DOI:
10.1039/D3SC01777J
(Edge Article)
Chem. Sci., 2023,
14, 7875-7886
Role of distal arginine residue in the mechanism of heme nitrite reductases†
Received
6th April 2023
, Accepted 14th June 2023
First published on 15th June 2023
Abstract
Heme nitrite reductases reduce NO2− by 1e−/2H+ to NO or by 6e−/8H+ to NH4+ which are key steps in the global nitrogen cycle. Second-sphere residues, such as arginine (with a guanidine head group), are proposed to play a key role in the reaction by assisting substrate binding and hydrogen bonding and by providing protons to the active site for the reaction. The reactivity of an iron porphyrin with a NO2− covalently attached to a guanidinium arm in its 2nd sphere was investigated to understand the role of arginine residues in the 2nd sphere of heme nitrite reductases. The presence of the guanidinium residue allows the synthetic ferrous porphyrin to reduce NO2− and produce a ferrous nitrosyl species ({FeNO}7), where the required protons are provided by the guanidinium group in the 2nd sphere. However, in the presence of additional proton sources in solution, the reaction of ferrous porphyrin with NO2− results in the formation of ferric porphyrin and the release of NO. Spectroscopic and kinetic data indicated that re-protonation of the guanidine group in the 2nd sphere by an external proton source causes NO to dissociate from a ferric nitrosyl species ({FeNO}6) at rates similar to those observed for enzymatic sites. This re-protonation of the guanidine group mimics the proton recharge mechanism in the active site of NiR. DFT calculations indicated that the lability of the Fe–NO bond in the {FeNO}6 species is derived from the greater binding affinity of anions (e.g. NO2−) to the ferric center relative to neutral NO due to hydrogen bonding and electrostatic interaction of these bound anions with the protonated guanidium group in the 2nd sphere. The reduced {FeNO}7 species, once formed, is not affected significantly by the re-protonation of the guanidine residue. These results provide direct insight into the role of the 2nd sphere arginine residue present in the active sites of heme-based NiRs in determining the fate of NO2− reduction. Specifically, the findings using the synthetic model suggest that rapid re-protonation of these arginine residues may trigger the dissociation of NO from the {FeNO}6, which may also be the case in the protein active site.
Introduction
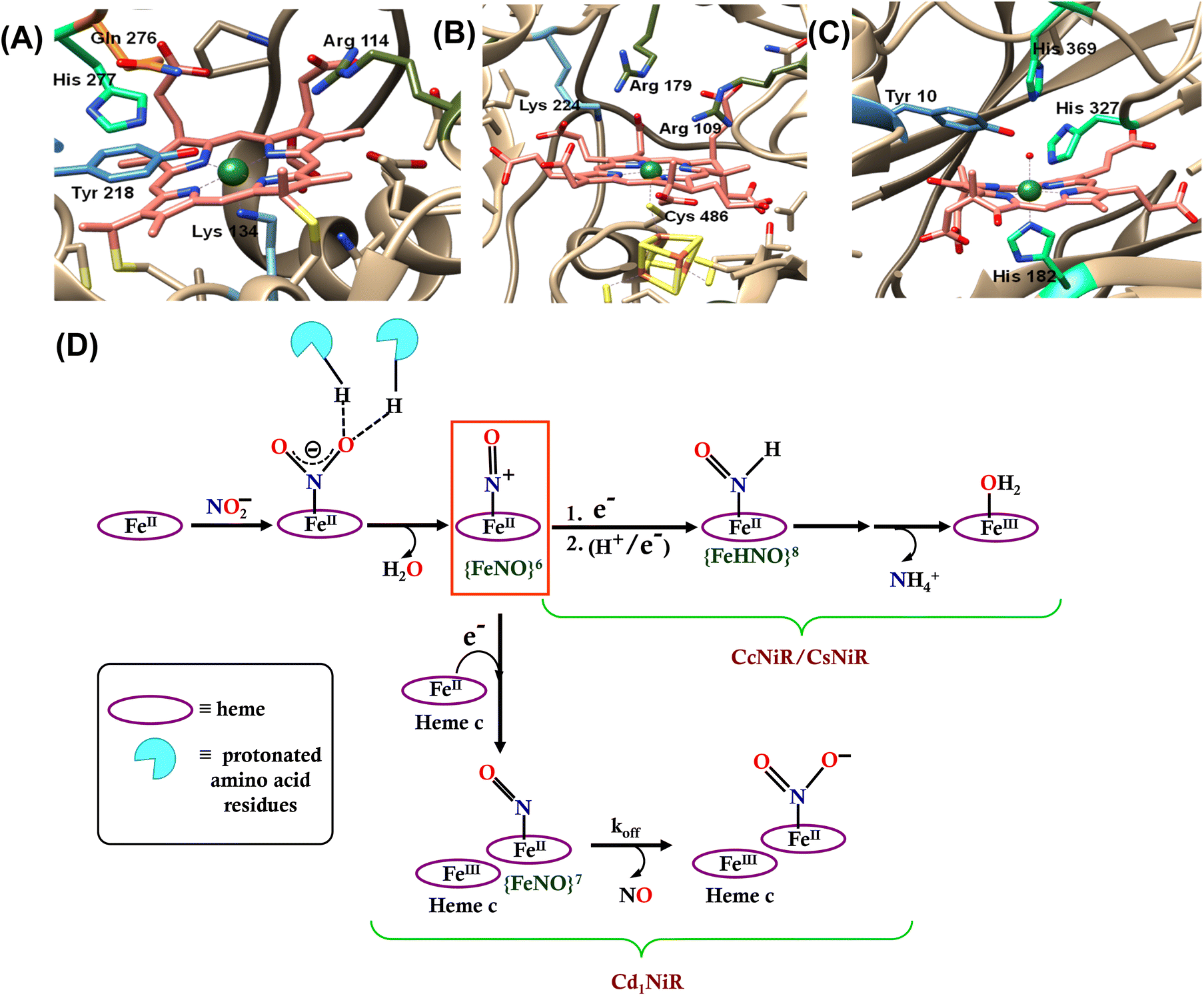
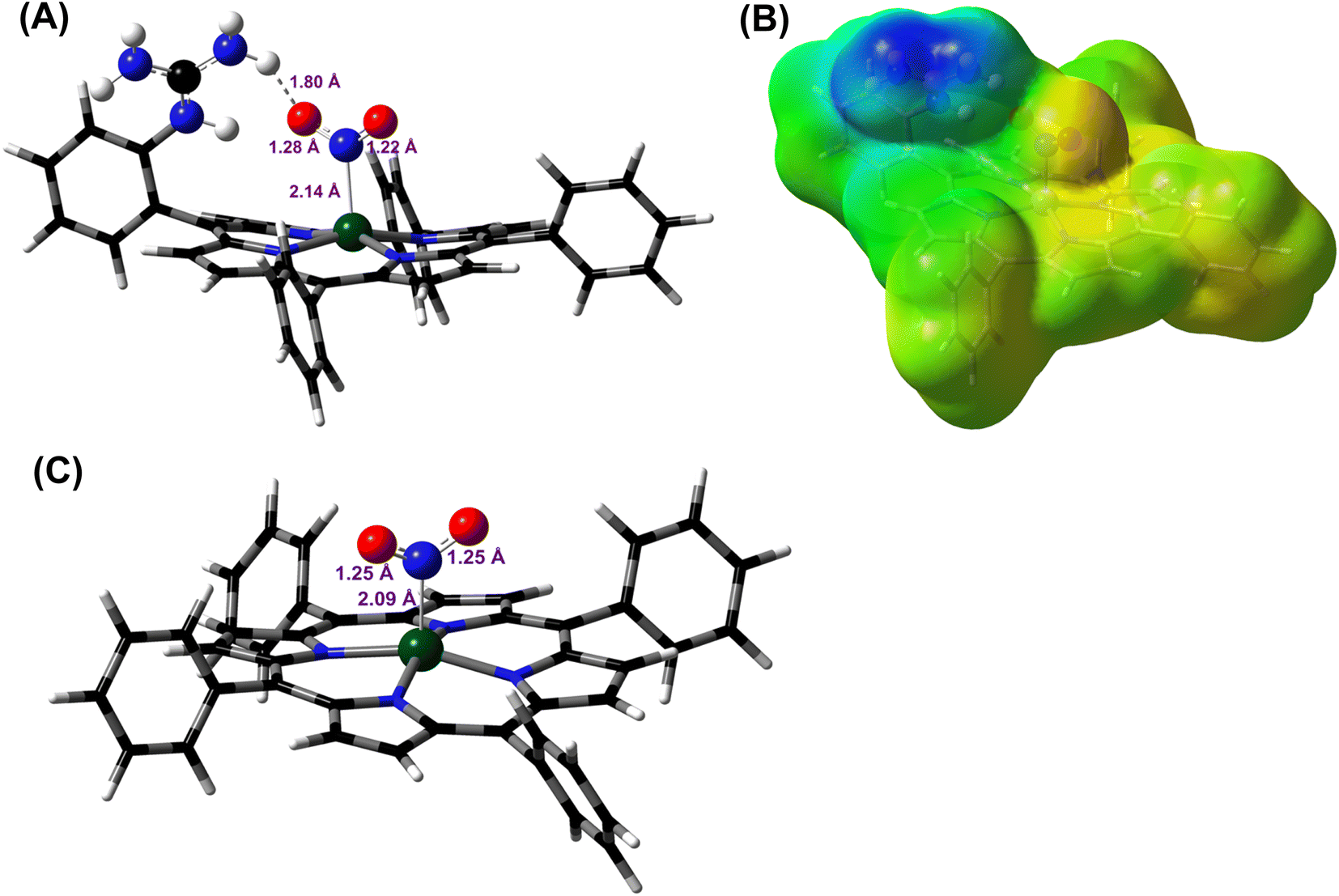
Nitrite (NO2−) is a key intermediate in the nitrogen biochemical cycle that involves oxidations and reductions of nitrogen oxides (NOx) by both aerobically and anaerobically respiring organisms during energy transduction.1–3 Reduction of nitrite can be dissimilatory, where nitrite is reduced to ammonium ion (NH4+) that later assembles into amino acids and is finally catalyzed by the multi-c hemes containing nitrite reductase enzyme (CcNiR, Fig. 1A). Assimilatory nitrite reduction generates NH4+ by the siroheme-containing nitrite reductase enzyme (CsNiR, Fig. 1B).3–12 Nitrite is also reduced to N2 in a denitrification pathway, where the first committed step is nitric oxide (NO˙) formation from nitrite and involves either the copper-dependent (CuNiR)13–16 or the heme-dependent (Cd1NiR, Fig. 1C)17–19 nitrite reductase enzyme. These heme-based nitrite reductases not only utilize different variations of heme cofactors, but also present different axial ligands and 2nd sphere amino acid residues at their distal sites (Fig. 1). In CcNiR, distal histidine and arginine residues anchor the approaching NO2− ion to the reduced iron center with strong hydrogen bonds, and the N–O bond cleavage is facilitated by the protons provided by these positively charged 2nd sphere residues.20–22 Two arginine and lysine residues play a similar role at the distal site of CsNiR.23,24 Cd1NiR also fashions two conserved distal histidine residues.17,25–29
 |
| | Fig. 1 The catalytic active site of (A) CcNiR (PDB file 2E80), (B) CsNiR (PDB file 2AKJ), and (C) Cd1NiR (PDB file 1NIR). (D) Bifurcation in the enzymatic pathway of nitrite reduction for different heme-dependent NiRs. | |
The mechanism of working of these heme-dependent nitrite reductases has been extensively investigated using different experimental and theoretical methods. These enzymes bind the NO2− molecule via the “nitro” mode to their ferrous state.17,28,30–37 In addition to the hydrogen bonding interaction of the bound nitrite with the distal amino acid residues, back-bonding from the ferrous iron to nitrite N-atom of the NO2− makes the N–O bond weaker.38–40 The initial N–O bond cleavage produces a ferric–NO complex or {FeNO}6 (Enmark–Feltham notation), which is the first iron-nitrosyl intermediate of the corresponding catalytic cycle.41 The protons required for the N–O bond cleavage, resulting in the elimination of water, are provided by the distal residues (arginine, histidine, lysine, etc.). From this point, the enzymatic pathways of different heme-dependent NiRs bifurcate. The CcNiR and CsNiR retain the bound NO to further reduce it into NH4+. To accomplish the reduction of NO, the enzyme's active sites “recharge” themselves with protons and electrons and utilize a proton-coupled electron transfer mechanism.42,43 On the other hand, the Cd1NiR reduces the {FeNO}6 complex by one electron to produce the ferrous–NO complex or {FeNO}7 (Enmark–Feltham notation) and releases NO with an exceptionally high NO dissociation rate, relative to other heme active sites and synthetic systems (Fig. 1D). Initially, NO has been proposed to be released from the {FeNO}6 complex,44 but substantial proof of kinetically competent NO dissociation from this complex is missing thus far.1,25,38,40,45–47 However, several experimental pieces of evidence suggest rapid NO release through the {FeNO}7 complex,44 despite its general high affinity for the reduced heme-Fe.25,35,38,39,48–54 The synergistic effect of the unique heme d1 structure and the network of the distal amino acid residues has been deemed important for facile NO dissociation from the {FeNO}7 complex of cd1NiR.35,39,48,50,53,55–57 Recent results using synthetic analogs of heme d1 have indicated that the structure of the porphyrin can indeed affect the stability of the Fe–NO bond in {FeNO}7 intermediates and, more specifically, electron-withdrawing peripheral substituents weakens the Fe–NO bond causing its dissociation.58 Further reduction of the bound NO to NH4+ requires a proton recharge mechanism which is actualized by re-protonating the arginine and lysine residues present in the distal site of the heme active sites of CsNiR and CcNiR which gets deprotonated during the reduction of NO2− to NO. The distal arginine residues are deemed important for substrate binding, as well as for N–O bond activation/cleavage and proton transfer to the active site. However, to date, the proposals regarding the roles of the crucial distal arginine residue have not been evaluated for NO2− reduction in synthetic systems.
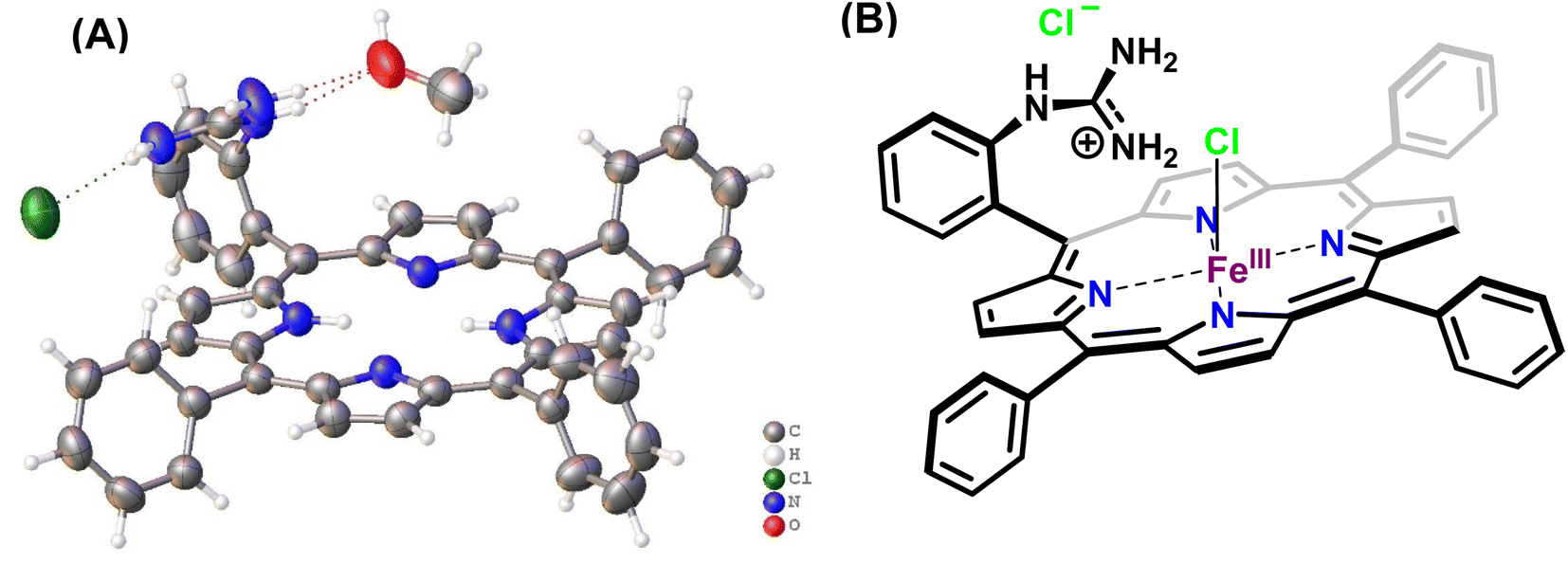
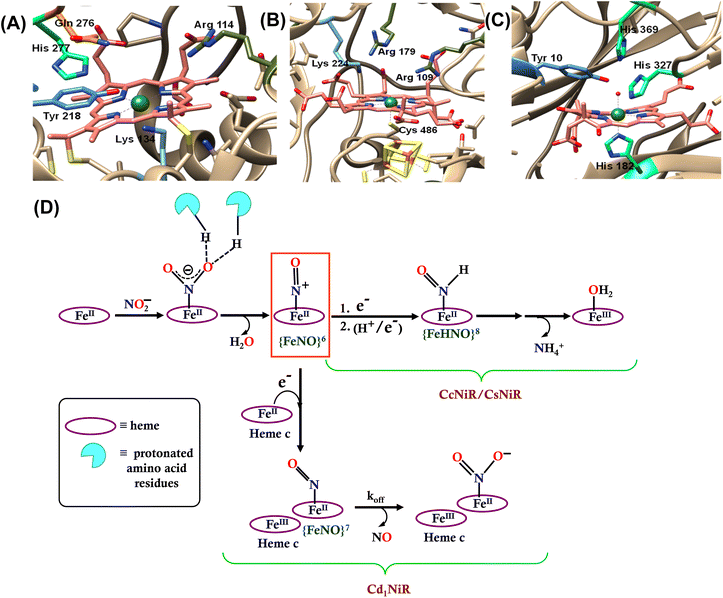
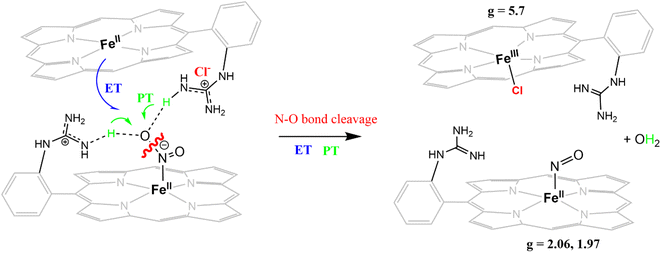
In this study, the reduction of NO2− ion was investigated using an iron complex of a guanidinium-armed porphyrin ligand, [MARGH]Cl (Fig. 2A, the exchangeable protons and counter ions are shown in bold). The crystal structure of the free [MARGH]Cl ligand exhibits that the pendant guanidinium group, attached covalently to the porphyrin, is protonated because of its high pKa and is counterbalanced by a chloride ion (Fig. 2B).59 This protonated guanidinium pendant group mimics the distal protonated amino acid groups in the catalytic active sites of heme-dependent NiRs. The [FeMARGH]Cl2 complex has been recently demonstrated to be able to translocate protons into the iron active site through the guanidine group, allowing them to act as peroxidases with very facile O–O bond heterolysis rates.59 Here, the ferrous porphyrin complex, ([FeIIMARGH]Cl), catalyzes the reduction of NO2− to NO without the necessity of external protons. The reduction of NO2− can lead to the formation of a {FeNO}7 complex or release NO from the {FeNO}6 intermediate depending on the protonation state of the distal guanidine group. These results suggest that “proton recharge” may trigger the dissociation of NO from the {FeNO}6 complex.
 |
| | Fig. 2 (A) Crystal structure of [MARGH]Cl ligand (Cl (green), O (red), C (grey), N (blue) and H (white)) and (B) schematic diagram of [FeMARGH]Cl2. | |
Results
Reactivity of [FeIIMARGH]Cl
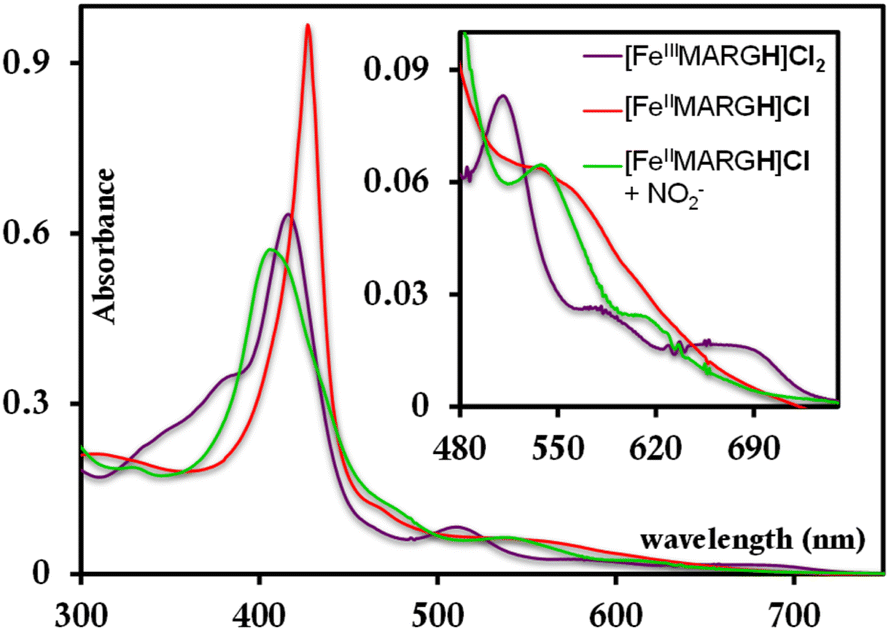
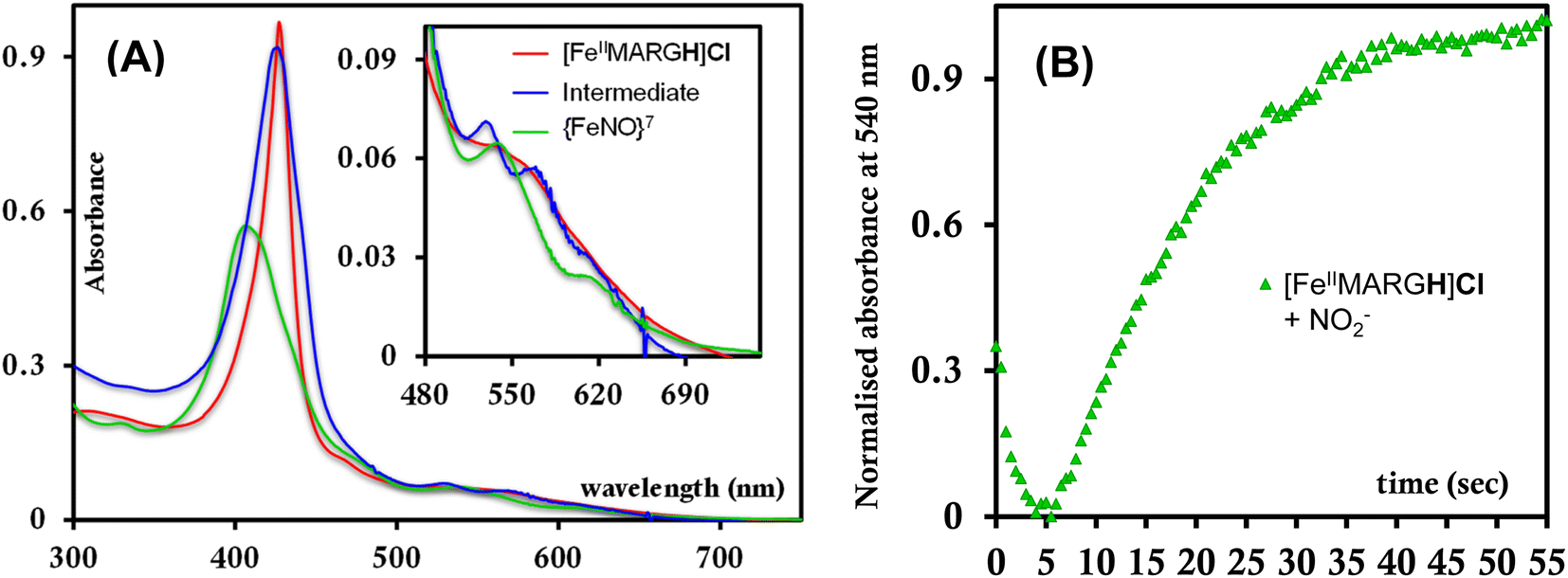
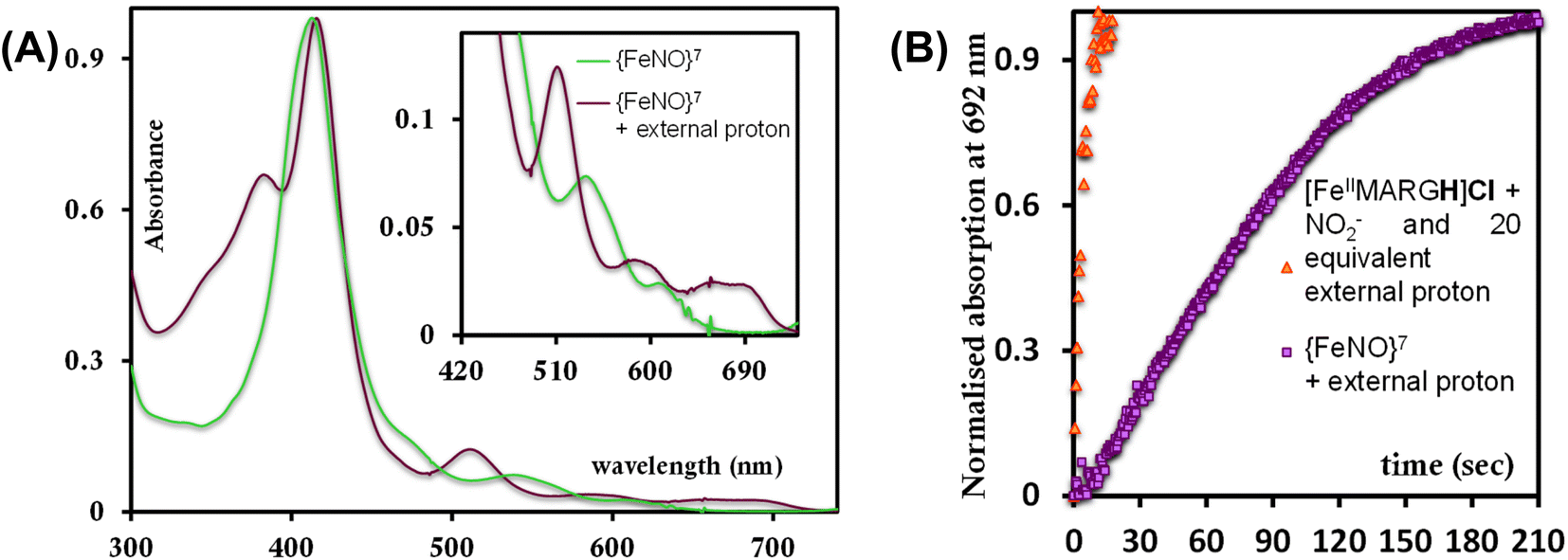
The [FeIIIMARGH]Cl2 is reduced in situ to the corresponding [FeIIMARGH]Cl complex and is characterized by its absorption bands at 428 nm (Soret) and 545 nm (Fig. 3, red). The Soret band shifts to 408 nm, and the Q band shifts to 540 nm (Fig. 3, green) when 5 equivalents of [NBu4]NO2 are added to [FeIIMARGH]Cl, indicating the formation of the corresponding ferrous nitrosyl complex, {FeNO}7. The νN–O of the {FeNO}7 complex is observed at 1680 cm−1 (Fig. S1†). A protonated guanidinium arm shows characteristic FTIR vibrations at 1727 cm−1 and 1780 cm−1 which disappear upon deprotonation (Fig. S2†). The FTIR data on the product from the reaction of [FeIIMARGH]Cl with [NBu4]NO2 shows a νN–O at 1680 cm−1 for {FeNO}7 species, but no peaks are observed corresponding to the protonated guanidinium group indicating that these protons are consumed during the reduction of NO2− by [FeIIMARGH]Cl to form a {FeNO}7 species.
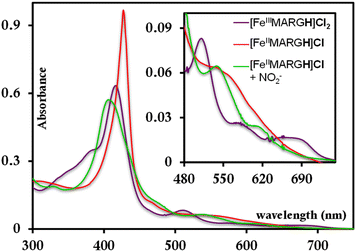 |
| | Fig. 3 Absorption spectra of [FeIIIMARGH]Cl2 (violet) and [FeIIMARGH]Cl (red). The green spectra correspond to the {FeNO}7 species generated in the reaction of [FeIIMARGH]Cl with [NBu4]NO2. | |
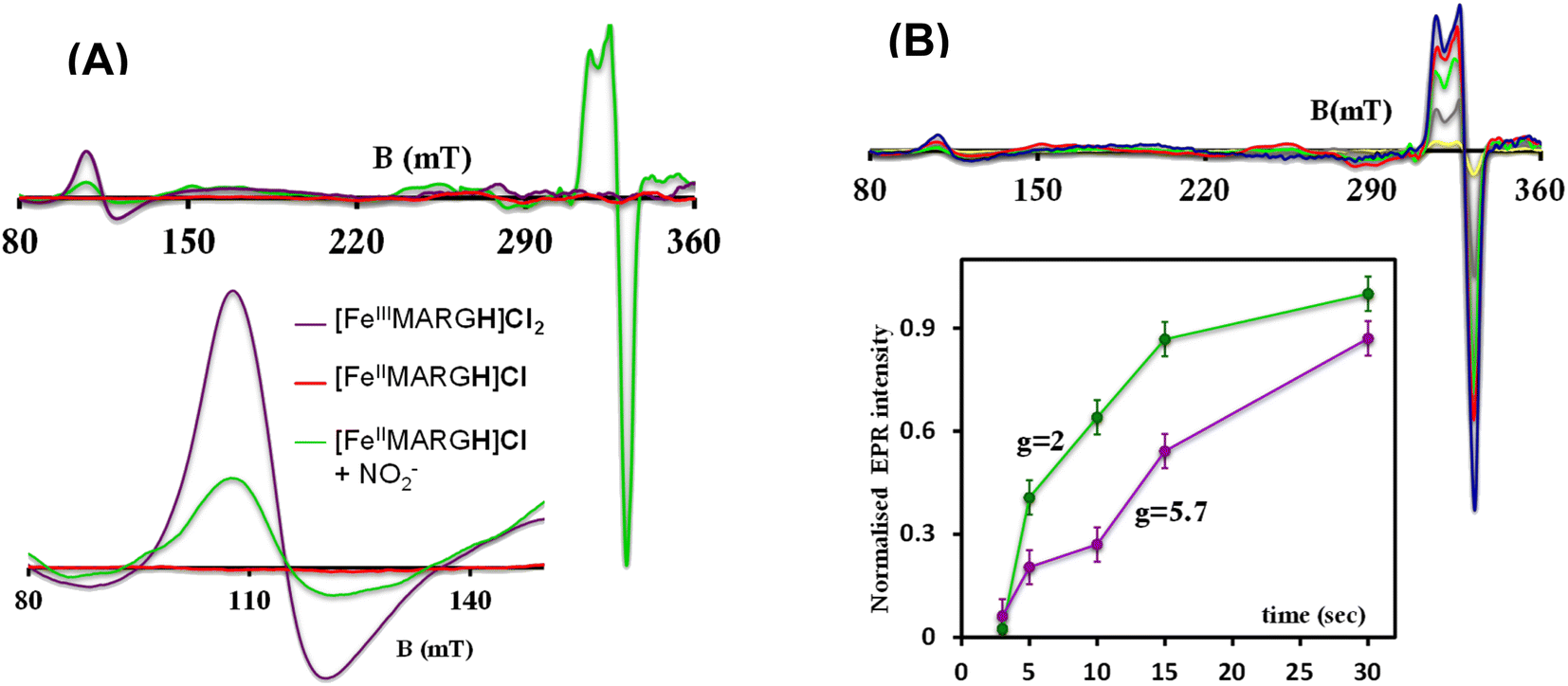
The X-band EPR spectrum of [FeIIIMARGH]Cl2 exhibits an axial signal at g = 5.7 (Fig. 4A, violet), while the [FeIIMARGH]Cl complex is EPR silent at 77 K (Fig. 4A, red). Addition of 5 equivalent of the [NBu4]NO2 solution to the ferrous complex results in two distinct EPR signals. First, a set of rhombic signals was observed with g = 2.06 and g = 1.97 representing the formation of a {FeNO}7 species and, second, an axial signal was observed at g = 5.7 indicating a ferric complex resulting from the oxidation of a free ferrous porphyrin (Fig. 4A, green). The axial EPR signal and lack of super-hyperfine structure on g3 suggests that the {FeNO}7 species is 6C likely co-ordinated weakly by a solvent molecule. Thus, a ferrous porphyrin with a protonated guanidium arm at the distal site reduces NO2− to generate a {FeNO}7 species (S = 1/2, g = 2.06, 1.97; νN–O = 1680 cm−1) and a ferric porphyrin (S = 5/2, g = 5.7) mimicking the reaction at the active site of Cd1NiR, where the protons needed, is provided by the distal residues and the electrons are provided by other heme sites. Here, the EPR data collected during the course of the reaction show the simultaneous formation of {FeNO}7 species and FeIII species (Fig. 4B). While the EPR signal of S = 5/2 Fe(III) species cannot be quantified at 77 K, spin quantification of the S = 1/2 {FeNO}7 signal relative to Cu(II) standard at 77 K indicated a 45% yield of the {FeNO}7 species, suggesting that 50% of the product was {FeNO}7 species and the remaining 50% was the Fe(III) porphyrin. Note that NO stays bound to the iron porphyrin even in the presence of 100 equivalents of NO2− in the solution. Thus, even though the mechanism of NO2− reduction by [FeIIMARGH]Cl resembles that of Cd1NiR, it is not a functional model of Cd1NiR as NO is not released from the reaction center. It is important to note that the corresponding FeTPP complex (which does not bear the protonated guanidinium arm) does not produce {FeNO}7 species under the same reaction conditions and remains unreacted (Fig. S3 and S4†). These observations suggest that the local proton source is pivotal for the cleavage of the N–O bond of the NO2− to result in the formation of {FeNO}7 species.
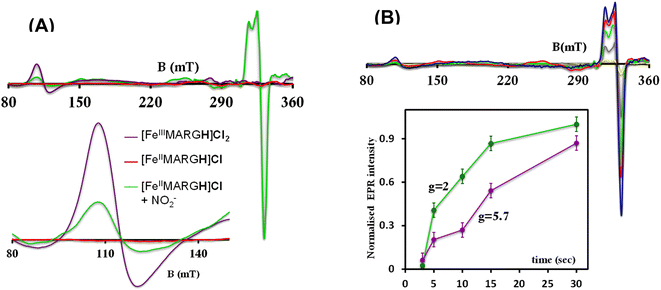 |
| | Fig. 4 (A) X-band EPR spectra of [FeIIIMARGH]Cl2 (violet) and [FeIIMARGH]Cl (red). The green spectra correspond to the {FeNO}7 species and the ferric porphyrin generated in the reaction of [FeIIMARGH]Cl with [NBu4]NO2. (B) The corresponding spectra in different time intervals upon addition of [NBu4]NO2 to [FeIIMARGH]Cl and a plot of normalized EPR intensities of the axial (violet) and rhombic (green) signals with time. | |
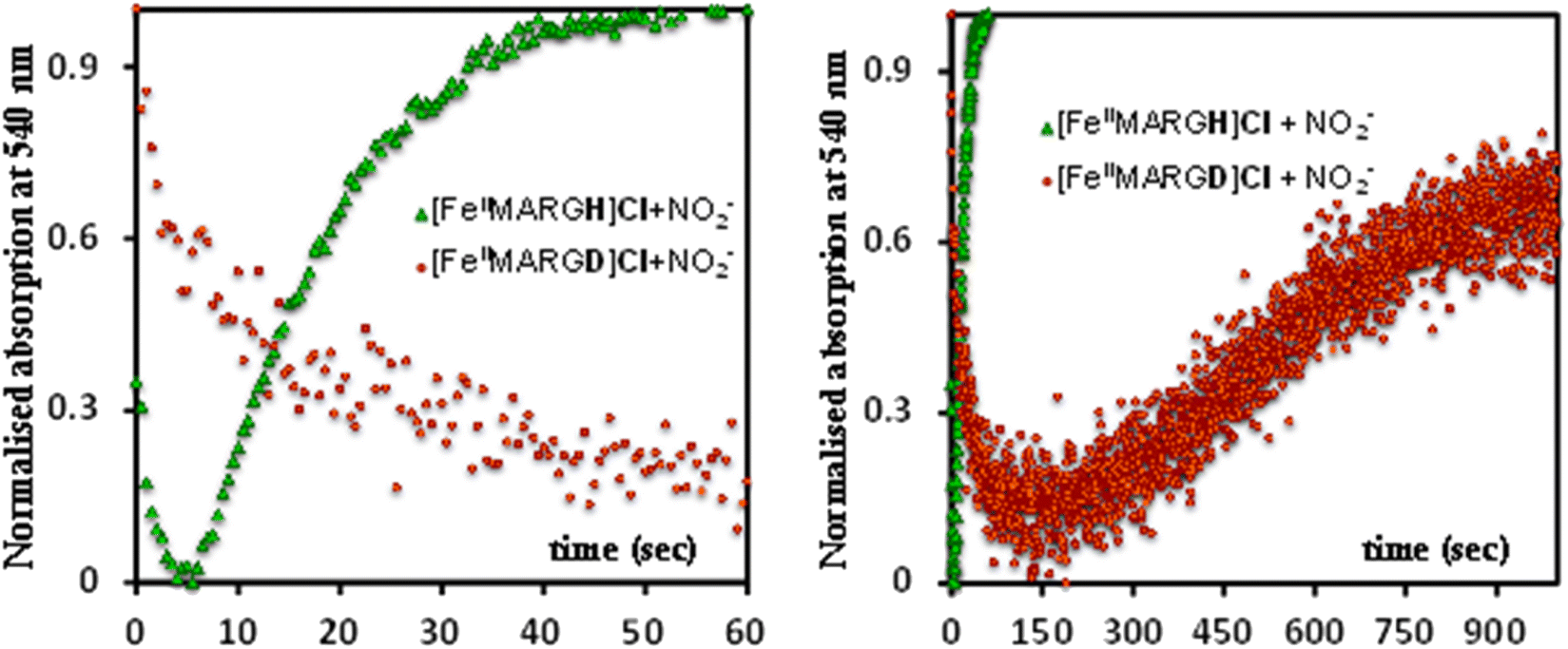
The formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 [{FeNO}7]:[FeIII] from FeII-porphyrin and NO2− requires binding of NO2− to the FeII-porphyrin followed by N–O bond cleavage to form a {FeNO}6 species and then the reduction of the {FeNO}6 species to the {FeNO}7 species by an electron transfer (ET) from another ferrous porphyrin present in the solution. Following the absorption spectra of the reaction of [FeIIMARGH]Cl and 100 equivalents of [NBu4]NO2 with time, at least one intermediate species was observed with Soret at 428 nm and Q bands at 531 nm and 569 nm (Fig. 5A, blue) which is distinct from both the reactant and the product. The kinetic trace followed at 540 nm, indicated that there are two steps in the reaction where the first step results in the formation of the intermediate, and the second step results in the formation of the {FeNO}7 species and is rate-determining (Fig. 5B). In EPR, the timescale (∼30 s) for the simultaneous formation of {FeNO}7 species and FeIII-porphyrin in a 1:1 distribution indicated that the second step corresponded to an ET from a free ferrous porphyrin in solution to the intermediate, which was also the rate-determining step of the reaction. Here, the intermediate could either be a FeII–NO2−, FeII–NO2H, or a {FeNO}6 species, and its exact determination is beyond the scope of this work. However, both the formation and decay of the intermediate showed a substantial H/D KIE (Fig. 6) indicating that the local protons are involved in the TS of formation, as well as in the decay of this intermediate. Since the reaction requires two protons for the N–O bond cleavage, and the second proton is obtained from another [FeIIMARGH]Cl, both the steps of N–O bond cleavage and bimolecular ET are likely coupled to this proton transfer resulting in a substantial KIE in the kinetics (Scheme 1). Logically, the concentration of local protons is likely to affect the rate of the reaction.
:1 [{FeNO}7]:[FeIII] from FeII-porphyrin and NO2− requires binding of NO2− to the FeII-porphyrin followed by N–O bond cleavage to form a {FeNO}6 species and then the reduction of the {FeNO}6 species to the {FeNO}7 species by an electron transfer (ET) from another ferrous porphyrin present in the solution. Following the absorption spectra of the reaction of [FeIIMARGH]Cl and 100 equivalents of [NBu4]NO2 with time, at least one intermediate species was observed with Soret at 428 nm and Q bands at 531 nm and 569 nm (Fig. 5A, blue) which is distinct from both the reactant and the product. The kinetic trace followed at 540 nm, indicated that there are two steps in the reaction where the first step results in the formation of the intermediate, and the second step results in the formation of the {FeNO}7 species and is rate-determining (Fig. 5B). In EPR, the timescale (∼30 s) for the simultaneous formation of {FeNO}7 species and FeIII-porphyrin in a 1:1 distribution indicated that the second step corresponded to an ET from a free ferrous porphyrin in solution to the intermediate, which was also the rate-determining step of the reaction. Here, the intermediate could either be a FeII–NO2−, FeII–NO2H, or a {FeNO}6 species, and its exact determination is beyond the scope of this work. However, both the formation and decay of the intermediate showed a substantial H/D KIE (Fig. 6) indicating that the local protons are involved in the TS of formation, as well as in the decay of this intermediate. Since the reaction requires two protons for the N–O bond cleavage, and the second proton is obtained from another [FeIIMARGH]Cl, both the steps of N–O bond cleavage and bimolecular ET are likely coupled to this proton transfer resulting in a substantial KIE in the kinetics (Scheme 1). Logically, the concentration of local protons is likely to affect the rate of the reaction.
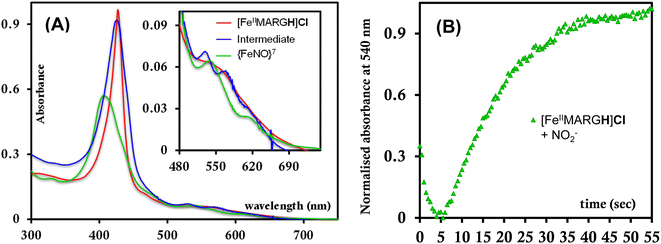 |
| | Fig. 5 (A) Intermediate absorption spectrum (blue) upon addition of [NBu4]NO2 to [FeIIMARGH]Cl (red) and the corresponding final spectrum of {FeNO}7 species (green). (B) Kinetic trace followed at 540 nm for the formation of {FeNO}7 complex upon addition of 100 equivalents of [NBu4]NO2 to [FeIIMARGH]Cl. | |
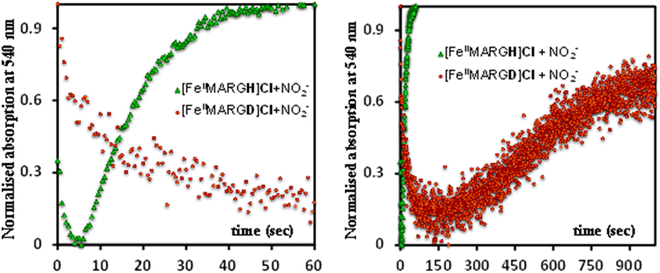 |
| | Fig. 6 Kinetic trace followed at 540 nm regarding the formation of {FeNO}7 complex upon addition of 100 equivalent of [NBu4]NO2 to [FeIIMARGH]Cl (green) and to [FeIIMARGD]Cl (orange). | |
 |
| | Scheme 1 A tentative description of the rate-determining ET step in the reduction of NO2− to {FeNO}7 species by [FeIIMARGH]Cl in the absence of external protons. The Cl− counter ion on the NO2− bound heme is not shown for clarity. | |
Reactivity of [FeIIMARGH]Cl with NO2− in the presence of an external proton
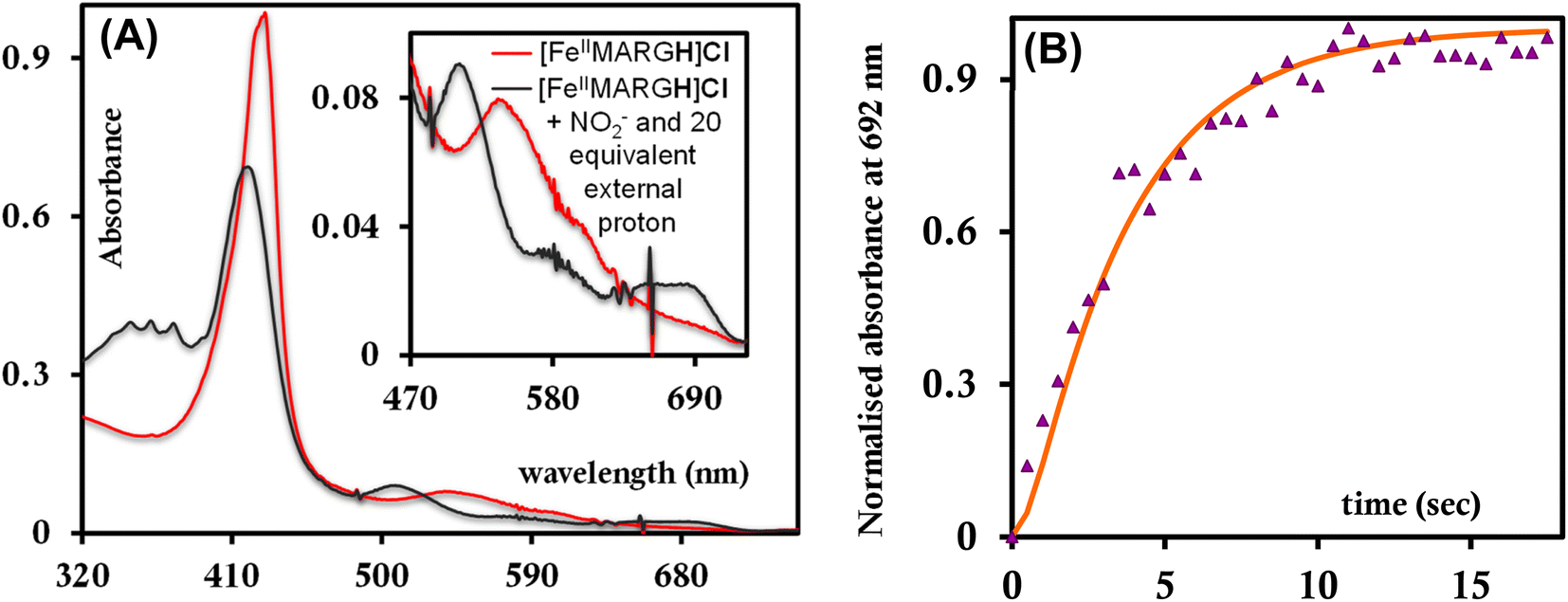
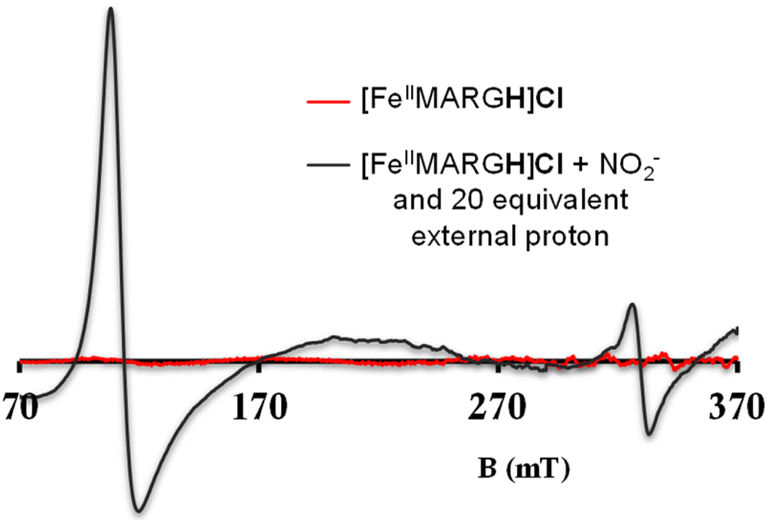
The NiRs require “proton recharge” in the enzyme as the protons in the active site are consumed during turnover.20 The distal residues get re-protonated either during the catalytic cycle, for example in CcNiR that requires a total of 8H+ to reduce NO2− to NH4+, or at the end of the catalytic cycle as is the case for Cd1NiR, which requires 2H+ for turnover. To enable the re-protonation of the pendant guanidine residue, 2,6-xylidinium chloride (as a source of the proton) was added to [FeIIMARGH]Cl simultaneously with 5 equivalents of [NBu4]NO2 at a ratio of [H+]/[NO2−] = 1. The kinetic trace of the reaction leading to the formation of {FeNO}7 species at 540 nm was significantly faster than that obtained without the addition of any external protons; also, the reaction no longer showed the intermediate species (Fig. S5†). Interestingly, the reaction completely changed when 2,6-xylidinium chloride was increased to 20 equivalents such that the [H+]/[NO2−] was 4. Here, the absorption spectrum rapidly changed to the FeIII–Cl species (Fig. 7A, grey) and no {FeNO}7 species were observed. A single axial signal at g = 5.7 in the corresponding EPR spectrum confirmed the formation of a high spin FeIII species (Fig. 8, grey), and the S = 1/2 signal for {FeNO}7 was almost absent. Note that, the formation of ferric porphyrin from the reaction of ferrous porphyrin and NO2− requires one equivalent of NO to be released. NO can be trapped using CoIITPP, which is EPR active, but forms an EPR-silent {Co(TPP)NO}7 species with NO. When the reaction mixture of 1 equivalent of [FeIIMARGH]Cl, 5 equivalents of [NBu4]NO2, and 20 equivalents of 2,6-xylidinium chloride solution was incubated with 1 equivalent CoIITPP, the corresponding absorption spectrum exhibited a Q band at 541 nm which is a characteristic of NO-bound CoIITPP complex, [CoTPP(NO)] (Fig. S6,† dark blue).60,61 Similarly, the characteristic EPR signal from 1 equivalent of CoIITPP was mostly diminished at the end of the reaction, confirming the formation of 1 equivalent of the diamagnetic [CoTPP(NO)] complex from the reaction of CoIITPP with the NO released (Fig. S7†). These results confirm that [FeIIMARGH]Cl reduces NO2− to NO in the presence of 20 equivalents of an external proton source, establishing this complex to be a functional model of Cd1NiR.
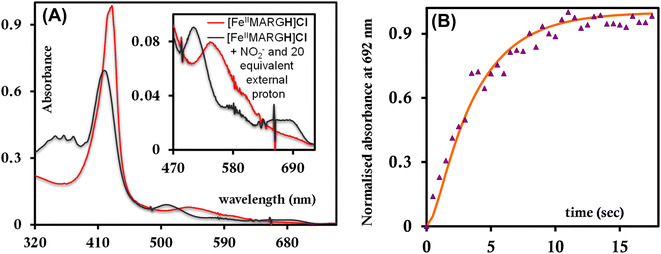 |
| | Fig. 7 (A) Absorption spectra of [FeIIMARGH]Cl (red) upon simultaneous addition of 5 equivalents of [NBu4]NO2 and 20 equivalents of 2,6-xylidinium chloride (dark grey). (B) Kinetic trace followed at 692 nm regarding the formation of FeIII–Cl species when nitrite and 20 equivalents of 2,6-xylidinium chloride are added to [FeIIMARGH]Cl simultaneously. | |
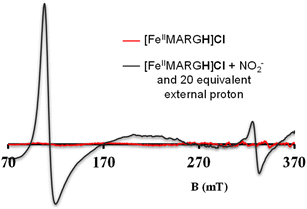 |
| | Fig. 8 X-band EPR spectra of [FeIIMARGH]Cl (red) upon simultaneous addition of 5 equivalents of [NBu4]NO2 and 20 equivalents of 2,6-xylidinium chloride (dark grey). | |
The 1st order rate of the reaction of [FeIIMARGH]Cl with [Nbu4]NO2 in the presence of 20 equivalents of 2,6-xylidinium chloride was obtained to be 0.29 ± 0.05 s−1 (Fig. 7B), and it was independent of the concentration of NO2− (Fig. S8†). The reaction did not show appreciable H/D KIE (Fig. S9†). The reaction with 2,6-xylidinium p-toluene sulfonate (different counter anion) yielded a ferric porphyrin species featuring different charge transfer bands (no Cl− counter-ion) with slightly lower rates, suggesting that NO dissociation is likely to be the rate-determining step (Fig. S10,† orange). Here, it is important to note that the reactivity of FeIITPP remains unaltered even if 20 equivalents of 2,6-xylidinium chloride solution are added to it simultaneously with [NBu4]NO2i.e., it produces a mixture of {FeNO}f7 species and FeIII–Cl of TPP species (Fig. S11,† blue). These results indicate that the fate of the reaction between [FeIIMARGH]Cl and NO2− in the presence of external protons depends mostly on the availability of protons. In the presence of an excess external proton source, the reaction does not produce the {FeNO}7 species, but produces a ferric porphyrin species and releases NO instead.
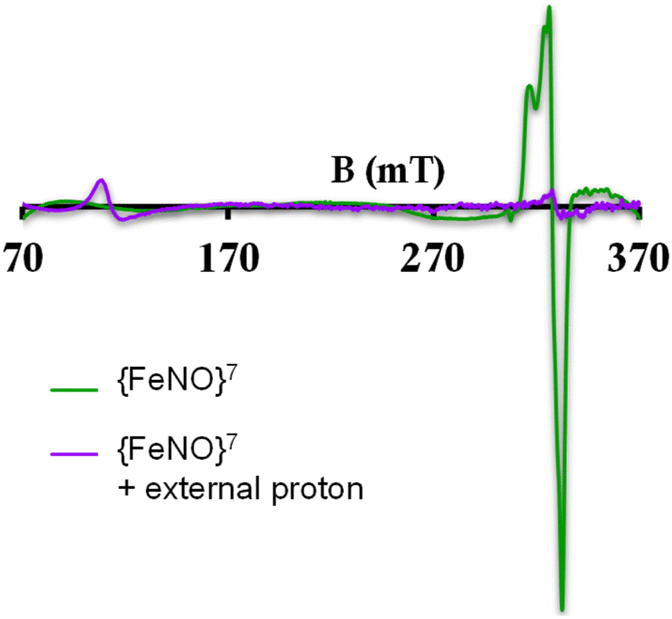
To further evaluate if a {FeNO}7 species of [MARGH]Cl porphyrin could be involved in the release of NO in the presence of an excess proton source, a {FeNO}7 complex of that porphyrin was generated by bubbling dry NO gas into an aprotic solution of [FeIIMARGH]Cl, which was confirmed by the corresponding absorption (Fig. 9A, green) and EPR spectra (Fig. 10, green), and 20 equivalents of 2,6-xylidinium chloride were added to it. A very slow reaction yielded a FeIII–Cl species (Fig. 9A, violet, and Fig. 10, purple, respectively). The corresponding EPR spectra showed that the intensity of the rhombic signal at g = 2 corresponding to the {FeNO}7 species gradually decays with a concomitant growth of the axial S = 5/2 high spin ferric porphyrin signal at g = 5.7 (Fig. S12, A and B†). However, the rate of this reaction, as monitored by the formation of FeIII–Cl species at 692 nm, was substantially slower than the rate of NO release from the reaction of [FeIIMARGH]Cl with [NBu4]NO2 in the presence of 20 equivalents of 2,6-xylidinium chloride solution (Fig. 9B). These data indicate that the presence of excess protons can induce the cleavage of the iron–nitrosyl bond of the {FeNO}7 complex of the [MARGH]Cl ligand to result in a ferric porphyrin species – albeit at a very slow rate. However, no free NO was detected here. Instead, analysis of headspace in gas chromatography (GC fitted with TCD) revealed that N2O gas was liberated. This suggests that, in the presence of excess protons, the {FeNO}7 species of the [MARGH]Cl ligand results in a ferric species and HNO, which eventually leads to the formation of N2O. Similar protonation of {FeNO}7 species leading to the liberation of N2O via an HNO intermediate is commonly observed in the active site of myoglobin62–64 and the synthetic heme d1 complexes.58,65,66 N2O is also frequently encountered as a product of NO2− reduction in cd1NiR under different assay conditions.67–69 A pure {FeNO}7 complex of TPP is also similarly generated. The {FeNO}7 species generated from FeIITPP do not show any change even with 20 equivalents of 2,6-xylidinium chloride solution at the same time scale indicating that the protonation of the corresponding {FeNO}7 species does not occur under the same reaction conditions. Similarly, NO saturated dry and degassed THF solution was added to an aprotic solution of [FeIIIMARGH]Cl2 till its corresponding EPR signal at g = 5.7 disappeared completely. The EPR silent spectrum was consistent with the formation of a diamagnetic {FeNO}6 species (Fig. S13†). The addition of 2,6-xylidinium chloride solution resulted in a regeneration of the starting ferric porphyrin complex with the axial signal at g = 5.7 (Fig. S13†) mirroring the reactivity observed during the reduction of NO2− by [FeIIMARGH]Cl in the presence of 20 equivalents of 2,6-xylidinium chloride in the solution.
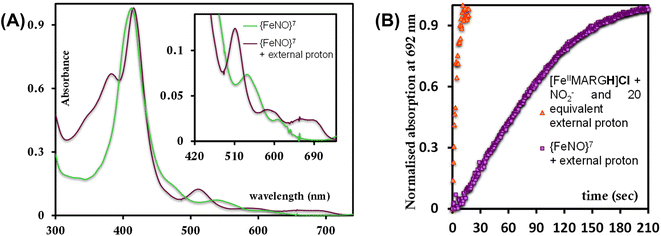 |
| | Fig. 9 (A) Absorption spectra of {FeNO}7 species of [MARGH]Cl (green) and the FeIII–Cl species generated upon addition of 2,6-xylidinium chloride to it (violet). (B) Kinetic trace at 692 nm when 2,6-xylidinium chloride is added to the {FeNO}7 complex (violet) and that of the reaction when 20 equivalents of 2,6-xylidinium chloride and 5 equivalents of [NBu4]NO2 are added simultaneously to [FeIIMARGH]Cl (orange). | |
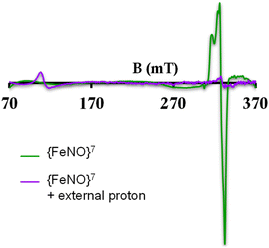 |
| | Fig. 10 X-band EPR spectra of {FeNO}7 species of [MARGH]Cl (green) and FeIII–Cl species upon addition of 2,6-xylidinium chloride to it (purple). | |
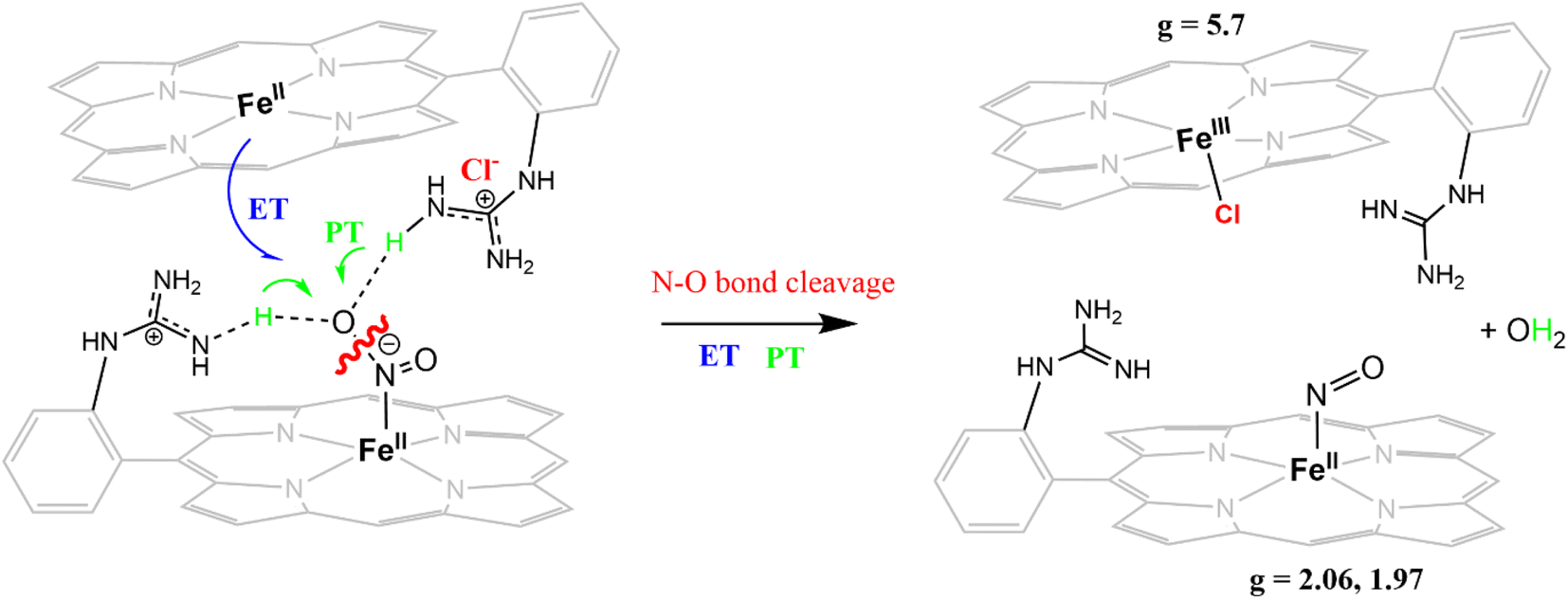
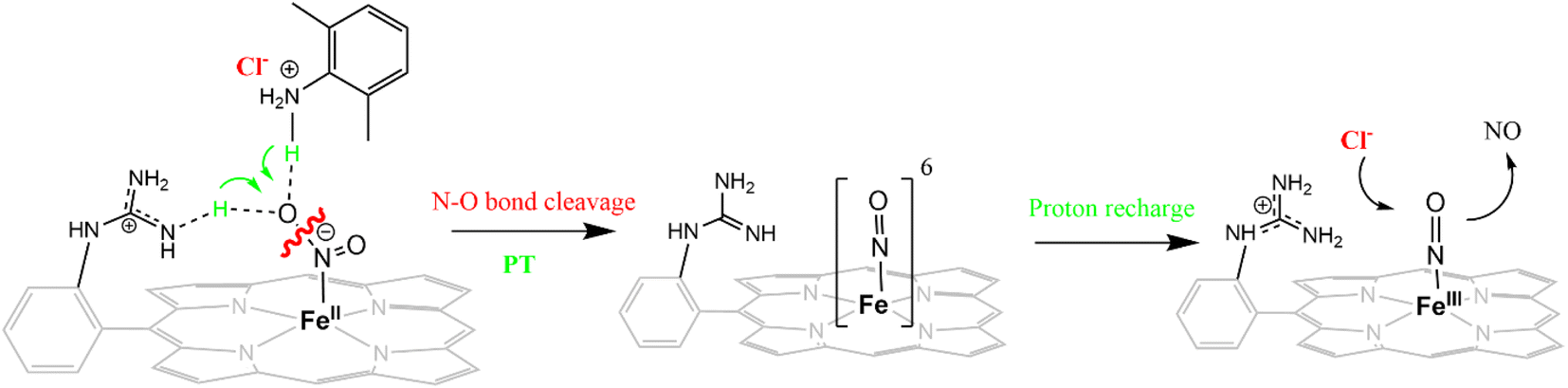
The reduction of NO2− to NO in the presence of 20 equivalents of 2,6-xylidinium chloride catalyzed by [FeIIMARGH]Cl likely follows a mechanism that involves a {FeNO}6 intermediate (Scheme 2). The rate of dissociation of NO from {FeNO}6 species is 0.29 ± 0.05 s−1, which is two orders of magnitude higher than the rate of NO dissociation observed for that of {FeNO}6 complex of TPP in the presence of halide anion.70 The binding of an anionic Cl− (counter anion) or NO2− (excess) ligand to the ferric center of the porphyrin in preference to a neutral NO ligand, driven by direct hydrogen bonding and electrostatic interaction of the anion with the protonated guanidinium pendant arm is likely to be the driving force behind NO dissociation from the {FeNO}6 species of [MARGH]Cl. Such protonated guanidinium pendant arm mimics the 2nd sphere amino acid residues (like arginine/lysine) in the catalytic active sites of heme NiRs which are protonated at physiological pH. Geometry-optimized density functional theory (DFT) calculations are used to gain insight into the effect of the protonated guanidinium pendant arm on the overall energy of the replacement of NO by NO2− in ferric porphyrins.
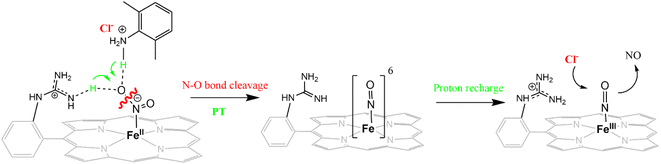 |
| | Scheme 2 Schematic representation of NO displacement from {FeNO}6 species of [MARGH]Cl by Cl− ligand upon simultaneous addition of 20 equivalents of 2,6-xylidinium chloride with [NBu4]NO2 to [FeIIMARGH]Cl complex. | |
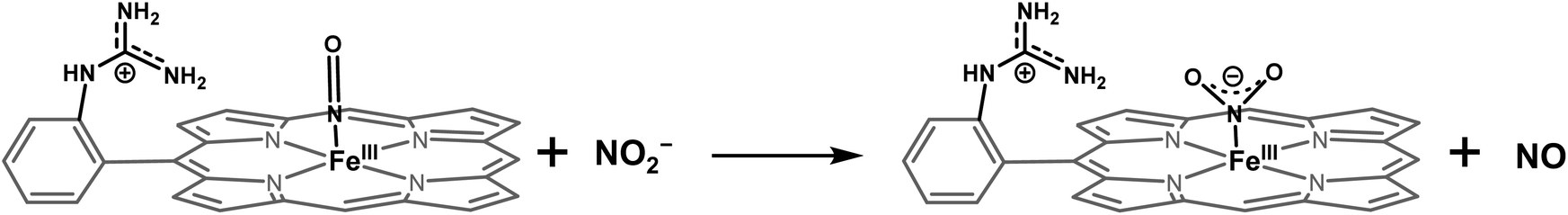
DFT optimized structures (BP86/6-311g*) are obtained for hypothetical models of {FeNO}6 species and FeIII–NO2−/FeIII–Cl species for the TPP ligand, as well as the [MARGH]Cl ligand, which carries a protonated guanidinium arm. The choice of NO2− along with Cl− is justified by the fact that the former is more relevant to the mechanism of heme NiRs. The reaction is presented in Scheme 3; the free energies (BP86/6-311 + g*) are calculated using a polarized continuum model (PCM) with THF as solvent. The ΔG of the reaction in the case of [MARGH]Cl was calculated to be 11.8 kcal mol−1, and the same reaction was calculated to be 20.3 kcal mol−1 in TPP. Thus, the pendant guanidinium arm lowers the energy of NO displacement by NO2− by ∼8.5 kcal mol−1. Similarly, the displacement of NO by Cl− was computed to be 16.3 kcal mol−1 in [MARGH]Cl relative to 22.9 kcal mol−1 in TPP. The optimized geometry of the NO2−-bound [FeIIIMARGH]NO2 showed a direct H-bonding interaction between the NO2− and the protonated guanidine group (Fig. 11A). Additionally, the electrostatic potential map of the structure shows positive potential on the protonated guanidinium residue and negative potential on the bound NO2− suggesting the presence of electrostatic attraction between the two centers (Fig. 11B). Both hydrogen bonding and electrostatic stabilization together resulted in an additional ∼8.5 kcal mol−1 stabilization for NO2− displacing NO in [MARGH]Cl relative to TPP.
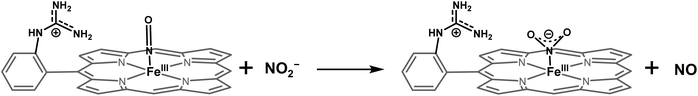 |
| | Scheme 3 Schematic representation of NO displacement from {FeNO}6 species of [MARGH]Cl by the π anisotropic NO2− ligand. | |
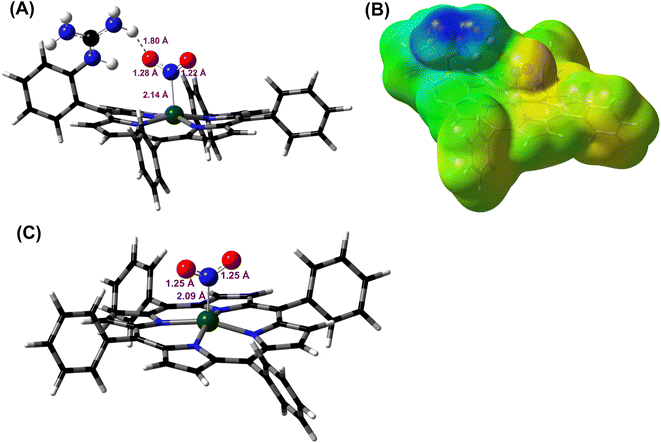 |
| | Fig. 11 (A) DFT optimized structure of the NO2− bound [FeIIIMARGH]Cl2 species. Fe: green, N: blue, O: red, H: off-white. Fe–N(NO2−), N–O, and the MARGH–O distances are shown here. (B) Electrostatic potential surface (blue: positive and red: negative) of the same species. This was generated using the cubegen utility of Gaussian 16 software with an iso value of 0.0004. (C) DFT optimized structure of the NO2− bound FeIIITPP species where Fe: green, N: blue, O: red, H: off-white. | |
Discussion
The FeTPP does not reduce NO2− in the absence of external protons but produces {FeNO}7 species when external protons are supplied. The {FeNO}7 complex generated is kinetically inert and does not release NO. In contrast, the [MARGH]Cl ligand framework allows the reduction of NO2− to {FeNO}7 species in the presence of 2nd sphere protonated guanidinium residue, which provides the necessary protons, and thus, it does not require any external protons. Importantly, [FeIIMARGH]Cl can release NO from the {FeNO}6 state when an external proton is present in excess (20 equivalents), in addition to the NO2− ion. The rate of NO dissociation (k = 29 ± 5× 10−2 s−1) under these conditions favorably compares with the values derived for NO dissociation in the NiR proteins (koff ≈ 10−1–10−2 s−1).39,54,71,72 Alternatively, the rate of NO dissociation from a {FeNO}6 complex of TPP has substantially slower rates (10−4–10−5 s−1) irrespective of the axial ligand.70 The rate of NO release from the {FeNO}6 complex of [MARGH]Cl is much faster due to more favorable binding of anions present in the reaction medium (e.g. Cl−, NO2−) relative to the neutral NO ligand in the presence of a protonated cationic guanidine in the second sphere. The key to facile NO release from {FeNO}6 species of [MARGH]Cl lies in recharging the distal guanidine (which gets deprotonated during the reduction of NO2− to NO) with external proton source at the end of turnover. In an enzyme active site, these protons may be obtained from the solution or stored in multiple such second sphere residues. This situation presents itself in both CcNiR and CsNiR enzymes, which reduce NO2− to NH4+, where the distal site contains protonated basic residues, such as arginine (multiple in CsNiR) and lysine. In CcNiR, the reduction of {FeNO}6 to {FeNO}7 competes with NO dissociation, when weak reductants are used, and the dissociation of NO from {FeNO}6 species is indeed observed at rates comparable to the rates observed here.73 However, with stronger reductants, the NO dissociation is avoided by rapid reduction of the {FeNO}6 species to the kinetically inert {FeNO}7 species, which is reasonably slow to react with a protonated guanidine present in the 2nd sphere. In CsNiR, the additional reducing equivalent present in the Fe4S4 cluster ensures rapid reduction of {FeNO}6 species produced after the N–O bond cleavage to {FeNO}7, which likely avoids NO dissociation before further reduction.
In the Cd1NiR active site, the protonation state of the distal histidine residues can affect the dynamic H-bonding network with NO of the iron–NO complex synergistically with the unique properties of the d1 heme structure to facilitate the release of NO from the {FeNO}7 intermediate.35,39,48,53,54 Previously, we and others have demonstrated that the structure of the heme can weaken the Fe–NO bond allowing NO dissociation from {FeNO}7 species, which is generally assumed to be kinetically inert, e.g., the {FeNO}7 complex of TPP. In this study, we have demonstrated that NO dissociation occurs from {FeNO}6 complex of [MARGH]Cl aided by the protonated distal guanidinium moiety and not from its {FeNO}7 species. This observation strongly indicates that while the heme structure is likely to contribute more to the lability of the Fe–NO bond in the {FeNO}7 intermediate of Cd1NiR, NO dissociation from {FeNO}6 species, on the other hand, is driven by the binding of counter anions to the ferric center of that species (Cl−/p-toluenesulfonate or NO2−) aided by the protonated distal residues.70,74 For [FeIIIMARGH]Cl2 porphyrin, these rates are higher than FeTPP by three orders of magnitude due to direct hydrogen bonding and electrostatic interactions of an anionic ligand with the protonated guanidinium moiety. The presence of the protonated guanidium moiety does not lead to NO dissociation from the {FeNO}7 species of [MARGH]Cl and a sluggish protonation is observed instead. Thus, the heme structure is the major factor resulting in the lability of the Fe–NO bond in the {FeNO}7 species, while the 2nd sphere protonated amino acid residue is the major factor contributing to the lability of the Fe–NO bond in the {FeNO}6 species.
Summary
An iron porphyrin complex, [FeIIIMARGH]Cl2, which possesses a covalently attached protonated pendant guanidinium arm, mimicking the arginine residues present in the active sites of NiR, can catalyze the reduction of NO2− to NO in the presence of excess external protons, whereas, in the absence of external protons, it is reduced to kinetically inert {FeNO}7 species. External protons present in the solution can “recharge” the distal deprotonated guanidine moiety of corresponding {FeNO}6 intermediate, facilitating the dissociation of NO from the ferric center of the complex by favoring the binding of anionic ligands, such as Cl−/NO2− assisted by hydrogen bonding and electrostatic interaction with the protonated guanidinium residue in the 2nd sphere. These results strongly suggest that the state of protonation of the distal guanidinium group of the iron porphyrin complex is crucial (a) to the reactivity of reducing NO2− to either NO (via {FeNO}6 intermediate) or N2O (via {FeNO}7 intermediate) and (b) in the release of NO, the product, from the {FeNO}6 intermediate by preferably binding NO2−, the substrate, during the reduction of nitrite making it a functional model of cd1NiR. Overall, the results presented here show that the 2nd sphere protonated arginine residue can be a major factor contributing to the lability of Fe–NO bond in the {FeNO}6 species formed in the active site of NiRs.
Data availability
Additional experimetal data, methods and optimised coordinates are available in the ESI file.†
Author contributions
A. D. designed the research. A. S. performed all the experiments. S. B. and A. S. synthesized the complexes. S. C. performed the theoretical calculations. A. S. and A. D. analysed the experimental results and wrote the manuscript. S. C. and A. D. analysed the theoritical results. All authors provided feedback on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is funded by the SERB Grant CRG/2021/000154. A. S. acknowledges the DST-INSPIRE fellowship of the Government of India. S. B. and S. C. acknowledge CSIR-SPM, Government of India and IACS Integrated PhD program, respectively, for their fellowship.
References
- R. S. Zajicek, M. L. Cartron and S. J. Ferguson, Probing the Unusual Oxidation/Reduction Behavior of Paracoccus pantotrophus Cytochrome cd1 Nitrite Reductase by Replacing a Switchable Methionine Heme Iron Ligand with Histidine, Biochemistry, 2006, 45(37), 11208–11216 CrossRef CAS PubMed.
- L. B. Maia and J. J. G. Moura, How Biology Handles Nitrite, Chem. Rev., 2014, 114(10), 5273–5357 CrossRef CAS PubMed.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, The Biologically Relevant Coordination Chemistry of Iron and Nitric Oxide: Electronic Structure and Reactivity, Chem. Rev., 2021, 121(24), 14682–14905 CrossRef CAS PubMed.
- C. Costa, J. J. Moura, I. Moura, M. Y. Liu, H. D. Peck, J. LeGall, Y. N. Wang and B. H. Huynh, Hexaheme nitrite reductase from Desulfovibrio desulfuricans. Mössbauer and EPR characterization of the heme groups, J. Biol. Chem., 1990, 265(24), 14382–14388 CrossRef CAS PubMed.
- P. Stach, O. Einsle, W. Schumacher, E. Kurun and P. M. Kroneck, Bacterial cytochrome c nitrite reductase: new structural and functional aspects, J. Inorg. Biochem., 2000, 79(1–4), 381–385 CrossRef CAS PubMed.
- S.-i. Kajie and Y. Anraku, Purification of a hexaheme cytochrome C552 from Escherichia coli K 12 and its properties as a nitrite reductase, Eur. J. Biochem., 1986, 154(2), 457–463 CrossRef CAS PubMed.
- C. Costa, A. Macedo, I. Moura, J. J. G. Moura, J. Le Gall, Y. Berlier, M. Y. Liu and W. J. Payne, Regulation of the hexaheme nitrite/nitric oxide reductase of Desulfovibrio desulfuricans, Wolinella succinogenes and Escherichia coli, FEBS Lett., 1990, 276(1–2), 67–70 CrossRef CAS PubMed.
- M. Rudolf, O. Einsle, F. Neese and P. M. H. Kroneck, Pentahaem cytochrome c nitrite reductase: reaction with hydroxylamine, a potential reaction intermediate and substrate, Biochem. Soc. Trans., 2002, 30(4), 649–653 CrossRef CAS PubMed.
- J. H. van Wonderen, B. Burlat, D. J. Richardson, M. R. Cheesman and J. N. Butt, The Nitric Oxide Reductase Activity of Cytochrome c Nitrite Reductase from Escherichia coli, J. Biol. Chem., 2008, 283(15), 9587–9594 CrossRef CAS PubMed.
- C. M. Silveira, S. Besson, I. Moura, J. J. G. Moura and M. G. Almeida, Measuring the Cytochrome c Nitrite Reductase Activity—Practical Considerations on the Enzyme Assays, Bioinorg. Chem. Appl., 2010, 2010, 634597 Search PubMed.
- M. Hirasawa, J. N. Tripathy, F. Sommer, R. Somasundaram, J.-S. Chung, M. Nestander, M. Kruthiventi, M. Zabet-Moghaddam, M. K. Johnson, S. S. Merchant, J. P. Allen and D. B. Knaff, Enzymatic properties of the ferredoxin-dependent nitrite reductase from Chlamydomonas reinhardtii. Evidence for hydroxylamine as a late intermediate in ammonia production, Photosynth. Res., 2010, 103(2), 67–77 CrossRef CAS PubMed.
- D. P. Hucklesby and E. J. Hewitt, Nitrite and hydroxylamine reduction in higher plants. Fractionation, electron donor and substrate specificity of leaf enzymes, principally from vegetable marrow (Cucurbita pepo L.), Biochem. J., 1970, 119(4), 615–627 CrossRef CAS PubMed.
- M. Kukimoto, M. Nishiyama, M. E. P. Murphy, S. Turley, E. T. Adman, S. Horinouchi and T. Beppu, X-ray Structure and Site-Directed Mutagenesis of a Nitrite Reductase from Alcaligenes Faecalis S-6: Roles of Two Copper Atoms in Nitrite Reduction, Biochemistry, 1994, 33(17), 5246–5252 CrossRef CAS PubMed.
- J. W. Godden, S. Turley, D. C. Teller, E. T. Adman, M. Y. Liu, W. J. Payne and J. LeGall, The 2.3 angstrom X-ray structure of nitrite reductase from Achromobacter cycloclastes, Science, 1991, 253(5018), 438 CrossRef CAS PubMed.
- R. W. Strange, F. E. Dodd, Z. H. L. Abraham, J. G. Grossmann, T. Brüser, R. R. Eady, B. E. Smith and S. S. Hasnain, The substrate-binding site in Cu nitrite reductase and its similarity to Zn carbonic anhydrase, Nat. Struct. Biol., 1995, 2(4), 287–292 CrossRef CAS PubMed.
- F. Jacobson, A. Pistorius, D. Farkas, W. De Grip, Ö. Hansson, L. Sjölin and R. Neutze, pH Dependence of Copper Geometry, Reduction Potential, and Nitrite Affinity in Nitrite Reductase, J. Biol. Chem., 2007, 282(9), 6347–6355 CrossRef CAS PubMed.
- D. Nurizzo, M.-C. Silvestrini, M. Mathieu, F. Cutruzzolà, D. Bourgeois, V. Fülöp, J. Hajdu, M. Brunori, M. Tegoni and C. Cambillau, N-terminal arm exchange is observed in the 2.15 A0 crystal structure of oxidized nitrite reductase from Pseudomonas aeruginosa, Structure, 1997, 5(9), 1157–1171 CrossRef CAS PubMed.
- V. Fülöp, J. W. B. Moir, S. J. Ferguson and J. Hajdu, The anatomy of a bifunctional enzyme: Structural basis for reduction of oxygen to water and synthesis of nitric oxide by cytochrome cd1, Cell, 1995, 81(3), 369–377 CrossRef PubMed.
- K. E. Hill and D. C. Wharton, Reconstitution of the apoenzyme of cytochrome oxidase from Pseudomonas aeruginosa with heme d1 and other heme groups, J. Biol. Chem., 1978, 253(2), 489–495 CrossRef CAS PubMed.
- D. Bykov and F. Neese, Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase: Insights from Density Functional Theory Studies, Inorg. Chem., 2015, 54(19), 9303–9316 CrossRef CAS PubMed.
- S. Hartshorne, D. J. Richardson and J. Simon, Multiple haem lyase genes indicate substrate specificity in cytochrome c biogenesis, Biochem. Soc. Trans., 2006, 34(1), 146–149 CrossRef CAS PubMed.
- W. Sun, M. Arese, M. Brunori, D. Nurizzo, K. Brown, C. Cambillau, M. Tegoni and F. Cutruzzolà, Cyanide Binding to cd1 Nitrite Reductase from Pseudomonas aeruginosa: Role of the Active-Site His369 in Ligand Stabilization, Biochem. Biophys. Res. Commun., 2002, 291(1), 1–7 CrossRef CAS PubMed.
- U. Swamy, M. Wang, J. N. Tripathy, S.-K. Kim, M. Hirasawa, D. B. Knaff and J. P. Allen, Structure of Spinach Nitrite Reductase: Implications for Multi-electron Reactions by the Iron−Sulfur:Siroheme Cofactor, Biochemistry, 2005, 44(49), 16054–16063 CrossRef CAS PubMed.
- S. Nakano, M. Takahashi, A. Sakamoto, H. Morikawa and K. Katayanagi, The reductive reaction mechanism of tobacco nitrite reductase derived from a combination of crystal structures and ultraviolet–visible microspectroscopy, Proteins: Struct., Funct., Bioinf., 2012, 80(8), 2035–2045 CAS.
- F. Cutruzzolà, K. Brown, E. K. Wilson, A. Bellelli, M. Arese, M. Tegoni, C. Cambillau and M. Brunori, The nitrite reductase from Pseudomonas aeruginosa: Essential role of two active-site histidines in the catalytic and structural properties, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(5), 2232 CrossRef PubMed.
- A. Jafferji, J. W. A. Allen, S. J. Ferguson and V. Fülöp, X-ray Crystallographic Study of Cyanide Binding Provides Insights into the Structure-Function Relationship for Cytochromecd 1 Nitrite Reductase from Paracoccus pantotrophus, J. Biol. Chem., 2000, 275(33), 25089–25094 CrossRef CAS PubMed.
- S. Rinaldo, G. Giardina, N. Castiglione, V. Stelitano and F. Cutruzzolà, The catalytic mechanism of Pseudomonas aeruginosa cd1 nitrite reductase, Biochem. Soc. Trans., 2011, 39(1), 195–200 CrossRef CAS PubMed.
- G. Ranghino, E. Scorza, T. Sjögren, P. A. Williams, M. Ricci and J. Hajdu, Quantum Mechanical Interpretation of Nitrite Reduction by Cytochrome cd1 Nitrite Reductase from Paracoccus pantotrophus, Biochemistry, 2000, 39(36), 10958–10966 CrossRef CAS PubMed.
- F. Cutruzzola, K. Brown, E. K. Wilson, A. Bellelli, M. Arese, M. Tegoni, C. Cambillau and M. Brunori, The nitrite reductase from Pseudomonas aeruginosa: essential role of two active-site histidines in the catalytic and structural properties, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(5), 2232–2237 CrossRef CAS PubMed.
- S. Kuznetsova, D. B. Knaff, M. Hirasawa, P. Sétif and T. A. Mattioli, Reactions of Spinach Nitrite Reductase with Its Substrate, Nitrite, and a Putative Intermediate, Hydroxylamine, Biochemistry, 2004, 43(33), 10765–10774 CrossRef CAS PubMed.
- K. M. Polyakov, K. M. Boyko, T. V. Tikhonova, A. Slutsky, A. N. Antipov, R. A. Zvyagilskaya, A. N. Popov, G. P. Bourenkov, V. S. Lamzin and V. O. Popov, High-Resolution Structural Analysis of a Novel Octaheme Cytochrome c Nitrite Reductase from the Haloalkaliphilic Bacterium Thioalkalivibrio nitratireducens, J. Mol. Biol., 2009, 389(5), 846–862 CrossRef CAS PubMed.
- O. Einsle, A. Messerschmidt, R. Huber, P. M. H. Kroneck and F. Neese, Mechanism of the Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase, J. Am. Chem. Soc., 2002, 124(39), 11737–11745 CrossRef CAS PubMed.
- E. P. Day, J. Peterson, J. J. Bonvoisin, L. J. Young, J. O. Wilkerson and L. M. Siegel, Magnetization of the sulfite and nitrite complexes of oxidized sulfite and nitrite reductases: EPR silent spin S = 1/2 states, Biochemistry, 1988, 27(6), 2126–2132 CrossRef CAS PubMed.
- L. J. Young and L. M. Siegel, On the reaction of ferric heme proteins with nitrite and sulfite, Biochemistry, 1988, 27(8), 2790–2800 CrossRef CAS PubMed.
- M. Radoul, D. Bykov, S. Rinaldo, F. Cutruzzolà, F. Neese and D. Goldfarb, Dynamic Hydrogen-Bonding Network in the Distal Pocket of the Nitrosyl Complex of Pseudomonas aeruginosa cd1 Nitrite Reductase, J. Am. Chem. Soc., 2011, 133(9), 3043–3055 CrossRef CAS PubMed.
- S. Kuznetsova, D. B. Knaff, M. Hirasawa, B. Lagoutte and P. Sétif, Mechanism of Spinach Chloroplast Ferredoxin-Dependent Nitrite Reductase: Spectroscopic Evidence for Intermediate States, Biochemistry, 2004, 43(2), 510–517 CrossRef CAS PubMed.
- J. Yi, L. M. Thomas and G. B. Richter-Addo, Distal Pocket Control of Nitrite Binding in Myoglobin, Angew. Chem., Int. Ed., 2012, 51(15), 3625–3627 CrossRef CAS PubMed.
- M. C. Silvestrini, M. G. Tordi, G. Musci and M. Brunori, The reaction of Pseudomonas nitrite reductase and nitrite. A stopped-flow and EPR study, J. Biol. Chem., 1990, 265(20), 11783–11787 CrossRef CAS PubMed.
- S. Rinaldo, A. Arcovito, M. Brunori and F. Cutruzzolà, Fast Dissociation of Nitric Oxide from Ferrous Pseudomonas aeruginosa cd1 Nitrite Reductase: A NOVEL OUTLOOK ON THE CATALYTIC MECHANISM, J. Biol. Chem., 2007, 282(20), 14761–14767 CrossRef CAS PubMed.
- D. Nurizzo, F. Cutruzzolà, M. Arese, D. Bourgeois, M. Brunori, C. Cambillau and M. Tegoni, Does the Reduction of c Heme Trigger the Conformational Change of Crystalline Nitrite Reductase?, J. Biol. Chem., 1999, 274(21), 14997–15004 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Principles of structure, bonding, and reactivity for metal nitrosyl complexes, Coord. Chem. Rev., 1974, 13(4), 339–406 CrossRef CAS.
- D. Bykov and F. Neese, Reductive activation of the heme iron–nitrosyl intermediate in the reaction mechanism of cytochrome c nitrite reductase: a theoretical study, JBIC, J. Biol. Inorg. Chem., 2012, 17(5), 741–760 CrossRef CAS PubMed.
- A. Freitag and E. Bock, Energy conservation in Nitrobacter, FEMS Microbiol. Lett., 1990, 66(1–3), 157–162 CrossRef CAS.
- L. E. Goodrich, S. Roy, E. E. Alp, J. Zhao, M. Y. Hu and N. Lehnert, Electronic Structure and Biologically Relevant Reactivity of Low-Spin {FeNO}8 Porphyrin Model Complexes: New Insight from a Bis-Picket Fence Porphyrin, Inorg. Chem., 2013, 52(13), 7766–7780 CrossRef CAS PubMed.
- B. A. Averill, Dissimilatory Nitrite and Nitric Oxide Reductases, Chem. Rev., 1996, 96(7), 2951–2964 CrossRef CAS PubMed.
- V. K. K. Praneeth, F. Paulat, T. C. Berto, S. D. George, C. Näther, C. D. Sulok and N. Lehnert, Electronic Structure of Six-Coordinate Iron(III)−Porphyrin NO Adducts: The Elusive Iron(III)−NO(radical) State and Its Influence on the Properties of These Complexes, J. Am. Chem. Soc., 2008, 130(46), 15288–15303 CrossRef CAS PubMed.
- S. J. George, J. W. A. Allen, S. J. Ferguson and R. N. F. Thorneley, Time-resolved Infrared Spectroscopy Reveals a Stable Ferric Heme-NO Intermediate in the Reaction of Paracoccus pantotrophus Cytochrome cd 1 Nitrite Reductase with Nitrite, J. Biol. Chem., 2000, 275(43), 33231–33237 CrossRef CAS PubMed.
- M. R. Cheesman, S. J. Ferguson, J. W. B. Moir, D. J. Richardson, W. G. Zumft and A. J. Thomson, Two Enzymes with a Common Function but Different Heme Ligands in the Forms as Isolated. Optical and Magnetic Properties of the Heme Groups in the Oxidized Forms of Nitrite Reductase, Cytochrome cd1, from Pseudomonas stutzeri and Thiosphaera pantotropha, Biochemistry, 1997, 36(51), 16267–16276 CrossRef CAS PubMed.
- K. A. Sam, M. J. F. Strampraad, S. de Vries and S. J. Ferguson, Very Early Reaction Intermediates Detected by Microsecond Time Scale Kinetics of Cytochrome cd1-catalyzed Reduction of Nitrite, J. Biol. Chem., 2008, 283(41), 27403–27409 CrossRef CAS PubMed.
- S. Rinaldo, K. A. Sam, N. Castiglione, V. Stelitano, A. Arcovito, M. Brunori, J. W. A. Allen, S. J. Ferguson and F. Cutruzzolà, Observation of fast release of NO from ferrous d1 haem allows formulation of a unified reaction mechanism for cytochrome cd1 nitrite reductases, Biochem. J., 2011, 435(1), 217–225 CrossRef CAS PubMed.
- C. D. Richter, J. W. A. Allen, C. W. Higham, A. Koppenhöfer, R. S. Zajicek, N. J. Watmough and S. J. Ferguson, Cytochrome cd 1, Reductive Activation and Kinetic Analysis of a Multifunctional Respiratory Enzyme, J. Biol. Chem., 2002, 277(5), 3093–3100 CrossRef CAS PubMed.
- K. Kobayashi, A. Koppenhöfer, S. J. Ferguson, N. J. Watmough and S. Tagawa, Intramolecular Electron Transfer from c Heme to d1 Heme in Bacterial Cytochrome cd1 Nitrite Reductase Occurs over the Same Distances at Very Different Rates Depending on the Source of the Enzyme, Biochemistry, 2001, 40(29), 8542–8547 CrossRef CAS PubMed.
- O. Farver, M. Brunori, F. Cutruzzolà, S. Rinaldo, S. Wherland and I. Pecht, Intramolecular Electron Transfer in Pseudomonas aeruginosa cd1 Nitrite Reductase: Thermodynamics and Kinetics, Biophys. J., 2009, 96(7), 2849–2856 CrossRef CAS PubMed.
- S. Rinaldo, M. Brunori and F. Cutruzzolà, Nitrite controls the release of nitric oxide in Pseudomonas aeruginosa cd1 nitrite reductase, Biochem. Biophys. Res. Commun., 2007, 363(3), 662–666 CrossRef CAS PubMed.
- F. Cutruzzolà, M. Arese, S. Grasso, A. Bellelli and M. Brunori, Mutagenesis of nitrite reductase from Pseudomonas aeruginosa: tyrosine-10 in the c heme domain is not involved in catalysis 1, FEBS Lett., 1997, 412(2), 365–369 CrossRef PubMed.
- M. Radoul, F. Centola, S. Rinaldo, F. Cutruzzolà, I. Pecht and D. Goldfarb, Heme d1 Nitrosyl Complex of cd1 Nitrite Reductase Studied by High-Field-Pulse Electron Paramagnetic Resonance Spectroscopy, Inorg. Chem., 2009, 48(9), 3913–3915 CrossRef CAS PubMed.
- S. E. J. Bowman and K. L. Bren, The chemistry and biochemistry of hemec: functional bases for covalent attachment, Nat. Prod. Rep., 2008, 25(6), 1118–1130 RSC.
- S. Amanullah and A. Dey, The role of porphyrin peripheral substituents in determining the reactivities of ferrous nitrosyl species, Chem. Sci., 2020, 11(23), 5909–5921 RSC.
- S. Bhakta, A. Nayek, B. Roy and A. Dey, Induction of Enzyme-like Peroxidase Activity in an Iron Porphyrin Complex Using Second Sphere Interactions, Inorg. Chem., 2019, 58, 2954–2964 CrossRef CAS PubMed.
- M. Kumar, N. A. Dixon, A. C. Merkle, M. Zeller, N. Lehnert and E. T. Papish, Hydrotris(triazolyl)borate complexes as functional models for Cu nitrite reductase: the electronic influence of distal nitrogens, Inorg. Chem., 2012, 51(13), 7004–7006 CrossRef CAS PubMed.
- T. Kurtikyan, H. AΠ, H. Harutyunyan, R. Ghazaryan and J. Goodwin, Spectral study of the nitrogen monoxide interaction with sublimed layers of meso-mono-4-pyridyl-tri-phenyl-and meso-mono-3-pyridyl-tri-phenyl-porphyrinatocobalt(II), J. Porphyrins Phthalocyanines, 2006, 10, 971 CrossRef CAS.
- R. Lin and P. J. Farmer, The HNO Adduct of Myoglobin: Synthesis and Characterization, J. Am. Chem. Soc., 2000, 122(10), 2393–2394 CrossRef CAS.
- C. E. Immoos, F. Sulc, P. J. Farmer, K. Czarnecki, D. F. Bocian, A. Levina, J. B. Aitken, R. S. Armstrong and P. A. Lay, Bonding in HNO-Myoglobin as Characterized by X-ray Absorption and Resonance Raman Spectroscopies, J. Am. Chem. Soc., 2005, 127(3), 814–815 CrossRef CAS PubMed.
- N. Lehnert, V. K. K. Praneeth and F. Paulat, Electronic structure of iron(II)–porphyrin nitroxyl complexes: Molecular mechanism of fungal nitric oxide reductase (P450nor), J. Comput. Chem., 2006, 27(12), 1338–1351 CrossRef CAS PubMed.
- V. Shafirovich and S. V. Lymar, Nitroxyl and its anion in aqueous solutions: Spin states, protic equilibria, and reactivities toward oxygen and nitric oxide, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(11), 7340 CrossRef CAS PubMed.
- J. L. Heinecke, C. Khin, J. C. M. Pereira, S. A. Suárez, A. V. Iretskii, F. Doctorovich and P. C. Ford, Nitrite Reduction Mediated by Heme Models. Routes to NO and HNO?, J. Am. Chem. Soc., 2013, 135(10), 4007–4017 CrossRef CAS PubMed.
- A. B. Hooper, A nitrite-reducing enzyme from Nitrosomonas europaea Preliminary characterization with hydroxylamine as electron donor, Biochim. Biophys. Acta, Bioenerg., 1968, 162(1), 49–65 CrossRef CAS PubMed.
- P. R. Alefounder and S. J. Ferguson, Electron transport-linked nitrous oxide synthesis and reduction by Paracoccusdenitrificans monitored with an electrode, Biochem. Biophys. Res. Commun., 1982, 104(3), 1149–1155 CrossRef CAS PubMed.
- P. Bessières and Y. Henry, Stoichiometry of nitrite reduction catalyzed by Pseudomonas aeruginosa nitrite-reductase, Biochimie, 1984, 66(4), 313–318 CrossRef PubMed.
- A. B. McQuarters, J. W. Kampf, E. E. Alp, M. Hu, J. Zhao and N. Lehnert, Ferric Heme-Nitrosyl Complexes: Kinetically Robust or Unstable Intermediates?, Inorg. Chem., 2017, 56(17), 10513–10528 CrossRef CAS PubMed.
- P. C. Ford and I. M. Lorkovic, Mechanistic Aspects of the Reactions of Nitric Oxide with Transition-Metal Complexes, Chem. Rev., 2002, 102(4), 993–1018 CrossRef CAS PubMed.
- C. E. Cooper, Nitric oxide and iron proteins, Biochim. Biophys. Acta, Bioenerg., 1999, 1411(2), 290–309 CrossRef CAS PubMed.
- S. Shahid, M. Ali, D. Legaspi-Humiston, J. Wilcoxen and A. A. Pacheco, A Kinetic Investigation of the Early Steps in Cytochrome c Nitrite Reductase (ccNiR)-Catalyzed Reduction of Nitrite, Biochemistry, 2021, 60(26), 2098–2115 CrossRef CAS PubMed.
- H. Fujii, D. Yamaki, T. Ogura and M. Hada, The functional role of the structure of the dioxo-isobacteriochlorin in the catalytic site of cytochrome cd1 for the reduction of nitrite, Chem. Sci., 2016, 7(4), 2896–2906 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details and additional supporting data are provided in the ESI and are available free of charge on the website http://www.pubs.acs.org/. See DOI: https://doi.org/10.1039/d3sc01777j |
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Snehadri
Bhakta
,
Samir
Chattopadhyay
,
Snehadri
Bhakta
,
Samir
Chattopadhyay