
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEstablishing PQ-ERA photoclick reactions with unprecedented efficiency by engineering of the nature of the phenanthraquinone triplet state†
Youxin
Fu
 a,
Georgios
Alachouzos
a,
Nadja A.
Simeth‡
a,
Mariangela
Di Donato
bc,
Michiel F.
Hilbers
d,
Wybren Jan
Buma
*de,
Wiktor
Szymanski
*af and
Ben L.
Feringa
*a
a,
Georgios
Alachouzos
a,
Nadja A.
Simeth‡
a,
Mariangela
Di Donato
bc,
Michiel F.
Hilbers
d,
Wybren Jan
Buma
*de,
Wiktor
Szymanski
*af and
Ben L.
Feringa
*a
aCentre for Systems Chemistry, Stratingh Institute for Chemistry, Faculty for Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bLENS (European Laboratory for Non-Linear Spectroscopy), Via N. Carrara 1, 50019 Sesto Fiorentino (FI), Italy
cICCOM-CNR, Via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy
dVan 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: W.J.Buma@uva.nl
eInstitute for Molecules and Materials, FELIX Laboratory, Radboud University, Toernooiveld 7c, 6525 ED Nijmegen, The Netherlands
fDepartment of Radiology, Medical Imaging Center, University of Groningen, University Medical Centre Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands. E-mail: w.szymanski@umcg.nl
First published on 21st June 2023
Abstract
The light-induced photocycloaddition of 9,10-phenanthrenequinone (PQ) with electron-rich alkenes (ERA), known as the PQ-ERA reaction, is a highly attractive photoclick reaction characterized by high selectivity, external non-invasive control with light and biocompatibility. The conventionally used PQ compounds show limited reactivity, which hinders the overall efficiency of the PQ-ERA reaction. To address this issue, we present in this study a simple strategy to boost the reactivity of the PQ triplet state to further enhance the efficiency of the PQ-ERA reaction, enabled by thiophene substitution at the 3-position of the PQ scaffold. Our investigations show that this substitution pattern significantly increases the population of the reactive triplet state (3ππ*) during excitation of 3-thiophene PQs. This results in a superb photoreaction quantum yield (ΦP, up to 98%), high second order rate constants (k2, up to 1974 M−1 s−1), and notable oxygen tolerance for the PQ-ERA reaction system. These results have been supported by both experimental transient absorption data and theoretical calculations, providing further evidence for the effectiveness of this strategy, and offering fine prospects for fast and efficient photoclick transformations.
1. Introduction
The use of photochemical activation1 procedures in click chemistry reactions has resulted in the emergence of a novel category of light-induced click reaction referred to as “photoclick chemistry”.2,3 As opposed to conventional click reactions, in which the level of spatial and temporal control over the reaction is often limited,4,5 photochemical reactions provide a high degree of both spatial and temporal control over the reaction, allowing it to occur precisely when needed.1 The photo-induced cycloaddition of tetrazole and alkene, reported by Lin and co-workers6 marked the beginning of a decade of significant advancements and since then various types of photoclick reactions have been explored.7–13 The advantages associated with photoclick reactions, including their temporal and spatial control, non-invasive use of light, high efficiency, and compatibility with various biological systems, make these transformations particularly well-suited for a broad range of applications. These include, but are not limited to, surface modification,14–16 3D printing,17,18 protein labeling,13,19,20 and bioimaging.11,21,22Among these photoclick reactions, the PQ-ERA reaction, originally developed by Schönberg and co-workers in the 1940s,23,24 is gaining significant attention due to its excellent kinetics and biocompatibility.11–13 This reaction involves photoinduced electron transfer (PeT) between an electron-rich alkene and a PQ, resulting in the formation of a 1,6-biradical intermediate that rapidly undergoes intramolecular radical recombination to form [4 + 2] cycloadducts.13 Recently we identified new functionalization handles and showed that using enamines as ERAs significantly enhances the reaction rate (up to 674 M−1 s−1) and photoreaction quantum yields (ΦP, up to 65%).12 Despite these advances, further improvement is necessary, in particular regarding efficiency and rate, to expand the applicability of this reaction in diverse fields such as material science, bioimaging, and bioconjugation. However, engineering photoactive molecules to modulate excited state properties remains highly challenging.
The mechanism of the PQ-ERA reaction involves photoexcitation of PQ with UV to blue light, resulting in population of the strongly allowed S2 (1ππ*) state, which undergoes internal conversion to the lowest excited S1 (1nπ*) singlet state. An almost quantitative intersystem crossing (ISC) process then takes place, leading to population of the two lowest triplet states, T1 (3ππ*) and T2 (3nπ*) (the former being the reactive state for the PQ-ERA reaction), which are close in energy.25–27 This is then followed by a [4 + 2] photocycloaddition reaction with an ERA.28 Thus, a better understanding of the properties of the PQ's triplet state and generating more 3ππ* triplet states is crucial for further optimization of the PQ-ERA photoclick reaction. Recent studies have been conducted showing that triplet energy levels can be influenced by substituents on the aromatic ring and in some aromatic ketones they can even be inverted. This phenomenon is known as “electronic state switching”, and it modifies the electronic character of the lowest triplet state, thereby affecting its reactivity.29–31
Building on this phenomenon, we present herein a strategy for boosting the reactivity of the triplet state of PQvia a simple thiophene substitution at the 3-position of the PQ scaffold (Scheme 1) to achieve an unprecedented enhancement of the efficiency of the PQ-ERA reaction. Our investigation shows that the thiophene substitution significantly increases the population of the reactive triplet state (3ππ*) upon excitation of 3-thiophenyl PQs, which then results in a higher photoreaction quantum yield (up to a remarkable value of 98%), higher second order rate constants (up to 1974 M−1 s−1), and notable oxygen tolerance for the PQ-ERA reaction system. These findings have been supported by both transient absorption experiments and theoretical calculations.
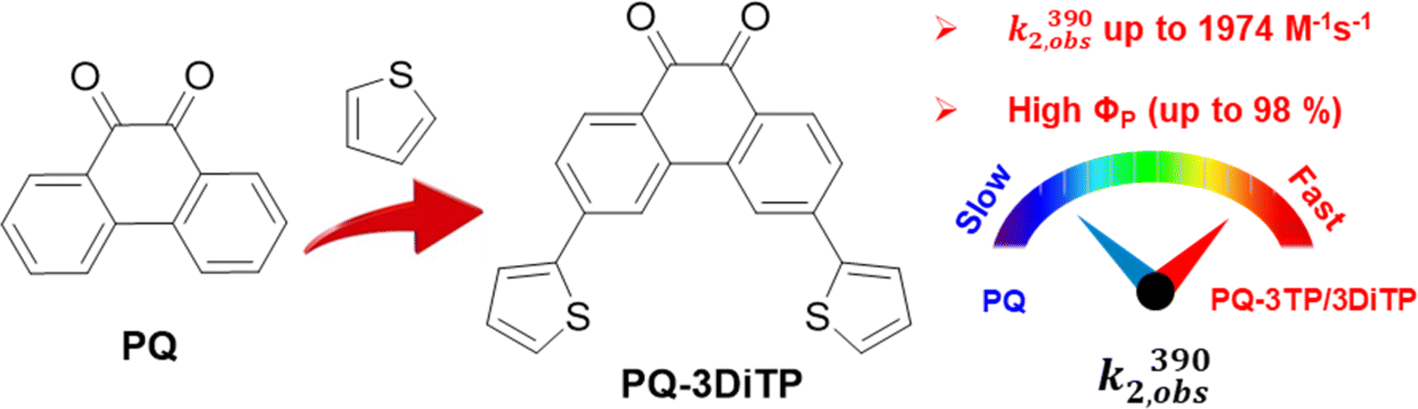 | ||
| Scheme 1 Comparison of photocycloaddition of 9,10-phenanthrenequinone (PQ) with electron-rich alkenes (ERAs). k2,obs(390) observed second-order reaction rate; and ΦP: photoreaction quantum yield. | ||
2. Results and discussion
2.1 Synthesis and photophysical evaluation of PQ derivatives
Based on our previous study,12 we assembled the two target compounds from commercially available 3-bromophenanthrene-9,10-dione (PQ-3Br), 3,6-dibromophenanthrene-9,10-dione (PQ-3DiBr), and thiophene boronic acid in a Suzuki–Miyaura cross-coupling reaction.32 Employing palladium-mediated cross-coupling and introducing the substituents via an arene extension, rather than by direct modifications on the PQ core, is synthetically straightforward and tolerates a broad set of substrates using the same protocol. We thus obtained PQ-3TP and PQ-3DiTP in satisfying yields (55% and 39%, respectively, Scheme S1, ESI†) employing Pd(PPh3)4 as catalyst.Next, both target molecules were characterized in terms of their electronic properties. Analysis of the UV-Vis absorption spectra of both the 3-thiophene and the 3,3′-dithiophene extended PQs revealed that the energetically lowest transition maximum, λmax, lies around 390 nm (Fig. S13†) and is hypsochromically shifted compared to unsubstituted PQ (λmax = 410 nm). This band is typically used to excite PQ derivates to trigger a PQ-ERA photoclick reaction.12
2.2 Photoclick reaction kinetics of PQs with PY
To test the performance of the newly synthesized PQ derivatives in a [4 + 2] photocycloaddition reaction, we irradiated a solution of each PQ (50 μM, MeCN, N2) with a 390 nm LED in the presence of a suitable reaction partner (Fig. 1A and B). We selected N-boc-2,3-dihydro-1H-pyrrole (PY, 10 eq., Fig. 1) as the ERA, as this cyclic enamine had shown the highest reactivity in our previous study.12 The progress of the reaction was monitored with UV-Vis spectroscopy providing the experimental rate constants kobs(390) of the photoclick process (0.25 s−1 and 0.295 s−1 for PQ-3TP and PQ-3DiTP, respectively, for detailed information see ESI, Section 4.2, Fig. S15 and S16†), which was significantly (>40 times) higher compared to the unsubstituted PQ_PY system (0.0062 s−1, Fig. S14†) under the same conditions. Next, we explored whether the system could also be triggered using light with a longer wavelength, since the absorption spectrum of thiophene-substituted PQs features a broad band ranging from 350 nm to 500 nm. We irradiated the PQs_PY mixtures with light of different wavelengths (365, 390, 420, and 445 nm) and, as expected, observed in all cases the formation of the photoclick products (Fig. S14–S16†). Important to notice is that the 3-position thiophene substituted PQs led to higher rate constants compared with the unsubstituted PQ under the same conditions (for detailed information see ESI, Section 4.2, Fig. S14–S16†). Finally, recognizing that thiophenes are known to be susceptible to side reactions, such as oxidation to radical cations in oxygenated solvents under prolonged light exposure, we assessed the PQ-ERA reaction substrate and product stability using the PQ-3TP_PY system as a representative model. We conducted the reaction in the presence of air with extended light irradiation lasting over 15 minutes. Gratifyingly, HPLC analysis revealed a clean reaction (Fig. S24†), strongly supporting the high selectivity of the reaction.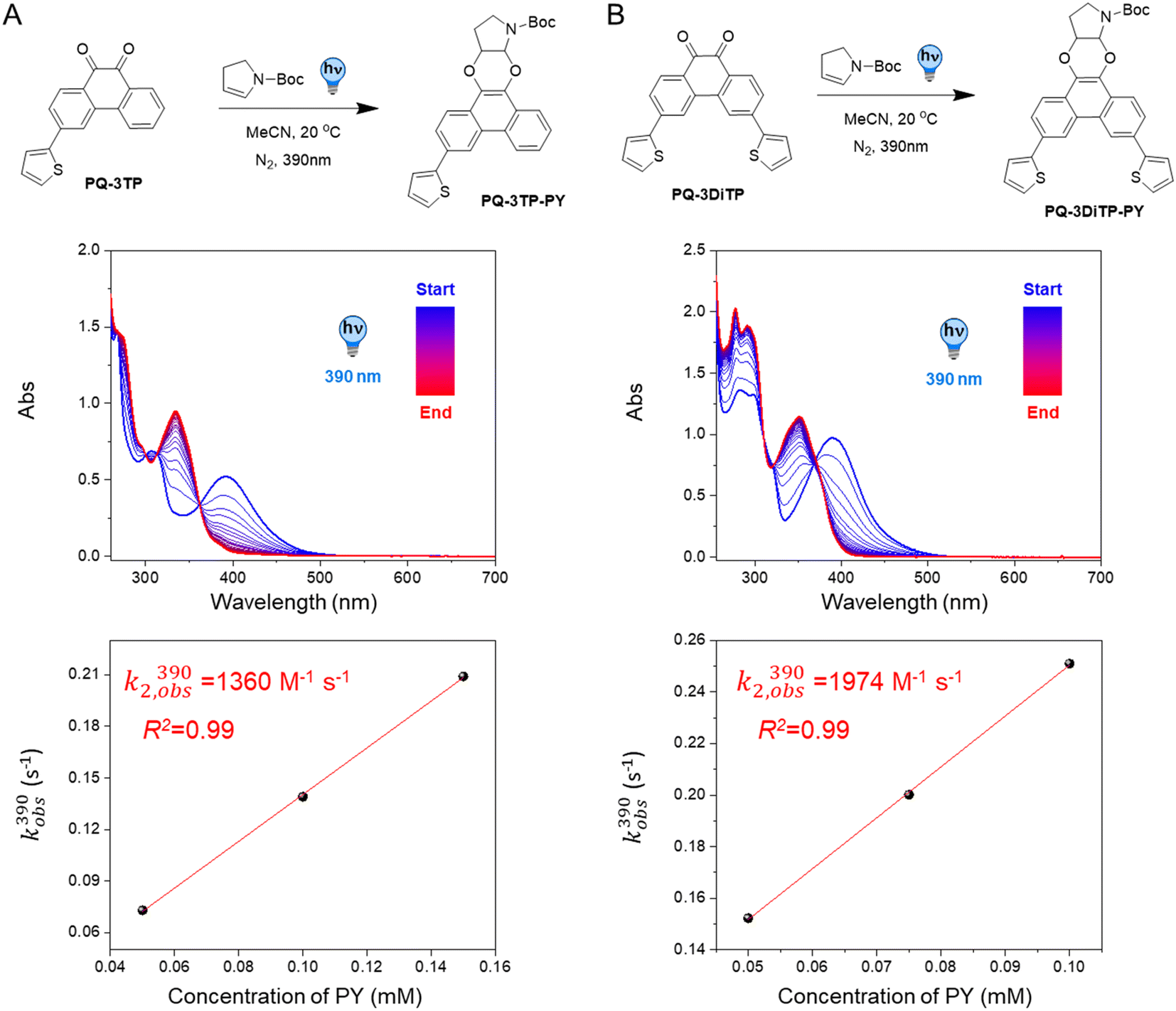 | ||
| Fig. 1 Kinetic analysis of PQ-ERA photocycloadditions. Reaction scheme, time-resolved UV-Vis absorption spectra, and plot of kobs(390) vs.PY concentration of (A) PQ-3TP (one of the possible isomers formed is shown as an example) and (B) PQ-3DiTP towards PY. For time-resolved UV-Vis absorption spectra, samples were irradiated by 390 nm LED under N2 atmosphere at 20 °C. The reaction was followed by UV-Vis absorption spectroscopy (sample interval 1 s, concentration: 50 μM PQs, 500 μM PY). The second-order rate constant k2,obs(390) were determined to be 1360 M−1 s−1 and 1974 M−1 s−1 for the PQ-3TP_PY and PQ-3DiTP_PY system, respectively, based on the slope of the fitted line. | ||
Next, we determined the second-order reaction rate k2,obs(390) of these photocycloaddition reactions (for details and further considerations see ESI Section 4.2, Fig. S17–S19†). The k2,obs(390) was found to be 1360 M−1 s−1 and 1974 M−1 s−1 for PQ-3TP_PY and PQ-3DiTP_PY, respectively (Fig. 1), which is more than 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 times higher than for the original PQ-VE1 (ethylene glycol vinyl ether) system (0.117 M−1 s−1)12 and over 160 times higher than the PQ–PY system (8.2 M−1 s−1, Fig. 2B and S17, for detailed information see ESI, Section 4.2†) under the same reaction conditions. In fact, this allows us to perform quantitative photoclick reactions in a matter of seconds. As such, it represents the fastest currently known PQ-ERA photoclick reaction.11–13
600 times higher than for the original PQ-VE1 (ethylene glycol vinyl ether) system (0.117 M−1 s−1)12 and over 160 times higher than the PQ–PY system (8.2 M−1 s−1, Fig. 2B and S17, for detailed information see ESI, Section 4.2†) under the same reaction conditions. In fact, this allows us to perform quantitative photoclick reactions in a matter of seconds. As such, it represents the fastest currently known PQ-ERA photoclick reaction.11–13
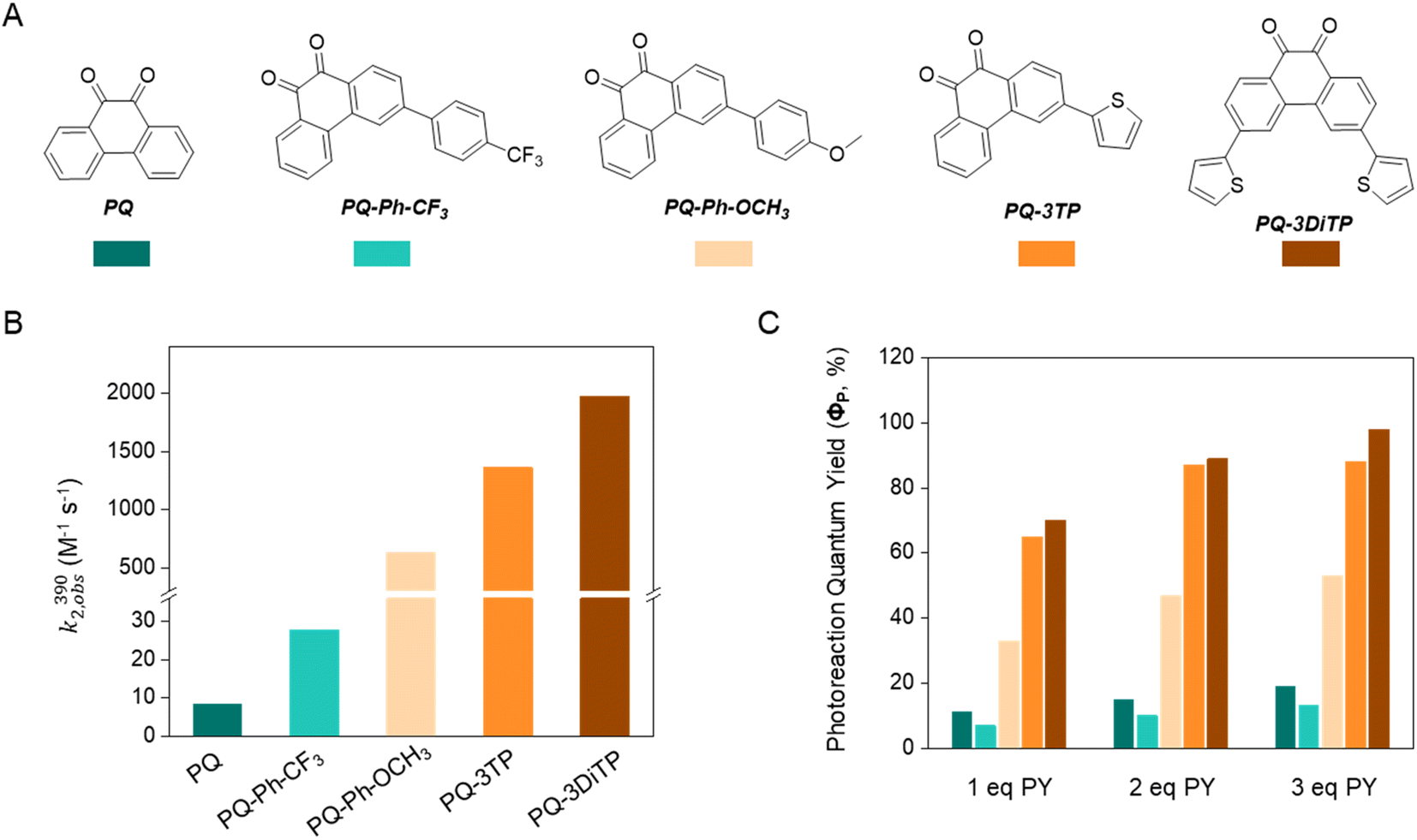 | ||
| Fig. 2 Overview of the second order reaction rate (k2) and photoreaction quantum yield (ΦP) of the PQ-ERA photoclick reaction system. (A) Structures of the investigated PQs, (B) k2,obs(390) and (C) ΦP of PQ-ERA photoclick reaction. | ||
Furthermore, in order to evaluate the generality of our 3-position thiophene substitution approach, we selected two additional ERAs, 4-oxo-3,4-dihydropyridine-1(2H)-carboxylate (PYD) and 3,4-dihydro-2H-pyran (DP), to react with both PQ and PQ-3TP. Analysis of the kinetic traces acquired through UV-Vis absorption spectroscopy (for details and further considerations see ESI Section 4.2, Fig. S20 and S21†) revealed that the photoclick reaction was significantly more efficient in the PQ-3TP system when compared to the PQ system. Specifically, under 390 nm light irradiation, the PQ-3TP system displayed kobs(390) values of 0.0059 s−1 and 0.092 s−1 for DP and PYD, respectively, whereas the PQ system exhibited kobs(390) values of 0.0003 s−1 and 0.0028 s−1 for DP and PYD, respectively. Finally, to evaluate if heteroaromatic compounds with a lower aromatic stabilization energy are able to work as ERAs in this reaction, we conducted reaction between PQ-3TP and an aromatic compound, specifically 1-methylindole, under the same conditions. As depicted in Fig. S22 (ESI, Section 4.2),† minimal changes were observed over a 15 minute period, indicating the absence of photoclick reactions between PQ-3TP and 1-methylindole upon 390 nm light irradiation, showing a general limitation of the electronic properties of ERAs in this reaction system. These findings serve as further evidence that the 3-position thiophene substitution strategy can enhance the photoclick reactivity of PQ towards a variety of ERAs.
These results are mirrored by the extraordinary high photoreaction quantum yield (ΦP) of these PQ-ERA reactions, which has been determined for reactions of PQ, PQ-Ph-CF3, PQ-Ph-OCH3, PQ-3TP, and PQ-3DiTP with different concentrations of PY (Fig. 2A). PQ-3TP and PQ-3DiTP exhibited an extremely high value of ΦP, reaching the up to 98% in the presence of 3 eq. of PY (Fig. 2C, for detailed information, see ESI, Section 4.3†). To the best of our knowledge, this is the highest ΦP observed for any photoclick reaction reported so far.3,12,33–36 Thus, the performance of the 3-position thiophene substituted PQ-3TP/PQ-3DiTP_PY system is markedly enhanced compared to our previously introduced PQs-VEs/PY systems (ΦP(PQs-VEs/PY) ranging from 0.6%12 to 53%, for details and further considerations see ESI Section 4.3, Fig. S25–S39†) as well as other reported photoclick reactions such as, for instance, o-naphthoquinone methides (oNQMs)-ene hetero-Diels–Alder cycloaddition (ΦP = 20%),36 photoinduced tetrazole–alkene cycloaddition (ΦP = 0.29% to 24%),22,35 and photoinduced sydnone–alkene/alkyne cycloadditions (ΦP = 10% to 25%).33,34
2.3 Effect of O2 and temperature on the PQ-ERA photoclick reaction
The photochemical characterization of the thiophene-PQ-ERA reaction reported so far shows that both PQ-3TP and PQ-3DiTP react significantly faster with PY compared to unsubstituted PQ and other PQ derivatives (Fig. 2B). Thus, we chose PQ-3TP as an ideal candidate to conduct a systematic and detailed study on the PQ-ERA photoclick reaction process (Fig. 3A). Previous reports showed that irradiation of PQs with 390 nm light in the presence of ERAs proceeds via the triplet state of PQs and results in the photocycloaddition of the two reactants, furnishing the PQ-ERA products.13,27,28 We examined the effect of molecular oxygen (O2), an important triplet state quencher,37,38 on the PQ-ERA reaction process.11 To probe the effect that O2 had on the PQ-ERA photocycloaddition, we sought to determine the kobs(390) of the PQ-3TP_PY photoclick reaction under both deoxygenated and non-deoxygenated conditions. As expected, in the absence of oxygen we found a higher kobs(390) compared to the non-degassed solutions (0.084 → 0.229 s−1 and 0.014 → 0.092 s−1 respectively, Fig. 3B), demonstrating the detrimental effect of O2 on this PQ-ERA reaction and corroborating the hypothesis of the involvement of triplet states. Nonetheless, our results indicate that, contrary to previous PQ-ERA photoclick systems,12 the PQ-3TP_PY photoclick reaction still proceeds also under aerobic conditions, marking an overall improvement in oxygen tolerance.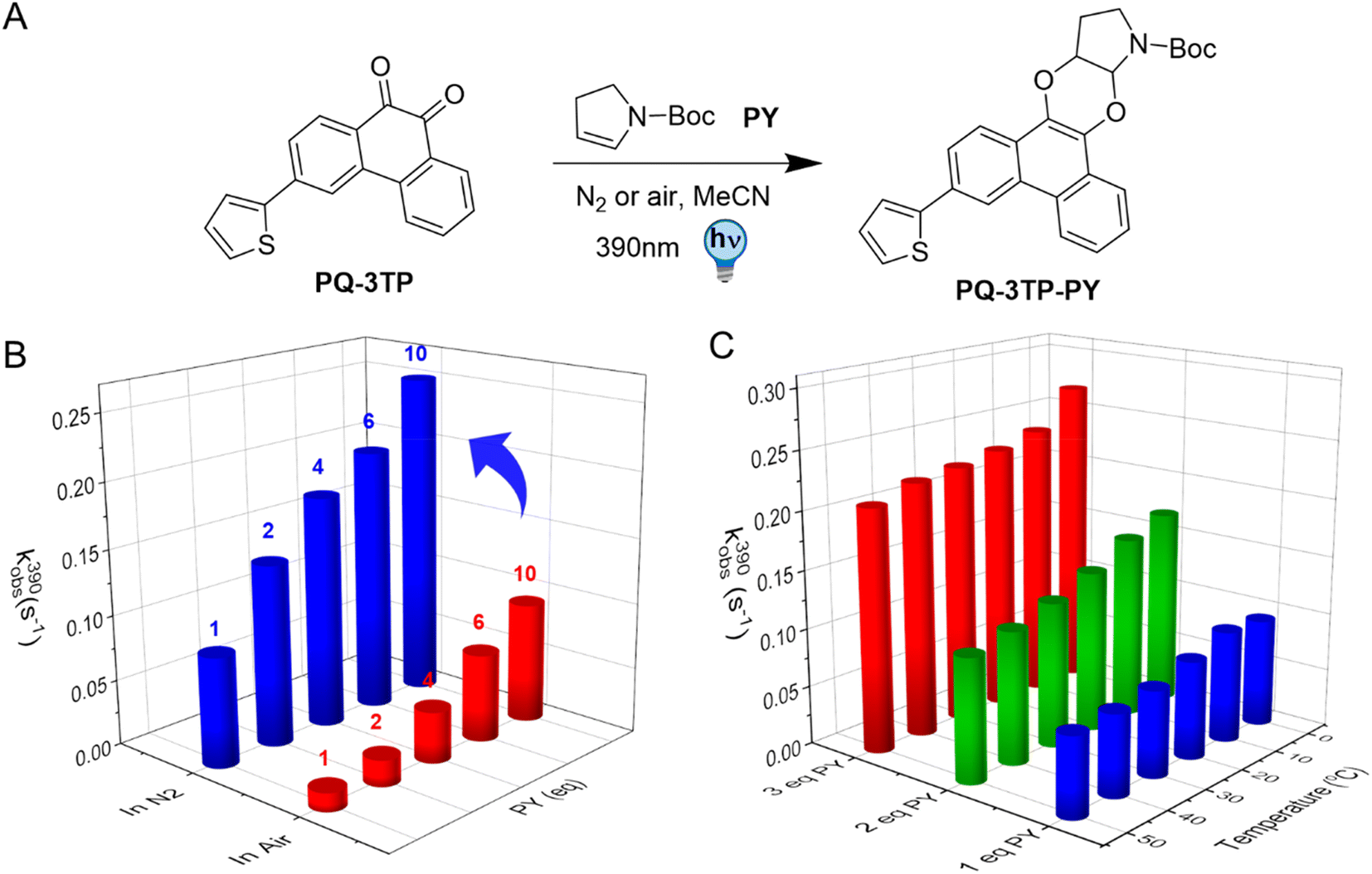 | ||
| Fig. 3 Kinetic analysis of PQ-3TP_PY photoclick reaction. (A) Reaction scheme (one of the possible isomers formed is shown as an example), (B) kinetic analysis of photocycloaddition between PQ-3TP (50 μM) with varying amounts of PY (1, 2, 4, 6, and 10 eq.) addition under both N2 (blue cylinders) and air (red cylinders) atmosphere at 20 °C. (C) Temperature dependence kinetic analysis of PQ-3TP_PY photoclick reaction in MeCN under different temperature 0 °C, 10 °C, 20 °C, 30 °C, 40 °C, and 50 °C respectively. For all of the measurements, the sample was irradiated with a 390 nm LED and followed over time by UV-Vis absorption spectroscopy (1 cm cuvette, 2.5 mL MeCN sample volume, sample interval 1 s). | ||
Also, previous studies have demonstrated that lowering the temperature may result in a prolonged triplet lifetime.39–41 Based on this, we hypothesized that changes in temperature would also have an effect on the PQ-3TP_PY photoclick reaction. To investigate this effect, the photoclick reactions were conducted over a temperature range between 0 °C to 50 °C, monitoring the reactions using UV-Vis spectroscopy to measure the experimental rate constants kobs(390) of the photoclick process. As shown in Fig. 3C, a clear enhancement of the photoclick reactivity was observed at lower temperatures. On the basis of quantum chemical calculations that will be discussed below, we attribute this enhancement to a reduced non-radiative decay rate of the reactive triplet state at lower temperatures leading to a longer lifetime.39,42
The results shown above indicate that the 3-position thiophene substitution on the PQ scaffold can bring about a marked improvement in its photoclick reactivity and that oxygen can be tolerated in the systems. Aerobic conditions result in slower photoclick reaction rates, but still allow the completion of the model reaction in less than two minutes. To gain a further fundamental understanding of these new phenomena, we next performed a detailed study on the electronically excited singlet and triplet states of PQs and their excited-state dynamics using femtosecond/nanosecond transient absorption spectroscopy and quantum chemical calculations.
2.4 Femtosecond/nanosecond transient absorption spectroscopy
We initially hypothesized that the improved reactivity of the 3-thiophene substituted PQs could in principle be attributed either to a higher triplet quantum yield or to a longer-lived triplet state. To further investigate the excited state dynamics of the newly synthetized PQs, the transient absorption spectra of these systems were measured both in the sub-picosecond to nanosecond and nanosecond to millisecond timescale. Previous studies demonstrated that unsubstituted PQ undergoes a rapid ISC, producing triplet states with a high quantum yield.29,43 The triplet absorption spectrum reported for PQ presents two positive bands at about 460 and 650 nm that have been attributed to two different triplet states with 3nπ* and 3ππ* nature reaching a fast equilibration.29Our measurements confirmed the occurrence of very fast ISC for PQ: indeed, upon exciting the molecule with 400 nm light, the transient spectrum attributed to the triplet state rises in about 9 ps. The decay of the absorption spectrum, as determined by ns transient absorption spectroscopy, is bi-exponential, with a major component decaying in 1.47 μs and a residual component with a lifetime of 40.5 μs (Fig. 5A, for detailed information, see ESI, Section 4.4 and 4.5, Fig. S40, S41 and S44†).
The transient absorption measurements were then repeated for the 3-substituted samples PQ-3TP (Fig. S42A†) and PQ-3DiTP (Fig. S43A†). Analogous to our analyses of the time-resolved spectra obtained for PQ, the recorded transient data have been analyzed using a global fit procedure and employing a sequential linear decay kinetic scheme from which the EADS (Evolution Associated Difference Spectra) and the rate constants describing the excited state dynamics are obtained (for detailed information, see ESI, Section 4.4, Fig. S42 and S43†). The EADS obtained for PQ-3TP and PQ-3DiTP, which are shown in Fig. 4, are qualitatively similar.
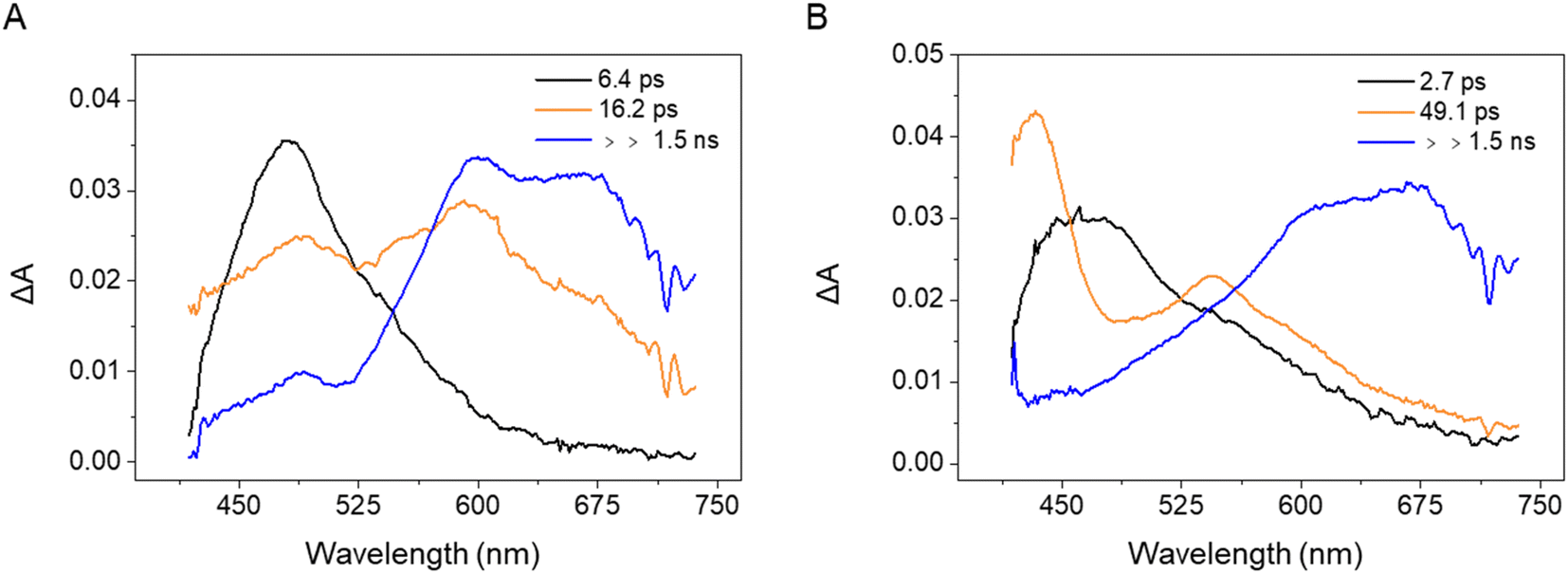 | ||
| Fig. 4 EADS obtained from a global analysis of the transient absorption data recorded for (A) PQ-3TP and (B) PQ-3DiTP in MeCN upon excitation at 400 nm. | ||
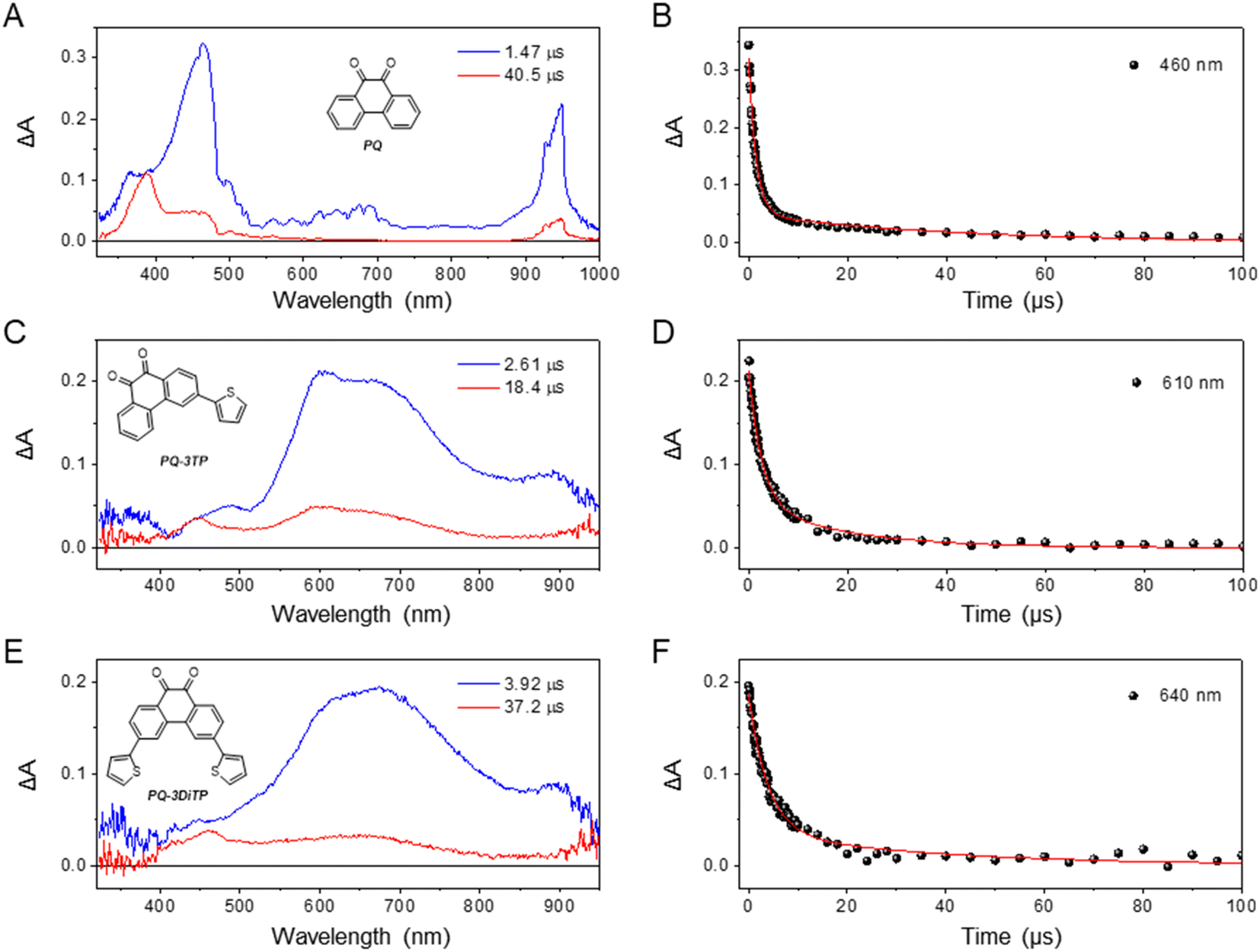 | ||
| Fig. 5 Evolution-associated difference spectrum (EADS) obtained from global analysis of ns transient absorption data recorded for (A) PQ, (C) PQ-3TP, and (E) PQ-3DiTP in degassed MeCN upon excitation at 400 nm. The fit of the decay of the absorption of the transient signal (red line) (B) PQ (at 460 nm), (D) PQ-3TP (at 610 nm), and (F) PQ-3DiTP (at 640 nm) as obtained from a global analysis of the transient absorption spectra. | ||
For both systems, the transient spectrum observed directly after excitation features a quite broad positive band peaked at about 470 nm, which, in analogy with observations made for PQ (Fig. S40†), can be assigned to excited state absorption (ESA) of the singlet state of the molecules (black EADS in Fig. 4A and B). The spectrum evolves within 6.4 ps in case of PQ-3TP to a double peaked structure (orange EADS, Fig. 4A). A further evolution is observed to occur in about 16 ps. On this timescale, the intensity of the band peaked at about 470 nm decays significantly, while a broad band extending from 580 to 700 nm rises in intensity. The lifetime of this spectral component is longer than the time range of the experiment, and, in analogy with what was observed for PQ, is assigned to a triplet state. For PQ-3DiTP, the initial EADS (Fig. 4B), assigned to a singlet excited state, evolves in about 2.7 ps towards the second component, which is characterized by an intense band at 430 nm and a second less intense band at 550 nm. This EADS then evolves in the final one on a time scale of about 49 ps. The shape of the long-living component for PQ-3DiTP is similar to the one observed for PQ-3TP and is therefore also assigned to a triplet state. The nature of the intermediate component is not clear: it could be attributed as a high-energy triplet state that rapidly decays into the lower-energy triplet, or to a ‘dark’ electronically excited singlet state, different from the state that initially is excited. The notable spectral difference between the first and second EADS makes it quite unlikely that the initial evolution is associated with a relaxation process within the state that is initially excited. The rapid evolution between the second and third component suggests that the more likely interpretation for the second EADS is that of a ‘dark’ singlet state from which ISC occurs. Such a conclusion is fully supported by the results of the quantum chemical calculations discussed below which show that – analogous to PQ – the ‘bright’ state that is excited is the S2 (1ππ*) state, while at lower excitation energies the ‘dark’ S1 (1nπ*) state is found (for detailed information, see the ESI, Section 5†). The ultrafast transient absorption spectra and relevant kinetic traces measured for PQ-3TP and PQ-3DiTP are shown in Fig. S42 and S43.† The analysis of these traces shows that the band peaked at about 630 nm, which is attributed to the triplet state, rises on a slightly faster timescale for PQ-3TP.
To confirm that the long-living components observed from the ultrafast measurements can be attributed to triplet states, we also measured the transient absorption spectra of both PQ-3TP and PQ-3DiTP using nanosecond (ns) laser excitation (for detailed information, see ESI, Section 4.5, Fig. S44–S46†). The EADS obtained from global analysis of the data recorded on the longer timescale are reported in Fig. 5, where for comparison also the EADS obtained from the global analysis of the PQ ns data are depicted. As can be observed, the spectrum obtained for both PQ-3TP and PQ-3DiTP immediately after excitation with a ns laser coincides with the long-living species observed in the ultrafast experiments, confirming its assignment in terms of a triplet state of both molecules. Similarly to what is observed for PQ (Fig. 5A and B), the triplet state decays bi-exponentially for both compounds with time constants of τ1 = 2.61 μs (blue component) and τ2 = 18.4 μs (red component) in case of PQ-3TP, and τ1 = 3.92 μs (blue component) and τ2 = 37.2 μs (red component) for PQ-3DiTP.
Some further comments should be made on these lifetimes. Firstly, only mono-exponential triplet decays have been reported for PQ so far. The observation of a bi-exponential decay for all three compounds should thus be rationalized. Secondly, for the ns transient absorption spectra reported for PQ-3TP and PQ-3DiTP in Fig. 5 and for PQ in Fig. S44,† excitation energies in the range of 0.2–0.4 mJ per pulse have been used, which are similar to those employed in previous transient absorption experiments on PQ.29 For PQ, we find indeed a fast decay component (τ1 = 1.5 μs) that reproduces the previously reported value.29 However, by repeating the measurements with different excitation energies, we noticed that both lifetimes increase upon lowering the excitation power, suggesting the occurrence of quenching at high irradiation energies. Furthermore, while the long-wavelength band (550–800 nm) is dominated by the fast decay component, the band around 450 nm predominantly decays on the longer time scale. In line with previous studies, we attribute this band to the formation of a ketyl radical, a side reaction product previously identified upon irradiation of PQs.25–27,44,45 We thus conclude that excitation of PQ, PQ-3TP and PQ-3DiTP leads to population of the lowest triplet state, which decays on a time scale of τ1, and the formation of a ketyl radical that disappears on a time scale of τ2. The observed quenching of the triplet is in line with previous studies of PQ in acetonitrile in which self-quenching was observed as well,46 while the disappearance of the ketyl radical is intrinsically bimolecular in nature. Finally, we point out that the triplet lifetimes of all compounds changed in a similar way with the excitation power, remaining on the same order of magnitude among each other. Further information about the power dependence of the triplet lifetimes can be found in the ESI, Section 4.6, Fig. S47–S49.†
Comparison of the blue EADS in Fig. 5 – attributed to the decay of the triplet state – of PQ with those of PQ-3TP and PQ-3DiTP shows significant differences. In the former case, the 400–500 nm band dominates the spectrum with a minor contribution from the broad band system spanning the 600–750 nm region, while for the latter the 400–500 nm band is nearly absent, and the spectrum is dominated by the 550–800 nm band system. This observation strongly suggests a population distribution involving two different triplet states in the three compounds with a different distribution for PQ compared to PQ-3TP and PQ-3DiTP. Such a conclusion nicely explains the differences in the experimentally observed photoclick quantum yield and rate constant (Fig. 2) and leads to the conclusion that the triplet state that is dominantly populated in PQ-3TP and PQ-3DiTP is much more reactive. In the following we will argue that this triplet state is the 3ππ* state.
2.5 Quantum chemical calculations on PQs
To further elucidate the observations made in the transient absorption studies, (Time-Dependent) Density Functional Theory (TD-DFT) calculations have been performed of vertical and adiabatic excitation energies of the lower-lying triplet states as well as of electronic absorption spectra predicted for these states.47–52 In these calculations, several levels of theory have been employed, giving rise to very similar results. Since the calculations at the M06-2X/6-31+G*/PCM(acetonitrile) level predict for PQ a 3nπ*–3ππ* energy gap closest to the value reported experimentally, we will in the following discuss the results obtained at that level. Further details are given in the ESI Section 5.†Table 1 presents the vertical and adiabatic excitation energies of the lowest 3nπ* and 3ππ* states of PQ, PQ-3TP and PQ-3DiTP. As can be seen, for vertical excitation the 3nπ* state is T1, but adiabatically the 3ππ* state becomes the lowest excited triplet state. Interestingly – and in agreement with the charge distribution and bonding characteristics of the n orbital and of the HOMO and LUMO orbitals – it is observed (i) that the energy of the 3nπ* state is hardly changed upon geometry relaxation, while the reorganization energy of the 3ππ* state is much larger, and (ii) that substitution on the 3 position does not change the vertical and adiabatic excitation energies of the 3nπ* state, while the 3ππ* state is lowered in energy.
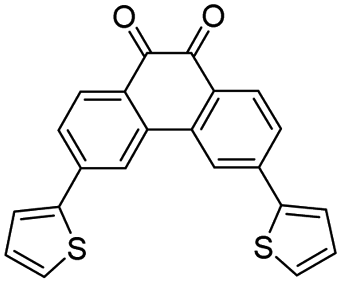
The electronic absorption spectra for the 3nπ* and 3ππ* states of PQ, PQ-3TP, and PQ-3DiTP are shown in Fig. 6. These spectra are quite informative, as they predict for PQ that the 3nπ* state only shows an appreciable absorption around 400 nm, while the 3ππ* state shows a slightly red-shifted band in the 400 nm region but also strong absorption bands in the 550–750 nm region. Similar observations are made for the predicted absorption spectra of the 3nπ* and 3ππ* states of PQ-3TP and PQ-3DiTP which only show appreciable absorption above 550 nm for the 3ππ* state. These observations strongly suggest that the band observed in this region in the experimental transient absorption spectra of these compounds (Fig. 5) is associated with the 3ππ* state and not with the 3nπ* state as assumed previously.29 In the ESI Section 5,† we expand further on this difference in conclusions. Based on the current calculations, it is predicted that the short-wavelength band detected in the ns transient absorption experiments can be attributed to absorption from both the 3nπ* and 3ππ* states, whereas the long-wavelength band can only be linked to absorption from the 3ππ* state. In combination with the calculated adiabatic excitation energies of the two states showing that the 3ππ* state is lowered in energy upon substitution, we therefore conclude that the photoclick reaction occurs in this state. Such a conclusion finds strong support from studies we have performed on 2,2′-substituted PQs substituted for which the 3ππ* state is both vertically as well as adiabatically the lowest excited triplet state with the 3nπ* state at much higher energies (∼15 kcal mol−1).54
 | ||
| Fig. 6 Calculated absorption spectra for the 3nπ* (up) to 3ππ* (down) states of (A) PQ, (B) PQ-3TP and, (C) PQ-3DiTP. Stick spectra have been convoluted with a Gaussian profile with a width of 0.1 eV for further comparison with the experiment. | ||
Summarizing all the experimental spectroscopic observations and the theoretical calculations, we conclude that in PQ and 3-thiophene-extended PQs the following sequence of events occurs. Excitation populates a strongly allowed 1ππ* state, which undergoes internal conversion on a ps time scale to the lower-lying 1nπ* state. Following El-Sayed's rules,55 this state has a strong spin–orbit coupling with the 3ππ* state. As a result, a fast and efficient intersystem crossing at a tens of ps timescale occurs that populates the 3ππ* state, that is the reactive state from which the photoclick reaction occurs. The difference in photoclick quantum yield and rate constant between PQ and 3-thiophene extended PQs results from a dominant participation of the 3nπ* state in the former.
3. Conclusion
In summary, we report how a simple substitution of the 3- and 3′-position of PQs with either one or two thiophene moieties results in an exceptionally high photoclick quantum yield (up to 98%), higher photoclick rate constants (up to 1974 M−1 s−1) and marked oxygen tolerance. We have also provided transient absorption data, strongly supporting the suggestion that the appropriate reactive triplet state that engages in the photocycloaddition with ERAs is preferentially populated during the excitation of 3-thiophenyl PQs. We envision that this novel ultrafast PQ-ERA photoclick reaction with 3-thiophenyl PQs is not limited to the chemistries shown here but will be advantageous in a wide range of applications, such as surface photo-patterning, labeling of biomacromolecules, and photochemical crosslinking. Studies along these lines are now ongoing in our laboratories.Data availability
All experimental and computational data associated with this article have been included in the main text and ESI.†Author contributions
Y. F., G. A., N. A. S., M. D. D., W. S., W. J. B., and B. L. F. conceived the project and designed phenanthrenequinone derivatives. B. L. F. and W. S. guided the research. Y. F. synthesized all phenanthrenequinone derivatives. G. A. and W. J. B. performed the TD-DFT calculations. Y. F. performed UV-Vis experiments. M. D. D. performed the femtosecond transient absorption spectroscopy measurements. Y. F., M. F. H., and W. J. B. performed the nanosecond transient absorption spectroscopy measurements. Y. F., G. A., N. A. S., M. D. D., W. S., W. J. B., and B. L. F. wrote the manuscript with support and contributions from all authors.Conflicts of interest
There are no competing conflicts of interests to declare.Acknowledgements
We thank Renze Sneep (University of Groningen) for his help with the HRMS measurements. We gratefully acknowledge the generous financial support from the Horizon 2020 Framework Program (ERC Advanced Investigator Grant No. 694345 to BLF), the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program No. 024.001.035 to BLF), and the European Molecular Biology Organization (EMBO LTF-232-2020 Postdoctoral Fellowship to GA). This research was supported by NWO domain TTW and Stryker European Operations Ltd. MDD gratefully acknowledges support from the European Union's Horizon 2020 Research and Innovation program under grant agreement no. 871124 Laserlab-Europe.References
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, 2006 Search PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390–7394 CrossRef CAS.
- C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18605–18611 CrossRef CAS.
- Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041–10047 CrossRef CAS PubMed.
- Y. Fu, N. A. Simeth, R. Toyoda, R. Brilmayer, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202218203 CrossRef CAS PubMed.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542–14546 CrossRef CAS PubMed.
- S. Arumugam, S. V. Orski, J. Locklin and V. V. Popik, J. Am. Chem. Soc., 2012, 134, 179–182 CrossRef CAS.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2012, 134, 8408–8411 CrossRef CAS PubMed.
- C. Xie, W. Sun, H. Lu, A. Kretzschmann, J. Liu, M. Wagner, H.-J. Butt, X. Deng and S. Wu, Nat. Commun., 2018, 9, 3842 CrossRef PubMed.
- R. Rizzo, D. Ruetsche, H. Liu and M. Zenobi-Wong, Adv. Mater., 2021, 33, 2102900 CrossRef CAS PubMed.
- S. J. Bailey, E. Hopkins, K. D. Rael, A. Hashmi, J. M. Urueña, M. Z. Wilson and J. Read de Alaniz, Angew. Chem., Int. Ed., 2023, e202301157 CAS.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860–4868 CrossRef CAS PubMed.
- C. Wang, H. Zhang, T. Zhang, X. Zou, H. Wang, J. E. Rosenberger, R. Vannam, W. S. Trout, J. B. Grimm, L. D. Lavis, C. Thorpe, X. Jia, Z. Li and J. M. Fox, J. Am. Chem. Soc., 2021, 143, 10793–10803 CrossRef CAS PubMed.
- G. S. Kumar, S. Racioppi, E. Zurek and Q. Lin, J. Am. Chem. Soc., 2022, 144, 57–62 CrossRef CAS PubMed.
- A. Schönberg and A. Mustafa, Nature, 1944, 153, 195 CrossRef.
- A. Schönberg and A. Mustafa, J. Chem. Soc., 1944, 387 RSC.
- Y. L. Chow and T. C. Joseph, Chem. Commun., 1968, 604 RSC.
- P. A. Carapellucci, H. P. Wolf and K. Weiss, J. Am. Chem. Soc., 1969, 91, 4635–4639 CrossRef CAS.
- M. B. Rubin and Z. Neuwirth-Weiss, J. Am. Chem. Soc., 1972, 94, 6048–6053 CrossRef CAS.
- J. Li, H. Kong, C. Zhu and Y. Zhang, Chem. Sci., 2020, 11, 3390–3396 RSC.
- V. R. Kumar, N. Rajkumar, F. Ariese and S. Umapathy, J. Phys. Chem. A, 2015, 119, 10147–10157 CrossRef CAS PubMed.
- K. F. Chang, M. Reduzzi, H. Wang, S. M. Poullain, Y. Kobayashi, L. Barreau, D. Prendergast, D. M. Neumark and S. R. Leone, Nat. Commun., 2020, 11, 4042 CrossRef CAS PubMed.
- H. Wu, Y. Zhou, L. Yin, C. Hang, X. Li, H. Ågren, T. Yi, Q. Zhang and L. Zhu, J. Am. Chem. Soc., 2017, 139, 785–791 CrossRef CAS PubMed.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- J. Gao, Q. Xiong, X. Wu, J. Deng, X. Zhang, X. Zhao, P. Deng and Z. Yu, Commun. Chem., 2020, 3, 29 CrossRef CAS PubMed.
- X. Zhang, X. Wu, S. Jiang, J. Gao, Z. Yao, J. Deng, L. Zhang and Z. Yu, Chem. Commun., 2019, 55, 7187–7190 RSC.
- S. Jiang, X. Wu, H. Liu, J. Deng, X. Zhang, Z. Yao, Y. Zheng, B. Li and Z. Yu, ChemPhotoChem, 2020, 4, 327–331 CrossRef CAS.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 5573–5579 CrossRef CAS PubMed.
- K. Kawaoka, A. U. Khan and D. R. Kearns, J. Chem. Phys., 1967, 46, 1842–1853 CrossRef CAS.
- O. L. J. Gijzeman, F. Kaufman and G. Porter, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 708 RSC.
- D. S. McClure, J. Chem. Phys., 1949, 17, 905–913 CrossRef CAS.
- S. H. Lin and R. Bersohn, J. Chem. Phys., 1968, 48, 2732–2736 CrossRef CAS.
- R. E. Kellogg and R. P. Schwenker, J. Chem. Phys., 1964, 41, 2860–2863 CrossRef CAS.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS.
- J. Guo, J. Dai, X. Peng, Q. Wang, S. Wang, X. Lou, F. Xia, Z. Zhao and B. Z. Tang, ACS Nano, 2021, 15, 20042–20055 CrossRef CAS PubMed.
- G. S. Hammond and P. A. Leermakers, J. Am. Chem. Soc., 1962, 84, 207–211 CrossRef CAS.
- A. Kuboyama, F. Kobayashi and S. Morokuma, Bull. Chem. Soc. Jpn., 1975, 48, 2145–2148 CrossRef CAS.
- D. E. Nicodem, R. S. Silva, D. M. Togashi and M. F. V. da Cunha, J. Photochem. Photobiol., A, 2005, 175, 154–158 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- R. S. Silva and D. E. Nicodem, J. Photochem. Photobiol., A, 2004, 162, 231–238 CrossRef CAS.
- Y. Fu, G. Alachouzos, N. A. Simeth, M. Di Donato, M. F. Hilbers, W. J. Buma, W. Szymanski and B. L. Feringa, submitted.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds, photophysical and chemical studies, details regarding the computational calculation. See DOI: https://doi.org/10.1039/d3sc01760e |
| ‡ Current address: Institute for Organic and Biomolecular Chemistry, Georg-August-University Göttingen, Tammanstraße 2, 37077 Göttingen, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |