
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium–rhenium and ruthenium–palladium supramolecular photocatalysts for photoelectrocatalytic CO2 and H+ reduction†‡
Joshua K. G.
Karlsson§
 a,
Florian J. R.
Cerpentier§
b,
Ralte
Lalrempuia
c,
Martin V.
Appleby
d,
James D.
Shipp
d,
Dimitri
Chekulaev
d,
Owen
Woodford
a,
Julia A.
Weinstein
d,
Mary T.
Pryce
*b and
Elizabeth A.
Gibson
*a
a,
Florian J. R.
Cerpentier§
b,
Ralte
Lalrempuia
c,
Martin V.
Appleby
d,
James D.
Shipp
d,
Dimitri
Chekulaev
d,
Owen
Woodford
a,
Julia A.
Weinstein
d,
Mary T.
Pryce
*b and
Elizabeth A.
Gibson
*a
aChemistry, Energy Materials Laboratory, School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, UK. E-mail: Elizabeth.Gibson@newcastle.ac.uk
bSchool of Chemical Sciences, Dublin City University, Dublin, Ireland. E-mail: mary.pryce@dcu.ie
cDepartment of Chemistry, School of Physical Sciences, Mizoram University, Aizawl, India
dDepartment of Chemistry, University of Sheffield, Sheffield, UK
First published on 15th June 2023
Abstract
Photoelectrocatalysis offers the opportunity to close the carbon loop and convert captured CO2 back into useful fuels and feedstocks, mitigating against anthropogenic climate change. However, since CO2 is inherently stable and sunlight is a diffuse and intermittent energy source, there are considerable scientific challenges to overcome. In this paper we present the integration of two new metal–organic photocatalysts into photocathodes for the reduction of CO2 using ambient light. The two molecular dyads contained a rhenium carbonyl or palladium-based catalytic centre bridged to a ruthenium bipyridyl photosensitizer functionalised with carboxylic acid groups to enable adsorption onto the surface of mesoporous NiO cathodes. The photocathodes were evaluated for photoelectrochemical reduction of CO2 to CO or H+ to H2 and the performances were compared directly with a control compound lacking the catalytic site. A suite of electrochemical, UV-visible steady-state/time-resolved spectroscopy, X-ray photoelectron spectroscopy and gas chromatography measurements were employed to gain kinetic and mechanistic insight to primary electron transfer processes and relate the structure to the photoelectrocatalytic performance under various conditions in aqueous media. A change in behaviour when the photocatalysts were immobilized on NiO was observed. Importantly, the transfer of electron density towards the Re–CO catalytic centre was observed, using time resolved infrared spectroscopy, only when the photocatalyst was immobilized on NiO and not in MeCN solution. We observed that photocurrent and gaseous photoproduct yields are limited by a relatively low yield of the required charge-separated state across the NiO|Photocatalyst interface. Nonetheless, the high faradaic efficiency (94%) and selectivity (99%) of the Re system towards CO evolution are very promising.
Introduction
For many decades, chemists have been trying to create molecular systems that mimic photosynthesis in order to capture the energy available in sunlight in chemical bonds. There are now an abundance of chromophores and catalysts,1–3 and various colloidal semiconductors that can drive the water splitting reaction or reduce CO2 to produce energy dense fuels such as H2, CO, HCO2H and hydrocarbons.4–8 These systems offer opportunities to tune the products with high selectivity and atom efficiency. Pioneering work into artificial photosynthesis and molecular photophysics from the 1970s and 1980s provided a wealth of information on the photophysics of such systems.9,10 The challenges of light-driven water-splitting and CO2 reduction using molecular systems in solution are well documented.11–14 These include the difficulty of driving multi-electron chemistry when each photon absorbed only provides a single pair of an electron and hole. Storing charges on the appropriate timescale to drive the chemistry, while avoiding decomposition pathways, is a major focus of current research. As more elaborate chromophores have been developed to resolve these challenges, detailed mechanistic insight has been gained from investigations into primary photo-induced electron transfer and energy transfer. These processes are highly sensitive to minor structural changes and modification of the local environment, adding another layer of complexity.15 At this point, for systems in solution, the thermodynamic and kinetic requirements for developing effective assemblies to separate redox pairs from light harvesting arrays are well-understood. Some headway has also been made towards developing catalysts from earth-abundant metals.16–20 A key issue still remains for molecular and colloidal artificial photosynthetic systems. That is, that the charge required to drive the oxidation or reduction reactions, which are usually performed with two separate photocatalysts, is usually provided by a sacrificial reagent. This requirement limits the sustainability of the process as well as generating reactive waste products.An alternative approach is to use a photoelectrochemical system where electrons liberated by the water oxidation reaction at the photoanode can then be consumed at the fuel forming reaction at the photocathode.21–26 Such systems perform with promising light-to fuel efficiency but in aqueous solutions, CO2 reduction competes with the kinetically more favourable hydrogen evolution reaction and selectivity is difficult to control.27–30 A challenge is finding a p-type semiconductor that is stable to corrosion and has an appropriate electronic band alignment with the desired electrochemical reactions to deliver a high photovoltage and photocurrent density.31 Photocathodes have been reported based on metal phosphides, chalcogenides, oxides, III–V, p-Si and metal free semiconductors.7,23,28,32 For example, Kang et al. demonstrated that photocathodes based on CuFeO2 and CuO binary films selectively produced formate from aqueous CO2 at ∼1% efficiency over 24 h, under AM 1.5G; 100 mW cm−2 simulated sunlight. However, the photocathode gradually degraded over 17 days.31 Zhang et al. reported a Cu2O-SnOx nanowire-based photocathode that produced syngas (CO and H2 with a ratio that could be adjusted with composition and applied bias) with a 11.61 mA cm−2 at −0.55 V versus RHE, and a total faradaic efficiency of 90.32% at −0.35 V versus RHE.33 p-Si decorated with catalytic metal nanoparticles (e.g. Cu, Ag, Au) has been used to convert CO2 to CO with faradaic efficiencies >80%, but parasitic light absorption can limit the overall solar to fuel efficiency. Gong et al. reported an amorphous silicon photocathode, protected by a thin layer of TiO2 and decorated with Au of variable thickness that enabled photocurrent densities of up to 6 mA m−2 and CO/H2 ratios that could be tuned between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:3.1 by changing the applied bias between −0.1 VRHE to 0.4 VRHE.34 Lewis et al. optimized a microstructured p-Si photocathode decorated with Cu and attained a photocurrent density of 30 mA cm−2 in 0.1 M KHCO3(aq) at −0.62 V vs. RHE under AM 1.5 simulated solar illumination. CO, CH4, and C2H4, were detected but the major product was hydrogen, with only 10% faradaic efficiency for carbon products.28
:2 and 1:3.1 by changing the applied bias between −0.1 VRHE to 0.4 VRHE.34 Lewis et al. optimized a microstructured p-Si photocathode decorated with Cu and attained a photocurrent density of 30 mA cm−2 in 0.1 M KHCO3(aq) at −0.62 V vs. RHE under AM 1.5 simulated solar illumination. CO, CH4, and C2H4, were detected but the major product was hydrogen, with only 10% faradaic efficiency for carbon products.28
A higher selectivity and lower parasitic absorption can be reached with molecular catalysts immobilized on semiconductor photocathodes.7 For example, Kumar et al. reported a p-type silicon photocathode coated with [Re(bipy-tBu)(CO)3Cl] that reduced CO2 to CO under non-aqueous conditions with a faradaic efficiency of 97%.35 Re(bpy)(CO)3Cl was immobilised via phosphonate linkers on the surface of a photocathode assembled from a nanostructured TiO scaffold deposited on TiO2-protected Cu2O photocathodes.36 Under non-aqueous conditions, the photocathode produced an initial photocurrent density of 2.5 mA cm−2 and faradaic yields for CO between 80 and 95%. The catalyst molecules exceed 70 turnovers during a 1.5 h test at −2.05 V vs. Fc, but there was a loss of activity. There are few reports of such high selectivity for CO under aqueous conditions, however.
In recent years, the wealth of knowledge gained from decades of research into dye-sensitized solar cells (DSSC) has been applied to optimizing photoelectrochemical (PEC) systems for both water-splitting and CO2 reduction.37–41 Photoelectrochemical systems based on dye-sensitized electrodes (DS-PEC)42–44 can be used to generate solar fuels, circumventing one of the main obstacles encountered by molecular photocatalytic systems in solution, namely the frequent need for a sacrificial electron and proton source other than water.45,46 Instead, the molecular chromophore and catalyst are immobilized on the surface of a transparent electrode and excitation of the chromophore drives a cascade of electron transfer reactions between the electrode and the catalyst.21,22,47,48 TiO2 is the optimum material for the photoanode. While a number of mesoporous materials were used for the photocathode, NiO is the most common choice due to its good transparency, p-type character, and stability under the conditions of the experiments.49–57 NiO is a well-understood p-type material with a valence band edge that lies sufficiently positive in energy to donate an electron to an excited molecular photosensitizer. It is typically chosen as the cathode material in photoelectrochemical systems due to its relatively good stability under a moderate pH range used and its optical band gap (3.6 eV) is sufficiently large that it does not compete for light absorption with the dyes and photocatalysts.58–61
While these systems are fairly straightforward to assemble, and they maintain the high atom efficiency of the catalyst while benefitting from the robust semiconductor scaffold, the mechanism is complicated and optimization of the system is not so simple. The dynamics of the light-driven processes determined via studies of analogous chromophores in dilute solution do not directly transfer to the equivalent DS-PEC device.62,63 Electron transfer from a dye/catalyst anchored to a semiconductor surface by the traditional strategy of an acid or phosphonate anchoring group leads to multiple competing recombination pathways, many similar to those seen with DSSCs.57,64–67 It is also now known that the chromophore's anchoring group itself can have a significant impact on charge-recombination.68 Recent work has begun to recognise the challenges in engineering suitable catalysts and sensitizers for DS-PEC, which involves a combination of device engineering, optimizing reaction conditions and dye/catalyst design.48,69–77 A key design feature in DS-PEC systems is the spatial separation of the catalytic centre from the semiconductor surface to slow down charge-recombination and maximise catalytic turnover. Mastering all these facets simultaneously and scaling up devices presents a major challenge.
CO2 reduction with DS-PEC has been investigated previously with devices incorporating a Ru(II)–Re(I) molecular photocatalyst. Ishitani and co-workers successfully demonstrated such a model system utilizing a phosphonic acid anchor for the RuRe catalyst on NiO where CO2 was reduced predominantly to CO with a MeCN electrolyte at an applied potential of −1.2 V vs. Ag/AgCl.78 To function effectively over 15 hours in an aqueous environment, a polymer layer was added on top of the catalyst sensitized NiO to prevent desorption.43,44 Optimization of this system led to remarkable stability over 100 hours and the total TON of the photocatalytic CO2 reduction exceeded 1200 (TONCO = 576, TONHCOOH = 695, TONH2 = 172).77
Previously we have shown that supramolecular photocatalysts containing ruthenium bipyridyl photosensitizers linked through a conjugated bridging ligand to a rhenium carbonyl or Pd catalytic centre drive photoelectrocatalytic reduction of CO2 to CO or H+ to H2 when integrated in NiO-based photocathodes. The Ru component enables the photocatalyst to absorb further into the red (towards 600 nm or beyond) than the Re complex alone (∼450 nm).79 A second advantage is that the absorption coefficient of the bimetallic system is higher than the Ru system alone, which means the NiO film can be quite thin to overcome the slow hole-transport.80 Co-immobilization of the photosensitizer and catalyst would lead to competition for binding sites at the surface and so the thickness of the NiO would need to be even thicker to immobilize the equivalent number of chromophores as the supramolecular system. The third advantage of the structure is to improve the photophysics. The supramolecular structure is intended to provide conjugation from the NiO through to the catalytic centre so that excitation of the photocatalyst leads directly to reduction. In other systems the catalyst has been electronically decoupled from the photosensitizer and several sequential electron transfer steps are required to reduce the catalyst. These processes compete with charge-recombination, which is prominent in NiO photoelectrodes, and can limit the overall efficiency.63
In our previous work, the photocatalysts were adsorbed on the NiO surface through ester groups, but only trace amounts of CO were produced and it was noted that carboxylic acids may be preferable for anchoring to metal oxide surfaces.63,80,81 In this paper, we consider two photoelectrocatalysts, one for CO2 reduction (PC1), one for H2 evolution (PC2), with carboxylic acid groups for NiO photocathodes. PC3 was included as a catalyst-free compound to benchmark the activity (Fig. 1). We also tested a Pt analogue of PC2, but this gave negligible photocurrent, so we have not included it here. This paper describes the optimization of reaction conditions, analysis of the electrochemical and photophysical properties in addition to chromatography measurements of gaseous products. Some of the major bottlenecks that we found in designing DS-PEC systems where the chromophore and catalyst are integrated in one molecule are discussed.
 | ||
| Fig. 1 Molecular structures of the photocatalysts used in the present study. [Ru(bpy)(dcb)(bpt)Re(CO)3Cl](PF6)[PC1], [Ru(bpy)(dcb)(bpt)PdCl2](PF6)2[PC2], [Ru(bpy)(dcb)(bpt)](PF6)2 [PC3], bpy = 2,2′-bipyridine, bpt = 3,5-di(pyridin-2-yl)-triazole, dcb = 2,2′-bipyridine-4,4′-dicarboxylic acid. PC1 is designed for CO2 reduction, PC2 is designed for H2 evolution and PC3 is a catalyst-free control. | ||
Results and discussion
Optical, electronic and electrocatalytic properties of the photocatalysts
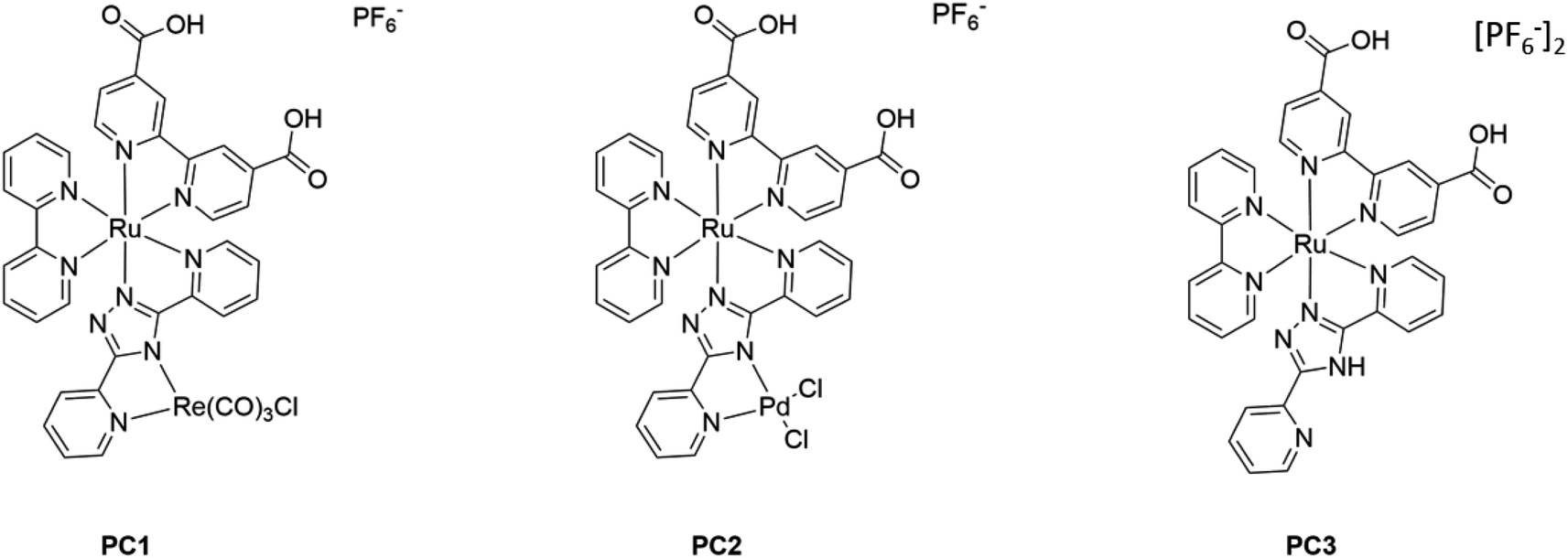
The photocatalysts were synthesized according to the procedures provided in the ESI‡ and were characterized by NMR, mass spectrometry and FTIR spectroscopy. Steady-state absorption and emission properties of the photocatalysts PC1–PC3 in solution are summarized in Fig. 2 and Table 1. The FTIR spectra of the ruthenium complexes in the 1650–2100 cm−1 region are shown in Fig. S28.‡ All complexes show a signal at 1735 cm−1 which is assigned to the CO stretch in the acid moiety on the dcb peripheral ligand. The IR spectrum for [Ru(dcb)(bpy)(bpt)Re(CO)3Cl](PF6) contains stretching vibration at 1735 (COOH), 1896, 1908, 1930, 2020 and 2036 cm−1. Based on the relative shapes of these stretches we assign these signals to a mixture of two different fac-Re(CO)3Cl complexes. Similar observations have been observed by other groups for Ru–Re assemblies, and attributed to isomers.82–84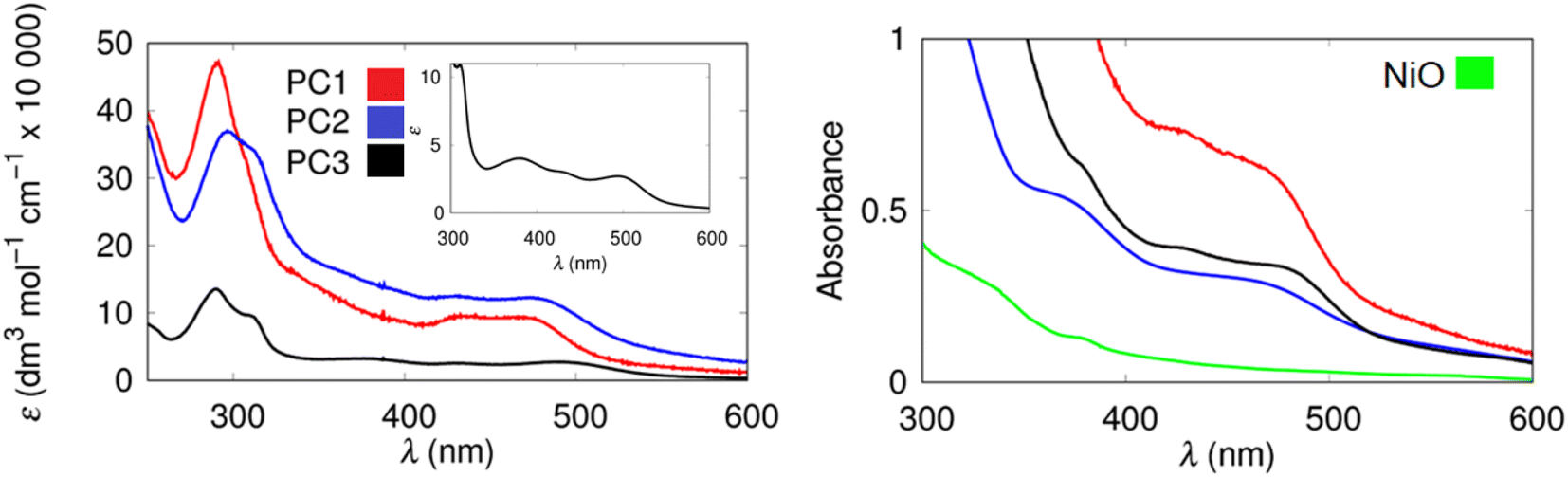 | ||
| Fig. 2 Left panel: steady-state absorption spectra of the photocatalysts in acetonitrile. Right panel: absorption spectra of the same systems when adhered to NiO electrodes following dye-bath deposition. | ||
| Compound | λ absmax (MeCN)/nm | λ fluormax (MeCN)/nm | λ absmax (NiO)/nm | E 00/eV (MeCN) | ε/M−1 cm−1 | Dye loading/mol cm−2 |
|---|---|---|---|---|---|---|
| PC1 | 470 | 671 | 470 | 2.05 | 9400 | 4.33 × 10−8 |
| PC2 | 474 | 665 | 467 | 1.98 | 12300 |
2.50 × 10−8 |
| PC3 | 490 | 720 | 480 | 1.99 | 2700 | 3.70 × 10−8 |
The chromophores developed in this study have absorption and emission properties analogous to other molecular systems developed around a ruthenium trisbipyridyl sensitizer, with metal-to-ligand charge transfer bands observed between 400 and 500 nm.63,85 As can be observed from Fig. S29,‡ the Re(Hbpt)(CO)3Cl catalyst has very limited absorbance in the visible region of the solar spectrum. The RuRe supramolecular assembly PC1, however, has a significantly extended absorbance range, which should enable this assembly to deliver improved photoelectrocatalytic activity compared to the Re(Hbpt)(CO)3Cl complex by itself.
The dyes were adsorbed on the NiO surface by immersing the electrodes in an acetonitrile solution containing 3 mM photocatalyst overnight. A 10–20 nm blue shift in the absorption spectra was observed for each photocatalyst, which is consistent with the behaviour of the ester equivalents.63 The loading of the photocatalyst on the freshly prepared surface of the NiO was quantified by desorbing the photocatalyst using 1 M NaOH solution and recording the UV-visible absorption spectrum (Table 1). The loading was approximately 25–50 nmol cm−2 (highest for PC1), which is much higher than the ester analogues (4–7 nmol cm−2).27,28,61,86 This means that the light harvesting efficiency is sufficient with a thin NiO film (1.5 μm). Weak fluorescence was readily observed in solution (see ESI‡), but this was very hard to detect reliably from the NiO surface. Therefore, fluorescence quenching could not be used effectively to characterize charge-transfer at the NiO|PC interface. Instead, transient absorption spectroscopy was employed (see below).
For charge-transfer to occur from the NiO to the photocatalyst following light absorption, the HOMO of the dye needs to be at more positive potential than the potential of the valence band edge, which is EVB = ca. −0.12 V vs. Fc for NiO in MeCN.87 To estimate the ample driving force for charge transfer at the NiO|PC interface, cyclic voltammetry (CV) for PC1–3 was performed and the data acquired is provided in the ESI.‡ The CVs were not as straightforward as for the ester moieties, which show three reversible reductions and a reversible oxidation, due to the non-innocent behaviour of the acid moieties,76 as the acids can undergo deprotonation following electrochemical reduction forming the carboxylates, causing irreversibility of the reduction process.
The CV for PC3 contains three reduction processes at −1.7, −2.0 and −2.2 V vs. Fc, which can be assigned to reduction of the three ligands. Based on previous reports, the reduction takes place in the order dcb > bpy > bpt.80,88,89 The first reduction appears to be irreversible while the second and third reduction have some reversible character. A fourth irreversible reduction at −2.8 V vs. Fc, may be assigned to a second reduction taking place on the bpt bridging ligand, as previously reported by Vos et al. for the Ru(bpy)2(bpt) complex. A reversible oxidation at ca. 0.75 V vs. Fc is assigned to the RuII/III oxidation. A small shouldering irreversible oxidation at 0.44 V was also observed for the Ru-ester analogue (Ru(dceb)2(bpt)) and can be assigned to residual Ru(dcb)(bpy)(Hbpt) in which the triazole moiety on the ruthenium complex is still protonated.90 No reductive signals were observed for this protonated species as the proton is lost under slight reductive conditions forming the deprotonated species which is consequently observed in the reductive region of the CV.
The CV for PC2 was more complicated than that of the Ru mononuclear species, PC3. The separate reduction processes observed between −1.7 and −2.2 V for PC3 were less resolved for PC2 and became fully irreversible. The reversible oxidation for the Ru centre was also shifted anodically to 0.85 V vs. Fc. A second irreversible oxidation was observed at 1.4 V vs. Fc, which relates to oxidation of the Pd centre. As for PC2, the CV for the RuRe analogue PC1 contained additional reductive and oxidative features compared to PC3. The reduction processes on the three ligands of the Ru complex also became irreversible and were observed at −1.8, −2.0 and −2.1 V. Reversibility was also partially lost for the RuII/III oxidation which is observed at 0.9 V vs. Fc. A second oxidation at 1.1 V is likely to be associated with the ReI/II oxidation.91
To determine the feasibility of CO2 reduction by the supramolecular assembly, PC1 and the corresponding Re(Hbpt)(CO)3Cl fragment were tested for electrocatalytic reduction of CO2. First, both catalysts were tested in a homogeneous system using DMF as the solvent and bubbled either with N2 (blank test) or CO2 (catalytic test), respectively. Both PC1 and the Re(bpt)(CO)3Cl complex showed a similar increase in current under a CO2 atmosphere with an Icat/I0 of 3.31 for the PC1 assembly and 3.04 for the Re(Hbpt)(CO)3Cl complex (Fig. S40 & S41‡). The observed current increase for Re(Hbpt)(CO)3Cl matches well with the value observed for the similar Re(Hph)(CO)3Cl (3.58) complex, which was previously reported for electrocatalytic CO2 reduction by Nguyen et al.92 The fairly small difference in catalytic activity between the Re(bpt)(CO)3Cl fragment and the supramolecular assembly PC1 is consistent with the limited influence of the Ru fragment on the Re catalytic centre as observed for the analogous supramolecular assembly utilising the bpt bridging ligand but with ester rather than acid groups on the peripheral bpy ligands.80
Kinetic experiments
Ultrafast UV-visible transient absorption spectroscopy (TA) and time-resolved infrared spectroscopy (TRIR) measurements were used to explore the excited state dynamics of PC1–3 in solution and when adsorbed on the NiO substrates. The spectra at selected time delays after excitation at 500 nm are shown in Fig. 3 and the results from global analysis of the data are provided in the ESI‡ with extracted time constants provided in Table 2. The spectra are consistent with the formation of a triplet metal-to-ligand charge transfer (MLCT) states.93 For PC3, a transient signal was observed below 430 nm, a bleach corresponding to the ground state absorption below 560 nm, and a broad transient absorption above 560 nm. The spectral features changed little within the timescale of the experiment (6 ns). Global analysis of the data revealed two components, τ1 ∼ 200 ps, and τ2 ≫ 1 ns.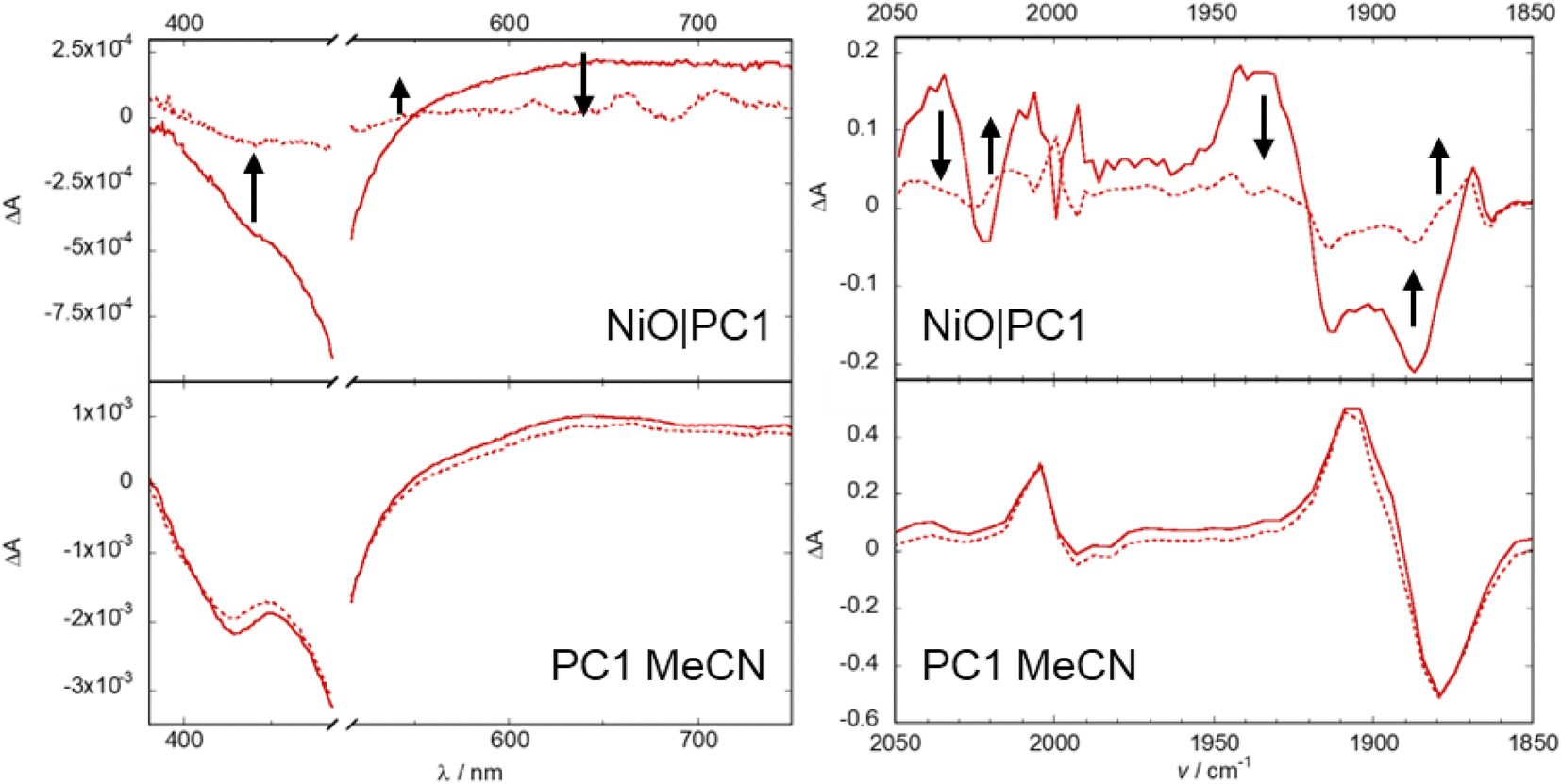 | ||
| Fig. 3 UV-visible transient absorption spectra (left) and time resolved infrared spectra (right) for PC1, 1 ps (solid line) and 1 ns (dotted line) following excitation at 500 nm in acetonitrile (bottom) and on a NiO thin film (top). The transient features at 1 ns spectrum for NiO/PC1 are imprints of the probe spectrum. | ||
| PC1 | PC2 | PC3 | |
|---|---|---|---|
| TAS in MeCN | τ 1 = 160 ps, and τ2 ≫ 1 ns | τ 1 = 35 ps, and τ2 = 980 ps, τ3 ≫ 1 ns | τ 1 = 200 ps, and τ2 ≫ 1 ns |
| TAS on NiO | τ 1 = 4.6 ps, τ2 = 110 ps, and τ3 ∼ 5.1 ns | τ 1 = 6.0 ps, τ2 = 120 ps, and τ3 ≫ 1 ns | τ 1 = 13 ps, and τ2 = 260, τ3 ≫ 1 ns |
| TRIR in MeCN | τ 1 = 130 ps, and τ2 ≫ 1 ns | τ 1 = 22 ps, and τ2 = 1.2 ns, τ3 ≫ 1 ns | τ 1 = 14 ps, and τ2 ≫ 1 ns |
| TRIR film | τ 1 = 2.4 ps, τ2 = 150 ps, and τ3 ≫ 1 ns | τ 1 = 7.0 ps, τ2 = 120 ps, and τ3 ≫ 1 ns | τ 1 = 7 ps, and τ2 = 81, τ3 ≫ 1 ns |
TA measurements of NiO|PC3 films revealed very different dynamics and global analysis revealed characteristically heterogeneous behaviour for dyes adsorbed on mesoporous semiconductor surfaces, which can be described by a minimum of three components. The two short-lived components were similar in shape and lifetime compared to PC3* in solution, τ1 = 13 ps, and τ2 = 260 ps, and these are likely to be associated with relaxation of the excited states and charge-transfer from the NiO to PC3* to form NiO+|PC3−. τ3 ≫ 1 ns, is assigned to the reduced photocatalyst (PC3−), with a weak transient absorption < 400 nm and >510 nm. Dietzek et al. showed that MLCT states localized on the carboxylic acid-functionalised bipyridine are unfavourable for charge-separation.94 In our previous work, we observed the presence of both charge-separated (NiO+|PC−) and excited states (NiO|PC*) for photocatalysts with diester-functionalised bipyridine ligands.62,63 In this case, for PC3, although most of the signal decays within 1 ns, there is evidence for successful charge-separation with the formation of NiO+|PC3−.
The transient absorption spectra for PC1 in MeCN and PC1|NiO followed the same trend as PC3 and PC3|NiO, which suggests that the Re-centre has little impact on the photophysics of the ruthenium complex. However, PC2 behaved differently. The excited states for PC2 in MeCN decayed mostly within the 6 ns window of the experiment. RuPd complexes typically have a low-lying triplet state which quenches the 3MLCT states. PC2|NiO contained spectral features and dynamics that were consistent with PC1|NiO and PC3|NiO, suggesting that charge-transfer from NiO to PC2* takes place, but the yield of the charge-separated state, NiO+|PC2− could be lower to that of the other systems because of the shorter lifetime of the excited state of the photocatalyst.
TRIR measurements provided information regarding the distribution of electron density in the excited state (Fig. 3 and S49–S53‡). The TRIR spectra for PC1 in MeCN are shown in Fig. 3 and S49.‡ Depletion of the ground state bands at 1735, 1880 and 1993 cm−1, together with formation of transient absorption bands at 1706, 1909 and 2004 cm−1 were observed within the laser pulse. The signals persisted beyond the 5 ns duration of the experiment, which is consistent with the properties of the ester analogue of PC1.28 Global fitting (ESI‡) of the TRIR data yielded two components but very little spectral change between species, with τ1 = 130 ps, and τ2 ≫ 1 ns, consistent with the TA data. The transient IR-absorption bands for the Re-CO shift to higher energy relative to the ground state bleach, consistently with electron density removed from the Re centre, resulting in a decrease in π-back bonding to the CO ligands. The transient IR-absorption of the ν(CO) of the bpy-C(O)OH ligand shifts to higher energy relative to the ground state bleach. This indicates that electron density moves towards the dcb ligands in the excited state rather than towards the catalytic centre. The TRIR spectra for PC2 and PC3 in MeCN (see ESI‡) are similar to those of PC1, showing a ground state bleach at 1735 cm−1 and a transient absorption at 1700 cm−1, consistent with the formation of a 3MLCT state. The lifetime of the excited state of PC2 (major component τ2 = 1.2 ns) was short relative to PC3 in MeCN (major component τ2 ≫ 1 ns). The efficiency of charge-injection could therefore be limited by the comparatively short excited-state lifetime. The results from the solution studies were consistent with our prior results for the ester derivatives.28
We also performed the TRIR experiments for the immobilised photocatalysts, NiO|PC. The data were affected by typical scattering from dye-sensitized films, but the key features were observed. For NiO|PC3 and NiO|PC2, there was no observable signal in the Re(CO) carbonyl region. It was expected that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch for the acid anchoring group would be absent in the IR spectra of photocatalysts adsorbed onto NiO.95 Instead, we observed a broad absorption across the fingerprint region corresponding to changes in electron density in the aromatic ligands following excitation. These features decayed on timescales broadly consistent with the transient absorption spectra. For NiO|PC1, we were able to observe the characteristic bleach and corresponding absorption for the CO ligands on the Re centre. Interestingly, the spectral shape changed during the duration of the experiment, unlike in the solution TRIR. The transient absorption shifts to lower wavenumber at increasing delay time after excitation and this is consistent with an increase in electron density on the Re centre, leading to increased π-back bonding to the CO ligands. This is consistent with the reduction of the photocatalyst and the shift in electron density towards the Re centre would favour catalysis. We attribute this difference to a change in the relative energy of the ligands when the photocatalysts are immobilised on NiO. An increase in the LUMO energy on binding to NiO would be consistent with the blue shift in the absorption spectrum observed (above).
O stretch for the acid anchoring group would be absent in the IR spectra of photocatalysts adsorbed onto NiO.95 Instead, we observed a broad absorption across the fingerprint region corresponding to changes in electron density in the aromatic ligands following excitation. These features decayed on timescales broadly consistent with the transient absorption spectra. For NiO|PC1, we were able to observe the characteristic bleach and corresponding absorption for the CO ligands on the Re centre. Interestingly, the spectral shape changed during the duration of the experiment, unlike in the solution TRIR. The transient absorption shifts to lower wavenumber at increasing delay time after excitation and this is consistent with an increase in electron density on the Re centre, leading to increased π-back bonding to the CO ligands. This is consistent with the reduction of the photocatalyst and the shift in electron density towards the Re centre would favour catalysis. We attribute this difference to a change in the relative energy of the ligands when the photocatalysts are immobilised on NiO. An increase in the LUMO energy on binding to NiO would be consistent with the blue shift in the absorption spectrum observed (above).
Photoelectrocatalysis
The photocurrent for NiO|PC1 was recorded under chopped light illumination in the presence of an electron acceptor in solution (10 mM tris(ethylenediamine)cobalt(III)) (see ESI‡). This provides an idealized case to test the photocathodic response where electrons are extracted from the photocathode. The observed current density of approx. 20 μA cm−2 confirmed that PC1 transfers charge from the NiO electrode to the electron acceptor in solution in the presence of light.Photoelectrochemical experiments were then performed with PC1|NiO without the electron acceptor but, instead, bubbling with CO2 or Ar to evaluate whether PC1 could drive photoelectrocatalysis. The photocurrents and evolved gasses were evaluated under a range of pH between 5 to 9.2. An ionic liquid, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide, was also tested due to its CO2-solvation ability.96,97 A summary of results for the evaluated systems is provided in Table 3. Similar current densities were recorded for each, under simulated 100 mW cm−2 intensity AM1.5 irradiation. Fig. 4 and S65–S73‡ illustrate the photocurrent observed in the devices tested.
| PC1|NiO @ −0.2 V vs. Ag/AgCl | pH 5 acetate (under CO2) | pH 6.6 50 mM NaHCO3 (under CO2) | pH 8 phosphate buffer (under CO2) | pH 9.2 carbonate buffer (under CO2) |
|---|---|---|---|---|
| J/μA cm−2 | 4.6 | 2.9 | 3.8 | 3.1 |
| η Far/% (CO) | 54.3 | 94.0 | 61.8 | 20.2 |
| η Far/% (H2) | 0.46 | 0.33 | 0.32 | 0.1 |
| [CO]/μmol h−1 cm−2 | 0.12 (±0.014) | 0.27 (±0.022) | 0.11 (±0.015) | 0.09 (±0.016) |
| [H2]/nmol h−1 cm−2 | 1.06 (±0.13) | 0.93 (±0.074) | 0.54 (±0.076) | 0.40 (±0.072) |
| TOF (CO) h−1 | 0.75 | 1.63 | 0.63 | 0.52 |
| TOF (H2) h−1 | 0.007 | 0.007 | 0.003 | 0.003 |
 | ||
| Fig. 4 Representative example of photocurrent in 0.1 M pH 5 acetate buffer under chopped light illumination for photoelectrocatalysis experiments of NiO films sensitized with photocatalysts. The applied bias was −0.2 V vs. Ag/AgCl. NiO, PC1|NiO and PC3|NiO are saturated with CO2, PC2|NiO is saturated with Ar. | ||
At the beginning of each experiment a characteristic spike in the dark current was recorded which has been associated with charging of the electrode surface.98 Results from PEC experiments under two different applied bias, −0.2 and −0.5 V vs. Ag/AgCl, are presented in the ESI (Fig. S65–S73‡). These potentials are more positive than the flat band potential of TiO2 that would typically serve as a photoanode.99 Control experiments with unsensitized NiO films confirmed that the majority of photocurrent generated arose from light absorption by the chromophore. Typical photocurrent densities were approximately 2–5 μA cm−2 for all systems in aqueous electrolytes where the applied bias was −0.5 V vs. Ag/AgCl. At a lower applied bias of −0.2 V vs. Ag/AgCl, the dark current for all systems was smaller and surface charging was less pronounced. The photocurrent density for PC2|NiO was remarkably lower than that attained with the ester analogue (ca. 40 μA cm−2).63 However, the photocurrent density for PC1|NiO was ca. 6 times higher than the ester analogue (1.3 μA cm−2).80 Both were higher than the catalyst-free control, PC3|NiO.
Bubbles formed on the electrode surface during extended periods of irradiation (AM1.5, 100 mW cm−2). Care was taken with gas sampling experiments to compare rigorously with appropriate control experiments so that any decomposition of the photocatalyst could be accounted for. Evolved gases from NiO|PC1, NiO|PC2 and NiO|PC3 and the NiO control were tested over a three-hour irradiation experiment in the three aqueous electrolytes. The headspace was periodically sampled, and the products were quantified by gas chromatography. This required occasional shaking of the cell to promote mixing. The electrolyte was tested for liquid products using ion chromatography, but no formate was detected. The results are provided in Table 3 and the ESI.‡ A very small amount of H2 was evolved from NiO|PC2. CO was the main product from NiO|PC1 and in pH 5 acetate buffer and −0.5 V vs. Ag/AgCl, CO was produced with a faradaic efficiency of approx. 91% and a turnover per catalytic centre of 5.4 in 3 hours. The turnover frequency for this system was equal to that reported by Ishitani et al. for a RuRe catalyst with a protective polymer layer.69 Hydrogen was also detected, but in very small amounts. To confirm the origin of the CO was the gas rather than decomposition of the photocatalyst, the electrolyte was purged with 13CO2, and gaseous products were analysed by mass spectrometry. The major product under both −0.5 V and −0.2 V vs. Ag/AgCl in pH 5 acetate buffer was 13CO (see ESI‡).
To further optimise the system, the electrolyte was replaced with pH 6.6 NaHCO3 solution. At −0.2 V, the average photocurrent density was 2.9 μA cm−2, the faradaic efficiency for CO was 94%, and a turnover of 4.88 per catalyst over 3 hours was attained. To our knowledge, this is the highest selectivity for CO production with a dye-sensitized photocathode. The summary of results for NiO|PC1 in different buffers is shown in Fig. 5.
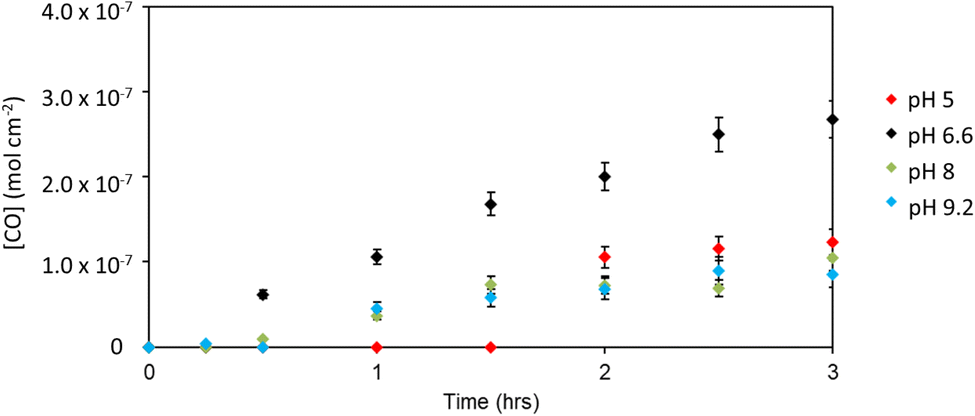 | ||
| Fig. 5 Evolved CO detected by gas chromatography for PC1|NiO thin films of 0.79 cm2 working area with an applied bias of −0.2 V vs. Ag/AgCl under 1 sun AM 1.5, (100 mW cm−2) irradiation in various pH buffer solutions as described earlier. Note the pH 6.6 is 50 mM NaHCO3 solution. | ||
Surface analysis
After the photoelectrocatalysis experiments, the UV-visible absorption spectra were re-recorded. There was an approx. 20% reduction in the absorption maxima for the three photocatalysts on average according to UV-vis absorption difference spectra. Some of this may be accounted for by the reduction of surface Ni3+ states during the reduction (see comment on current spikes above), but it appears that some of the photocatalyst was lost during the reaction. To further characterize the surface of the photocathode before and after the experiments, X-ray photoelectron spectroscopy was used.The results from the X-ray photoelectron spectroscopy provided evidence the photocatalysts were indeed deposited on the NiO surface and enabled us to determine changes/losses of chromophore after a standard photocatalysis reaction. No noticeable change in the oxidation state of the key elements was determined for our experiments, but there was a slight decrease in signal intensity following photocatalysis for 15 minutes (note these were different samples from the same batch). PC2 was observed to be less stable than PC1, with some loss of ruthenium after the photoelectrocatalysis experiments (Fig. S108 ESI‡). This is consistent with the relatively poor faradaic efficiency for PC2 as compared to PC1 (Table 3).
Conclusions
In summary, we have evaluated the photoelectrocatalytic performance of two supramolecular photocatalysts with carboxylic acid anchoring groups, integrated in NiO photocathodes for CO2 reduction and hydrogen evolution. The RuRe compound PC1, performed better than the ester analogue previously reported.80 In contrast, the RuPd compound PC2 was significantly less active for hydrogen generation than the ester analogue.63 The main reason for the change in behaviour is attributed to the difference in lifetime of the excited state. This is much shorter in the case of PC2, which could limit light-induced charge transfer at the NiO|PC2 interface. The NiO|PC1 system is, however, a promising, highly selective system for dye-sensitized NiO photocathodes for CO2 reduction. Further tuning of PC2 could improve the photocurrent and TON. For example, modifying the bridging ligand to promote charge transfer towards the catalytic metal and away from NiO. The absorption coefficients of the photocatalysts are quite low compared to state-of-the-art photosensitizers in NiO-based DSSCs, which are typically organic systems rather than coordination complexes. A hybrid system with an organic chromophore may lead to much higher light harvesting efficiency and, consequently, better performance. Finally, the solubility of CO2 in the solvent should be greater to support the higher photocurrent densities and further work on alternatives to MeCN are necessary.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Harwell XPS (The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195) and Nexus at Newcastle University. Transient Absorption Spectroscopy and Time-resolved Infrared Spectroscopy was performed at the Lord Porter Ultrafast Laser Spectroscopy Laboratory at the University of Sheffield. Joshua Karlsson and Elizabeth Gibson thank Newcastle University, the Engineering and Sciences Research Council (EPSRC) and the North East Centre for Energy Materials EP/R021503/1 for funding. Mary T. Pryce and Florian Cerpentier thank the Sustainable Energy Authority of Ireland under the SEAI National Energy Research, Development and Demonstration Funding Programme 2018 (Grant numbers 18/RDD/282) for funding. RL would like to thank the European Commission, for a Marie Sklodowska−Curie Fellowship (No. 799778). JAW, MVA, JDS, DC thank the University of Sheffield for support, the EPSRC for funding, and Grantham Center for Sustainable Futures for PhD studentships to JDS and MVA. We thank Karina Scurupa Machado for her valuable assistance in performing GC-MS experiments for isotope labelling studies.References
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268 RSC.
- W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed.
- R. Abe, K. Shinmei, K. Hara and B. Ohtani, Chem. Commun., 2009, 3577 RSC.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795 RSC.
- Y. Yang, S. Ajmal, X. Zheng and L. Zhang, Sustainable Energy Fuels, 2018, 2, 510–537 RSC.
- Y. Liu, F. Wang, Z. Jiao, S. Bai, H. Qiu and L. Guo, Electrochem. Energy Rev., 1918, 5, 5 CrossRef.
- J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M.-C. Richoux, Coord. Chem. Rev., 1982, 44, 83–126 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- T. Kowacs, L. O'Reilly, Q. Pan, A. Huijser, P. Lang, S. Rau, W. R. Browne, M. T. Pryce and J. G. Vos, Inorg. Chem., 2016, 55, 2685–2690 CrossRef CAS PubMed.
- Y. Yamazaki, K. Ohkubo, D. Saito, T. Yatsu, Y. Tamaki, S. Tanaka, K. Koike, K. Onda and O. Ishitani, Inorg. Chem., 2019, 58, 11480–11492 CrossRef CAS PubMed.
- N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2020, 142, 274–282 CrossRef CAS PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- H. Takeda, Y. Monma and O. Ishitani, ACS Catal., 2021, 11, 11973–11984 CrossRef CAS.
- G. Segev, J. Kibsgaard, C. Hahn, Z. J. Xu, W.-H. Cheng, T. G. Deutsch, C. Xiang, J. Z. Zhang, L. Hammarström, D. G. Nocera, A. Z. Weber, P. Agbo, T. Hisatomi, F. E. Osterloh, K. Domen, F. F. Abdi, S. Haussener, D. J. Miller, S. Ardo, P. C. McIntyre, T. Hannappel, S. Hu, H. Atwater, J. M. Gregoire, M. Z. Ertem, I. D. Sharp, K.-S. Choi, J. S. Lee, O. Ishitani, J. W. Ager, R. R. Prabhakar, A. T. Bell, S. W. Boettcher, K. Vincent, K. Takanabe, V. Artero, R. Napier, B. R. Cuenya, M. T. M. Koper, R. van de Krol and F. Houle, J. Phys. D: Appl. Phys., 2022, 55, 323003 CrossRef.
- D. H. Nam, J. Z. Zhang, V. Andrei, N. Kornienko, N. Heidary, A. Wagner, K. Nakanishi, K. P. Sokol, B. Slater, I. Zebger, S. Hofmann, J. C. Fontecilla-Camps, C. B. Park and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 10595–10599 CrossRef CAS PubMed.
- M. S. Prévot and K. Sivula, J. Phys. Chem. C, 2013, 117, 17879–17893 CrossRef.
- H. Pang, T. Masuda and J. Ye, Chem.–Asian J., 2018, 13, 127–142 CrossRef CAS PubMed.
- S. Mandal, D. Ghosh and P. Kumar, J. Mater. Chem. A, 2022, 10, 20667–20706 RSC.
- P. Ding, T. Jiang, N. Han and Y. Li, Mater. Today Nano, 2020, 10, 100077 CrossRef.
- E. Kalamaras, M. M. Maroto-Valer, M. Shao, J. Xuan and H. Wang, Catal. Today, 2018, 317, 56–75 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- Y. Liu, S. Bai, F. Wang and Y. Chen, Environ. Chem. Lett., 2022, 20, 1169–1192 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- P. Wen, H. Li, X. Ma, R. Lei, X. Wang, S. M. Geyer and Y. Qiu, J. Mater. Chem. A, 2021, 9, 3589–3596 RSC.
- U. Kang and H. Park, J. Mater. Chem. A, 2017, 5, 2123–2131 RSC.
- J. H. Kim, H. E. Kim, J. H. Kim and J. S. Lee, J. Mater. Chem. A, 2020, 8, 9447–9482 RSC.
- Y. Zhang, D. Pan, Y. Tao, H. Shang, D. Zhang, G. Li and H. Li, Adv. Funct. Mater., 2022, 32, 2109600 CrossRef CAS.
- C. Li, T. Wang, B. Liu, M. Chen, A. Li, G. Zhang, M. Du, H. Wang, S. F. Liu and J. Gong, Energy Environ. Sci., 2019, 12, 923–928 RSC.
- B. Kumar, J. M. Smieja and C. P. Kubiak, J. Phys. Chem. C, 2010, 114, 14220–14223 CrossRef CAS.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Grätzel, in Materials for Sustainable Energy, Macmillan Publishers Ltd, UK, 2010, pp. 26–32 Search PubMed.
- A. B. Muñoz-García, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- M. D. Kärkäs, O. Verho, E. v. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- A. Charisiadis, E. Giannoudis, Z. Pournara, A. Kosma, V. Nikolaou, G. Charalambidis, V. Artero, M. Chavarot-Kerlidou and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2021, 2021, 1122–1129 CrossRef CAS.
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199, 1–7 CrossRef CAS.
- D. Saito, Y. Yamazaki, Y. Tamaki and O. Ishitani, J. Am. Chem. Soc., 2020, 142, 19249–19258 CrossRef CAS PubMed.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- M. Shizuno, K. Kato, S. Nishioka, T. Kanazawa, D. Saito, S. Nozawa, A. Yamakata, O. Ishitani and K. Maeda, ACS Appl. Energy Mater., 2022, 5, 9479–9486 CrossRef CAS.
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- F. Kuttassery, H. Kumagai, R. Kamata, Y. Ebato, M. Higashi, H. Suzuki, R. Abe and O. Ishitani, Chem. Sci., 2021, 12, 13216–13232 RSC.
- Y. Ueda, H. Takeda, T. Yui, K. Koike, Y. Goto, S. Inagaki and O. Ishitani, ChemSusChem, 2015, 8, 439–442 CrossRef CAS PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS.
- D. Dini, Y. Halpin, J. G. Vos and E. A. Gibson, Coord. Chem. Rev., 2015, 304–305, 179–201 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef CAS.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D’Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. la Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- A. A. Cullen, K. Heintz, L. O’Reilly, C. Long, A. Heise, R. Murphy, J. Karlsson, E. Gibson, G. M. Greetham, M. Towrie and M. T. Pryce, Front. Chem., 2020, 8, 584060 CrossRef CAS PubMed.
- N. Põldme, L. O'Reilly, I. Fletcher, J. Portoles, I. V. Sazanovich, M. Towrie, C. Long, J. G. Vos, M. T. Pryce and E. A. Gibson, Chem. Sci., 2019, 10, 99–112 RSC.
- T. Daeneke, A. J. Mozer, Y. Uemura, S. Makuta, M. Fekete, Y. Tachibana, N. Koumura, U. Bach and L. Spiccia, J. Am. Chem. Soc., 2012, 134, 16925–16928 CrossRef CAS PubMed.
- U. B. Cappel, S. M. Feldt, J. Schöneboom, A. Hagfeldt and G. Boschloo, J. Am. Chem. Soc., 2010, 132, 9096–9101 CrossRef CAS PubMed.
- J. Wiberg, T. Marinado, D. P. Hagberg, L. Sun, A. Hagfeldt and B. Albinsson, J. Phys. Chem. C, 2009, 113, 3881–3886 CrossRef CAS.
- M. Braumüller, M. Schulz, M. Staniszewska, D. Sorsche, M. Wunderlin, J. Popp, J. Guthmuller, B. Dietzek and S. Rau, Dalton Trans., 2016, 45, 9216–9228 RSC.
- L. Zhang, X. Yang, S. Li, Z. Yu, A. Hagfeldt and L. Sun, Sol. RRL, 2020, 4, 1900436 CrossRef CAS.
- R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS PubMed.
- K. L. Materna, A. M. Beiler, A. Thapper, S. Ott, H. Tian and L. Hammarström, ACS Appl. Mater. Interfaces, 2020, 12, 31372–31381 CrossRef CAS PubMed.
- T.-T. Li, B. Shan and T. J. Meyer, ACS Energy Lett., 2019, 4, 629–636 CrossRef CAS.
- A. Charisiadis, E. Glymenaki, A. Planchat, S. Margiola, A.-C. Lavergne-Bril, E. Nikoloudakis, V. Nikolaou, G. Charalambidis, A. G. Coutsolelos and F. Odobel, Dyes Pigm., 2021, 185, 108908 CrossRef CAS.
- J. Huang, B. Xu, L. Tian, P. B. Pati, A. S. Etman, J. Sun, L. Hammarström and H. Tian, Chem. Commun., 2019, 55, 7918–7921 RSC.
- D. Wang, Y. Wang, M. D. Brady, M. V. Sheridan, B. D. Sherman, B. H. Farnum, Y. Liu, S. L. Marquard, G. J. Meyer, C. J. Dares and T. J. Meyer, Chem. Sci., 2019, 10, 4436–4444 RSC.
- S. Bold, J. Massin, E. Giannoudis, M. Koepf, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, ACS Catal., 2021, 11, 3662–3678 CrossRef CAS.
- J.-S. Lee, D.-I. Won, W.-J. Jung, H.-J. Son, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2017, 56, 976–980 CrossRef CAS PubMed.
- R. Kamata, H. Kumagai, Y. Yamazaki, M. Higashi, R. Abe and O. Ishitani, J. Mater. Chem. A, 2021, 9, 1517–1529 RSC.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Chem. Soc. Rev., 2020, 49, 765 RSC.
- F. J. R. Cerpentier, J. Karlsson, R. Lalrempuia, M. P. Brandon, I. V. Sazanovich, G. M. Greetham, E. A. Gibson and M. T. Pryce, Front. Chem., 2021, 9, 795877 CrossRef CAS PubMed.
- A. A. Seddon, J. K. G. Karlsson, E. A. Gibson, L. O'Reilly, M. Kaufmann, J. G. Vos and M. T. Pryce, Johnson Matthey Technol. Rev., 2022, 66, 21–31 CrossRef CAS.
- Z.-Y. Bian, K. Sumi, M. Furue, S. Sato, K. Koike and O. Ishitani, Dalton Trans., 2009, 983–993 RSC.
- R. Sahai, D. P. Rillema, R. Shaver, S. Van Wallendael, D. C. Jackman and M. Boldaji, Inorg. Chem., 1989, 28, 1022–1028 CrossRef CAS.
- S. Van Wallendael, R. J. Shaver, D. P. Rillema, B. J. Yoblinski, M. Stathis and T. F. Guarr, Inorg. Chem., 1990, 29, 1761–1767 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 CrossRef CAS.
- L. Li, E. A. Gibson, P. Qin, G. Boschloo, M. Gorlov, A. Hagfeldt and L. Sun, Adv. Mater., 2010, 22, 1759–1762 CrossRef CAS PubMed.
- E. A. Gibson, L. le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485–6493 CrossRef CAS PubMed.
- D. Mulhern, S. Brooker, H. Görls, S. Rau and J. G. Vos, Dalton Trans., 2006, 51–57 RSC.
- W. R. Browne, C. M. O'Connor, H. P. Hughes, R. Hage, O. Walter, M. Doering, J. F. Gallagher and J. G. Vos, J. Chem. Soc., Dalton Trans., 2002, 4048–4054 RSC.
- R. Hage, A. H. J. Dijkhuis, J. G. Haasnoot, R. Prins, J. Reedijk, B. E. Buchanan and J. G. Vos, Inorg. Chem., 1988, 27, 2185–2189 CrossRef CAS.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- P. N. Nguyen, T.-B.-N. Dao, T. T. Tran, N.-A. T. Tran, T. A. Nguyen, T.-D. L. Phan, L. P. Nguyen, V. Q. Dang, T. M. Nguyen and N. N. Dang, ACS Omega, 2022, 7, 34089–34097 CrossRef CAS PubMed.
- J. K. McCusker, Acc. Chem. Res., 2003, 36, 876–887 CrossRef CAS PubMed.
- M. Bräutigam, J. Kübel, M. Schulz, J. G. Vos and B. Dietzek, Phys. Chem. Chem. Phys., 2015, 17, 7823–7830 RSC.
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738–4748 CrossRef CAS.
- T. C. Lourenço, M. F. C. Coelho, T. C. Ramalho, D. van der Spoel and L. T. Costa, Environ. Sci. Technol., 2013, 47, 7421–7429 CrossRef PubMed.
- A. M. Schilderman, S. Raeissi and C. J. Peters, Fluid Phase Equilib., 2007, 260, 19–22 CrossRef CAS.
- A. D. Taggart, J. M. Evans, L. Li, K. J. Lee, J. L. Dempsey, Y. Kanai and J. F. Cahoon, ACS Appl. Energy Mater., 2020, 3, 10702–10713 CrossRef CAS.
- G. Rothenberger, D. Fitzmaurice and M. Gratzel, J. Phys. Chem., 1992, 96, 5983–5986 CrossRef CAS.
Footnotes |
| † Archived raw data for this study can be found at https://doi.org/10.25405/data.ncl.21617889.v1. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and data for electrochemical measurements (linear sweeps, chronoamperometry and cyclic voltammetry), characterization data for the photocatalyst materials, XPS data, information on gas chromatography experiments and detailed transient infrared absorption spectroscopy results. See DOI: https://doi.org/10.1039/d3se00442b |
| § These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2023 |