
The structural evolution of poly(ethylene terephthalate) oligomers produced via glycolysis depolymerization†
Joshua
Moncada
 and
Mark D.
Dadmun
*
and
Mark D.
Dadmun
*
Department of Chemistry, University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: dad@utk.edu
First published on 13th February 2023
Abstract
Polymeric materials have become an integral part of our society, and their high demand has created a large quantity of polymers that end up in the waste stream. For instance, poly(ethylene terephthalate) (PET) is widely used in a broad range of applications, where the chemical recycling of PET is of growing interest. Most methods focus on the complete depolymerization of PET to the monomer, however pushing the equilibrium reaction to the monomer is time- and energy-intensive. We hypothesize that by intercepting intermediates in the depolymerization, telechelic oligomers can be captured that can also be used as reactants to produce value-added goods. To this end, the effect of reaction type, catalyst loading, reaction time, and temperature on the evolution of the product chain structure and yield of the glycolysis depolymerization of PET is studied. For a heterogeneous reaction at lower temperatures (165 °C), the rate of depolymerization is sufficiently slow to offer access to a broad range of molecular weight products (3000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Daltons) at a high yield (nearly 100%). At higher heterogeneous reaction temperatures (175 and 185 °C), the reaction rate increases, producing oligomers of a narrower molecular weight range (2000–5000 Daltons) with significant loss of the original PET, up to 40%, as water soluble products. In the heterogeneous reaction, little change was observed when altering the catalyst loading at higher temperatures, but lower temperatures and decreased catalyst loading produce accessible higher molecular weight oligomers. Homogeneous catalysis of the glycolysis reactions increases the rate of depolymerization, such that it is difficult to isolate oligomers with Mn > 1000 Daltons. The oligomers from heterogeneous reactions were used as reactants to form block copolymers with ethylene glycol, exemplifying their use as precursors in the production of value-added materials. These experiments, therefore, offer crucial insight into how reaction conditions can be readily tuned to produce target telechelic oligomers of PET.
000 Daltons) at a high yield (nearly 100%). At higher heterogeneous reaction temperatures (175 and 185 °C), the reaction rate increases, producing oligomers of a narrower molecular weight range (2000–5000 Daltons) with significant loss of the original PET, up to 40%, as water soluble products. In the heterogeneous reaction, little change was observed when altering the catalyst loading at higher temperatures, but lower temperatures and decreased catalyst loading produce accessible higher molecular weight oligomers. Homogeneous catalysis of the glycolysis reactions increases the rate of depolymerization, such that it is difficult to isolate oligomers with Mn > 1000 Daltons. The oligomers from heterogeneous reactions were used as reactants to form block copolymers with ethylene glycol, exemplifying their use as precursors in the production of value-added materials. These experiments, therefore, offer crucial insight into how reaction conditions can be readily tuned to produce target telechelic oligomers of PET.
Introduction
Polymeric materials are used in many aspects of society, as they are integral to the industrial, agricultural, and residential sectors. With this demand, polymer production has risen, which in turn leads to problems at their end of life, as most plastics end up in landfills or the environment. For instance, the Environmental Protection Agency (EPA) estimates that the U.S. produced ∼35000000 U.S. tons of plastics in 2018.1 Of this large amount of polymers produced, poly(ethylene terephthalate) (PET) accounts for about 15% of the produced materials and polyethylene 42%. Moreover, these two polymers make up a large portion of the recycled polymer materials reported by the EPA.1
There are a few methods that decrease the plastic waste burden, such as incineration or recycling.2 Incineration is often the easiest and most cost-effective but comes with the production of greenhouse gases and other undesirable byproducts, making this method suboptimal for several different types of polymers. Recycling falls into two main categories, mechanical and chemical recycling. Mechanical recycling is a method where the polymer is physically converted into usable precursors that can be reprocessed to produce new products. Mechanical recycling is a common method used in recycling plastics, but there are many factors that decrease the quality and value of the recycled materials. A major issue is that mechanical recycling results in degradation of the polymer every time it is recycled. This is due to the repeated melting and processing of the materials that leads to degradation of the polymer chains, and in turn decreases the mechanical properties of the resulting materials.3–7 The inclusion of additives during original fabrication and contaminants from waste streams also make this recycling method more difficult. Mechanical recycling of PET waste into usable bottles requires pristine, transparent, raw materials, however these materials can only be recycled about six times, after which the degradation renders the materials unusable.3,8
Another recycling method that can overcome the shortcomings of mechanical recycling is chemical recycling. Chemical recycling depolymerizes the used polymer back to useable precursors (usually monomer) that can be repolymerized to synthesize new materials. There are quite a few reactions that can be used to chemically recycle PET due to the presence of the ester group in the backbone, which is susceptible to reactions with different functional groups. The most common method is glycolysis, in which a glycol reacts with the ester linkage of PET to shorten the polymer chain and produce monomeric precursors that can be repolymerized to form PET.9–18 A common example of this reaction is the reaction of PET with ethylene glycol, which depolymerizes PET to bis(2-hydroxyethyl) terephthalate (BHET). BHET can be used as a monomer to repolymerize PET. Other reactants, and therefore reactions, can be employed, some of which include hydrolysis, methanolysis, or aminolysis, where each reaction will form a slightly different precursor that can be utilized to form new materials.3,9,19 One drawback to the chemical recycling of PET is the solubility of the polymer itself. PET is a highly recalcitrant material, which will not dissolve in most organic solvents. Thus, severe conditions are often required to realize these depolymerization reactions, such as harsh solvents and/or high temperatures.20 These reactions also require a catalyst for high conversion to the monomer, where organometallic catalysts are commonly employed, with the most prominent being zinc, lead, cobalt, or manganese acetates.10–12,21–23 Further studies have examined replacing these catalysts, as separation of the catalyst from the final product is often difficult. Alternative catalysts include nanoparticles, ionic liquids, light metal salts, zeolites, organocatalysts, and deep-eutectic solvents.13–15,24–26 Most recently, the use of protic ionic salts as catalysts in the depolymerization of PET has removed the metal component of the catalyst, resulting in products that are more benign.20,27
Blending polymers together provides an additional pathway to use polymers at their end of life. Unfortunately, most polymers do not thermodynamically mix with each other. Due to the low entropy of mixing, these blends will usually phase separate, which will create materials that have inferior properties to the original materials.28–30 Compatibilization is a method to improve the properties of phase separated polymer blends where incorporation of an interfacial modifier may improve the interface between the two polymers in the blend, creating a more robust material.31–33 Copolymers that consist of monomers that are identical to either blend homopolymer are common compatibilizers, where block copolymers show significant improvement in the properties of the resulting blends. The formation of these block copolymers can occur before they are added to a blend, or they may be formed by an in situ reaction that occurs in the blend, known as reactive compatibilization.34–45 For the block copolymers that are synthesized before they are added to the blends, the copolymer will migrate to the interface between the polymers during processing and mediate the interactions between the two polymers, stabilizing the blend. Reactive compatibilizers perform similarly, but are formed in situ in the blend, where each block contains mutually reactive groups such as carboxylic acids and epoxies, or carboxylic acid and amines. As these two functional homopolymers migrate to the blend’s interface, the groups react to form new bonds and create the copolymers only at the biphasic interface.
The simplest form of reactive compatibilizers are diblock copolymers, but through the addition of telechelic monomers, multiblock copolymers can be formed.46–57 These multiblock copolymers (MBCPs) have shown the ability to further improve the interfacial adhesion relative to diblock copolymers. This is mainly due to the compatibilizers crossing the interface multiple times, creating a “stitch” across the interface. Eastwood and Dadmun examined the incorporation of MBCPs as compatibilizers for blends of polystyrene and poly(methyl methacrylate).50 This study examined various block copolymer architectures of compatibilizers that were composed of styrene and methyl methacrylate. Their work found that the compatibilizers of pentablock architecture demonstrated the best reinforcement of the blend, followed by triblock, diblock, heptablock and then random copolymers. The study concludes that the ability to cross the interface multiple times increased the interfacial adhesion, but a critical molecular weight is required for the block lengths to adequately entangle with the homopolymers of the blend.
Blending of PET and HDPE is a potential method of recycling these materials. The production of useful materials from cheap sources can give new life to discarded materials. As these polymers are immiscible, there have been a number of studies that have examined the compatibilization of PET and HDPE blends using different block copolymer compatibilizers.58–68 A common compatibilizer is the styrene–ethylene-co-butene–styrene (SEBS) triblock copolymer, where studies have shown improvements in the ductility of the compatibilized blend at higher loadings of the compatibilizer (>10%). Other compatibilizers that have been examined include linear low-density PE (LLDPE) or low-density PE (LDPE) with reactive groups, such as maleic anhydride, glycidyl, or amino functional groups that can react with PET in situ during the processing of the blends. Todd et al. used tert-butyloxycarbonyl protected amino-telechelic PE (Boc-ATPE) as a compatibilizer for blends of PET/HDPE (90/10 wt%).59 The Boc-ATPE was added during melt mixing of PET and PE, where Boc-ATPE was converted to ATPE and the unprotected amino groups react with PET via ester aminolysis to form MBCPs of PET-b-PE. Even at low loadings (0.5 wt% ATPE), the authors saw a reduction in the size of the HDPE phase, as well as an increase in the elongation at break when compared to unmodified blends. Downsides to this reactive compatibilization scheme included that relatively long mixing times (∼10 minutes) were needed to form the compatibilizer, and the by-products of the deprotection of Boc-ATPE (carbon dioxide and isobutylene) form voids and defects during the melt mixing of the blends. Similarly, Nomura et al. synthesized multiblock copolymers composed of PET and PE via a coupling method using terephthaloyl chloride as a linker between hydroxyl terminated PE and PET.65 The incorporation of this pre-made compatibilizer into the PET/HDPE (80/20 wt%) blends at low loadings (i.e., 0.5 wt%) showed a reduction in size of the HDPE domains in the PET matrix, as well as a 30 times increase in the strain at break compared to neat blends. These two studies demonstrate the high potential of MBCPs to compatibilize PET/HDPE blends.
Though strides have been made to understand the compatibilization of PET and HDPE blends, all previous studies have used newly synthesized materials as compatibilizers. Though just a small amount of compatibilizer is needed, this still adds to the already high quantity of plastic materials produced. Chemically recycling polymers into usable compatibilizers or reactive precursors for compatibilization could overcome this hurdle as it would not increase the amount of produced polymers. This manuscript presents the results of a study that begins to address this concern, where the structural evolution of the products during the depolymerization of PET is monitored. Understanding this evolution of the chain structure offers an opportunity to intercept the intermediates of the depolymerization process that can be used to synthesize value-added products, including compatibilizers.
This will be achieved by monitoring the products from the glycolysis of PET as a function of reaction time, where the product oligomers should have hydroxyl functional groups on both ends that can react to form potential compatibilizers. The impact of reaction type (homogeneous vs. heterogeneous catalysis), temperature and catalyst loading on the progress of the depolymerization reaction of the PET is examined to offer insight to obtain targeted molecular weight oligomers at a significant yield. The products of the glycolysis depolymerization reaction will be used as reactants in reactive processing to determine their suitability in forming block copolymer compatibilizers. The results of this study will therefore offer researchers insight into the progression of the products in the glycolysis depolymerization of PET and how the products of this reaction may be used to form value-added materials.
Experimental
PET used in these experiments was obtained from commercial soft drink bottles that were washed with water, cut into flakes (4 cm × 4 cm) and dried in a vacuum oven at 85 °C overnight before use. Ethylene glycol, N-methyl-2-pyrrolidone (NMP) and zinc acetate dihydrate (Zn(Ac)2) were purchased from Fisher Scientific and used without further purification. Poly(ethylene glycol) (4000 g mol−1) was purchased from Sigma-Aldrich and used without further purification. 1,1,1,2,2,2-Hexafluoroisopropanol (HFIP) was purchased from Oakwood Chemical and deuterated chloroform (CDCl3) was purchased from Cambridge Isotope Labs, and both were used without further purification.The heterogeneous glycolysis reactions were adapted from procedures outlined in the literature.10,20 The general reaction includes the addition of dried PET (1 gram) to a vial with ethylene glycol (2.4 mL) and heating to 185, 175, or 165 °C. Once the temperature equilibrated, the catalyst (zinc acetate; molar ratios of 200:1 or 100:1 PET:Zn(Ac)2) was added to the vial and the reaction proceeded where vials were removed from the oil bath at 10 minute intervals over 80 minutes. As the stirring speed influences the glycolysis reactions, a constant speed of 600 rpm was used in all samples.11 Upon removal of the vials from the oil bath, they were quenched in room temperature water to stop the glycolysis depolymerization reaction. Subsequently, deionized water was added to the vials (15 mL) and heated to 90 °C for 20 minutes to remove all water-soluble reactants and products in the vial. The contents of the vial were filtered and the solid was washed with 10 mL of hot DI water to ensure complete removal of water-soluble materials. The recovered products of the reaction were then dried in a vacuum oven at 65 °C for 12 hours before weighing.
The homogeneous glycolysis reaction was adapted from procedures outlined in the literature.69 Five grams of dry PET was added to a round bottom flask with 48 mL of NMP, and 12 mL of EG. The flask was heated to 165 °C and 55 mg of Zn(Ac)2 was added to the flask. A 3 mL aliquot was taken every 2 minutes until 10 minutes had elapsed. Each aliquot was analyzed in a similar manner to the products from the heterogeneous reactions.
The mass and molecular weight of the products of the depolymerization reaction were determined, where NMR is used to determine the molecular weight of PET. Representative spectra of the starting PET material (Fig. S1†), 100:1 PET:Zn(Ac)2 heterogeneous glycolysis products (Fig. S2–S25†), and the homogeneous reactions (Fig. S26–S28†) are found in the ESI.† In this NMR experiment, the initial and depolymerized PET were dissolved in HFIP at a concentration of 20 mg mL−1. The samples were agitated for 2 days to completely dissolve PET. This PET solution (50 μL) was added to 800 μL of CDCl3 in an NMR tube. Proton NMR analysis was performed on a Jeol JM-ECZS 400 MHz. NMR data were analyzed using MestReNova 14.2.3. The baseline was manually corrected. The number average molecular weight (Mn) was calculated using the method outlined by Falkenstein et al.70 In this procedure, the NMR spectrum of PET has two sharp singlets that correspond to the four protons from the terephthalate ring and the four proton oxyethylene units at ∼8.1 and ∼4.7 ppm, respectively. A weak signal near 4.0 ppm corresponds to the methylene protons of the α adjacent to the hydroxyl end group of the PET chains. Assuming each PET chain has one hydroxyl chain end on average, the degree of polymerization (DP) is calculated from the ratio of the average integration of the peaks at 8.1 and 4.7 ppm and the hydroxyl end group peak at 4.0 ppm, then dividing by 4 (number of protons per repeat unit for the terephthalate or oxyethylene signals) to get the DP. Doing this for the starting materials gave a DP of 127 and 112, which corresponds to a Mn of 24400 and 21500 g mol−1. This result was verified by a commercial size exclusion chromatography analysis of the starting materials by PolyAnalytik (in the ESI as Fig. S29†).
The products of the 200:1 PET:Zn(Ac)2 heterogeneous depolymerization reaction at 175 °C for 20 minutes (Mn = 4900 g mol−1) were examined as reactants in the synthesis of block copolymers. In this reaction, the product of the depolymerization was added to a reaction flask with polyethylene glycol as described in the literature.71 Following the literature procedure, the flask was sealed and the atmosphere was replaced with nitrogen. The flask was then heated to 280 °C with stirring for 30 minutes. After the reaction was complete, the flask was cooled to room temperature where a brown solid was present. The solid was dissolved in HFIP and the product of this reaction was crashed out in water to remove any unreacted PEG. The material was filtered and dried in a vacuum oven at 60 °C overnight. Analysis of the product was performed using NMR and DSC. The NMR analysis mimics those described above for the pure PET materials. DSC analysis was performed on a TA Instruments Q-2000, with a heat ramp of 10 °C min−1 with a range from −85–285 °C. The samples were heat cycled once to remove thermal history and the second scan was used for analysis.
Results and discussion
Evolution of the chain structure during heterogeneous glycolysis of PET
A primary goal of this experiment is to elucidate the evolution of the chain structure during the glycolysis of PET. Fig. 1 plots the mass fraction of depolymerized PET from the heterogeneous catalysis reaction that is recovered after washing with water as a function of reaction time. Inspection of this figure shows that the amount of depolymerized PET that is recovered at any given reaction time decreases with an increase in reaction temperature, which is consistent with a faster depolymerization reaction at higher temperatures. It is interesting that at 165 °C, nearly all of the mass of the original PET is recovered even after 80 minutes. This implies that the depolymerization reaction is sufficiently slow to create oligomeric products that are suitably large that they are insoluble in water. On the other hand, when the reaction is run at 185 °C, as much as 35–40% of the original PET is not recovered at later reaction times (70–80 minutes). This indicates that a significant portion of PET has been depolymerized to create water soluble products under these conditions.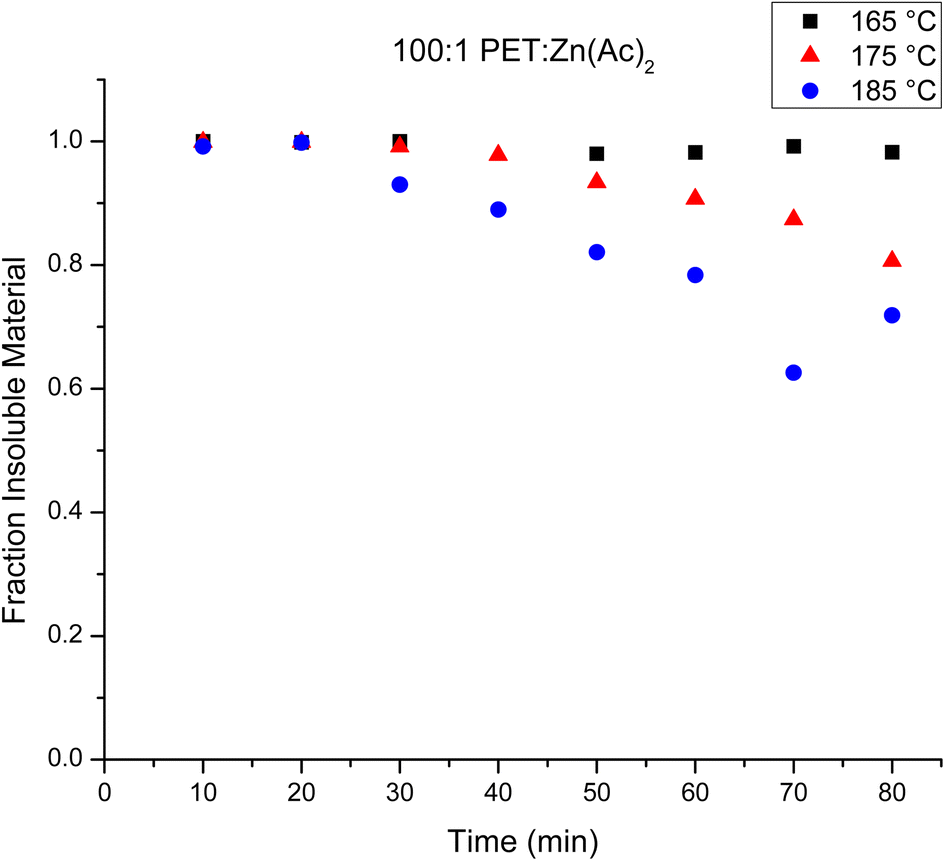 | ||
| Fig. 1 Fraction of water insoluble materials over the observed time for the heterogeneous reactions at 165 °C (black squares), 175 °C (red triangles) and 185 °C (blue circles). | ||
The number average molecular weights (Mn) of the products of the heterogeneous depolymerization reaction using a catalyst loading of 100:1 PET:Zn(Ac)2 are determined with NMR. The Mn of the products of the depolymerization reaction are plotted in Fig. 2 as a function of reaction time for the three reaction temperatures. Fig. 2 shows that the average chain length of the depolymerization products decreases rapidly in the first 10 minutes, but then gradually decreases further as the reaction proceeds. To account for a modest difference in the original molecular weight of the starting PET material, the relative change in the molecular weight of the depolymerization products as a function of reaction time is determined by dividing the Mn of the depolymerization product at different reaction times by the starting Mn. This normalized progress of the decrease in chain length during the depolymerization is plotted in Fig. 3 as a function of reaction time for the three reaction temperatures. Inspection of this figure further documents the acceleration of the depolymerization reaction with an increase in temperature, where the reactions at 185 and 175 °C reach a minimum chain that is ca. 10% of the original chain length after ∼40 minutes of reaction time, while the chain length of the products of the heterogeneous reaction at 165 °C does not level off at longer reaction times. It is interesting that the limiting molecular weight that is accessible from water washing at long reaction times is independent of the reaction temperature.
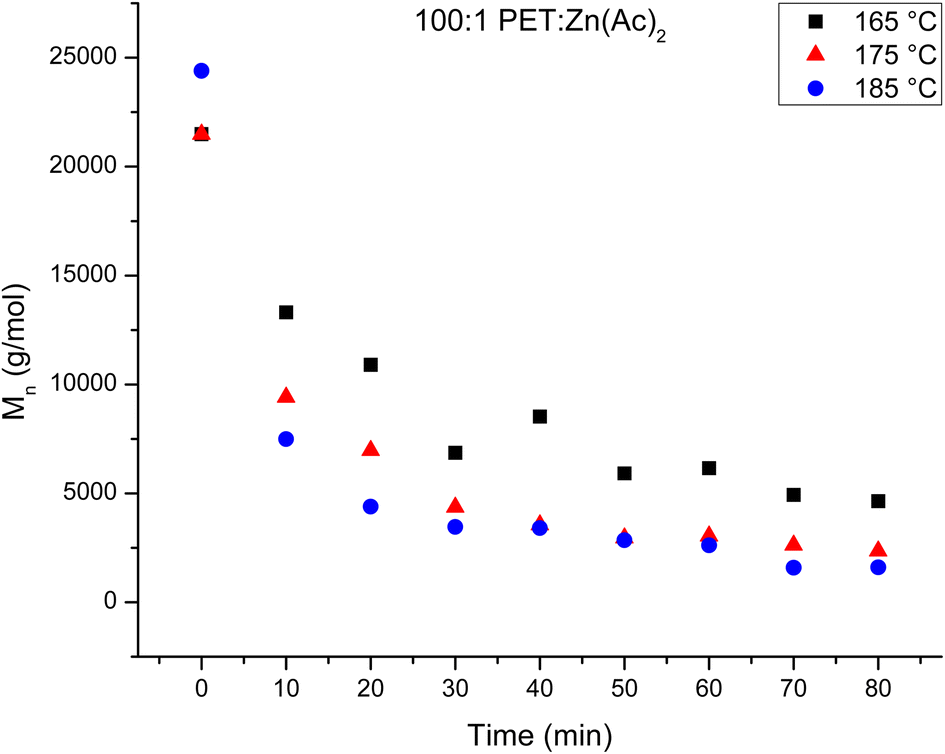 | ||
| Fig. 2 Decrease in Mn of PET as a function of glycolysis reaction time at different temperatures for the heterogeneous reactions. The reaction conditions are PET:Zn(Ac)2 of 100:1 at 165 °C (black squares), 175 °C (red triangles) and 185 °C (blue circles). | ||
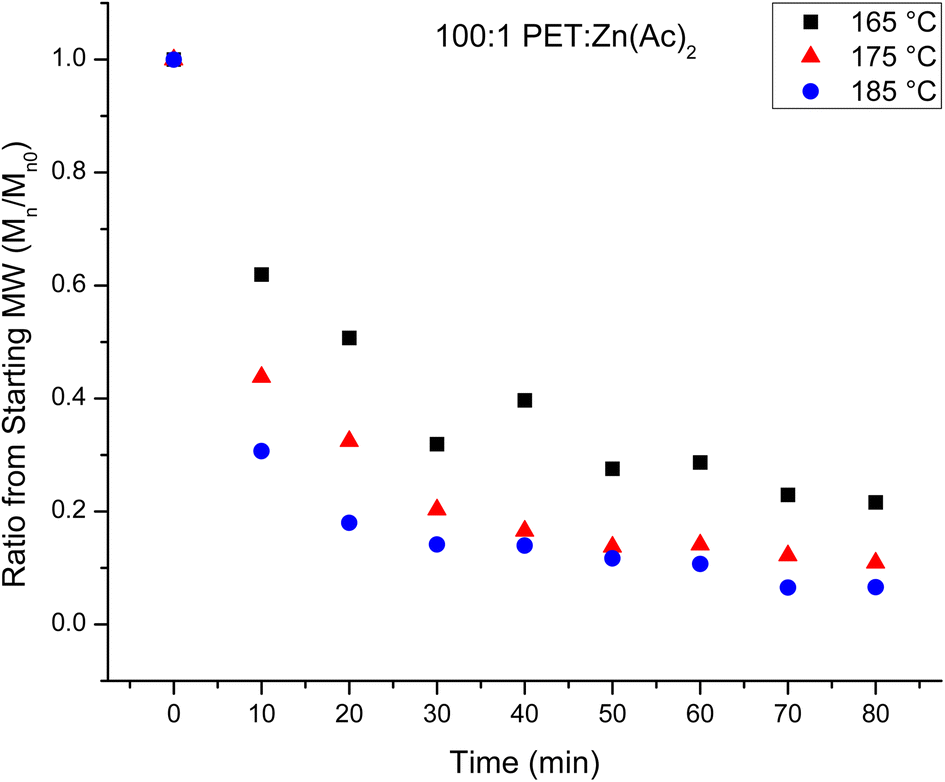 | ||
| Fig. 3 The normalized decrease in Mn as a function of glycolysis reaction time at different temperatures for the heterogeneous glycolysis. The reaction conditions are PET:Zn(Ac)2 of 100:1 at 165 °C (black squares), 175 °C (red triangles) and 185 °C (blue circles). | ||
The loading of catalyst impacts the rate of reaction, where it has been shown that higher loadings of catalyst result in faster depolymerization of PET.10 The impact of catalyst loading on the evolution of depolymerization products from the heterogeneous reaction was examined, where the catalyst loading was halved to 200:1 PET:Zn(Ac)2. The results were analyzed in a similar fashion to the 100:1 PET:Zn(Ac)2 samples. Fig. 4 plots the mass fraction of depolymerized PET that is recovered after washing with water as a function of reaction time for the three different temperatures. Halving the amount of catalyst appears to slow the heterogeneous reaction at 165 and 175 °C, as the amount of recovered PET remains relatively unchanged over the observed reaction time. At 185 °C, the rate of the reaction appears faster than that of the reaction using 100:1 PET:Zn(Ac)2 catalyst loading. For instance, the amount of PET recovered after 60 minutes is about 60% of the original PET in the 200:1 reaction, while ca. about 75–80% of the original PET is recovered after 60 minutes in the 100:1 reaction. Fig. 5 plots the Mn of the depolymerized PET as a function of reaction time at the three temperatures for the 200:1 catalyst loading. The variation in the amount of recovered PET as a function of reaction time can be correlated to this decrease in chain length with reaction. For the 165 °C and 175 °C reactions, the Mn of the recovered PET never decreases below 3000 g mol−1. Thus, all of the products are insoluble in water, and all of the depolymerized PET is recoverable over the observed reaction time. For the 185 °C reaction, the Mn of the recovered PET is ∼2900 g mol−1 after 40 minutes of reaction. Thus, further depolymerization creates shorter PET chains that become soluble in water and the product is lost during the water wash.
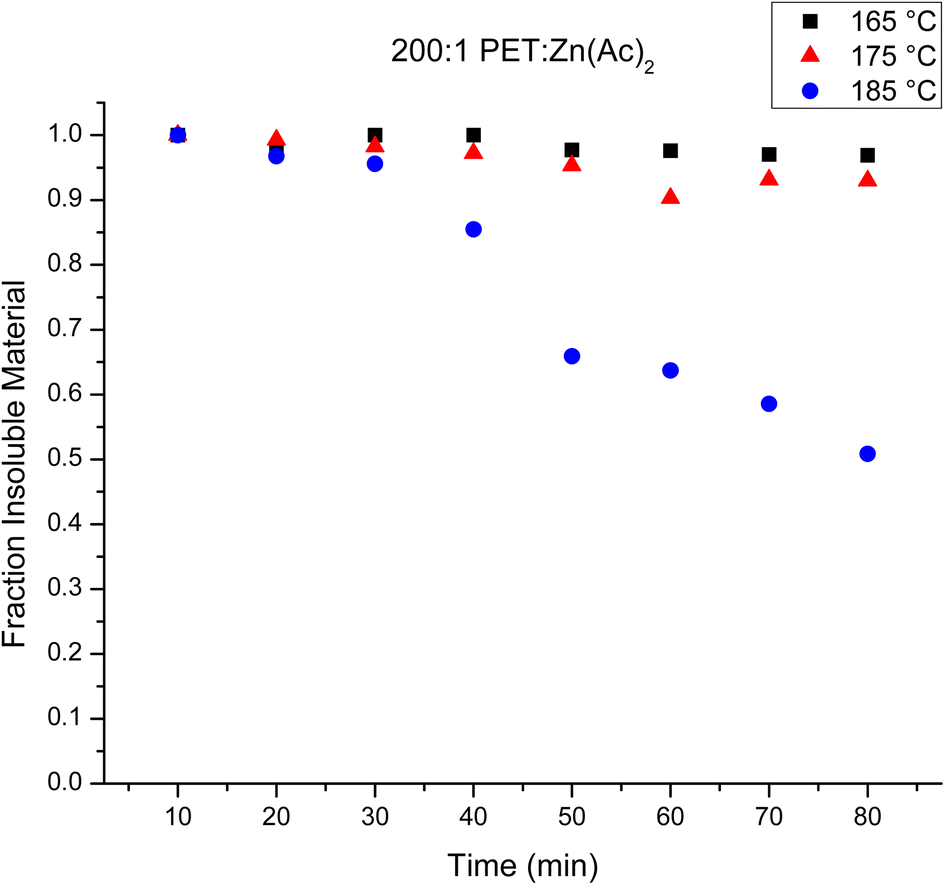 | ||
| Fig. 4 Mass fraction of water insoluble products from the heterogeneous reaction as a function of the observed reaction time using 200:1 PET:Zn(Ac)2 loading at 165 °C (black squares), 175 °C (red triangles) and 185 °C (blue circles). | ||
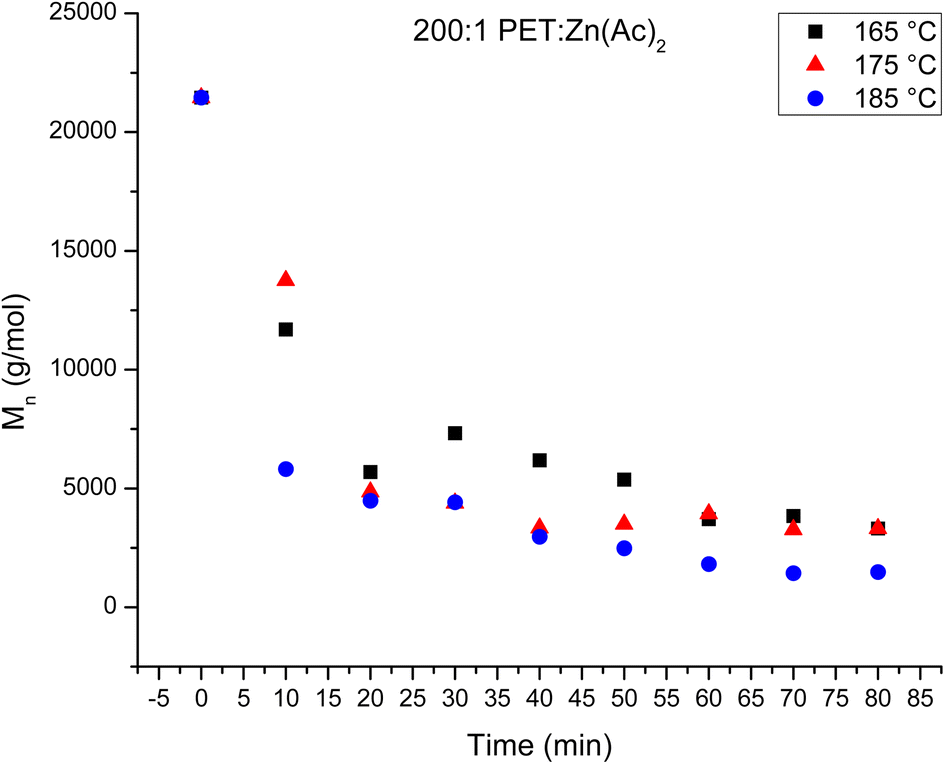 | ||
| Fig. 5 Decrease in Mn of the depolymerized PET as a function of reaction time at different temperatures for the heterogeneous glycolysis reaction. Reaction conditions are 200:1 PET:Zn(Ac)2 catalyst loading at 165 °C (black squares), 175 °C (red triangles) and 185 °C (blue circles). | ||
This initial analysis suggests that lowering the catalyst counterintuitively increases the reaction rate of the heterogeneous reaction. To critically evaluate this interpretation, Fig. 6 plots the normalized decrease in chain length as a function of reaction time for the two different catalyst loadings for all three temperatures to account for the variation in the original PET chain length. At 165 °C, there is a significant difference in the change in the chain length, where the higher catalyst loading reduces the chain length at a much faster rate than the lowest catalyst loading. As the temperature increases, this variation with catalyst loading is dampened, indicating that there is little impact on the rate of the depolymerization reaction by changing the catalyst loading at higher temperatures. Thus, it appears that monitoring the decrease in the normalized chain length with the reaction time is the most sensitive parameter to elucidate the impact of catalyst loading on the heterogeneous reaction progress.
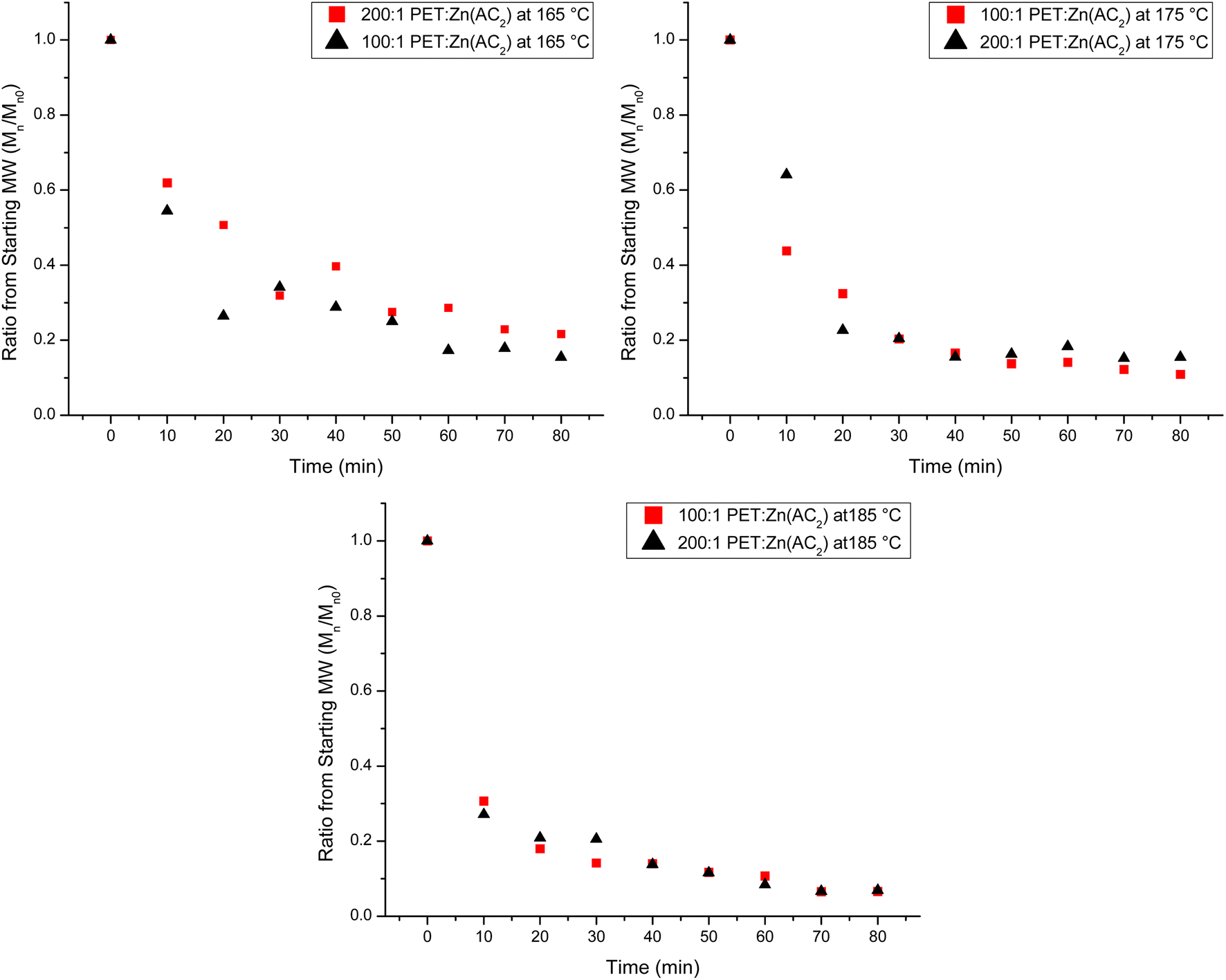 | ||
| Fig. 6 Normalized decrease in Mn as a function of reaction time for both catalyst loadings (100:1 (red squares) and 200:1 (black triangles) PET:Zn(Ac2)) at 165 °C (top-left), 175 °C (top-right), and 185 °C (bottom). | ||
Evolution of the chain structure during homogeneous glycolysis of PET
The homogeneous catalysis of the glycolysis of PET has also been studied, where previous work showed that dissolving the PET speeds up the reaction significantly. In a study by Liu et al., 82% of PET was converted to BHET at a reaction time of one minute using solvents such as dimethyl sulfoxide or NMP at 180 °C.69 On lowering the temperature, the reaction slows and the chain evolution may be observed in a similar fashion to the heterogeneous reactions. Thus, the homogeneous depolymerization of PET was monitored at 165 °C, which is the lowest temperature that produced a homogeneous solution. Fig. 7 plots the mass fraction of depolymerized PET that is recovered from the aliquots after washing with water as a function of reaction time. Comparison of these results to those of the heterogeneous reactions shows that PET depolymerizes much more quickly, with approximately 50% yield in 4 minutes. This decrease is accompanied by rapid chain depolymerization of PET, as shown in Fig. 8. Fig. 8 plots the Mn of the depolymerized PET from the homogeneous reaction as a function of reaction time. The homogeneous reaction leads to the rapid depolymerization of the PET chain in a matter of minutes. For instance, after two minutes of reaction, the molecular weight of the recovered PET is under 1000 g mol−1. This molecular weight does not change significantly with further reaction time to 4 and 6 minutes. After 8 and 10 minute reactions, the products exhibited no distinguishable end group peaks in the NMR spectra. The use of homogeneous reaction conditions, therefore, leads to the quick depolymerization of PET, even at lower temperatures, and these rapid changes do not allow for the interception of telechelic intermediates that can be used to produce value-added products.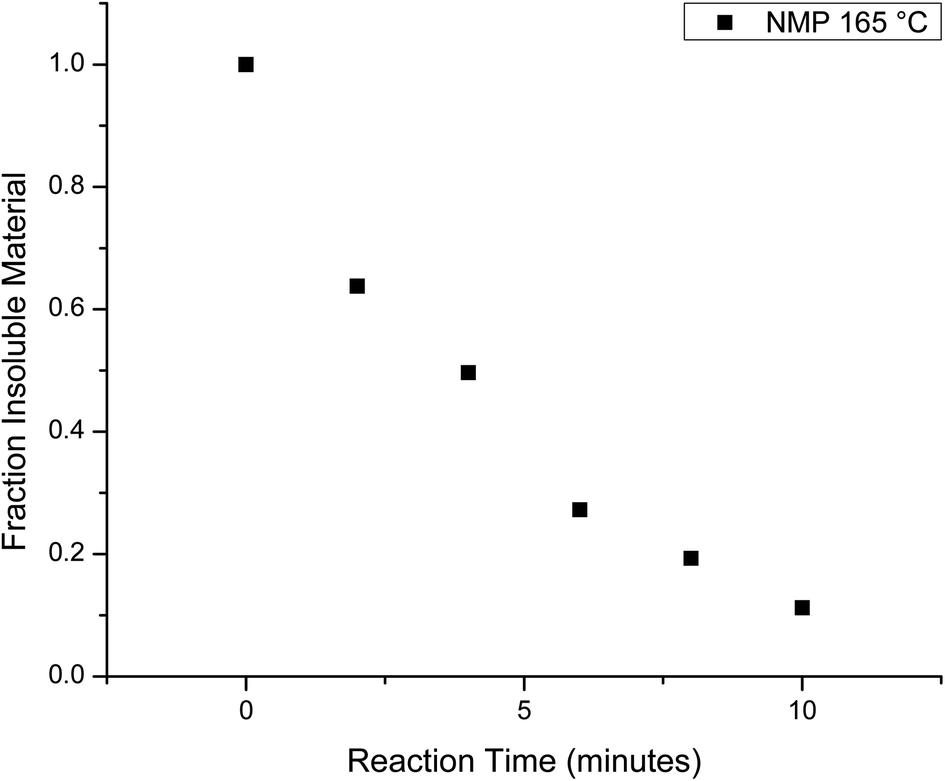 | ||
| Fig. 7 Mass fraction of water insoluble products as a function of the observed reaction for the homogeneous glycolysis in NMP at 165 °C. | ||
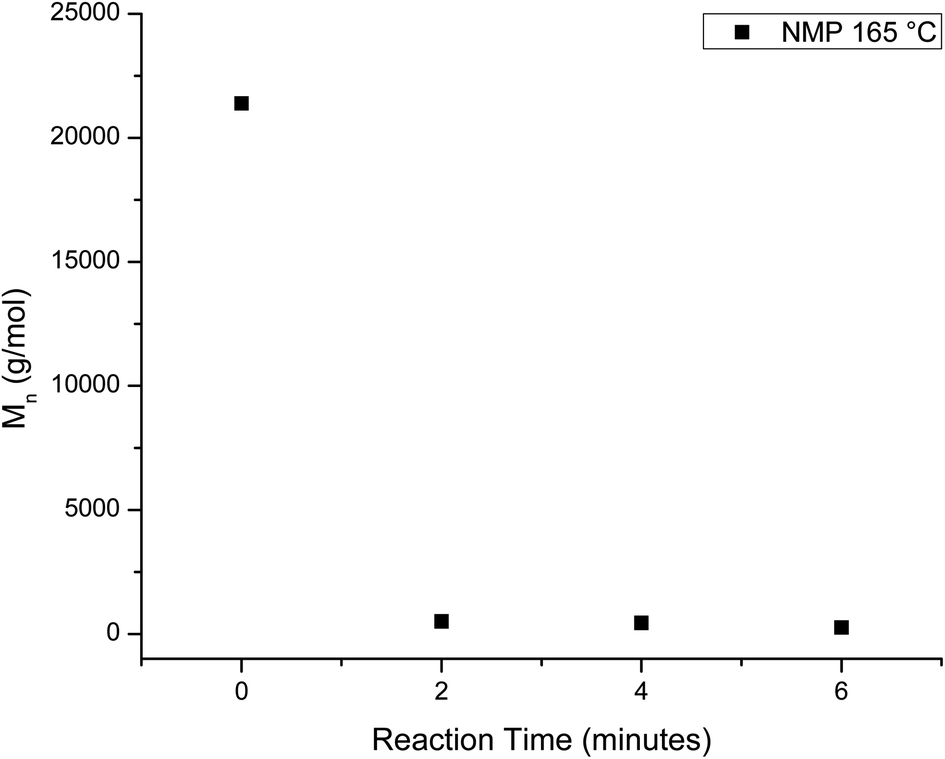 | ||
| Fig. 8 Decrease in Mn of the depolymerized PET as a function of reaction time for the homogeneous glycolysis in NMP at 165 °C. | ||
Block copolymer synthesis
We are interested in developing an understanding of how the intermediate products of the depolymerization reaction may be used as precursors to value-added products. One target is to use the telechelic oligomers that emerge from the depolymerization as reactants in the production of block copolymers. There are many reactions that can produce PET copolymers, but a method that can be readily scaled up is preferable. One approach that fits this criterion is the melt-mixing of PET telechelics with other polymers to form blocky copolymers. In our studies, we will examine the ability of the telechelic products of the depolymerization reaction to react with polyethylene glycol (PEG) to form blocky copolymers. The proposed melt-mixed reaction between PET and PEG is outlined in Fig. 9. This reaction has some similarity to the glycolysis depolymerization reaction, where the –OH of PEG will react with the carbonyl, replacing ethylene glycol in the PET backbone and inserting a PEG block.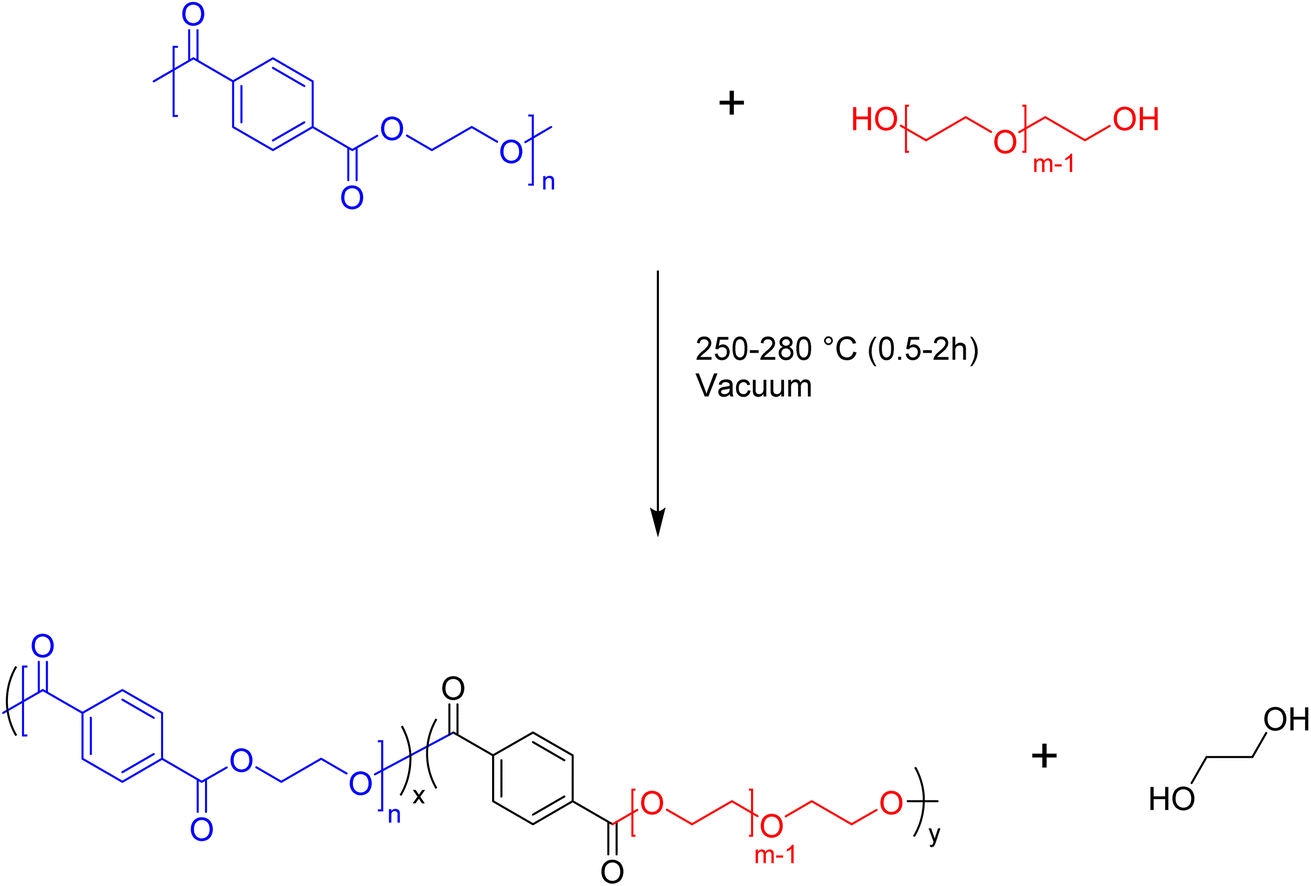 | ||
| Fig. 9 Reaction scheme for the synthesis of PET-b-PEG. | ||
The products of the reaction were analyzed using both NMR and DSC to evaluate the success of the proposed reaction. The NMR spectra of the PET oligomers from the 200:1 PET:Zn(Ac)2 heterogeneous depolymerization at 175 °C for 20 minutes, PEG starting materials and the product of the melt-mixed reaction are presented in Fig. 10. The spectrum for the product of the melt-mixed reaction exhibits not only peaks for PET at 8 and 4.7 ppm, but also the peaks for PEG at 3.6 and 3.8 ppm, which indicate incorporation of PEG blocks in the product.71 The recovery procedure of this product includes dissolution in HFIP, precipitation in water, and further heating and washing with hot water to remove residual, unreacted PEG. Thus, we expect that the peak at 3.6 ppm indicates the incorporation of PEG into a copolymer. To further verify the successful formation of the blocky copolymer during the melt-mixing, DSC curves of the starting materials and product are shown in Fig. 11. The DSC curve of the PET starting materials shows a melting peak near 246 °C, while that of PEG shows a melting peak at ∼60 °C. The DSC curve of the product of the melt-mixing shows two glass transitions, one at −48 °C and another at 7 °C, and two melt peaks, at 37 °C and 193 °C. These results are consistent with previous work that synthesized PEG/PET copolymers.72 Thermal analysis of the PET/PEG copolymers synthesized in these previous studies found two melting temperatures, one at ∼37 °C that is ascribed to the melting of the PEG block and one at ∼245 °C that is assigned to the melting of the PET block. The melting transition of the PEG block in our products is similar to that of the previous work, however, our melting peak for the PET block is lower than that reported by Hu et al. We ascribe this to incorporation of more PEG in our blocky copolymers than in those produced by Hu et al., where the copolymers synthesized by Hu et al. incorporate ca. 25% PEG. As our melt-mixed reaction involved a ∼1:1 molar ratio of PET:PEG in the melt-mixed reaction, we expect that the resulting block copolymers will incorporate more PEG. The decrease in the melting temperature of our PET block at ∼193 °C is consistent with the disruption of the PET domains due to the presence of more PEG blocks.71
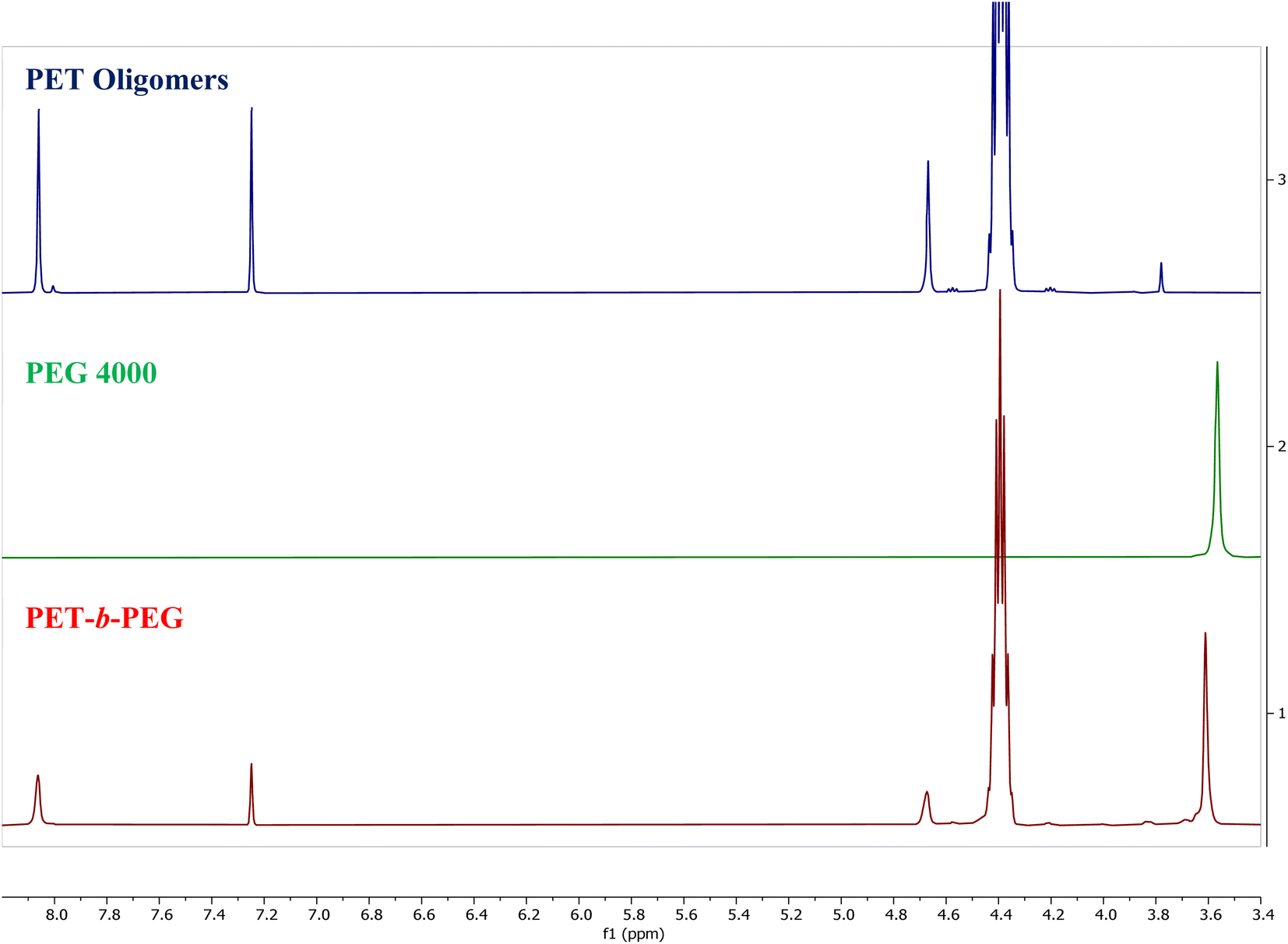 | ||
| Fig. 10 NMR spectra of PET oligomers from the 200:1 PET:Zn(Ac)2 heterogeneous glycolysis reaction at 175 °C for 20 minutes, PEG, and the melt mixed product. | ||
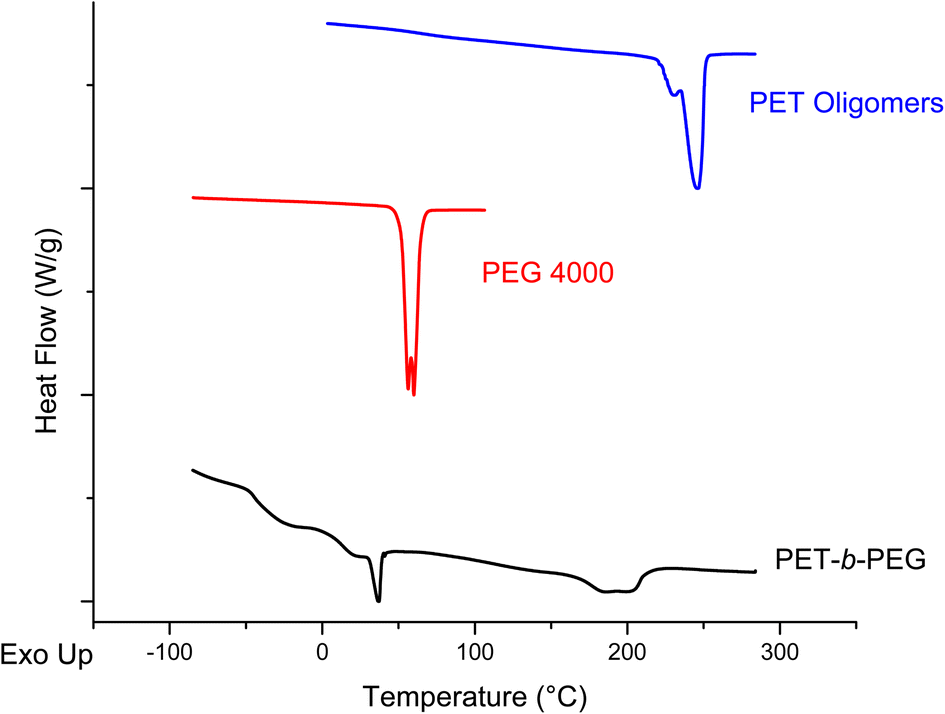 | ||
| Fig. 11 DSC thermograms for PET oligomers from the 200:1 PET:Zn(Ac)2 heterogeneous glycolysis reaction at 175 °C for 20 minutes (top trace), PEG 4000 (middle trace) and the PET-b-PEG product (bottom trace) from melt mixing the two materials. | ||
To rule out the presence of a PET/PEG blend in the melt-mixed product, the DSC curve of a dry blend of pure PET and PEG was also obtained. This DSC curve is shown in Fig. 12, showing the melt peaks of the two pure components. The melt peak of PEG is at 55 °C and that of PET presents at ∼250 °C. These curves are very similar to the sum of the DSC curves of the individual components, verifying that there is no reaction between these materials during the DSC experiment. Comparing the black trace for the melt mixed product in Fig. 11 and the curve in Fig. 12 shows drastic differences, further supporting that the product of the melt mixed reaction is a blocky copolymer and not an unreacted blend of the two reactants.
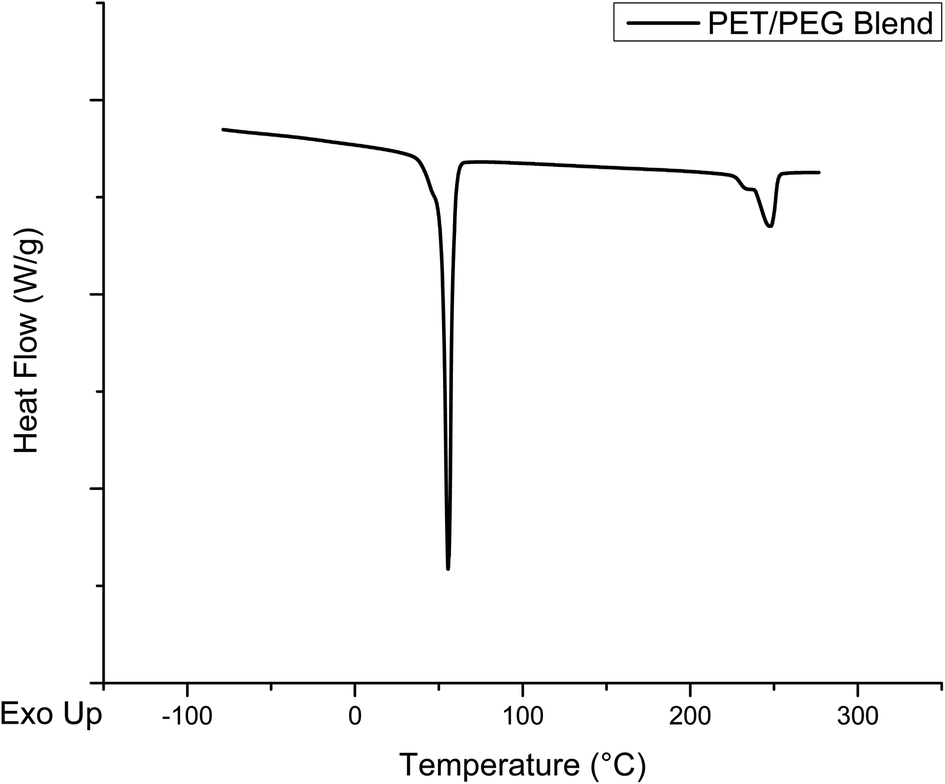 | ||
| Fig. 12 DSC thermogram of a blend of glycolyzed PET material and PEG 4000. | ||
The results presented above elucidate the evolution of the chain structure during the heterogeneous depolymerization of PET by glycolysis catalyzed by zinc acetate. From these experiments, the effects of catalyst loading, reaction time, and temperature on the evolution of the chain structure and percent of recovered PET are revealed. Altering the temperature appears to have the largest impact on the evolution of chain length of the products and yield. Decreasing the catalyst loading shows insignificant variation in the decrease in chain length with reaction time for higher temperatures, but at 165 °C, the decreased catalyst loading provides avenues to extract oligomers of higher Mn than when using the higher catalyst loading. Tuning these parameters in conjunction with the reaction time offers pathways to produce telechelic oligomers with target molecular weights using heterogeneous conditions. The homogeneous glycolysis reaction in a solvent, such as NMP, leads to rapid depolymerization of PET. The oligomers produced from the heterogeneous reaction can then be used in the production of block copolymers. These oligomers can be mixed with a broad range of diols, where transesterification reactions produce blocky copolymers that may be useful in the compatibilization of PET blends.
Conclusion
This work monitors the evolution of the chain structure in the depolymerization of PET by glycolysis. The impact of catalyst loading, reaction type, reaction time, and temperature on the evolution of product chain length and yield was established. These results show that for the heterogeneous reaction, at lower temperatures (165 °C), the rate is sufficiently slow to access a broad range of molecular weight products (3000–10000 Daltons) in high yield (nearly 100%). At higher temperatures (175 and 185 °C) the reaction is significantly faster, such that the resulting oligomers produced are of a narrower molecular weight range (2000–5000 Daltons) with significant loss of original PET, up to 40%, as water soluble products. Lowering the catalyst loading results in little change to the reaction products at higher temperatures, but produces higher molecular weight oligomers at lower temperatures. The use of NMP as a solvent speeds up the homogeneous reaction significantly, where the depolymerization occurred so quickly that it is difficult to intercept oligomers of usable molecular weights. The products from the heterogeneous reactions are further utilized in reactions to produce blocky copolymers. Through melt mixing the depolymerized PET with PEG, blocky copolymers were successfully synthesized. These experiments reveal the impact of temperature, catalyst loading, and reaction time on the properties of the oligomeric products of glycolysis, and how these parameters can be tuned to produce target oligomers of PET. These materials can then be used to produce blocky copolymers, which may prove useful as compatibilizers of PET blends.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation, Polymers Program, Division of Materials Research (DMR-2104982).References
- US EPA, O. Plastics: Material-Specific Data. US EPA, https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/plastics-material-specific-data, accessed 2020-09-17.
- S. J. Huang, Polymer Waste Management–Biodegradation, Incineration, and Recycling, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32(4), 593–597, DOI:10.1080/10601329508010272.
- D. Damayanti and H.-S. Wu, Strategic Possibility Routes of Recycled PET, Polymers, 2021, 13(9), 1475, DOI:10.3390/polym13091475.
- K. Kaiser, M. Schmid and M. Schlummer, Recycling of Polymer-Based Multilayer Packaging: A Review, Recycling, 2018, 3(1), 1, DOI:10.3390/recycling3010001.
- R. L. Smith, S. Takkellapati and R. C. Riegerix, Recycling of Plastics in the United States: Plastic Material Flows and Polyethylene Terephthalate (PET) Recycling Processes, ACS Sustainable Chem. Eng., 2022, 10(6), 2084–2096, DOI:10.1021/acssuschemeng.1c06845.
- J. Hopewell, R. Dvorak and E. Kosior, Plastics Recycling: Challenges and Opportunities, Philos. Trans. R. Soc., B, 2009, 364(1526), 2115–2126, DOI:10.1098/rstb.2008.0311.
- Z. O. G. Schyns and M. P. Shaver, Mechanical Recycling of Packaging Plastics: A Review, Macromol. Rapid Commun., 2021, 42(3), 2000415, DOI:10.1002/marc.202000415.
- Plastic has a problem; is chemical recycling the solution?. Chemical & Engineering News, https://cen.acs.org/environment/recycling/Plastic-problem-chemical-recycling-solution/97/i39, accessed 2022-06-13.
- Q. F. Yue, C. X. Wang, L. N. Zhang, Y. Ni and Y. X. Jin, Glycolysis of Poly(Ethylene Terephthalate) (PET) Using Basic Ionic Liquids as Catalysts, Polym. Degrad. Stab., 2011, 96(4), 399–403, DOI:10.1016/j.polymdegradstab.2010.12.020.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Chemical Recycling of Post-Consumer PET Wastes by Glycolysis in the Presence of Metal Salts, Polym. Degrad. Stab., 2010, 95(6), 1022–1028, DOI:10.1016/j.polymdegradstab.2010.03.007.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Kinetics of Catalytic Glycolysis of PET Wastes with Sodium Carbonate, Chem. Eng. J., 2011, 168(1), 312–320, DOI:10.1016/j.cej.2011.01.031.
- C.-H. Chen, C.-Y. Chen, Y.-W. Lo, C.-F. Mao and W.-T. Liao, Studies of Glycolysis of Poly(Ethylene Terephthalate) Recycled from Postconsumer Soft-Drink Bottles. I. Influences of Glycolysis Conditions, J. Appl. Polym. Sci., 2001, 80(7), 943–948, DOI:10.1002/app.1174.
- M. Imran, K. G. Lee, Q. Imtiaz, B.-K. Kim, M. Han, B. G. Cho and D. H. Kim, Metal-Oxide-Doped Silica Nanoparticles for the Catalytic Glycolysis of Polyethylene Terephthalate, J. Nanosci. Nanotechnol., 2011, 11(1), 824–828, DOI:10.1166/jnn.2011.3201.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Glycolysis of Poly(Ethylene Terephthalate) Catalyzed by the Lewis Base Ionic Liquid [Bmim][OAc], Ind. Eng. Chem. Res., 2014, 53(48), 18443–18451, DOI:10.1021/ie503677w.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Deep Eutectic Solvents as Highly Active Catalysts for the Fast and Mild Glycolysis of Poly(Ethylene Terephthalate)(PET), Green Chem., 2015, 17(4), 2473–2479, 10.1039/C4GC02401J.
- J.-W. Chen and L.-W. Chen, The Glycolysis of Poly(Ethylene Terephthalate), J. Appl. Polym. Sci., 1999, 73(1), 35–40, DOI:10.1002/(SICI)1097-4628(19990705)73:1<35::AID-APP4>3.0.CO;2-W.
- G. Colomines, J.-J. Robin and G. Tersac, Study of the Glycolysis of PET by Oligoesters, Polymer, 2005, 46(10), 3230–3247, DOI:10.1016/j.polymer.2005.02.047.
- A. Sheel and D. Pant, 4-Chemical Depolymerization of PET Bottles via Glycolysis, in Recycling of Polyethylene Terephthalate Bottles, ed. Thomas S., Rane A., Kanny K., Vk A. and Thomas M. G., Plastics Design Library, William Andrew Publishing, 2019, pp. 61–84, DOI:10.1016/B978-0-12-811361-5.00004-3.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, Current Developments in Chemical Recycling of Post-Consumer Polyethylene Terephthalate Wastes for New Materials Production: A Review, J. Cleaner Prod., 2019, 225, 1052–1064, DOI:10.1016/j.jclepro.2019.04.019.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Organocatalysed Depolymerisation of PET in a Fully Sustainable Cycle Using Thermally Stable Protic Ionic Salt, Green Chem., 1396, 20(6), 1205–1212, 10.1039/C7GC03396F.
- Y. Chujo, H. Kobayashi and Y. Yamashita, Synthesis of Aromatic Dicarboxyl-Terminated Poly(Methyl Methacrylate) Macromonomers, J. Polym. Sci., Part A: Polym. Chem., 1989, 27(6), 2007–2014, DOI:10.1002/pola.1989.080270621.
- Y. Yang, Y. Lu, H. Xiang, Y. Xu and Y. Li, Study on Methanolytic Depolymerization of PET with Supercritical Methanol for Chemical Recycling, Polym. Degrad. Stab., 2002, 75(1), 185–191, DOI:10.1016/S0141-3910(01)00217-8.
- U. R. Vaidya and V. M. Nadkarni, Unsaturated Polyester Resins from Poly(Ethylene Terephthalate) Waste. 1. Synthesis and Characterization, Ind. Eng. Chem. Res., 1987, 26(2), 194–198, DOI:10.1021/ie00062a003.
- L. Bartolome, M. Imran, B. G. Cho, W. A. Al-Masry and D. H. Kim, Recent Developments in the Chemical Recycling of PET, in Material Recycling – Trends and Perspectives, IntechOpen, 2012, DOI:10.5772/33800.
- S. R. Shukla, V. Palekar and N. Pingale, Zeolite Catalyzed Glycolysis of Poly(Ethylene Terephthalate) Bottle Waste, J. Appl. Polym. Sci., 2008, 110(1), 501–506, DOI:10.1002/app.28656.
- Z. Fehér, J. Kiss, P. Kisszékelyi, J. Molnár, P. Huszthy, L. Kárpáti and J. Kupai, Optimisation of PET Glycolysis by Applying Recyclable Heterogeneous Organocatalysts, Green Chem., 2022, 24(21), 8447–8459, 10.1039/D2GC02860C.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, Selective Chemical Upcycling of Mixed Plastics Guided by a Thermally Stable Organocatalyst, Angew. Chem., Int. Ed. Engl., 2021, 60, 6710–6717, DOI:10.1002/anie.202014860.
- N. C. Liu and W. E. Baker, Reactive Polymers for Blend Compatibilization, Adv. Polym. Technol., 1992, 11(4), 249–262, DOI:10.1002/adv.1992.060110403.
- D. Feldman, Polyblend Compatibilization, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 42(5), 587–605, DOI:10.1081/MA-200056331.
- C. Koning, M. Van Duin, C. Pagnoulle and R. Jerome, Strategies for Compatibilization of Polymer Blends, Prog. Polym. Sci., 1998, 23(4), 707–757, DOI:10.1016/S0079-6700(97)00054-3.
- H. E. H. Meijer, P. J. Lemstra and P. H. M. Elemans, Structured Polymer Blends, Makromol. Chem., Macromol. Symp., 1988, 16(1), 113–135, DOI:10.1002/masy.19880160109.
- R. Fayt, P. Hadjiandreou and P. Teyssie, Molecular Design of Multicomponent Polymer Systems. VII. Emulsifying Effect of Poly(Ethylene–b–Styrene) Copolymer in High-Density Polyethylene/Polystyrene Blends, J. Polym. Sci., Polym. Chem. Ed., 1985, 23(2), 337–342, DOI:10.1002/pol.1985.170230209.
- Immiscible Polymer Blends, https://www.pslc.ws/macrog/iblend.htm, accessed 2021-04-22.
- L. Leibler, Emulsifying Effects of Block Copolymers in Incompatible Polymer Blends, Makromol. Chem., Macromol. Symp., 1988, 16(1), 1–17, DOI:10.1002/masy.19880160103.
- S. H. Anastasiadis, I. Gancarz and J. T. Koberstein, Compatibilizing Effect of Block Copolymers Added to the Polymer/Polymer Interface, Macromolecules, 1989, 22(3), 1449–1453, DOI:10.1021/ma00193a074.
- J. Noolandi, Multiblock Copolymers as Polymeric Surfactants: Are “Pancakes” Better than “Dumbbells”, Macromol. Theory Simul., 1992, 1(5), 295–298, DOI:10.1002/mats.1992.040010503.
- W. Hu, J. T. Koberstein, J. P. Lingelser and Y. Gallot, Interfacial Tension Reduction in Polystyrene/Poly(Dimethylsiloxane) Blends by the Addition of Poly(Styrene-b-Dimethylsiloxane), Macromolecules, 1995, 28(15), 5209–5214, DOI:10.1021/ma00119a007.
- B. Crist and A. R. Nesarikar, Coarsening in Polyethylene-Copolymer Blends, Macromolecules, 1995, 28(4), 890–896, DOI:10.1021/ma00108a014.
- U. Sundararaj and C. W. Macosko, Drop Breakup and Coalescence in Polymer Blends: The Effects of Concentration and Compatibilization, Macromolecules, 1995, 28(8), 2647–2657, DOI:10.1021/ma00112a009.
- C. W. Macosko, P. Guégan, A. K. Khandpur, A. Nakayama, P. Marechal and T. Inoue, Compatibilizers for Melt Blending: Premade Block Copolymers, Macromolecules, 1996, 29(17), 5590–5598, DOI:10.1021/ma9602482.
- P. Teyssié, R. Fayt and R. Jérôme, Study and Control of Polymer Blends Morphology and Related Properties, Makromol. Chem., Macromol. Symp., 1988, 16(1), 41–56, DOI:10.1002/masy.19880160105.
- C. Creton, E. J. Kramer, C. Y. Hui and H. R. Brown, Failure Mechanisms of Polymer Interfaces Reinforced with Block Copolymers, Macromolecules, 1992, 25(12), 3075–3088, DOI:10.1021/ma00038a010.
- H. K. Jeon, J. Zhang and C. W. Macosko, Premade vs. Reactively Formed Compatibilizers for PMMA/PS Melt Blends, Polymer, 2005, 46(26), 12422–12429, DOI:10.1016/j.polymer.2005.10.125.
- K. Cho, O. A. Tae, S. R. Hyun and H. S. Kyung, Mechanical Effects According to the Type of Poly(Styrene-Co-Methyl Methacrylate) Copolymers at Polystyrene/Poly(Methyl Methacrylate) Interfaces, Polymer, 1996, 37(21), 4849–4852, DOI:10.1016/S0032-3861(96)00356-4.
- M. Dadmun, Effect of Copolymer Architecture on the Interfacial Structure and Miscibility of a Ternary Polymer Blend Containing a Copolymer and Two Homopolymers, Macromolecules, 1996, 29(11), 3868–3874, DOI:10.1021/ma951500t.
- M. Sikka, N. N. Pellegrini, E. A. Schmitt and K. I. Winey, Modifying a Polystyrene/Poly(Methyl Methacrylate) Interface with Poly(Styrene-Co-Methyl Methacrylate) Random Copolymers, Macromolecules, 1997, 30(3), 445–455, DOI:10.1021/ma961302h.
- C.-A. Dai, K. D. Jandt, D. R. Iyengar, N. L. Slack, K. H. Dai, W. B. Davidson, E. J. Kramer and C.-Y. Hui, Strengthening Polymer Interfaces with Triblock Copolymers, Macromolecules, 1997, 30(3), 549–560, DOI:10.1021/ma960396s.
- M. J. Arlen and M. D. Dadmun, The Reinforcement of Polystyrene and Poly(Methyl Methacrylate) Interfaces Using Alternating Copolymers, Polymer, 2003, 44, 6883–6889 CrossRef CAS.
- A. C. Balazs, C. P. Siemasko and C. W. Lantman, Monte Carlo Simulations for the Behavior of Multiblock Copolymers at a Penetrable Interface, J. Chem. Phys., 1991, 94(2), 1653–1663, DOI:10.1063/1.460715.
- E. A. Eastwood and M. D. Dadmun, Multiblock Copolymers in the Compatibilization of Polystyrene and Poly(Methyl Methacrylate) Blends: Role of Polymer Architecture, Macromolecules, 2002, 35(13), 5069–5077, DOI:10.1021/ma011701z.
- E. A. Eastwood and M. D. Dadmun, Compatibilization of Poly(Vinyl Chloride) and Polyolefin Elastomer Blends with Multiblock/Blocky Chlorinated Polyethylenes, Polymer, 2002, 43(25), 6707–6717, DOI:10.1016/S0032-3861(02)00639-0.
- P. Guegan, C. W. Macosko, T. Ishizone, A. Hirao and S. Nakahama, Kinetics of Chain Coupling at Melt Interfaces, Macromolecules, 1994, 27(18), 4993–4997, DOI:10.1021/ma00096a022.
- C. A. Orr, A. Adedeji, A. Hirao, F. S. Bates and C. W. Macosko, Flow-Induced Reactive Self-Assembly, Macromolecules, 1997, 30(4), 1243–1246, DOI:10.1021/ma961574k.
- J. S. Schulze, J. J. Cernohous, A. Hirao, T. P. Lodge and C. W. Macosko, Reaction Kinetics of End-Functionalized Chains at a Polystyrene/Poly(Methyl Methacrylate) Interface, Macromolecules, 2000, 33(4), 1191–1198, DOI:10.1021/ma9911344.
- C. P. O'Brien, J. K. Rice and M. D. Dadmun, Reactive Processing with Difunctional Oligomers to Compatibilize Polymer Blends, Eur. Polym. J., 2004, 40(7), 1515–1523, DOI:10.1016/j.eurpolymj.2004.01.023.
- E. Ashcraft, H. Ji, J. Mays and M. Dadmun, A Novel Reactive Processing Technique: Using Telechelic Polymers To Reactively Compatibilize Polymer Blends, ACS Appl. Mater. Interfaces, 2009, 1(10), 2163–2173, DOI:10.1021/am900333y.
- E. Ashcraft, H. Ji, J. Mays and M. Dadmun, Grafting Polymer Loops onto Functionalized Nanotubes: Monitoring Grafting and Loop Formation, Macromol. Chem. Phys., 2011, 212(5), 465–477, DOI:10.1002/macp.201000557.
- N. K. Kalfoglou, D. S. Skafidas, J. K. Kallitsis, J.-C. Lambert and L. Van der Stappen, Comparison of Compatibilizer Effectiveness for PET/HDPE Blends, Polymer, 1995, 36(23), 4453–4462, DOI:10.1016/0032-3861(95)96853-Z.
- A. D. Todd, R. J. McEneany, V. A. Topolkaraev, C. W. Macosko and M. A. Hillmyer, Reactive Compatibilization of Poly(Ethylene Terephthalate) and High-Density Polyethylene Using Amino-Telechelic Polyethylene, Macromolecules, 2016, 49(23), 8988–8994, DOI:10.1021/acs.macromol.6b02080.
- B. Boutevin, J. M. Lusinchi, Y. Pietrasanta and J. Robin, J. Improving Poly(Ethylene Terephthalate)/High-Density Polyethylene Blends by Using Graft Copolymers, Polym. Eng. Sci., 1996, 36(6), 879–884, DOI:10.1002/pen.10475.
- S. S. Dagli and K. M. Kamdar, Effects of Component Addition Protocol on the Reactive Compatibilization of HDPE/PET Blends, Polym. Eng. Sci., 1994, 34(23), 1709–1719, DOI:10.1002/pen.760342302.
- S. Kim, C. E. Park, J. H. An, D. Lee and J. Kim, The Effect of Functional Group Content on Poly(Ethylene Terephthalate)/High Density Polyethylene Blends Compatibilized with Poly(Ethylene-Co-Acrylic Acid), Polym. J., 1997, 29(3), 274–278, DOI:10.1295/polymj.29.274.
- J. M. Lusinchi, B. Boutevin, N. Torres and J. Robin, J. In Situ Compatibilization of HDPE/PET Blends, J. Appl. Polym. Sci., 2001, 79(5), 874–880, DOI:10.1002/1097-4628(20010131)79:5<874::AID-APP120>3.0.CO;2-B.
- S. Mbarek, M. Jaziri, Y. Chalamet and C. Carrot, Effect of the Viscosity Ratio on the Morphology and Properties of PET/HDPE Blends with and without Compatibilization, J. Appl. Polym. Sci., 2010, 117(3), 1683–1694, DOI:10.1002/app.32050.
- K. Nomura, X. Peng, H. Kim, K. Jin, H. J. Kim, A. F. Bratton, C. R. Bond, A. E. Broman, K. M. Miller and C. J. Ellison, Multiblock Copolymers for Recycling Polyethylene–Poly(Ethylene Terephthalate) Mixed Waste, ACS Appl. Mater. Interfaces, 2020, 12(8), 9726–9735, DOI:10.1021/acsami.9b20242.
- Y. Pietrasanta, J.-J. Robin, N. Torres and B. Boutevin, Reactive Compatibilization of HDPE/PET Blends by Glycidyl Methacrylate Functionalized Polyolefins, Macromol. Chem. Phys., 1999, 200(1), 142–149, DOI:10.1002/(SICI)1521-3935(19990101)200:1<142::AID-MACP142>3.0.CO;2-W.
- M. Pracella, L. Rolla, D. Chionna and A. Galeski, Compatibilization and Properties of Poly(Ethylene Terephthalate)/Polyethylene Blends Based on Recycled Materials, Macromol. Chem. Phys., 2002, 203(10–11), 1473–1485, DOI:10.1002/1521-3935(200207)203:10/11<1473::AID-MACP1473>3.0.CO;2-4.
- M. Pluta, Z. Bartczak, A. Pawlak, A. Galeski and M. Pracella, Phase Structure and Viscoelastic Properties of Compatibilized Blends of PET and HDPE Recyclates, J. Appl. Polym. Sci., 2001, 82(6), 1423–1436, DOI:10.1002/app.1980.
- B. Liu, X. Lu, Z. Ju, P. Sun, J. Xin, X. Yao, Q. Zhou and S. Zhang, Ultrafast Homogeneous Glycolysis of Waste Polyethylene Terephthalate via a Dissolution-Degradation Strategy, Ind. Eng. Chem. Res., 2018, 57(48), 16239–16245, DOI:10.1021/acs.iecr.8b03854.
- P. Falkenstein, D. Gräsing, P. Bielytskyi, W. Zimmermann, J. Matysik, R. Wei and C. Song, UV Pretreatment Impairs the Enzymatic Degradation of Polyethylene Terephthalate, Front. Microbiol., 2020, 11, 689, DOI:10.3389/fmicb.2020.00689.
- Z. Qian, S. Li, Y. He and X. Liu, Synthesis and in Vitro Degradation Study of Poly(Ethylene Terephthalate)/Poly(Ethylene Glycol) (PET/PEG) Multiblock Copolymer, Polym. Degrad. Stab., 2004, 83(1), 93–100, DOI:10.1016/S0141-3910(03)00229-5.
- J. Hu, H. Yu, Y. Chen and M. Zhu, Study on Phase-Change Characteristics of PET-PEG Copolymers, J. Macromol. Sci., Part B: Phys., 2006, 45(4), 615–621, DOI:10.1080/00222340600770210.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07467b |
| This journal is © The Royal Society of Chemistry 2023 |