
Nonionic oligo(ethylene glycol)-substituted viologen negolytes for aqueous organic redox flow batteries†
Yanxin
Yao
a,
Wanzhen
Ma
b,
Jiafeng
Lei
a,
Zengyue
Wang
a,
Yi-Chun
Lu
 *a and
Lei
Liu
*b
*a and
Lei
Liu
*b
aElectrochemical Energy and Interfaces Laboratory, Department of Mechanical and Automation Engineering, The Chinese University of Hong Kong, Shatin, N.T. 999077, Hong Kong SAR, China. E-mail: yichunlu@mae.cuhk.edu.hk
bCollege of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China. E-mail: liulei@ahnu.edu.cn
First published on 30th March 2023
Abstract
Viologen derivatives are attractive active materials for negolytes in aqueous organic redox flow batteries (AORFBs) owing to their fast kinetics and suitable reduction potentials. However, viologens with alkyl substitutions only (e.g., methyl viologen and ethyl viologen) suffer from poor stability and the insoluble nature of the second-electron product limits their energy density. A key to enabling durable and high-capacity viologen-based AORFBs lies in the molecular engineering of viologens. Here we demonstrated a series of oligo(ethylene glycol) (OEG) substituted viologens with improved cycling stability via the steric-hindrance effect and enhanced water solubility of the second-electron product. Electrochemical and symmetric cell investigations validated the high reversibility of tri(ethylene glycol) groups modified viologen (Vi-OEG3) and its improved solubility of the second-electron products in neutral aqueous solutions. When paired with a ferrocene-based posolyte, a full flow cell with Vi-OEG3 showed a much reduced capacity decay rate of 0.00253% per day or 0.000105% per cycle compared with ethyl viologen. This work expands the molecular design of viologens and shows an attractive viologen-based AORFB for low-cost and large-scale renewable energy storage systems.
10th anniversary statementJournal of Materials Chemistry A (JMCA) is an international platform for high quality research on materials chemistry for energy and sustainability applications. This year marks its 10th year anniversary of serving the large materials/chemistry community. Energy and sustainability are pressing global issues that need interdisciplinary research and innovations, which is in line with the mission of JMCA. JMCA has been dedicated to promoting and nurturing young scientists with Emerging Investigators Issues. The first JMCA paper from my research group was published in the themed collection of “Emerging Investigators 2017”. I am looking forward to reading new Emerging Investigators articles especially from my former PhD students/postdocs as independent principal investigators. JMCA connects all of us in materials chemistry and it's my honour to be part of this community. |
Introduction
Low-cost and reliable energy storage systems are essential to the transition from fossil fuels to clean and renewable energy resources (e.g., wind and solar) globally.1–3 Aqueous redox flow batteries (ARFBs) have drawn tremendous attention as large-scale energy storage systems owing to their superior safety and advantage in decoupling power and energy (for all flow batteries).4 However, the widespread practical application of traditional inorganic ARFBs, such as all-vanadium RFBs and zinc-based RFBs, is challenged by issues including high materials cost and scalability, respectively.5–7Aqueous organic RFBs (AORFBs) provide opportunities for low-cost large-scale energy storage systems by employing water-soluble organic compounds as active materials, which are composed of earth-abundant elements (e.g., C, H, O, N, and S) with the potential to be prepared sustainably and inexpensively on large scales.8,9 In addition, thanks to the structural tunability of organic redox compounds, rational properties such as solubility, redox potential, and chemical/electrochemical stability could be manipulated by the function-oriented molecular engineering.10
Viologen is one of the most attractive organic active materials for negolytes due to its suitable redox potential (e.g., methyl viologen (MV), −0.45 V vs. SHE) and fast kinetics.11 However, it has been reported that the colored radical cation Vi˙+ can undergo a bimolecular process forming redox-inactive viologen dimer and can be easily oxidized by oxygen.12,13 These degradation routes are irreversible, resulting in a capacity loss in full cell cycling. By replacing the alkyl substitutions with ionic ones (such as quaternary ammonium, phosphonate, and sulphonate) in the viologens, stable radical cation Vi˙+ can be achieved for the intended AORFB applications. Jin et al. demonstrated a phosphonate group-substituted viologen, 1,1′-bis(3-phosphonopropyl)-[4,4′-bipyridine]-1,1′diium (BPP-Vi), which enabled a near pH-neutral AORFB with a low capacity decay rate (0.016% per day or 0.00069% per cycle over ∼13 days) when paired with potassium ferri-/ferrocyanide as posolyte.14 Bis(3-trimethylammonio)propyl viologen tetrachloride (BTMAP-Vi) was also reported as one of the most stable viologen derivatives by introducing positively-charged quaternary ammonium groups to the viologen core.15,16 The enhanced charge repulsion between viologen molecules and the repulsion between viologen and anion exchange membrane (AEM) mitigate the radical dimerization and crossover of redox-active materials, thus improving the cycling stability. However, the introduction of the electron-withdrawing quaternary ammonium groups elevates the redox potential of BTMAP-Vi to −0.36 V vs. SHE, leading to lower cell voltage and energy density. Recently, we proposed a new “molecular spectator” approach to suppress the dimerization of two viologen radicals by exploiting α-cyclodextrin (α-CD) as the molecular spectator. A stable cycling with a volumetric capacity of 21.4 A h Lnegolyte−1 (80% capacity utilization) was achieved over 500 cycles (26 days).17
The second-electron utilization of viologens in aqueous batteries is limited by the insolubility of the charge-neutral MV0 in water. Rational molecular engineering of introducing functional groups and extending the π-conjugation framework are two effective directions to access the two-electron storage of viologens.18 Viologen derivatives with hydrophilic functional groups, such as ammonium and hydroxyl groups, were designed to increase the water solubility of the second-reduction product.16,19 Meanwhile, extending the π-conjugation framework by introducing external groups (e.g., thiazolo[5,4-d]thiazole (TTz), a phenylene group) between the two pyridinium rings was demonstrated to achieve two-electron storage of viologens.20,21
Poly(ethylene glycol)s (PEGs), commonly used as polyelectrolyte in batteries and solubilizing agents for pharmaceuticals, are inert within a large electrochemical potential window. A series of PEGylated polymers bearing a viologen unit were designed and synthesized with large molar mass and viscosity, resulting in low energy density and power of a corresponding aqueous batteries by decreasing the ion mobility and efficiency.22 Another PEGylated viologen with 12 oxyethylene units (PEG12-V) was designed as negolyte for non-aqueous RFBs.23 Besides common limitations of non-aqueous RFBs, e.g., low current density (∼2 mA cm−2) and flammability of non-aqueous solvents, the PEG12-V/Fc full cell in acetonitrile demonstrated a capacity decay rate of 21.6% per day over 185 hours. Recently, a PEGylated viologen with four positive charges (BTMAE-Vi) was synthesized and showed high water solubility of 3.4 M.24 However, the high-concentration full cell test was conducted at 0.5 M and cycled for less than 2 days. In summary, the cycling stability of ethylene glycol functioned viologens are still challenging and more efforts are required to improve the cycling performance at full cell level.
Here, we develop an alternative strategy to design water-miscible viologens as active materials in negolyte for AORFBs, applying nonionic oligo(ethylene glycol) (OEG) substitutions in viologens. The resulting viologens bearing OEG side chains enable superior cycling stability of the first-electron redox reaction and accessibility of the second-electron energy storage, benefiting from the bulky and hydrophilic substitutions. The electrochemical properties and cycling stability of the one-electron storage were studied and compared with the control viologen (ethyl viologen, EV) possessing short and hydrophobic substitutions. Coupled with a ferrocyanide-based posolyte, a flow cell operated with the first-electron redox reaction of Vi-OEG3, a viologen with tri(ethylene glycol) substitutions, demonstrated stable cycling with a capacity decay rate of 0.00253% per day or 0.000105% per cycle (6 days). In addition, the accessibility of the second-electron reduction of Vi-OEG3 was revealed by cyclic voltammetry (CV) and rotating ring disk electrode (RRDE) analysis, resulting in higher cell voltage and higher available volumetric capacity in full cell cycling compared with EV. This work provides a facile and effective strategy to stabilize the viologen radicals and realize the two-electron storage of viologens for AORFBs applications.
Results and discussion
For practical application in AORFBs, viologens are required to be highly stable under cycling in high concentrations. Thus, we designed a series of OEG-based viologens to implement steric-hindrance effect on the charged viologens (forming radical cations), aiming to improve the cycling stability with the help of bulky hydrated OEG side chains. Meanwhile, OEG chain, well-known for its hydrophilicity, is commonly used to increase molecules/polymers' solubility in water or polar organic solvents.25 The introduction of hydrophilic OEG chains enabled both high solubility and two-electron storage of the resulting OEG-based viologens.Fig. 1a showed the synthetic routes of Vi-OEGx, which involve a simple one-step reaction between 4,4′-bipyridine and bromide-functionalized OEG. The length of OEG side chains can be easily tuned by using bromide-functionalized OEG with different lengths to afford Vi-OEG2 (1,1′-bis(2-(2-methoxyethoxy)ethyl)-[4,4′-bipyridine]-1,1′-diium bromide), Vi-OEG3 (1,1′-bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-[4,4′-bipyridine]-1,1′-diium bromide), and Vi-OEG4 (1,1′-di(2,5,8,11-tetraoxatridecan-13-yl)-[4,4′-bipyridine]-1,1′-diium bromide). The structure and purity of Vi-OEGx were characterized by NMR spectroscopy. As shown in Fig. 1c and d, the aromatic protons ascribed to the 4,4′-bipyridinium unit appear at ∼8.5 and 9.1 ppm, and the characteristic protons from OEG substitutions can be found at ∼4.9, 4.1, 3.4–3.7 and 3.3 ppm. These results combined with 13C NMR and 1H NMR spectra with exact peak positions and integrals suggest the successful synthesis of targeted viologens with OEG side chains (Fig. S1 and S2, ESI†). After purification, Vi-OEG2 and Vi-OEG3 are yellow and pale-yellow powders; however, with the increase of OEG chains' length in viologens, Vi-OEG4 becomes very sticky (Fig. 1b and S3, ESI†). Therefore, we mainly focus on Vi-OEG2 and Vi-OEG3 in the following studies.
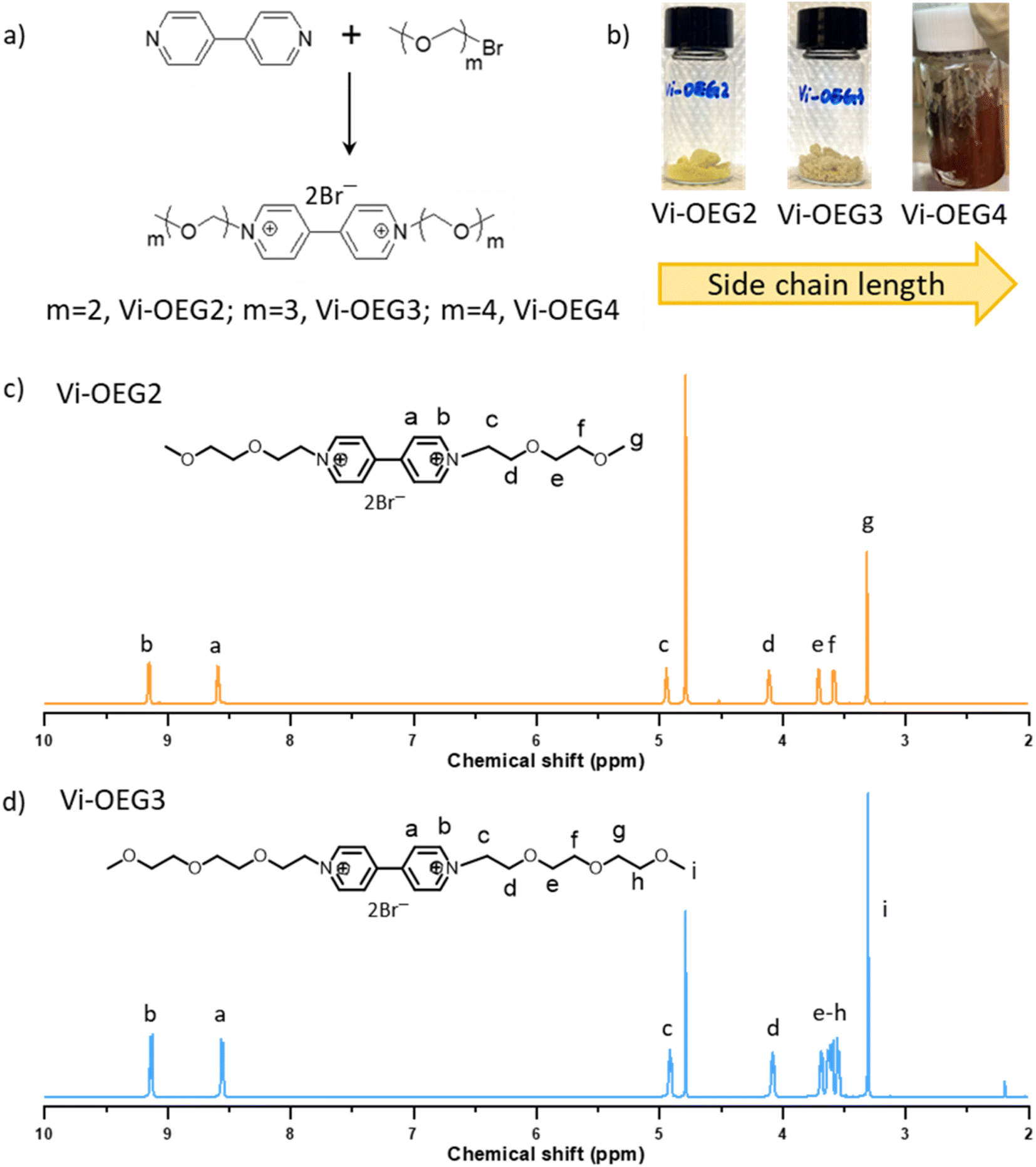 | ||
| Fig. 1 (a) Synthesis of OEG-based viologens through a simple one-step method. (b) Digital photos of OEG-based viologens. 1H NMR of (c) Vi-OEG2 and (d) Vi-OEG3. | ||
As expected, OEG-based viologens exhibited high solubilities in water (1.6 M for Vi-OEG2 and 1.4 M for Vi-OEG3, Table 1). The introduction of OEG side chains slightly increases the viscosity and reduces the conductivity of the electrolytes with and without 1 M NaBr (Fig. S4, ESI†). Reversible CV peaks with a 54 mV-peak separation were obtained for Vi-OEG2 and Vi-OEG3. The equilibrium potential of Vi-OEG3 is ∼−0.55 V vs. SCE, which is 10 mV more positive than that of Vi-OEG2 (Fig. 2a). Due to the electron-donating nature of OEG substitutions, OEG-based viologens have more negative potentials than the well-studied BTMAP-Vi, which enable a higher full cell voltage. In addition, as shown in Fig. S5 (ESI†), the CV curves of OEG-based viologens remained stable after 100 scan cycles, indicating their high redox stability and reversibility. The linear relationship between the peak currents and the square root of the scanning rates reveals that the reversible oxidation and reduction are limited by the diffusion process. The above electrochemical measurements demonstrated high reversibility and cycling stability of Vi-OEGx with relatively negative potentials.
| Sample | Solubility (M) | D (cm2 s−1) | k 0 (cm s−1) |
|---|---|---|---|
| a All materials were synthesized in bromide form. Solubility was tested in DI water. b The solubility of EV was adapted from ref. 17. c The solubility of BTMAP-Vi in bromide form was adapted from ref. 16. | |||
| EV | 2.81b | 5.67 × 10−6 | 1.42 × 10−2 |
| BTMAP-Vi | 1.6c | 3.94 × 10−6 | 1.06 × 10−2 |
| Vi-OEG2 | 1.6 | 4.24 × 10−6 | 1.04 × 10−2 |
| Vi-OEG3 | 1.4 | 3.97 × 10−6 | 1.04 × 10−2 |
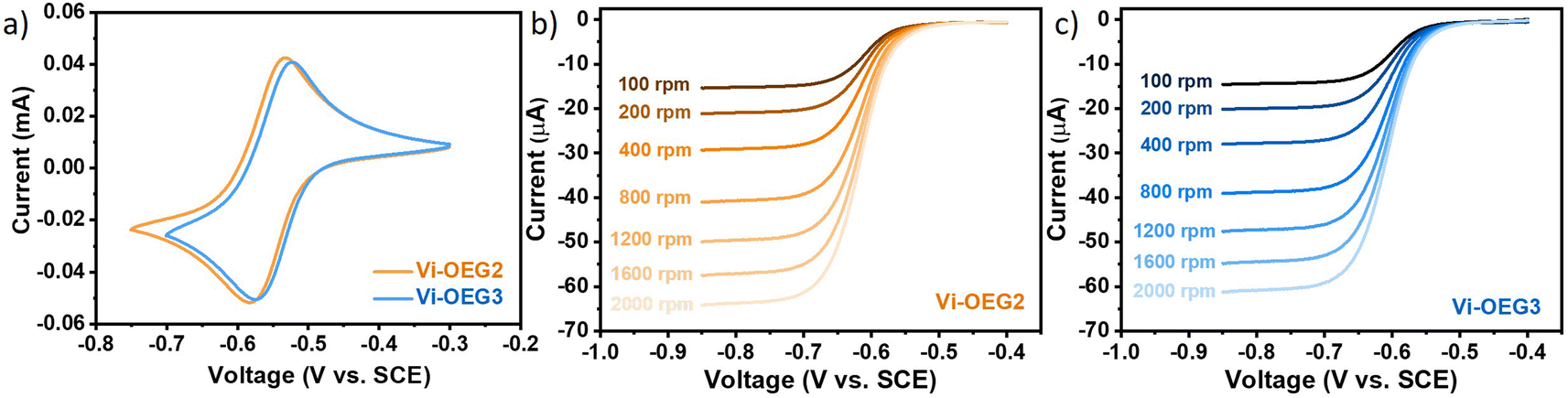 | ||
| Fig. 2 (a) Cyclic voltammograms of Vi-OEG2 and Vi-OEG3. Condition: 5 mM viologens in 0.5 M NaBr aqueous electrolyte at a scan rate of 50 mV s−1. Linear sweep voltammograms of (b) Vi-OEG2 and (c) Vi-OEG3. Condition: 1 mM viologens in 0.5 M NaCl aqueous electrolyte at a scan rate of 5 mV s−1. | ||
We also performed CV measurements to access the reversibility of the second-electron reaction of Vi-OEGx. As shown in Fig. S6 (ESI†), since neutral EV0 is insoluble in water, the utilization of EV's second redox couple is inaccessible, which is further confirmed by the sharp oxidation peak at −0.92 V vs. SCE. And the oxidation peak at a more positive potential of −0.85 V vs. SCE could be attributed to the chemical disproportionation of EV dimer, which is typical for alkyl-substituted viologens.26 Replacing alkyl substitutions with water-miscible tri(ethylene glycol) side chains could significantly improve the reversibility of [Vi-OEG3]˙+/0 redox couple owing to the high solubility of the charge-neutral [Vi-OEG3]0. There are two reversible reduction and oxidation processes for Vi-OEG3 with two separated redox potentials at about −0.55 V and −0.95 V vs. SCE, which are stable over 100 cycles of scanning, indicating the high redox stability and reversibility of the two-electron redox reactions of Vi-OEG3 (Fig. S7, ESI†). However, the reversibility of the second reduction and oxidation process of Vi-OEGx depends on the length of OEG chains. The charge-neutral [Vi-OEG2]0 could in part deposit on the electrode as evidenced by the sharp oxidation peak (Fig. S6, ESI†) associated with the second-electron redox reaction, resulting in the unavailable two-electron storage of Vi-OEG2 in AORFBs.
To further understand the reduction kinetics of OEG-based viologens, we performed linear sweep voltammetry (LSV) with a glassy carbon rotating-disk electrode, sweeping negatively at a scan rate of 5 mV s−1 with rotation rates increasing from 100 to 2000 rpm. LSV plots and the derived Levich plots of Vi-OEG2 and Vi-OEG3 are shown in Fig. 2b, c and S8 (ESI†). The diffusion coefficients were calculated by the Levich equation according to the corresponding slopes from the linear relationships between limiting currents and the square root of rotation speeds. In addition, by using Nicholson's method, we estimated the electron transfer rate constant of Vi-OEG2 and Vi-OEG3. The calculated diffusion coefficients and rate constants of the first-electron transfer reaction are summarized in Table 1. Note that both Vi-OEG2 and Vi-OEG3 exhibit fast diffusion and possess large rate constants comparable with other organic compounds used in AORFB applications (e.g., EV and BTMAP-Vi, Fig. S9 and S10, ESI†).16,27 In addition, the RRDE experiment in Fig. S11 and S12 (ESI†) revealed that the second-electron redox reaction of Vi-OEG3 exhibits fast kinetics with high reversibility, which is in strong contrast to EV with passivation features.28
We firstly examine the stability of the first-electron redox reaction of OEG-based viologens where the oxidated radical cations tend to dimerize, leading to the unrecoverable loss of capacity. The unbalanced symmetric cell configuration is applied to probe the intrinsic stability of the material of interest.5,29 For better comparison, the symmetric static cell assembly process and testing parameters were the same as our previous report, in which the control cell with EV showed a decay rate of 0.31% per day over 20 days of cycling.17 As shown in Fig. 3, static symmetric cell employing Vi-OEG3 demonstrated a reduced capacity decay rate (0.047% per day) compared to Vi-OEG2 (0.173% per day) for 19 days, indicating that the increase in the length of OEG chain could enhance the cycling stability. These results supported that the OEG side chains in Vi-OEGx molecules effectively suppress the dimerization of viologen radical cations and improved the cycling stability.
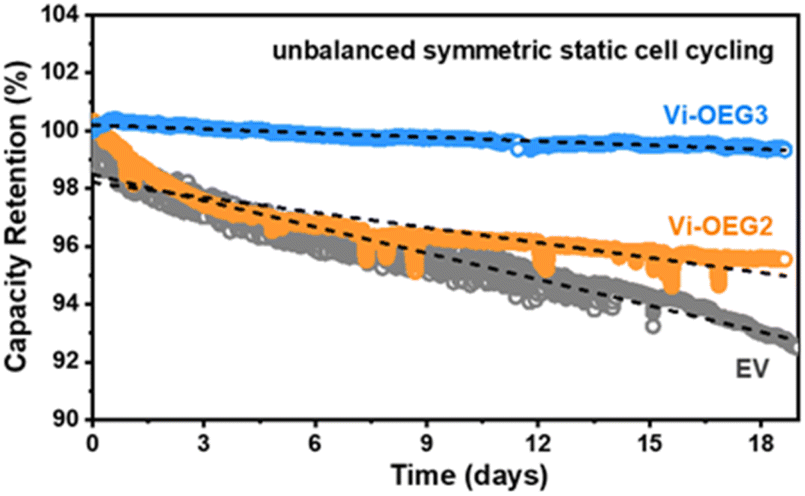 | ||
| Fig. 3 Capacity retention of unbalanced symmetric static cell employing EV, Vi-OEG2 and Vi-OEG3. The decay rates were calculated based on the slope of the fitted lines (black dash). The EV data was adapted from ref. 17. | ||
We constructed a full flow cell coupled with N-(ferrocenylmethyl)-N,N-dimethyl-N-ethylammonium bromide (FcNEBr), a commonly used stable and low-crossover (with permeability of 3.85 × 10−9 cm2 s−1) ferrocene derivative posolyte (Fig. 4a). As shown in Fig. S13 (ESI†), the equilibrium potential of FcNEBr is ∼0.4 V vs. SCE, demonstrating a 0.9 V cell voltage of the 0.5 M Vi-OEG3/FcNEBr flow cell. The rate performance was tested within a voltage range of 0.4–1.15 V at a constant flow rate of 50 mL min−1 by operating at increasing the current densities from 20 mA cm−2 to 50 mA cm−2 and then followed by 30 mA cm−2. Compared with 20 mA cm−2, the capacity retention was 95%, 83%, and 58% at 30 mA cm−2, 40 mA cm−2, and 50 mA cm−2, respectively. And energy efficiency (EE) decreased from 80.35% at 20 mA cm−2 to 56.64% at 50 mA cm−2. The coulombic efficiency (CE) remained above 99.6% for all rates. The cycling performance of this flow cell was performed at 30 mA cm−2 within a specific voltage range (0.4–1.15 V) for 138 cycles (∼6 days), delivering a stable volumetric capacity of 12 A h Lnegolyte−1 (Fig. 4b–e). Post-cell characterization of the electrolytes was conducted by CV (Fig. S14, ESI†) and NMR (Fig. S15, ESI†). No noticeable structural changes of the electrolytes were observed before and after cycling and no crossover was observed. We summarized the testing parameters and cycling performance of some reported viologen-based AORFBs in Table S1 (ESI†). This superior cycling stability further confirmed the dimerization suppression effectiveness of OEG chain substitution. Moreover, thanks to the bulky OEG chains, the permeabilities through a commercial-available AMV anion exchange membrane of Vi-OEGx were significantly reduced compared with that of EV. Specifically, when tested in a flow cell configuration, the permeability of Vi-OEG3 was calculated to be 3.89 × 10−9 cm2 s−1, which is ten times less than that of EV (1.72 × 10−8 cm2 s−1).
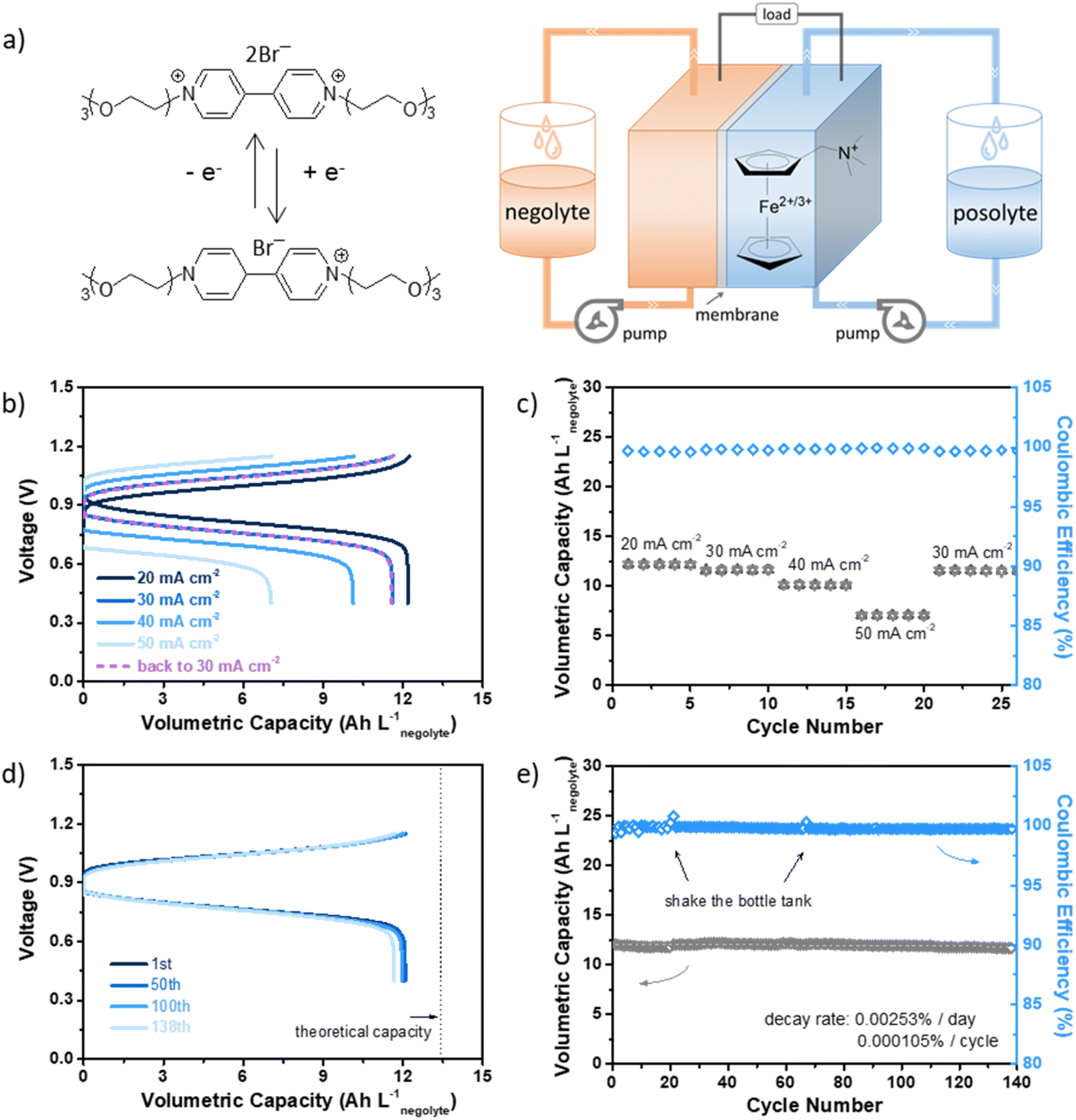 | ||
| Fig. 4 (a) The schematic representation of the Vi-OEG3/FcNEBr redox flow cell. (b) Rate performance, and (c) volumetric capacity and coulombic efficiency as a function of cycle number from 20 to 50 mA cm−2 with increments of 10 mA cm−2 and back to 30 mA cm−2 at the end. (d) The galvanostatic voltage profile of the specific cycle (1st, 50th, 100th, and 138th) and (e) cycling retention of volumetric capacity and coulombic efficiency at 30 mA cm−2. Conditions: the electrolytes comprise 5 mL 0.5 M Vi-OEG3 in 1 M NaBr and 10 mL 0.4 M FcNEBr in 1 M NaBr using AMV as the membrane with a flow rate of 50 mL min−1. The flow cell was cycling between 0.4 V and 1.15 V. | ||
The capability of the two-electron storage of Vi-OEG3 were also demonstrated in full cells. As shown in Fig. S16,† the Vi-OEG3/FcNEBr cell shows obvious higher cell voltage and volumetric capacity compared to the EV/FcNEBr cell, further confirming the accessibility of the second-electron reaction of Vi-OEG3. However, the superiority in cycling stability is not significant. The long-term cycling stability (with a low capacity decay rate of ≤ 0.01% per day, nearly 80% capacity retention after five years)30 of the two-electron OEG-based viologens is yet to be achieved. We hypothesize that the low redox potential of the second-electron reduction favors side reactions (e.g., HER),31 leading to irreversible capacity loss in full cell cycling. Further works focusing on electrolyte additives and carbonaceous electrodes with low HER activity is on-going to verify the hypothesis.
Conclusions
In summary, we synthesized a series of viologens bearing bulky and hydrophilic OEG chains for AORFBs application. This design strategy enables the dimerization suppression of viologen radical cations via steric hindrance and significantly improved the cycling stability of viologens. Symmetric static cells with a capacity decay rate of 0.173% per day and 0.047% per day were observed for Vi-OEG2 and Vi-OEG3 within 19 days, much lower than EV. Benefiting from the hydrophilic nature of OEG groups, Vi-OEG3 are shown to store two electrons reversibly with lower redox potential. This work expands the molecular design of viologens and shows an attractive viologen-based AORFB for low-cost and large-scale renewable energy storage systems.Experimental section
Chemicals and materials preparation
All chemicals were purchased from TCI Chemical, Energy Chemical, or RCI Labscan and used as received without further purification. The commercial anion exchange membrane (AMV, Selemion, Japan) was obtained from Selemion, Japan. Activated graphite felt (GFD 4.6 EA) was used as received from SGL Carbon GmbH, Germany. Ethyl viologen (EV) in bromide form and N-(ferrocenylmethyl)-N,N-dimethyl-N-ethylammonium bromide (FcNEBr) were prepared as our previous work.17 1,1′-Bis[3-(trimethylamonium)propyl]-4,4′-bipyridinium tetrabromide (BTMAP-Vi) was prepared as reported.16Measurements and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 c mol−1, electrode area A = 0.1257 cm2, active material concentration C0 = 1.0 × 10−3 M, D is the diffusion coefficient, and kinematic viscosity ν = 0.009 cm2 s−1 for 0.5 M NaCl solution. A plot of overpotentials versus log10(ik) was constructed where ik is the kinetic current for the reduction reactions, and the intercept of the fitted line of the current collected at each overpotential over the square root of the rotation rate gave the value of ik. The X-intercept of the fitted Tafel plot affords the log of the exchange current i0, which is equal to FAC0k0, and gives the electron transfer rate constant (k0).
485 c mol−1, electrode area A = 0.1257 cm2, active material concentration C0 = 1.0 × 10−3 M, D is the diffusion coefficient, and kinematic viscosity ν = 0.009 cm2 s−1 for 0.5 M NaCl solution. A plot of overpotentials versus log10(ik) was constructed where ik is the kinetic current for the reduction reactions, and the intercept of the fitted line of the current collected at each overpotential over the square root of the rotation rate gave the value of ik. The X-intercept of the fitted Tafel plot affords the log of the exchange current i0, which is equal to FAC0k0, and gives the electron transfer rate constant (k0).
The working electrode used in RRDE tests was a GC ring/GC disk electrode (ALS CO., Ltd, Japan), consisting of a PEEK embedded 4 mm diameter GC disk surrounded by a GC ring with internal and external diameters of 5 mm and 7 mm. The three-electrode systems used in both LSV and RRDE were bubbled with argon for 30 minutes before each CV experiment, followed by gas blanket during the entire test to prevent contamination from ambient air.
Symmetric and full cell tests
For static cells, 400 μL negolyte and 500 μL posolyte were used to ensure that the capacity was limited by the negative side. For flow cells, apart from the basic components, i.e., activated SGL graphite felt and Selemion AMV anion exchange membrane, a peristaltic pump was also required to circulate the electrolytes at a set-up flow rate (e.g., 50 mL min−1 in this work). When operating with the first-electron redox reaction, the full cell cycling was demonstrated in the voltage range of 0.4–1.1 V for static mode and 0.4–1.15 V for flow mode. When accessing the two-electron energy storage, a higher upper cut-off voltage was set to be 1.8 V. The charge/discharge cycling was performed on a battery testing system, either LAND Battery Testing System (CT2001A) or an Arbin Instrument, at room temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Research Grants Council (RGC) of the Hong Kong Administrative Region, China, under CUHK 14308622.References
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- N. D. Popovich, D. Rajagopal, E. Tasar and A. Phadke, Nat. Energy, 2021, 6, 1017–1025 CrossRef.
- J. Huang, S. T. Boles and J.-M. Tarascon, Nat. Sustain., 2022, 5, 194–204 CrossRef.
- R. Feng, X. Zhang, V. Murugesan, A. Hollas, Y. Chen, Y. Shao, E. Walter, P. N. Wellala Nadeesha, L. Yan, M. Rosso Kevin and W. Wang, Science, 2021, 372, 836–840 CrossRef CAS PubMed.
- Y. Yao, J. Lei, Y. Shi, F. Ai and Y.-C. Lu, Nat. Energy, 2021, 6, 582–588 CrossRef.
- Z. Yuan, Y. Yin, C. Xie, H. Zhang, Y. Yao and X. Li, Adv. Mater., 2019, 31, 1902025 CrossRef CAS PubMed.
- L. Li, S. Kim, W. Wang, M. Vijayakumar, Z. Nie, B. Chen, J. Zhang, G. Xia, J. Hu, G. Graff, J. Liu and Z. Yang, Adv. Energy Mater., 2011, 1, 394–400 CrossRef CAS.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed.
- J. M. Cameron, C. Holc, A. J. Kibler, C. L. Peake, D. A. Walsh, G. N. Newton and L. R. Johnson, Chem. Soc. Rev., 2021, 50, 5863–5883 RSC.
- T. Liu, X. Wei, Z. Nie, V. Sprenkle and W. Wang, Adv. Energy Mater., 2016, 6, 1501449 CrossRef.
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef CAS.
- C. D. Clark, J. D. Debad, E. H. Yonemoto, T. E. Mallouk and A. J. Bard, J. Am. Chem. Soc., 1997, 119, 10525–10531 CrossRef CAS.
- S. Jin, E. M. Fell, L. Vina-Lopez, Y. Jing, P. W. Michalak, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2020, 10, 2000100 CrossRef CAS.
- E. S. Beh, D. De Porcellinis, R. L. Gracia, K. T. Xia, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2017, 2, 639–644 CrossRef CAS.
- C. DeBruler, B. Hu, J. Moss, X. Liu, J. Luo, Y. Sun and T. L. Liu, Chem, 2017, 3, 961–978 CAS.
- L. Liu, Y. Yao, Z. Wang and Y.-C. Lu, Nano Energy, 2021, 84, 105897 CrossRef CAS.
- Q. Chen, Y. Lv, Z. Yuan, X. Li, G. Yu, Z. Yang and T. Xu, Adv. Funct. Mater., 2022, 32, 2108777 CrossRef CAS.
- W. Liu, Y. Liu, H. Zhang, C. Xie, L. Shi, Y.-G. Zhou and X. Li, Chem. Commun., 2019, 55, 4801–4804 RSC.
- S. Hu, T. Li, M. Huang, J. Huang, W. Li, L. Wang, Z. Chen, Z. Fu, X. Li and Z. Liang, Adv. Mater., 2021, 33, 2005839 CrossRef CAS PubMed.
- J. Luo, B. Hu, C. Debruler and T. L. Liu, Angew. Chem., Int. Ed., 2018, 57, 231–235 CrossRef CAS PubMed.
- T. Janoschka, S. Morgenstern, H. Hiller, C. Friebe, K. Wolkersdörfer, B. Häupler, M. D. Hager and U. S. Schubert, Polym. Chem., 2015, 6, 7801–7811 RSC.
- J. Chai, A. Lashgari, Z. Cao, C. K. Williams, X. Wang, J. Dong and J. J. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 15262–15270 CrossRef CAS PubMed.
- K. Peng, P. Sun, Z. Yang and T. Xu, Batteries Supercaps, 2023, 6, e202200426 CAS.
- B. Meng, H. Song, X. Chen, Z. Xie, J. Liu and L. Wang, Macromolecules, 2015, 48, 4357–4363 CrossRef CAS.
- E. E. Engelman and D. H. Evans, J. Electroanal. Chem., 1993, 349, 141–158 CrossRef CAS.
- C. DeBruler, B. Hu, J. Moss, J. Luo and T. L. Liu, ACS Energy Lett., 2018, 3, 663–668 CrossRef CAS.
- W. Wang, N.-C. Lai, Z. Liang, Y. Wang and Y.-C. Lu, Angew. Chem., Int. Ed., 2018, 57, 5042–5046 CrossRef CAS PubMed.
- M.-A. Goulet and M. J. Aziz, J. Electrochem. Soc., 2018, 165, A1466–A1477 CrossRef CAS.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
- J. Luo, B. Hu, M. Hu, Y. Zhao and T. L. Liu, ACS Energy Lett., 2019, 4, 2220–2240 CrossRef CAS.
- B. Hu, M. Hu, J. Luo and T. L. Liu, Adv. Energy Mater., 2022, 12, 2102577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09177a |
| This journal is © The Royal Society of Chemistry 2023 |