
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of ubiquitinated proteins for biochemical and functional analysis
Julia
Kriegesmann
 and
Ashraf
Brik
*
and
Ashraf
Brik
*
Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa, Israel. E-mail: abrik@technion.ac.il
First published on 30th August 2023
Abstract
Ubiquitination plays a crucial role in controlling various biological processes such as translation, DNA repair and immune response. Protein degradation for example, is one of the main processes which is controlled by the ubiquitin system and has significant implications on human health. In order to investigate these processes and the roles played by different ubiquitination patterns on biological systems, homogeneously ubiquitinated proteins are needed. Notably, these conjugates that are made enzymatically in cells cannot be easily obtained in large amounts and high homogeneity by employing such strategies. Therefore, chemical and semisynthetic approaches have emerged to prepare different ubiquitinated proteins. In this review, we will present the key synthetic strategies and their applications for the preparation of various ubiquitinated proteins. Furthermore, the use of these precious conjugates in different biochemical and functional studies will be highlighted.
 Julia Kriegesmann | Julia Kriegesmann received her MSc in Chemical Biology from the TU Dortmund, Germany in 2016 and her PhD in Chemistry from the University of Vienna, Austria, in 2021 under the supervision of Prof. Dr Christian F. W. Becker. Julia is currently a postdoctoral researcher in the group of Prof. Ashraf Brik at the Technion – Israel Institute of Technology, where she is working on the chemical ubiquitination of proteins. |
 Ashraf Brik | Ashraf Brik is a professor at the Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, holding the Jordan and Irene Tark Academic Chair. He received his BSc in Chemistry from the Ben-Gurion University of the Negev and MSc in Chemistry from the Technion. He obtained his PhD in Chemistry in 2001 from the Technion. From 2002 to 2006, he was a Research Associate at the Scripps Research Institute. In 2007, he joined the Department of Chemistry in the Ben-Gurion University as an assistant professor and was promoted to full professor in 2012. In 2015, Prof. Brik moved to Technion. |
Introduction
Ubiquitination
Ubiquitination is one of the most important posttranslational modifications (PTMs), which influences a wide range of cellular processes, such as protein degradation by the proteasome, DNA damage response and intracellular trafficking.1,2 Ubiquitination is catalyzed by three enzymes known as the E1 ubiquitin (Ub) activating enzymes, E2 Ub conjugating enzymes and E3 Ub protein ligases (Fig. 1). In a first step, Ub, a highly conserved protein of 76 amino acids, is activated by the E1 enzyme in an ATP-dependent manner to form a thioester intermediate, which is then transferred to the E2 enzyme. Afterwards, Ub is transferred to the substrate protein by E3 ligases.3,4 Notably, there are only two E1 enzymes, 30–40 E2 enzymes and several hundred E3 ligases.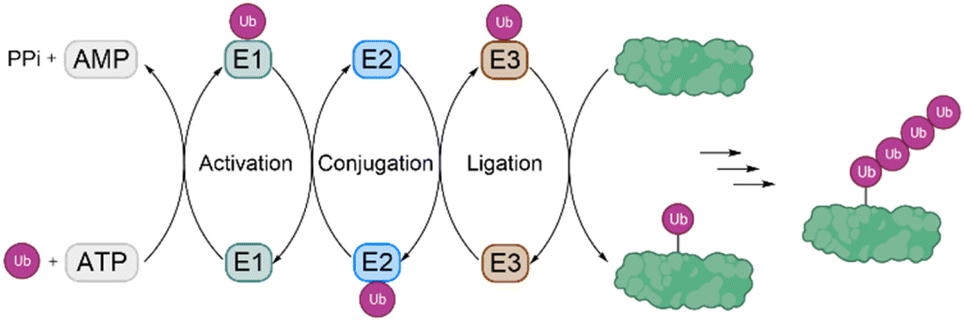 | ||
| Fig. 1 The process of ubiquitination is catalysed by the three enzymes E1, E2 and E3. Ubiquitination starts by activation of Ub by E1, followed by transfer to E2 and finally to the substrate protein by an E3 ligase. A subset of E3 ligases accepts Ub on an active site thiol before transferring it to substrates, while most of the E3 ligases only position the substrate lysine close to the E2–Ub conjugate without involving thioester formation with Ub. | ||
In this process, the C-terminus carboxy group of Ub is attached to the ε-amine of a Lys residue or the N-terminal amine5 and to a lesser extent to the side chain of Ser/Thr/Cys6 of the substrate protein. Ub can be attached as a single moiety or as a polymeric chain in which several Ub moieties are linked internally through isopeptide bonds. Based on the linkage type, the formed Ub chains adopt different conformations. This leads to a great variety of signals within cells, since all the seven lysine residues within Ub (K6, K11, K27, K29, K33, K48, K63) and Met1 can be involved in ubiquitination (Fig. 2).7 If the same residue is modified during elongation, the Ub chains are called homotypic, whereas different linkages lead to mixed chains. If a single Ub is modified with multiple Ubs, this leads to branched chains.1,5 Notably, all possible linkages have been detected in cells.8,9 Ubiquitination is a reversible PTM, in which a family of enzymes known as deubiquitinases (DUBs) removes Ub or Ub chains, which stops or edits the respective signal in cells.10,11
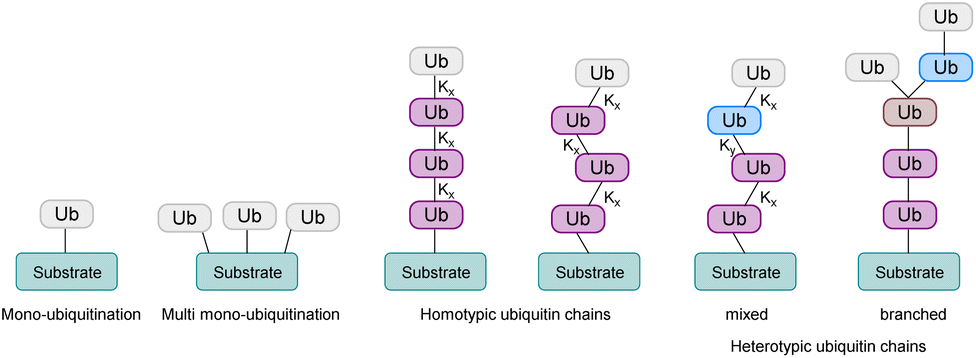 | ||
| Fig. 2 Ubiquitination of a substrate protein can either be mono- or polyubiquitination. Based on the linkages within the Ub chain, polyubiquitination can lead to homotypic or heterotypic chains, which can either be mixed or branched. x and y stand for the position of the lysine residue that is linked to the next Ub moiety. | ||
As ubiquitination and deubiquitination play important roles in many cellular signaling pathways that are relevant for human health and disease, understanding the great details of this signal is extremely important for basic research and for the development of novel therapeutics for various diseases, such as cancer, among several others.
Chemical synthesis of proteins
To study the role and mechanism of PTMs such as ubiquitination in great details, the modified protein must be prepared in homogeneous form and workable quantities. The generation of defined modified conjugates by enzymatic methods is challenging, especially in the context of ubiquitination, as most of the E3 ligases are promiscuous and ubiquitinate target proteins either on several lysine residues or lead to mono-as well as polyubiquitination.In the last two decades, several synthetic and semisynthetic strategies have been developed to generate homogeneously modified proteins containing either the native bond or unnatural linkages between the respective modification and the substrate. These methods rely on protein expression, chemical synthesis or semisynthesis (Fig. 3) and have been reviewed elsewhere.12–18 Genetic code expansion has been used to express proteins containing for example an azide and alkyne functionality in order to link the Ub and the protein substrate using Cu-catalyzed azide–alkyne cycloaddition (CuAAC).19 Ligation methods such as Native Chemical Ligation (NCL) have been proven to be a very suitable tool for chemically synthesizing proteins, by linking peptide segments to form the polypeptide chains. In NCL, often a peptide containing a C-terminal thioester moiety is ligated with another segment bearing an N-terminal cysteine residue to form the native bond at the ligation site.20
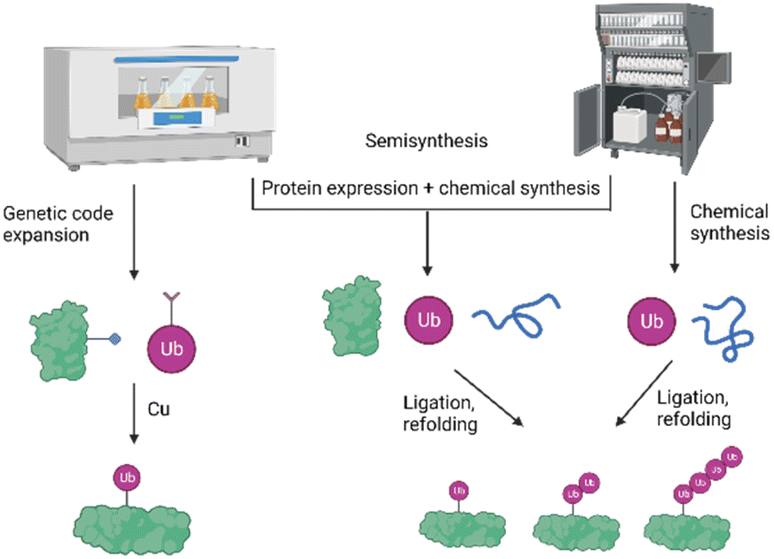 | ||
| Fig. 3 Schematic presentation of the different strategies for the preparation of ubiquitinated proteins based on protein expression, chemical and semisynthetic strategies. | ||
Chemical synthesis of ubiquitinated proteins
While there are several excellent reviews about the chemistry and biology of ubiquitin signaling,21–24 in this review we will focus only on the different strategies for the preparation of ubiquitinated proteins, containing native or unnatural linkages. We will also emphasize the studies that have been performed with these conjugates to shed light on interesting biochemical, structural and functional aspects of the Ub system.Synthesis of monoubiquitinated proteins containing a native isopeptide linkage and their biological implications
Different PTMs of histones such as acetylation, methylation, phosphorylation and ubiquitination have been described, which can in general either disrupt contacts between nucleosomes or recruit non-histone proteins. This regulates different processes such as transcription, DNA repair and replication.26 Crosstalk between these PTMs either on the same histone or within different histones provides an additional layer of regulation and specificity as the activity of the enzyme for the second PTM is often controlled by the first modification.27
Ubiquitination of histones produces the most representative functional interplays with other histone PTMs as it provides steric bulk and interacting surfaces.28 Ubiquitination-dependent histone crosstalk can be classified into three types: in the first case, histone ubiquitination promotes the installation or removal of the second modification by increasing the binding affinity of an enzyme for the nucleosome. In the second case, the Ub on the histone directly interacts with the enzymes to restrict the active conformations, but without increasing the binding affinities of enzymes for the nucleosome. In the last case, ubiquitination leads to structural rearrangements of an enzyme to ease auto-inhibition or activating activity of the catalytic subunit.29 Decoding the role of histone PTMs is important to understand fundamental processes of epigenetic regulation in health and diseases.
It is known that monoubiquitination at K120 in human histone H2B (H2BK120-Ub) plays important roles in transcriptional elongation and trans-tail histone H3 methylation.30 The proteins hSet1, a member of the MLL protein family, and hDot1L have been shown to methylate the H3 residues K4 and K79, respectively, in a H2BK120-Ub-dependent manner.31,32 Chromosomal translocation of the MLL protein, which results in its fusion with protein partners such as AF-10, have been shown to mistarget hDot1L activity to a subset of hox genes. This leads to hypermethylation and overexpression of these genes and causes acute myeloid or acute lymphoblastic leukemias.33
Understanding how H2B-Ub stimulates trans-tail hDot1L activity would not only enhance our understanding of the role of this modification in epigenetic control mechanisms but could also lead to new therapeutic strategies to target leukemias mediated by hDot1L activity.34,35
In 2008, the Muir group aimed to shed light on the role of H2BK120-Ub in H3 K79 methylation.32 The group developed a strategy for the synthesis of homogeneously ubiquitinated H2B based on expressed protein ligation (EPL) of three different fragments (Fig. 4A). EPL is used to link recombinant and synthetic polypeptides by an amide bond, one containing a C-terminal thioester and the other one an N-terminal cysteine. As no native cysteine is present in H2B and Ub, two traceless ligation strategies were needed to prepare native H2BK120-Ub. For the first ligation, the group used a photolytically removable thiol-bearing ligation auxiliary linked to the K120 side chain of H2B. To exclude double ubiquitination during ligation with the recombinant Ub thioester, the cysteine of this H2B fragment was protected with the photolabile S-(o-nitrobenzyl) group. After removal of the auxiliary and the cysteine protecting group, the ubiquitinated H2B fragment was ligated to the recombinant thioester H2B fragment. A final desulfurization step gave the native H2BK120-Ub.
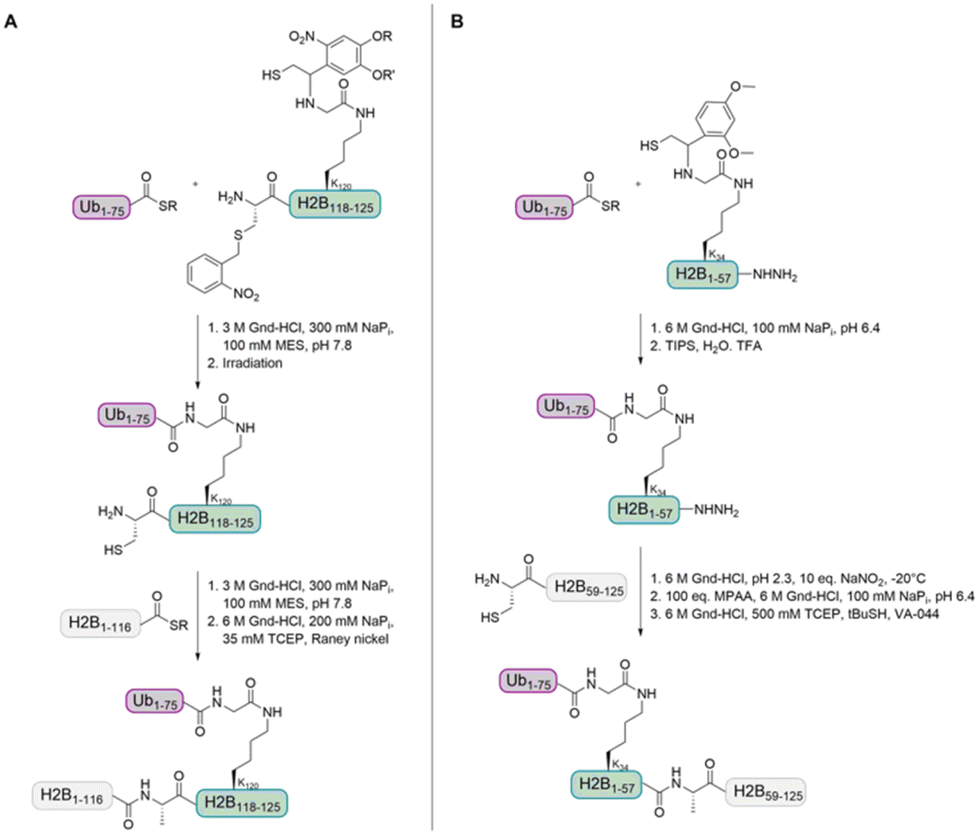 | ||
| Fig. 4 Two different auxiliary-based ligation strategies for the preparation of ubiquitinated H2B. (A) The Muir group used an H2B fragment with a photolytically removable auxiliary containing a nitrobenzyl-protected cysteine that was linked to the Ub-thioester. After cysteine deprotection, the second H2B thioester fragment was ligated. (B) The Liu group used an H2B fragment with an acid-labile auxiliary and a C-terminal hydrazide. After ligation to the Ub-thioester, the hydrazide was activated and ligated with the second H2B cysteine fragment. | ||
The H2BK120-Ub was incorporated into histone octamers with recombinant H2A, H3 and H4 proteins and these were used to reconstitute mononucleosomes. With these nucleosomes, the effect of H2BK120-Ub on Dot1 methyltransferase activity was investigated by a 3H-SAM methyltransferase assay. Methyltransferase activity was only detected in mononucleosomes containing H2BK120-Ub and not in unmodified nucleosomes. This can be explained by the proximity of H3 K79 and H2B K120, which builds the structural basis for this crosstalk as was revealed by cryo-electron microscopy (EM) studies.36,37 Docking studies have shown that the catalytic domain of hDot1L is located adjacent to the H2B ubiquitination.38
As H2B ubiquitination is correlated with increased levels of di- and trimethylation of H3 K79 in humans39 and yeast,40 respectively, the group aimed to determine the degree of methylation occurring in their assays. In unmodified nucleosomes some monomethylation and no di- and trimethylation was observed, whereas mononucleosomes containing H2BK120-Ub showed mono- and dimethylation. No trimethylation was observed, which is in accordance with analysis of H3 K79 methylation in human cell lines.41
Recently, Liu and co-workers compared Dot1L activities stimulated by histone H2B ubiquitinated at K34 and K120 (H2BK34-Ub and H2BK120-Ub) nucleosomes. The group prepared the natively ubiquitinated histone variants by chemical protein synthesis and auxiliary-mediated site-specific ubiquitination (Fig. 4B).29 The acid-labile auxiliary was linked to the lysine side chain (K34 or K120) of the H2B fragment containing a C-terminal hydrazide. After ligation to the Ub thioester and auxiliary removal, the hydrazide was oxidized and converted to a thioester, which allowed the ligation with the second H2B cysteine fragment. Desulfurization gave the native ubiquitinated H2B variants. These proteins were reconstituted into octamers with recombinantly expressed unmodified H2A, H3 and H4 and the methyltransferase activities of Dot1L were compared. Ubiquitinated H2B variants stimulated the catalytic activity of Dot1L on H3 K79 methylation. The activity on H2BK34-Ub and H2BK120-Ub nucleosomes was four- and seven-fold higher than on unmodified nucleosomes, respectively.
Next, the group used cryo-EM to determine the structure of Dot1L bound to the H2BK34-Ub nucleosome and to elucidate the molecular details of Dot1L stimulation by H2BK34-Ub. Notably, K79 of H3 was mutated to norleucine, which was known to increase the binding affinity between the methyltransferase and a nucleosome and traps the enzyme in an active state in a SAM-dependent manner.42 These structural investigations in combination with site-directed mutagenesis of Dot1L, maleimide footprinting and Ub displacement revealed the biochemical and structural basis for the crosstalk between histone H2B K34 ubiquitination and methylation of H3 K79 by Dot1L. This study revealed that H2BK34-Ub restricts the orientation of Dot1L, without any direct Ub-Dot1L interaction. It induces a nucleosome distortion, which orients Dot1L on the disk face of the nucleosome and positions its catalytic pocket to face H3 K79, leading to stimulation of Dot1L. The stimulation of Dot1L by H2BK34-Ub by reshaping the nucleosome core structure to accommodate the activity of histone-modifying enzymes, represents a new mode of trans-histone crosstalk, which may also account for other histone crosstalks.29
Notably, the Brik group previously prepared histone H2B monoubiquitinated at K34 by convergent chemical synthesis.43 This method also allowed the synthesis of doubly modified H2B with Ub and N-acetylglucosamine.44 The glycosylation of H2B on S112 (H2BS112-GlcNAc) was found to promote its ubiquitination on K120 (ref. 45) and the preparation of H2BS112-GlcNAc-K120-Ub will allow to understand the mechanism behind this. Interestingly, the group compared the total chemical synthesis of four H2B variants (monoubiquitinated at K34 or K120, glycosylation at S112 and doubly modified H2B at S112 and K120) from four fragments by convergent and one-pot approaches. This can be used as a guideline when selecting the most efficient approach for the preparation of complex protein targets.
In the context of PTM crosstalks, experiments with oligonucleosomes reconstituted from homo sapiens histones and containing uniformly ubiquitinated H2A at K119 (H2AK119-Ub), purified from mammalian cells, have shown that H2A K119 ubiquitination inhibits the activities of several H3 K36-specific methyltransferases, leading to negative regulation of H3 K36 methylation levels.46 However, it remained unclear if the inhibition was caused by the Ub present in the same or the neighbouring nucleosome in folded oligonucleosomes.
Therefore, the Rhodes and Liu groups prepared monoubiquitinated histone H2A at K119 (H2AK119-Ub) by genetic incorporation of azidonorleucine in combination with auxiliary-assisted NCL and compared the enzymatic activity of NSD2 and SETD2 (methyltransferases for H3 K36-specific di- and tri-methylation, respectively) on unmodified and ubiquitinated nucleosome core particles.47 The levels of di- and tri-methylation were significantly decreased on ubiquitinated nucleosome core particles (NCPs). In order to confirm that this was indeed caused by Ub, the group removed Ub from H2AK119-Ub by USP21 treatment, which restored the levels of di- and tri-methylation. Furthermore, the group confirmed the inhibitory role of H2A ubiquitination on H3 K36 methylation by the specific methyltransferases in mononucleosomes reconstituted from Xenopus laevis histones, which explains the observation that H2A ubiquitination and H3 K36 methylation rarely coexist in vivo.46
Although mostly the crosstalk of PTMs within different histones was investigated, an example for the preparation of one histone containing two PTMs was also reported. The Li group developed the total chemical synthesis of H3 containing K56 acetylation and K122 ubiquitination (H3K56-Ac/K122-Ub).48 Combining standard hydrazide-based NCL with auxiliary-mediated ligation for site-specific ubiquitination led to the desired product on the tens of milligrams scale.
The critical and rate-limiting step in nucleosome assembly is believed to be the deposition of (H3–H4)2 tetramers,49 which has been speculated to be facilitated by the site-specific acetylation and ubiquitination of histone H3. With this doubly modified H3 variant the group aimed to investigate this hypothesis, but during reconstitution of the three tetramers (H3, H3K56-Ac and H3K56-Ac/K122-Ub) with unmodified H4, the experiment with the ubiquitinated variant failed. It was suggested that chaperones are required to place H3K56-Ac/K122-Ub in an appropriate conformation to prevent nonspecific interactions in the formation of (H3–H4)2 tetramers.
Another important aspect in the context of histones is histone deubiquitination, which is involved in DNA damage repair, gene activation inhibition and chromosome condensation.50 Aberration on deubiquitination is highly associated with human diseases such as cancer, but also with aging and infertility.51 Therefore, studying DUB-mediated deubiquitination is of current interest in medicine.52
While most of the histone-associated DUBs act on a variety of substrates,53 also some site-selective histone-associated DUBs have been identified. USP51 was found to be able to cleave H2A with ubiquitination at K13 and K15 (H2AK13-Ub and H2AK15-Ub), but not K119 (H2AK119-Ub) in vivo.54
In order to study the mechanism and selectivity of USP51, the Liu group prepared H2A ubiquitinated at K13, K15 or K119 by their auxiliary-based ligation strategy.55 In contrast to the in vivo results, USP51 did not favour H2A ubiquitinated at K13 and K15 against ubiquitination at K119 in vitro. Interestingly, H2AK119-Ub was cleaved more quickly than H2AK13/15-Ub. These results suggest that other factors such as other histone-modifications or competitive reader proteins that bind to H2AK119-Ub might influence the selectivity and activity of USP51.
H2B is deubiquitinated by the Spt-Ada-Gcn5 acetyltransferase (SAGA) coactivator, which contains a subcomplex consisting of the four proteins Ubp8, Sgf11, Sus1 and Sgf37, also known as the SAGA DUB module.56–58 Deubiquitination of H2B was found to be inhibited by phosphorylation of Y57 in histone H2A.59 To study this interaction, the Wolberger and Brik groups prepared phosphorylated histone H2A (H2AY57-P) by total chemical synthesis and ubiquitinated histone H2B by semisynthesis.60 The histones H2BK120-Ub, H3, H4 and the unmodified H2A or H2AY57-P were reconstituted into octamers and the ability of the yeast SAGA DUB module to deubiquitinate H2B was compared for both cases. In nucleosomes containing H2AY57-P, the deubiquitination showed a 30-fold reduction compared to the nucleosomes containing unmodified H2A. This confirms the inhibitory effect of H2AY57-P on H2BK120-Ub deubiquitination. This is in accordance with investigations of the crystal structure of the SAGA DUB module bound to ubiquitinated nucleosomes,61 which will be described in the section related to ubiquitinated proteins containing unnatural linkages.
In order to address this problem, the Brik and Lashuel groups devised a synthetic strategy to prepare homogeneously monoubiquitinated forms of α-Syn by using the δ-mercaptolysine strategy (Fig. 5).65 The latter was installed to prompt transthioesterification with a Ub thioester followed by an S–N acyl transfer step to form the modified isopeptide linkage between the Ub and lysine of the substrate.
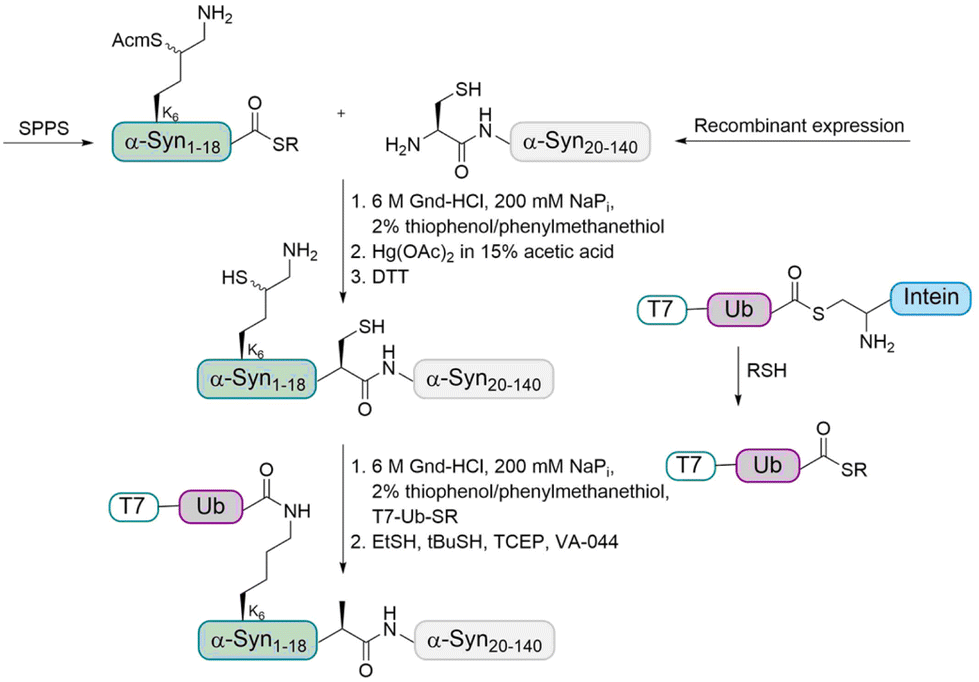 | ||
| Fig. 5 Synthesis strategy for the preparation of ubiquitinated α-Syn using δ-mercaptolysine. The N-terminal fragment of α-Syn containing Acm-protected δ-mercaptolysine at position K6 with C-terminus thioester was first linked to the recombinantly expressed C-terminal fragment containing a cysteine. After Acm removal, the Ub thioester was ligated followed by desulfurization to give the desired ubiquitinated α-Syn. | ||
As the sequence of α-Syn does not contain Cys residues, A19 was chosen as ligation site and the desulfurization of the δ-mercaptolysine after ligation led to the native monoubiquitinated protein. In more details, the fragment α-Syn19–140 containing an N-terminal Cys was expressed in E. coli and the α-Syn1–18 thioester containing a δ-mercaptolysine protected with Acm (acetamidomethyl) at position K6 was prepared using Boc SPPS. These two fragments were linked by NCL under denaturing conditions. After purification, the Acm protecting group was removed and the free δ-mercaptolysine containing polypeptide was ligated with the Ub thioester, which was followed by desulfurization to give the desired monoubiquitinated α-Syn in the purified form as confirmed by different methods.
In order to determine the effect of ubiquitination on α-Syn fibril formation, the groups compared the fibril formation of monoubiquitinated α-Syn at K6 and the wildtype (wt) protein in a thioflavin T (ThT) assay and with transmission electron microscopy (TEM). The α-Syn wt was found to form extensive fibrillar structures, whereas monoubiquitination at K6 inhibited fibril formation, suggesting that ubiquitination could occur after fibrilization. The data obtained during this study contradict the results of other studies that indicated enhancement of α-Syn aggregation by ubiquitination in vitro and in cell cultures.66 Notably, in these studies heterogenous mixtures of unmodified α-Syn and α-Syn ubiquitinated at different lysine residues have been used. This further highlights the importance of the preparation of homogeneously ubiquitinated proteins. Furthermore, it allows to investigate the crosstalk between different PTMs. In this case, the group aimed to explore the effect of ubiquitination at K6 on α-Syn phosphorylation at S87 and S129 by three different kinases, which was found not to significantly influence the extent of α-Syn phosphorylation.
The Brik group aimed to prepare ester- and isopeptide-linked ubiquitinated α-globin (modified at T127 and K127, respectively) in order to compare their behaviour with various DUBs.68 Only a few examples for the incorporation of an ester unit into proteins have been reported.69–71 The Brik group reported the first total chemical synthesis of a ubiquitinated protein, which contains an ester-linked Ub unit (Fig. 6). In detail, the group divided HA-α-globin in four fragments that were prepared by SPPS. The fragment containing the ester linkage at position T127 (Ub46–76–α-globin120–150) was also prepared via standard SPPS up to position 128. Afterwards, the group had to overcome the problem of cyclization at two different synthesis steps. After coupling of Fmoc-Thr-OH, the next amino acid was coupled to prevent an intramolecular attack of the Thr free amine on the ester bond. The ester bond was formed by coupling allyloxycarbonyl (Alloc) protected glycine to the free Thr hydroxyl group. After finishing the synthesis of the backbone peptide, the Alloc protecting group of the branched glycine was removed and the following Arg–Gly was coupled as a dipeptide to prevent intramolecular diketopiperazine formation. The remaining peptide was synthesized using standard SPPS. Ligations of the four fragments followed by desulfurization gave the final HA-α-globin-Ub analogue.
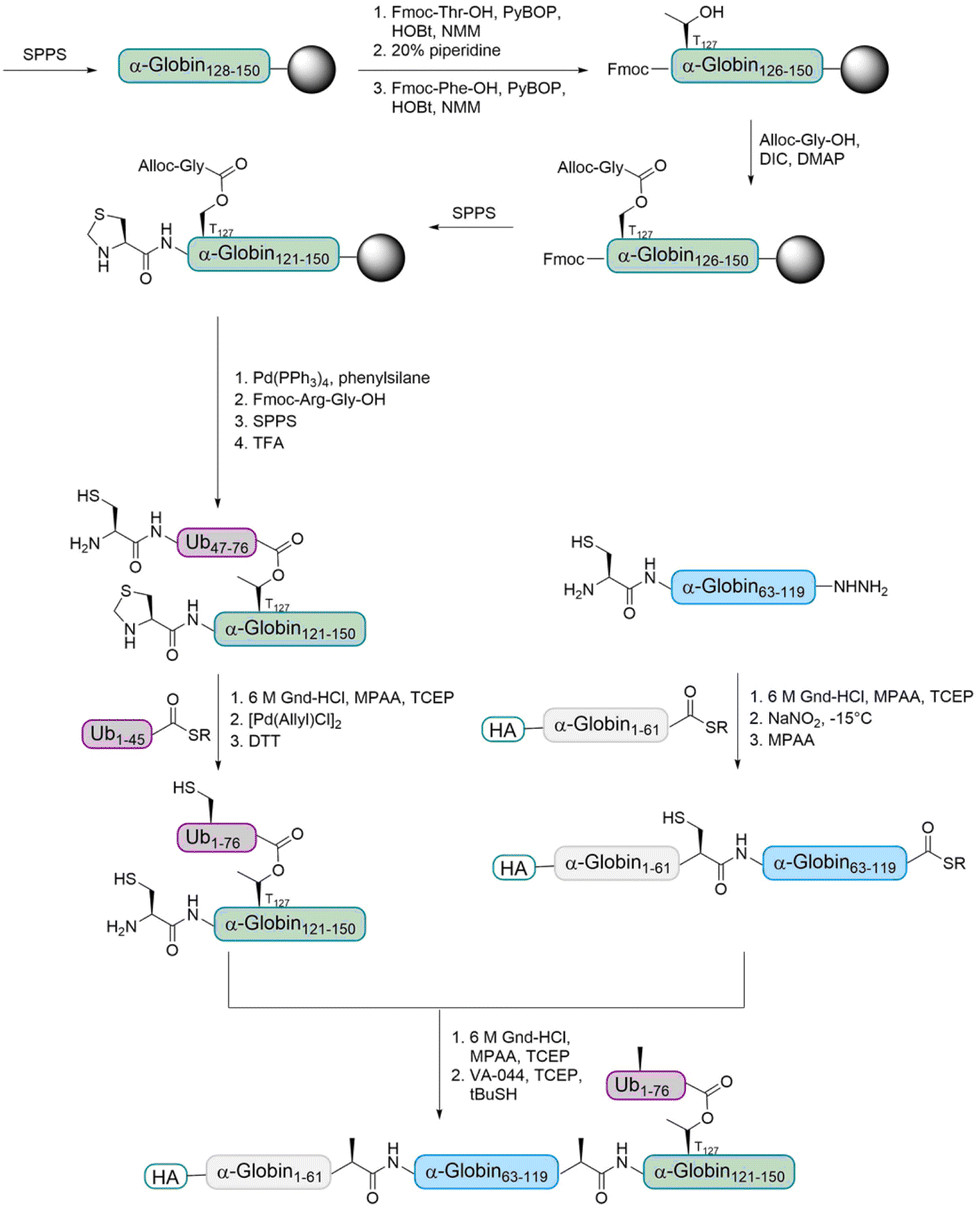 | ||
| Fig. 6 Synthesis strategy for the preparation of ubiquitinated α-globin containing an ester bond. The α-globin121–150 fragment was synthesized by SPPS and introduction of an Alloc-protected glycine onto a tyrosine side chain led to the desired ester bond between Ub and α-globin. | ||
With both HA-α-globin-Ub variants in hand, the group compared the recognition and stability of the ester- and isopeptide linkage in DUBs cleavage assays. Therefore, the variants were incubated with different purified DUBs and the cleavage efficiency was evaluated. A detailed comparison of the cleavage efficiency by USP2 showed that the isopeptide linked HA-α-globin-Ub undergoes faster cleavage than the ester-linked variant. After a short incubation time, over 70% of the isopeptide variant underwent cleavage, while it was only around 30% for the ester-linked variant. Analysis with USP15 showed comparable differences in the cleavage of the two variants, but it was faster for both compared to USP2. Importantly, these findings show, for the first time, that DUBs can cleave an ester bond between Ub and another protein.
Okamoto and co-workers developed a one-pot ligation strategy, which enabled the introduction of several PTMs into the HP1α protein, including ubiquitination, phosphorylation, citrullination and acetylation.73 In the context of HP1α ubiquitination, they focused on the K154 ubiquitination site which has been proposed to promote the degradation of HP1α through the autophagy pathway to enable efficient DNA repair. As K154 is located at the chromo shadow domain, which is responsible for the self-dimerization of HP1α, the group suggested that steric hindrance caused by the Ub disturbs the dimerization.
Synthesis of polyubiquitinated proteins containing a native isopeptide linkage and their biological implications
In addition to monoubiquitinated proteins, polyubiquitinated proteins containing a natural linkage between the substrate protein and the Ub chain have been synthesized, however to a lesser extent.For the first time, the synthesis of diubiquitinated histone H2A (H2AKX-di-Ub) was recently established by the Liu group74 to investigate its interaction with p53-binding protein 1 (53BP1), which is a critical regulator of cellular response to DNA double strand breaks (DSBs). 53BP1 binds to an NCP containing dimethylated H4 at K20 and ubiquitinated H2A. 53BP1 was considered as a specific reader of H2AK15-Ub,75 but as the Ub ligase RNF168 ubiquitinates H2A at K15 and K13 without selectivity and introduces polyubiquitination, this raised the question whether 53BP1 in addition to these events, is also a reader of H2AK13-Ub or diubiquitinated H2A. To answer this question, the Liu group synthesized ubiquitinated H2A through convergent ligation combined with desulfurization.74
The four synthetic H2A variants (mono- and di-ubiquitinated at K13 or K15) were incorporated into histone octamers and nucleosomes with recombinant H2B, H3 and chemically synthesized histone H4 dimethylated at K20 (H4K20-di-Me). In order to investigate the interaction between 53BP1 and the modified NCPs, the group performed pull-down experiments with GST-53BP1 fusion proteins.75 53BP1 selectively bound to NCPs containing H2AK15-Ub, but not H2AK13-Ub and these interactions were only detected between 53BP1 and H4K20-di-Me-containing NCPs, consistent with previous studies.75,76 Furthermore, 53BP1 was found to bind to H2AK13-di-Ub and H2AK15-di-Ub in the presence of H4K20-di-Me. The group proposed a binding model in which the 53BP1 and NCPs containing diubiquitinated H2A interact via the hydrophobic patch, H4K20-di-Me and the nucleosome acidic patch.
For the preparation of α-Syn, the α-Syn30–140 fragment, containing an N-terminal Cys, was recombinantly expressed in E. coli, while the α-Syn1–29 thioester peptide containing a δ-mercaptolysine at K12 was chemically synthesized. Ligation of these fragments gave α-Syn bearing the δ-mercaptolysine. This allowed the introduction of the tetra-Ub chain via two sequential ligation steps of di-Ub-thioester (Fig. 7). Desulfurization followed by purification via gel-eluted liquid fraction entrapment electrophoresis led to the desired tetra-ubiquitinated α-Syn1–140 in high homogeneity.
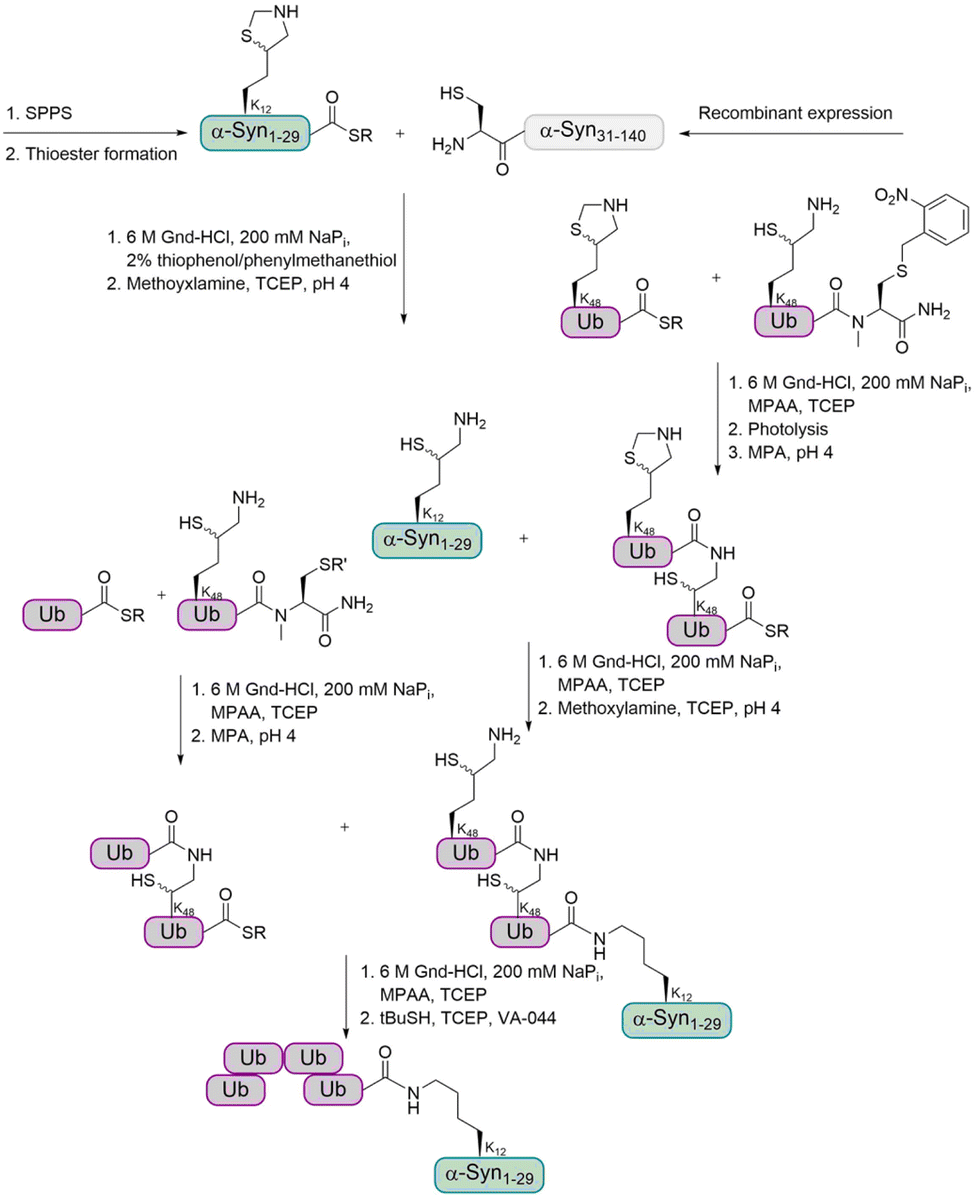 | ||
| Fig. 7 Synthetic strategy for the preparation of tetraubiquitinated α-Syn based on the use of δ-mercaptolysine. The ligation of two di-Ub fragments to the α-Syn, followed by desulfurization gave the natively tetraubiquitinated protein. | ||
In order to investigate the effect of ubiquitination on the stability of α-Syn and its degradation, the mono-, di- and tetra-ubiquitinated α-Syn variants were incubated in crude cell extract. The results clearly showed that di- and tetraubiquitinated α-Syn are more resistant to DUBs than monoubiquitinated α-Syn and are degraded by the proteasome.
In a next step, the aggregation of wt and tetraubiquitinated α-Syn were compared to find out about the effect of Ub chain length on α-Syn fibril formation. While monoubiquitination was found to stabilize monomeric α-Syn, tetraubiquitination led to the formation of nonfibrillar aggregates. These findings suggest that ubiquitination of α-Syn inhibits fibril formation and probably occurs after fibril formation. This possibly suggests that ubiquitination by the longer chains is an active cellular response to disassociate these aggregates and promote clearance of α-Syn fibrils by degradation by the proteasome.
The researchers have also studied the interplay of ubiquitination and phosphorylation and showed that the effect of phosphorylation at Y125 on α-Syn aggregation is dependent on the length of the poly-Ub chain. Monoubiquitinated α-Syn was found not to aggregate upon phosphorylation at Y125, while tetraubiquitinated α-Syn exhibited higher aggregation propensity. It has been hypothesized that the combination of tetraubiquitination and phosphorylation at Y125 induces conformational changes that lead to the formation of an aggregation-prone structure. Notably, when α-Syn is either ubiquitinated or phosphorylated, this does not influence its aggregation.
Cyclin B1 is a known substrate for ubiquitin-dependent 26S proteasome degradation, but potentially a substrate for both 20S and 26S proteasomes. It has a disordered N-terminal region, which contains 15 lysine residues that can be modified by Ub.81
The chemical synthesis of homogenously ubiquitinated proteins allows a comparison of 20S and 26S proteasomes with regard to their substrate selection and peptide-product generation. Therefore, the Brik and Glickman groups prepared mono-, di- and tetraubiquitinated cyclin B1 by using δ-mercaptolysine assisted ubiqutination.82 The Ub units were linked via K48 and attached to K64 of cyclin B1. To facilitate the tracking of specific Ub units in the chain, the proximal Ub in all chains was tagged with Myc peptide and the distal Ub with Flag.
In vitro, unmodified cyclin B1 was proteolyzed faster by purified 20S proteasomes than by 26S proteasomes. In contrast, tetra-Ub-cyclin B1 was proteolyzed faster by 26S proteasomes. The rate of cyclin B1 degradation by the 20S proteasome was proportionate to the number of Ub units attached to the same substrate. For the 26S proteasome, the inverse behavior was observed.
Interestingly, proteolysis by the 26S proteasome led to Ub recycling whereas in the 20S proteasome the Ub was proteolyzed along with its attached target protein. While the 26S proteasome showed a distinct cleavage pattern and generated longer peptides, the 20S proteasome showed greater flexibility to access potential cleavage sites and generated longer peptides. Ub units facilitated the degradation of a tagged substrate by the 26S proteasome by binding to Ub receptors. This was not the case for the 20S proteasome as it lacks Ub receptors.
The samples were incubated in rabbit fraction II, a crude cell extract containing all the UPS components required for conjugation and degradation, including E1, most of the E2s and E3s, the proteasome and a broad array of DUBs. As also observed during the studies with cyclin B1, the degradation efficiency was proportionate to the Ub chain length. The distal Ub moiety was removed by DUBs and reconjugated to other substrates in the extract. In contrast to this, the proximal Ub moiety was degraded with the substrate. For mono-Ub HA-α-globin, the Ub moiety was removed rapidly, leading to nearly no degradation of the substrate, while the tetra-Ub was found to be an effective degradation signal (Fig. 8).
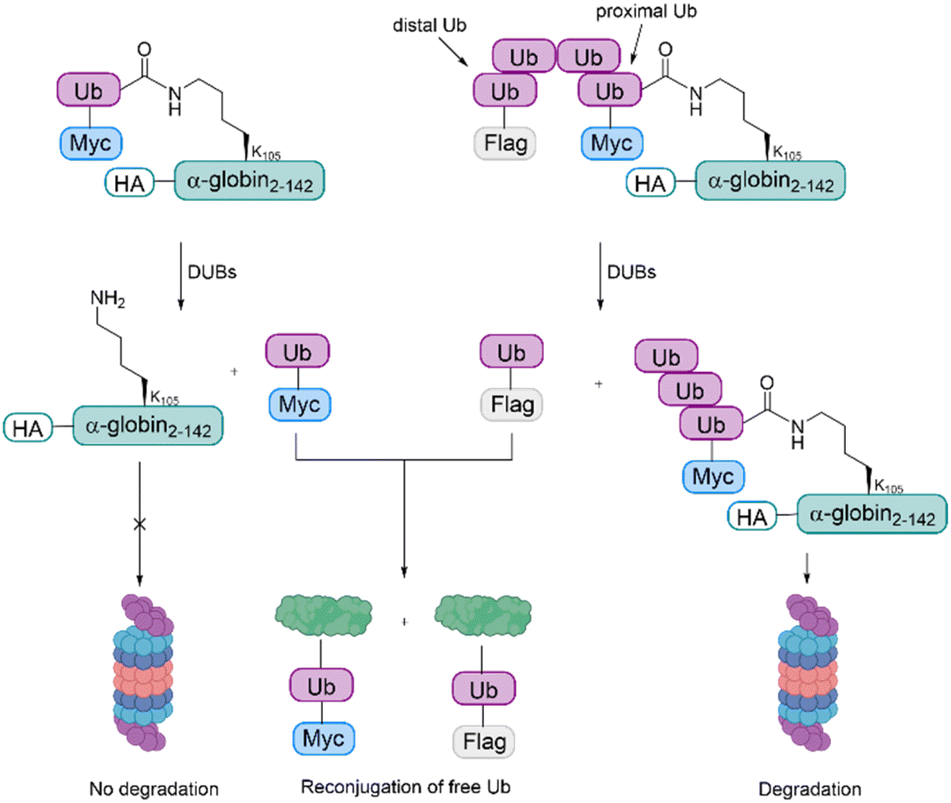 | ||
| Fig. 8 Investigation of ubiquitinated α-globin variants. For the monoubiquitinated α-globin, the Ub gets cleaved by DUBs, making degradation by the proteasome impossible. In contrast, for the tetraubiquitinated α-globin, the distal Ub gets cleaved by DUBs, but the remaining Ub chain is still a sufficient degradation signal. The free Ub moieties are reconjugated to other protein substrates. | ||
These findings suggest that proximal Ub moieties are necessary for securing the association of the substrate with the proteasome during the proteolytic process, whereas the distal Ub moieties are important for protecting the proximal moieties from premature deubiquitination. Furthermore, these studies highlight the importance of the entire repertoire of cellular DUBs in regulating the degradation of proteasomal substrates, as the mono- and tetra-Ub HA- α-globin were similarly degraded when using purified 26S proteasome for the experiments.
Synthesis of ubiquitinated proteins with unnatural linkages and their biological implications
In addition to the synthesis of native Ub conjugates, various strategies for the preparation of Ub conjugates containing unnatural linkages have been developed.Histone H2B. As described previously, ubiquitination of K120 in histone H2B plays an important role for the methylation of histone H3. Examination of the nucleosome structure revealed that several lysine residues in the C-terminal helix of H2B are solvent exposed. H2B K125 in humans85 as well as H2B K111 in yeast86 (analogous to K108 in mammals) have been reported to undergo ubiquitination in vivo, but their possible biological roles and effect on hDot1L are unknown. The Muir group aimed to investigate the crosstalk between ubiquitination and methylation. They wanted to study if the ability of H2B ubiquitination to stimulate H3 K79 methylation by hDot1L is strictly dependent on the Ub attachment site. Therefore, they envisioned the incorporation of a suitable sulfur-containing linker as a replacement of the isopeptide linkage and chose to use a disulfide-directed strategy for site specific ubiquitination of histones.87 This method is well suited especially in the context of histones, as there is only one cysteine residue in the four core mammalian histones (C110 in H3) and the mutation of this residue to alanine has no major effect on nucleosome function.88 Furthermore, Ub does not contain a cysteine residue.
For this semisynthetic strategy (Fig. 9), the fusion construct Ub-GyrA was expressed in E. coli and after purification was incubated overnight with cysteamine to yield the free Ub containing a C-terminal aminoethanethiol linker. The H2B K120C mutant was expressed in E. coli and the cysteine was activated with 2,2′-dithiobis(5-nitropyridine) (DTNP). Incubation of the activated H2B K120C mutant with two-fold molar excess of Ub at pH 6.9 for 1 h gave the desired disulfide linked monoubiquitinated H2B (H2B-Ub(ss)). The resulting linkage between Ub and H2B was around 2.4 Å longer than the isopeptide linkage and due to the disulfide bond, no reducing agents could be used in biochemical assays. Therefore, the group examined if these changes have any effect on biochemical activity, especially on hDot1L stimulation. Therefore, they prepared octamers consisting of the core histones H2A and H4, the H3 C110A mutant and either H2B, native H2B-Ub or H2B-Ub(ss). The H3 C110A mutant was used to preclude disulfide exchange with the H2B-Ub(ss) during octamer formation.
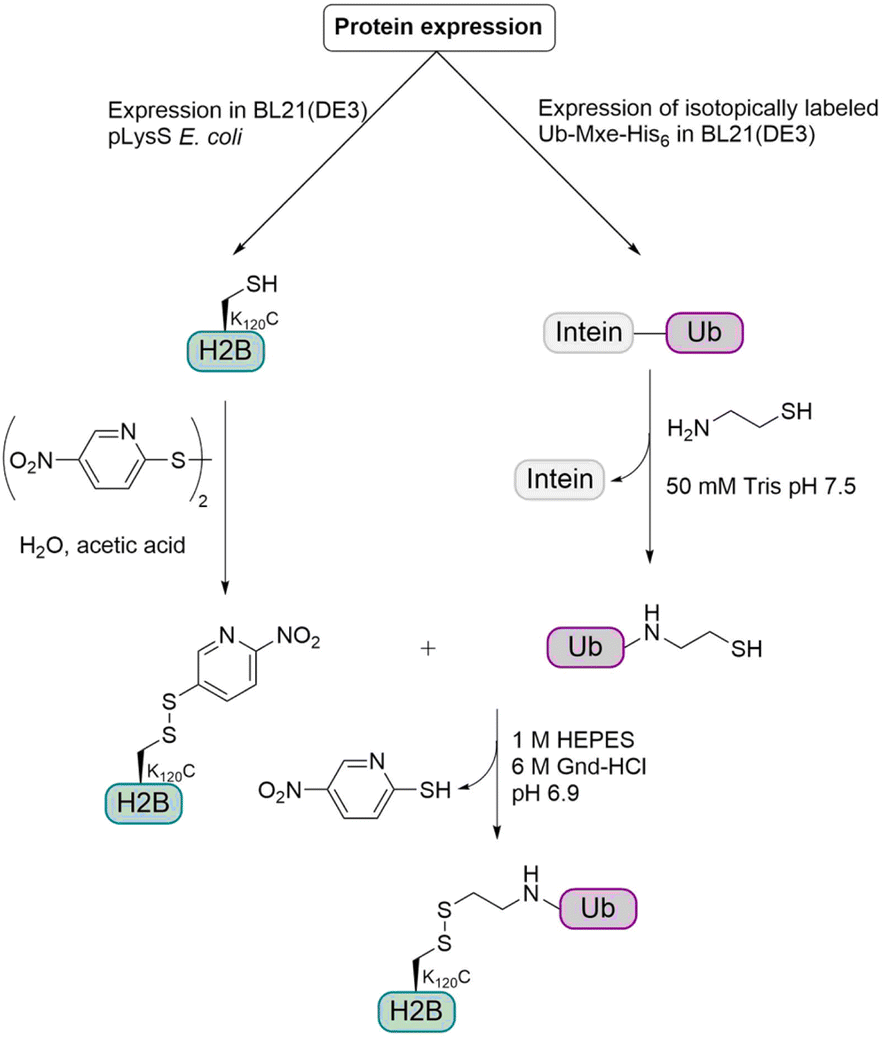 | ||
| Fig. 9 Semisynthetic strategy for the preparation of monoubiquitinated histone H2B containing a disulfide linkage. The histone H2B and the Ub were expressed in E. coli. Modification of the Ub with a thiol and activation of the H2B cysteine with DTNP allowed the formation of a disulfide bond. | ||
Notably, mononucleosomes reconstituted with H2B ubiquitinated at the positions 108 and 116 migrated faster than the other ubiquitinated mononucleosomes during native gel electrophoresis, indicating that Ub influences the structure and/or surface charge in a position-dependent manner. Structure–activity relationship studies narrowed down the region of the Ub surface required to stimulate hDot1L activity. Furthermore, it was revealed that this stimulatory effect is not strictly dependent on the Ub attachment point.
Chromatin compaction can be regulated by histone PTMs as described in the previous section.89 Acetylation of histone H4 (H4-Ac) at position K16 for example leads to fiber decompaction90 whereas its trimethylation at position K20 results in increased folding.91 It has been suggested that H2B ubiquitination increases nucleosome and DNA access to downstream factors by locally open the chromatin structure.92 But as no detailed analysis of the specific effect of H2B ubiquitination on chromatin structure has been reported, the Muir group aimed to chemically synthesize H2B-Ub and investigate the conformation and accessibility of ubiquitinated and unmodified chromatin fibers in solution. The H2B-Ub was prepared by the disulfide-directed methodology for the site-specific ubiquitination at K120.93 The group devised a method based on homo-FRET between nucleosomes that directly reports on internucleosomal distance changes in equilibrium. This allowed them to show that divalent cation-induced chromatin fiber compaction involves conformationally heterogenous intermediates. Interestingly, H4-Ac and H2B-Ub showed different effects. H4-Ac affects compaction throughout the folding transition by H4 tail binding to the H2A acidic patch.94 This leads to a reduction of closely interacting nucleosomes and prevents full fiber folding because of counteracting electrostatic repulsion. In contrast to this, H2B-Ub interferes with later stages of compaction. While transient interactions between nucleosomes are not impaired, upon further compaction the regular fiber packing is impaired, which may lead to fiber instability and local unfolding.
It was hypothesized that specific interactions between Ub and the nucleosomal surface might be required to prevent escape of Ub from the interface between nucleosomes during compaction, as the similar-sized protein Hub1 could not substitute for Ub in impairing chromatin folding. In addition, ubiquitination of H2A at the opposite site of the nucleosomal surface does not hinder fiber compaction.95 Therefore, it remains unclear how exactly Ub can influence chromatin fiber compaction.
In order to gain insights into the mechanism of chromatin decompaction, the Muir group used a hydrogen–deuterium exchange strategy coupled with NMR spectroscopy to map the parts of Ub responsible for structural effects on chromatin.96 The group prepared ubiquitinated H2B at position K120C histone via the disulfide strategy.87 Previous studies have suggested that features on the Ub surface are responsible for decompaction.93 The current study confirmed this hypothesis by revealing that the amino acids E16 and E18, within the acidic patch on Ub, are essential for decompaction as they mediate electrostatic interactions with the basic histone proteins. Interestingly, Ub-nucleosome interactions seem to be Mg2+ dependent. The addition of Mg2+ brings the surfaces of neighboring nucleosomes closer that are too far apart for electrostatic interactions in the absence of Mg2+. Besides Ub-nucleosome interactions, also Ub–Ub contacts occur in the chromatin environment, and they are important for the solubilization of chromatin polymers by preventing the establishment of a close interface between nucleosomes.96
Monoubiquitinated H2B at K120 in human (and K123 in yeast) also plays multiple roles in transcription activation97 and has been shown to be deubiquitinated by the SAGA DUB module.57 The crystal structure of the DUB module bound to ubiquitinated nucleosomes was determined by the Wolberger and Brik groups.61 Two DUB module heterotetramers are bound to a Xenopus laevis NCP containing two copies of H2B which is ubiquitinated at K120 via a non-hydrolyzable dichloroacetone linkage. The crystal structure shows the interaction of an arginine cluster on the Sgf11 zinc finger domain with the acidic patches in H2A and H2B. Furthermore, the Ubp8 catalytic domain provides additional contacts with H2B as well as with the conjugated Ub.
The groups investigated the deubiquitination of H2B by the DUB module in the presence and absence of FACT (facilitates chromatin transcription), which is a histone chaperone that mediates H2A/H2B dimer clearance and nucleosome reassembly.98 The DUB module can deubiquitinate H2B from intact as well as disassembled nucleosomes, suggesting that it could target H2B at different stages of nucleosome dis- and reassembly during transcription.
Histone H2A. In a previous study, the Liu group observed that 53BP1 is a potential reader of H2A K15 monoubiquitination as well as H2A K13 polyubiquitination.74,99 The group further aimed to investigate the interplay of multiple histone PTMs by using a synthetic ubiquitinated and phosphorylated histone variant H2AX. This synthetic target was prepared by expressed protein hydrazinolysis and auxiliary-mediated protein ligation.100 The Ub was introduced using the 1,3-dibromoacetone strategy (Fig. 10).101 With these histone variants in hand, they investigated how H2AX phosphorylation affects the recruitment of 53BP1, in the presence and absence of methylated H4 K20 and ubiquitinated H2AX K15.
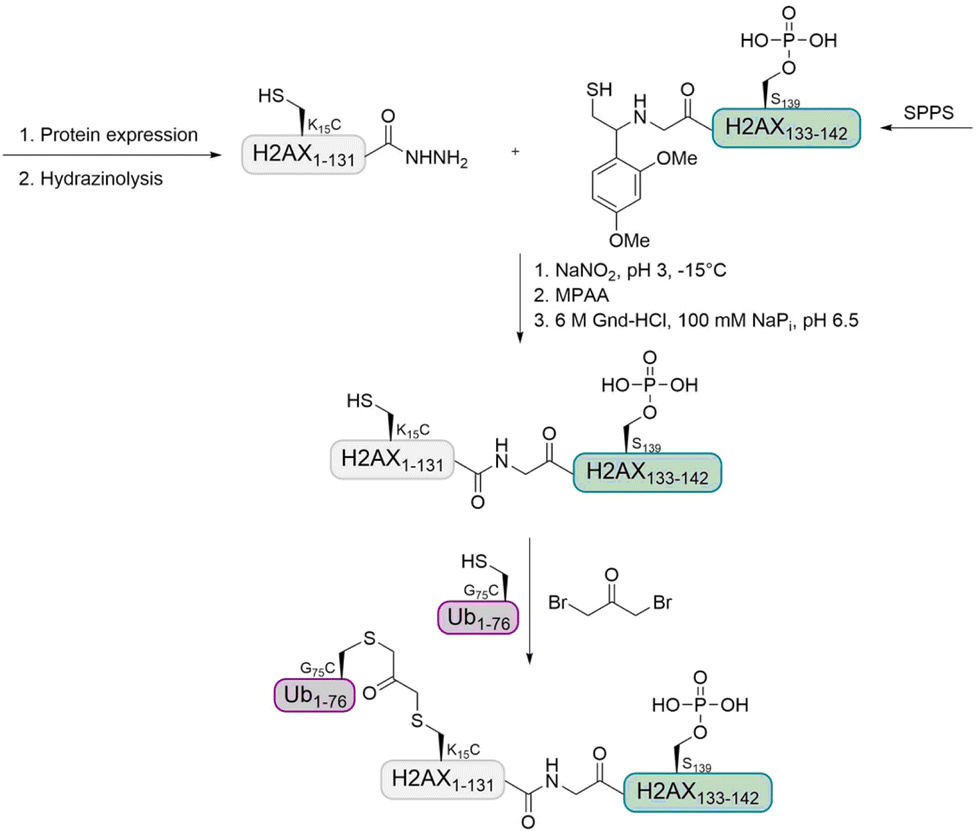 | ||
| Fig. 10 Strategy for the preparation of ubiquitinated and phosphorylated histone H2AX using auxiliary-mediated ligation and a 1,3-dibromoacetone linkage. | ||
Histone H2AX is phosphorylated by ATM kinase during the initial phase of DSB repair. Besides recruiting a host of DNA damage response factors including 53BP1 to damage foci, the phosphorylated H2AX acts as a signal to trigger further modifications on neighboring nucleosomes, such as di-methylation of H4 at K20 (H4K20-di-Me) and ubiquitination of H2A or H2AX at K15. The trivalent H2AX-nucleosomes (bearing H4K20-di-Me, H2AXK15-Ub and H2AXS139-P) and bivalent H2A-nucleosomes (bearing H4K20-di-Me and H2AK15-Ub) can recruit 53BP1, while 53BP1 does not favor the trivalent H2AX-nuleosomes over the bivalent H2A-nucleosomes. Interestingly, phosphorylation can significantly enhance the binding to unmodified NCP or NCPs, containing only ubiquitination or methylation, but not the one having both modifications. This could explain why phosphorylation is only involved in the initial phase of DSB repair, where it triggers ubiquitination and methylation to the nucleosomes.
Histone H1. Marx and co-workers prepared H1 Ub conjugates by genetic code expansion and Cu-catalyzed alkyne azide cycloaddition (Fig. 11) to characterize the ubiquitination-dependent cellular interactome for H1.102 Azidohomoalanine (Aha) is a methionine analogue that can replace its natural counterpart in an appropriate Met auxotrophic E. coli strain. The group introduced Aha at the C-terminus of Ub, by substituting the codon for the C-terminal Gly by the ATG Met coding triplet. An alkyne functionality was incorporated into the histone H1, via a pyrrolysine analogue (Plk). These analogues can be introduced in response to an amber stop codon UAG by a pyrrolysyl-tRNA synthetase/tRNA pair from Methanosarcina barkeri in E. coli.
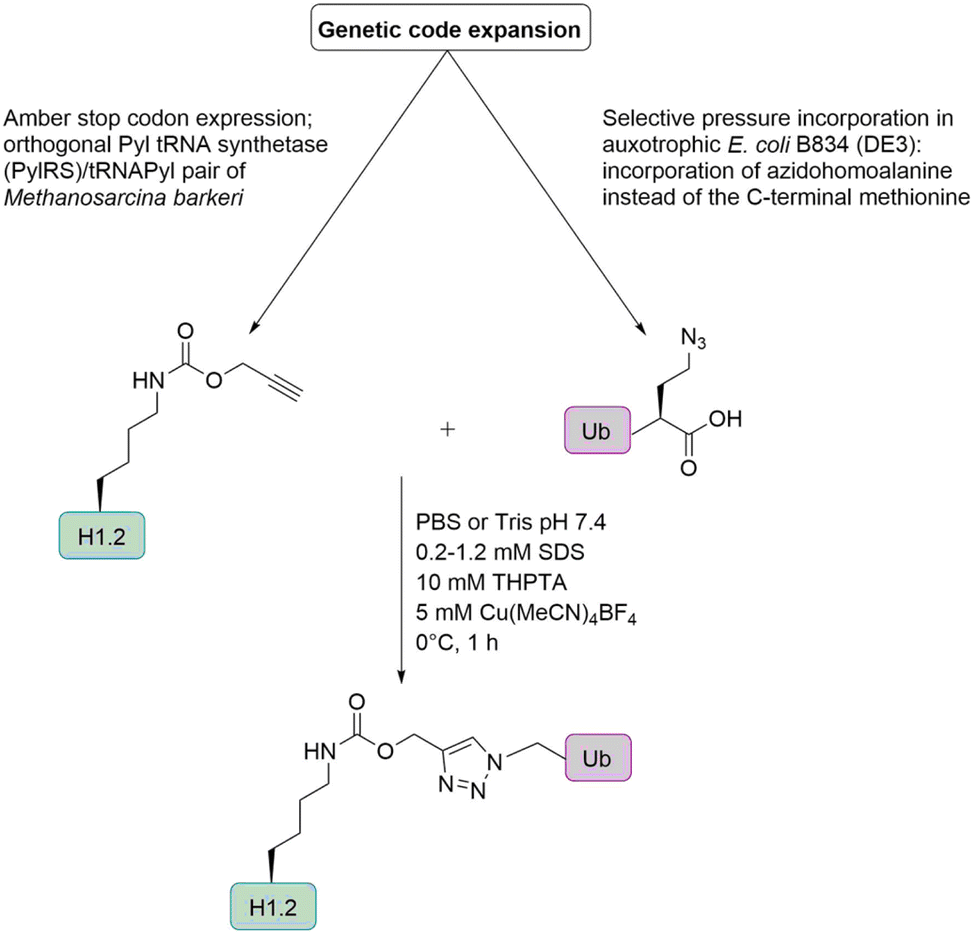 | ||
| Fig. 11 Semisynthetic strategy for the preparation of ubiquitinated histone H1 based on genetic code expansion in combination with CuAAC. | ||
In the context of histone H1, Ub was introduced at the positions K17, K64 and K206, leading to three monoubiquitinated H1 variants (H1KX-Ub) that were used in an affinity purification-mass spectrometry-based approach to identify Ub-dependent interaction partners of H1. Although most of the interactors bind to unmodified as well as ubiquitinated H1, around 20% of the proteins interacted in a Ub-specific manner. Most of the identified Ub-specific interaction partners are enzymes involved in protein modification, which suggests that ubiquitination of H1 regulates cellular functions by direct protein–protein interactions. H1K64-Ub preferentially interacts with a subset of DUBs such as USP15, USP13 and UCHL5. It also modulates the interaction with the deacetylase SIRT1 in vitro, which suggests that ubiquitination of H1 counteracts transcriptional repression. Furthermore, it affects and modulates condensate formation of H1.
While it has been shown that binding of unmodified H1 induces the nucleosome to adopt a more compact and rigid conformation as well as to promote more condensed chromatin structures, the ubiquitination of H1, especially at position K64 relaxes chromatin and results in a more open conformation. Overall, ubiquitination of histone H1 promotes a transcriptionally active state.
Notably, the group also used this method to prepare monoubiquitinated proliferating cell nuclear antigen103 and monoubiquitinated tumor suppressor p53.104 However, as the presence of copper affected the structural integrity of p53, as previously shown for other zinc-binding proteins,105 the group switched their strategy to oxime ligation as it does not require any metal ions.
α-Synuclein. In Parkinson's disease α-Syn aggregates into toxic oligomers and fibrils as we described in the previous section. In a previous study, K6-Ub α-Syn was synthesized using a semisynthetic strategy based on EPL.65 Monomeric K6-Ub α-Syn was shown to resist fibril formation when compared to the unmodified protein. As this strategy is synthetically demanding, the Pratt group used disulfide-directed ubiquitination to generate site-specifically ubiquitinated analogs representing all nine known Ub modification sites (K6, K10, K12, K21, K23, K32, K34, K46, K96).106 The group studied the effect of Ub on structure and aggregation of α-Syn by circular dichroism (CD), ThT fluorescence and TEM. The CD spectrum of α-Syn wt corresponded with a random-coil structure and the Ub showed a mixture of random-coils, α-helices and β-sheets, which are found in the correctly folded Ub structure.107 The CD spectra of the nine ubiquitinated α-Syn variants reflect a combination of the α-Syn and Ub spectra.
The effect of Ub on fibril formation was investigated by ThT assay. While unmodified α-Syn showed a rapid increase in fibril formation, ubiquitination of α-Syn had differential and site-dependent effects on α-Syn aggregation. α-Syn ubiquitination at the positions K10 and K23 led to similar levels of fibrils as the unmodified protein, but with moderate inhibition of the formation kinetics. In contrast to this, K6, K12 and K21 ubiquitination led to moderate inhibition of fibril formation. For the ubiquitinated α-Syn variants at the positions K32, K34, K43 and K96, which are all located in its middle part, no fibril formation was observed, suggesting a strong inhibitory effect. These results were also confirmed with TEM measurements after 5 days of incubation. Furthermore, it was observed that ubiquitination at K96 might promote the formation of oligomers. Notably, the obtained data is consistent with the region of α-Syn building the core of the fiber. Ubiquitination that occurs within this core region (K32, K34 and K43) completely blocked fibrillization, while ubiquitination at the N-terminus (K6, K10 and K12) did not prevent fibril formation completely.
In a similar study, the Pratt group investigated the influence of ubiquitination on α-Syn linked with the bisthioacetone (BTA) linkage.108 Interestingly, the obtained results for α-Syn BTA-ubiquitinated at K23 were not in agreement with the same α-Syn variant linked via a disulfide bond. Using the disulfide approach, the group observed that ubiquitination at K23 inhibited the kinetics of aggregation but not the formation of fibers,106 while fibrillization was blocked by ubiquitination at K23. Under mild aggregation conditions (concentration of 50 μM, agitation in a thermomixer), both inhibited α-Syn aggregation completely. However, when these conjugates were subjected to harsher aggregation conditions (concentration of 100 μM, stir-bar agitation), the disulfide-linked variant showed less inhibition, consistent with previous experiments. This highlights that different analogues of the isopeptide bond could result in different experimental outcomes and careful control must be performed.
Liu and co-workers developed the cysteine-aminoethylation-assisted chemical ubiquitination for the generation of mono- and diubiquitinated histone H2A.99 Here, an N-alkylated 2-bromoethylamine derivative was introduced at the cysteine side chain of a recombinantly expressed histone. Auxiliary-mediated NCL with recombinant hydrazides led to the ubiquitinated histone. This strategy enabled the preparation of multi-milligram quantities of homogeneously ubiquitinated histones (1.5–6 mg), since both the histones and Ub are obtained by recombinant expression.
In this study, histone H2A with monoubiquitination at K119, mono- and diubiquitination at K13 and K15 as well as histone H2B with monoubiquitination at K34 were prepared and tested for the molecular recognition of reader proteins and hydrolytic processing by DUBs. The recognition properties of NCPs containing ubiquitinated histone H2A with the interacting protein 53BP1 were similar to the natural counterparts. When treating H2AK13C-Ub with the two DUBs, YUH1 and UCHL1, it was observed that they can recognize and hydrolyze sulfur-containing isopeptide bonds.
α- and β-Globin. Preparation of tetraubiquitinated proteins containing a native linkage has only been reported by the Brik group so far, using the δ-mercaptolysine strategy.77,82,83 As the introduction of the unnatural δ-mercaptolysine can be challenging in some cases, the group also aimed to develop a more straightforward method to link the tetra-Ub to the target protein. Therefore, they envisioned to use cysteine to link the target protein to a Ub chain having an electrophilic moiety.
In a first step, the group aimed to conjugate the Ub chain to cysteine C104 of α-globin via a disulfide bond (Fig. 12A).114 This allowed the preparation of mono-, di-, tri- and tetraubiquitinated α-globin. Since the reducible disulfide linkage is not well suited for all biochemical studies, the group searched for alternative linkages. To achieve this, the group investigated the use of a stable thioether linkage. They used α-bromo acetamide and maleimide, containing an ethylamine tail, to facilitate the attachment to the Ub chain (Fig. 12B). As the number of atoms within the linkage increased, the last glycine residue (G76) of the proximal Ub was omitted. Interestingly, this method was found to allow only the preparation of mono- and diubiquitinated α-globin, but not tri- and tetraubiquitinated variants.
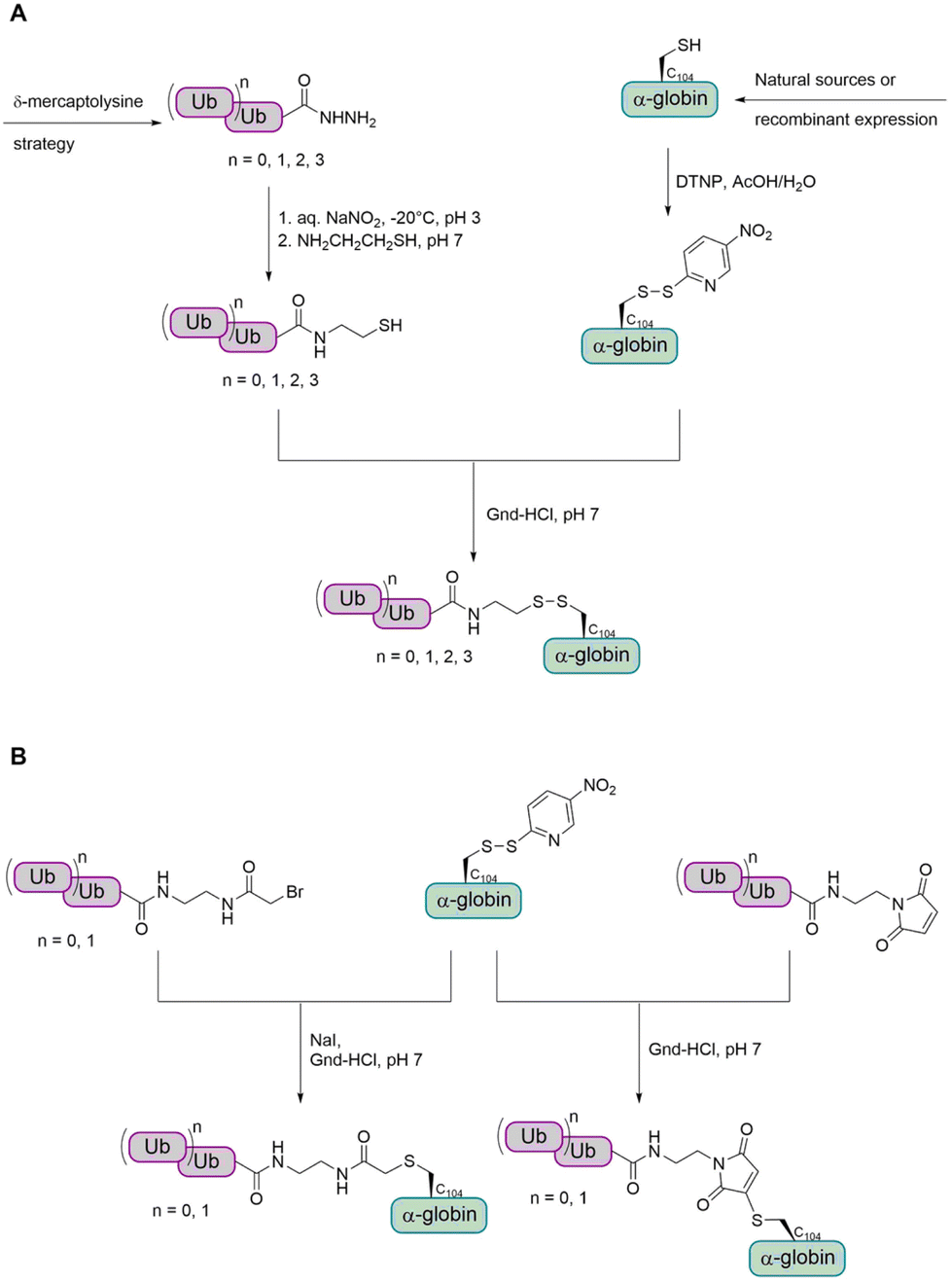 | ||
| Fig. 12 Preparation of ubiquitinated α-globin variants with different cysteine-linkages. (A) The Ub was linked to the α-globin via a reducible disulfide bond. (B) The Ub was linked to the α-globin via a stable thioether bond. | ||
As the strategy for thioether conjugation was not high yielding for di-Ub and not possible for longer chains, the group searched for a new method to obtain polyubiquitinated proteins with a stable linkage. The group reported a new strategy for the chemical synthesis of Ub conjugates containing an oxime linkage, as a replacement of the native isopeptide bond between the Ub chain and the substrate.79 This was achieved by reversing the previous approach through switching the cysteine residue to an electrophilic aldehyde group, while modifying the Ub C-terminus with a nucleophilic oxyamino. The oxyamino moiety was introduced by treating Ub-thioester with 1,2-bisaminoxy ethane.115 α-Globin was treated with chloroacetaldehyde to add an acetaldehyde moiety to the C104. The oxime ligation between the α-globin and Ub was completed within 15 min. However, when trying the ligation under the same conditions with K48-linked di-Ub, the N–O bond was cleaved to a considerable amount. Therefore, the group decided to perform the oxime ligation between α-globin and Ub first, followed by adding the Ub moieties by NCL to obtain di-, tri-, and tetraubiquitinated α-globin (Fig. 13A).
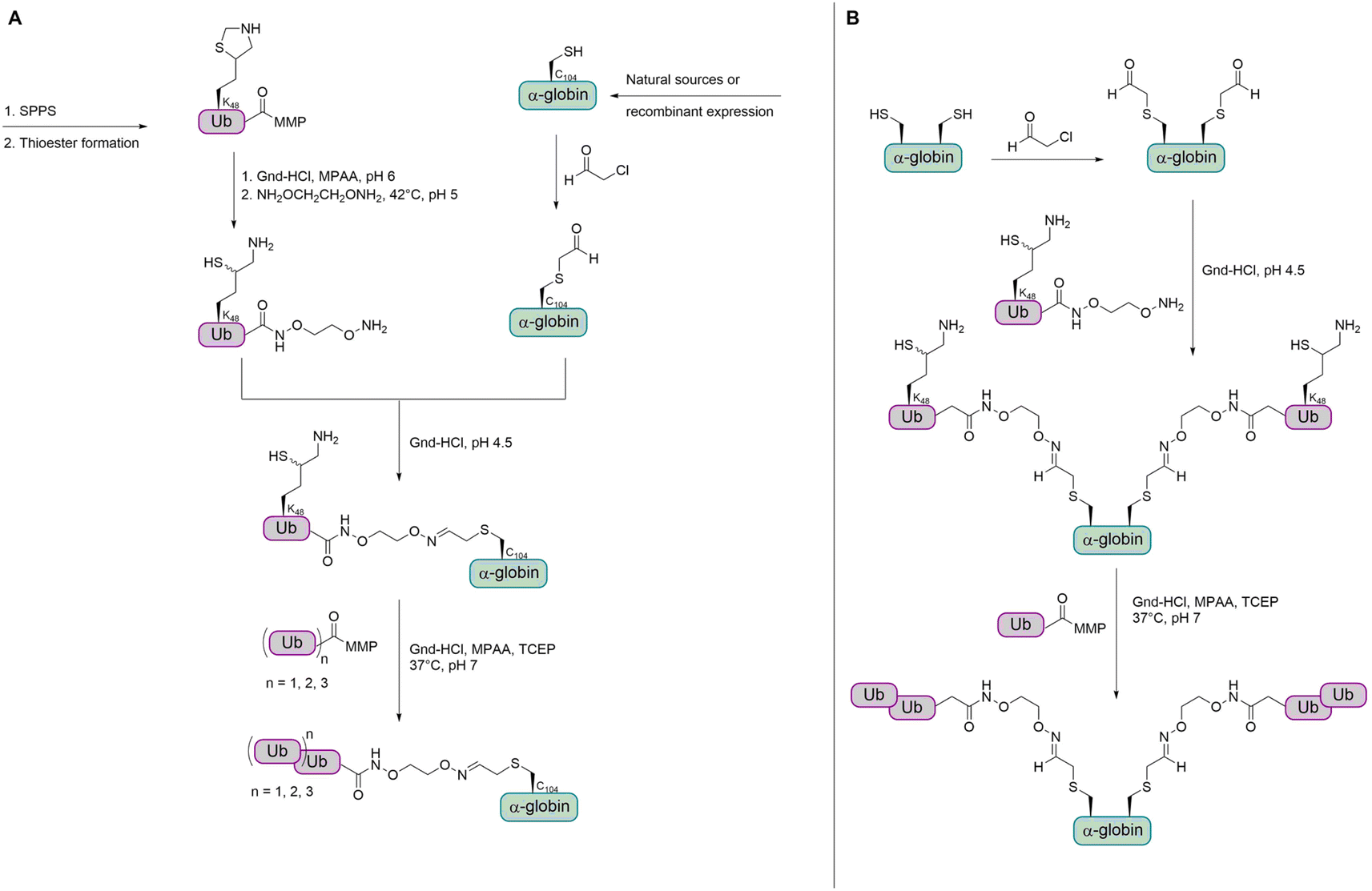 | ||
| Fig. 13 Strategy for the preparation of ubiquitinated α- and β-globin containing an oxime linkage. (A) Mono-, di-, tri- and tetraubiquitination of α-globin. (B) Bis mono- and diubiquitination of β-globin. MMP = methyl 3-mercaptopropionate. | ||
While having a DUB-stable oxime bond between the α-globin and the first ubiquitin, the other Ub moieties were linked by isopeptide bonds bearing the thiol handle, due to instability of the oxime bond during the desulfurization step. It has been shown that the thiol handle in unanchored Ub chains does not interfere with DUBs or binding to Ub binding domains.116
In order to confirm that the oxime linkage between the Ub and globin is stable in the presence of DUBs and to investigate deubiquitination and degradation in general, ubiquitinated globin variants were treated with human 26S proteasome. No degradation or deubiquitination was observed for the mono-Ub-α-globin and di-Ub-α-globin was trimmed to mono-Ub-α-globin (∼50%). Tri-Ub-α-globin was degraded to around 25% and mono- and di-Ub-α-globin was generated to an extent of around 10% and 50%, respectively. In the case of tetra-Ub-α-globin, 70% were degraded and only traces of deubiquitination could be observed. Therefore, the group wanted to test how dividing the tetra-Ub into two di-Ub modifications would affect the reaction outcome.
The group then aimed to synthesize a substrate modified with Ub at two sites and chose β-globin as it contains two cysteines (C94 and C113). Bis(mono-/di-Ub)-β-globin was prepared as described for the synthesis of modified α-globin (Fig. 13B). The degradation of bis(di-Ub)-β-globin was with 25% much lower than for the tetra-Ub-α-globin, although both proteins were modified with four Ub units in total. Instead of degradation, both Ub chains were trimmed and resulted in bis(mono-Ub)-β-globin as the main product.
Conclusions
In the last two decades, various strategies for the chemical synthesis and semisynthetic preparation of different ubiquitinated proteins were developed. This assisted many groups to shed light on the different roles of ubiquitination on various biochemical, structural and functional aspects.While most of the strategies focus on the preparation of monoubiquitinated proteins, examples on the synthesis of proteins having longer chains e.g. tetra-Ub are rare. Therefore, the community still needs to further expand these methods to prepare more complex systems having different Ub chains with different lengths and linkage types. This can be perhaps achieved by developing new synthetic strategies and/or using the existing chemical tools combined with enzymatic methods. Since when applying the E1–E3 enzymatic machinery, the specific position for modification in the target protein and the length of the Ub chain are difficult to control, the Lang and Bode groups for example have used different enzymes to install ubiquitin onto target proteins.117–119
Furthermore, the preparation of ubiquitinated proteins builds a new basis for the development of activity-based probes as the Brik group and others have shown in different examples.120–123 These activity-based probes are important tools that can for example trap DUBs, which allows profiling of their activities. In general, they can be used to answer fundamental questions and shed light on processes involving ubiquitination and deubiquitination in health and diseases. Finally, with the novel existing methods for the delivery of Ub–protein conjugates,124–128 these synthetic ubiquitinated proteins should also be studied in live cells. This will allow to investigate the influence of ubiquitination on their localization and fate as well as the interactions with other proteins. Compared to in vitro assays, different behaviours could be expected due to the native environment and their interactome that could affect their function.
Author contributions
J. K. wrote the original draft and A. B. reviewed and edited the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J. K. thanks the Alexander von Humboldt foundation for funding. A. B. holds The Jordan and Irene Tark Academic Chair. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant agreement no. 831783). Some of the figures were prepared with https://www.https://www.biorender.com.References
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- A. C. O. Vertegaal, Chem. Rev., 2011, 111, 7923–7940 CrossRef CAS PubMed.
- A. L. Haas and I. A. Rose, J. Biol. Chem., 1982, 257, 10329–10337 CrossRef CAS PubMed.
- M. H. Glickman and A. Ciechanover, Physiol. Rev., 2002, 82, 373–428 CrossRef CAS PubMed.
- R. Yau and M. Rape, Nat. Cell Biol., 2016, 18, 579–586 CrossRef CAS PubMed.
- I. Dikic and B. A. Schulman, Nat. Rev. Mol. Cell Biol., 2023, 24, 273–287 CrossRef CAS PubMed.
- C. M. Pickart and D. Fushman, Curr. Opin. Chem. Biol., 2004, 8, 610–616 CrossRef CAS PubMed.
- J. Peng, D. Schwartz, J. E. Elias, C. C. Thoreen, D. Cheng, G. Marsischky, J. Roelofs, D. Finley and S. P. Gygi, Nat. Biotechnol., 2003, 21, 921–926 CrossRef CAS.
- P. Xu, D. M. Duong, N. T. Seyfried, D. Cheng, Y. Xie, J. Robert, J. Rush, M. Hochstrasser, D. Finley and J. Peng, Cell, 2009, 137, 133–145 CrossRef CAS PubMed.
- K. R. Love, A. Catic, C. Schlieker and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 697–705 CrossRef CAS PubMed.
- F. E. Reyes-Turcu and K. D. Wilkinson, Chem. Rev., 2009, 109, 1495–1508 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- A. Brik, S. Ficht and C. H. Wong, Curr. Opin. Chem. Biol., 2006, 10, 638–644 CrossRef CAS PubMed.
- Z. Chen and P. A. Cole, Curr. Opin. Chem. Biol., 2015, 28, 115–122 CrossRef CAS PubMed.
- C. Hanna, J. Kriegesmann, L. Dowman, C. Becker and R. J. Payne, Angew. Chem., Int. Ed., 2021, e202111266 Search PubMed.
- M. Holt and T. Muir, Annu. Rev. Biochem., 2015, 84, 265–290 CrossRef CAS PubMed.
- T. Bilbrough, E. Piemontese and O. Seitz, Chem. Soc. Rev., 2022, 51, 5691–5730 RSC.
- P. Siman and A. Brik, Org. Biomol. Chem., 2012, 10, 5684–5697 RSC.
- J. Chen and Y. H. Tsai, J. Mol. Biol., 2022, 434, 167424 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- C. E. Weller, M. E. Pilkerton and C. Chatterjee, Biopolymers, 2014, 101, 144–155 CrossRef CAS.
- K. F. Witting, M. P. C. Mulder and H. Ovaa, J. Mol. Biol., 2017, 429, 3388–3394 CrossRef CAS PubMed.
- S. M. Mali, S. K. Singh, E. Eid and A. Brik, J. Am. Chem. Soc., 2017, 139, 4971–4986 CrossRef CAS PubMed.
- W. Gui, G. A. Davidson and Z. Zhuang, RSC Chem. Biol., 2021, 2, 450–467 RSC.
- R. J. Burgess and Z. Zhang, Nat. Struct. Mol. Biol., 2013, 20, 14–22 CrossRef CAS.
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef CAS PubMed.
- W. Fischle, Y. Wang and C. D. Allis, Curr. Opin. Cell Biol., 2003, 15, 172–183 CrossRef CAS.
- B. Zhu, Y. Zheng, A. D. Pham, S. S. Mandal, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Mol. Cell, 2005, 20, 601–611 CrossRef CAS PubMed.
- H. Ai, M. Sun, A. Liu, Z. Sun, T. Liu, L. Cao, L. Liang, Q. Qu, Z. Li, Z. Deng, Z. Tong, G. Chu, X. Tian, H. Deng, S. Zhao, J. Bin Li, Z. Lou and L. Liu, Nat. Chem. Biol., 2022, 18, 972–980 CrossRef CAS.
- V. M. Weake and J. L. Workman, Mol. Cell, 2008, 29, 653–663 CrossRef CAS.
- J. Kim, M. Guermah, R. K. McGinty, J. S. Lee, Z. Tang, T. A. Milne, A. Shilatifard, T. W. Muir and R. G. Roeder, Cell, 2009, 137, 459–471 CrossRef CAS.
- R. K. McGinty, J. Kim, C. Chatterjee, R. G. Roeder and T. W. Muir, Nature, 2008, 453, 812–816 CrossRef CAS.
- Y. Okada, Q. Feng, Y. Lin, Q. Jiang, Y. Li, V. M. Coffield, L. Su, G. Xu and Y. Zhang, Cell, 2005, 121, 167–178 CrossRef CAS.
- K. M. Bernt, N. Zhu, A. U. Sinha, S. Vempati, J. Faber, A. V. Krivtsov, Z. Feng, N. Punt, A. Daigle, L. Bullinger, R. M. Pollock, V. M. Richon, A. L. Kung and S. A. Armstrong, Cancer Cell, 2011, 20, 66–78 CrossRef CAS PubMed.
- C. W. Chen and S. A. Armstrong, Exp. Hematol., 2015, 43, 673–684 CrossRef CAS.
- E. J. Worden, N. A. Hoffmann, C. W. Hicks and C. Wolberger, Cell, 2019, 176, 1490–1501 CrossRef CAS PubMed.
- M. I. Valencia-Sánchez, P. De Ioannes, M. Wang, N. Vasilyev, R. Chen, E. Nudler, J. P. Armache and K. J. Armache, Mol. Cell, 2019, 74, 1010–1019 CrossRef.
- J. Min, Q. Feng, Z. Li, Y. Zhang and R. M. Xu, Cell, 2003, 112, 711–723 CrossRef CAS PubMed.
- J. Kim, S. B. Hake and R. G. Roeder, Mol. Cell, 2005, 20, 759–770 CrossRef CAS.
- M. D. Shahbazian, K. Zhang and M. Grunstein, Mol. Cell, 2005, 19, 271–277 CrossRef CAS.
- B. A. Garcia, S. B. Hake, R. L. Diaz, M. Kauer, S. A. Morris, J. Recht, J. Shabanowitz, N. Mishra, B. D. Strahl, C. D. Allis and D. F. Hunt, J. Biol. Chem., 2007, 282, 7641–7655 CrossRef CAS.
- H. Jayaram, D. Hoelper, S. U. Jain, N. Cantone, S. M. Lundgren, F. Poy, C. D. Allis, R. Cummings, S. Bellon and P. W. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6282–6287 Search PubMed.
- P. Siman, S. V. Karthikeyan, M. Nikolov, W. Fischle and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 8059–8063 CrossRef CAS PubMed.
- M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374–12378 CrossRef CAS PubMed.
- R. Fujiki, W. Hashiba, H. Sekine, A. Yokoyama, T. Chikanishi, S. Ito, Y. Imai, J. Kim, H. H. He, K. Igarashi, J. Kanno, F. Ohtake, H. Kitagawa, R. G. Roeder, M. Brown and S. Kato, Nature, 2011, 480, 557–560 CrossRef CAS PubMed.
- G. Yuan, B. Ma, W. Yuan, Z. Zhang, P. Chen, X. Ding, L. Feng, X. Shen, S. Chen, G. Li and B. Zhu, J. Biol. Chem., 2013, 288, 30832–30842 CrossRef CAS.
- X. Bi, R. Yang, X. Feng, D. Rhodes and C. F. Liu, Org. Biomol. Chem., 2016, 14, 835–839 RSC.
- Y. K. Qi, Q. Q. He, H. S. Ai, J. Guo and J. Bin Li, Chem. Commun., 2017, 53, 4148–4151 RSC.
- K. Luger, Chromosome Res., 2006, 14, 5–16 CrossRef CAS PubMed.
- J. Cao and Q. Yan, Front. Oncol., 2012, 2, 1–9 Search PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS.
- J. A. Harrigan, X. Jacq, N. M. Martin and S. P. Jackson, Nat. Rev. Drug Discovery, 2018, 17, 57–77 CrossRef CAS.
- J. I. Belle and A. Nijnik, Int. J. Biochem. Cell Biol., 2014, 50, 161–174 CrossRef CAS.
- Z. Wang, H. Zhang, J. Liu, A. Cheruiyot, J. H. Lee, T. Ordog, Z. Lou, Z. You and Z. Zhang, Genes Dev., 2016, 30, 946–959 CrossRef CAS.
- H. Ai, Y. Guo, D. Sun, S. Liu, Y. Qi, J. Guo, Q. Qu, Q. Gong, S. Zhao, J. Li and L. Liu, ChemBioChem, 2019, 20, 221–229 CrossRef CAS PubMed.
- K. W. Henry, A. Wyce, W. S. Lo, L. J. Duggan, N. C. T. Emre, C. F. Kao, L. Pillus, A. Shilatifard, M. A. Osley and S. L. Berger, Genes Dev., 2003, 17, 2648–2663 CrossRef CAS PubMed.
- N. L. Samara, A. B. Datta, C. E. Berndsen, X. Zhang, T. Yao, R. E. Cohen and C. Wolberger, Science, 2010, 328, 1025–1029 CrossRef CAS.
- A. Köhler, E. Zimmerman, M. Schneider, E. Hurt and N. Zheng, Cell, 2010, 141, 606–617 CrossRef PubMed.
- H. Basnet, X. B. Su, Y. Tan, J. Meisenhelder, D. Merkurjev, K. A. Ohgi, T. Hunter, L. Pillus and M. G. Rosenfeld, Nature, 2014, 516, 267–271 CrossRef CAS.
- M. Jbara, S. K. Maity, M. Morgan, C. Wolberger and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 4972–4976 CrossRef CAS.
- M. T. Morgan, M. Haj-Yahya, A. E. Ringel, P. Bandi, A. Brik and C. Wolberger, Science, 2016, 351, 725–728 CrossRef CAS.
- M. G. Spillantini, T. D. Bird and B. Ghetti, Brain Pathol., 1998, 8, 387–402 CrossRef CAS PubMed.
- H. A. Lashuel, Curr. Opin. Chem. Biol., 2021, 64, 67–75 CrossRef CAS PubMed.
- D. M. Sampathu, B. I. Giasson, A. C. Pawlyk, J. Q. Trojanowski and V. M. Y. Lee, Am. J. Pathol., 2003, 163, 91–100 CrossRef CAS PubMed.
- M. Hejjaoui, M. Haj-Yahya, K. S. A. Kumar, A. Brik and H. A. Lashuel, Angew. Chem., Int. Ed., 2011, 50, 405–409 CrossRef CAS PubMed.
- R. Rott, R. Szargel, J. Haskin, V. Shani, A. Shainskaya, I. Manov, E. Liani, E. Avraham and S. Engelender, J. Biol. Chem., 2008, 283, 3316–3328 CrossRef CAS PubMed.
- K. Cadwell and L. Coscoy, Science, 2005, 309, 127–130 CrossRef CAS PubMed.
- H. Sun, R. Meledin, S. M. Mali and A. Brik, Chem. Sci., 2018, 9, 1661–1665 RSC.
- G. S. Beligere and P. E. Dawson, J. Am. Chem. Soc., 2000, 122, 12079–12082 CrossRef CAS.
- Y. Sohma, Q. X. Hua, J. Whittaker, M. A. Weiss and S. B. H. Kent, Angew. Chem., Int. Ed., 2010, 49, 5489–5493 CrossRef CAS PubMed.
- Y. C. Huang, Y. M. Li, Y. Chen, M. Pan, Y. T. Li, L. Yu, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2013, 52, 4858–4862 CrossRef CAS PubMed.
- F. Burlina, A. B. M. Abdel-Aal, R. Raz, I. Pinzuti, G. Papageorgiou, J. Li, R. Antrobus, S. R. Martin, S. Kunzelmann, B. Stieglitz and J. Offer, Commun. Chem., 2019, 2, 1–10 CrossRef CAS.
- N. Kamo, T. Kujirai, H. Kurumizaka, H. Murakami, G. Hayashi and A. Okamoto, Chem. Sci., 2021, 12, 5926–5937 RSC.
- J.-B. Li, Y.-K. Qi, Q.-Q. He, H.-S. Ai, S. Liu, J.-X. Wang, J.-S. Zheng, L. Liu and C. Tian, Cell Res., 2018, 28, 257–260 CrossRef.
- A. Fradet-Turcotte, M. D. Canny, C. Escribano-Díaz, A. Orthwein, C. C. Y. Leung, H. Huang, M. C. Landry, J. Kitevski-Leblanc, S. M. Noordermeer, F. Sicheri and D. Durocher, Nature, 2013, 499, 50–54 CrossRef CAS.
- M. D. Wilson, S. Benlekbir, A. Fradet-Turcotte, A. Sherker, J. P. Julien, A. McEwan, S. M. Noordermeer, F. Sicheri, J. L. Rubinstein and D. Durocher, Nature, 2016, 536, 100–103 CrossRef CAS.
- M. Haj-Yahya, B. Fauvet, Y. Herman-Bachinsky, M. Hejjaoui, S. N. Bavikar, S. V. Karthikeyan, A. Ciechanover, H. A. Lashuel and A. Brik, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17726–17731 CrossRef CAS.
- D. Finley, Annu. Rev. Biochem., 2009, 78, 477–513 CrossRef CAS.
- S. K. Singh, I. Sahu, S. M. Mali, H. P. Hemantha, O. Kleifeld, M. H. Glickman and A. Brik, J. Am. Chem. Soc., 2016, 138, 16004–16015 CrossRef CAS.
- T. Mayor, M. Sharon and M. H. Glickman, Am. J. Physiol.: Cell Physiol., 2016, 311, C793–C804 CrossRef PubMed.
- H. Yamano, C. Tsurumi, J. Gannon and T. Hunt, EMBO J., 1998, 17, 5670–5678 CrossRef CAS PubMed.
- I. Sahu, S. M. Mali, P. Sulkshane, C. Xu, A. Rozenberg, R. Morag, M. P. Sahoo, S. K. Singh, Z. Ding, Y. Wang, S. Day, Y. Cong, O. Kleifeld, A. Brik and M. H. Glickman, Nat. Commun., 2021, 12, 6173 CrossRef CAS.
- H. Sun, S. M. Mali, S. K. Singh, R. Meledin, A. Brik, Y. T. Kwon, Y. Kravtsova-Ivantsiv, B. Bercovich and A. Ciechanover, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7805–7812 CrossRef CAS PubMed.
- H. Sun and A. Brik, Acc. Chem. Res., 2019, 52, 3361–3371 CrossRef CAS PubMed.
- N. Minsky and M. Oren, Mol. Cell, 2004, 16, 631–639 CrossRef CAS PubMed.
- F. Geng and W. P. Tansey, Mol. Biol. Cell, 2008, 19, 3616–3624 CrossRef CAS PubMed.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS PubMed.
- M. D. Simon, F. Chu, L. R. Racki, C. C. de la Cruz, A. L. Burlingame, B. Panning, G. J. Narlikar and K. M. Shokat, Cell, 2007, 128, 1003–1012 CrossRef CAS PubMed.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41–45 CrossRef CAS.
- M. Shogren-Knaak, H. Ishii, J. M. Sun, M. J. Pazin, J. R. Davie and C. L. Peterson, Science, 2006, 311, 844–847 CrossRef CAS.
- X. Lu, M. D. Simon, J. V. Chodaparambil, J. C. Hansen, K. M. Shokat and K. Luger, Nat. Struct. Mol. Biol., 2008, 15, 1122–1124 CrossRef CAS PubMed.
- K. Robzyk, J. Recht and M. A. Osley, Science, 2000, 287, 501–504 CrossRef CAS PubMed.
- B. Fierz, C. Chatterjee, R. K. McGinty, M. Bar-Dagan, D. P. Raleigh and T. W. Muir, Nat. Chem. Biol., 2011, 7, 113–119 CrossRef CAS PubMed.
- P. J. J. Robinson, W. An, A. Routh, F. Martino, L. Chapman, R. G. Roeder and D. Rhodes, J. Mol. Biol., 2008, 381, 816–825 CrossRef CAS PubMed.
- L. J. M. Jason, S. C. Moore, J. Ausió and G. Lindsey, J. Biol. Chem., 2001, 276, 14597–14601 CrossRef CAS PubMed.
- G. T. Debelouchina, K. Gerecht and T. W. Muir, Nat. Chem. Biol., 2017, 13, 105–110 CrossRef CAS.
- G. Fuchs and M. Oren, Biochim. Biophys. Acta, 2014, 1839, 694–701 CrossRef CAS PubMed.
- M. Nune, M. T. Morgan, Z. Connell, L. McCullough, M. Jbara, H. Sun, A. Brik, T. Formosa and C. Wolberger, eLife, 2019, 8, e40988 CrossRef.
- G. C. Chu, M. Pan, J. Li, S. Liu, C. Zuo, Z. Bin Tong, J. S. Bai, Q. Gong, H. Ai, J. Fan, X. Meng, Y. C. Huang, J. Shi, H. Deng, C. Tian, Y. M. Li and L. Liu, J. Am. Chem. Soc., 2019, 141, 3654–3663 CrossRef CAS PubMed.
- H. Ai, G. C. Chu, Q. Gong, Z. Bin Tong, Z. Deng, X. Liu, F. Yang, Z. Xu, J. Bin Li, C. Tian and L. Liu, J. Am. Chem. Soc., 2022, 144, 18329–18337 CrossRef CAS PubMed.
- G. C. Chu, R. Zhao, X. Wu, J. Shi and Y. M. Li, J. Org. Chem., 2020, 85, 15631–15637 CrossRef CAS PubMed.
- E. Höllmüller, S. Geigges, M. L. Niedermeier, K. M. Kammer, S. M. Kienle, D. Rösner, M. Scheffner, A. Marx and F. Stengel, Nat. Commun., 2021, 12, 1–15 CrossRef PubMed.
- S. Eger, B. Castrec, U. Hübscher, M. Scheffner, M. Rubini and A. Marx, ChemBioChem, 2011, 12, 2807–2812 CrossRef CAS PubMed.
- A. Julier, V. Radtke, A. Marx and M. Scheffner, ChemBioChem, 2022, 23, e202100659 CrossRef CAS.
- Y. Kim, S. H. Kim, D. Ferracane, J. A. Katzenellenbogen and C. M. Schroeder, Bioconjugate Chem., 2012, 23, 1891–1901 CrossRef CAS PubMed.
- F. Meier, T. Abeywardana, A. Dhall, N. P. Marotta, J. Varkey, R. Langen, C. Chatterjee and M. R. Pratt, J. Am. Chem. Soc., 2012, 134, 5468–5471 CrossRef CAS PubMed.
- J. Jenson, G. Goldstein and E. Breslow, Biochim. Biophys. Acta, Proteins Proteomics, 1980, 624, 378–385 CAS.
- Y. E. Lewis, T. Abeywardana, Y. H. Lin, A. Galesic and M. R. Pratt, ACS Chem. Biol., 2016, 11, 931–942 CrossRef CAS.
- J. S. Thrower, L. Hoffman, M. Rechsteiner and C. M. Pickart, EMBO J., 2000, 19, 94–102 CrossRef CAS PubMed.
- S. C. Boutet, M. H. Disatnik, L. S. Chan, K. Iori and T. A. Rando, Cell, 2007, 130, 349–362 CrossRef CAS PubMed.
- A. Guterman and M. H. Glickman, J. Biol. Chem., 2004, 279, 1729–1738 CrossRef CAS PubMed.
- N. Shabek, Y. Herman-Bachinsky, S. Buchsbaum, O. Lewinson, M. Haj-Yahya, M. Hejjaoui, H. A. Lashuel, T. Sommer, A. Brik and A. Ciechanover, Mol. Cell, 2012, 48, 87–97 CrossRef CAS PubMed.
- N. V. Dimova, N. A. Hathaway, B. H. Lee, D. S. Kirkpatrick, M. L. Berkowitz, S. P. Gygi, D. Finley and R. W. King, Nat. Cell Biol., 2012, 14, 168 CrossRef CAS PubMed.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS PubMed.
- L. Yi, H. Sun, Y. W. Wu, G. Triola, H. Waldmann and R. S. Goody, Angew. Chem., Int. Ed., 2010, 49, 9417–9421 CrossRef CAS PubMed.
- N. Haj-Yahya, M. Haj-Yahya, C. A. Castañeda, L. Spasser, H. P. Hemantha, M. Jbara, M. Penner, A. Ciechanover, D. Fushman and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 11149–11153 CrossRef CAS PubMed.
- M. Fottner, A. D. Brunner, V. Bittl, D. Horn-Ghetko, A. Jussupow, V. R. I. Kaila, A. Bremm and K. Lang, Nat. Chem. Biol., 2019, 15, 276–284 CrossRef CAS PubMed.
- M. Fottner, J. Heimgärtner, M. Gantz, R. Mühlhofer, T. Nast-Kolb and K. Lang, J. Am. Chem. Soc., 2022, 144, 13118–13126 CrossRef CAS PubMed.
- R. Hofmann, G. Akimoto, T. G. Wucherpfennig, C. Zeymer and J. W. Bode, Nat. Chem., 2020, 12, 1008–1015 CrossRef CAS.
- R. Meledin, S. M. Mali, O. Kleifeld and A. Brik, Angew. Chem., Int. Ed., 2018, 57, 5645–5649 CrossRef CAS PubMed.
- M. Jbara, S. Laps, M. Morgan, G. Kamnesky, G. Mann, C. Wolberger and A. Brik, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- H. Ai, Z. Tong, Z. Deng, J. Tian, L. Zhang, M. Sun, Y. Du, Z. Xu, Q. Shi, L. Liang, Q. Zheng, J. Bin Li, M. Pan and L. Liu, Chem, 2023, 9, 1221–1240 CAS.
- P. Gong, G. A. Davidson, W. Gui, K. Yang, W. P. Bozza and Z. Zhuang, Chem. Sci., 2018, 9, 7859–7865 RSC.
- G. Mann, P. Sadhu and A. Brik, ChemBioChem, 2022, 23, e202200122 CrossRef CAS PubMed.
- G. Mann, G. Satish, P. Sulkshane, S. Mandal, M. H. Glickman and A. Brik, Chem. Commun., 2021, 57, 9438–9441 RSC.
- G. Mann, P. Sadhu and A. Brik, Acc. Chem. Res., 2022, 55, 2055–2067 CrossRef CAS PubMed.
- S. Mandal and A. Brik, Chem. Commun., 2022, 58, 8782 RSC.
- A. Saha, S. Mandal, J. V. V. Arafiles, J. Gómez-González, C. P. R. Hackenberger and A. Brik, Angew. Chem., Int. Ed., 2022, 61, e202207551 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |