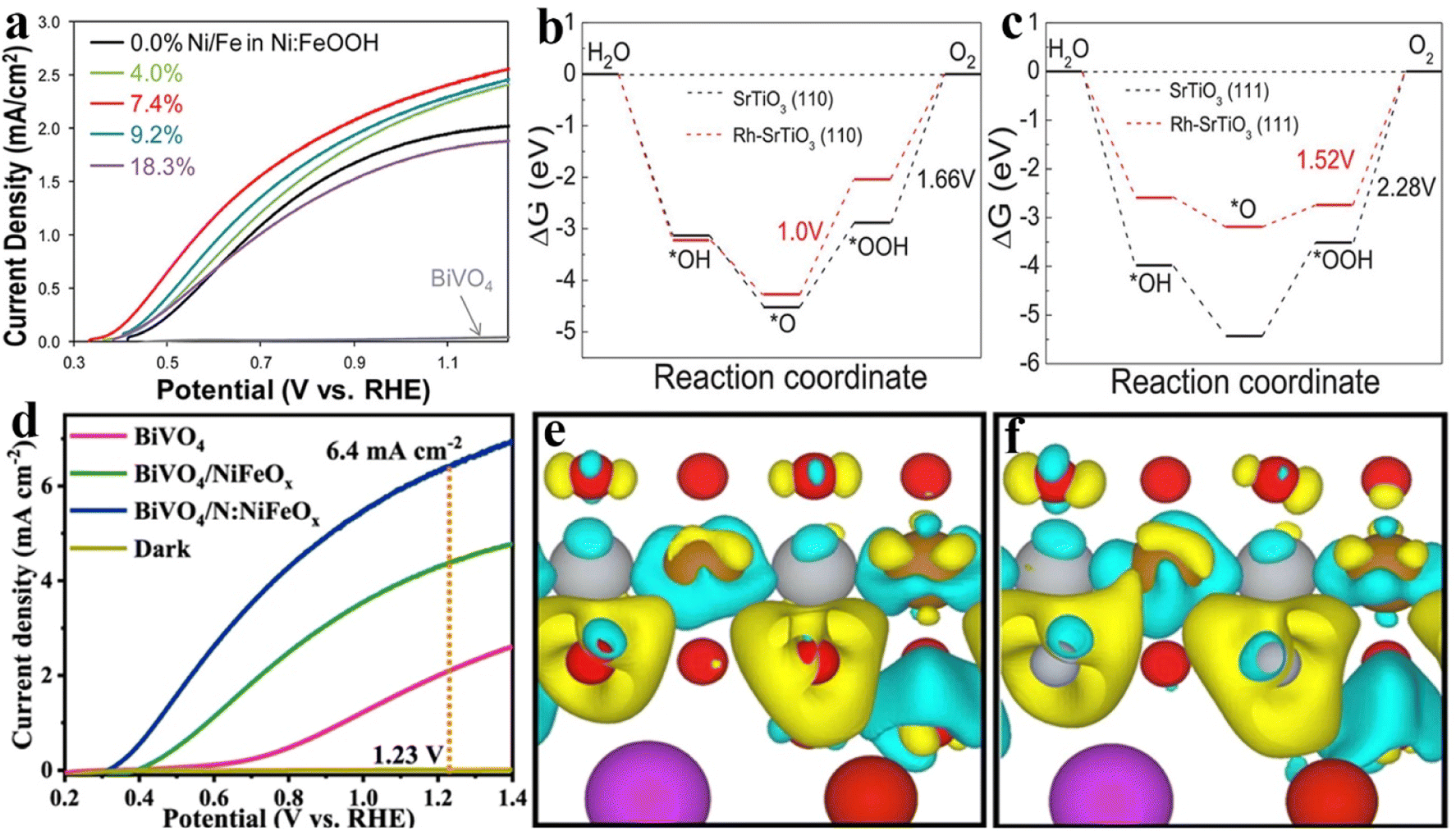 | ||
| Fig. 6 (a) Photocurrent density versus potential curves of BiVO4 planar films with and without Ni:FeOOH (Ni/Fe ratios vary between 0.0 and 18.3% estimated from XPS data) in a 0.5 M potassium phosphate electrolyte buffered to pH 7 under AM 1.5G illumination. Reproduced with permission.94 Copyright 2016, American Chemical Society. (b) and (c) The calculation results of standard Gibbs free energy diagrams for the OER over the (110) and (111) facets of SrTiO3 or Rh–SrTiO3 at U = 1.23 V. Reproduced with permission.95 Copyright 2019, Wiley-VCH. (d) Linear-sweep voltammograms of BiVO4, BiVO4/NiFeOx, and BiVO4/N:NiFeOx photoanodes at 1.23 V vs. RHE in 0.5 M K3BO3 (pH = 9.5) electrolyte. (e) and (f) Schematic of charge density differences (yellow and cyan represent charge accumulation and depletion, respectively; the cut-off of the density-difference isosurface is 0.01 Å−3) of BiVO4/NiFeOx (e) and BiVO4/N:NiFeOx (f). Reproduced with permission.101 Copyright 2021, Springer Nature. | ||
In addition, many non-metallic elements exhibited excellent advantages as dopants in the modification of OER catalysts. Owing to the fluorine (F) element’s strongest electronegativity, F incorporation into OER catalysts could induce the positive charge densities on the metal sites to elevate OER activity. As well, the N–metal bonds have higher electrical conductivity than O–metal bonds, thereby the N element is generally selected as a heteroatom to dope metal oxide OER catalysts. For instance, the F doped FeOOH brings a higher photocurrent density and water oxidation activity than undoped FeOOH in an Fe2O3 or WO3 photoanode system, and both of them could decrease the lower overpotential and facilitate hole transfer.99,100 Recently, we reported that N doping could activate NiFeOx catalysts (N:NiFeOx) for promoting the oxygen evolution activity and stability of BiVO4 photoanodes. As shown in Fig. 6d, the photocurrent density of BiVO4/N:NiFeOx (6.4 mA cm−2 at 1.23 VRHE) is much higher than that of BiVO4/NiFeOx (4.4 mA cm−2 at 1.23 VRHE) and the pristine BiVO4 photoanode (2.1 mA cm−2 at 1.23 VRHE). Because of the partial substitution of O sites in NiFeOx catalysts by N atoms, the Fe and Ni sites possess enriched electron densities. Thus, the electron-enriched Ni sites conversely donated electrons to V sites for restraining V5+ dissolution, and the electron-enriched Fe sites attract photo-generated holes from BiVO4 surfaces. In addition, the charge density difference calculation was used to reveal the change in electron densities in Fe and Ni sites after incorporation of N atoms. As shown in Fig. 6e and f, the increased electron densities could be observed at Fe and Ni sites after N doping, which further explains the crucial role of N-incorporation in regulating the electronic structures of NiFeOx.101 Zhu and co-workers demonstrated that N-doping into a cobalt–iron oxide (CFO) catalyst could boost the PEC performances of BiVO4 due to the enhancement of electrical conductivity and reaction activity. By comparing this with CFO with oxygen vacancies, they suggest that N dopants may be a more effective defect to improve the activity of OER catalysts for PEC water oxidation.102 Different from the doping-enhanced properties of OER catalysts by altering the electron structure, Kim et al. reported that S-doping not only improved the catalyst activity of the FeOOH catalyst, but also produced FeOxSy to form a heterojunction with the Fe2O3 photoanode, resulting in an internal electric field for facilitating charge separation and transfer.103
From the above element-doping studies, it is difficult to propose a conventional selection guideline to construct OER catalysts for further enhanced activity, either for metallic elements or non-metallic elements. Nonetheless, a number of general considerations can be highlighted in the doping studies of OER catalysts, such as whether doping causes unintentional damage to the underlying photoelectrode materials, whether doping will lead to changes in the morphology of the original OER catalyst, and whether doping may result in the creation of new species in the OER catalyst. These are important for understanding and elucidating the inner mechanisms of accelerated charge separation, transfer and utilization by element-doping. Besides, doping design and performance optimization of OER catalysts need to be investigated together with photoanodes in PEC water splitting, rather than considering their OER catalytic activity in isolation.
4.4 Morphology regulation
Semiconductor nanomaterials with a well-defined morphology will exhibit specific properties due to their structural characteristics; thereby morphology design is considered one of the effective routes to fabricate high-performance materials. Especially in the field of photo(electro)catalysis, morphological regulation offers significant advantages, such as high electrical conductivity, abundant active sites, excellent stability, and outstanding light absorption. More specifically, zero-dimensional (0-D) morphology, such as single atoms, clusters, quantum dots and small size nanoparticles, features large surface state density and significant quantum size effects, which could facilitate visible and infrared light absorption and active site fabrication.104,105 One-dimensional (1-D) morphology, including nanowires, nanofibers, and nanotubes, has the advantages of high aspect ratios, high conductivity, and good chemical stability, attracting a great deal of application in improving catalytic performances.106 As well, two-dimensional (2D) morphology with large specific surface areas, short carrier migration distances, and many active surface atoms exhibits critical roles in adsorbing reactant molecules and transporting photogenerated electrons and holes.107 Three-dimensional (3-D) shapes are generally created by stacking basic units of lower dimensional forms, which possess excellent properties such as high surface area, excellent accessibility to active sites, and enhanced mass transport and diffusion, owing to their porous channel characteristics.108Therefore, the construction of OER catalysts with unique morphology features may be an important route to promote the PEC water splitting activity. For example, owing to the up-conversion fluorescence effects, 0-D carbon quantum dots were utilized as OER catalysts to capture longer wavelength irradiation for increasing the spectrum absorption ability of the photoanode and generate short wavelength light to excite the photoanode for producing more electron–hole pairs.109,110 Chang and co-workers reported that Fe2O3 decorated with Ni(OH)2 quantum dots would exhibit a higher photocurrent density than that decorated with large sized Ni(OH)2 nanoparticles, illustrating that the smaller size would be much more favourable for hole transfer between the electrolyte and Fe2O3 photoanode.111 For the OER catalyst with 2-D morphology, Li et al. reported the Co-incorporated hydroxyapatite (Co–HAP) nanoarchitecture for boosting the PEC water splitting performances of the Fe2O3 photoanode. The 2-D nanostructure of Co–HAP allows for efficient charge transfer and water absorption, and the abundant Lewis acidic and Lewis basic sites on the surfaces could facilitate the activation/dissociation of water molecules.112 Luo et al. deposited 2-D thin Ti3C3Tx flakes on the top of the BiVO4 photoanode to facilitate the charge transfer and reduce the charge recombination, resulting in promoted PEC water oxidation activity.113 As distinguished from freestanding 2-D morphology on the photoanode surface, most 2-D structural OER catalysts are tightly adhered to the electrode surface by using a photo-absorbing semiconductor as a support skeleton, which usually wraps around the entire photoanode in the form of ultra-thin structured nanolayers. For instance, Zhang and co-workers designed an OER catalyst TANF by means of phenolic ligand (tannic acid, TA) coordination of Ni and Fe ions. As shown in Fig. 7a and b, compared to traditional OER catalysts with island growth, TANF consists of 2-D networks by connecting TA molecules with metal ions, resulting in an intact and uniform thin-film on the photoanode surface. As a result, the Mo:BiVO4 photoanode decorated with 2-D TANF exhibits a higher photocurrent density of 5.1 ± 0.13 mA cm−2 at 1.23 VRHE than that modified by other OER catalysts (ferrihydrite and cobalt phosphate) with island morphology.114 Abdi et al. reported that a MnOx modified BiVO4 photoanode will achieve an optimum photocurrent when the 2-D MnOx catalyst has a thickness of about 4 nm. The effects of MnOx thickness on the PEC performances were investigated and are shown in Fig. 7c–e. More specifically, the energy band bending at the BiVO4–MnOx interface would increase with increasing MnOx thickness, leading to a lower surface majority carrier concentration, reduced surface recombination, and higher photocurrent. Nevertheless, thick MnOx films would induce charge shunting between the BiVO4 surface and the conductive substrate owing to the direct contact between MnOx and the substrate.115 It can be inferred from this study that the suitable thickness of 2-D morphology OER catalysts on a semiconductor surface is an important parameter for determining its OER activity; the ultra-thin configuration (about 1–4 nm) may be a feasible choice. For example, NiFeOx with a thickness of ∼3 nm on the Fe2O3 photoanode offered more effective PEC water oxidation activity than that with a thickness of 0.6 nm or 5 nm.116 Compared with thick NiOOH catalysts, ultra-thin NiOOH exhibits more efficient OER catalytic activity in BiVO4 photoanode systems.117
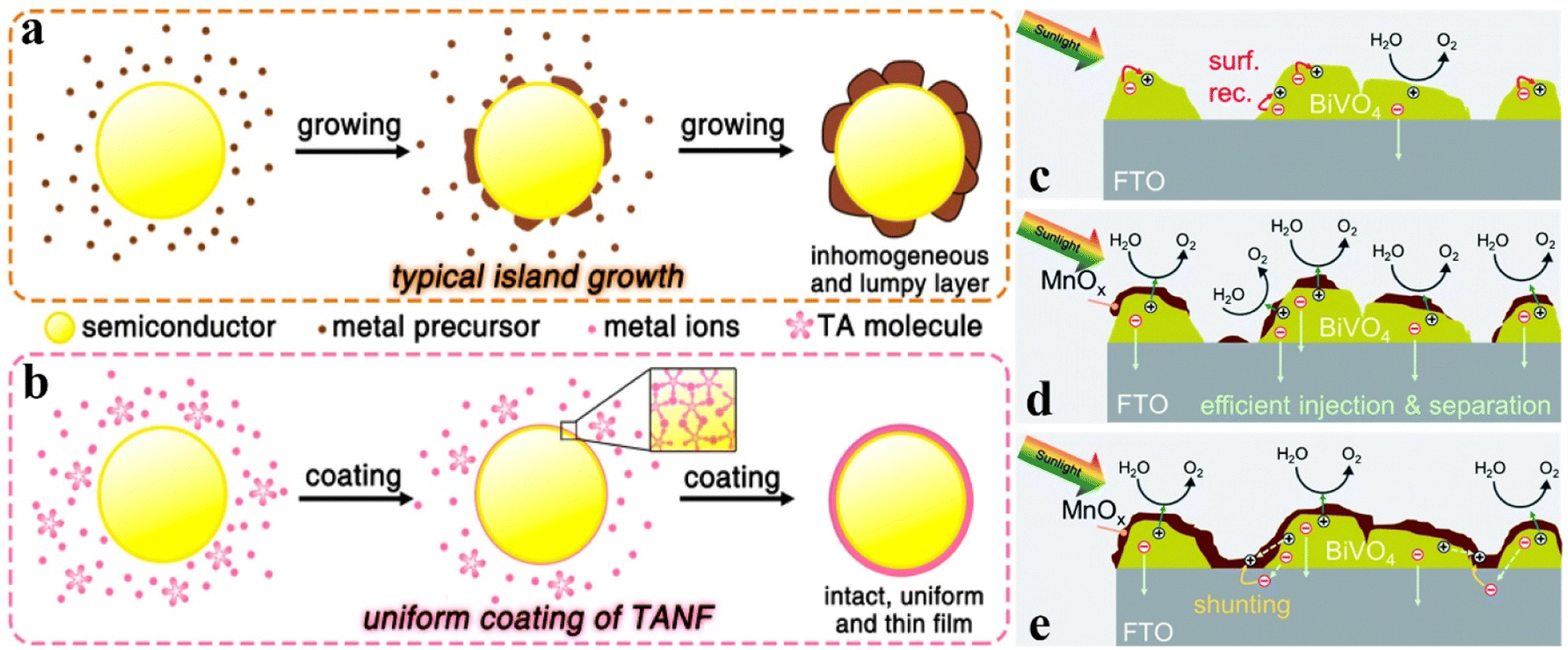 | ||
| Fig. 7 (a) and (b) Schematic illustrations of the growth procedure for (a) island growth of traditional cocatalysts and (b) coating of TANF on the surface of the semiconductor. Reproduced with permission.114 Copyright 2018, American Chemical Society. (c) In bare BiVO4, charge transfer from the surface of BiVO4 is limited by surface recombination (red arrows). (d) When a thin MnOx film is deposited on the surface of BiVO4 (up to 4 nm), a discontinuous layer is formed. At the interface, the band bending is enhanced, and the surface recombination is suppressed. (e) Depositing a thicker MnOx film (>4 nm) leads to a full coverage, even at the exposed FTO surface. Direct contact between MnOx and FTO results in shunting pathways. Reproduced with permission.115 Copyright 2020, Royal Society of Chemistry. | ||
Besides, OER catalysts with 3-D morphology are often applied in photoanode construction for efficient water oxidation. Ye et al. assembled Sn3O4 nanosheets on TiO2 nanorod arrays to obtain 3-D morphology configurations, which could facilitate light capture and a large contact area with the electrolyte. Accordingly, a three-fold enhanced photocurrent density was obtained for this 3-D Sn3O4/TiO2 photoanode.118 Similar to the thickness regulation of a 2-D structure, the 3-D morphology of Sn3O4 could be rationally adjusted by varying the precursor concentrations. Under suitable conditions, both large surface area and good charge transport properties could be achieved.119 Moreover, 3-D OER catalysts could also address the shortcomings of insufficient light absorption, limited loading and severe carrier recombination in light-harvesting semiconductors. Yang and co-workers displayed a decoupling strategy for constructing 3-D OER catalysts of a pore-spanning crisscross conducting polymer (CP) by the in situ cogrowth of FeOx nanoparticles and a CP network (CP-FeOx). Owing to the large space and electron blocking layer of the CP matrix, this 3-D CP-FeOx catalyst could effectively boost the PEC water oxidation performances of different nanostructured photoanodes such as TiO2 nanorod arrays, WO3 nanosheet arrays and planar TiO2 nanoparticle films.120 Therefore, morphological regulation of OER catalysts should be a promising strategy for further improving the PEC water splitting activity.
4.5 Multi-component synergy
Because of their crucial roles in PEC water splitting, it is desirable to develop ideal OER catalysts that present the outstanding features of promoting charge separation/catalyst activity, hindering surface recombination/photo-corrosion, and enhancing wettability. However, considering the intrinsic physical/chemical properties of OER catalysts, such as lattice or energy level match, coverage area ratios and the light shielding effect, it is difficult to construct such a perfect OER catalyst in practical photoanode systems. In addition to maximizing the inherent functions of the individual OER catalyst, use of multiple OER catalyst hybrids has become an effective route to acquire the desired OER performances.121 The most representative study is the use of the dual-OER catalysts of FeOOH and NiOOH for enhancing the photocurrent density of BiVO4 photoanodes. The prepared BiVO4/FeOOH junction could effectively reduce interface recombination, whereas the overlying NiOOH is conducive to create a more favourable Helmholtz layer potential drop between the OER catalyst and electrolyte. Thereby, improved surface electron–hole separation and enhanced PEC activity were achieved for this BiVO4/FeOOH/NiOOH photoanode with an optimized OER catalyst structure.122 Since then, coupling two or more OER catalysts has been extensively employed to construct high-performance “OER catalysts/semiconductor” photoanode systems. For example, ferrihydrite (Fh) and Co3O4 nanoparticles were deposited on Ta3N5 to construct the Co3O4/Fh/Ta3N5 photoanode for PEC water splitting, and the hierarchical structures of Ta3N5 and OER catalysts were displayed using a high-resolution transmission electron microscopy (HRTEM) image (Fig. 8a). As shown in Fig. 8b, Ta3N5 has exhibited an enhanced photocurrent density after single Fh or Co3O4 loading, while the photostability of Co3O4/Ta3N5 is far below that of the Fh/Ta3N5 photoanode. Interestingly, the dual-decorated Ta3N5 photoanode not only achieved a high photocurrent value similar to that of Co3O4/Ta3N5, but also maintained the same outstanding stability as Fh/Ta3N5. From the charge injection efficiency calculations and the charge storage capacity analysis (Fig. 8c), it was found that the Co3O4 nanoparticles and Fh layer exhibit accelerated surface water oxidation reactivity and increased hole storage ability for improving PEC activity and stability of the Ta3N5 photoanode, respectively.123 As shown in Fig. 8d–f, their group also constructed Ti–Fe2O3/Ni(OH)2/IrO2 photoanodes by coupling Ni(OH)2 and IrO2 OER catalysts. Owing to the synergetic effect of the improved interfacial charge transfer of Ni(OH)2 and highly efficient hole utilization of IrO2, this Fe2O3 photoanode achieved a significantly improved photocurrent density.124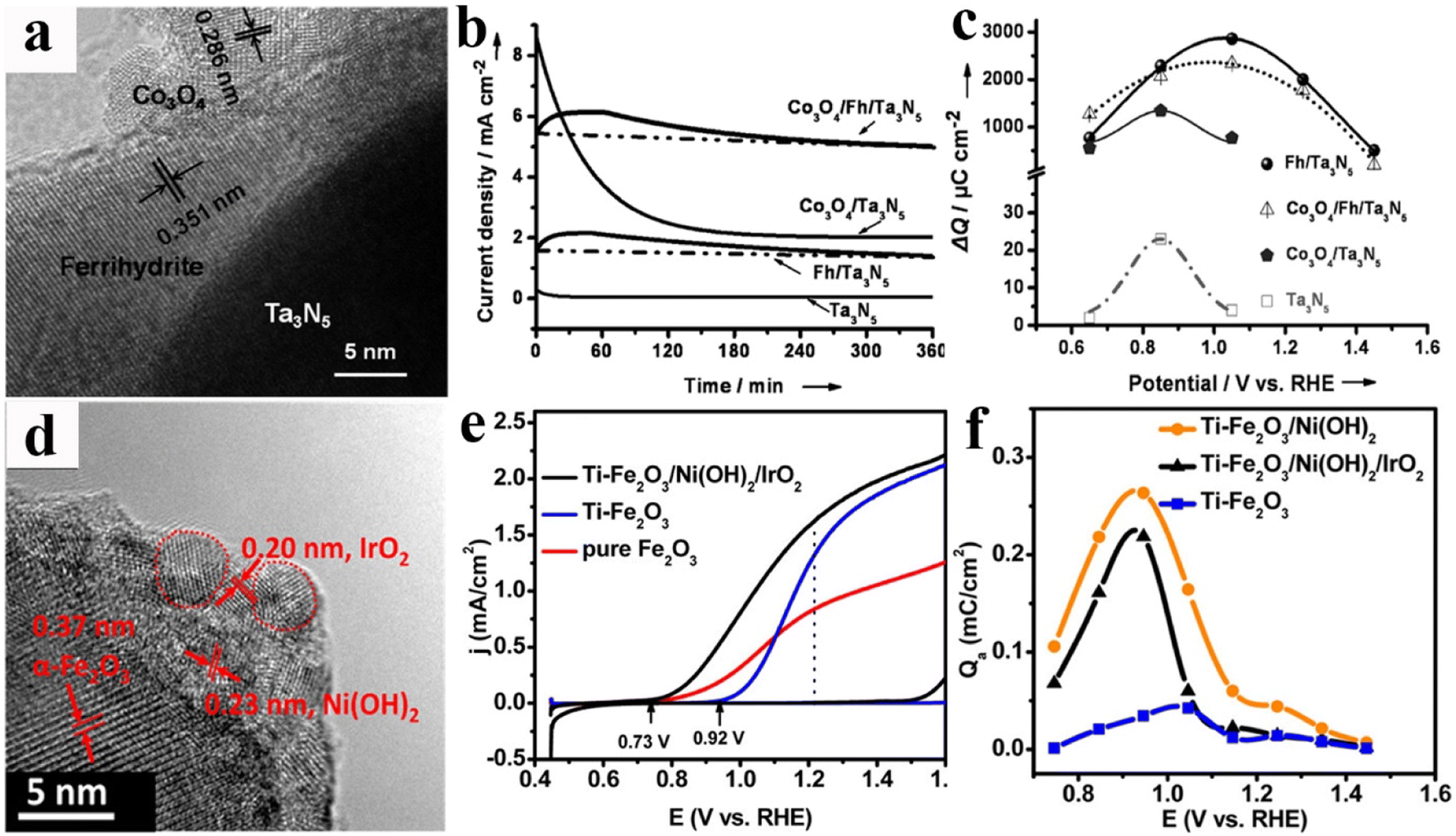 | ||
| Fig. 8 (a) HRTEM image of a Co3O4/Fh/Ta3N5 film. (b) Chronoamperometry measurements of the above photoanodes at 1.23 V vs. RHE under AM 1.5G simulated sunlight in 1 M NaOH aqueous solution. (c) Charge storage versus potential curves of Ta3N5, Fh/Ta3N5, Co3O4/Ta3N5, and Co3O4/Fh/Ta3N5 photoanodes. Reproduced with permission.123 Copyright 2014, Wiley-VCH. (d) HRTEM image of the Ti–Fe2O3/Ni(OH)2/IrO2 photoanode. (e) Current–voltage curves of the electrodes of pure Fe2O3, Ti–Fe2O3 (blue), and Ti–Fe2O3/Ni(OH)2/IrO2 photoanodes. (f) Accumulated charge in the spike at different potentials. Reproduced with permission.124 Copyright 2015, American Chemical Society. | ||
Metal oxides and metal hydroxides are the most common OER catalysts for enhancing PEC water oxidation performances. Zou et al. reported that AgOx and NiOx could synergistically enhance the bulk phase charge separation and surface charge transfer of thin BiVO4 photoanodes for promoting PEC water splitting, and the relevant kinetic and thermodynamic function analyses have been performed. For the composite of AgOx and NiOx catalysts, the author considered that one component of the AgOx/NiOx composite may stabilize the high valence states of the other metal ions, and the high valence states of Ag or Ni ions are responsible for the construction of surface active sites and Schottky-type electrode–electrolyte junctions with a high barrier height.125 For the low hole utilization efficiency of Si-based electrodes, Oh and co-workers designed a double-layer CoOx catalyst decorated n-Si photoanode with a photocurrent density of 3.5 mA cm−2 without a buried junction, which could also maintain a saturating current density of 32.5 mA cm−2 without noticeable degradation during 12 h. Although Co3O4 possesses excellent water oxidation activity, it has a significant valence band offset with n-Si, which would block hole transport from n-Si to electrolyte. However, the less active CoO could efficiently extract holes from n-Si due to its suitable energy band structure. Therefore, the double-layered CoO and Co3O4 on Si-based electrodes could synergistically eliminate this competitive correlation for achieving high efficiency and stable PEC water splitting performances.126
Besides, C-based materials are also selected for coupling with metal-based OER catalysts in PEC water splitting owing to the high conductivity and tailoring effect of the electronic structure for surface active sites. Gong et al. reported the synergistic accelerated water oxidation of carbon nanodots and Co3O4 nanoclusters in the Fe2O3 photoanode. As shown in Fig. 9a, the C/Co3O4–Fe2O3 photoanode exhibited a photocurrent density of 1.48 mA cm−2 at 1.23 VRHE, which is 78% higher than that of the pristine Fe2O3 photoanode accompanied by an obvious 60 mV cathodic shift of onset potential. Compared with the four-electron water oxidation reaction on fast-reaction sites of Co3O4, the formed H2O2 on slow-reaction sites was further oxidized to molecular O2 on C nanodots, which results in a kinetically beneficial two-step-two-electron reaction for promoting the oxygen evolution (Fig. 9b).127 Our group has coupled carbon quantum dots (CDs) with phosphating NiCo (NiCo–P) catalysts on the Fe2O3 photoanode surface to construct an dual-modified photoanode of Fe2O3/NiCo–P(CDs), which exhibited an outstanding photocurrent density of 4.26 mA cm−2 at 1.23 VRHE in 1 M NaOH electrolyte (Fig. 9c). As shown in Fig. 9d, CDs could effectively enhance the interfacial interactions between Fe2O3 and the NiCo–P catalyst. Specifically, the CDs could facilitate the electron transfer from the Co sites of NiCo–P to the Fe sites of Fe2O3, and improve the hole extraction from the Fe sites of Fe2O3 to the electron-enriched Ni2+ sites of NiCo–P, resulting in reduced charge recombination and enhanced water oxidation activity.128
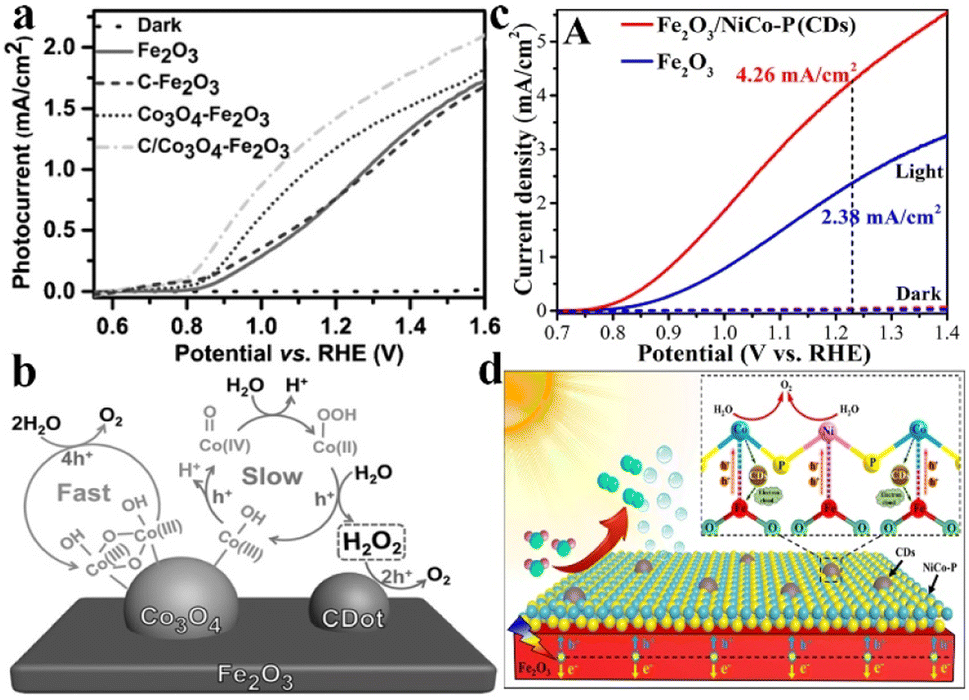 | ||
| Fig. 9 (a) Current–voltage curves of Fe2O3, C–Fe2O3, Co3O4–Fe2O3, and C/Co3O4–Fe2O3 photoanodes. (b) Schematic illustration of the fast/slow reaction processes on the Co3O4 OER catalyst and the two-step-two-electron reaction pathway for photocatalytic water oxidation on the C/Co3O4–Fe2O3 photoanode. Reproduced with permission.127 Copyright 2016, Wiley-VCH. (c) The LSV curves of Fe2O3 and Fe2O3/NiCo–P(CDs) photoelectrodes. (d) Schematic illustrations of charge separation and transfer on the Fe2O3/NiCo–P(CDs) photoanode. Reproduced with permission.128 Copyright 2022, Royal Society of Chemistry. | ||
Recently, because of the advantages of facile preparation, excellent activities and adjustable structures, many molecule catalysts have been widely used as OER catalysts for decreasing the reaction barrier and improving water oxidation performances. However, the low electrical conductivity requires that the molecule catalysts couple with other conductive catalysts to improve charge transfer capability. For example, Yu et al. demonstrated an innovative BiVO4–N/C–CoPOM photoanode for PEC water oxidation. Owing to the accelerated hole transfer of N-doped carbon (N/C) and reduced energy barrier of cobalt polyoxometalate (CoPOM), the photocurrent density of BiVO4 photoanodes has been increased up to 3.30 mA cm−2 at 1.23 VRHE, which is a 5.4-fold enhancement over that of the pristine BiVO4 photoanode.129 Du et al. demonstrated the deposition of molecular cobaloxime and Ni-(oxy)hydroxide/borate (Ni-OEC) on the BiVO4 photoanode, achieving a superior photocurrent density of 5.1 mA cm−2 at 1.23 VRHE under AM 1.5G illumination.130 Qiu et al. reported the in situ formation of NiOOH on the Ni2P surface to construct a core–shell structure for PEC water oxidation, and the coupling of the high electrical conductivity of Ni2P nanocrystals with high oxygen evolution activity of NiOOH for guaranteeing the enhancement of OER activity of the hematite photoanode.131 These studies illustrate the versatility and importance of the multi-component synergy of OER catalysts for the design and construction of PEC photoanodes. Furthermore, the representative PEC performances of typical photoanodes were briefly described here. To our knowledge, Ta3N5,132 BiVO4,87 Fe2O3,133 and TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 134 have achieved the optimum photocurrent densities of about 12.5 mA cm−2, 6.7 mA cm−2, 6.0 mA cm−2 and 3.2 mA cm−2 at 1.23 VRHE under AM 1.5G solar irradiation after being modified with AlOx–Fh–Co3O4, FeNiPOx, TiO2–CoPi and CQDs, respectively. Thereby, OER catalysts as well as their structural optimizations should be crucial in the design and construction of high efficiency photoanode systems.
134 have achieved the optimum photocurrent densities of about 12.5 mA cm−2, 6.7 mA cm−2, 6.0 mA cm−2 and 3.2 mA cm−2 at 1.23 VRHE under AM 1.5G solar irradiation after being modified with AlOx–Fh–Co3O4, FeNiPOx, TiO2–CoPi and CQDs, respectively. Thereby, OER catalysts as well as their structural optimizations should be crucial in the design and construction of high efficiency photoanode systems.
5. Conclusions and outlook
The water oxidation half-reaction in PEC water splitting is crucial to determine the ultimate solar-to-hydrogen conversion efficiency. During the past few decades, remarkable progress has been achieved in the exploration and utilization of OER catalysts for constructing high efficiency and stable semiconductor photoanode systems. In this review, we have summarized the structure optimization of OER catalysts, and discussed the design principles, specific strategies and the corresponding mechanisms for further improving their catalytic performances. The intention of loading an OER catalyst on a semiconductor photoanode was mainly focused on improving PEC conversion efficiency and stability. Therefore, its possible adverse influences such as the light blocking effect, charge transfer recombination, poor surface coverage, interface mismatches, and insufficient active sites should also be considered. Although photoanodes have achieved significant improvements in surface charge separation, photogenerated hole transfer and PEC stability after OER catalyst decoration, the fact is that a state-of-the-art “OER catalyst/semiconductor” photoanode system cannot satisfy a commercial solar-to-chemical energy conversion efficiency of >10%. This means that there are still many challenges in the future to explore more suitable semiconductor light-harvesting materials and better OER catalysts. Based on the topics discussed in this review, the following challenges and opportunities are highlighted for developing highly efficient OER catalysts for PEC water splitting.5.1 Studying the inner mechanism of PEC water oxidation
To further construct efficient and reliable OER catalyst modified semiconductor photoanode systems, the definition of the active sites of the water oxidation reaction and the investigation of the O–O bond formation process remain the research focus, which will provide clear guidance for the design of appropriate OER catalysts and the selection of suitable activation methods. To date, except for the precise analysis of morphology, phase, spectrum, element, and valence state, the development of in situ characterization techniques, including in situ X-ray absorption spectroscopy (XAS), transient absorption spectroscopy (TAS) etc., is very important. For example, Smith et al. performed in situ XAS experiments at the V K-edge to identify the effect of photocharging treatment on the BiVO4 photoanode, demonstrating that bulk V always preserves the +5 oxidation state.135 Melo and co-workers investigated the charge carrier dynamics of Fe2O3 by TAS, revealing the significant electron–hole recombination within the first few hundred picoseconds and smaller amplitude of longer-lived charge carriers.136 These detection techniques could provide accurate probing of the electronic structure of catalysts or the charge transfer dynamics in the excited states. More importantly, in situ irradiation X-ray photoelectron spectroscopy (SIXPS) could directly observe the interatomic electron transfer, providing a visual analysis of charge separation and transport under excitation states.137 Furthermore, theoretical calculations are able to provide a corresponding reference for the determination of adsorption sites and the extrapolation of reaction pathways. Therefore, multi-perspective in situ characterization and theoretical calculations are essential to revealing the water oxidation reaction mechanism, guiding the selection and modification of OER catalysts.5.2 Addressing the interface issues between the OER catalyst and semiconductor photoanode
Although OER catalysts, particularly transition metal (Fe, Co, Ni, etc.)-based materials, have achieved significant progress in PEC photoanode fabrication, some existing issues still restrict their universality and efficiency in practical applications. The interface recombination between OER catalysts and photoanodes severely restricts the efficiency of charge transfer. In addition to the construction of interfacial interlayers (hole storage layers, passivation layers, etc.), selective interfacial bonding has the more outstanding advantage for eliminating the interfacial energy barriers, which allows rapid hole transfer by the chemical bonding of the OER catalyst and photoanode. The conflict between surface sites and conductivity also needs to be kept in mind. It is highly desirable that OER catalysts not only exhibit a low resistance but also have sufficient active centres, which may be accomplished by structure modulation, heteroatom introduction, or active site reconstruction. Moreover, as most metal-based catalysts are only stable in alkaline electrolyte, the stability of OER catalysts should be considered for the future application of PEC water splitting in harsh solution environments and under solar irradiation.5.3 Reducing the cost of manufacturing OER catalysts
The cost of OER catalysts, including the raw material, the preparation method, and the output proportion, are the most fundamental considerations. Although noble metal (Ru, Ir, etc.)-based materials are the best choice as OER catalysts for achieving superior oxygen evolution activity, their high cost has greatly limited their large-scale applications. Recently, earth-abundant transition metal-based materials with relatively low cost have been successfully applied to the water oxidation reaction. For example, dual-transition metal oxides and hydroxyl oxides (FeNiOx, FeNiOOH, etc.) exhibited excellent OER catalytic activity and stability on different semiconductor (BiVO4, Fe2O3, etc.) photoanodes by a simple solution impregnation process, which may be more competitive as OER catalysts for PEC water oxidation. Besides, owing to their easily adjustable structure, well-defined active sites, and high adhesion, molecular catalysts have great potential to become more effective OER catalysts, once the shortcomings in conductivity and stability are addressed.5.4 Developing the advanced devices of PEC water splitting
Recently, a traditional PEC system integrated with a solar cell (PEC-PV) for water splitting application has also been reported, which could be operated without the assistance of an external power source. For example, Shen et al. reported a core/shell TiO2@BiVO4 photoanode in combination with a CH3NH3PbI3-based perovskite solar cell configuration, achieving an overall solar-to-hydrogen (STH) efficiency of 1.24%.138 Li and his colleagues designed a PEC-PV system consisting of a transparent CdIn2S4/In2S3 photoanode and a perovskite solar cell. Owing to the ultrahigh charge separation efficiency induced by the bulk-heterojunction of CdIn2S4 and In2S3, this system delivered a STH conversion efficiency of 3.3%.139 Note that the efficiency of STH of the PEC-PV device is determined by the photocurrent density and stability of the photoanode system. Compared to conventional PEC configuration, this advanced hybrid PEC-PV device offers a new insight into the design and construction of solar-driven PEC water splitting.In summary, it is highly desirable to design and construct an efficient, stable, inexpensive and environmentally friendly “OER catalyst/semiconductor” photoanode system by combining many aspects such as the novel reaction device, photoanode material and electrolyte solution. Although some important achievements have been discussed here, many challenges and problems still need to be conquered. From the results of the research achieved so far, it is clear that with persistent hard work, commercial applications of solar-driven PEC water splitting will eventually be realized.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21832005 and 22002175); the Science and Technology Planning Project of Chengguan District of Lanzhou City (2021RCCX0018); the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA21061011); the Low Carbon Catalysis and Carbon Dioxide Utilization National Key Laboratory Self-Deployment Project supported by the LICP (2022000126).References
- M. Asif and T. Muneer, Renewable Sustainable Energy Rev., 2007, 11, 1388–1413 CrossRef.
- K. Kaygusuz, Renewable Sustainable Energy Rev., 2012, 16, 1116–1126 CrossRef.
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Sivula and R. Van De Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- C. Jiang, S. J. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- X. Lu, S. Xie, H. Yang, Y. Tong and H. Ji, Chem. Soc. Rev., 2014, 43, 7581–7593 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- J. A. Seabold and K.-S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef CAS PubMed.
- E. M. P. Steinmiller and K.-S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20633–20636 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- D. E. Schipper, Z. Zhao, A. P. Leitner, L. Xie, F. Qin, M. K. Alam, S. Chen, D. Wang, Z. Ren, Z. Wang, J. Bao and K. H. Whitmire, ACS Nano, 2017, 11, 4051–4059 CrossRef CAS PubMed.
- I. Poli, U. Hintermair, M. Regue, S. Kumar, E. V. Sackville, J. Baker, T. M. Watson, S. Eslava and P. J. Cameron, Nat. Commun., 2019, 10, 2097 CrossRef PubMed.
- M. A. Butler and D. S. Ginley, J. Mater. Sci., 1980, 15, 1–19 CrossRef CAS.
- Z. Wang and L. Wang, Chin. J. Catal., 2018, 39, 369–378 CrossRef CAS.
- C. Xia, H. Wang, J. K. Kim and J. Wang, Adv. Funct. Mater., 2021, 31, 2008247 CrossRef CAS.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, P. A. Glans, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, Chem. Mater., 2014, 26, 5365–5373 CrossRef CAS.
- M. Mishra and D.-M. Chun, Appl. Catal., A, 2015, 498, 126–141 CrossRef CAS.
- Y. Kuang, Q. Jia, H. Nishiyama, T. Yamada, A. Kudo and K. Domen, Adv. Energy Mater., 2016, 6, 1501645 CrossRef.
- C. Zhen, R. Chen, L. Wang, G. Liu and H.-M. Cheng, J. Mater. Chem. A, 2016, 4, 2783–2800 RSC.
- H. Wang, Y. Xia, H. Li, X. Wang, Y. Yu, X. Jiao and D. Chen, Nat. Commun., 2020, 11, 3078 CrossRef CAS PubMed.
- T. Higashi, H. Nishiyama, V. Nandal, Y. Pihosh, Y. Kawase, R. Shoji, M. Nakabayashi, Y. Sasaki, N. Shibata, H. Matsuzaki, K. Seki, K. Takanabe and K. Domen, Energy Environ. Sci., 2022, 15, 4761–4775 RSC.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- W. Guo, C. Xu, X. Wang, S. Wang, C. Pan, C. Lin and Z. L. Wang, J. Am. Chem. Soc., 2012, 134, 4437–4441 CrossRef CAS PubMed.
- F. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 818–841 RSC.
- C. Liu, Y. Yang, J. Li, S. Chen, W. Li and X. Tang, Chem. Eng. J., 2017, 326, 603–611 CrossRef CAS.
- H. Zhang, D. Li, W. J. Byun, X. Wang, T. J. Shin, H. Y. Jeong, H. Han, C. Li and J. S. Lee, Nat. Commun., 2020, 11, 4622 CrossRef PubMed.
- G. Dong, Y. Zhang, W. Wang, L. Wang and Y. Bi, Energy Technol., 2017, 5, 1912–1918 CrossRef CAS.
- I. S. Cho, C. H. Lee, Y. Feng, M. Logar, P. M. Rao, L. Cai, D. R. Kim, R. Sinclair and X. Zheng, Nat. Commun., 2013, 4, 1723 CrossRef PubMed.
- C. Li, M. Chen, Y. Xie, J. Jian, H. Wang and L. Jia, J. Power Sources, 2022, 528, 231242 CrossRef CAS.
- W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS PubMed.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- J.-B. Pan, S. Shen, L. Chen, C.-T. Au and S.-F. Yin, Adv. Funct. Mater., 2021, 31, 2104269 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- J. Zhang, J. Cui and S. Eslava, Adv. Energy Mater., 2021, 11, 2003111 CrossRef CAS.
- S. Wang, D. Cui, W. Hao and Y. Du, Energy Fuels, 2022, 36, 11394–11403 CrossRef CAS.
- H. Wu, H. L. Tan, C. Y. Toe, J. Scott, L. Wang, R. Amal and Y. H. Ng, Adv. Mater., 2020, 32, 1904717 CrossRef CAS PubMed.
- J. G. Mavroides, D. I. Tchernev, J. A. Kafalas and D. F. Kolesar, Mater. Res. Bull., 1975, 10, 1023–1030 CrossRef CAS.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- Y. Kuang, T. Yamada and K. Domen, Joule, 2017, 1, 290–305 CrossRef CAS.
- G. Dong, Y. Zhang and Y. Bi, J. Mater. Chem. A, 2017, 5, 20594–20597 RSC.
- Y. Wang, W. Tian, C. Chen, W. Xu and L. Li, Adv. Funct. Mater., 2019, 29, 1809036 CrossRef.
- L. Meng, J. He, X. Zhou, K. Deng, W. Xu, P. Kidkhunthod, R. Long, Y. Tang and L. Li, Nat. Commun., 2021, 12, 5247 CrossRef CAS PubMed.
- H. Wang, Y. Xia, N. Wen, Z. Shu, X. Jiao and D. Chen, Appl. Catal., B, 2022, 300, 120717 CrossRef CAS.
- S. Ye, C. Ding, M. Liu, A. Wang, Q. Huang and C. Li, Adv. Mater., 2019, 31, 1902069 CrossRef CAS PubMed.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS PubMed.
- L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 RSC.
- B.-Y. Cheng, J.-S. Yang, H.-W. Cho and J.-J. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20032–20039 CrossRef CAS PubMed.
- S. G. Shim, J. Tan, H. Lee, J. Park, J. Yun, Y. S. Park, K. Kim, J. Lee and J. Moon, Chem. Eng. J., 2022, 430, 133061 CrossRef CAS.
- X. Chang, T. Wang, P. Zhang, J. Zhang, A. Li and J. Gong, J. Am. Chem. Soc., 2015, 137, 8356–8359 CrossRef CAS.
- D. Jiang, Q. Yue, S. Tang, L. Zhang, L. Zhu and P. Du, J. Catal., 2018, 366, 275–280 CrossRef CAS.
- X. Fan, B. Gao, T. Wang, X. Huang, H. Gong, H. Xue, H. Guo, L. Song, W. Xia and J. He, Appl. Catal., A, 2016, 528, 52–58 CrossRef CAS.
- W. Li, D. He, S. W. Sheehan, Y. He, J. E. Thorne, X. Yao, G. W. Brudvig and D. Wang, Energy Environ. Sci., 2016, 9, 1794–1802 RSC.
- C. Zachäus, F. F. Abdi, L. M. Peter and R. Van De Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- G. Dong, H. Hu, L. Wang, Y. Zhang and Y. Bi, J. Catal., 2018, 366, 258–265 CrossRef CAS.
- X. Ren, Y. Ji, Y. Zhai, N. Yuan, J. Ding, Y. Li, J. Yan and S. F. Liu, J. Energy Chem., 2021, 60, 512–521 CrossRef CAS.
- S. Wang, T. He, J.-H. Yun, Y. Hu, M. Xiao, A. Du and L. Wang, Adv. Funct. Mater., 2018, 28, 1802685 CrossRef.
- D. Chen, Z. Liu and S. Zhang, Appl. Catal., B, 2020, 265, 118580 CrossRef CAS.
- M. Liao, J. Feng, W. Luo, Z. Wang, J. Zhang, Z. Li, T. Yu and Z. Zou, Adv. Funct. Mater., 2012, 22, 3066–3074 CrossRef CAS.
- R. Chen, C. Zhen, Y. Yang, X. Sun, J. T. S. Irvine, L. Wang, G. Liu and H.-M. Cheng, Nano Energy, 2019, 59, 683–688 CrossRef CAS.
- J.-B. Pan, B.-H. Wang, J.-B. Wang, H.-Z. Ding, W. Zhou, X. Liu, J.-R. Zhang, S. Shen, J.-K. Guo, L. Chen, C.-T. Au, L.-L. Jiang and S.-F. Yin, Angew. Chem., Int. Ed., 2021, 60, 1433–1440 CrossRef CAS PubMed.
- Z. Kang, Z. Sun, Y. Zang, S. Wan, Y.-Z. Zheng and X. Tao, Chem. Eng. J., 2022, 431, 133379 CrossRef CAS.
- W. L. Kwong, C. C. Lee and J. Messinger, J. Phys. Chem. C, 2016, 120, 10941–10950 CrossRef CAS.
- L. Wang and F.-S. Xiao, ChemCatChem, 2014, 6, 3048–3052 CrossRef CAS.
- R. Xu, D. Zhu, K. Du, D. Cui, H. Feng, W. Hao, D. Tian and Y. Du, Mater. Today Energy, 2022, 25, 100961 CrossRef CAS.
- P. Yue, H. She, L. Zhang, B. Niu, R. Lian, J. Huang, L. Wang and Q. Wang, Appl. Catal., B, 2021, 286, 119875 CrossRef CAS.
- H. Sun, W. Hua, Y. Li and J.-G. Wang, Chem. Eng. J., 2022, 427, 131004 CrossRef CAS.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef.
- Z. Kang, X. Lv, Z. Sun, S. Wang, Y.-Z. Zheng and X. Tao, Chem. Eng. J., 2021, 421, 129819 CrossRef CAS.
- J. Wang, Z. Zhang, H. Song, B. Zhang, J. Liu, X. Shai and L. Miao, Adv. Funct. Mater., 2021, 31, 2008578 CrossRef CAS.
- B. Zhang, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Angew. Chem., Int. Ed., 2018, 57, 2248–2252 CrossRef CAS PubMed.
- T. Zhou, J. Wang, Y. Zhang, C. Zhou, J. Bai, J. Li and B. Zhou, Chem. Eng. J., 2022, 431, 133414 CrossRef CAS.
- G. Dong, H. Hu, X. Huang, Y. Zhang and Y. Bi, J. Mater. Chem. A, 2018, 6, 21003–21009 RSC.
- B. Zhang, X. Huang, H. Hu, L. Chou and Y. Bi, J. Mater. Chem. A, 2019, 7, 4415–4419 RSC.
- Z. Zhou, F. Wang, P. Liang, L. Yang, Y. Yu, L. Li, Y. Guo and S. Wu, ACS Appl. Energy Mater., 2022, 5, 8999–9008 CrossRef CAS.
- S. Wang, T. He, J.-H. Yun, Y. Hu, M. Xiao, A. Du and L. Wang, Adv. Funct. Mater., 2018, 28, 1802685 CrossRef.
- Y. Gao, Z. Tian, H. Zhu, H. Xue, L. Ma, Y. Dai, W. Zhao, X. Li, N. Li and L. Ge, ACS Appl. Energy Mater., 2021, 4, 14649–14661 CrossRef CAS.
- H. Sun, W. Hua, S. Liang, Y. Li and J.-G. Wang, J. Colloid Interface Sci., 2022, 611, 278–286 CrossRef CAS PubMed.
- Y. Yang, P. Gao, Y. Wang, L. Sha, X. Ren, J. Zhang, Y. Chen, T. Wu, P. Yang and X. Li, Nano Energy, 2017, 33, 29–36 CrossRef CAS.
- F. Niu, Q. Zhou, Y. Han, R. Liu, Z. Zhao, Z. Zhang and K. Hu, ACS Catal., 2022, 12, 10028–10038 CrossRef CAS.
- Y. An, J. Hao, C. Lin, S. Zhang, K. Zhang and Y. Min, ACS Appl. Mater. Interfaces, 2022, 14, 42134–42143 CrossRef CAS PubMed.
- X. Cheng, G. Dong, Y. Zhang, C. Feng and Y. Bi, Appl. Catal., B, 2020, 267, 118723 CrossRef CAS.
- B. Zhang, X. Huang, Y. Zhang, G. Lu, L. Chou and Y. Bi, Angew. Chem., Int. Ed., 2020, 59, 18990–18995 CrossRef CAS PubMed.
- Z. Zhang, X. Huang, B. Zhang and Y. Bi, Energy Environ. Sci., 2022, 15, 2867–2873 RSC.
- X. Zhang, P. Zhai, Y. Zhang, Y. Wu, C. Wang, L. Ran, J. Gao, Z. Li, B. Zhang, Z. Fan, L. Sun and J. Hou, J. Am. Chem. Soc., 2021, 143, 20657–20669 CrossRef CAS PubMed.
- C. Du, B. Yan, Z. Lin and G. Yang, J. Mater. Chem. A, 2020, 8, 207–217 RSC.
- G. Dong, X. Huang and Y. Bi, Angew. Chem., Int. Ed., 2022, 61, e202204271 CAS.
- M. Tayebi and B.-K. Lee, Catal. Today, 2021, 361, 183–190 CrossRef CAS.
- J. Wang, S. Xin, Y. Xiao, Z. Zhang, Z. Li, W. Zhang, C. Li, R. Bao, J. Peng, J. Yi and S. Chou, Angew. Chem., Int. Ed., 2022, 61, e202202518 CAS.
- L. K. Dhandole, T. S. Koh, P. Anushkkaran, H.-S. Chung, W.-S. Chae, H. H. Lee, S. H. Choi, M. Cho and J. S. Jang, Appl. Catal., B, 2022, 315, 121538 CrossRef CAS.
- L. Cai, J. Zhao, H. Li, J. Park, I. S. Cho, H. S. Han and X. Zheng, ACS Energy Lett., 2016, 1, 624–632 CrossRef CAS.
- Y. Zhang, Y. Li, D. Ni, Z. Chen, X. Wang, Y. Bu and J.-P. Ao, Adv. Funct. Mater., 2019, 29, 1902101 CrossRef.
- H.-J. Ahn, K.-Y. Yoon, M.-J. Kwak and J.-H. Jang, Angew. Chem., Int. Ed., 2016, 55, 9922–9926 CrossRef CAS PubMed.
- K.-Y. Yoon, H.-J. Ahn, M.-J. Kwak, S.-I. Kim, J. Park and J.-H. Jang, J. Mater. Chem. A, 2016, 4, 18730–18736 RSC.
- H. Li, M. Yin, X. Li and R. Mo, ChemSusChem, 2021, 14, 2331–2340 CrossRef CAS PubMed.
- J. Deng, Q. Zhang, K. Feng, H. Lan, J. Zhong, M. Chaker and D. Ma, ChemSusChem, 2018, 11, 3783–3789 CrossRef CAS PubMed.
- Y. Li, Q. Mei, Z. Liu, X. Hu, Z. Zhou, J. Huang, B. Bai, H. Liu, F. Ding and Q. Wang, Appl. Catal., B, 2022, 304, 120995 CrossRef CAS.
- B. Zhang, S. Yu, Y. Dai, X. Huang, L. Chou, G. Lu, G. Dong and Y. Bi, Nat. Commun., 2021, 12, 6969 CrossRef CAS PubMed.
- J. Lin, X. Han, S. Liu, Y. Lv, X. Li, Y. Zhao, Y. Li, L. Wang and S. Zhu, Appl. Catal., B, 2023, 320, 121947 CrossRef CAS.
- N. D. Quang, P. C. Van, S. Majumder, J.-R. Jeong, D. Kim and C. Kim, J. Colloid Interface Sci., 2022, 616, 749–758 CrossRef PubMed.
- J. He, P. Liu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 6835–6871 RSC.
- X. Wang, G. Sun, N. Li and P. Chen, Chem. Soc. Rev., 2016, 45, 2239–2262 RSC.
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC.
- D. Qin, Y. Zhou, W. Wang, C. Zhang, G. Zeng, D. Huang, L. Wang, H. Wang, Y. Yang, L. Lei, S. Chen and D. He, J. Mater. Chem. A, 2020, 8, 19156–19195 RSC.
- M.-H. Sun, S.-Z. Huang, L.-H. Chen, Y. Li, X.-Y. Yang, Z.-Y. Yuan and B.-L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
- Z. Liang, H. Hou, Z. Fang, F. Gao, L. Wang, D. Chen and W. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 19167–19175 CrossRef CAS PubMed.
- X. Huang, L. Yang, S. Hao, B. Zheng, L. Yan, F. Qu, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2017, 4, 537–540 RSC.
- J. Rong, Z. Wang, J. Lv, M. Fan, R. Chong and Z. Chang, Chin. J. Catal., 2021, 42, 1999–2009 CrossRef CAS.
- R. Chong, Y. Du, Z. Chang, Y. Jia, Y. Qiao, S. Liu, Y. Liu, Y. Zhou and D. Li, Appl. Catal., B, 2019, 250, 224–233 CrossRef CAS.
- D. Yan, X. Fu, Z. Shang, J. Liu and H. Luo, Chem. Eng. J., 2019, 361, 853–861 CrossRef CAS.
- Y. Shi, Y. Yu, Y. Yu, Y. Huang, B. Zhao and B. Zhang, ACS Energy Lett., 2018, 3, 1648–1654 CrossRef CAS.
- R. Irani, P. Plate, C. Höhn, P. Bogdanoff, M. Wollgarten, K. Höflich, R. van de Krol and F. F. Abdi, J. Mater. Chem. A, 2020, 8, 5508–5516 RSC.
- S. Seenivasan, S. Adhikari and D.-H. Kim, Chem. Eng. J., 2021, 422, 130137 CrossRef CAS.
- H. Luo, C. Liu, Y. Xu, C. Zhang, W. Wang and Z. Chen, Int. J. Hydrogen Energy, 2019, 44, 30160–30170 CrossRef CAS.
- L. Zhu, H. Lu, D. Hao, L. Wang, Z. Wu, L. Wang, P. Li and J. Ye, ACS Appl. Mater. Interfaces, 2017, 9, 38537–38544 CrossRef CAS PubMed.
- G. Dong, X. Cheng and Y. Bi, Sol. RRL, 2021, 5, 2000449 CrossRef CAS.
- H. Lin, X. Long, Y. An, D. Zhou and S. Yang, Nano Lett., 2019, 19, 455–460 CrossRef CAS PubMed.
- S. Zhong, Y. Xi, S. Wu, Q. Liu, L. Zhao and S. Bai, J. Mater. Chem. A, 2020, 8, 14863–14894 RSC.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- G. Liu, J. Shi, F. Zhang, Z. Chen, J. Han, C. Ding, S. Chen, Z. Wang, H. Han and C. Li, Angew. Chem., Int. Ed., 2014, 53, 7295–7299 CrossRef CAS PubMed.
- Z. Wang, G. Liu, C. Ding, Z. Chen, F. Zhang, J. Shi and C. Li, J. Phys. Chem. C, 2015, 119, 19607–19612 CrossRef CAS.
- Y. Hu, Y. Wu, J. Feng, H. Huang, C. Zhang, Q. Qian, T. Fang, J. Xu, P. Wang, Z. Li and Z. Zou, J. Mater. Chem. A, 2018, 6, 2568–2576 RSC.
- S. Oh, S. Jung, Y. H. Lee, J. T. Song, T. H. Kim, D. K. Nandi, S.-H. Kim and J. Oh, ACS Catal., 2018, 8, 9755–9764 CrossRef CAS.
- P. Zhang, T. Wang, X. Chang, L. Zhang and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 5851–5855 CrossRef CAS PubMed.
- B. Zhao, C. Feng, X. Huang, Y. Ding and Y. Bi, J. Mater. Chem. A, 2022, 10, 2813–2818 RSC.
- K. Fan, H. Chen, B. He and J. Yu, Chem. Eng. J., 2020, 392, 123744 CrossRef CAS.
- T. Wang, H. Cao, J. Wu, M. G. Haghighi, R. Sedghi and P. Du, J. Phys. Chem. C, 2022, 126, 11042–11050 CrossRef CAS.
- X. Cao, P. Wen, R. Ma, Y. Liu, S. Sun, Q. Ma, P. Zhang and Y. Qiu, Chem. Eng. J., 2022, 449, 137792 CrossRef CAS.
- Y. Zhao, H. Xie, W. Shi, H. Wang, C. Shao and C. Li, J. Energy Chem., 2022, 64, 33–37 CrossRef CAS.
- T. H. Jeona, G. Moona, H. Parkb and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef.
- M. Feng, Y. Liu, N. Wei, S. Ma, Z. Li, H. Li, S. Chen, J. Liu and D. Wang, J. Mater. Chem. A, 2018, 6, 18293–18303 RSC.
- B. J. Trześniewski, I. A. Digdaya, T. Nagaki, S. Ravishankar, I. Herraiz-Cardona, D. A. Vermaas, A. Longo, S. Gimenez and W. A. Smith, Energy Environ. Sci., 2017, 10, 1517–1529 RSC.
- J. Pina, P. Dias, C. Serpa, J. Azevedo, A. Mendes and J. S. S. Melo, J. Phys. Chem. C, 2021, 125, 8274–8284 CrossRef CAS.
- X. Liu, G. Dong, S. Li, G. Lu and Y. Bi, J. Am. Chem. Soc., 2016, 138, 2917–2920 CrossRef CAS PubMed.
- X. Zhang, B. Zhang, K. Cao, J. Brillet, J. Chen, M. Wang and Y. Shen, J. Mater. Chem. A, 2015, 3, 21630–21636 RSC.
- L. Meng, M. Wang, H. Sun, W. Tian, C. Xiao, S. Wu, F. Cao and L. Li, Adv. Mater., 2020, 32, 2002893 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |