
Covalent organic framework and hydrogen-bonded organic framework for solar-driven photocatalysis
Wei-Kang
Qin
ab,
Chen-Ho
Tung
 ab and
Li-Zhu
Wu
*abc
ab and
Li-Zhu
Wu
*abc
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: lzwu@mail.ipc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cNew Cornerstone Science Laboratory, Shenzhen, 518054, P. R. China
First published on 16th March 2023
Abstract
The newly emerging porous crystalline materials, covalent organic framework (COF) and hydrogen-bonded organic framework (HOF), are characterized by their large surface area, considerable structural diversity, and functional tailorability. Combining these properties, they are considered to have the potential for photocatalysis. This review summarizes their advantages and compatibility, particularly focusing on design strategies to overcome the existing issues of poor charge conductivity and deficient active sites. With representative examples of solar-driven hydrogen (H2) evolution and carbon dioxide (CO2) reduction, the exploration and expectation of such functional porous materials are presented.
 Wei-Kang Qin | Wei-Kang Qin obtained his B.E. degree from the Zhejiang University in 2015 and M.E. degree from the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (CAS), in 2019. Currently, he is a PhD candidate at Technical Institute of Physics and Chemistry, CAS, under the supervision of Prof. Li-Zhu Wu and Prof. Chen-Ho Tung. His research interests mainly focus on functional organic frameworks and photocatalytic CO2 reduction. |
 Chen-Ho Tung | Prof. Chen-Ho Tung graduated from the University of Science and Technology of China in 1963 and was awarded his PhD degree in 1983 from Columbia University in New York City under the supervision of Prof. Nicholas J. Turro. He was elected as an academician of CAS in 1999 and is currently a professor at Technical Institute of Physics and Chemistry, CAS. His research interests include photochemical reactions, photoinduced electron transfer and energy transfer in supramolecular systems, and photocatalytic water splitting. |
 Li-Zhu Wu | Prof. Li-Zhu Wu received her BS degree from the Lanzhou University in 1990 and PhD from the Institute of Photographic Chemistry of Chinese Academy of Sciences in 1995 under the supervision of Prof. Chen-Ho Tung. After one-year postdoctoral stay at University of Hong Kong with Prof. Chi-Ming Che, she was appointed as a professor at Technical Institute of Physics and Chemistry, the Chinese Academy of Sciences. She was elected as an academician of Chinese Academy of Sciences in 2019 and a Fellow of The World Academy of Sciences (TWAS) in 2021. Her research interests are focused on photochemistry research, including artificial photosynthesis, visible light catalysis for organic transformation, and photoinduced electron transfer, energy transfer and chemical reactions in supramolecular systems. |
10th anniversary statementPhotocatalysis is acknowledged as a significant field with a particular relationship to energy conversion and storage, sustainability and health. As a high-quality journal dedicated to discovering new molecules and materials and studying their properties and mechanisms, Journal of Materials Chemistry A has published numerous works about novel porous materials and corresponding applications as effective catalysts for solar energy conversion, serving as a vibrant hub for the advancement of high-performance organic frameworks. Therefore, the future practical utilization of porous framework materials deserves special attention. We sincerely congratulate the 10th anniversary of the Journal of Materials Chemistry A and heartfeltly appreciate the opportunity to publish our review in the journal. |
Introduction
Since the 18th century, the utilization of fossil energy for industrial development has been increasing with the growth in economy and improvement in living standards. Accompanied by continuous hazards in natural environments, such as the greenhouse effect, acid rain, and atmospheric contamination, it is, therefore, urgent to explore renewable energy for sustainable society. Solar energy is appealing as the most potential energy source in the future. Using solar energy, the natural photosynthesis occurring in the chloroplasts of green plants guides the conversion of sunlight to chemical energy. Various artificial application domains, including H2 evolution, CO2 reduction, and photoinduced organic synthesis, have been developed. Throughout the application scenarios of photocatalysis, it is known that the essence of an appropriate catalyst is to drive a reaction. Under light irradiation, taking photo-redox catalysis as an example, photoexcited carriers (electrons and holes) within the photocatalyst are generated by harvesting the energy of photons and then the charges transfer, followed by the subsequent redox reaction with the substrate at the active sites of the catalyst.1 However, it is often necessary to confront the limitation of photocatalytic reactions present in the inherent properties of catalysts, such as high electron/hole recombination and poor light absorption capacity. Moreover, photocatalysis is restricted by the high energy barrier for activating reactants by a catalyst and product selectivity resulting from ambiguous chemical conversion pathways.2 Therefore, the exploration of smart materials for photocatalysis is a prerequisite for achieving the utilization of solar energy for chemical transformation.Over the past few decades, polymer semiconductors, such as g-C3N4, conjugated polymers, and porous organic frameworks, have become famous owing to their unique physicochemical properties and optoelectronic features. Porous crystalline materials, encompassing a metal organic framework (MOF), covalent organic framework (COF), and hydrogen-bonded organic framework (HOF), have been developing rapidly since the first report of MOF conception proposed by Yaghi in the 1990s. Recently, the reticular materials of COF and HOF have received widespread attention (Fig. 1b). As shown in Fig. 1a, both are constitutive of organic monomers with multiple binding groups for generating covalent bonds (COF) or hydrogen bonds (HOF) with other synthons.3 The network structures present two-dimensional or three-dimensional periodic construction, in which the π–π stacking interaction among the organic monomers contributes to stabilizing the porous backbones.4 Owing to the various building blocks, topologies, and organic compositions, COFs and HOFs feature high surface area,5 rich structural diversity,6–9 and tuneable functionality.10–13 These merits provide the frameworks to be candidates for promising photocatalysis.
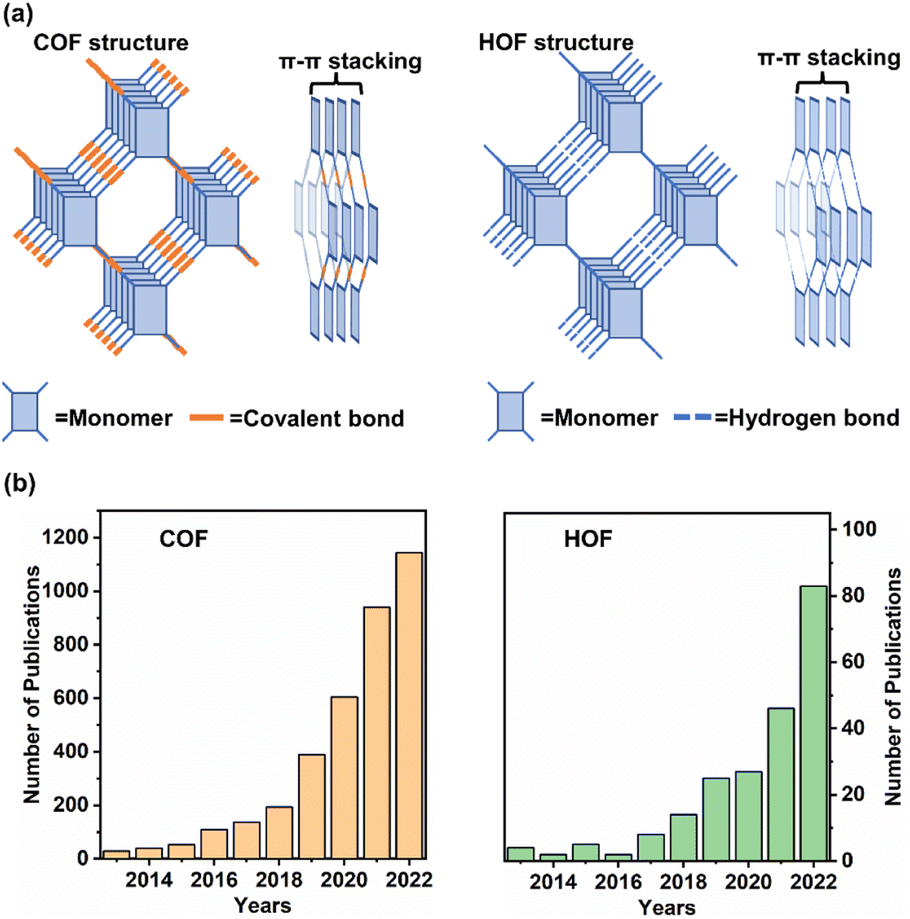 | ||
| Fig. 1 (a) Structures of two-dimensional COF and HOF constructed by organic monomers. (b) The numbers of publications related to COF and HOF in the last 10 years. | ||
Owing to the inherent characteristics of organic crystalline frameworks, the compatibility and superiority of COFs and HOFs utilized for photocatalysis are as follows: (i) excellent porosity:14 the organic frameworks with periodic arrangement possess well-defined regular pores and channels, which provide diffusion paths for reactants and products of catalytic reactions. (ii) Dispersed active sites: intrinsic active sites, such as Lewis acid or the post-introduced catalytic species, can disperse evenly on the backbone of frameworks. The uniform active sites and internal periodic photosensitized units shorten the migration distance of the photoexcited charge carriers, thereby improving catalytic efficiency. (iii) Adaptable photo-response ability: by rational design of structural units and connection types to modulate the energy band structures of COF and HOF, a light absorption range and adjustable photo-redox capacity can be achieved, which is essential for collecting solar energy.15,16 Herein, we review the latest progress of COF and HOF used for photocatalysis. Considering the representative photocatalytic H2 evolution and CO2 reduction, the improvement of chemical robustness, charge transfer and reaction at active sites is discussed. For a clear description of the merits within the internal structures, the structure–activity relationship is analysed to highlight the applicability and advantages of COF and HOF as photocatalysts. With the introduction of extensive photocatalytic examples, the challenges and prospects of a promising organic framework are also proposed for photochemical synthesis and conversion.
Respective structural features of COF and HOF
The inherent features of tailorable structures, such as low density, high surface area, tuneable pores, and diverse functionalities, highlight COF and HOF for photocatalysis. These two kinds of porous materials have their respective properties in various aspects. COFs have excellent stability for maintaining the network in the face of harsh chemical environments and exhibit various physical and chemical properties with diverse connection linkages, including B–O, imine linkage, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and triazine.17 Rational selections of constructed units and connection types enable COF colour diversity from orange to dark, which is beneficial for light harvesting at different irradiation conditions.18 Furthermore, the extended π-conjugated systems and abundant interplanar π–π stacking interaction can improve π–electron delocalization in the COF, thereby facilitating the photoexcited carriers transfer.
C, and triazine.17 Rational selections of constructed units and connection types enable COF colour diversity from orange to dark, which is beneficial for light harvesting at different irradiation conditions.18 Furthermore, the extended π-conjugated systems and abundant interplanar π–π stacking interaction can improve π–electron delocalization in the COF, thereby facilitating the photoexcited carriers transfer.
HOFs exhibit their unique characteristics in high crystallinity, sustainability and practicability. Because of the flexible and spontaneous hydrogen bonds,19 organic building blocks tend to fabricate highly crystalline networks through self-assembly.20 Based on the diverse design of topologies and monomers, the high crystallinity of HOFs provides a more precise structure and pore information characterized by powder or single crystal X-ray diffraction, which is helpful for the definite exploration of structure–activity relationships in photocatalysis. Moreover, HOFs possess the capacity for structural regeneration through easy solvent retreatment, allowing for sustainable utilization under practical conditions.
Based on existing studies, however, COFs and HOFs usually exhibit low catalytic activity due to the low carrier transfer efficiency and severe charge recombination.23 The pure organic composition of COF and HOF lacks active sites for the activation of reactants,24 which are generally required for photocatalytic reactions. Therefore, it is important to rationally construct stable and active materials by efficiently utilizing structural advantages and an efficacious solution to the drawbacks.
Strategy for the construction of COFs and HOFs for efficient photocatalysis
Improvement of stability
Photocatalysts are required for continuous and stable performance, which means that the photocatalysts exhibit sufficient photochemical stability under irradiation and exhibit excellent thermal stability under reaction conditions. Through covalent bonds with high binding energy, the stability of the COFs is better than that of other frameworks, such as MOFs and HOFs.25 However, the design of high COFs depends on those covalent bonds with reversibility, which are often unstable under harsh conditions.26,27 For example, the reversible boroxine and boronate ester linkages are also widely utilized in the construction of the COF crystals.28 However, their hydrolysis causes structural fragility in moisture circumstances.27 The most frequently used covalent bond, imine, also exhibits insufficient stability in an acidic environment.27,29 To fabricate promising porous crystal photocatalysts, COFs still need a better contrivable strategy for robust networks with the prerequisite of high crystallinity. The intuitional way is to select the covalent linkages with stronger robustness, such as CC29–31 and dioxin bonds,32–34 whereas these strong linkages make it challenging to generate crystalline COFs because of their irreversibility and hyperthermal formation conditions. Liu et al. reported an aza-Diels–Alder cycloaddition reaction for the transformation of the COF linkage from imine to aromatic heterocycle (Fig. 2b). Through the addition of phenylacetylene, the internal aryl imine reacts with arylalkyne, yielding a six-membered nitrogen heterocycle. The obtained quinoline-linked COFs, MF-1 series, maintain high crystallinity and show excellent robustness to resist acidic, basic, and redox environments.21
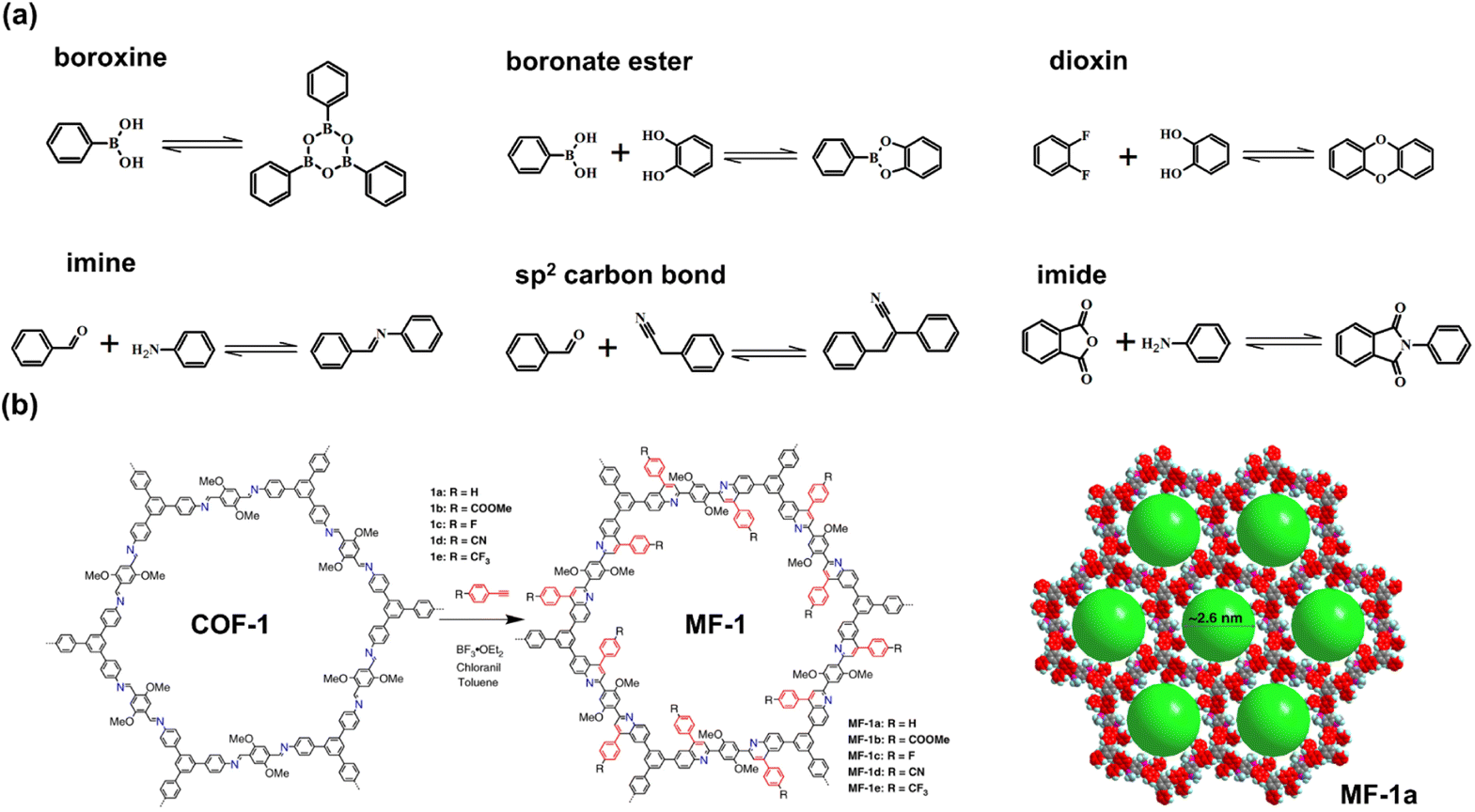 | ||
| Fig. 2 (a) The types of covalent linkages of COFs; (b) post-synthetic modification (PSM) of COF from imine linkage to aromatic heterocycle by an aza-Diels–Alder cycloaddition reaction. Reprinted with permission from ref. 21. Copyright 2018, Nature Publishing Group. | ||
The synthetic methodology for robust HOFs resembles that of COFs, and the quantity of hydrogen bonds is important to HOF structures. For multiple hydrogen bonding, the organic monomer should be decorated with more than three functional groups.35 Carboxyl acid is usually chosen as a connection, whereas its deprotonation nature in alkaline conditions restricts the chemical stability of HOFs.36 Therefore, nitrogenous groups are applied for generating N–H⋯H or other mixed hydrogen bonds, such as pyrazole,37 diamino triazine,3,38–40 and guanine,41 to promote the robustness of HOFs in non-acid conditions. As a kind of weak interaction, hydrogen bonds have difficulty in solely stabilizing the crystal framework with permanent porosity.22 Hence, it is vital to introduce π–π stacking interaction for maintaining HOF structures using appropriate organic building blocks with large π conjugated systems.42 Cao and coworkers reported an ultra-robust HOF structure consisting of 1,3,6,8-tetrakis (p-benzoic acid) pyrene named PFC-1 (Fig. 3b), which exhibits excellent porosity and stability for photodynamic therapy. For PFC-1, the π–π interaction provided by the central pyrene stacking contributes a lot to the stability of the framework.22
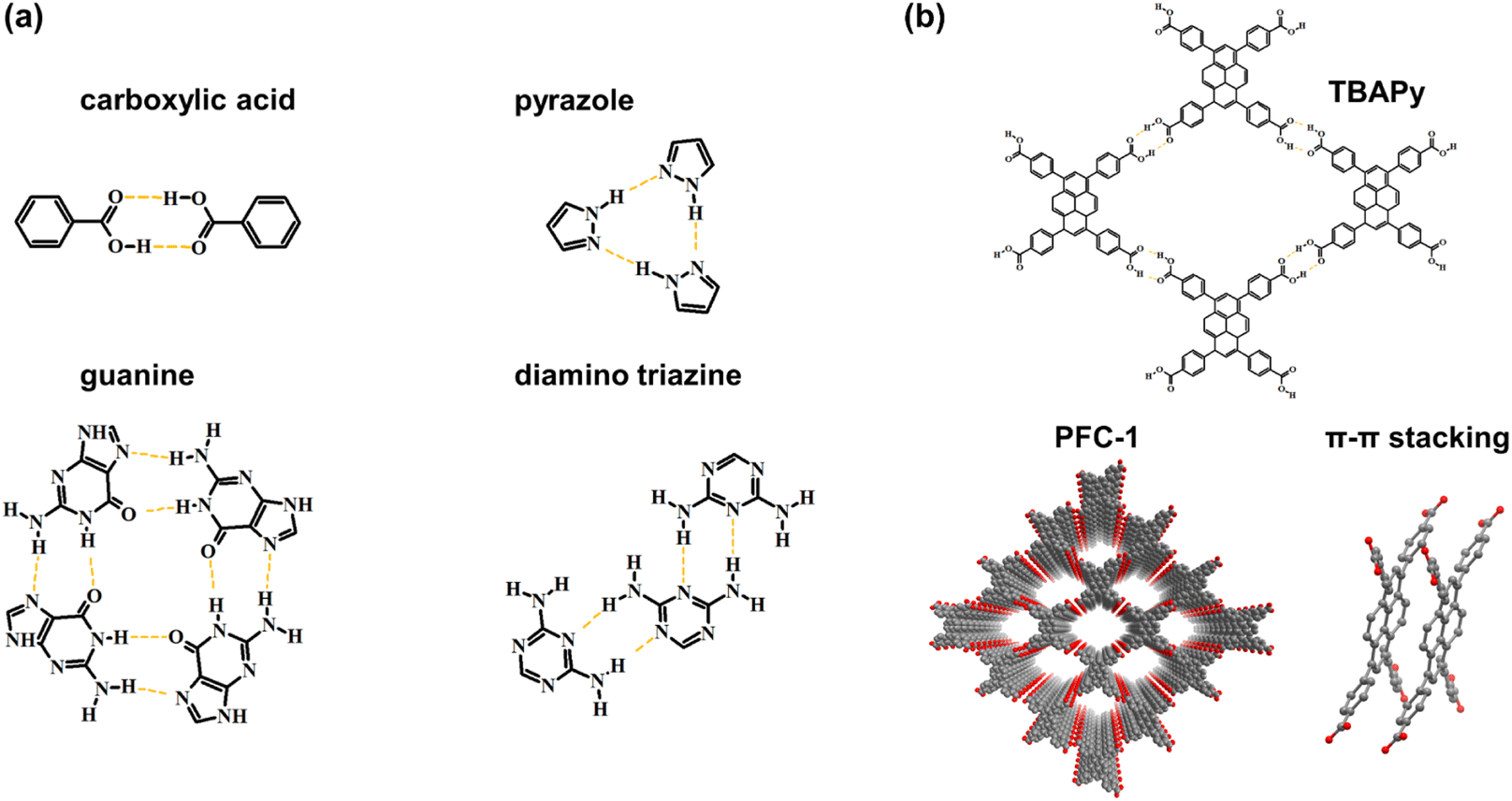 | ||
| Fig. 3 (a) Some functional groups for generating hydrogen bonds; (b) the monomer, structure and stacking mode of PFC-1. | ||
Light harvesting and charge transfer
The rational selection of monomers is the key factor for fabricating COF photocatalysts with suitable energy bands. The band structure with conduction band (CB) and valence band (VB) location should not only satisfy the requirement for light harvesting and charge transfer but also cover the redox potential of a specific photocatalytic reaction.26 Through the conjugation of π bonds and the inductive effect of various substituent groups, the molecular energy level structure with HOMO and LUMO can be modulated for effective visible light absorption. For covalent linkages of COF structure, the connection of sp2 π conjugation of monomers can be further extended, resulting in the enhancement of π electron delocalization, thereby promoting the photoexcited carrier transfer of frameworks. Moreover, the overlaps of π conjugation decrease the π → π* transition energy, endowing the structure with a broader absorption range of visible light. However, the imine linkage exhibits weak characteristics in the π-delocalization for the high polarization of N.45,46 Instead, the sp2 carbon-linked COF can expand the π conjugation and facilitate intermolecular electron transfer.30,47By suitable integration of electron donor and acceptor molecules into the skeleton of COF, the rearrangement of molecular orbital within the donor–acceptor (D–A) system decreases the energy gap and promotes the separation of photoexcited electrons and holes.14,46,48,49 Typically, the large π conjugated aromatic ring, such as pyrene and perylene, serves as electron donor units.14 Yang et al. integrated electron-deficient thiazolo[5,4-d]thiazole molecule to construct effective D–A coupling COF (Fig. 4a). The efficient charge separation of the COF exhibits activity in light-driven H2 evolution.43 In addition to π conjugation, vertical π–π stacking configuration can also enhance the charge transfer, especially for two-dimensional COF with the parallel stacking mode of donor-on-donor and acceptor-on-acceptor.50 Taking porphyrin-based COF as an example, Wang et al. reported a series of 2D COF with the AA stacking of metallized porphyrin (Fig. 4b). Revealed by the photocatalytic H2 evolution experiment and theoretical calculation, the porphyrin ligand stacking chain and central metal (Zn, Co, and Ni) chain are regarded as the transfer channels of electrons and holes, respectively, thus preventing charge recombination. In contrast to various center metal ions, Zn2+ porphyrin chelate exhibits the best charge separation efficiency. The steady valence electron configuration (3d10) further suppresses recombination by the confinement of the ligand-to-metal charge transfer (LMCT) process.44
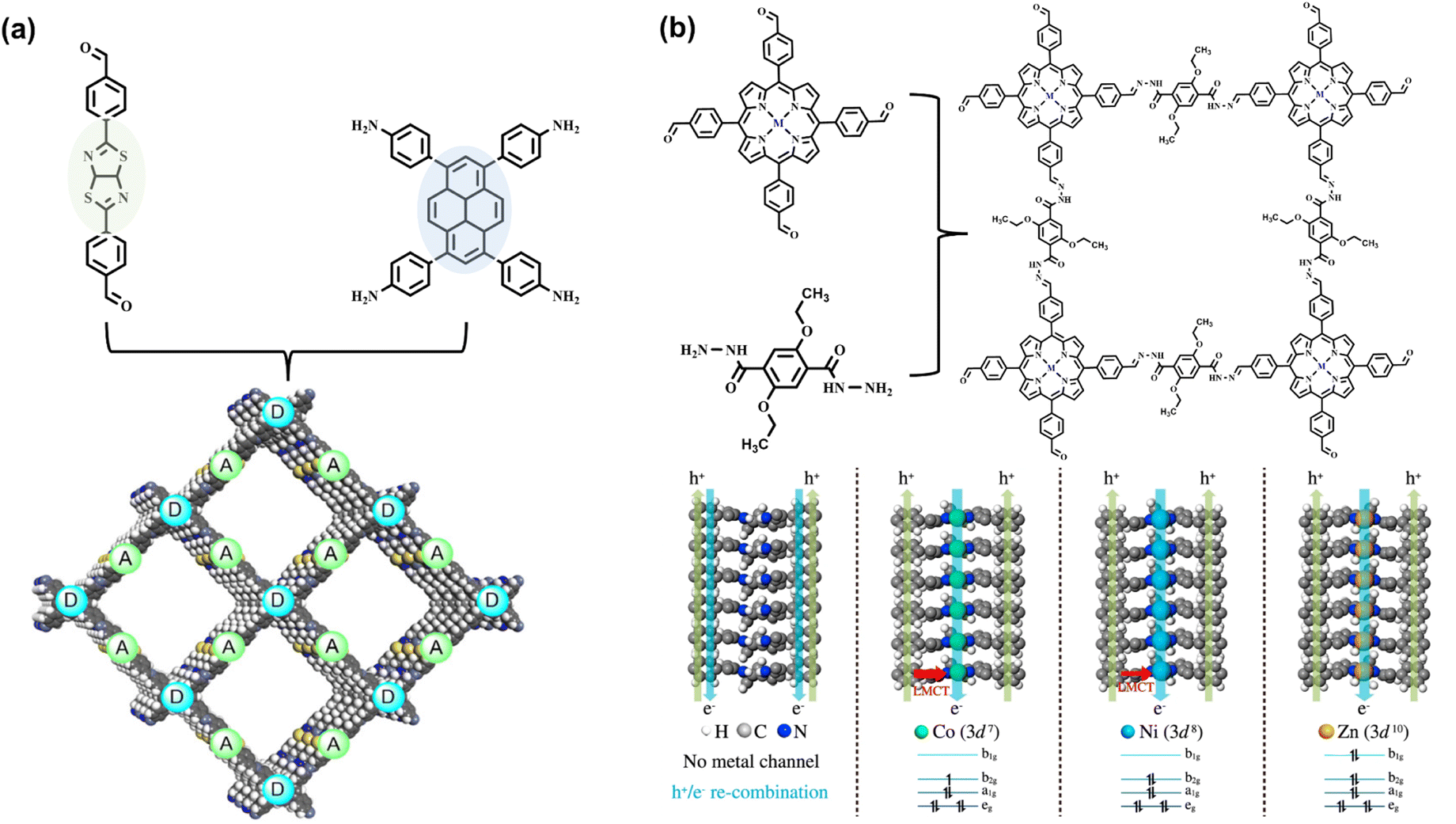 | ||
| Fig. 4 (a) The D–A conjugated COF comprising pyrene and thiazolo[5,4-d]thiazole moieties. Adapted with permission from ref. 43. Copyright 2020, Wiley-VCH. (b) The structure of metallized porphyrin-based COFs and the simulated charge transfer pathway of electrons and holes within the stacking metal porphyrin column. Adapted with permission from ref. 44. Copyright 2021, Nature Publishing Group. | ||
For HOFs, their structure tunability and function diversity allow them to improve their performance by applying corresponding functional building blocks. PFC-1 materials constructed by large π conjugated pyrene components show significant light absorption ability.22 Similarly, the incorporation of conductive molecules into the skeletons of HOFs can lead to the enhancement of charge transfer in the framework. Based on the hydrogen bonding and stacking molecules, tetrathiafulvalene (TTF), HOF-110 exhibits potential regarding rapid charge conduction. Owing to the strong π–electron donor characteristic and dynamic TTF/TTF˙+ variation, the stacking dimerized TTF molecules significantly affect the electron transfer (Fig. 5a).51 Because the monomer comprises four naphthoic acid groups around the TTF core, the result also confirms the significance of π–π stacking interaction for charge transfer. Compared to benzene rings, naphthalene rings provide a stronger combination interaction owing to the larger stacking area,52 resulting in the stabilization of the TTF-on-TTF column in more efficient vertical charge conduction (Fig. 5b). Based on the high reversibility of hydrogen bonds, HOFs tend to form highly crystalline materials, and the relationship among the charge conduction and internal hydrogen-bonding connections, as well as stacking modes, can be characterized and therefore ideal for deeper exploration.
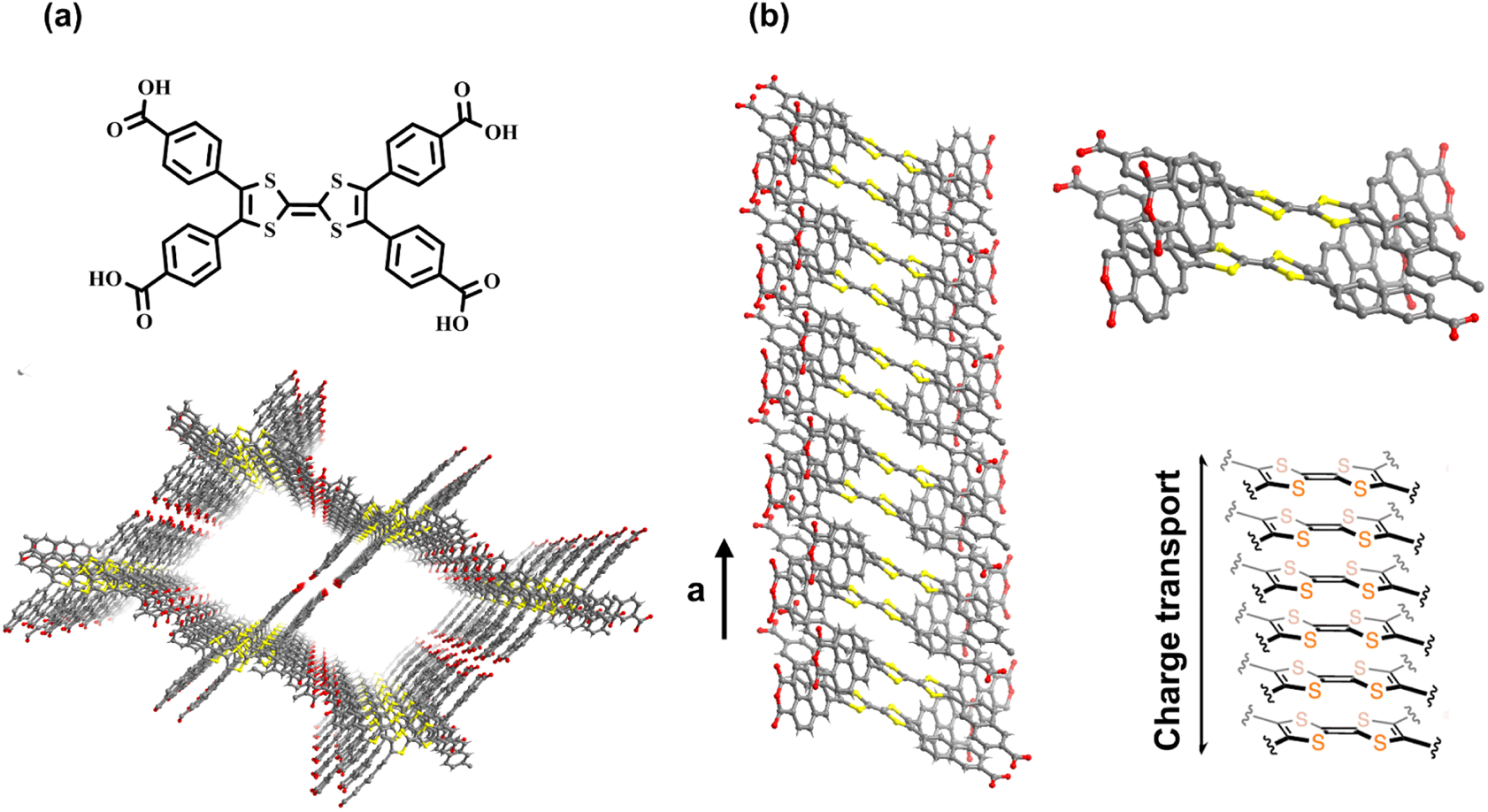 | ||
| Fig. 5 (a) Structure of HOF constructed by TTF derivate; (b) the strong π–π stacking interaction provided by naphthalene rings and the ABAB stacking mode of TTF groups. Adapted with permission from ref. 51. Copyright 2021, American Chemical Society. | ||
Integration of catalytic active sites
In photo-redox catalysis, it is important for surface active sites (such as coordinatively unsaturated metal sites) to decrease the activation energy of the catalytic reaction and chemisorb the substrate molecules. Taking CO2 photoreduction as an example, the adsorbed CO2 accepts one electron from a photosensitizer to form the active chemisorbed intermediate (such as *COOH) on the metal active sites.53 However, most COFs and HOFs showcase metal-free composition features. Thus, the integration of metal elements must proceed by rational design at the levels of monomers, linkages, and topologies. Moreover, the incorporation of metal active sites into an organic framework facilitates the delocalization of conjugated organic molecules.Metal-macrocyclic complexes, such as metal porphyrin and phthalocyanine, are ideal for the metallization of an organic framework.54–61 Chelated by an internal coordinated N atom, the transition metal ions can be anchored in the central position of rings. The highly π-conjugated macrocycle exhibits excellent light absorption ability. The π electron delocalization and multiple coordination bonding result in effective electron transfer to the metal center, promoting photocatalytic efficacy.62 Based on the D–A system, the electron accumulation of central active sites can be significantly improved when coupled with strong electron donors, such as pyrene, TTF,63 and metal-free porphyrin.54 Lan et al. reported the metal porphyrin molecules as the building blocks for synthesizing a series of metallized COFs, named TTCOF-M (Fig. 6a). The internal TTF molecules exhibit light harvesting ability, as well as electron transfer to metal sites; therefore, the dual-component COF has photocatalytic activity in CO2-to-CO reduction reaction.64 In addition to metal macrocycle molecules, linear bidentate ligands, such as bipyridine, are also suitable for coupling metal ions.65,66 Through post-synthetic modification (PSM), transition metal species are dispersedly anchored on the skeleton of COF. The dispersed metal sites can meet the reactant in the pores, and the charge transfer path is significantly shortened from the photoexcited units of the COF to the catalytic sites. Compared to the metal macrocyclic complex, it seems more flexible and efficient for the PSM metallization of COF. Consequently, Fu et al. reported a 2D pyridine-based COF post-decorated by [Re(CO)5Cl] compounds. The metallized composite shows high activity in CO2 photoreduction and CO product selectivity (Fig. 6b).67 In this COF, pyrene moieties play an essential role as photosensitizer, while metallized units, bipyridine moieties, act as uniformly distributed active sites that accept the transferred photoelectrons and leave the photoexcited holes in pyrene, thereby promoting charge separation.
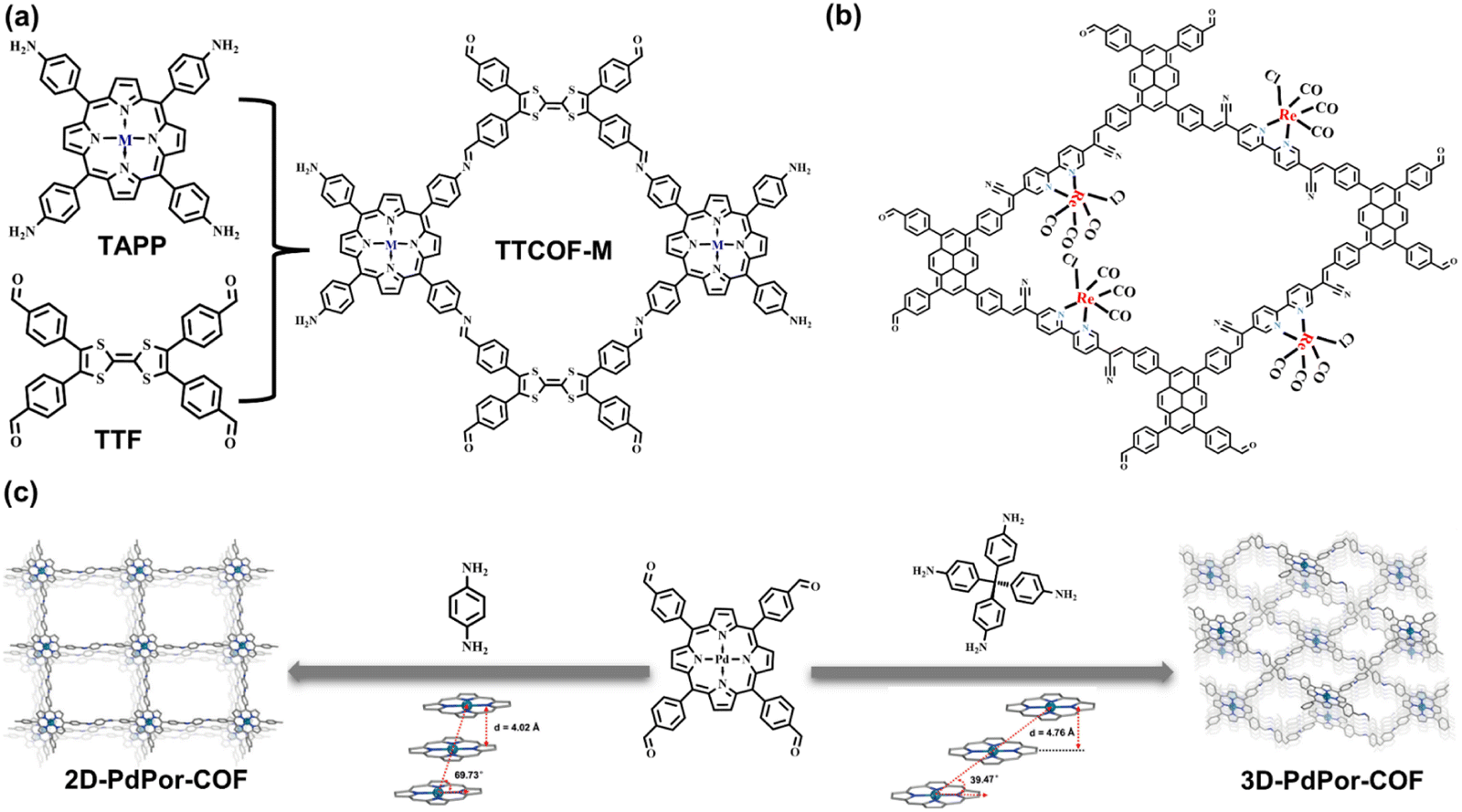 | ||
| Fig. 6 (a) Monomers and connection of TTCOF-M. Adapted with permission from ref. 64. Copyright 2019, Wiley-VCH. (b) Post-synthetic metallization of bipyridine-based COF. Adapted with permission from ref. 67. Copyright 2020, Royal Society of Chemistry. (c) The monomers, topologies, and π–π stacking degrees of the 2D and 3D porphyrin-based COFs. Adapted with permission from ref. 68. Copyright 2019, Wiley-VCH. | ||
Reticular chemistry is applied to the rational arrangement of structural units to form organic frameworks in predictable topologies that rely on the geometrical configuration and connection numbers of building blocks. Owing to the importance of open active sites, 3D topologies may be more applicable for catalysis because of the high connectivity and robust porous structures. As mentioned, there are strong π–π interactions within the 2D framework, and the face-to-face stacking mode of close 2D sheets results in better charge transfer. However, the embedded catalytic sites are usually buried among the stacking molecules, especially for COFs comprising metal macrocyclic complexes. In contrast, the molecular interaction in 3D structures depends more on multi-directional covalent bonds instead of interlayer π–π stacking to support the framework. Hence, the internal backbone has better pore connectivity, which is more beneficial for the contact and coordination between the active sites and reactants. Despite the unclear structure–activity relationship of 3D frameworks, their unique features have been reported.69,70 For example, Wang and coworkers discussed the performance difference between 2D sql and 3D pts topologies, with the same covalent bonding type and active Pd porphyrin moiety (Fig. 6c). The 3D structure exhibits efficient superoxide radical anion formation because more open porphyrin sites yield higher activity in the photoinduced oxidation of thioanisole to methyl phenyl sulfoxide.68
To explore metallized HOF in photocatalysis, macrocyclic compounds decorated by multiple hydrogen-bonded donor/acceptor functional groups are applied to build porous structures with permanent porosity. With strong π–π interaction, porphyrin derivatives have been used for constructing robust HOF with catalytic functions.40,71 Based on metal-free porphyrin HOF,72 Liu et al. first synthesized the porphyrin-based HOF with various chelated central metal ions, including Zn2+, Cu2+, Co2+, and Ni2+ (Fig. 7a).73 Characterized by single-crystal X-ray diffraction, the four metallized porphyrin-based HOFs exhibit ABAB stacking modes with slightly different π–π stacking degrees. The N2 adsorption measurements confirm excellent porosity, with more than 1600 m2 g−1 of the calculated Brunauer–Emmett–Teller (BET) surface area. Through the gas–solid experiments, the decorated Cu/Co/Ni HOFs show activity in CO2-to-CO reduction, in which the CoII porphyrin HOF, named PFC-72-Co, exhibits optimal performance with an efficiency of 14.7 μmol g−1 h−1. Similarly, such HOF materials were used for electrocatalysis by Li et al., and PFC-72-Co exhibits the best activity in two-electron O2 reduction.74 According to theoretical calculations, the activation energy barrier of the Co sites for adsorbing gas O2 molecules to generate the *OOH intermediate is lower than that of other metal ions. In the two cases, both the photocurrent and electrochemical impedance spectroscopy reveal that the Co-anchored reticular HOFs have better charge transfer ability, which may be responsible for high activity in photo- or electro-induced reduction reactions.
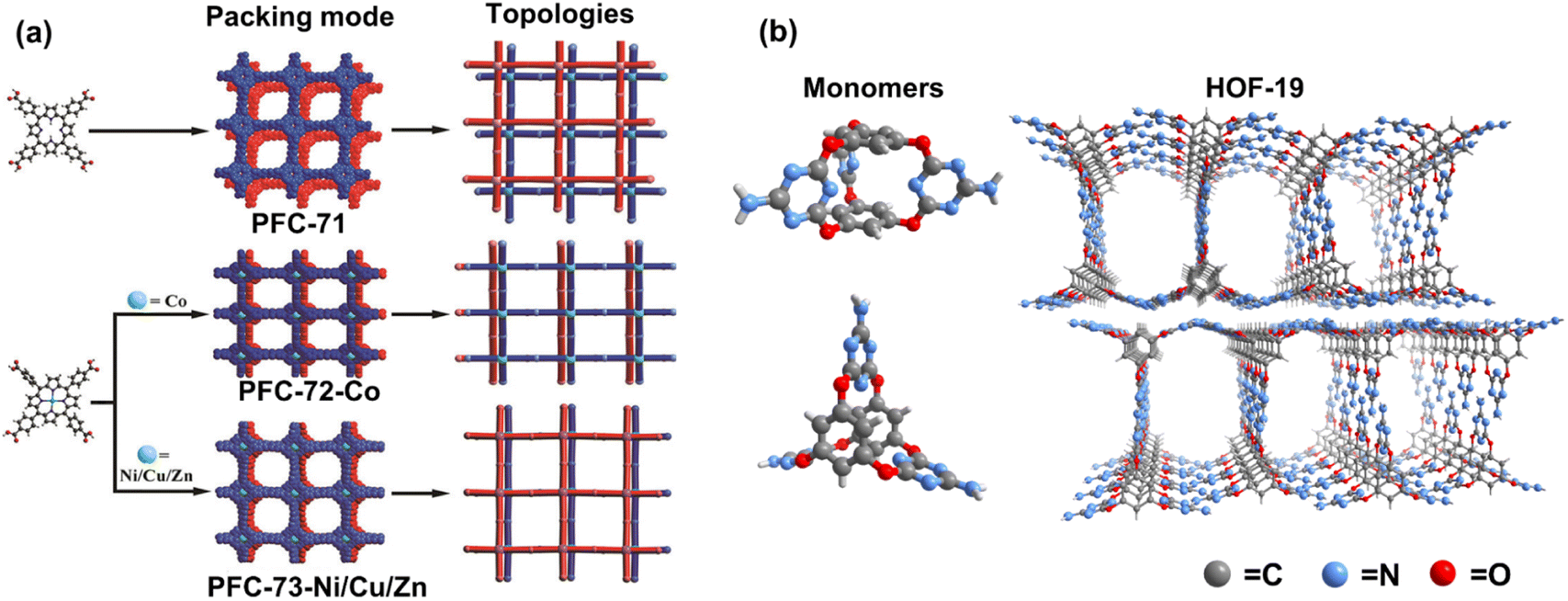 | ||
| Fig. 7 (a) The monomers, topologies, and π–π stacking modes of PFC-71, PFC-72, and PFC-73. Reprinted with permission from ref. 73. Copyright 2022, Wiley-VCH. (b) 3D molecular conformation of monomers and the structure of HOF-19. Adapted with permission from ref. 75. Copyright 2019, American Chemical Society. | ||
Owing to synthetic flexibility, solvent affinities, and internal connection sites, post-synthetic modification is the appropriate way to introduce active guests into HOFs. Chen et al. reported the integration of PdII species in the nitrogenous carboxyl-based HOF material, HOF-19 (Fig. 7b).75 Following recrystallization, palladium acetate is added into the acetone solution of dispersed HOF-19. The Pd2+ ions can be uniformly dispersed in the pores of HOF-19 with high loading. The formed composite HOF-19⊃Pd(II) is highly active in the Pd-induced Suzuki–Miyaura coupling reaction. In addition to post-synthetic decoration, in situ synthesis of HOF composites based on active species is also available.76,77 Cao et al. demonstrated that mechanochemical means, such as ball milling, can synthesize HOFs. By solvent-assisted grinding of HOF precursor and as-prepared Pd nanoparticles, the latter can be effectively loaded onto the prepared HOFs.78 Evidently, an efficient and easy way can be applied for further exploration of HOF metallization.
Advanced COFs and HOFs for photocatalysis
The rapid growth of photocatalysis in recent years has introduced advanced materials to achieve efficient solar energy conversion under mild conditions. Taking examples of photocatalytic H2 evolution and CO2 reduction, the merits of photocatalysts can be clearly reflected by these reactions owing to their widespread applications, accessibility of reactants, and clean fuel products. Photocatalytic water splitting and CO2 reduction are promising approaches to sustainable transformation. The photo-redox half-reaction of H2 and O2 evolution via water splitting is shown in eqn (1) and (2):| 2H+ + 2e− → H2 | (1) |
| H2O + 2h+ → 1/2O2 + 2H+ | (2) |
The photocatalytic H2 evolution driven by COF or HOF catalyst starts with the light harvesting of the photosensitizer to generate electron–hole pairs. Following the charge separation, H2 formation occurs on the active site with the involvement of two electrons, and the photoexcited holes promote the O2 evolution. For more efficient charge separation and utilization, a sacrificial electron donor is often added into the reaction system to consume the photo-generated holes instead of water as a reducing agent.79 Therefore, the light absorption and charge separation of COF and HOF materials correlate with the activity of photocatalytic H2 evolution.
Similarly, the light harvesting and charge separation ability of photocatalysts is essential to achieve CO2 conversion. In contrast to H2 evolution, CO2 molecules are highly stable, so the activation of CO2 to generate CO2˙− intermediate is more difficult.80 Moreover, the interaction of reactants with catalysts as well as the formed intermediate on the active sites determine the activity and selectivity of CO2 photoreduction. Taking CO2-to-CO conversion as an example, the pathway from CO2 molecules to the CO product is shown in eqn (3)–(5):81
| CO2 + e− → CO2˙− | (3) |
| CO2˙− + H+ → *COOH | (4) |
| *COOH + e− + H+ → CO + H2O | (5) |
In photocatalysis reaction systems, it is vital to suppress the side reaction on the active sites for the expected products with high selectivity. Moreover, the activity and selectivity of CO2 reduction indicate the properties of catalytic sites for COF and HOF photocatalysis. The representative studies of COFs and HOFs in the photocatalytic application of H2 evolution as well as CO2 reduction are discussed below, and the specific performance parameters of catalysis are shown in Table 1.
| Photocatalysts | Light source | Reaction medium | Production rate (μmol g−1 h−1) | AQY | Ref. |
|---|---|---|---|---|---|
| COF-alkene | >420 nm | TEOA; Pt (3 wt%); H2O | H2: 2330 | 6.7% (420 nm) | 82 |
| COF-TtaTfa | >420 nm | AA; H2PtCl6 (8 wt%); H2O | H2: 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
1.43% (450 nm) | 83 |
| BtCOF150 | >400 nm | TEOA; Pt (1 wt%); H2O | H2: 750 | 0.2% (420 nm) | 84 |
| CYANO-CON | >420 nm | AA; Pt (1 wt%); H2O | H2: 134200 |
82.6% (450 nm) | 49 |
| TpPa-COF-(CH3)2 | >420 nm | SA; Pt (3 wt%); PBS buffer | H2: 8330 | — | 85 |
| BT-PyBuOH | 380–780 nm | AA; Pt (2 wt%); MeOH/H2O | H2: 11285 |
4.88% (420 nm) | 86 |
| Ni(OH)2-2.5%/TpPa-2 | >420 nm | SA; PBS buffer | H2: 1895.99 | — | 92 |
| Ni-Py-COF | >420 nm | AA; H2PtCl6 (8 wt%); H2O | H2: 13231 |
4.28% (420 nm) | 93 |
| PFC-1 | >400 nm | AA; K2PtCl4 (8 wt%); H2O | H2: 31200 |
1.97% (400 nm) | 88 |
| PFC-42 | >400 nm | TEOA; Pt (1.54 wt%); H2O | H2: 9733 | 5.56% (420 nm) | 90 |
| COF-367-CoIII | >380 nm | TEA; MeCN | HCOOH: 93.0 selectivity: 82.8% | — | 62 |
| CoPor-DPP-COF | >420 nm | [Ru(bpy)3]Cl2·6H2O; TIPA; MeCN/H2O | CO: 10200 selectivity: 82% |
— | 94 |
| Fe SAS/Tr-COF | >420 nm | [Ru(bpy)3]Cl2·6H2O; TEOA; MeCN/H2O | CO: 980.3 selectivity: 96.4% | 3.17% (420 nm) | 95 |
| TCOF-MnMo6 | 400-800 nm | H2O vapor | CO: 37.25 selectivity: ca. 100% | 0.0067% (400 nm) | 96 |
| PFC-72-Co | >400 nm | TEOA vapor; H2O vapor | CO: 14.7 | — | 73 |
| PFC-45/Cu2O@CP | >400 nm | H2O vapor | CO: 11.81 selectivity: 99% | — | 89 |
| PFC-58-30 | >400 nm | H2O vapor | HCOOH: 29.8 | — | 99 |
| HOF-25-Re | >420 nm | [Ru(bpy)3]Cl2·6H2O; TIPA; MeCN | CO: 3030 selectivity: 93% | 0.18% (420 nm) | 41 |
| HOF-25-Ni@GO-10 | >420 nm | [Ru(bpy)3]Cl2·6H2O; TIPA; MeCN/H2O | CO: 24323 selectivity: 96.3% |
— | 100 |
Improving light harvesting and charge separation ability
Owing to the π-conjugated bonding and D–A system on the photoinduced charge transfer, Yu et al. utilized three types of covalent bonds, alkene, imine, and imide linkages, to combine 1,3,5-triphenylbenzene moieties (Fig. 8a).82 It is believed that the π-conjugated system with alkene linkage is more beneficial to π delocalization, and the electron-withdrawing cyan functionality in connection with the electron-donating 1,3,5-triphenylbenzene moiety can generate the D–A heterojunction. The synthesis of the COF-alkene is based on the Knoevenagel condensation reaction, in which the linkage was cyano-substituted sp2 carbon bonds. The COF-alkene exhibits the best photocatalytic H2 evolution rate of 2330 μmol g−1 h−1, with loading Pt as catalytic active sites, while COF-imide and COF-imine only show negligible activities of 34 μmol g−1 h−1 and 12 μmol g−1 h−1, respectively. Through kinetic analysis reflected by femtosecond transient absorption (fs-TA) at 700 nm, the longest excited state absorption decay of COF-alkene supports the most efficient charge separation. Theoretical calculations of HOMO and LUMO energy levels within the linkage sections of COFs reveal that the alkene linkage with cyano-substituent has the smallest energy gap, which also provides a rational explanation for the high charge transfer efficiency and catalytic activity of COF-alkene.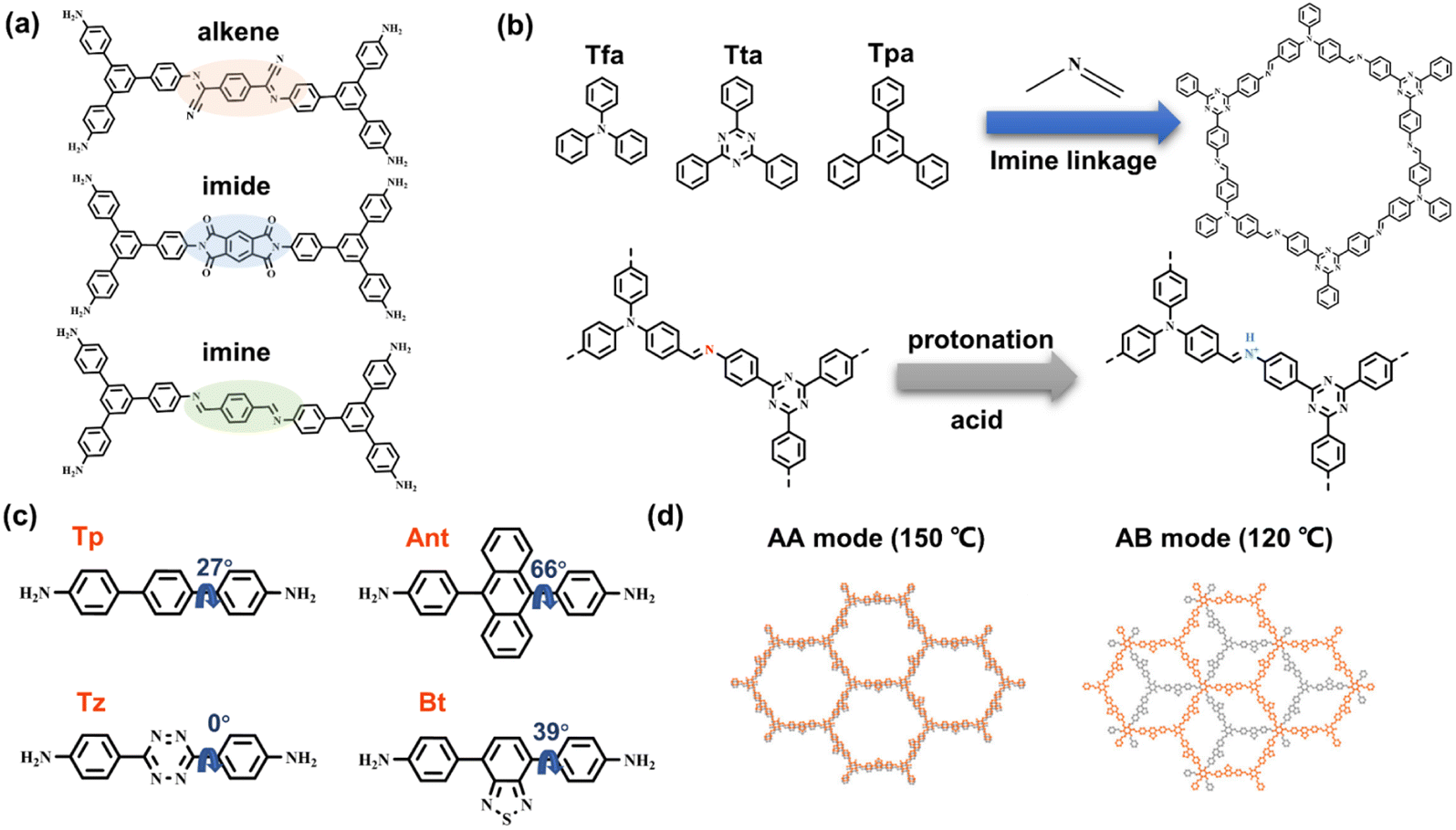 | ||
| Fig. 8 (a) Alkene, imine, and imide linkages within the structure of COFs. Adapted with permission from ref. 82. Copyright 2020, Wiley-VCH. (b) Schematic showing the protonation of the imine linkage in a series of 2D COFs. Adapted with permission from ref. 83. Copyright 2021, Wiley-VCH. (c) Various building blocks with different torsional angles and the generated 2D COFs; (d) different π–π stacking modes determined by respective synthesized temperatures. Reprinted with permission from ref. 84. Copyright 2021, American Chemical Society. | ||
Although the relatively low π electron delocalization of the imine group does not benefit the charge transfer, Thomas et al. demonstrated that the protonated imine linkage can promote delocalization, which is responsible for imine-linked COF with the acidic sacrificial electron donors in photocatalytic H2 evolution.83 Three organic monomer knots, 2,4,6-Tris(4-aminophenyl)triazine (Tta), Tris(4-formylphenyl)-amine (Tfa), and 1,3,5-triphenylbenzene (Tpa), are utilized for building 2D hcb COFs with imine linkage (Fig. 8b). In an acidic solution, the protonated N atoms of imine can be characterized by FT-IR measurement. The disappearance of stretching vibration peaks of CN bonds at 1623 cm−1 and the new emerging peaks at 1791 cm−1 agree with the formation of CNH+, accompanied by darkening the color. Through the in situ EPR analysis, the signal of acidic COFs significantly increases, in line with the enhanced charge transfer. The authors propose that the integration of protons improves imine π conjugation. The more planar molecular conformation favors electron transfer and charge separation. Moreover, more effective binding with protons promotes the reduction of protons to H2. The combination of Tta and Tfa moieties can form D–A systems, leading COF-TtaTfa to 20.7 mmol g−1 h−1 activity in H2 evolution with a Pt cocatalyst in an acidic sacrificial agent environment.
In comprehensive consideration of molecular conformation, synergistic charge transfer, and stacking modes, Ghosh et al. construct a series of 2D β-ketoenamine-linked COFs based on 1,3,5-triformylphloroglucinol (TH) building block.84 With 4,4′′-diamino-substituted p-terphenyl (Tp), electron-donating anthracene (Ant), electron-deficient benzothiadiazole (Bt) and tetrazine (Tz), COFs with different components exhibit different band energies (Fig. 8c). The BtCOF and TzCOF have small energy gaps, whereas the electron driving force of the latter is insufficient for proton reduction, thus showing no activity in subsequent H2 evolution. Owing to the large torsional angle of the building block, no AA mode can be formed for the AntCOF. The monomers of BtCOF, TzCOF, and TpCOF can be arranged as AA parallel stacking at a synthetic temperature of 150 °C, while they tend to form AB stacking mode at 120 °C (Fig. 8d). The BtCOF synthesized at 150 °C exhibits the best H2 evolution activity, suggesting the significance of light harvesting, charge transfer, and porosity in constructing an efficient COF catalyst.
The properties of COF structures can be significantly adjusted by the proper introduction of functional groups on skeletons. Li et al. reported CYANO-COF comprising 1,3,5-triformylphloroglucinol (Tp) and 4,4′-diamino-[1,1′-biphenyl]-3,3′-dicarbonitrile (BD-CYANO) (Fig. 9a).49 Because of the electron-withdrawing groups of cyano substituents, the ketene–cyano (D–A) pair could be constructed by Schiff-base condensation reaction, which contributes to a rapid charge transfer. The nano-sized COF material, CYANO-CON, is prepared by ball milling. Compared to the nano COF without cyano groups, CYANO-CON, with the more efficient charge separation, exhibits a higher photocatalytic performance of H2 evolution (134200 μmol g−1 h−1) and excellent apparent quantum efficiency (82.6%) under 450 nm light. Zhang et al. synthesized a series of TpPa-COF-X materials using various substituent groups (X = –H, –(CH3)2, and –NO2) (Fig. 9b).85 In the photocatalytic H2 evolution experiment, the H2-generated rates of COFs follow the trend of TpPa-COF-(CH3)2 (8.33 mmol g−1 h−1) > TpPa-COF (1.56 mmol g−1 h−1) > TpPa-COF-NO2 (0.22 mmol g−1 h−1), which agrees with the electron-donating ability of CH3 > H > NO2. Measured by electrochemical impedance spectroscopy (EIS), photocurrents, and photoluminescence spectra, TpPa-COF-(CH3)2 has the enhanced efficiency of charge transfer with the incorporation of the CH3 group. Therefore, the authors believe that the electron-donating functional groups are beneficial for the inner carrier transport ability of photocatalysts.
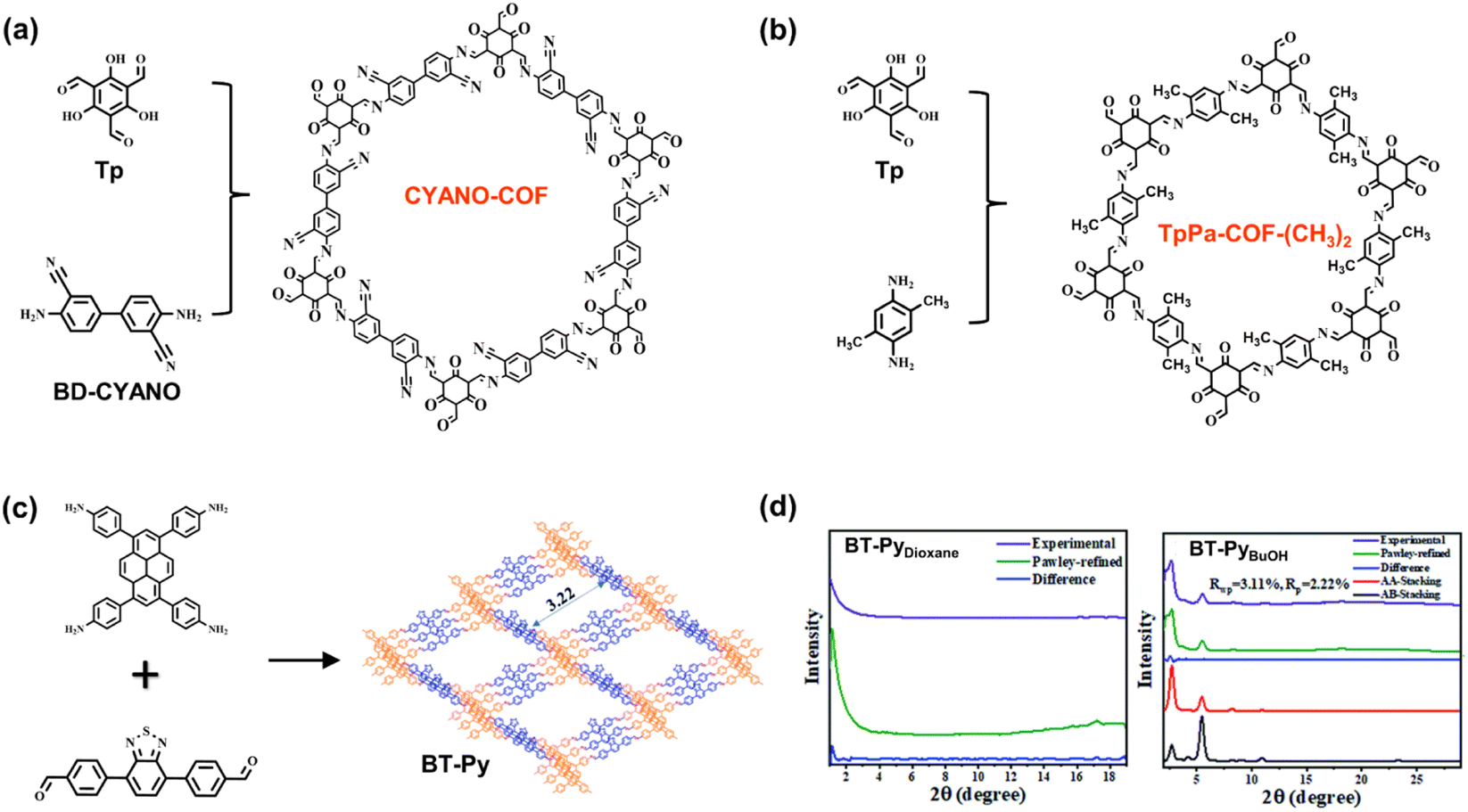 | ||
| Fig. 9 (a) The monomers and structures of CYANO-COF. Adapted with permission from ref. 49. Copyright 2022, Nature Publishing Group. (b) The monomers and structures of TpPa-COF-(CH3)2. Adapted with permission from ref. 85. Copyright 2019, Wiley-VCH. (c) The monomers and structure of BT-Py COF; (d) the respective XRD spectra of BT-PyDioxane and BT-PyBuOH. Reprinted with permission from ref. 86. Copyright 2022, Royal Society of Chemistry. | ||
The ordered arrangement of monomers in the crystalline structures determines the properties of COFs as porous materials. High crystallinity affects charge transfer due to the π–π stacking and synergetic effect of various compositions and contributes to the accessibility of internal active sites with better porosity and dispersity. Therefore, the improvement in crystallinity seems to be the proper way to enhance the photocatalytic performance of COFs. Chou et al. explored the relationships between the solubility and crystallinity of the imine-linked COF BT-Py (Fig. 9c).86 In the synthetic process of BT-Py COF, the dissolution of monomers promoted crystal nucleation and growth, and the high polarity and strong hydrogen bonding ability enhanced the growth rate of the crystalline phase. Compared to the binary solvent of mesitylene/p-dioxane, the mixture of o-DCB/n-BuOH and o-DCB/EtOH with higher polarity provided better solvent systems for COF preparation. In contrast to BT-PyDioxane, BT-PyEtOH and BT-PyBuOH exhibit excellent crystallization (Fig. 9d), and the latter has a large surface area with the highest N2 adsorption. The long-range order of BT-PyBuOH facilitates the charge transfer and suppresses the electron/hole recombination as well as charge trapping at the defects. Furthermore, excellent porosity favors the exposure of the internal active species. Therefore, the resulting BT-PyBuOH material demonstrates the high performance of photocatalytic H2 evolution with a generation rate of 11285 μmol g−1 h−1.
For the well-organized structure of HOF, the regular arrangement not only contributes to light harvesting and charge transfer but also provides high connectivity for mass transfer to the internal active sites. Owing to the strong π stacking interaction of center-conjugated planar pyrene-based HOF materials, the HOF-100 series,87 including PFC-1, have shown sufficient stability in chemical and high-temperature environments. PFC-1 is characteristic of photoexcited electron generation. Active species Pt or planar [Co(qpy)(OH2)2]2+ molecule, as electron injection units and reduction catalysis sites, are loaded in PFC-1. The well-loading active species and highly ordered arrangement of pyrene photosensitizer units can consume the photoexcited electrons for H2 evolution. Accordingly, outstanding activity is achieved at a rate of 31.2 mmol g−1 h−1 for Pt loading, as well as 14.8 mmol g−1 h−1 for [Co(qpy)(OH2)2]2+ loading.88
Moreover, Liu et al. selected a tridentate carboxyl-based ligand conjugated with the C3N4 moiety to synthesize crystalline HOF, PFC-42 (Fig. 10a), followed by the post integration of Pt nanoparticles. Compared with the Pt-loading amorphous polymer, g-C3N4, in the gas–solid phase for H2 evolution,90 the composite PFC-42 Pt exhibits much higher activity, with a rate of 11.32 mmol g−1 h−1. Under the same conditions with the same Pt loading, uncrystallized monomers and poorly crystalline HOF treated by acid show low catalytic activity, indicating the significance of the crystalline structure. Revealed by photocurrent, electrochemical impedance spectroscopy (EIS), and fluorescence tests, effective light absorption, rapid charge transfer, and efficient mass transport determine the excellent performance of HOF-based catalysts. This research provides insight into the utilization of crystalline HOF in photocatalysis.
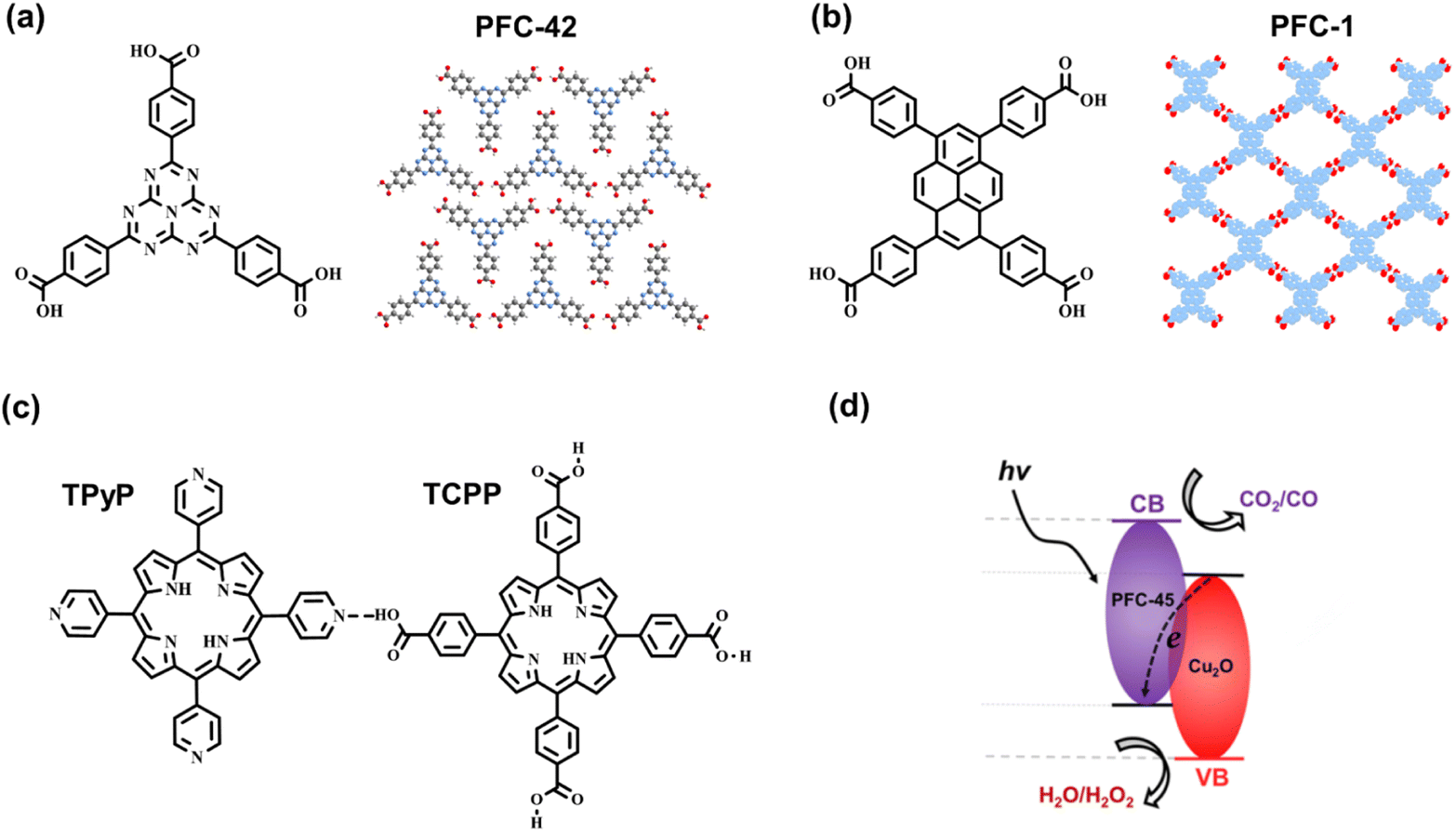 | ||
| Fig. 10 The monomers and hydrogen-bonded structures of (a) PFC-42 and (b) PFC-1. (c) The monomers and hydrogen bond of PFC-45; (d) the band structure of PFC-45/Cu2O. Reprinted with permission from ref. 89. Copyright 2022, American Chemical Society. | ||
Guided by hydrogen bonding with low formation energy and high reversibility,91 Cao et al. applied in situ electrophoretic deposition (EPD) to synthesize PFC-45/Cu2O film deposited on carbolic paper (CP) electrode.89 The multicomponent HOF, PFC-45, comprises two categories of porphyrin derivates substituted by carboxyl and pyridine (Fig. 10c). The intrinsic defects of the deprotonation of carboxyl lead to the negative surface charge of PFC-45, so the corresponding nano particles (NPs) can be driven into the electrode under the electric field. By further improving EPD, the mixed PFC-45/Cu2O NPs can be loaded onto CP to form the PFC-45/Cu2O@CP film. Owing to the incorporation of n-type PFC-45 and p-type Cu2O, p–n heterojunction forms on the interface, which facilitates the separation of excited electrons and holes. In the Z-scheme charge separation, the electrons in the CB of Cu2O can combine with the holes in the VB of the PFC-45 (Fig. 10d), suppressing the inherently excited charge recombination. The electrons in the CB of PFC-45 with negative enough potential can participate in the CO2-to-CO reduction.
Integrating efficient active sites
To incorporate active sites for H2 evolution, the easy solution is to load Pt as the electron sinks and proton combined sites. However, it is important to introduce low-cost transition metal elements into COF structures from an economical and sustainable perspective. Through simple post-synthetic decoration, Zhang et al. prepared a series of nickel hydroxide-modified COF (Ni(OH)2-X%/TpPa-2) (Fig. 11a).92 Measured by H2 evolution photocatalysis, the obtained composite Ni(OH)2-2.5%/TpPa-2 enhanced an optimal H2 generation rate of 1895.99 μmol g−1 h−1. The introduced Ni-based species can act as active sites for efficient catalytic reactions.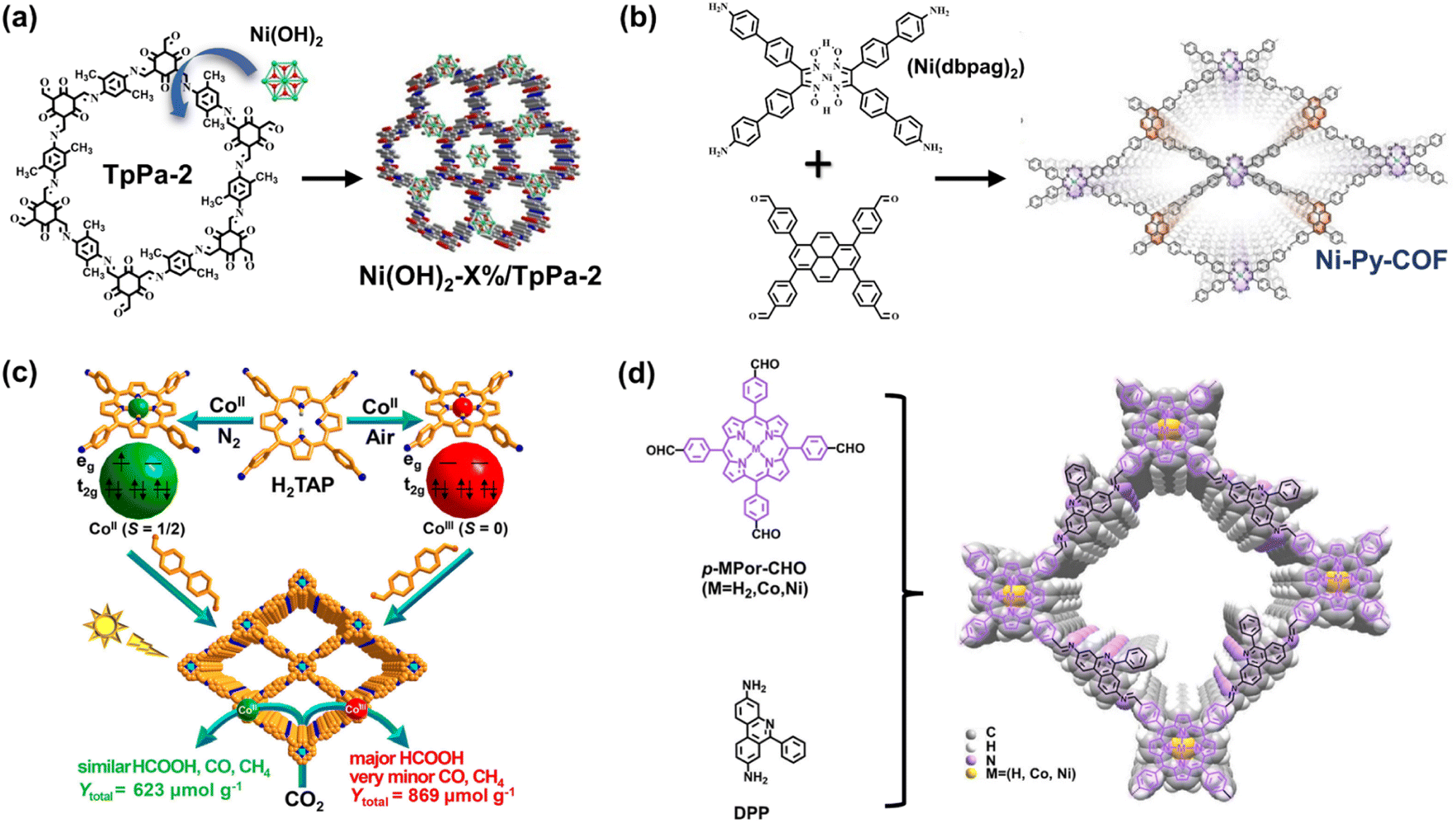 | ||
| Fig. 11 (a) Schematic illustration of the synthesis of Ni(OH)2-X%/TpPa-2 composite materials. Adapted with permission from ref. 92. Copyright 2020, Elsevier. (b) Ni-Py-COF generated from the Ni(dbpag)2 and pyrene moieties. Adapted with permission from ref. 93. Copyright 2022, Wiley-VCH. (c) The various electron spin states of the center metal ions and the respective photocatalysis performance of CoII/CoIII porphyrin COFs. Reprinted with permission from ref. 62. Copyright 2020, American Chemical Society. (d) The structure and monomers of the porphyrin-based COF with enlarged interlayered spacing. Adapted with permission from ref. 94. Copyright 2022, American Chemical Society. | ||
Metal macrocyclic compounds and bipyridine derivative complexes satisfy the requirements at the molecule level. Lan et al. utilized nickel glyoximate (Ni(dbpag)2) as a building block to prepare COF with a new metallization module.93 By combining the efficient photosensitizer with a strong electron-donating block, such as pyrene, nickel glyoximate units exhibit higher photocatalytic activity (Fig. 11b). Owing to the electron enrichment on the metal sites, the Ni-Py-COF exhibits a catalytic H2 evolution rate of 626 μmol g−1 h−1 without a Pt cocatalyst. The synergetic effect of light harvesting cores (pyrene), stable π–π stacking interaction, and electron-rich active Ni sites improve catalytic performance, further extending the research direction of the metal within the backbone.
For optimal catalytic efficiency and product selectivity, it is essential to modulate the catalytic microenvironment of COF structures. As mentioned above, porphyrin derivatives are appropriate for the metallization of COF. Adjusted by center metal elements and substituted functional groups, porphyrin species exhibit various catalytic properties in CO2 reduction. In a recent report, Jiang et al. found that product selectivity can be affected by the valence electron spin states of porphyrin central metal ions.62 Through preparation in N2 and air atmosphere, the center metal Co ions of porphyrin-based COF-367-Co exist in CoII and CoIII states, respectively, and the latter exhibits high activity and selectivity in photocatalytic reduction from CO2 to HCOOH. In particular, COF-367-CoIII has a higher photocurrent response, lower charge-transfer resistance, and decreased photoluminescence intensity, revealing an enhanced charge transfer ability. Moreover, the various spin states of chelated Co ions (Fig. 11c) determine the electron distribution/orientation of the 3d orbital, thus remarkably affecting the interaction of Co with adsorbates. Theoretical calculations indicate that the coupling strengths between CoIII with CO2, or HCOOH, are higher than those of CoII, and the decreased activation energy of hydrogenation steps in the process of HCOOH formation on CoIII sites explains the higher selectivity of the HCOOH reduction product.
Owing to the strong π–π stacking interaction along the molecular symmetric axis, the vertical dense packing of molecules existing in many 2D COFs might be prevented from the accessibility of metal sites. Wang et al. implemented an interesting strategy for constructing a 2D porphyrin-based COF with enlarged inter-layer spacing by 3,8-diamino-6-phenyl-phenanthridine (DPP).94 The large steric hindrance between the layers caused by the phenyl substitute of DPP effectively hinders the axial π–π stacking of porphyrin, resulting in an interlayered spacing approaching 6 Å (Fig. 11d). With stable AA stacking modes, incompact layer-by-layer stacking renders the metal site accessible to the guest molecules, thereby improving the mass transfer of the reaction. Moreover, a BET surface area of 1021 m2 g−1 measured by the N2 adsorption experiment suggests that the permanent porous structure is still maintained in such an open framework for CO2 reduction. The feasibility of enlarging the interlayer space for efficient COF photocatalysts has been proven by catalytic activity at a rate of 10200 μmol g−1 h−1 to generate CO products.
Furthermore, the rational incorporation of linear bidentate ligands into COFs can achieve metallization by post-synthetic modification. Hou et al. selected an ace-naphthenequinone monomer to generate imine linkages, and the imine-substituted blocks provide bidentate coordination sites for metal elements (Fig. 12a), such as Fe, Co, Ni, Zn, Cu, Mn, and Ru.95 X-ray absorption fine structure spectroscopy revealed no metal–metal bonds within the COF (Fig. 12b), suggesting the atomical dispersion of metal sites. The integration of uniformed metal species provides Lewis acid sites for CO2 chemisorption and improves electron delocalization through chelation interaction. Consequently, the metallized COFs exhibit high activity for photocatalytic CO2-to-CO reduction with a generation rate of 980.3 μmol g−1 h−1.
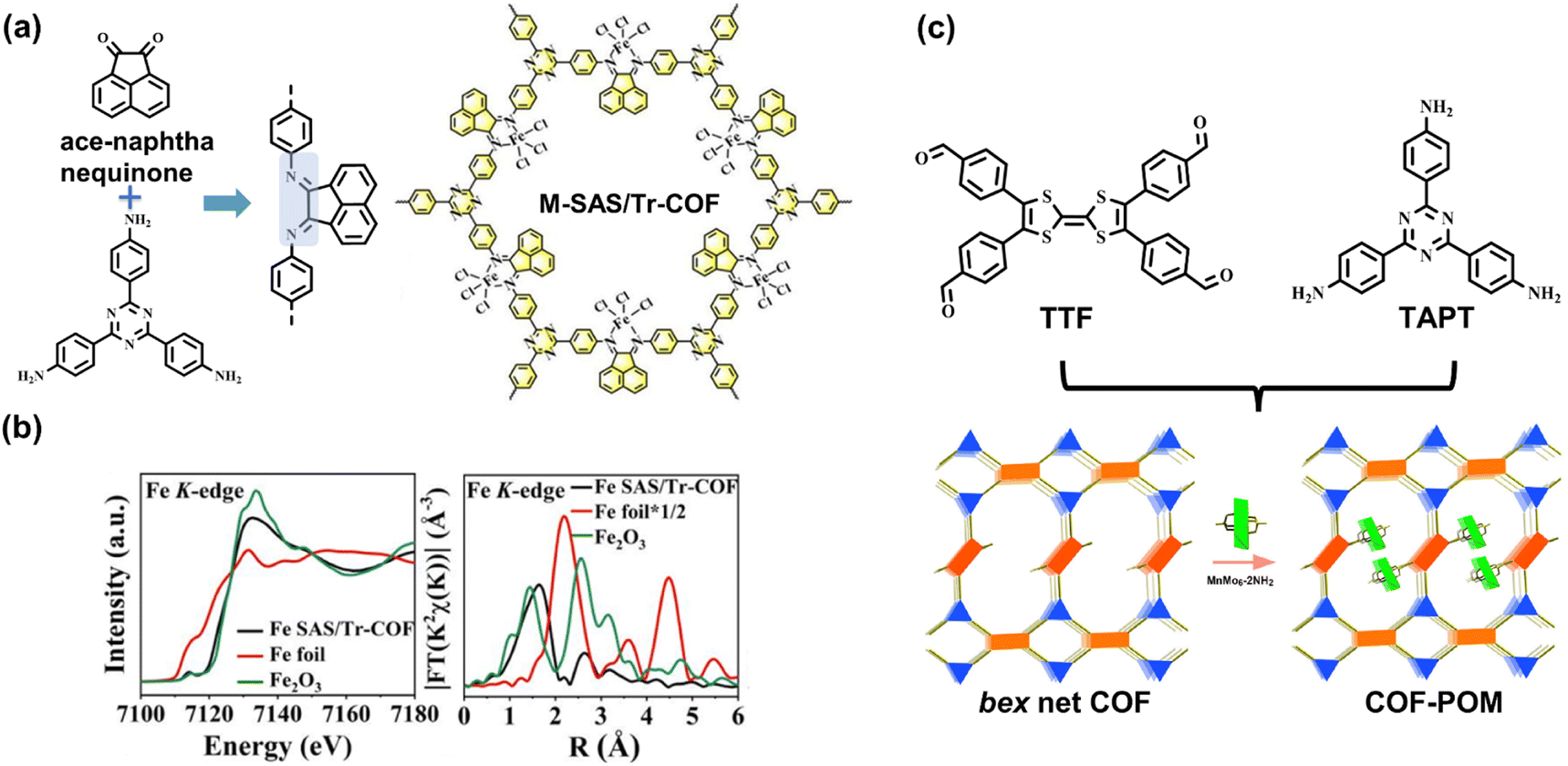 | ||
| Fig. 12 (a) The metallization method of ace-naphthenequinone-based COF; (b) the normalized XANES and Fourier-transformed (FT) EXAFS spectra of the Fe K-edge. Adapted with permission from ref. 95. Copyright 2022, American Chemical Society. (c) The [4 + 3] bex COF network comprises TTF and TAPT blocks. Adapted with permission from ref. 96. Copyright 2022, American Chemical Society. | ||
The reticular chemistry of topology structures can be introduced into the goal-directed synthesis of COF. Most syntheses of the topologies are based on the entire consumption of connection sites by generating covalent bonds, such as the [3 + 3], [4 + 4], and [4 + 2] combinations. Lan et al. explored various COFs with unusual bex topology referring to the [4 + 3] combination.96 There are inherent uncondensed aldehyde functionalities periodically placed on the skeleton, which further connect with the post-introduced polyoxometalates, MnMo6–2NH2 for generating homodispersed single-cluster sites. Based on the covalent bonding with the internal periodical binding groups, such loaded polyoxometalates can be embedded in the skeleton to effectively avoid the deintercalation and agglomeration of the internal metal sites. In the subsequent gas–solid CO2 reduction photocatalysis, the bex network COF integrated with the single-cluster polyoxometalates exhibits high activity of 37.25 μmol g−1 h−1 with ca. 100% selectivity. As demonstrated in Fig. 12c, the TTF moieties and embedded metal sites comprise D–A systems so that the photoexcited electrons can transfer to the LUMO of polyoxometalates, resulting in high charge separation efficiency. Theoretical calculations indicate that CO2 molecules are chemisorbed with the addition of one electron, and then the main intermediate *COOH species are formed, followed by the breaking of C–O bonds and the desorption of CO molecules.97,98
For HOF materials, metal porphyrin molecules have been demonstrated to be the appropriate building blocks. In the porphyrin-based HOF with a single monomer composition, however, the porphyrin units play the roles of photosensitizer and catalytic sites, and all molecules exhibit similar photo-response behavior that is not beneficial for charge separation. Considering that CO2 molecules are only chemisorbed on partial metal sites, photoexcited electrons participate only in the rapid redox of metal ions instead of adsorbed CO2. Therefore, the separation of the photosensitizer and catalyst into different building blocks may improve the utilization efficiency of electrons and holes in photocatalysis. In terms of intermolecular hydrogen bonding interaction, Cao et al. combined Cu porphyrin and metal-free porphyrin moieties to construct HOF, PFC-58-X series (X% present the metallized molecule ratios, Fig. 13a).99 The structure of PFC-58, which consists of Cu porphyrin, is precisely simulated by the single crystal characterization, and the identical PXRD patterns reveal that the metalloporphyrin content does not affect the construction. Note that PFC-58-30 (30% Cu porphyrin) exhibits the highest activity for CO2-to-HCOOH conversion. Theoretical calculations suggest that the rational match between stacking electron donors and acceptors provides a charge transfer pathway from the former to the latter. Therefore, the electron distribution of chelated Cu ions significantly increases, and excited holes are left in metal-free porphyrin, which enhances charge separation and promotes the reduction ability of active sites. The combination of metallized and metal-free porphyrin is similar to the D–A conjunction in the HOF structure.
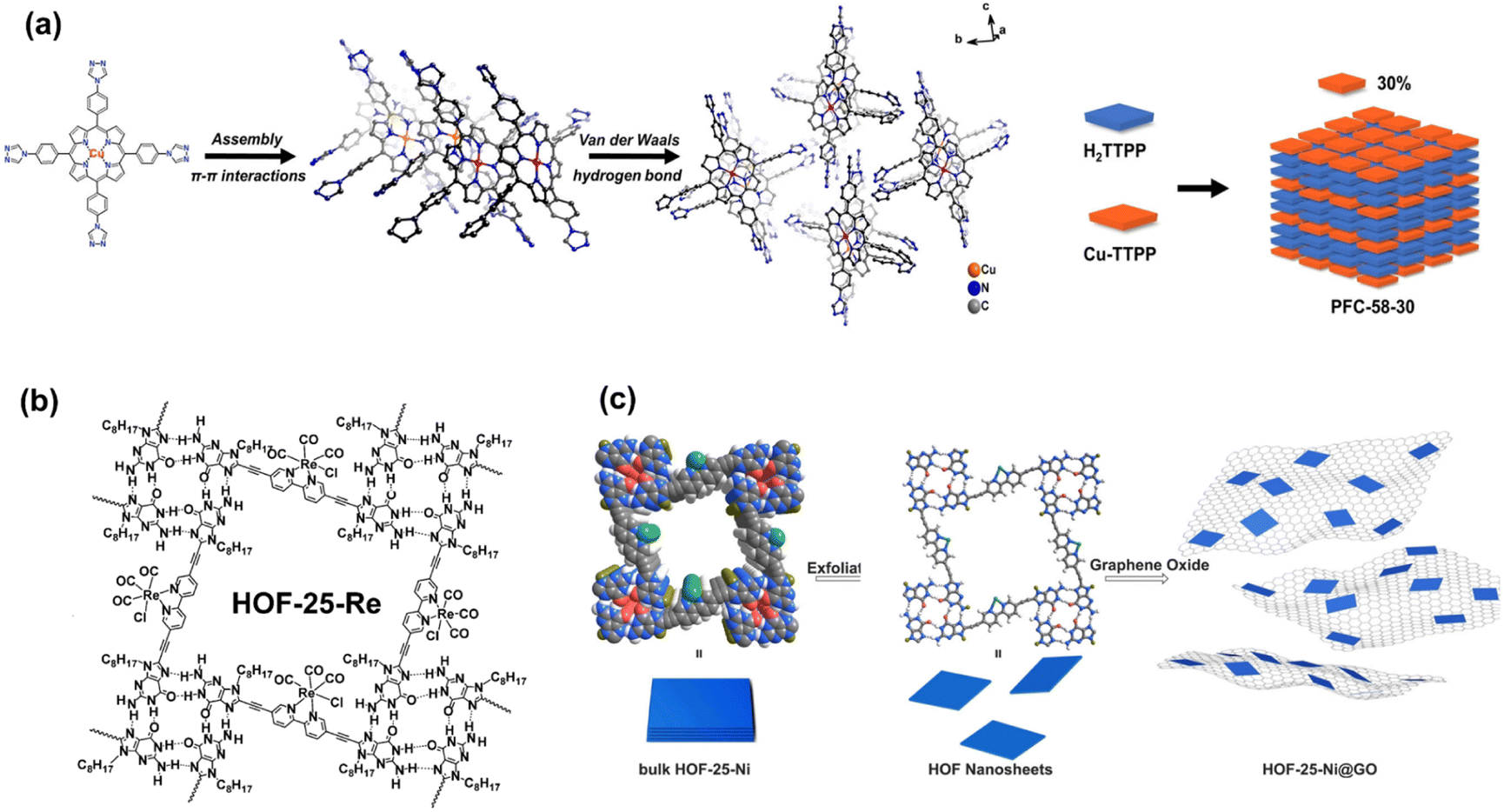 | ||
| Fig. 13 (a) Construction of PFC-58 and schematic show of the PFC-58 series with 30% metallized molecule ratios. Reprinted with permission from ref. 99. Copyright 2022, Wiley-VCH. (b) Composition of HOF-25-Re. Reprinted with permission from ref. 41. Copyright 2021, Wiley-VCH. (c) Illustration of the synthesis of HOF-25-Ni 2D nanosheets and HOF-25-Ni@GO for photocatalytic CO2 reduction. Reprinted with permission from ref. 100. Copyright 2022, Wiley-VCH. | ||
The mentioned building blocks, pyrene and porphyrin, possess large planar π conjugation, can generate stable stacking interaction to maintain the structure of HOFs. Considering that multiple hydrogen bonds can supply stable interaction for locking monomers, the large planar π conjugated aromatic rings and functional groups with abundant hydrogen-bonding sites can be integrated into the synthons of HOFs, which can self-assemble to generate highly steady porous structures. Inspired by such a design strategy, Chen et al. utilized a 2,2′-bipyridine-derived building block with hydrogen-bonded G-quadruplexes for the synthesis of HOF-25 with excellent chemical stability (Fig. 13b), especially in the alkaline environment.41 Owing to the bidentate-coordinated units, simple post-synthetic modification can achieve the metallization of the structure. Therefore, Re(CO)5Cl-loading HOF-25-Re with strong robustness, high porosity, a clear charge transfer pathway, and uniform metal sites possesses the potential for high performance of catalysis. However, within the liquid–solid system of CO2 photoreduction, HOF-25-Re exhibits lower activity compared to Re(bpy)(CO)3Cl homogenous catalyst. With the addition of photosensitizer [Ru(bpy)3]Cl2·6H2O, the system yields a high conversion rate of 3030 μmol g−1 h−1 for CO generation.
Based on HOF-25, Jiang et al. further improved the photocatalysis performance by preparing 2D nano material.100 The thin 2D layer structure can be achieved as a consequence of weak-interlayered interactions compared to in-plane combination binding. In HOF-25-Ni, multiple hydrogen bonds generated by G-quadruplexes make it possible to exfoliate materials into 2D nanosheets by breaking layer-by-layer stacking. In contrast to bulk materials, nanosheets exhibit a higher surface area, enhanced charge conductivity, and size-dependent properties. To avoid self-agglomeration, the as-synthesized HOF-25-Ni nanosheets adhere to GO by the strong π–π stacking interaction (Fig. 13c). Moreover, the GO base can accelerate internal electron transfer. As a validation, the photocatalytic CO2 reduction experiment indicates an excellent CO generation rate of 24323 μmol g−1 h−1 for HOF-25-Ni@GO nanosheets, which is significantly higher than that of the above bulk HOF-25-Re.
Table 1 summarizes the recent progress of H2 evolution and CO2 reduction by COFs and HOFs, which exhibit excellent photocatalytic activity and applicability. Compared with the characteristics and applications, the respective advantages and drawbacks of COFs and HOFs can be precisely tracked. In the practical applications of photocatalysis, COFs have demonstrated superior robustness, which makes COFs applicable in harsh reaction environments, such as acidic, alkaline, and strong irradiation conditions. With the long-term photocatalysis test, COF materials can therefore maintain excellent cycle performance. After the reaction, the crystallinity and BET surface area of COFs may be slightly reduced, but no change in the framework backbone can be spotted by specific measurements, demonstrating the stable structures of porous materials. Moreover, the tunability of conjugated linkages contributes to the in-plane π delocalization, improving the charge mobility for the high performance of photocatalysis. As for HOFs, in addition to the high crystallinity, the features of fabrication diversity and easy processability make it possible to improve photocatalytic activity by methods such as electrophoretic deposition and post-synthetic modification. HOFs also show the stable performance of recycle photocatalytic reaction without obvious reducing activity, and the PXRD spectra with the maintained intensity of peaks prove enough stability of HOFs in the proper photocatalytic reaction systems. Notably, the loss of crystallinity and photocatalytic performance can be recovered after a simple recrystallization procedure, which is beneficial for practical utilization.41 Even though the current studies of HOFs in the field of photocatalysis are still limited, their unique properties can be further explored to achieve higher activity and stability.
Conclusion and perspective
As blossom categories of novel functional porous framework materials, COF and HOF have shown huge potential in photocatalysis, which is enabled by their excellent porosity, high crystallinity, flexible structural tunability, and compositional diversity. Over the past several years, the necessity of performance-oriented fabrication with a rational ordered arrangement of various functionalized building blocks has been achieved. However, intrinsic poor charge conductivity and deficient active sites have been restricting catalytic application, referring to the involvement of photo-electrons/holes. Precise molecular engineering design and reticular chemical synthesis are considered research directions to promote inherent features and overcome disadvantages. Because of the expectation of a promising porous catalyst, there are still some knotty challenges and continuous arguments waiting for further innovations in the construction of COF and HOF.Modulation of connection bonding for the balance of crystallinity and robustness
Covalent linkages with high reversibility are beneficial to crystalline COFs, but they have relatively weak stability. The opposite is usually true for strong covalent bonds. However, HOFs usually present highly crystalline structures due to hydrogen bonding, which exhibits remarkable formation reversibility. Under harsh conditions, stable structures are necessary for catalytic reactions. A well-organized arrangement of monomers yields excellent porosity and effective charge transfer, facilitating a more precise characterization of the catalysis microenvironment. Furthermore, based on the exact structures of the organic frameworks, the structure–property-activity relationship can be explored for improvement. For the synthesis of COFs, it is important to achieve joint improvement in high crystallinity, strong steadiness, and effective π conjugation extension. For HOFs, to promote stability, multiple hydrogen-bonded and π–π stacking interactions must be enhanced for more robust framework fabrication.Precise modification based on molecular engineering
It is important to promote light harvesting, charge transfer, and catalytic ability through rational design at the molecular level. Referring to the effect of precise decoration on photocatalytic efficiency and product selectivity, the guidance for photocatalysis needs further studies, especially for HOFs in molecular engineering fields, such as the D–A system and π–π interaction. To this end, more comprehensive data from computational simulation and machine learning should help in the structural and molecular design and synthesis of multifunctional COFs and HOFs.Reticular design for more topologies
Reticular chemistry with various topologies creates diversity for the construction of COF and HOF networks. In general, the accessibility of metal sites chelated within the organic ligands for reactants can be enhanced in 3D topologies owing to the open porous structures. In contrast, the high degree overlap of planar π conjugation in 2D topologies can promote interlayered charge transfer but largely bury the metal active sites. Despite limited control studies on the dimensions of topologies, it is believed that the suitable construction of COF and HOF 3D structures with high connectivity, strong stability, and unburied active sites is beneficial in promoting photocatalytic efficiency.Optimization of practical application domains
For higher economic value and more efficient utilization of solar energy, current photocatalysis of COFs and HOFs should be extended into more challenging reaction systems, such as overall water splitting and multi-carbon conversion of CO2, which demonstrate higher energy barriers. Considering overall water splitting, for instance, the photogenerated electrons and holes are involved in the photo-redox half-reaction of H2 and O2 evolution, in which the efficient separation and rapid transfer of the photoexcited charges to active sites should be achieved. For the fabrication of COFs and HOFs, it is necessary to develop a more effective strategy, such as Z-scheme heterojunction formation,101 for achieving charge separation and strong photo-redox stability. Owing to the high-intensity proportion of long-wavelength irradiation of sunlight, the extended solar absorption in the red and near-infrared ranges contributes to the further conversion of solar energy to chemical energy.COFs and HOFs should be established in terms of the respective features of the materials. For COFs, more elaborate structural modeling based on computational simulation is required for the precise analysis of properties and performances, which contributes to better catalytic efficiency. Research on COF-based catalysts concentrates more on the combination of various building blocks to motivate the synergetic effect of the internal structural units. Based on the robust in-plane covalent linkage, it is worthwhile to synthesize the 2D nano materials of COFs by breaking the interlayered weak interactions. For HOFs, precise knowable crystal information helps understand the structure–activity relationship. However, the instability of hydrogen bonds is challenging for the accurate fabrication of HOF with a predictable network. Therefore, the design and synthesis of functional HOFs need further expansion. Furthermore, the synthesis of multicomponent HOF should be developed to achieve the synergistic effect of building blocks.
In conclusion, HOF and COF, as novel crystalline porous materials, have become new members of the photocatalysis family. Currently, the utilization of HOFs and COFs is still in the preliminary stage. We expect deeper exploration and exploitation to create more opportunities for such novel porous materials in photocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Ministry of Science and Technology of China (2021YFA1500800, 2022YFA1502900 and 2022YFA0911900), the National Natural Science Foundation of China (22088102, 21933007, and 22193013), and New Cornerstone Science Foundation.Notes and references
- Z.-B. Fang, T.-T. Liu, J. Liu, S. Jin, X.-P. Wu, X.-Q. Gong, K. Wang, Q. Yin, T.-F. Liu, R. Cao and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- H. J. Freund and R. P. Messmer, Surf. Sci., 1986, 172, 1–30 CrossRef CAS.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- I. Hisaki, Y. Suzuki, E. Gomez, Q. Ji, N. Tohnai, T. Nakamura and A. Douhal, J. Am. Chem. Soc., 2019, 141, 2111–2121 CrossRef CAS PubMed.
- H. Wang, B. Li, H. Wu, T. L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862–4871 CrossRef CAS PubMed.
- X. Zhang, L. Li, J. X. Wang, H. M. Wen, R. Krishna, H. Wu, W. Zhou, Z. N. Chen, B. Li, G. Qian and B. Chen, J. Am. Chem. Soc., 2020, 142, 633–640 CrossRef CAS.
- H. Wang, H. Wu, J. Kan, G. Chang, Z. Yao, B. Li, W. Zhou, S. Xiang, J. Cong-Gui Zhao and B. Chen, J. Mater. Chem. A, 2017, 5, 8292–8296 RSC.
- T. Banerjee, F. Haase, S. Trenker, B. P. Biswal, G. Savasci, V. Duppel, I. Moudrakovski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2019, 10, 2689 CrossRef PubMed.
- Y. Chen, D. Yang, B. Shi, W. Dai, H. Ren, K. An, Z. Zhou, Z. Zhao, W. Wang and Z. Jiang, J. Mater. Chem. A, 2020, 8, 7724–7732 RSC.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, Nat. Commun., 2019, 10, 3074 CrossRef PubMed.
- I. Hisaki, J. Inclusion Phenom. Macrocyclic Chem., 2020, 96, 215–231 CrossRef CAS.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- Y.-P. Zhang, H.-L. Tang, H. Dong, M.-Y. Gao, C.-C. Li, X.-J. Sun, J.-Z. Wei, Y. Qu, Z.-J. Li and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 4334–4340 RSC.
- A. F. M. El-Mahdy, A. M. Elewa, S. W. Huang, H. H. Chou and S. W. Kuo, Adv. Opt. Mater., 2020, 8, 2000641 CrossRef CAS.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- F. M. Zhang, J. L. Sheng, Z. D. Yang, X. J. Sun, H. L. Tang, M. Lu, H. Dong, F. C. Shen, J. Liu and Y. Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 12106–12110 CrossRef CAS PubMed.
- L. J. Karas, C. H. Wu, R. Das and J. I. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, 1477 Search PubMed.
- Y. Yang, L. Li, R. B. Lin, Y. Ye, Z. Yao, L. Yang, F. Xiang, S. Chen, Z. Zhang, S. Xiang and B. Chen, Nat. Chem., 2021, 13, 933–939 CrossRef CAS.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- Q. Yin, P. Zhao, R. J. Sa, G. C. Chen, J. Lu, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS PubMed.
- M. Zhang, M. Lu, Z. L. Lang, J. Liu, M. Liu, J. N. Chang, L. Y. Li, L. J. Shang, M. Wang, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 6500–6506 CrossRef CAS.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS.
- Y. Zhi, Z. Wang, H. L. Zhang and Q. Zhang, Small, 2020, 16, e2001070 CrossRef PubMed.
- W. Huang, W. Luo and Y. Li, Mater. Today, 2020, 40, 160–172 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- E. L. Spitler, J. W. Colson, F. J. Uribe-Romo, A. R. Woll, M. R. Giovino, A. Saldivar and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 2623–2627 CrossRef CAS PubMed.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- E. Q. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. H. Chen and D. L. Jiang, Science, 2017, 357, 673–676 CrossRef CAS.
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS.
- M. Lu, M. Zhang, C. G. Liu, J. Liu, L. J. Shang, M. Wang, J. N. Chang, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 4864–4871 CrossRef CAS PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS.
- Y. Yue, P. Cai, K. Xu, H. Li, H. Chen, H. C. Zhou and N. Huang, J. Am. Chem. Soc., 2021, 143, 18052–18060 CrossRef CAS PubMed.
- Y. F. Han, Y. X. Yuan and H. B. Wang, Molecules, 2017, 22, 266 CrossRef.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS.
- T. H. Chen, I. Popov, W. Kaveevivitchai, Y. C. Chuang, Y. S. Chen, O. Daugulis, A. J. Jacobson and O. S. Miljanic, Nat. Commun., 2014, 5, 5131 CrossRef CAS.
- P. Li, Y. He, H. D. Arman, R. Krishna, H. Wang, L. Weng and B. Chen, Chem. Commun., 2014, 50, 13081–13084 RSC.
- J. Liu, L. Li, W. Niu, N. Wang, D. Zhao, S. Zeng and S. Chen, ChemElectroChem, 2016, 3, 1116–1123 CrossRef CAS.
- W. Yang, B. Li, H. Wang, O. Alduhaish, K. Alfooty, M. A. Zayed, P. Li, H. D. Arman and B. Chen, Cryst. Growth Des., 2015, 15, 2000–2004 CrossRef CAS.
- B. Yu, L. Li, S. Liu, H. Wang, H. Liu, C. Lin, C. Liu, H. Wu, W. Zhou, X. Li, T. Wang, B. Chen and J. Jiang, Angew. Chem., Int. Ed., 2021, 60, 8983–8989 CrossRef CAS.
- I. Hisaki, S. Nakagawa, N. Tohnai and M. Miyata, Angew. Chem., Int. Ed., 2015, 54, 3008–3012 CrossRef CAS PubMed.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X. Y. Gao, L. Gao, L. Qiao, X. B. Li, L. Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- Q. Yang, M. Luo, K. Liu, H. Cao and H. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS.
- L. Wang, L. Zhang, B. Lin, Y. Zheng, J. Chen, Y. Zheng, B. Gao, J. Long and Y. Chen, Small, 2021, 17, e2101017 CrossRef PubMed.
- S. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505–4511 RSC.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- H.-Y. Yu, J.-S. Wang, F.-Y. Xie, Q. Yang, Y. Chen, L. Zhao, Y. Li and W.-J. Ruan, Chem. Eng. J., 2022, 445, 136713 CrossRef CAS.
- K. O. Kirlikovali, S. Goswami, M. R. Mian, M. D. Krzyaniak, M. R. Wasielewski, J. T. Hupp, P. Li and O. K. Farha, ACS Mater. Lett., 2021, 4, 128–135 CrossRef.
- I. Hisaki, N. Q. Emilya Affendy and N. Tohnai, CrystEngComm, 2017, 19, 4892–4898 RSC.
- C. Du, X. Wang, W. Chen, S. Feng, J. Wen and Y. A. Wu, Mater. Today Adv., 2020, 6, 100071 CrossRef.
- J. Yuan, S. Chen, Y. Zhang, R. Li, J. Zhang and T. Peng, Adv. Mater., 2022, 34, e2203139 CrossRef PubMed.
- L. Zou, R. Sa, H. Zhong, H. Lv, X. Wang and R. Wang, ACS Catal., 2022, 12, 3550–3557 CrossRef CAS.
- M. D. Zhang, D. H. Si, J. D. Yi, S. S. Zhao, Y. B. Huang and R. Cao, Small, 2020, 16, e2005254 CrossRef PubMed.
- Y. Yue, H. Li, H. Chen and N. Huang, J. Am. Chem. Soc., 2022, 144, 2873–2878 CrossRef CAS PubMed.
- X. Wang, M. Bahri, Z. Fu, M. A. Little, L. Liu, H. Niu, N. D. Browning, S. Y. Chong, L. Chen, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2021, 143, 15011–15016 CrossRef CAS PubMed.
- M. Wang, M. Wang, H. H. Lin, M. Ballabio, H. Zhong, M. Bonn, S. Zhou, T. Heine, E. Canovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2020, 142, 21622–21627 CrossRef CAS PubMed.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS.
- M. Zhang, J. N. Chang, Y. Chen, M. Lu, T. Y. Yu, C. Jiang, S. L. Li, Y. P. Cai and Y. Q. Lan, Adv. Mater., 2021, 33, e2105002 CrossRef PubMed.
- Y. N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H. L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- Q. Wu, M. J. Mao, Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Small, 2021, 17, e2004933 CrossRef PubMed.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton, L. Liu, X. Wu, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS.
- Y. Xiang, W. Dong, P. Wang, S. Wang, X. Ding, F. Ichihara, Z. Wang, Y. Wada, S. Jin, Y. Weng, H. Chen and J. Ye, Appl. Catal., B, 2020, 274, 119096 CrossRef CAS.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- Y. Meng, Y. Luo, J. L. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 CrossRef CAS PubMed.
- F. Jin, E. Lin, T. Wang, D. Yan, Y. Yang, Y. Chen, P. Cheng and Z. Zhang, Chem, 2022, 8, 1–17 Search PubMed.
- Z. Z. Gao, Z. K. Wang, L. Wei, G. Yin, J. Tian, C. Z. Liu, H. Wang, D. W. Zhang, Y. B. Zhang, X. Li, Y. Liu and Z. T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 1404–1411 CrossRef CAS PubMed.
- X.-T. He, Y.-H. Luo, Z.-Y. Zheng, C. Wang, J.-Y. Wang, D.-L. Hong, L.-H. Zhai, L.-H. Guo and B.-W. Sun, ACS Appl. Nano Mater., 2019, 2, 7719–7727 CrossRef CAS.
- Q. Yin, J. Lü, H.-F. Li, T.-F. Liu and R. Cao, Cryst. Growth Des., 2019, 19, 4157–4161 CrossRef CAS.
- Q. Yin, E. V. Alexandrov, D. H. Si, Q. Q. Huang, Z. B. Fang, Y. Zhang, A. A. Zhang, W. K. Qin, Y. L. Li, T. F. Liu and D. M. Proserpio, Angew. Chem., Int. Ed., 2022, 61, e202115854 CrossRef CAS PubMed.
- X. Zhao, Q. Yin, X. Mao, C. Cheng, L. Zhang, L. Wang, T. F. Liu, Y. Li and Y. Li, Nat. Commun., 2022, 13, 2721 CrossRef CAS PubMed.
- B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- P. Tholen, C. A. Peeples, R. Schaper, C. Bayraktar, T. S. Erkal, M. M. Ayhan, B. Cosut, J. Beckmann, A. O. Yazaydin, M. Wark, G. Hanna, Y. Zorlu and G. Yucesan, Nat. Commun., 2020, 11, 3180 CrossRef CAS PubMed.
- W. Liang, F. Carraro, M. B. Solomon, S. G. Bell, H. Amenitsch, C. J. Sumby, N. G. White, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 14298–14305 CrossRef CAS PubMed.
- W. K. Qin, D. H. Si, Q. Yin, X. Y. Gao, Q. Q. Huang, Y. N. Feng, L. Xie, S. Zhang, X. S. Huang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202202089 CAS.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- A. G. A. Mohamed, E. Zhou, Z. Zeng, J. Xie, D. Gao and Y. Wang, Adv. Sci., 2022, 9, 2104138 CrossRef CAS PubMed.
- C. Mo, M. Yang, F. Sun, J. Jian, L. Zhong, Z. Fang, J. Feng and D. Yu, Adv. Sci., 2020, 7, 1902988 CrossRef CAS PubMed.
- J. Yang, A. Acharjya, M. Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Gruneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomacker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS.
- J.-L. Sheng, H. Dong, X.-B. Meng, H.-L. Tang, Y.-H. Yao, D.-Q. Liu, L.-L. Bai, F.-M. Zhang, J.-Z. Wei and X.-J. Sun, ChemCatChem, 2019, 11, 2313–2319 CrossRef CAS.
- A. M. Elewa, A. F. M. EL-Mahdy, A. E. Hassan, Z. Wen, J. Jayakumar, T.-L. Lee, L.-Y. Ting, I. M. A. Mekhemer, T.-F. Huang, M. H. Elsayed, C.-L. Chang, W.-C. Lin and H.-H. Chou, J. Mater. Chem. A, 2022, 10, 12378–12390 RSC.
- K. Ma, P. Li, J. H. Xin, Y. Chen, Z. Chen, S. Goswami, X. Liu, S. Kato, H. Chen, X. Zhang, J. Bai, M. C. Wasson, R. R. Maldonado, R. Q. Snurr and O. K. Farha, Cell Rep. Phys. Sci., 2020, 1, 10024 Search PubMed.
- N. Zhang, Q. Yin, S. Guo, K.-K. Chen, T.-F. Liu, P. Wang, Z.-M. Zhang and T.-B. Lu, Appl. Catal., B, 2021, 296, 120337 CrossRef CAS.
- A. A. Zhang, Y. L. Li, Z. B. Fang, L. Xie, R. Cao, Y. Liu and T. F. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 21050–21058 CrossRef CAS PubMed.
- T. Li, B.-T. Liu, Z.-B. Fang, Q. Yin, R. Wang and T.-F. Liu, J. Mater. Chem. A, 2021, 9, 4687–4691 RSC.
- J. F. Feng, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 22392–22396 CrossRef CAS PubMed.
- H. Dong, X.-B. Meng, X. Zhang, H.-L. Tang, J.-W. Liu, J.-H. Wang, J.-Z. Wei, F.-M. Zhang, L.-L. Bai and X.-J. Sun, Chem. Eng. J., 2020, 379, 122342 CrossRef CAS.
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y. Q. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204326 CAS.
- X. Wang, X. Ding, T. Wang, K. Wang, Y. Jin, Y. Han, P. Zhang, N. Li, H. Wang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 41122–41130 CrossRef CAS PubMed.
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed.
- M. Lu, M. Zhang, J. Liu, T. Y. Yu, J. N. Chang, L. J. Shang, S. L. Li and Y. Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- T. Kong, Y. Jiang and Y. Xiong, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- A. A. Zhang, D. Si, H. Huang, L. Xie, Z. B. Fang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202203955 CAS.
- B. Yu, T. Meng, X. Ding, X. Liu, H. Wang, B. Chen, T. Zheng, W. Li, Q. Zeng and J. Jiang, Angew. Chem., Int. Ed., 2022, e202211482 CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
| This journal is © The Royal Society of Chemistry 2023 |