
Encapsulated C12A7 electride material enables a multistep electron transfer process for cross-coupling reactions†
Bo
Dai‡
a,
Zichuang
Li‡
a,
Miao
Xu‡
b,
Jiang
Li
c,
Yangfan
Lu
 d,
Jiantao
Zai
a,
Liuyin
Fan
e,
Sang-Won
Park
f,
Masato
Sasase
c,
Masaaki
Kitano
*c,
Hideo
Hosono
*c,
Xin-Hao
Li
a,
Tian-Nan
Ye
*a and
Jie-Sheng
Chen
*a
d,
Jiantao
Zai
a,
Liuyin
Fan
e,
Sang-Won
Park
f,
Masato
Sasase
c,
Masaaki
Kitano
*c,
Hideo
Hosono
*c,
Xin-Hao
Li
a,
Tian-Nan
Ye
*a and
Jie-Sheng
Chen
*a
aFrontiers Science Center for Transformative Molecules, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: ytn2011@sjtu.edu.cn; chemcj@sjtu.edu.cn
bState Key Laboratory of Space Power-Sources Technology, Shanghai Institute of Space Power-Sources, Shanghai 200245, China
cMDX Research Center for Element Strategy, International Research Frontiers Initiative, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: kitano.m.aa@m.titech.ac.jp; hosono@mces.titech.ac.jp
dNational Engineering Research Center for Magnesium Alloys, College of Materials Science and Engineering, Chongqing University, Chongqing 400030, China
eStudent Innovation Center, Shanghai Jiao Tong University, Shanghai 200240, China
fDepartment of Chemical and Materials Engineering, University of Suwon, Hwaseong, Gyeonggi 18323, Republic of Korea
First published on 27th January 2023
Abstract
The electronic structures of active sites fundamentally determine the catalytic performance in chemical reactions and are also crucial for obtaining a detailed understanding of charge transport and reaction mechanisms. In this study, the regulation of the electronic structure of active metal Pd can be achieved through a multi-step electron transfer process formed by a synergy of [Ca24Al28O64]4+(e−)4 (C12A7:e−) electride and conductive graphene (Gr). The composite catalytic system (Pd/Gr/C12A7:e−) significantly facilitates the transfer of electrons from electron-rich Pd active sites to aryl halides in Suzuki-coupling reactions, which enables superior catalytic performance with TOFs above 20 times higher than well-studied negatively charged Pd catalysts. No catalytic degradation was observed even after impregnating the catalyst in water because of the well-protected C12A7:e− electride by Gr. The present efficient catalyst can further trigger various carbon–carbon cross-coupling reactions with high activities. These results provide significant advantages for expanding the potential applications of electride materials, thereby allowing precise control of the electronic structure of the active sites and aiding in tuning the reaction conditions using a simple method.
10th anniversary statementWe would like to congratulate on the upcoming 10th anniversary celebration of the Journal of Materials Chemistry A. In the discipline of materials and chemistry, the Journal of Materials Chemistry A has a significant influence that cannot be ignored. Many excellent opinions and articles have been published in the journal, playing a decisive role in the profession. As materials chemistry researchers, we feel honoured to publish articles in the Journal of Materials Chemistry A. In the past ten years, measurable progress on the research of materials chemistry in which the future of energy and sustainability is embodied. Electride materials are regarded as one category of promising emergent materials that have more advantages than traditional catalytic materials because of their unique electronic properties that can accelerate or limit specific chemical reactions. At present, there are few cases in which electride materials are used in industrial catalysis because of their chemical instability. Our research goal is to solve this problem and to develop stable electride composite materials that can be successfully put into use in the near future through continuous technological breakthroughs. |
1. Introduction
Cross coupling is of fundamental significance in the formation of a carbon–carbon bond and is an important class of reaction in organic synthesis.1–4 It was initially investigated using homogeneous catalysts with electron-rich Pd active sites by introducing electron-donating ligands that were designed to reduce the high energy barrier of the reactants associated with the rate-determining step and further enhance the catalytic activity.5–7 However, the combination of unrecyclable Pd species and potentially toxic ligands, e.g., Pd-based phosphine complex has raised tremendous environmental concerns amid increasing demands for sustainability in the chemical industry.8,9 Thus, heterogeneous catalysts have a distinct advantage in their facile recyclability without employing ligand promoters.10–15 Although many studies on heterogeneous catalysts have focused on the design of electron-rich Pd active sites by introducing multi-metallic components or organic modifiers, the occupation of Pd sites by other elements always results in inferior catalytic activity.16,17 Herein, we focus on an approach that utilizes metal–support interactions, in which the electronic structure of the active metal can be effectively controlled by the support material that favours the enhancement of catalytic activity.18–23[Ca24Al28O64]4+(e−)4 (C12A7:e−) electride materials, which accommodate anionic electrons in their periodically distributed lattice, have been attracting considerable attention because their low work function feature enables strong electron donation ability.24–30 One of their most successful applications is in ammonia synthesis, in which an electron transfer from C12A7:e− electride to the antibonding orbitals of N2 facilitates N2 dissociation, and the overall activation energy of the reaction is suppressed.31–34 However, C12A7:e− electride is typically prepared by a solid-state method, resulting in a small surface area of a few square meters per gram. Such a low surface area leads to a relatively low dispersion of the loaded active metals, and thus, only the exposure of limited active sites. Moreover, the moisture sensitivity of C12A7:e− severely restricts further applications in aqueous reactions.35,36 Therefore, it is highly desired to achieve C12A7:e− electride with high stability towards moisture while retaining its intrinsic electronic structure. Graphene (Gr)-based materials have been well-studied as water or gas filters,37,38 and thus, we are inspired to combine C12A7:e− electride with Gr to form an encapsulated nano-structure.
Herein, we report the successful preparation of the Pd/Gr/C12A7:e− catalyst and its high catalytic activity towards cross-coupling reactions. In this composite material, the C12A7:e− electride surface was coated with Gr layers, which are highly dispersed with nanometric Pd clusters, to achieve high chemical stability towards moisture. Negatively charged Pd was achieved through a multistep electron transfer process, in which electrons can be donated from interior C12A7:e−via highly conductive Gr to the external Pd active species owing to the work function gap between C12A7:e− (ΦWF = 2.4 eV) and Pd (ΦWF = 5.1 eV).39,40 The electron-rich Pd active sites enable the aryl halide activation, thereby reducing the apparent activation energy of the Suzuki-coupling reaction by ca. 30%; therefore, the reaction could proceed under mild conditions. Impressively, Pd/Gr/C12A7:e− showed no degradation in catalytic activity even after the impregnation of water, which demonstrates the chemical protection of Gr. These results demonstrate the effectiveness of combining an electride and traditional active metal, and they reveal a simple and effective approach to improving catalytic activity and stability.
2. Results and discussion
The overall procedure for the synthesis of the catalyst is illustrated in Fig. S1.† Nanometric Pd clusters were first deposited on the Gr surface using Na2PdCl4 as a Pd precursor and an ultraviolet (UV) light-induced reduction process. Both Gr and Pd/Gr could be dispersed uniformly in ethanol for a long time (Fig. S1 left†), which suggests the thin nature of the Gr layers. The C12A7:e− electride was fabricated by a conventional solid-phase reaction (see Experimental section), and the surface area was around 1.0 m2 g−1. Subsequently, C12A7:e− powder was mixed with fresh Pd/Gr in the solvent and stirred for 12 h. After vacuum drying and annealing in an Ar atmosphere at 250 °C, Pd/Gr encapsulated C12A7:e− (Pd/Gr/C12A7:e−) was obtained. It is noteworthy to mention that Pd/Gr/C12A7:e− is easily collected because it settles to the bottom of the solvent (Fig. S1 left†), which indicates the formation of Pd/Gr and C12A7:e− electride composite with increased material density. Fig. 1a and b show that the scanning electron microscopy (SEM) images of Pd/Gr/C12A7:e− reveal that the C12A7:e− particles were well encapsulated by Pd/Gr multi-layers and each element was uniformly distributed throughout the particles. The aberration-corrected high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) image depicted in Fig. S2† shows that Pd clusters are uniformly dispersed on the Gr surface with a small average particle size of 1.5 nm.When electronic conductive materials are in contact, a charge transfer is allowed to balance the chemical potentials of each material. For the Gr/C12A7:e− heterostructure, an inorganic C12A7:e− electride with a low work function of 2.4 eV could be regarded as an electron donor (Fig. S3†) for Gr with a higher work function of 5.0 eV, resulting in an electron transfer from C12A7:e− to Gr (Fig. 1c),39,40 which could be confirmed by Raman spectroscopy (Fig. 1d). In the Raman spectra, the invariant D-band of Gr with and without C12A7:e− suggests a physical interaction between C12A7:e− and Gr. Comparatively, a blue shift of the G-band appeared after the addition of C12A7:e− electride. Regarding the results on carrier-injected carbon nanotubes,41,42 this blue shift should be attributed to the lattice distortion of Gr evoked by the accepted electrons from C12A7:e− and the Fermi level shift induced by electron–phonon coupling. Moreover, the C 1s peak of X-ray photoelectron spectroscopy (XPS) of Pd/Gr/C12A7:e− and Pd/Gr further show that the addition of C12A7:e− up-shifted the binding energies of sp3-hybridized carbon atoms, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH and CO32− over Gr layers, which means that electron transfer is mainly performed on the defect sites and functional groups of Gr (Fig. S4 and Table S1†). The CC peak remains unchanged, indicating no chemisorption process or modification of the graphitic structure. In combination with the Raman shift and up-shifted C 1s peaks in the XPS spectra, it is reasonable to conclude that the electrons can be effectively transferred to the Gr surface.
O, COOH and CO32− over Gr layers, which means that electron transfer is mainly performed on the defect sites and functional groups of Gr (Fig. S4 and Table S1†). The CC peak remains unchanged, indicating no chemisorption process or modification of the graphitic structure. In combination with the Raman shift and up-shifted C 1s peaks in the XPS spectra, it is reasonable to conclude that the electrons can be effectively transferred to the Gr surface.
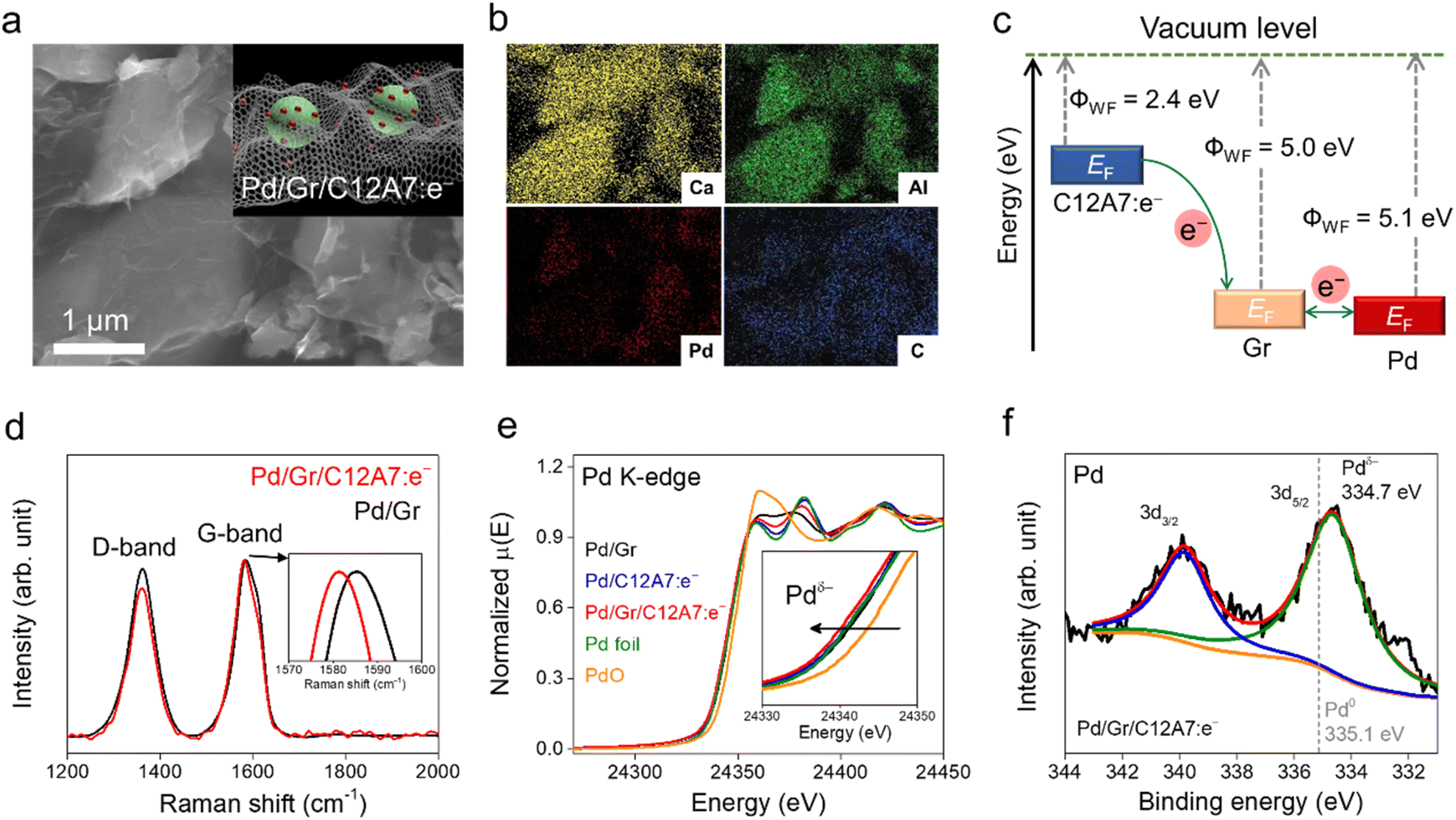 | ||
| Fig. 1 Representative (a) SEM image and (b) corresponding elemental mapping of the Pd/Gr/C12A7:e− composite catalyst. Inset (a) schematic illustration of the structure of Pd/Gr/C12A7:e−. (c) Comparison of the Fermi level in the C12A7:e− electride, Gr and Pd metal. Blue arrows indicate the direction of electron transfer from C12A7:e− to Gr and Pd metals. (d) Raman spectra of Pd/Gr/C12A7:e− and Pd/Gr. The G-mode shift is magnified in the inset. (e) Pd K-edge XANES spectra of Pd in Pd/Gr/C12A7:e−. The data of Pd/Gr, Pd/C12A7:e−, Pd foil and PdO are also shown as references, which indicate the negatively charged Pd state in Pd/Gr/C12A7:e−. The Pd K-edge shift is magnified in the inset. (f) XPS spectra of Pd 3d in Pd/Gr/C12A7:e−. The grey dashed line represents the binding energy of the reference Pd metal with a zero valence state. | ||
Given that Gr (ΦWF = 5.0 eV) and Pd (ΦWF = 5.1 eV) exhibit comparable work functions, the electron could easily flow between the Gr and loaded Pd cluster. Negatively charged Pd species are thus achieved, which was confirmed by X-ray absorption near-edge structure (XANES) measurement. The absorption edge for Pd species in Pd/Gr/C12A7:e− was located at a lower energy relative to that of Pd foil (Fig. 1e), implying negatively charged Pd species. However, no energy shift was detected for Pd in Pd/Gr without the presence of C12A7:e− electride. It is noteworthy that Pd on C12A7:e− was also in metallic form, which could be attributed to the uneven charge distribution at large Pd particles on C12A7:e− without the dispersion of Gr (Fig. S5†). As the surface reaction of the catalytic process, we then investigated the surface properties of the catalyst using XPS. Compared with the zero valence state of Pd (335.1 eV), the obviously lower energy shift of the Pd 3d XPS peaks in Pd/Gr/C12A7:e− implies electron-rich Pd species (334.7 eV) (Fig. 1f). These observations combined with the XANES analysis results indicate the key role of the C12A7:e− electride as an electron donor in the modification of the electron density of highly dispersed Pd clusters on Gr.
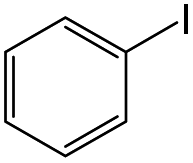
We initially tested the possibility of using the negatively charged Pd active sites of the electride composite material in organic catalysis to promote the Suzuki coupling of iodobenzene with phenylboronic acid as a model reaction. In Fig. 2a, Pd/Gr/C12A7:e− showed high catalytic performance with a reaction rate of 60.0 mmol g−1 h−1 at room temperature, and its turnover frequency (TOF) value was estimated to be 1413.3 h−1 based on the total amount of Pd, which is in the orders of magnitude larger than the Pd/Gr (5 wt% Pd, 42.4 h−1), Pd/C12A7:e− (1 wt% Pd, 71.0 h−1) and Pd/C12A7:O2− (1 wt% Pd, 43.5 h−1) reference catalysts (Fig. 2a, Table S2†). Impressively, in terms of TOFs, Pd/Gr/C12A7:e− even outperforms our previously reported negatively charged Pd catalysts (Pd/ZrC, ZrPd3 and Y3Pd2) and other benchmarked commercial heterogeneous Pd catalysts (Lindlar catalyst from TCI, 5 wt% Pd; Pd/Al2O3 from Sigma-Aldrich, 5 wt% Pd; Pd/C from Sigma-Aldrich, 10 wt% Pd) 20-fold under the same reaction conditions (Table S3†). The lower reaction rates of Pd/C12A7:e− (1 wt% Pd) and Pd/C12A7:O2− (1 wt% Pd) are attributed to the limited number of exposed active sites owing to the large Pd particle size (Fig. S5 and S6†). It is noteworthy to mention that the activity of Pd/C12A7:e− (0.2 wt% Pd, 1335.6 h−1) is comparable to that of Pd/Gr/C12A7:e− when the Pd particle size was reduced (Fig. S7, Table S2†), indicating an electron donation effect on nanosized Pd particles by C12A7:e−, which could also be confirmed by the same energy location of Pd 3d XPS spectra (Fig. S7b†). Additionally, with a similar Pd particle size (Fig. S8†), the lower activity of the Pd/Gr sample is mainly attributed to the absence of C12A7:e−.
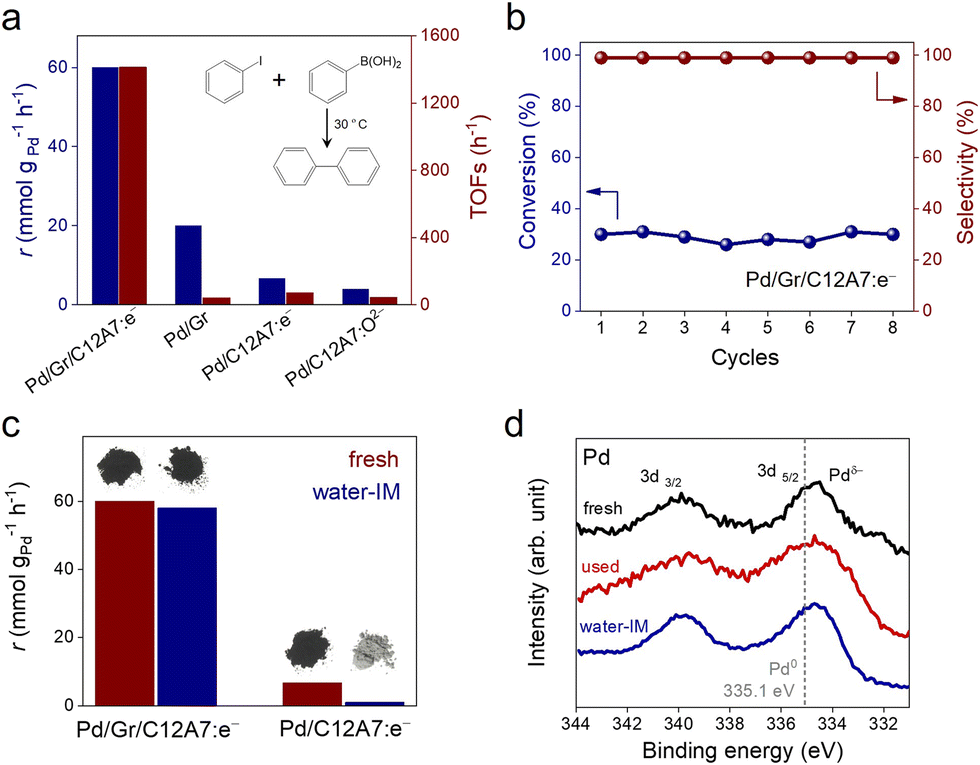 | ||
| Fig. 2 (a) Reaction rates and TOFs for the coupling of iodobenzene and phenylboronic acid over Pd/Gr/C12A7:e−, Pd/Gr, Pd/C12A7:e− and Pd/C12A7:O2− catalysts. (b) Recycling experiment for the coupling reaction over Pd/Gr/C12A7:e−. (c) Reaction rates over fresh and water-impregnated (IM) Pd/Gr/C12A7:e− and Pd/C12A7:e− for the coupling of iodobenzene and phenylboronic acid with photographs of the corresponding catalyst powders. (d) XPS spectra of Pd 3d over fresh, used and water IM Pd/Gr/C12A7:e− catalysts. The grey dashed line represents the binding energy of the reference Pd metal with a zero valence state. | ||
In addition to the high catalytic performance for the Suzuki coupling reactions, the stable recyclability of the Pd/Gr/C12A7:e− catalyst should be noted. Pd/Gr/C12A7:e− catalyst can be recycled at least 8 times under both low (Fig. 2b) and high conversion (Fig. S9†) levels. After the reaction, the crystal structure remained largely unchanged (Fig. S10†). HAADF-STEM observation clearly revealed that the Pd particle size and morphology remained unchanged after recycling (Fig. S11†). XPS measurements showed that the Pd species remained negatively charged, i.e., the surface electronic structure is highly stable (Fig. 2d). The hot filtration experiment indicated that the reaction proceeds only in the presence of Pd/Gr/C12A7:e−, and no more coupling products could be produced after the removal of the Pd/Gr/C12A7:e− catalyst (Fig. S12†). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements also showed that the Pd species in the filtrate was below the detection limit (0.007 ppm). These results for the used Pd/Gr/C12A7:e− catalyst demonstrated the robustness of the Pd active sites and confirmed the excellent stability of this catalyst.
Moisture sensitivity is one of the most serious challenges to the practical application of C12A7:e− electride, which has always undergone a transformation from C12A7:e− to Ca and Al hydrates with the release of the anionic electron, which significantly reduces its carrier density.43 However, Pd/Gr/C12A7:e− was found to be robust against water. A Suzuki coupling reaction using water-impregnated Pd/Gr/C12A7:e− powder as a catalyst was conducted without changing to any other conditions. The obtained coupling rate was almost identical to that of the fresh catalyst, and the color of the samples did not change (Fig. 2c). Decomposition of the catalyst, such as by the generation of Ca and Al hydrates, was not identified from X-ray diffraction (XRD) measurements after the impregnation in water (Fig. S10†). Based on XPS measurements, the partially negatively charged Pd was unaffected by the water impregnation, which demonstrates the robustness of the Pd/Gr/C12A7:e− catalyst (Fig. 2d). In contrast, the catalytic activity of the Pd/C12A7:e− catalyst was obviously degraded after impregnation in water, and the colour of the sample changed from black to light grey, associated with the transformation to the related Ca and Al hydrates (Fig. 2c). This degradation can be attributed to the absence of a protective layer of Gr, which results in the progressive release of anionic electrons from C12A7:e−.
To further investigate the electron donation effect of C12A7:e−, the controlled electron concentration (Ne) of C12A7:e− was investigated. C12A7:e− with different Ne were obtained by treatment with Ti metal at different temperatures (Fig. 3). Fig. 3a shows the UV-vis absorption spectra for the synthesized powders of C12A7:e− or C12A7:O2− with various Ne. There is no adsorption peak of the C12A7:O2− sample (orange line) in the visible region, and the corresponding 3.5 eV absorption edge is attributed to the excitation between the energy level of encaged O2− ions and the cage conduction band (CCB).44 Moreover, a broad absorption peak at around 2.3–2.7 eV (2.3 eV, 2.5 eV and 2.7 eV for green, blue and red lines, respectively) appeared in C12A7:e−, which can be attributed to an intra-cage transition of electrons confined in the cages. The other absorption peak below 2 eV is ascribed to an inter-cage transition as charge transfer occurs from an electron-trapped cage to a neighboring vacant cage.45 Therefore, it can be deduced that O2− ions in the cages of C12A7:O2− are substituted by electrons, thus forming C12A7:e−. Based on our previous studies, the adsorption peak position (Esp) and electron concentration (Ne) are related to the following equation: 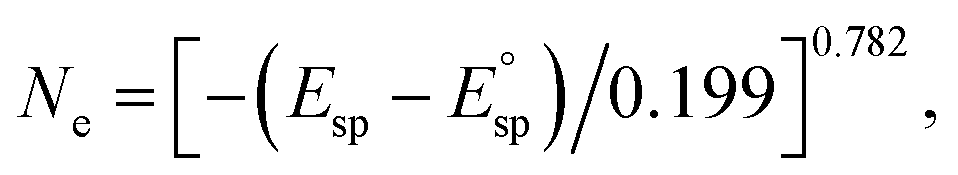 and one typical case is that
and one typical case is that 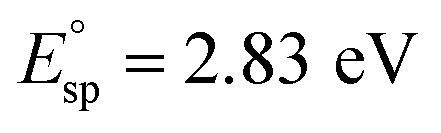 represents Ne ≈ 1.0 × 1018 cm−3.46 Thus, the electron concentrations of the above C12A7:e− samples can be calculated to be 1.2 × 1020, 1.1 × 1021, and 2.2 × 1021 cm−3, respectively (Fig. 3b). Accordingly, the sample color changed from white to green to black with increased Ne (Fig. 3b).
represents Ne ≈ 1.0 × 1018 cm−3.46 Thus, the electron concentrations of the above C12A7:e− samples can be calculated to be 1.2 × 1020, 1.1 × 1021, and 2.2 × 1021 cm−3, respectively (Fig. 3b). Accordingly, the sample color changed from white to green to black with increased Ne (Fig. 3b).
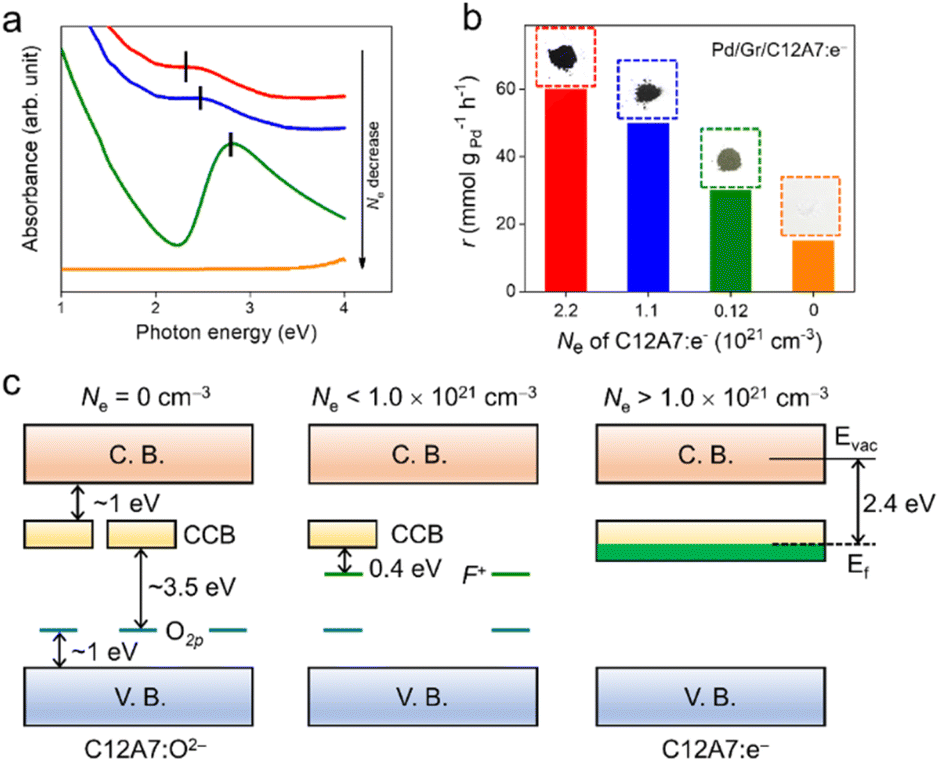 | ||
| Fig. 3 (a) The UV-vis absorption spectra for the synthesized powders of C12A7:e− or C12A7:O2− with various electron concentrations (Ne). The electron concentrations corresponding to the red, blue, green and orange lines are 2.2 × 1021, 1.1 × 1021, 1.2 × 1020 and 0 cm−3. (b) Reaction rates over the Pd/Gr/C12A7:e− catalyst as a function of the electron concentration of C12A7:e−, and corresponding photographs of the synthesized powders of C12A7:e− with various Ne. (c) Scheme of the electronic structures of C12A7:O2− and C12A7:e− with various Ne. | ||
Fig. 3c shows the electronic structures of C12A7:e− or C12A7:O2− with various ranges of Ne. For C12A7:O2− with Ne = 0 cm−3, the 2p orbitals of the framework O2− ions and 4s orbitals of the framework Ca2+ ions contributed to the valence band (VB) and conduction band (CB), respectively. In contrast to framework O2− ions, the energy level of the encaged O2− ions is located slightly above the top of the VB of the cage framework. In addition, the electron tunneling among the three-dimensionally connected cages enables the formation of the CCB, which is located at ∼1.0 eV below the bottom of CB.47 In the case of Ne < 1.0 × 1021 cm−3, the cage-encaged electrons with low density formed an F+-like center, and the energy level is located at 0.4 eV below the CCB. When the Ne is higher than 1.0 × 1021 cm−3, the cage-trapped electrons occupy the CCB, raising the Fermi level of C12A7:e− to 0.5 eV above the CCB minimum. Therefore, an intrinsic low work function property was achieved over such a unique electron-occupied CCB electronic structure. The high electron concentration leading to a low work function property enables high electron donation power for C12A7:e−. The catalytic activity of Pd/Gr/C12A7:e− provides information on the critical electron concentration (Ne) of C12A7:e− as a support. Impressively, the reaction rates were observed to increase monotonically with the Ne of C12A7:e− (Fig. 3b). This observation suggests that the catalytic activity of this reaction is directly proportional to the electron donation ability of the support. Accordingly, the measured apparent activation energy (Ea) decreases from 83.0 kJ mol−1 to 58.7 kJ mol−1 with increasing Ne (Fig. S13†), which should be related to the electron transfer from C12A7:e−via surface Pd active sites to aryl halide substrates.
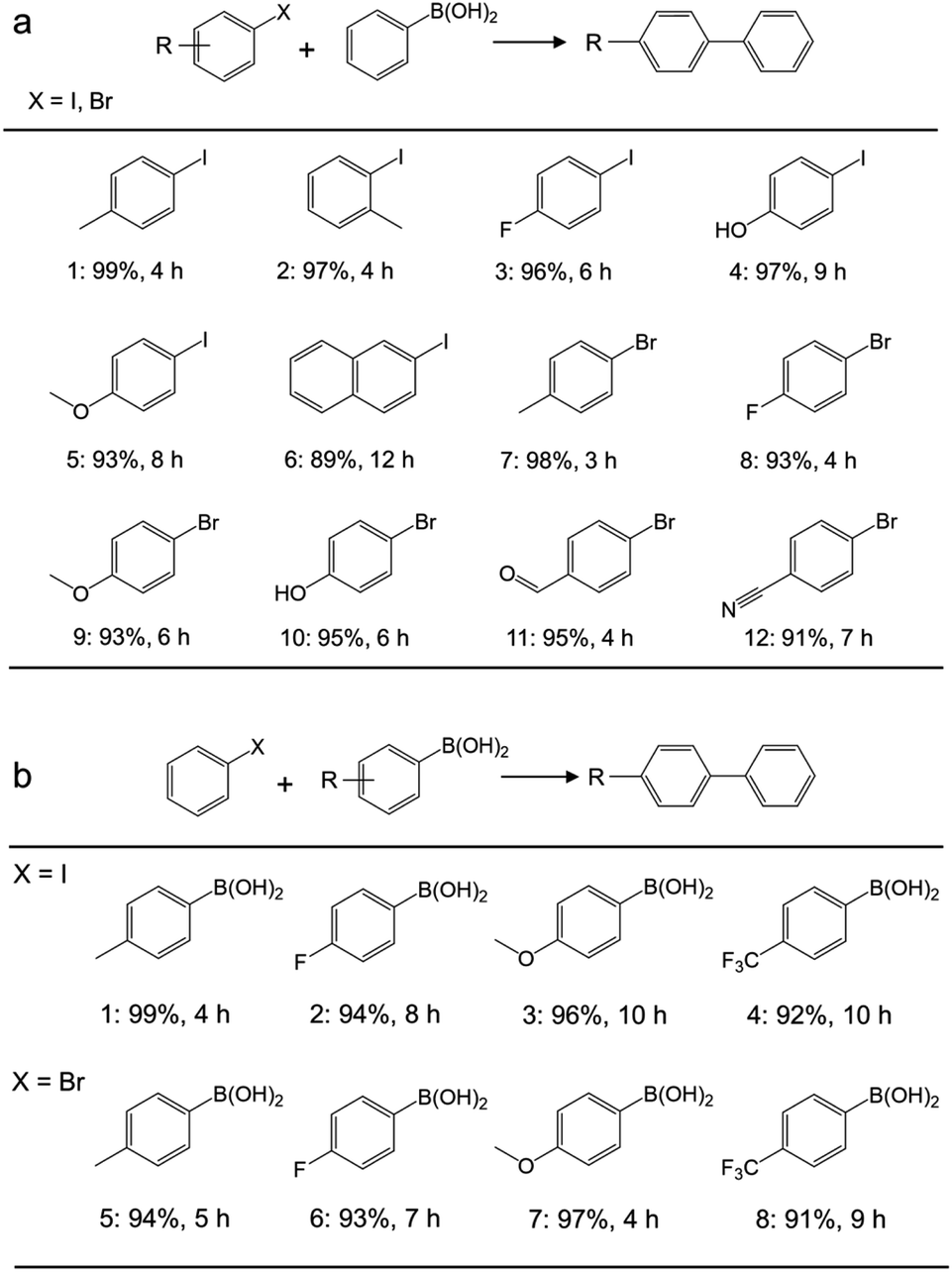
Application of the catalyst was then extended to various aryl halides and boronic acids to verify the general activity of the Pd/Gr/C12A7:e− catalyst towards Suzuki coupling reactions. Table 1, (a) shows that all the tested aryl halides with a range of functional groups could be converted to the corresponding coupled products in high yields, regardless of whether the functional groups were electron-donating or electron-withdrawing. Next, different types of boronic acids were further exploited as coupling partners (Table 1, (b)). The results confirmed the general applicability of the Pd/Gr/C12A7:e− catalyst with high selectivity and functional group tolerance. In addition, the coupling reaction of chlorobenzene with boronic acids was also investigated (Table S4†), and the diphenyl production activity was poor, which may be ascribed to the much higher bonding energy of C–Cl (346 kJ mol−1) than that of C–Br (290 kJ mol−1) and C–I (228 kJ mol−1).48
| a Reaction conditions: Pd (0.47 mol% relative to organohalide), 0.5 mmol organohalide, 0.8 mmol arylboronic acid, 1.5 mmol K2CO3, 5 mL ethanol, iodides 30 °C, bromides 60 °C. The yields given below the structure were determined using GC and GC-MS. |
|---|
|
Next, Arrhenius plots were plotted to investigate the apparent activation energies (Ea) of the Suzuki coupling over the Pd/Gr/C12A7:e−, Pd/Gr and Pd/C12A7:O2− catalysts. The Ea of Pd/Gr/C12A7:e− was estimated to be 58.7 kJ mol−1 of the coupling reaction of iodobenzene and phenylboronic acid (Fig. 4a), which shows a ca. 27.4% and 29.3% reduction compared with those of Pd/Gr (80.9 kJ mol−1) and Pd/C12A7:O2− (83.0 kJ mol−1), respectively (Fig. 4c). Moreover, the Ea follows the same trend as the iodobenzene replaced by bromobenzene (Fig. 4b), in which the calculated Ea of Pd/Gr/C12A7:e− (70.8 kJ mol−1) is ca. 31.0% and 28.6% less than those of Pd/Gr (102.6 kJ mol−1) and Pd/C12A7:O2− (99.1 kJ mol−1), respectively (Fig. 4c). These results suggest that the Suzuki coupling reaction is promoted in the presence of C12A7:e−. Kinetic reaction orders were estimated by changing the concentration of aryl halides and phenylboronic acid over Pd/Gr/C12A7:e−. Fig. 4d shows that both reaction rates are sensitive to the concentration of aryl halides but independent of phenylboronic acid. These results imply that the activation of aryl halides controlled the overall reaction process over Pd/Gr/C12A7:e−, which is well accepted for both homogeneous and heterogeneous Pd-based catalysts in Suzuki coupling reactions, i.e., the oxidative addition of the aryl halide is the rate-determining step for the catalytic cycle.49–52 From this kinetic analysis, the smaller Eas and reaction orders reveal that the enhanced activity of Pd/Gr/C12A7:e− over that for the Pd/Gr catalyst exclusively originates from the promoted activation of the aryl halides.
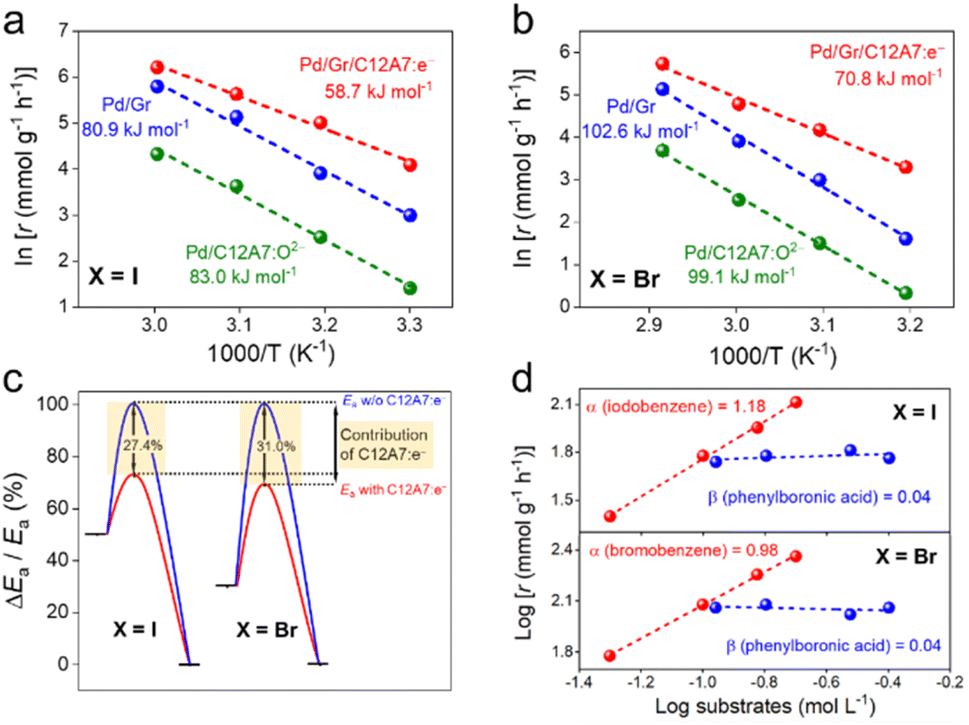 | ||
| Fig. 4 Arrhenius plots of Suzuki cross-coupling reactions performed using (a) iodobenzene and (b) bromobenzene as substrates over Pd/Gr/C12A7:e− and Pd/Gr catalysts, respectively. Reaction conditions: Pd (0.47 mol% relative to organohalide), 0.5 mmol organohalide, 0.8 mmol arylboronic acid, 1.5 mmol K2CO3 and 5 mL solvent. (c) Apparent activation energies (Ea) calculated for Suzuki cross-coupling reactions over catalysts with and without C12A7:e−. (d) Reaction rate dependence on the organohalide and arylboronic acid concentration over the Pd/Gr/C12A7:e− catalyst. | ||
To further understand the activation behaviours of aryl halides, DFT calculations and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) spectra were also performed. As illustrated in Fig. 5a and b, obvious electron-deficient and rich regions are constructed at the interface between C12A7:e− and Pd. The elevated electron density of the Pd sites enables electron transfer (0.15 e−) to the adsorbed iodobenzene substrate, thereby weakening the C–I bond with 9.5% elongation (Fig. 5c). Here, the reaction barriers for the oxidative addition of aryl halide were also calculated over Pd/C12A7:e− and Pd (111). The activation process of the iodobenzene molecule proceeds with an energy barrier of 0.21 eV on Pd/C12A7:e−, much lower than that of 0.58 eV on Pd (111) (Fig. 5d). The tendency of the suppressed activation energy agrees well with the experimental observations, further suggesting that the activation step of aryl halide on Pd/C12A7:e− is remarkably enhanced.
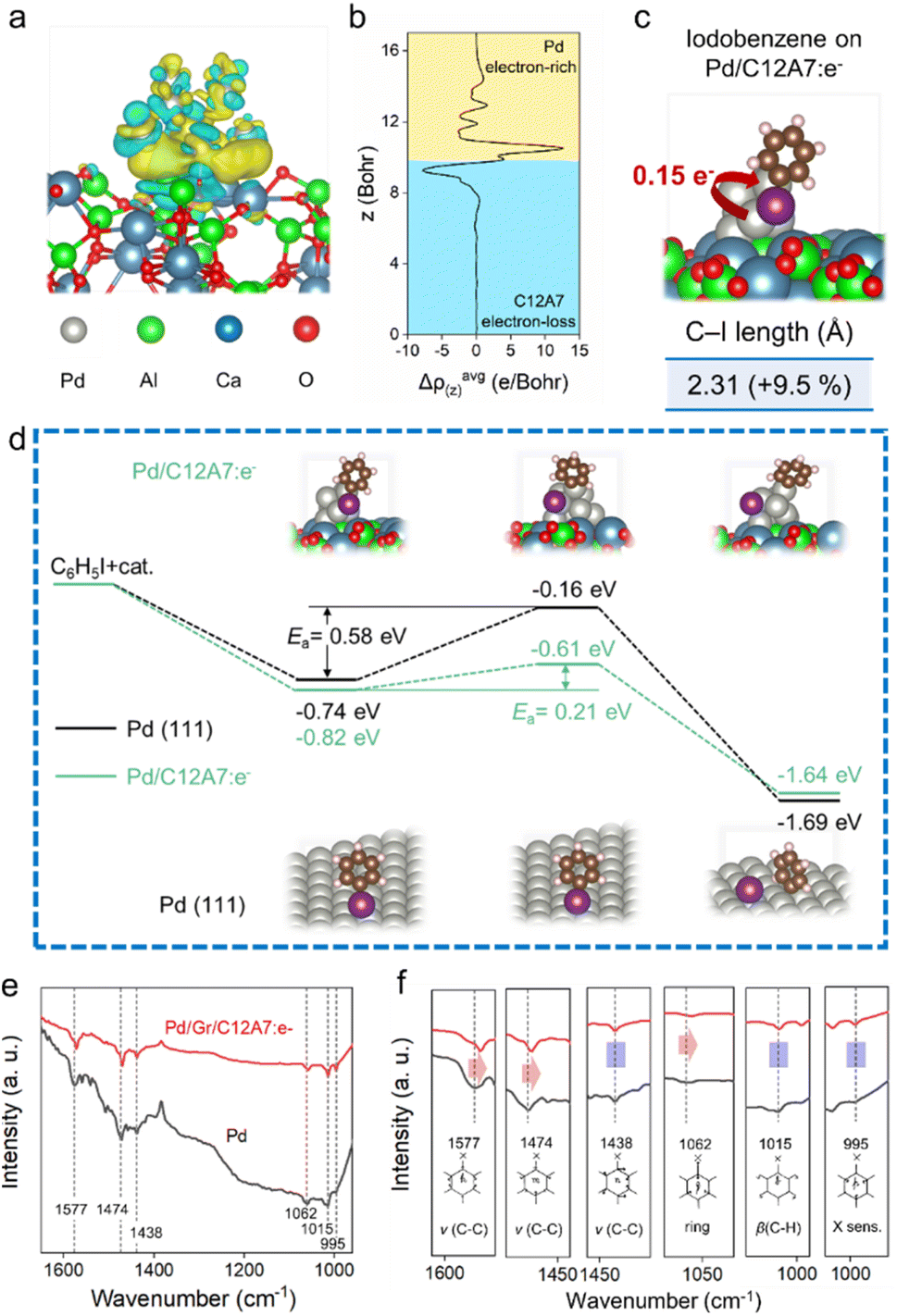 | ||
| Fig. 5 Depictions of the electronic interaction between catalysts and aryl halides. (a) 3D electron density isosurface map and (b) planar-averaged electron density difference Δρ(z) of Pd/C12A7:e−. The yellow and cyan areas indicate electron accumulation and depletion, respectively. (c) Adsorption of iodobenzene on Pd/C12A7:e−. A view of the system from the size and C–I bond lengths is shown at the bottom. (d) Energy profiles from DFT studies of the activation of iodobenzene on Pd/C12A7:e− and Pd (111) surfaces. (e) DRIFTS spectra for the adsorption of iodobenzene onto Pd and Pd/Gr/C12A7:e−. (f) Enlarged DRIFTS spectra emphasize the redshift of the C–I bond-related vibration on Pd/Gr/C12A7:e−. | ||
Next, the adsorption behaviour of iodobenzene is experimentally evaluated using DRIFT spectroscopy (Fig. 5e). For Pd (111), the black line shows bands in the range of 1400–1600 cm−1, associated with C–C stretching in aromatic vibration (νC–C).53 In the low-frequency region, the IR vibrational bands of halogen-sensitive vibration (995 cm−1), out-of-plane C–H deformation (β(C–H): 1015 cm−1), and trigonal ring breathing (1062 cm−1) can be observed. Notably, if the catalyst changed to Pd/Gr/C12A7:e−, the IR vibrational peaks of 1577 and 1474 cm−1 associated with the admixture of C–I deformation show a significant redshift compared to the pure Pd case (Fig. 5f). Additionally, a similar redshift was detected for the C–I stretching vibration (1062 cm−1, Fig. 5f). The redshift phenomenon of the C–I bond-related vibration indicates a carbon halogen bond-weakening process. These results agree with the DFT calculation results, in which the low work function C12A7:e− electride generally acts as an electron-donating ligand and improves electron donation to the antibonding orbitals of the aryl halides through the surface Pd active sites, which is responsible for the weakening of the carbon halogen bond, the rate-determining step of the reaction. The results of the kinetic analysis suggest the importance of the electronic effect between C12A7:e− and the highly dispersed active Pd species in terms of electron density modification, which results in the significant enhancement of the catalytic activity of Pd active sites towards the cross-coupling reaction.
Owing to the high activity of the activation of aryl halides over Pd/Gr/C12A7:e−, we became interested in testing the catalyst for various C–C couplings, such as Sonogashira, Stille, Hiyama and Heck coupling reactions. As shown in Table 2, each of the tested iodobenzene and coupling partners, such as phenylacetylene, tributylphenylstannane, phenylethylene, and trimethoxyphenylsilane, could be converted to the corresponding coupled products in a high yield. The results confirmed the versatile applicability of the Pd/Gr/C12A7:e− catalyst for various C–C cross-coupling reactions.
| Entry | Aryl halides | Coupling partner | Product | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: Pd (0.47 mol% relative to organohalide); 0.5 mmol iodobenzene, 0.8 mmol coupling partner, 1.5 mmol K2CO3. The yields given below the structure were determined using GC and GC-MS. b 5 mL ethanol, 60 °C. c 5 mL DMF, 120 °C. | |||||
| 1 |
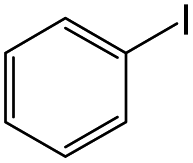
|
|
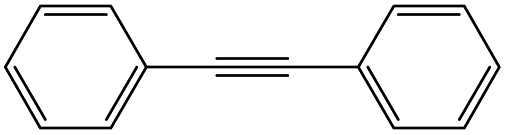
|
12 | 93 |
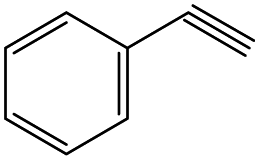
| 2 |
|
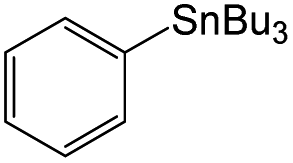
|
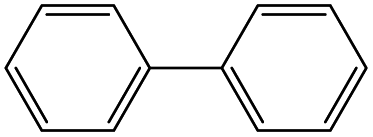
|
18 | 91 |
| 3 |
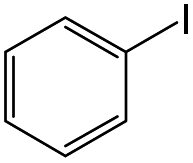
|
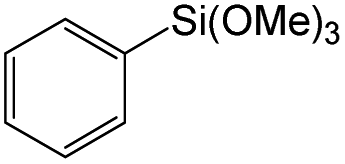
|
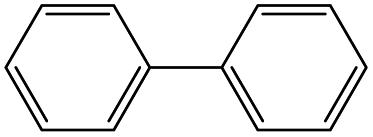
|
24 | 82 |
| 4 |
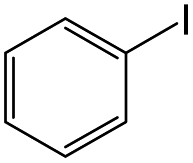
|
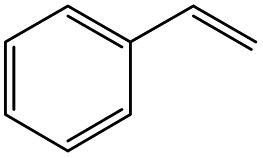
|
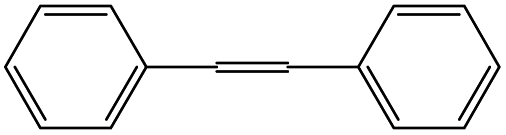
|
24 | 88 |
3. Conclusions
In summary, we successfully designed and prepared an electride composite material Pd/Gr/C12A7:e−, in which nanometric Pd clusters are dispersed on a Gr support with encapsulated C12A7:e− electride. The intermediate Gr layer serves as an excellent electron transport medium to promote electron transfer from the internal C12A7:e− electride to external Pd active sites and as a protective layer for the C12A7:e− electride against water. The obtained Pd/Gr/C12A7:e− catalyst hosts both highly dispersed and partially negatively charged Pd active sites, allowing the cross-coupling reaction to proceed with reduced activation energies. Mechanistic studies demonstrated that the strong electron donation ability of the C12A7:e− electride enabled negatively charged Pd sites through a multistep electron transfer process to promote the activation of aryl halides, which is the rate-determining step for the investigated cross-coupling reactions. Pd/Gr/C12A7:e− could also be easily separated and recycled for more than eight cycles without a significant loss of activity. The present efficient catalyst can further trigger various carbon–carbon cross-coupling reactions, such as Sonogashira, Stille, Hiyama and Heck coupling, with high activities.4. Experimental section
4.1 Sample preparation
The Gr powder used in this study was purchased from Sigma Aldrich, Ltd. (USA). Prior to deposition, the Gr powder was pre-treated at 400 °C for 5 h in a vacuum (ca. 1 × 10−4 Pa) to remove the water and oxygen adsorbed on the surface. It is noteworthy that the thickness of the commercial Gr was less than three layers based on the instructions of the product. The treated Gr powder was dispersed in a solution of Na2PdCl4 in ethanol and continuously stirred under UV irradiation (Xe lamp parallel light source system). After UV treatment for 30 min, the sample was centrifuged, washed three times with ethanol and water, and then dried in a vacuum oven to obtain Pd/Gr. C12A7:O2− (C12A7) powders were synthesized by a conventional solid-state reaction of CaCO3 and α-Al2O3 with a molar ratio of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 at 1300 °C for 20 h in an air atmosphere. Pd loading was conducted using the same method used for Gr. C12A7:e− electride was prepared by the reaction of C12A7:O2− and Ti metal at 900–1100 °C. Typically, the C12A7:O2− powder was sealed in a quartz tube with Ti metal under a vacuum of ∼10−3 torr. Then, the sealed tube was thermally annealed at temperatures of 900–1100 °C for 24–72 h. Adjusting the temperature and duration of the Ti-treatment replaces all or some free oxygen ions with electrons by the reaction, Ti (surface) + xO2− (cage) → TiOx (surface) + 2xe− (cage), which forms electron-doped C12A7. The C12A7:e− samples with various electron concentrations of 1.2 × 1020, 1.1 × 1021 and 2.2 × 1021 cm−3 are obtained by the heat treatment of C12A7:O2− under temperatures of 900, 1000, and 1100 °C, respectively. Then, the obtained C12A7:e− powder was mixed with fresh Pd/Gr (weight ratio of C12A7:e− and Pd/Gr of 10:1) and stirred overnight in super dry ethanol. After drying in a vacuum and annealing in an Ar atmosphere at 250 °C for 6 h, Pd/Gr encapsulated C12A7:e− (Pd/Gr/C12A7:e−) was obtained.
:7 at 1300 °C for 20 h in an air atmosphere. Pd loading was conducted using the same method used for Gr. C12A7:e− electride was prepared by the reaction of C12A7:O2− and Ti metal at 900–1100 °C. Typically, the C12A7:O2− powder was sealed in a quartz tube with Ti metal under a vacuum of ∼10−3 torr. Then, the sealed tube was thermally annealed at temperatures of 900–1100 °C for 24–72 h. Adjusting the temperature and duration of the Ti-treatment replaces all or some free oxygen ions with electrons by the reaction, Ti (surface) + xO2− (cage) → TiOx (surface) + 2xe− (cage), which forms electron-doped C12A7. The C12A7:e− samples with various electron concentrations of 1.2 × 1020, 1.1 × 1021 and 2.2 × 1021 cm−3 are obtained by the heat treatment of C12A7:O2− under temperatures of 900, 1000, and 1100 °C, respectively. Then, the obtained C12A7:e− powder was mixed with fresh Pd/Gr (weight ratio of C12A7:e− and Pd/Gr of 10:1) and stirred overnight in super dry ethanol. After drying in a vacuum and annealing in an Ar atmosphere at 250 °C for 6 h, Pd/Gr encapsulated C12A7:e− (Pd/Gr/C12A7:e−) was obtained.
4.2 Procedure for catalytic reactions
The cross-coupling reactions were carried out in a 25 mL stainless steel autoclave equipped with a magnetic stirrer. In a typical reaction, 0.5 mmol aryl halides, 0.8 mmol coupling partners (arylboronic acids, phenylacetylene, tributylphenylstannane, trimethoxyphenylsilane, phenylethylene), 1.5 mmol K2CO3, and Pd (0.47 mol% relative to aryl halides) catalysts were mixed in 5 mL solvent. The autoclave was then flushed three times with Ar, and the reaction was performed at a temperature range of 30 to 70 °C. The products were analyzed by gas chromatography (GC), and gas chromatography-mass spectrometry (GC-MS) was used to further confirm the coupling product. The TOF was calculated based on the reaction rate at a low conversion level derived from the total number of Pd atoms used in the catalyst.To check whether the coupling reaction over Pd/Gr/C12A7:e− catalyst is a heterogeneous reaction, the catalyst was removed from the reaction mixture by hot filtration after 1 h of reaction, and the filtrate proceeded under the same reaction condition. For the stability test, each coupling reaction of aryl halides and phenylboronic acid was conducted under the same conditions. After finishing each coupling reaction, the catalyst powder was separated via centrifugation, followed by washing three times with ethanol and water to remove the organic and inorganic residues. Subsequently, the catalyst powder was allowed to dry in a vacuum at room temperature, weighed, and reused in the next run. For the moisture-resistant test, Pd/Gr/C12A7:e− powder was impregnated in water for 1 h at room temperature. The catalyst powder was then separated via centrifugation, followed by the removal of water and drying under a vacuum overnight.
4.3 Sample characterization
The crystal structure was analyzed using XRD (D8 Advance, Bruker) with monochromate Cu Kα radiation (λ = 0.15418 nm). XPS (ESCA-3200, Shimadzu) measurements were performed using Mg Kα radiation at <10−6 Pa (8 kV bias voltage was applied to the X-ray source). The morphology of the sample was evaluated by aberration-corrected HAADF-STEM (JEM ARM-200F, JEOL) at 200 kV. Before the measurement, the powder sample was dispersed in super dry ethanol and dropwise added onto a holey carbon film with copper mesh. For the scanning electron microscopy (SEM) test, the sample powders were dispersed onto an Al-stub and examined in SE and backscatter modes using a JSM-7600F (JEOL) equipped with an energy dispersive X-ray spectrometer (EDX). The diameters of the Pd particles were estimated by counting the sizes of more than 100 particles for each sample. X-ray absorption fine structure (XAFS) measurements were carried out on the BL-12C beamline. To obtain the monochromatized X-ray beam, a Si (111) double-crystal monochromator was used. The corresponding spectra were recorded in transmission mode by diluting the samples in BN powder. The Pd content was determined using ICP-AES (ICPS-8100, Shimadzu). To evaluate the Brunauer–Emmett–Teller (BET) surface area, nitrogen sorption measurements were conducted using an adsorption analyzer (BELSORP-mini II, BEL, Japan). Prior to measurements, the catalysts were degassed in a vacuum at 150 °C for 12 h.4.4 DFT calculations
All DFT calculations were conducted using the Vienna Ab initio Simulation Package (VASP).54,55 The generalized gradient approximation method with the Perdew–Burke–Ernzerhof (PBE)56 exchange–correlation functional was used to manage the electron exchange and related energy; the core electrons were described using the projector augmented wave (PAW) method.57,58 The plane wave basis kinetic energy with a cut-off value of 450 eV was employed to describe the valence electrons. A mesh of 2 × 2 × 1 was used for the k-point sampling obtained from the gamma center. Furthermore, in the case of the system, the unit cell was 11.79 × 11.87 × 25 Å3, and the thickness of the vacuum layer was 20 Å. All models were fully optimized until the energy and forces converged to 1 × 10−5 eV and 0.0257 eV Å−1, respectively. For the transition state (TS) calculation, the parameters of CI-NEB59,60 are kept the same as the structure relaxation. The calculations of the binding energy of the intermediates were conducted using the following equation:| ΔE*x = Esys − Eslab − Ex, |
Author contributions
J.-S. C. proposed the idea behind the research and T.-N. Y. supervised the project. B. D., M. X., Y. L., J. L., J. Z., L. F., X.-H. L. and M. K. performed the synthesis, characterization and catalytic measurements. Z. L. and S.-W. P. conducted the model construction and DFT calculations. M. S. conducted STEM measurements. T.-N. Y., M. K., H. H. and J.-S. C. co-wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22105122, 22275121), the Science and Technology Commission of Shanghai Municipality (21PJ1407400), Shanghai Municipal Science and Technology Major Project and the Open Foundation Commission of Shaoxing Research Institute of Renewable Energy and Molecular Engineering (JDSX2022038). A part of this work was also supported by Element Strategy Initiative to Form Core Research Center (Grant Number JPMXP0112101001) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, Kakenhi Grants-in-Aid (JP17H06153, JP22H00272) from the Japan Society for the Promotion of Science (JSPS) and JST FOREST Program (JPMJFR203A).Notes and references
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- C. C. C. J. Seechurn, M. O. Kitching, J. Colacot and T. V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- B. W. Glasspoole and C. M. Crudden, Nat. Chem., 2011, 3, 912–913 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed.
- S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- Á. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS PubMed.
- T. N. Ye, Y. F. Lu, Z. Xiao, J. Li, T. Nakao, H. Abe, Y. Niwa, M. Kitano, T. Tada and H. Hosono, Nat. Commun., 2019, 10, 5653 CrossRef PubMed.
- Y. F. Lu, T. N. Ye, S. W. Park, J. Li, M. Sasase, H. Abe, Y. Niwa, M. Kitano and H. Hosono, ACS Catal., 2020, 10, 14366–14374 CrossRef CAS.
- B. Sun, L. Ning and H. C. Zeng, J. Am. Chem. Soc., 2020, 142, 13823–13832 CrossRef CAS PubMed.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed.
- B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS PubMed.
- T. Noël, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 5943–5946 CrossRef PubMed.
- Ö. Metin, S. F. Ho, C. Alp, H. Can, M. N. Mankin, M. S. Gültekin, M. Chi and S. Sun, Nano Res., 2013, 6, 10–18 CrossRef.
- Y. Wu, D. Wang, P. Zhao, Z. Niu, Q. Peng and Y. Li, Inorg. Chem., 2011, 50, 2046–2048 CrossRef CAS PubMed.
- T. N. Ye, J. Li, M. Kitano, M. Sasase and H. Hosono, Chem. Sci., 2016, 7, 5969–5975 RSC.
- X. H. Li, M. Baar, S. Blechert and M. Antonietti, Sci. Rep., 2013, 3, 1743 CrossRef.
- W. Ke, T. Cui, Q. Yu, M. Wang, L. Lv, H. Wang, Z. Jiang, X. H. Li and J. Chen, Nano Res., 2018, 11, 874–881 CrossRef CAS.
- T. N. Ye, J. Li, M. Kitano and H. Hosono, Green Chem., 2017, 19, 749–756 RSC.
- Z. Chen, E. Vorobyeva, S. Mitche, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed.
- H. Hosono and M. Kitano, Chem. Rev., 2021, 121, 3121–3185 CrossRef CAS PubMed.
- S. Matsuishi, Y. Toda, M. Miyakawa, K. Hayashi, T. Kamiya, M. Hirano, I. Tanaka and H. Hosono, Science, 2003, 301, 626–629 CrossRef CAS PubMed.
- K. Lee, S. W. Kim, Y. Toda, S. Matsuishi and H. Hosono, Nature, 2013, 494, 336–340 CrossRef CAS PubMed.
- Y. F. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2016, 138, 3970–3973 CrossRef CAS PubMed.
- T. N. Ye, Y. F. Lu, J. Li, T. Nakao, H. S. Yang, T. Tada, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2017, 139, 17089–17097 CrossRef CAS PubMed.
- J. Z. Wu, Y. T. Gong, T. Inoshita, D. C. Fredrickson, J. J. Wang, Y. F. Lu, M. Kitano and H. Hosono, Adv. Mater., 2017, 29, 1700924 CrossRef PubMed.
- J. Z. Wu, J. Li, Y. T. Gong, M. Kitano, T. Inoshita and H. Hosono, Angew. Chem., Int. Ed., 2019, 58, 825–829 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- S. Kanbara, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, J. Am. Chem. Soc., 2015, 137, 14517–14524 CrossRef CAS PubMed.
- F. Hayashi, Y. Toda, Y. Kanie, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2013, 4, 3124–3130 RSC.
- H. Buchammagari, Y. Toda, M. Hirano, H. Hosono, D. Takeuchi and K. Osakada, Org. Lett., 2007, 9, 4287–4289 CrossRef CAS PubMed.
- S. M. Kim, H. S. Yoo, H. Hosono, J. W. Yang and S. W. Kim, Sci. Rep., 2015, 5, 10366 CrossRef PubMed.
- J. Bong, T. Lim, K. Seo, C. Kwon, J. H. Park, S. K. Kwak and S. Ju, Sci. Rep., 2015, 5, 14321 CrossRef CAS PubMed.
- W. Wei, S. Yang, H. Zhou, I. Lieberwirth, X. Feng and K. Müllen, Adv. Mater., 2013, 25, 2909–2914 CrossRef CAS PubMed.
- Y. Toda, H. Yanagi, E. Ikenaga, J. J. Kim, M. Kobata, S. Ueda, T. Kamiya, M. Hirano, K. Kobayashi and H. Hosono, Adv. Mater., 2007, 19, 3564–3569 CrossRef CAS.
- J. M. Beebe, V. B. Engelkes, L. L. Miller and C. D. Frisbie, J. Am. Chem. Soc., 2002, 124, 11268–11269 CrossRef CAS PubMed.
- Y. N. Gartstein, A. A. Zakhidov and R. H. Baughman, Phys. Rev. Lett., 2002, 89, 045503 CrossRef PubMed.
- M. M. Menamparambath, J. Park, H. Yoo, S. P. Patole, J. Yoo, S. W. Kim and S. Baik, Nanoscale, 2014, 6, 8844–8851 RSC.
- C. K. Park, Cem. Concr. Res., 1998, 28, 1357–1362 CrossRef CAS.
- K. Hayashi, P. V. Sushko, D. M. Ramo, A. L. Shluger, S. Watauchi, I. Tanaka, S. Matsuishi, M. Hirano and H. Hosono, J. Phys. Chem. B, 2007, 111, 1946–1956 CrossRef CAS PubMed.
- S. Matsuishi, S. W. Kim, T. Kamiya, M. Hirano and H. Hosono, J. Phys. Chem. C, 2008, 112, 4753–4760 CrossRef CAS.
- S. Matsuishi, T. Nomura, M. Hirano, K. Kodama, S. Shamoto and H. Hosono, Chem. Mater., 2009, 21, 2589–2591 CrossRef CAS.
- P. V. Sushko, A. L. Shluger, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2007, 129, 942–951 CrossRef CAS PubMed.
- A. Leyva-Perez, J. Oliver-Meseguer, P. Rubio-Marqués and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 11554–11559 CrossRef CAS PubMed.
- F. Wang, C. Li, H. Chen, R. Jiang, L. Sun, Q. Li, J. Wang, J. C. Yu and C. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed.
- X. Zhang, Z. Sun, B. Wang, Y. Tang, L. Nguyen, Y. Li and F. F. Tao, J. Am. Chem. Soc., 2018, 140, 954–962 CrossRef CAS PubMed.
- S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793–5801 CrossRef CAS PubMed.
- Q. Xiao, S. Sarina, A. Bo, J. Jia, H. Liu, D. P. Arnold, Y. Huang, H. Wu and H. Zhu, ACS Catal., 2014, 4, 1725–1734 CrossRef CAS.
- D. H. Whiffen, J. Chem. Soc., 1956, 273, 1350–1356 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 128, 9901–9904 CrossRef.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08899a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |