
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and metalation of polycatechol nanohoops derived from fluorocycloparaphenylenes†
Ashlyn A.
Kamin
 a,
Tara D.
Clayton
b,
Claire E.
Otteson
b,
Paige M.
Gannon
a,
Sebastian
Krajewski
a,
Werner
Kaminsky
a,
Ramesh
Jasti
b and
Dianne J.
Xiao
*a
a,
Tara D.
Clayton
b,
Claire E.
Otteson
b,
Paige M.
Gannon
a,
Sebastian
Krajewski
a,
Werner
Kaminsky
a,
Ramesh
Jasti
b and
Dianne J.
Xiao
*a
aDepartment of Chemistry, University of Washington, Seattle, Washington 98195, USA. E-mail: djxiao@uw.edu
bDepartment of Chemistry and Biochemistry, Materials Science Institute, Knight Campus for Accelerating Scientific Impact, University of Oregon, Eugene, Oregon 97403, USA
First published on 6th September 2023
Abstract
Due to their unique topology and distinct physical properties, cycloparaphenylenes (CPPs) are attractive building blocks for new materials synthesis. While both noncovalent interactions and irreversible covalent bonds have been used to link CPP monomers into extended materials, a coordination chemistry approach remains less explored. Here we show that nucleophilic aromatic substitution reactions can be leveraged to rapidly introduce donor groups (–OR, –SR) onto polyfluorinated CPP rings. Demethylation of methoxide-substituted CPPs produces polycatechol nanohoop ligands that are readily metalated to produce well-defined, multimetallic CPP complexes. As catechols are recurring motifs throughout coordination chemistry and dynamic covalent chemistry, the polycatechol nanohoops reported here open the door to new strategies for the bottom-up synthesis of atomically precise CPP-based materials.
Introduction
Cycloparaphenylenes ([n]CPPs, where n is the number of phenylene units) are a class of highly strained macrocyclic molecules with radially oriented π systems and unique optoelectronic properties.1–5 Ever since their initial synthesis in 2008,6 the potential to elongate these “carbon nanohoops” into homochiral carbon nanotubes and other extended materials has captivated the imagination of researchers.1,7 In 2013, [n]CPP templates (n = 9, 12) were suggested to be competent seeds for the growth of carbon nanotubes with tunable diameters.8 Recently, spurred by new advances in the synthetic chemistry of CPPs,4,9–11 functionalized CPPs have also been explored as monomers for the synthesis of novel polymers via cross-coupling12,13 and ring-opening metathesis polymerization strategies.14 However, despite tremendous strides in CPP-based materials chemistry, the initial vision of using CPPs for the bottom-up synthesis of well-defined nanotubular structures has yet to be fully realized. For example, CPP-templated carbon nanotubes still display a distribution of diameters, chiralities, and wall thicknesses.8 Similarly, CPP-based polymers are conformationally disordered and amorphous.An alternative and less-explored approach to the construction of extended materials from CPPs is via their employment as polytopic ligands. The installation of metal-binding substituents on the CPP backbone would enable coordination chemistry approaches to CPP elongation and polymerization, leading to novel CPP-based metal complexes and extended metal–organic materials. Furthermore, through the appropriate choice of metal and ligand, it should be possible to preserve the extended π-conjugation observed in carbon nanotubes.15,16 Ultimately, such a route may enable the construction of atomically precise metal–organic analogues of single-walled carbon nanotubes.
While the use of CPPs as topologically unique ligands is a tantalizing prospect, our current understanding of CPP-based coordination chemistry remains relatively limited. Thus far, only two metal-coordinating motifs have been established in CPPs: (1) the η6-coordination of metals to CPP phenylene rings, and (2) the chelation of metal cations to CPP-embedded 2,2′-bipyridyl groups. Organometallic fragments such as M(CO)3 (M = Cr, Mo, W)17 and [M(Cp)]+ (M = Ru)18,19 have been shown to bind CPP phenylene rings in a face-on fashion, generating η6-complexes. In addition, 2,2′-bipyridyl-embedded CPPs have been shown to chelate first- and second-row transition metals (M = Fe2+, Ru2+, Pd2+).20,21 In the case of Pd2+, a nanohoop dimer was crystallized, with each Pd2+ center bound by two separate bipyridyl-embedded CPP rings.20 Due to steric clash between the ortho-hydrogens of the coordinated bipyridine ligands, the Pd2+ center displays a distorted square-planar geometry and the CPP rings adopt a trans conformation. This steric strain may make it difficult to achieve extended tubular structures with bipyridine-based CPP motifs alone.
Taken together, these prior examples have established CPPs and their substituted derivatives as competent ligands. However, the continued exploration of new polytopic ligand designs is needed to realize the full promise of CPP-based metal–organic materials. Here, we illustrate how nucleophilic aromatic substitution (SNAr) reactions can be leveraged to rapidly introduce donor atoms (–OR, –SR) onto polyfluorinated [12]CPP rings. A simple two-step SNAr alkoxylation–deprotection sequence produces polytopic, catechol-based nanohoop ligands in excellent overall yields (67–79%). Finally, we show that catechol-containing CPPs readily bind transition metals such as ruthenium, generating discrete di- and hexa-ruthenium CPP complexes. Given the rich redox chemistry, electronic properties, and magnetic behavior of extended metal–catecholates,22 the new chemistry reported here represents an important step towards realizing well-defined and multifunctional CPP-based materials.
Results and discussion
Alkoxylation and thiolation of polyfluorinated arenes
Established methods for the late-stage modification of unfunctionalized CPPs are rare, and often suffer from limited scope and low yields.17,23 We hypothesized that a nucleophilic aromatic substitution strategy to derivatize fluorinated CPPs may be more effective, as it circumvents the site-selectivity challenges associated with the modification of unfunctionalized CPPs. Such a route is particularly attractive given the plethora of polyfluorinated CPPs available. Cycloparaphenylenes bearing fluorine substituents are one of the most widely reported classes of functionalized CPPs (Fig. 1a).24–28 This is largely due to the chemical inertness of the C–F bonds, which provide minimal complications during the challenging macrocycle formation and aromatization stages of CPP synthesis. Fluorine substituents have proven amenable to both the reductive aromatization25–27 and metal-mediated reductive elimination routes24,28 for synthesizing CPPs, the latter of which has been demonstrated with both Ni- and Au-based macrocyclic precursors.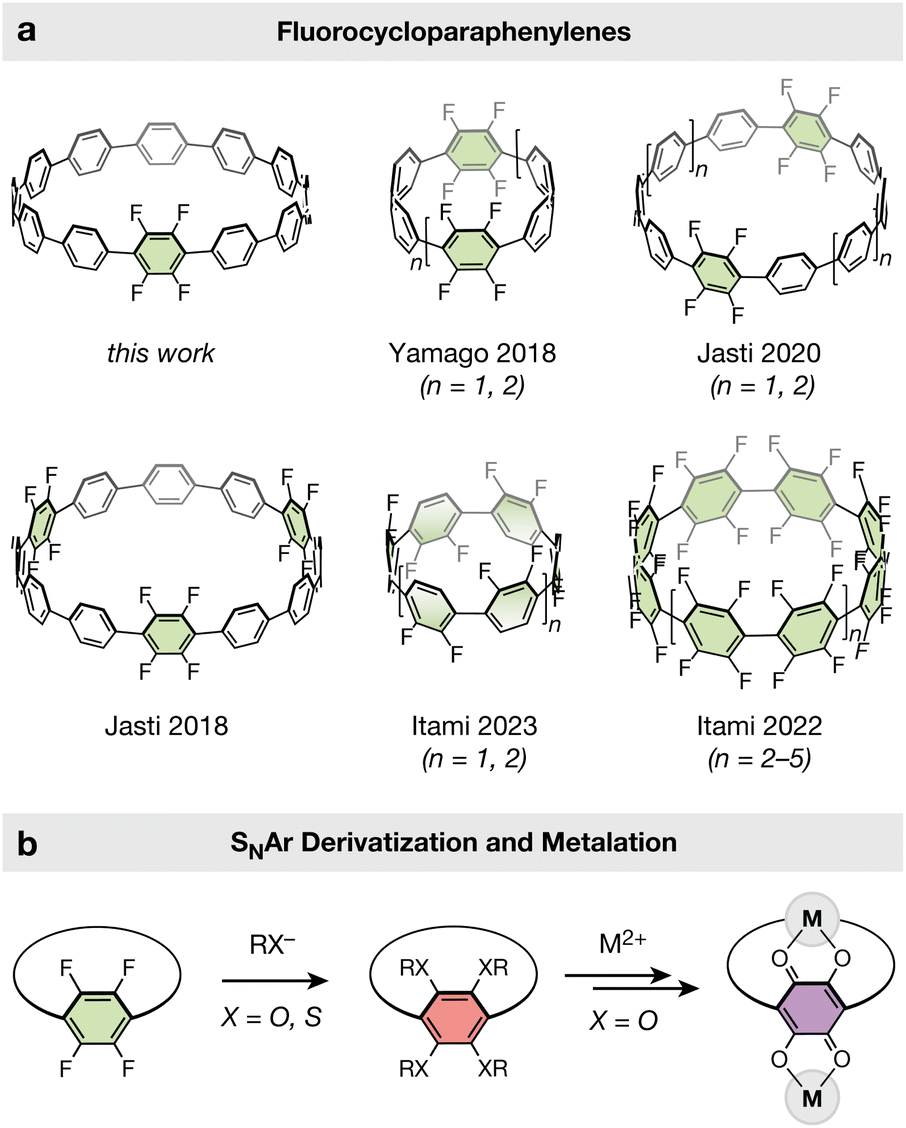 | ||
| Fig. 1 (a) Fluorine-substituted CPPs previously reported in the literature alongside the tetrafluorinated [12]CPP reported in this work. (b) Generalized late-state modification of a fluorinated CPP via nucleophilic aromatic substitution and subsequent metalation. | ||
Yamago and coworkers recently demonstrated that SNAr can be an effective tool for the late-stage modification of polyfluorinated alkene-inserted carbon nanohoops using pyrollide nucleophiles.29 Similarly, Itami and coworkers demonstrated the use of SNAr with pyrollide nucleophiles on half-fluorinated CPPs.24 We hypothesized that fluorinated CPPs could be similarly functionalized with oxygen and sulfur-based nucleophiles (Fig. 1b), affording rapid access to a diversity of new CPPs bearing alkoxy and alkylthiol substituents. Methoxy-functionalized CPPs are challenging targets for de novo CPP synthesis due to the destabilization of key cyclohexadiene intermediates by electron donation from the methoxy groups,30–34 and no alkylthiol-bearing CPPs have been reported to date.
To achieve high overall yields in polysubstitution reactions, near-quantitative yield must be observed for each individual substitution event. Because there are only a few literature reports of the exhaustive substitution of polyfluoroarenes with methoxide nucleophiles,35–38 we first optimized our synthetic conditions on a model tetrafluorinated terphenyl substrate. The tetramethoxylation of 1,2,4,5-tetrafluoro-3,6-diphenylbenzene using sodium methoxide was carefully optimized to minimize the presence of partially substituted intermediates and reaction byproducts. Solvent mixtures containing MeOH severely hindered reaction progress, but neat N,N′-dimethyl-2-imidazolidinone (DMI) or neat N-methyl-2-pyrrolidone (NMP) were found to be acceptable solvents for this reaction. Aliquots collected over 48 h and monitored by GC-MS showed that the percentage of the desired tetramethoyxylated product in the reaction mixture reached a maximum at 16 h at 80 °C. At higher temperatures, we observed partial in situ demethylation of the methoxy substituents, resulting in reaction byproducts containing one hydroxyl group. With our final optimized conditions (16 equiv. NaOMe, 80 °C, 16 h, neat NMP or DMI), the desired 1,2,4,5-tetramethoxy-3,6-diphenylbenzene could be obtained in 86% yield.
Late-stage modification of polyfluorinated [12]CPP
To test whether our optimized SNAr conditions would translate to a cyclic system, we first synthesized a new tetrafluorinated [12]CPP, 4F-CPP, which bears all four fluorine substituents on a single phenylene unit (Scheme 1). 4F-CPP was prepared using the same reductive aromatization approach employed by Jasti and coworkers in the synthesis of prior fluorinated CPPs such as 12F-CPP.26,27 Suzuki–Miyaura coupling between two curved precursors (compounds S1 and S2, see ESI†) produced the intermediate fluorinated macrocycle, 4F-MC-OTES, in 31% yield. Subsequent desilylation with tetrabutylammonium fluoride (TBAF) and acetic acid followed by reductive aromatization using H2SnCl4 gave the desired molecule, 4F-CPP (23% yield over two steps).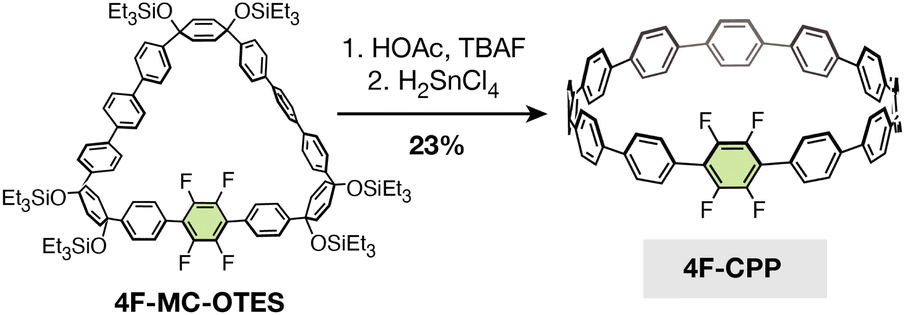 | ||
| Scheme 1 Synthesis of the tetrafluorinated [12]CPP, 4F-CPP. | ||
Excitingly, treating a solution of 4F-CPP in DMI with 16 equiv. of sodium methoxide at 80 °C in a nitrogen-filled glovebox produced the desired 4MeO-CPP in 87% yield (Fig. 2a). Successful methoxylation was confirmed by 1H NMR, 13C NMR, and high-resolution mass spectrometry (see ESI† for details). In addition, single crystals of both 4F-CPP and 4MeO-CPP suitable for X-ray crystallography were obtained by vapor diffusion of n-heptane into a solution of the CPP in trichloroethylene (Fig. 2b and c).
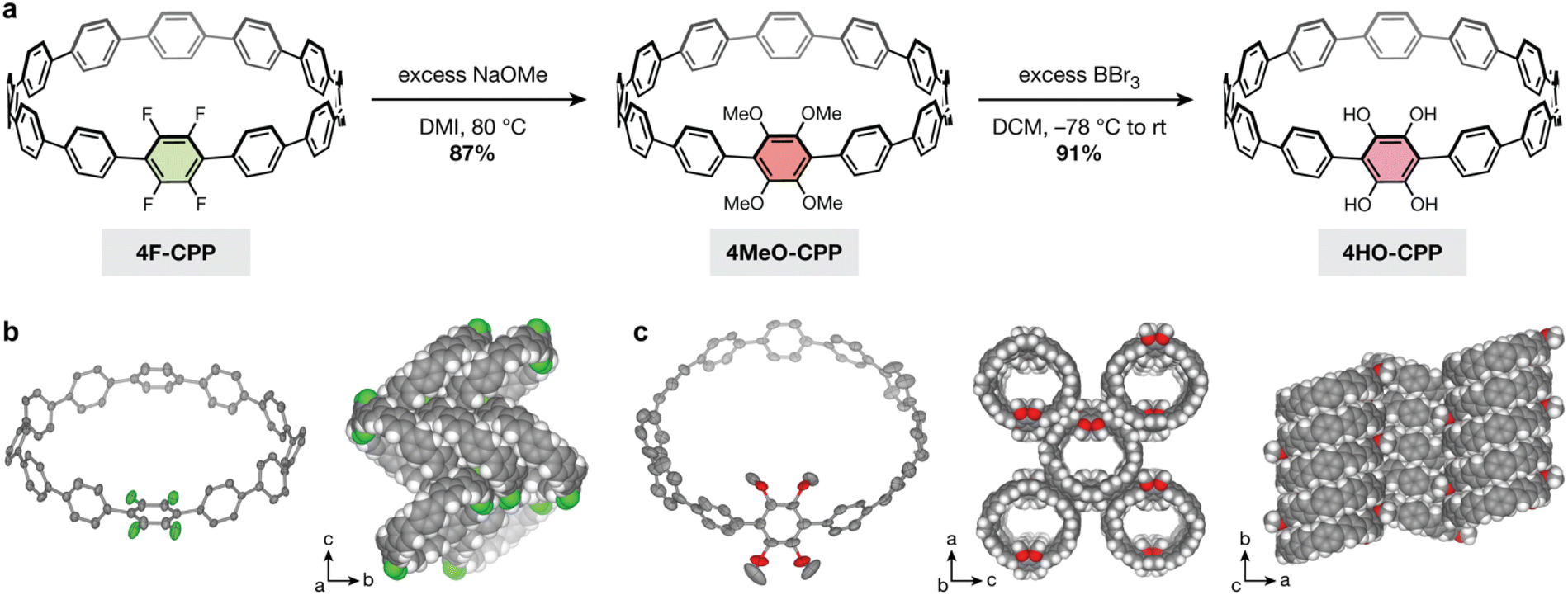 | ||
| Fig. 2 (a) Synthesis of the dicatechol-substituted [12]CPP, 4HO-CPP, via nucleophilic aromatic substitution followed by demethylation. (b) Single crystal structure and packing of 4F-CPP; the tetrafluorophenylene unit is disordered over two positions, as can be seen in the packing diagram. (c) Single crystal structure and packing of 4MeO-CPP. For all structures, thermal ellipsoids are drawn at the 50% probability level. Solvent molecules and select protons are omitted for clarity. F, O, C and H atoms are represented by green, red, gray, and white, respectively. | ||
The single-crystal structure of 4F-CPP reveals a herringbone packing structure with no individually resolved solvent molecules. The position of the tetrafluorophenylene unit within the CPP ring is disordered, giving rise to eight fluorinated positions, each with a 50% fluorine and proton site occupancy. While this herringbone packing matches the packing structure of unsubstituted [12]CPP,39 it stands in contrast to the tubular packing observed in more heavily fluorinated CPPs, which can be driven by noncovalent C–H⋯F hydrogen bonding and arene–perfluoroarene interactions.27
Unexpectedly, and unlike 4F-CPP, the single-crystal structure of 4MeO-CPP reveals a nearly tubular packing structure with one trichloroethylene molecule per CPP. Additional solvent embedded within each ring is highly disordered and could not be individually resolved. To minimize steric interactions between ortho substituents, the dihedral angles between the tetramethoxyphenylene unit and the adjacent phenylene rings average 51(7)°. This value is much larger than the values observed between the bare phenylenes (∠avg = 28(14)°) and between the phenylenes in unsubstituted [12]CPP (∠avg = 28(15)°).39
In order to employ our tetrasubstituted CPP as a ligand, it is necessary to remove the methyl groups to yield free catechol moieties. Deprotection of the aryl methyl ethers was successfully carried out by treating 4MeO-CPP with excess boron tribromide, yielding the desired dicatechol-substituted [12]CPP, 4HO-CPP, in 91% yield (79% over two steps from 4F-CPP). Due to the redox-active nature of catechols, the workup for this reaction was performed inside a nitrogen-filled glovebox to prevent ligand oxidation. The removal of all methyl groups was confirmed by the disappearance of the resonance at 3.50 ppm in the 1H NMR spectrum. Peaks at m/z = 975.3471 and 973.3321 were observed by high-resolution ESI-MS immediately following removal of 4HO-CPP from the glovebox. The first of these peaks is consistent with 4HO-CPP in the as-synthesized form (calculated m/z = 975.3469 for [M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −H]−), while the second peak corresponds to the oxidation of the dicatechol-substituted phenylene units to the corresponding 2,5-dihydroxy-1,4-benzoquinone form (calculated m/z = 973.3312 for [M−H]−).
−H]−), while the second peak corresponds to the oxidation of the dicatechol-substituted phenylene units to the corresponding 2,5-dihydroxy-1,4-benzoquinone form (calculated m/z = 973.3312 for [M−H]−).
In order to demonstrate that our SNAr method can be extended to more heavily fluorinated CPPs, we next carried out SNAr on a dodecafluorinated [12]CPP, 12F-CPP (Fig. 3a). The gram-scale synthesis of 12F-CPP was previously reported by Jasti and coworkers.26,27 The reaction of 12F-CPP with excess sodium methoxide in NMP was carried out at 80 °C in a nitrogen-filled glovebox. The desired dodecamethoxy-substituted product, 12MeO-CPP, was recovered in good yield (Fig. 3a) and fully characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (see ESI† for details).
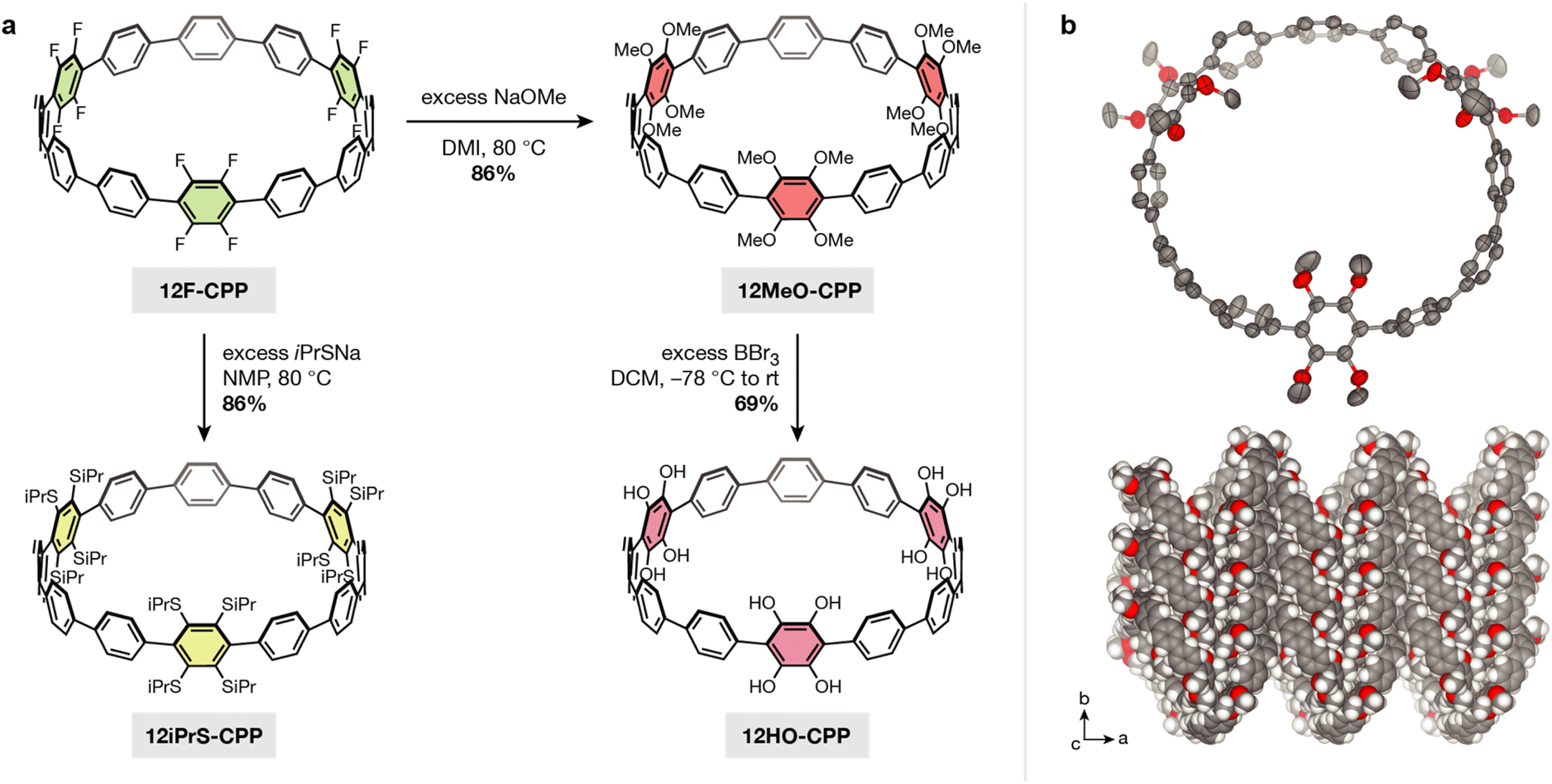 | ||
| Fig. 3 (a) Synthesis of methoxy and 2-propanethiol-substituted [12]CPPs via nucleophilic aromatic substitution. The methoxy substituents can be deprotected to yield the corresponding free catechols. (b) Single crystal structure and packing of 12MeO-CPP. Thermal ellipsoids are drawn at the 50% probability level. Solvent molecules and select protons are omitted for clarity. O, C, and H atoms are represented by red, gray, and white, respectively. | ||
We note that, under the reaction conditions required to achieve complete methoxylation of the 12F-CPP substrate, it is difficult to avoid partial demethylation of the methoxy groups into hydroxyl groups. This can be reversed by subsequent addition of methyl iodide to give pure 12MeO-CPP in 86% yield. Because the fully deprotected 12HO-CPP is our desired target, we typically proceeded forward without the addition of methyl iodide. In this case, if both 12MeO-CPP as well as partially demethylated species are taken into account, then the overall yield increases to 97%.
Diffraction-quality single crystals of 12MeO-CPP were grown via slow evaporation out of a mixture of acetone and MeOH (Fig. 3b). 12MeO-CPP adopts a herringbone-type packing with two acetone molecules embedded within each CPP. The difference in CPP packing between 12MeO-CPP and 4MeO-CPP suggests that the tubular packing in 4MeO-CPP is not a result of electronic effects from the methoxy groups, but may be a result of either the steric profile of the molecule or solvent-driven intermolecular interactions. Both the tubular and herringbone packing observed in 4MeO-CPP and 12MeO-CPP, respectively, differ from the brick-wall style packing observed in dodecamethoxy-substituted [6]CPP, which is the only other methoxy-substituted CPP with a reported crystal structure.32 With regard to torsion in the CPP backbone, the three methoxy-substituted phenylenes in 12MeO-CPP display a wide range of dihedral angles relative to the adjacent phenylenes, measuring 47(6)°, 51(7)°, and 69(9)°, respectively. The remaining unsubstituted phenylene units display much smaller dihedral angles relative to each other (∠avg = 29(4)°), well within the range observed in unsubstituted [12]CPP.
Reacting 12MeO-CPP with excess boron tribromide afforded 12HO-CPP, a [12]CPP containing six catechol moieties, in 69% yield (67% over two steps from 12F-CPP). This compound is extremely air-sensitive, and partial oxidation was observed even when the reaction and workup were performed entirely under a nitrogen atmosphere. As such, resolving the peak multiplicity in the 1H NMR was not possible (see ESI† for details). However, the disappearance of the methoxy CH3 protons at 3.53 ppm unequivocally confirmed the removal of all methyl substituents. High-resolution ESI-MS of 12HO-CPP immediately following removal from the glovebox showed prominent peaks at m/z = 1143.2779 and 1141.2622. The first of these peaks is consistent with the positively charged potassium adduct of 12HO-CPP in the as-synthesized state (calculated m/z = 1143.2777 for [M + K]+), while the second peak corresponds to the loss of two protons induced by the rapid oxidation of one of the dicatechol-substituted phenylene units (calculated m/z = 1141.2621 for [M + K]+). This behavior is consistent with the oxidation under ESI-MS conditions that we observed in 4HO-CPP.
Given the success of SNAr on fluorinated CPPs with methoxide nucleophiles, we hypothesized that similar reactions could be replicated with stronger sulfur-based nucleophiles. Indeed, the reaction of 12F-CPP with excess sodium 2-propanethiolate in DMI at 80 °C afforded the corresponding [12]CPP with twelve isopropylthiol substituents, 12iPrS-CPP (Fig. 3a). The product was recovered in 86% yield and fully characterized by 1H NMR, 13C NMR, and high-resolution ESI-MS (see ESI† for details). To our knowledge, 12iPrS-CPP represents the first CPP functionalized with alkylthiol substituents.
Coordination chemistry of polycatechol CPP ligands
As prototypical redox non-innocent ligands, catecholates readily traverse multiple redox states (catecholate, o-semiquinone, and o-quinone) when bound to metal cations.40 The rich redox behavior of metal–catecholates has been shown to confer attractive catalytic, magnetic, and electronic properties,41–43 making them exciting building blocks for metal–organic materials. In addition to unusual metal–organic architectures, the unique cyclic topology of polycatechol CPP ligands also provides an opportunity to study how strained, curved π-systems alter the redox behavior and metal-binding ability of catechol groups. Before investigating the more complex cyclic CPP systems, we first focused our attention on a simpler linear terphenyl model ligand, 1,2,4,5-tetrahydroxy-3,6-diphenylbenzene (H4Ph2dhbq) (Fig. 4a). Our lab has previously studied the coordination chemistry of H4Ph2dhbq and its oxidized derivative, 2,5-dihydroxy-3,6-dipheny-1,4-benzoquinone (H2Ph2dhbq), in the context of 1D iron chains.43 Here, we have focused our studies on ruthenium, as half-sandwich Ru(II) arene complexes are known to bind R2dhbq2− ligands to form stable and soluble diamagnetic complexes that are readily characterized by 1H NMR.44–47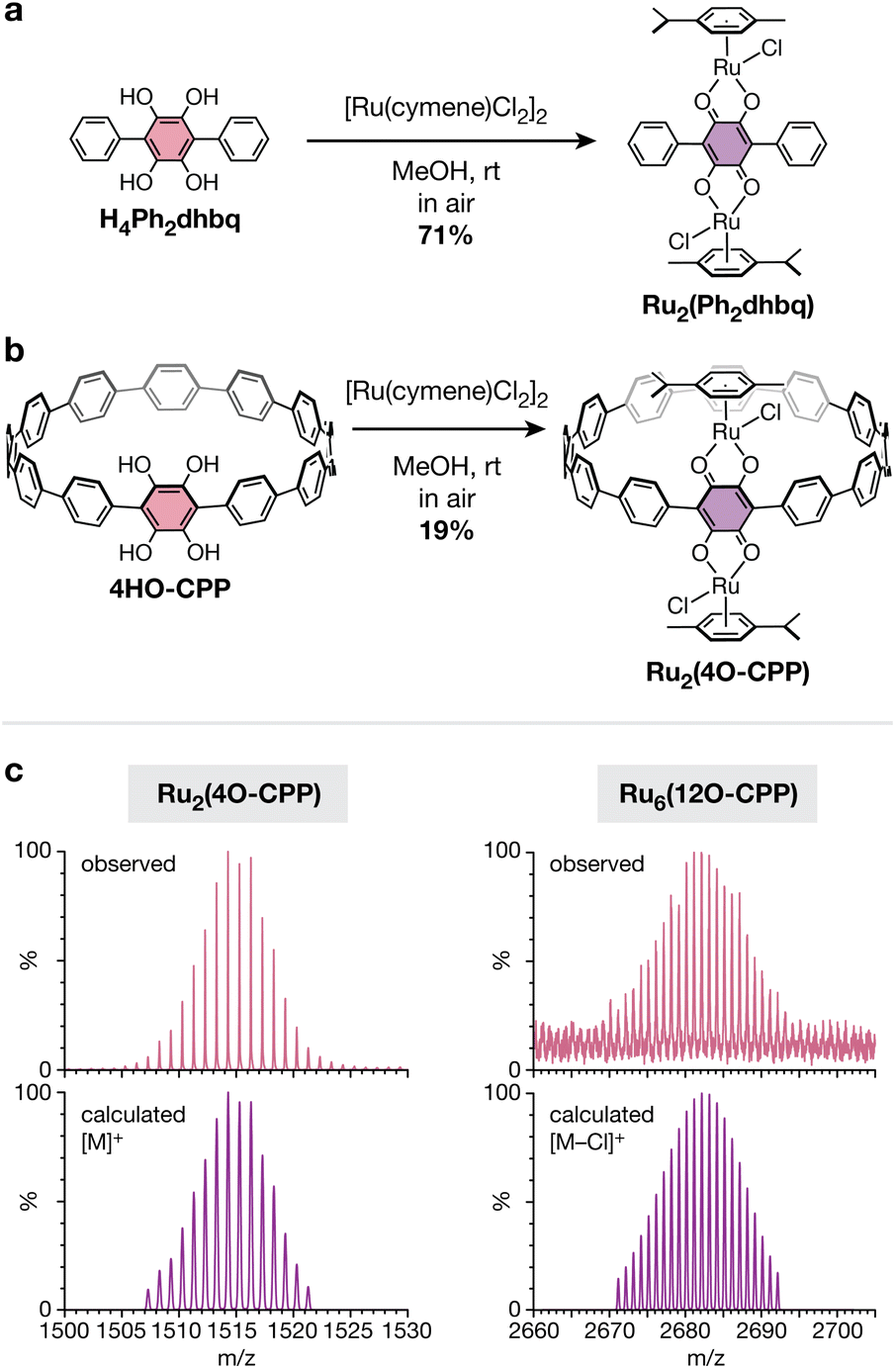 | ||
| Fig. 4 (a) Synthesis of the diruthenium complex Ru2(Ph2dhbq) from the reduced form of the model ligand, H4Ph2dhbq. (b) Synthesis of the diruthenium complex Ru2(4O-CPP) from the ligand 4HO-CPP. (c) Observed (top, pink) versus simulated (bottom, purple) high-resolution MALDI-TOF spectra of the polyruthenium complexes Ru2(4O-CPP) and Ru6(12O-CPP). | ||
While the reaction between [Ru(p-cymene)Cl2]2 and H2Ph2dhbq has been reported to yield the diruthenium complex Ru2(p-cymene)2(Ph2dhbq)Cl2 (Ru2(Ph2dhbq)),47 it was not known if similar reactivity would be observed with the fully reduced, dicatechol form of the ligand. Excitingly, we found that the reaction between H4Ph2dhbq and [Ru(p-cymene)Cl2]2 in air produced the identical diruthenium complex in 71% yield (Fig. 4a). The 1H NMR and ESI-MS spectra matched the previously reported data for this compound. In addition, single crystals suitable for X-ray diffraction were obtained for the first time through slow diffusion of MeOH into DCM, unambiguously confirming that the desired bimetallic product was obtained (Fig. 5a).
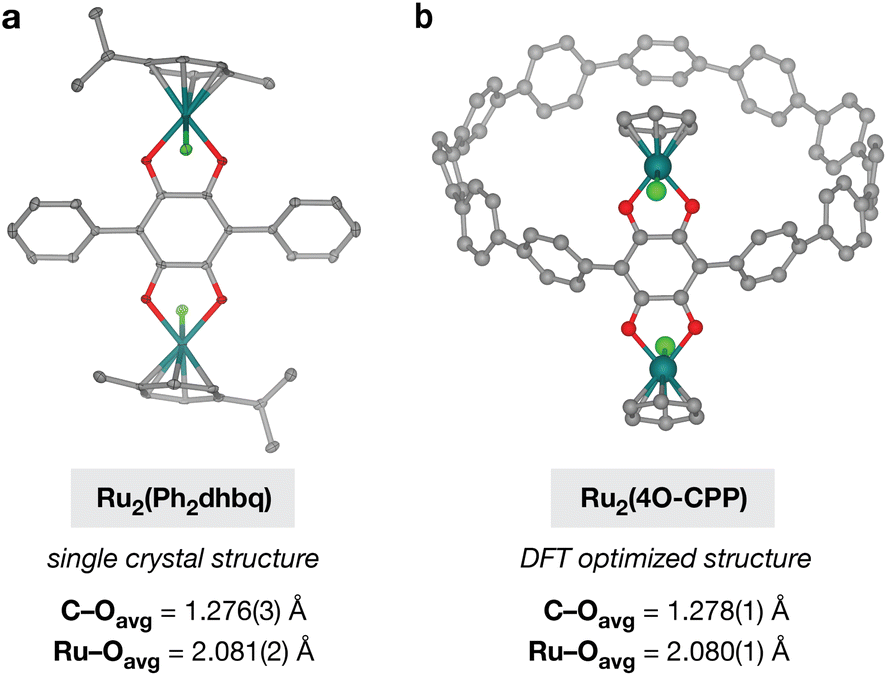 | ||
| Fig. 5 (a) Average C–O and Ru–O bond lengths observed in the single crystal structure of Ru2(Ph2dhbq). Thermal ellipsoids are rendered at the 50% probability level. (b) Average C–O and Ru–O bond lengths observed in the optimized structure of Ru2(4O-CPP) obtained via DFT using the B3LYP hybrid functional with a split basis set of LANL2DZ for Ru and 6-31G* for all other atoms. η6-Cymene ligands on Ru were modeled as η6-benzene. For both parts, Ru, Cl, O, and C atoms are represented by dark green, light green, red, and gray, respectively, and protons are omitted for clarity. | ||
After confirming that the linear H4Ph2dhbq ligand undergoes successful in situ oxidation and Ru(II) metalation, we next turned our attention to the cyclic analogue, 4HO-CPP. The slow diffusion of air into methanolic solutions of 4HO-CPP and excess [Ru(p-cymene)Cl2]2 produced a mixture of products from which the desired compound, Ru2(p-cymene)2(4O-CPP)Cl2 (Ru2(4O-CPP), Fig. 4b) could be purified and isolated in 19% yield (see ESI† for details). The use of MeOH as the solvent appeared to be critical. Alternative solvent systems containing solvents such as dimethylformamide (DMF) and CHCl3 led to either worse yields or no reaction. The lower yields observed with 4HO-CPP relative to H4Ph2dhbq suggest that catechols in curved π-systems may have more complex redox behavior than their linear counterparts. However, this difference in yield may also be exacerbated by the smaller reaction scale and need for chromatographic purification of Ru2(4O-CPP). More work is needed to fully understand these differences, and studies along this vein are underway.
The 1H NMR spectrum of Ru2(4O-CPP) confirms its diamagnetic nature (Fig. S17†), suggesting that, as in the model complex, the metals centers are in the +2 oxidation state and the 4HO-CPP ligand is oxidized to the 2,5-dihydroxy-1,4-benzoquinone form and doubly deprotonated (4O-CPP2−). The m/z values and corresponding isotopic envelopes observed by high-resolution MALDI-TOF (Fig. 4c) and ESI-MS (Fig. S30†) are consistent with the predicted spectra for [M]+ and [M−Cl]+, respectively.
While exhaustive metalation studies with 12HO-CPP were hindered by the poor solubility and oxidative sensitivity of the ligand, preliminary experiments suggest that 12HO-CPP can be similarly metalated to achieve a hexaruthenium CPP complex. A mixture of 12HO-CPP and excess [Ru(p-cymene)Cl2]2 was dissolved in 1:1 DMF:MeOH and stirred in air for 48 h at room temperature. After solvent removal and MeOH washing, a dark red solid with moderate solubility in chlorinated organic solvents was obtained in yields of up to 54%. The formation of the desired complex, Ru6(p-cymene)6(12O-CPP)Cl6 (Ru6(12O-CPP)), was confirmed via high-resolution MALDI-TOF (Fig. 4c). The resonances in the 1H NMR appear consistent with the formation of Ru6(12O-CPP). However, the peaks are broad and poorly resolved, preventing the accurate assessment of peak integrations and multiplicities. The observed peak broadness may be due to hindered rotation of the p-cymene ligands and CPP phenylene rings, as the hexaruthenium complex is significantly more sterically congested than Ru2(4O-CPP). Similar peak broadening has been observed in other CPPs that are densely decorated with bulky substituents.24 Variable temperature 1H NMR experiments in tetrachloroethane-d2 show that peak shape improves significantly with increasing temperature, however the peaks and their multiplicities are still not fully resolved at 120 °C (Fig. S19†).
DFT structural analysis
As we were unable to isolate single crystals of the Ru–CPP complexes for structural analysis by X-ray crystallography, geometry optimizations of Ru2(4O-CPP) and Ru6(12O-CPP) were performed using density functional theory (DFT). All calculations were carried out at the B3LYP level of theory with the LANL2DZ basis set on Ru atoms and 6-31G* basis on all other atoms. For the ease of calculations, the η6-cymene ligands were modeled as η6-benzene.Analysis of multiple structural isomers of Ru2(4O-CPP) suggests that the lowest energy isomer contains two Cl atoms in a trans configuration on the Ru centers (Fig. 5b). This is consistent with the single crystal structure of Ru2(Ph2dhbq) where the two Cl atoms are also oriented in the trans configuration (Fig. 5a). Additional details and analysis of the structure of Ru6(12O-CPP) can be found in the ESI.†
Comparison between the lowest-energy DFT optimized structure of Ru2(4O-CPP) and the single crystal structure of Ru2(Ph2dhbq) shows that the bond lengths match closely. The average C–O bond lengths in the optimized structure of Ru2(4O-CPP) are 1.278(1) Å, very similar to the C–O bond lengths of 1.274(3) and 1.278(3) Å observed in the single crystal structure of Ru2(Ph2dhbq). In addition, the optimized structure of Ru2(4O-CPP) displays an average Ru–O bond length of 2.080(1) Å, very similar to the bond lengths of 2.070(2) and 2.092(2) Å observed in the single crystal structure of Ru2(Ph2dhbq).
Conclusions
In conclusion, we have shown that nucleophilic aromatic substitution is a highly efficient strategy for the late-stage diversification of fluorinated CPPs, and provides a route for the synthesis of new CPP-based ligand platforms. Both alkoxide and alkylthiolate nucleophiles can be used, providing rapid access to electron-rich, densely substituted CPP rings that cannot be easily obtained through existing methods. An important outcome of our SNAr approach is the ability to convert the current repertoire of fluorinated CPPs into a diverse library of redox-active and topologically unique polycatechol ligands. The catechol-substituted CPPs reported here, 4HO-CPP and 12HO-CPP, can be readily synthesized from their fluorinated counterparts in just two steps with excellent overall yields (up to 79%). In addition to their interesting coordination chemistry, polycatechol nanohoops are also versatile partners for dynamic covalent chemistry, enabling the construction of new CPP-based organic polymers and covalent organic frameworks.Both 4HO-CPP and 12HO-CPP can be metalated with half-sandwich Ru(II) arene fragments in the presence of air to produce well-defined dinuclear and hexanuclear compounds, respectively. In principle, it should be possible to replace the chloride bound to each Ru(II) center with other ligands.48 As such, these compounds represent the first examples of multimetallic CPP complexes that are primed for further extension. Given the widespread use of dinuclear arene ruthenium complexes in the construction of metallacycles and metallaprisms,44–46 we anticipate that these metalated CPP complexes will serve as an exciting new synthon in supramolecular chemistry.
Finally, the preliminary metalation studies outlined here have already revealed intriguing similarities and differences between linear and curved polycatechol ligands. The metalation of 4HO-CPP proceeds in significantly lower yields than the linear analogue, suggesting that curvature may induce more complicated redox behavior and/or coordination chemistry. On the other hand, close analysis of DFT-optimized structures shows that the metal–dioxolene linkages in linear and curved systems display few, if any, structural differences. These structural studies are encouraging and suggest that cyclic polycatechol ligands may be amenable to forming extended structures, much like their more well-studied linear counterparts.41 Further work to understand the coordination chemistry of polycatechol CPP ligands, particularly towards the construction of extended metal–organic architectures, is under way.
Data availability
Additional experimental data supporting this article are included in the ESI.†Author contributions
AAK and DJX designed the research and wrote the manuscript; AAK carried out the synthesis and characterization of all species, with the exception of the fluorinated cycloparaphenylenes, which were carried out by TDC; PMG, SK, and WK collected and refined the crystallographic data; AAK performed the DFT calculations. All authors interpreted the results and contributed to reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Initial cycloparaphenylene substitution studies were supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021966 (DJX), and subsequent metalation studies were supported by Arnold and Mabel Beckman Foundation through a Beckman Young Investigator Award (DJX). The synthesis and characterization of fluorinated cycloparaphenylenes was supported by NSF Award Number CHE-2102567 (RJ). AAK is supported by an NSF graduate research fellowship and a University of Washington (UW) Clean Energy Institute graduate fellowship. The X-ray facility of the UW Department of Chemistry is supported by NSF Award Number CHE-0840520. The NMR facility at the UW Department of Chemistry is supported by NIH Award Number S10OD030224-01A1. This work utilized the computational infrastructure of the Hyak supercomputer system at the UW. This research also utilized resources of the Advanced Light Source (ALS), which is a DOE Office of Science User Facility under Contract Number DE-AC02-05CH11231; we thank Drs Simon Teat and Nick Settineri for their assistance with the collection of synchrotron single crystal X-ray diffraction data at ALS beamline 12.2.1.Notes and references
- H. Omachi, Y. Segawa and K. Itami, Synthesis of Cycloparaphenylenes and Related Carbon Nanorings: A Step toward the Controlled Synthesis of Carbon Nanotubes, Acc. Chem. Res., 2012, 45(8), 1378–1389, DOI:10.1021/ar300055x
.
- E. R. Darzi and R. Jasti, The Dynamic, Size-Dependent Properties of [5]–[12]Cycloparaphenylenes, Chem. Soc. Rev., 2015, 44(18), 6401–6410, 10.1039/C5CS00143A
- M. R. Golder and R. Jasti, Syntheses of the Smallest Carbon Nanohoops and the Emergence of Unique Physical Phenomena, Acc. Chem. Res., 2015, 48(3), 557–566, DOI:10.1021/ar5004253
- E. J. Leonhardt and R. Jasti, Emerging Applications of Carbon Nanohoops, Nat. Rev. Chem, 2019, 3(12), 672–686, DOI:10.1038/s41570-019-0140-0
- Y. Segawa, D. R. Levine and K. Itami, Topologically Unique Molecular Nanocarbons, Acc. Chem. Res., 2019, 52(10), 2760–2767, DOI:10.1021/acs.accounts.9b00402
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, Synthesis, Characterization, and Theory of [9]-, [12]-, and [18]Cycloparaphenylene: Carbon Nanohoop Structures, J. Am. Chem. Soc., 2008, 130(52), 17646–17647, DOI:10.1021/ja807126u
- R. Jasti and C. R. Bertozzi, Progress and Challenges for the Bottom-up Synthesis of Carbon Nanotubes with Discrete Chirality, Chem. Phys. Lett., 2010, 494(1–3), 1–7, DOI:10.1016/j.cplett.2010.04.067
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Initiation of Carbon Nanotube Growth by Well-Defined Carbon Nanorings, Nat. Chem., 2013, 5(7), 572–576, DOI:10.1038/nchem.1655
- Y. Segawa, A. Yagi, K. Matsui and K. Itami, Design and Synthesis of Carbon Nanotube Segments, Angew. Chem., Int. Ed., 2016, 55(17), 5136–5158, DOI:10.1002/anie.201508384
- Y. Segawa, A. Yagi and K. Itami, Chemical Synthesis of Cycloparaphenylenes, Phys. Sci. Rev., 2017, 2(1), 20160102, DOI:10.1515/psr-2016-0102
- D. Wu, W. Cheng, X. Ban and J. Xia, Cycloparaphenylenes (CPPs): An Overview of Synthesis, Properties, and Potential Applications, Asian J. Org. Chem., 2018, 7(11), 2161–2181, DOI:10.1002/ajoc.201800397
- Q. Huang, G. Zhuang, M. Zhang, J. Wang, S. Wang, Y. Wu, S. Yang and P. Du, A Long π-Conjugated Poly(Para-Phenylene)-Based Polymeric Segment of Single-Walled Carbon Nanotubes, J. Am. Chem. Soc., 2019, 141(48), 18938–18943, DOI:10.1021/jacs.9b10358
- G. M. Peters, G. Grover, R. L. Maust, C. E. Colwell, H. Bates, W. A. Edgell, R. Jasti, M. Kertesz and J. D. Tovar, Linear and Radial Conjugation in Extended π-Electron Systems, J. Am. Chem. Soc., 2020, 142(5), 2293–2300, DOI:10.1021/jacs.9b10785
- R. L. Maust, P. Li, B. Shao, S. M. Zeitler, P. B. Sun, H. W. Reid, L. N. Zakharov, M. R. Golder and R. Jasti, Controlled Polymerization of Norbornene Cycloparaphenylenes Expands Carbon Nanomaterials Design Space, ACS Cent. Sci., 2021, 7(6), 1056–1065, DOI:10.1021/acscentsci.1c00345
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, New Porous Crystals of Extended Metal-Catecholates, Chem. Mater., 2012, 24(18), 3511–3513, DOI:10.1021/cm301194a
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, High Electrical Conductivity in Ni3 (2,3,6,7,10,11-Hexaiminotriphenylene)2, a Semiconducting Metal–Organic Graphene Analogue, J. Am. Chem. Soc., 2014, 136(25), 8859–8862, DOI:10.1021/ja502765n
- N. Kubota, Y. Segawa and K. Itami, η6-Cycloparaphenylene Transition Metal Complexes: Synthesis, Structure, Photophysical Properties, and Application to the Selective Monofunctionalization of Cycloparaphenylenes, J. Am. Chem. Soc., 2015, 137(3), 1356–1361, DOI:10.1021/ja512271p
- E. Kayahara, V. K. Patel, A. Mercier, E. P. Kündig and S. Yamago, Regioselective Synthesis and Characterization of Multinuclear Convex-Bound Ruthenium-[n]Cycloparaphenylene (N = 5 and 6) Complexes, Angew. Chem., Int. Ed., 2016, 55(1), 302–306, DOI:10.1002/anie.201508003
- K. Ypsilantis, T. Tsolis and A. Garoufis, Interactions of (η5-CpRu)-[12]Cycloparaphenylene Full-Sandwich Complexes with 9-Methylguanine, Inorg. Chem. Commun., 2021, 134, 108992, DOI:10.1016/j.inoche.2021.108992
- J. M. Van Raden, S. Louie, L. N. Zakharov and R. Jasti, 2,2′-Bipyridyl-Embedded Cycloparaphenylenes as a General Strategy To Investigate Nanohoop-Based Coordination Complexes, J. Am. Chem. Soc., 2017, 139(8), 2936–2939, DOI:10.1021/jacs.7b00359
- M. J. Heras Ojea, J. M. Van Raden, S. Louie, R. Collins, D. Pividori, J. Cirera, K. Meyer, R. Jasti and R. A. Layfield, Spin-Crossover Properties of an Iron(II) Coordination Nanohoop, Angew. Chem., Int. Ed., 2021, 60(7), 3515–3518, DOI:10.1002/anie.202013374
- M. L. Mercuri, F. Congiu, G. Concas and S. A. Sahadevan, Recent Advances on Anilato-Based Molecular Materials with Magnetic and/or Conducting Properties, Magnetochemistry, 2017, 3(2), 17, DOI:10.3390/magnetochemistry3020017
- E. Kayahara, R. Qu and S. Yamago, Bromination of Cycloparaphenylenes: Strain-Induced Site-Selective Bis-Addition and Its Application for Late-Stage Functionalization, Angew. Chem., Int. Ed., 2017, 56(35), 10428–10432, DOI:10.1002/anie.201704982
- H. Shudo; M. Kuwayama; Y. Segawa; A. Yagi and K. ItamiHalf-Substituted Fluorocycloparaphenylenes with High Symmetry: Synthesis, Properties and Derivatization to Densely Substituted Carbon Nanorings. ChemRxiv, 2023, DOI: DOI:10.26434/chemrxiv-2023-zsvnt.
- S. Hashimoto, E. Kayahara, Y. Mizuhata, N. Tokitoh, K. Takeuchi, F. Ozawa and S. Yamago, Synthesis and Physical Properties of Polyfluorinated Cycloparaphenylenes, Org. Lett., 2018, 20(18), 5973–5976, DOI:10.1021/acs.orglett.8b02715
- E. J. Leonhardt, J. M. Van Raden, D. Miller, L. N. Zakharov, B. Alemán and R. Jasti, A Bottom-Up Approach to Solution-Processed, Atomically Precise Graphitic Cylinders on Graphite, Nano Lett., 2018, 18(12), 7991–7997, DOI:10.1021/acs.nanolett.8b03979
- J. M. Van Raden, E. J. Leonhardt, L. N. Zakharov, A. Pérez-Guardiola, A. J. Pérez-Jiménez, C. R. Marshall, C. K. Brozek, J. C. Sancho-García and R. Jasti, Precision Nanotube Mimics via Self-Assembly of Programmed Carbon Nanohoops, J. Org. Chem., 2020, 85(1), 129–141, DOI:10.1021/acs.joc.9b02340
- H. Shudo, M. Kuwayama, M. Shimasaki, T. Nishihara, Y. Takeda, N. Mitoma, T. Kuwabara, A. Yagi, Y. Segawa and K. Itami, Perfluorocycloparaphenylenes, Nat. Commun., 2022, 13(1), 3713, DOI:10.1038/s41467-022-31530-x
- T. Terabayashi, E. Kayahara, Y. Zhang, Y. Mizuhata, N. Tokitoh, T. Nishinaga, T. Kato and S. Yamago, Synthesis of Twisted [n]Cycloparaphenylene by Alkene Insertion, Angew. Chem., Int. Ed., 2023, 62(2), e202214960, DOI:10.1002/anie.202214960
- S. Cui, G. Zhuang, J. Wang, Q. Huang, S. Wang and P. Du, Multifunctionalized Octamethoxy-[8]Cycloparaphenylene: Facile Synthesis and Analysis of Novel Photophysical and Photoinduced Electron Transfer Properties, Org. Chem. Front., 2019, 6(11), 1885–1890, 10.1039/C9QO00372J
- D. Lu, G. Zhuang, H. Jia, J. Wang, Q. Huang, S. Cui and P. Du, A Novel Symmetrically Multifunctionalized Dodecamethoxy-Cycloparaphenylene: Synthesis, Photophysical, and Supramolecular Properties, Org. Chem. Front., 2018, 5(9), 1446–1451, 10.1039/C8QO00033F
- N. Narita; Y. Kurita; K. Osakada; T. Ide; Y. Tsuchido and H. KawaiA Dodecamethoxy [6]Cycloparaphenylene Consisting Entirely of Hydoroquinone Ether Units: Redox Properties and Host-Guest Complexation. ChemRxiv, 2022, DOI: DOI:10.26434/chemrxiv-2022-0c1s7.
- P. Della Sala, C. Talotta, T. Caruso, M. De Rosa, A. Soriente, P. Neri and C. Gaeta, Tuning Cycloparaphenylene Host Properties by Chemical Modification, J. Org. Chem., 2017, 82(18), 9885–9889, DOI:10.1021/acs.joc.7b01588
- H. Chen, M. Shao, H. Li, H. Liu, W.-M. Wei, R.-H. Zheng, M. Song, R. Liu and D. Lu, Modular Synthesis, Racemization Pathway, and Photophysical Properties of Asymmetrically Substituted Cycloparaphenylenes, New J. Chem., 2022, 46(35), 16670–16674, 10.1039/D2NJ03166C
- T. F. Mikhailovskaya, A. G. Makarov, N. Yu. Selikhova, A. Yu. Makarov, E. A. Pritchina, I. Yu. Bagryanskaya, E. V. Vorontsova, I. D. Ivanov, V. D. Tikhova, N. P. Gritsan, Y. G. Slizhov and A. V. Zibarev, Carbocyclic Functionalization of Quinoxalines, Their Chalcogen Congeners 2,1,3-Benzothia/Selenadiazoles, and Related 1,2-Diaminobenzenes Based on Nucleophilic Substitution of Fluorine, J. Fluorine Chem., 2016, 183, 44–58, DOI:10.1016/j.jfluchem.2016.01.009
- M. Tashiro, H. Fujimoto, A. Tsuge, S. Mataka and H. Kobayashi, Metacyclophanes and Related Compounds. 23. Preparation of Fluorinated [2,2]Metacyclophanes, J. Org. Chem., 1989, 54(8), 2012–2015, DOI:10.1021/jo00269a051
- M. Jarman and R. McCague, Octafluorotoluene as a Reagent for the Selective Protection of Alcoholic and Phenolic Functions. Synthesis and Cleavage of Perfluorotolyl and Other Perfluoroaryl Ethers of Steroids and Other Model Compounds, J. Chem. Res., 1985, 114–115 CAS
- J. Hellberg, E. Dahlstedt and M. E. Pelcman, Synthesis of Annulated Dioxins as Electron-Rich Donors for Cation Radical Salts, Tetrahedron, 2004, 60(40), 8899–8912, DOI:10.1016/j.tet.2004.07.017
- Y. Segawa, S. Miyamoto, H. Omachi, S. Matsuura, P. Šenel, T. Sasamori, N. Tokitoh and K. Itami, Concise Synthesis and Crystal Structure of [12]Cycloparaphenylene, Angew. Chem., Int. Ed., 2011, 50(14), 3244–3248, DOI:10.1002/anie.201007232
- C. G. Pierpont and R. M. Buchanan, Transition Metal Complexes of O-Benzoquinone, o-Semiquinone, and Catecholate Ligands, Coord. Chem. Rev., 1981, 38(1), 45–87, DOI:10.1016/S0010-8545(00)80499-3
- S. Kitagawa and S. Kawata, Coordination Compounds of 1,4-Dihydroxybenzoquinone and Its Homologues. Structures and Properties, Coord. Chem. Rev., 2002, 224(1), 11–34, DOI:10.1016/S0010-8545(01)00369-1
- V. Lyaskovskyy and B. de Bruin, Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions, ACS Catal., 2012, 2(2), 270–279, DOI:10.1021/cs200660v
- A. A. Kamin, I. P. Moseley, J. Oh, E. J. Brannan, P. M. Gannon, W. Kaminsky, J. M. Zadrozny and D. J. Xiao, Geometry-Dependent Valence Tautomerism, Magnetism, and Electrical Conductivity in 1D Iron–Tetraoxolene Chains, Chem. Sci., 2023, 14, 4083–4090, 10.1039/D2SC06392A
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, The “Complex-in-a-Complex” Cations [(Acac)2M⊂Ru6(p-IPrC6H4Me)6(Tpt)2(Dhbq)3]6+: A Trojan Horse for Cancer Cells, Angew. Chem., Int. Ed., 2008, 47(20), 3773–3776, DOI:10.1002/anie.200800186
- J. Mattsson, P. Govindaswamy, A. K. Renfrew, P. J. Dyson, P. Štěpnička, G. Süss-Fink and B. Therrien, Synthesis, Molecular Structure, and Anticancer Activity of Cationic Arene Ruthenium Metallarectangles, Organometallics, 2009, 28(15), 4350–4357, DOI:10.1021/om900359j
- B. Therrien, Transporting and Shielding Photosensitisers by Using Water-Soluble Organometallic Cages: A New Strategy in Drug Delivery and Photodynamic Therapy, Chem.–Eur. J., 2013, 19(26), 8378–8386, DOI:10.1002/chem.201301348
- A. Garci, J.-P. Mbakidi, V. Chaleix, V. Sol, E. Orhan and B. Therrien, Tunable Arene Ruthenium Metallaprisms to Transport, Shield, and Release Porphin in Cancer Cells, Organometallics, 2015, 34(16), 4138–4146, DOI:10.1021/acs.organomet.5b00555
- A. Garci, G. Gupta, C. Dalvit and B. Therrien, Investigating the Formation Mechanism of Arene Ruthenium Metallacycles by NMR Spectroscopy, Eur. J. Inorg. Chem., 2014, 2014(33), 5651–5661, DOI:10.1002/ejic.201402639
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2264974–2264977. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03561a |
| This journal is © The Royal Society of Chemistry 2023 |