
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic covalent self-assembly and self-sorting processes in the formation of imine-based macrocycles and macrobicyclic cages†
Zhaozheng
Yang
 ab,
Ferran
Esteve
b,
Cyril
Antheaume
b and
Jean-Marie
Lehn
*ab
ab,
Ferran
Esteve
b,
Cyril
Antheaume
b and
Jean-Marie
Lehn
*ab
aLehn Institute of Functional Materials (LIFM), Sun Yat-Sen University, 510006 Guangzhou, China
bLaboratoire de Chimie Supramoléculaire, Institut de Science et d'Ingénierie Supramoléculaires (ISIS), Université de Strasbourg, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: lehn@unistra.fr
First published on 2nd June 2023
Abstract
Investigating the self-assembly and self-sorting behaviour of dynamic covalent organic architectures makes possible the parallel generation of multiple discrete products in a single one pot procedure. We here report the self-assembly of covalent organic macrocycles and macrobicyclic cages from dialdehyde and polyamine components via multiple [2 + 2] and [3 + 2] polyimine condensations. Furthermore, component self-sorting processes have been monitored within the dynamic covalent libraries formed by these macrocycles and macrobicyclic cages. The progressive assembly of the final structures involves intermediates which undergo component selection and self-correction to generate the final thermodynamic constituents. The homo-self-sorting observed seems to involve entropic factors, as the homoleptic species present a higher symmetry than the competing heteroleptic ones. This study not only emphasizes the importance of an adequate design of the components of complex self-sorting systems, but also verifies the conjecture that systems of higher complexity may generate simpler outputs through the operation of competitive self-sorting.
Introduction
The active development of dynamic covalent chemistry (DCC) offers a powerful tool to chemists for generating well-defined discrete organic architectures from a mixture of components connected by reversible covalent bonds.1 In particular, it has provided novel perspectives relating to previous studies of dynamic macrocyclic and macrobicyclic architectures assembled through the formation of multiple reversible imine bonds between amines and carbonyl components, such as tetraimino-macrocycles and the hexaimino macrobicyclic cryptand-type molecular cages.2 As is the case for their non-dynamic macrobicyclic analogues, the intramolecular cavity of such well-defined structures may lead to applications such as sensing3 and catalysis.4 However, from the perspective of their synthetic methodology, the dynamically assembled structures offer new features such as self-sorting and adaptation.5 Self-sorting systems based on the dynamic covalent libraries (DCovLs) generated by DCC operate by incorporation, decorporation and exchange of components following the agonistic and antagonistic relationships between the constituents they build up.6The selective formation of specific architectures along the array of many possible reactions occurring within the DCovL via self-sorting of the components is of especially interest. The generation of such well-defined architectures by component self-sorting from multi-component DCovLs involves competition between the formation of: (i) discrete homoleptic self-sorted constituents (homo-self-sorting);6 (ii) mixed component heteroleptic self-sorted constituents (hetero-self-sorting);7 (iii) mixed constituents containing different components (scrambling).8 The challenge is to control the system in such a way, that the increase in molecular complexity (increase in the number of components and of reacting groups on a single component) will not result in totally random connections from scrambled condensations or polymer formation due to the similarity of bond energies. To meet this challenge, it is crucial to decipher the self-sorting mechanism and to take advantage of it. Thus, tracking the formation of products and understanding the conversion of intermediates are of particular interest and will be helpful for the prediction of self-sorting outcomes.
Recently, we reported the concurrent formation of two representative architectures of different cyclic order, tetraimino macrocycles and hexaimino macrobicyclic cages, from DCovLs of a diamine, a triamine and dialdehydes.6e These studies revealed the occurrence of kinetic switching via component exchange in the course of the formation of macrocycles and macrobicyclic cages by self-sorting. The present work focuses on the multiple imine condensation processes and multi-component covalent self-sorting mechanisms taking place in such macrocycles and macrobicyclic cages. It examines correlations between the selection of specific components/building blocks of different shape and size and the structural features of the resulting species. A significant feature of these self-sorting processes resides in the self-correction behaviour that occurs based on reversible imine condensations. Finally, a [4 × 2] six-component DCovL of higher order complexity was set up by an appropriate choice of DCovL precursors, four different dialdehydes and two amines (a triamine and a diamine). In this case, only specific imine architectures, one macrobicyclic cage and three agonistic macrocycles, were produced via the self-sorting processes.
Results and discussion
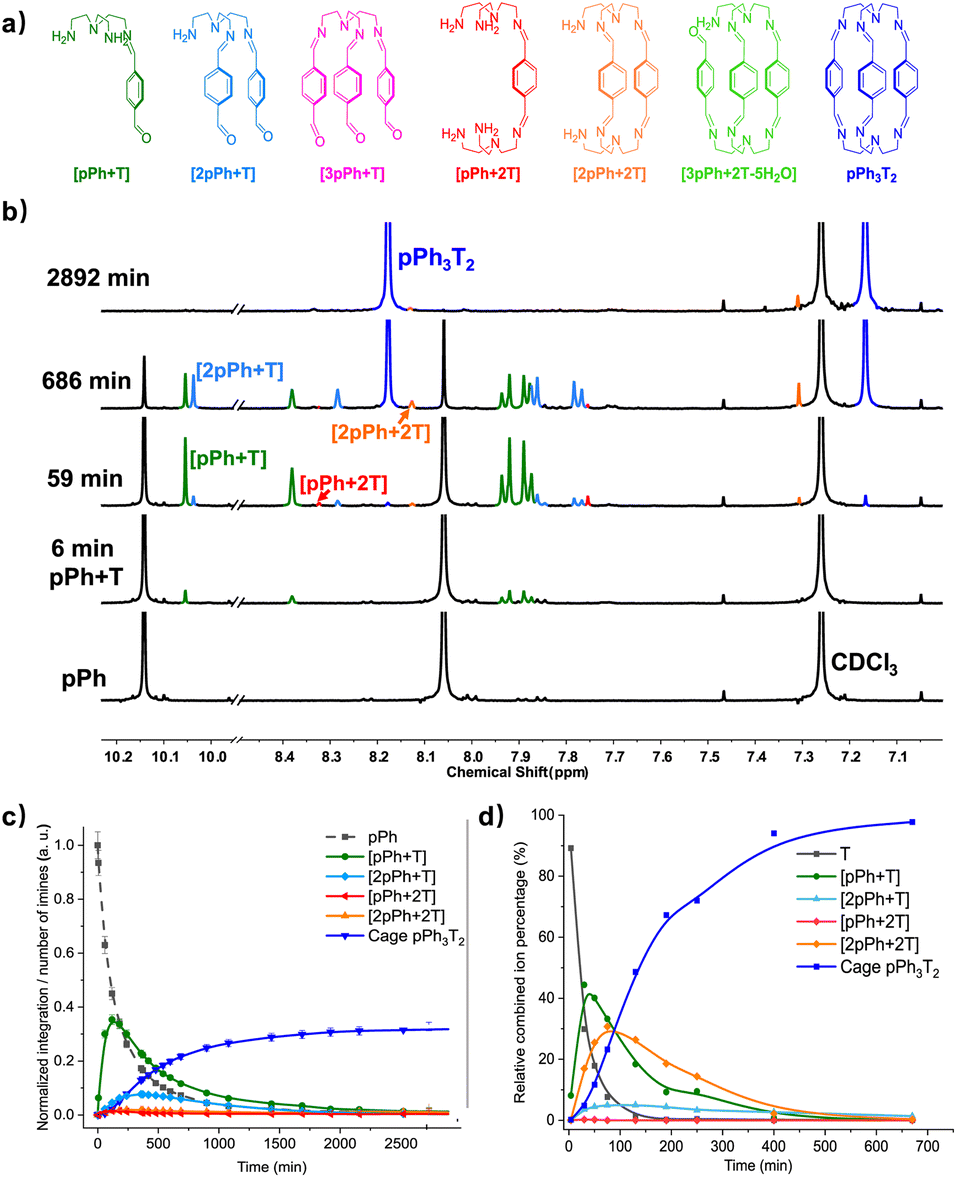
Previous work has described the synthesis and characterization of macrocyclic and macrobicyclic cryptand-type structures, as well as their self-sorting behaviour.6e,9 These studies have now been extended to the investigation of the formation mechanisms and component selection features of homoleptic macrocycles and macrobicyclic cages in imine-based dynamic covalent systems. To this end, the formation of the D2h-symmetric tetraimino macrocycles and D3h-symmetric hexaimino macrobicycles shown in Scheme 1 from their dialdehyde and diamine or triamine components were monitored by 1H NMR (in CDCl3 at 23–25 °C) and by HRMS (in 50/50 (v/v) CHCl3/MeOH at 25 °C).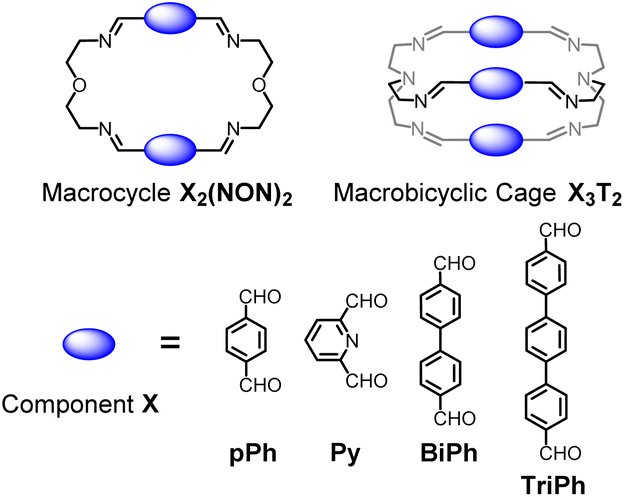 | ||
| Scheme 1 Molecular structures of the dialdehyde components (bottom) and of the homoleptic imine-based constituents: the [2 + 2] macrocycles and the [3 + 2] macrobicyclic cages (top) studied in the present work. The macrocycles TriPh2(NON)2 and the macrobicycle TriPh3T2 are new compounds. The macrocycles pPh2(NON)2,10Py2(NON)2,11BiPh2(NON)2,6e and the macrobicycles pPh3T2,12Py3T2,2aBiPh3T2,9a have been previously reported in the literature as indicated. | ||
The time evolution curves shown in the figures below depend on the experimental conditions (see ESI Page S5†). As also observed previously, due to the formation of a variety of intermediates, the consumption of aldehyde components is much faster than the appearance of the final products. Both the 1H NMR spectra and the HRMS spectra were recorded immediately after addition of 2 equiv. 2,2′-oxybis(ethylamine) (NON) into 2 equiv. dialdehyde solution or 2 equiv. tris(2-aminoethyl)amine (T) into 3 equiv. dialdehyde solution. The reaction rates can be estimated by the time of 50% dialdehyde consumption (tC1/2) and the time of 50% product formation (tF1/2). The results obtained are given in the ESI (see the NMR monitoring sections in ESI, Pages S7–S25†). The molecular structures of pPh2(NON)2, BiPh2(NON)2 and TriPh3T2 were confirmed by single-crystal X-ray crystallography (Fig. 1; see details in ESI, Fig. S63–S65 and Table S9†).‡
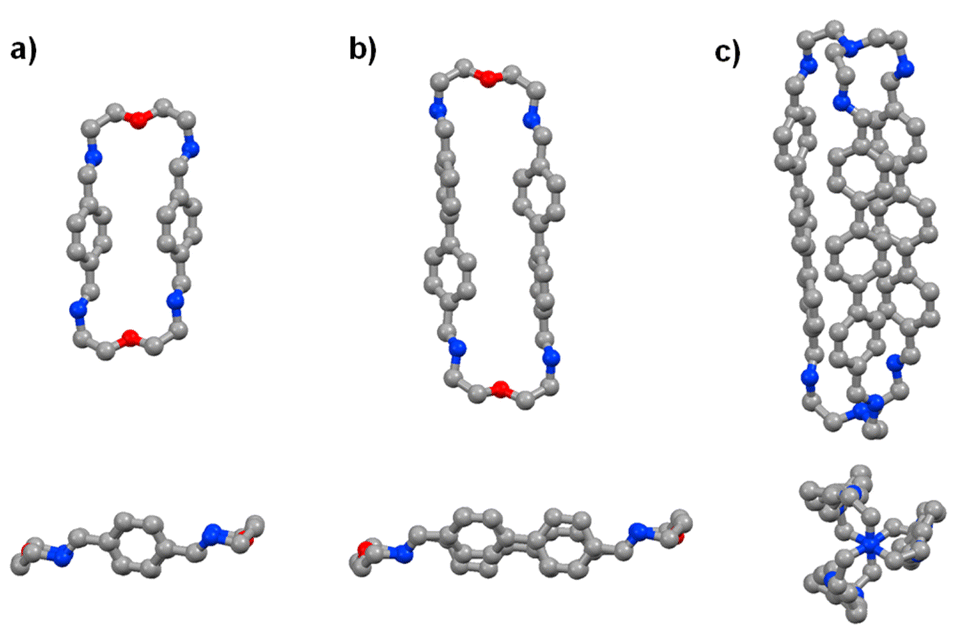 | ||
| Fig. 1 Single-crystal X-ray structures of the macrocycles pPh2(NON)2 and BiPh2(NON)2 as well as of the macrobicyclic cage TriPh3T2 (side and front/axial views). Colour scheme: carbon (grey), nitrogen (blue), oxygen (red). Hydrogen, H2O and CHCl3 molecules are omitted for clarity, see details in ESI, Fig. S63–S65.† | ||
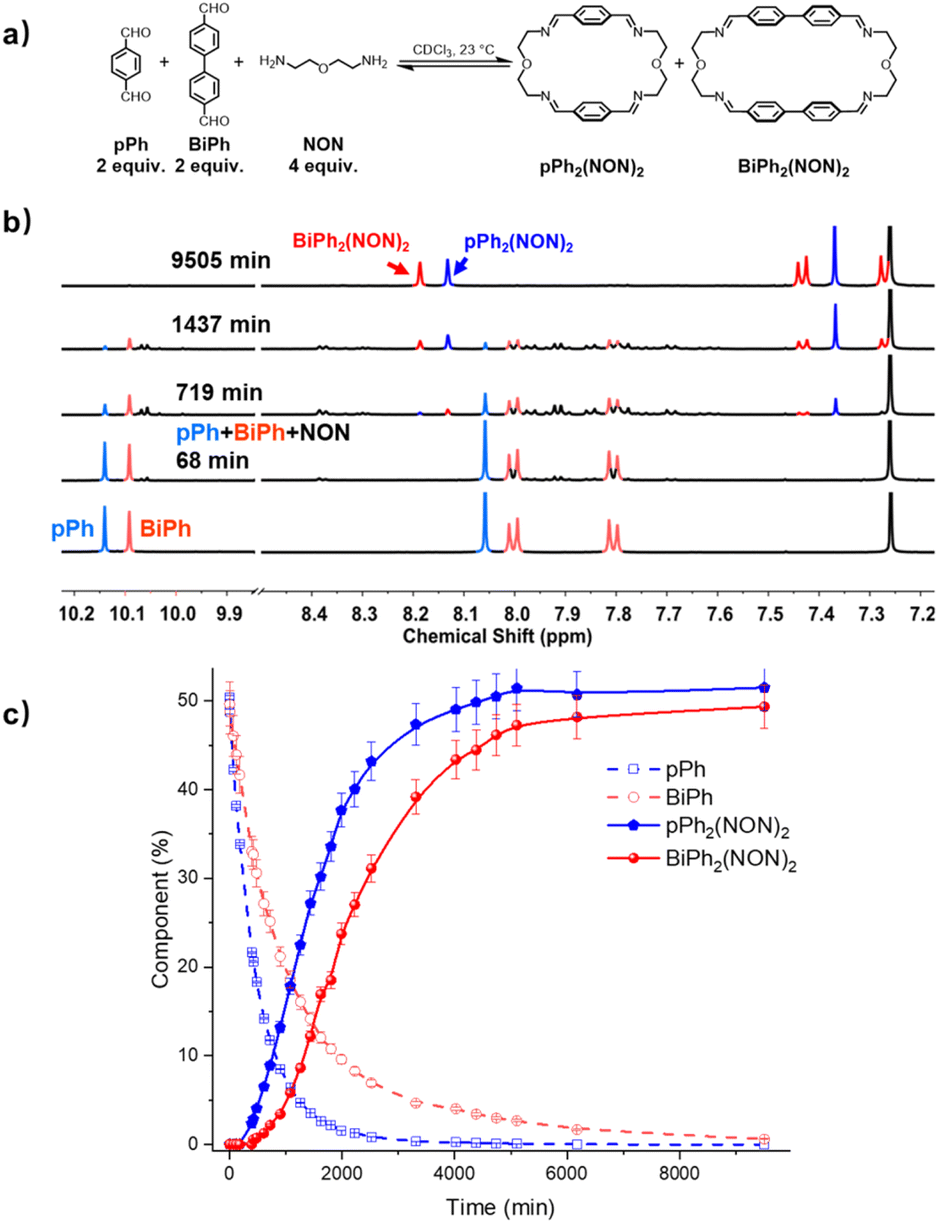
Formation and characterization of the homoleptic macrocycles pPh2(NON)2 and BiPh2(NON)2 from their components as a function of time in imine-based dynamic covalent systems
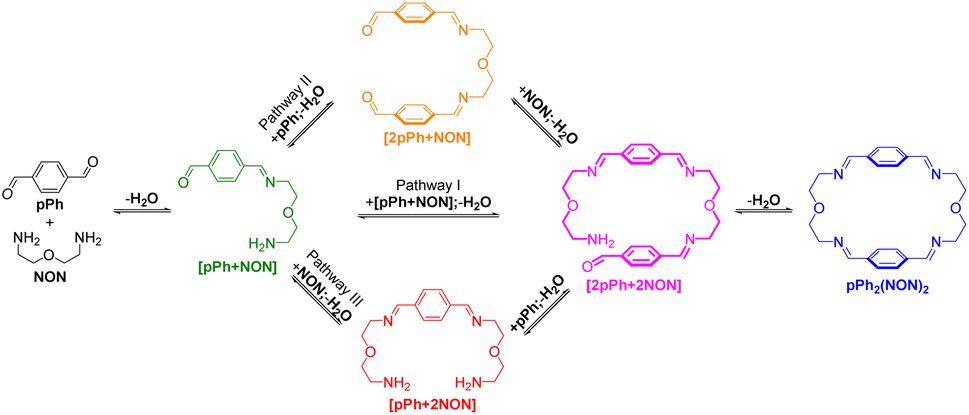
The multiple condensation reactions involved in the formation of compounds containing several imine groups from their carbonyl and amine components lead to the generation of numerous transient intermediates which greatly complicates the 1H NMR investigation of the reaction course. In order to minimize the analysis difficulties, we first studied the macrocycle pPh2(NON)2 formation from terephthalaldehyde (pPh) and 2,2′-oxybis(ethylamine) (NON), as there is only a singlet 1H NMR signal in the aromatic area of the dialdehyde pPh. Throughout the measurement, signals due to several metastable intermediates were observed. Four possible acyclic intermediates may be considered, with different dialdehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diamine compositions: [pPh + NON], [pPh + 2NON], [2pPh + NON], [2pPh + 2NON] (Fig. 2a), and could be identified from their 1H NMR signals according to their different structural features. For example, the [1 + 1] unsymmetrical acyclic [pPh + NON] intermediate should form first and show one aldehyde signal, one imine signal, two peaks in the aromatic area and four peaks in aliphatic area. These spectroscopic characteristics coincide with the green peaks in Fig. 2b. The orange peaks are those of symmetrical [2pPh + NON]. For the remaining red signals in Fig. 2b, two singlets at 8.33 ppm and 7.78 ppm can then be assigned to the symmetrical [pPh + 2NON]. Ultimately, all these intermediates are converted into the macrocycle pPh2(NON)2 (Fig. 2b, blue). The evolution of the reaction mixture is plotted in Fig. 2c. It can be seen that the intermediate [pPh + NON] markedly increased with time from 7 to 900 min to reach 30% of the maximum possible amount and then smoothly decreased, while smaller quantities of [2pPh + NON] and [pPh + 2NON] showed a similar but somewhat slower time dependence.
:diamine compositions: [pPh + NON], [pPh + 2NON], [2pPh + NON], [2pPh + 2NON] (Fig. 2a), and could be identified from their 1H NMR signals according to their different structural features. For example, the [1 + 1] unsymmetrical acyclic [pPh + NON] intermediate should form first and show one aldehyde signal, one imine signal, two peaks in the aromatic area and four peaks in aliphatic area. These spectroscopic characteristics coincide with the green peaks in Fig. 2b. The orange peaks are those of symmetrical [2pPh + NON]. For the remaining red signals in Fig. 2b, two singlets at 8.33 ppm and 7.78 ppm can then be assigned to the symmetrical [pPh + 2NON]. Ultimately, all these intermediates are converted into the macrocycle pPh2(NON)2 (Fig. 2b, blue). The evolution of the reaction mixture is plotted in Fig. 2c. It can be seen that the intermediate [pPh + NON] markedly increased with time from 7 to 900 min to reach 30% of the maximum possible amount and then smoothly decreased, while smaller quantities of [2pPh + NON] and [pPh + 2NON] showed a similar but somewhat slower time dependence.
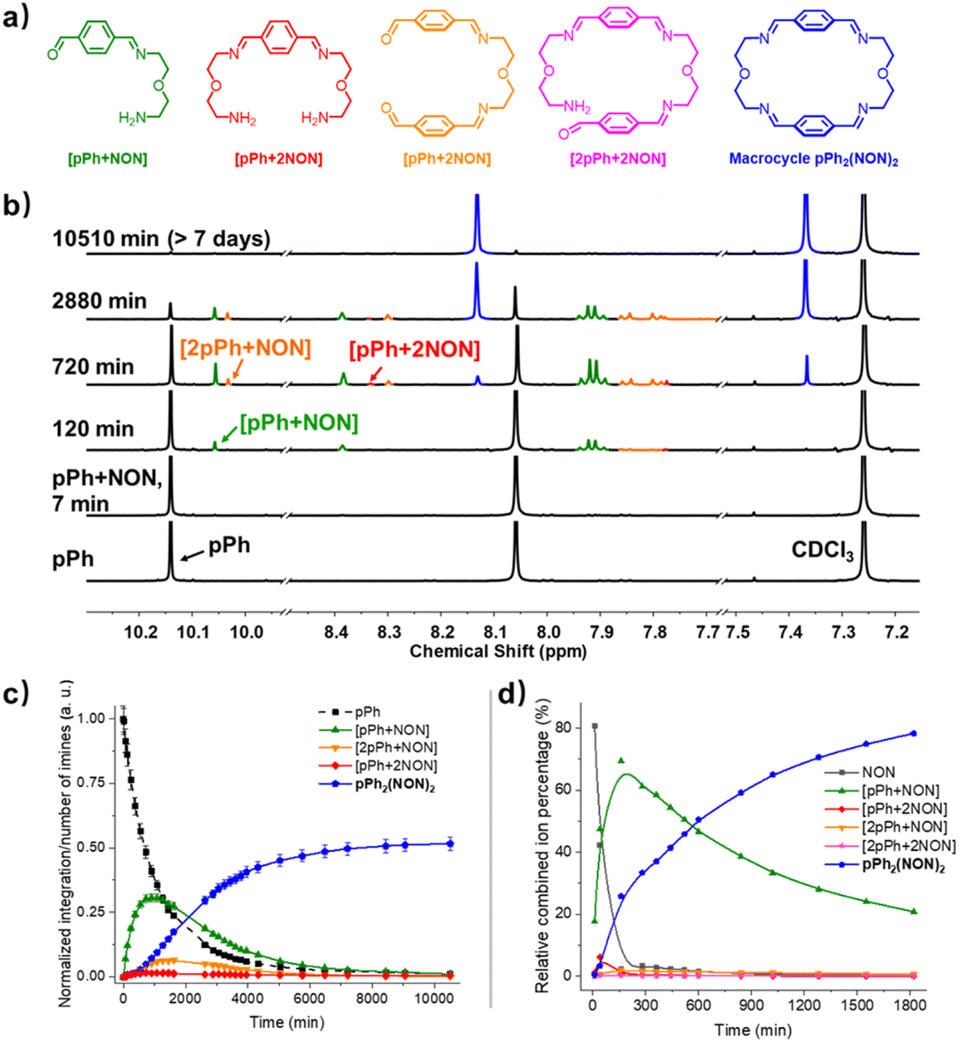 | ||
| Fig. 2 (a) Chemical structures of the different intermediates and of pPh2(NON)2, (b) evolution of the 1H NMR spectra (500 MHz, CDCl3, 23 °C) of a 1:1 mixture of pPh and NON ([pPh]0 = [NON]0 = 3.6 mM) showing the formation of three intermediates and of the final macrocycle pPh2(NON)2 after 7 min, 120 min, 720 min, 2880 min and 10510 min. (c) 1H NMR monitoring of the evolution of the species generated during pPh2(NON)2 formation from a 1:1 mixture of pPh and NON as a function of time over 10510 min. Error in 1H-NMR signal integration: ±5%. (d) HRMS-ESI monitoring of the species generated during the formation of the macrocycle pPh2(NON)2 from a 1:1 mixture of pPh and NON ([pPh]0 = [NON]0 = 2 mM, 50/50 (v/v) CHCl3/MeOH, 25 °C) over 1820 min. The spectra and kinetic curves are coloured in accordance with the molecular structures. NB: the relative combined ion percentage is obtained by the ratio of combined ion count of each species to the sum of combined ion counts for all species at each time point. These data do not provide quantitative information about the relative amounts of each species identified by its mass, but, taken separately, they display the evolution of a given identified species during the course of the reaction. The curves are added to guide the eye. | ||
The evolution of the reaction mixture was also monitored by HRMS, providing additional information on the identification of intermediates. All the possible intermediates were identified, including [2pPh + 2NON], indiscernible in the 1H NMR spectra. The evolution of their m/z peak intensities over time is plotted in Fig. 2d. Both the 1H NMR and HRMS curves for the macrocycle product show an induction period, consistent with its formation involving passage through at least one intermediate.6e A similar behaviour was found for the formation of BiPh2(NON)2 as indicated by the corresponding 1H NMR and HRMS data. [BiPh + NON] and [2BiPh + NON] compositions were detected and identified by 1H NMR spectroscopy (Fig. S9–S12†) and HMRS (Fig. S16 and S17†).
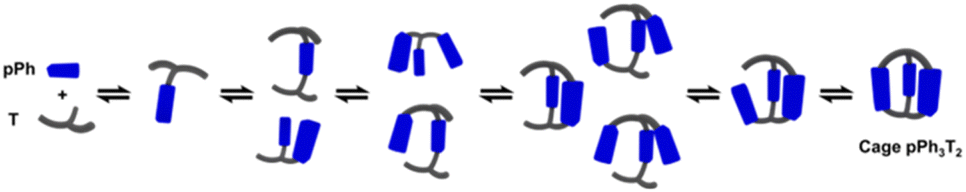
A possible self-assembly mechanism of such [2 + 2] covalent organic macrocycles can be proposed as shown in Scheme 2. Thus, in the case of pPh2(NON)2 for example, initial formation of the [1 + 1] intermediate [pPh + NON] is followed by generation of [2pPh + 2NON] along three pathways, (i) (main process): one-step dimerization of two [pPh + NON] intermediates; (ii) and (iii): condensation with an additional component (pPh or NON) to form intermediates [1 + 2] and [2 + 1]; finally, intramolecular macrocyclization yielding macrocycle pPh2(NON)2. At the beginning, with only pPh and NON present, only [pPh + NON] can form. If then it reacts further with itself or with pPh or NON separately at a rate similar to that of its formation, then it should start to generate [2pPh + 2NON] (which undergoes a rapid intramolecular cyclization) along with [2pPh + NON] and [pPh + 2NON]. The latter two then have to react with free NON and pPh respectively to give [2pPh + 2NON] and thus the macrocycle by cyclization but at relatively slow rates because the concentrations of free pPh and NON are now quite low compared to the initial values controlling the formation of [pPh + NON].
 | ||
| Scheme 2 Stepwise [2 + 2] imine condensation processes showing the intermediates on the way to the formation of the imine-based dynamic covalent macrocycle pPh2(NON)2. | ||
Formation and characterization of the homoleptic macrobicyclic cages pPh3T2 and BiPh3T2 from their components as a function of time in imine-based dynamic covalent systems
The assembly of cage pPh3T2 was monitored by 1H NMR spectroscopy and HRMS. In the 1H NMR spectra, signals from several intermediates were observed. As there may be nine possible intermediates for the generation of a cage, it is not surprising that the spectra are more complicated than in the case of the macrocycles.Throughout the 1H NMR, several metastable intermediates, for instance [pPh + T], [2pPh + T], [pPh + 2T], and [2pPh + 2T] could be identified (Fig. 3a, for assignment of 1H NMR see ESI Fig. S20, S23 and S24†). The 1H NMR and HRMS kinetic monitoring (Fig. 3b and c) showed that all the intermediates were first found to increase and then decrease together with the formation of the final macrobicyclic product. The intermediates [2pPh + T] and [2pPh + 2T] reached their maximum amount later than [pPh + T], indicating a sequential formation of intermediates with increased stoichiometry along the reaction toward the final macrobicyclic cage pPh3T2. Scheme 3 illustrates all the imine species possibly involved in the dynamic self-assembling process (nine intermediates as well as two reagents and a cage product), although some of them were not detected by HRMS-ESI.
 | ||
| Fig. 3 (a) Chemical structures of the different intermediates and of pPh3T2. (b) Evolution of the 1H NMR spectra (500 MHz, CDCl3, 23 °C) of a 3:2 mixture of pPh (3.6 mM) and T showing the formation of intermediates [pPh + T], [2pPh + T], [pPh + 2T], and [2pPh + 2T] together with that of the final macrobicyclic cage pPh3T2. (c) 1H NMR monitoring of the evolution of the species generated during cage pPh3T2 formation as a function of time over 2892 min. Error in 1H-NMR signal integration: ±5%. (d) HRMS-ESI kinetic evolution of the species generated during the formation of the macrocycle pPh3T2 from pPh (2 mM) and T (50/50 CHCl3/MeOH, r. t). The spectra and kinetic curves are coloured in accordance with the molecular structures. NB: the relative combined ion percentage is obtained by the ratio of combined ion count of each species to the sum of combined ion counts for all species at each time point. These data do not provide quantitative information about the relative amounts of each species identified by its mass, but, taken separately, they display the evolution of a given identified species during the course of the reaction. The curves are added to guide the eye. | ||
 | ||
| Scheme 3 Stepwise [3 + 2] imine condensation processes showing the intermediates corresponding to an increase in number of imine connections on the way to the formation of a dynamic covalent macrocyclic cage. | ||
A similar behaviour was found for the formation of cage BiPh3T2 (at a slower rate) involving also a range of intermediates of different stoichiometries (see ESI Fig. S25–S31, S38 and S39†).
Among all these products, the macrobicyclic cages form faster than their corresponding macrocycles with diamine NON despite the fact that the macrobicycles require six imine condensations and macrocycles only four, a feature possibly related to the higher probability of intramolecular reaction in the first case.6e
Self-correction in the self-sorting processes leading to the formation of homoleptic macrocycles
The homo-self-sorting behaviour was investigated for a three-component mixture of two dialdehydes pPh, BiPh of different lengths and NON in a 2:2:4 ratio. Fig. 4a represents the corresponding 1H NMR spectra showing a progressive formation of new molecules which were assigned to different intermediates depending on the formation sequence. The consumption of pPh and BiPh as well as the formation of the two macrocycles, are plotted versus reaction time in Fig. 4b. The formation of macrocycle pPh2(NON)2 was faster than that of BiPh2(NON)2, which was also related to the faster consumption of pPh than of BiPh. The formation of pPh2(NON)2, BiPh2(NON)2 in the self-sorting pPh:BiPh:NON system is faster than that observed for their separate formations (see ESI, Fig. S40†). This may be due to the doubled initial concentration of NON in the self-sorting system. The composition of the system was 51% of pPh2(NON)2, 49% of BiPh2(NON)2 and less than 1% of unreacted BiPh at equilibrium (after 160 h; see Fig. 4).§ The final spectrum indicated the high-fidelity homo-self-sorting and confirmed the absence of heteroleptic macrocycles. During the dynamic self-assembly, one observes several signals belonging to neither the identified intermediates of pPh/NON reaction nor those of BiPh/NON reaction. They may be due to several heteroleptic intermediates (for instance, [pPh + BiPh + NON], [pPh + BiPh + 2NON]) which then disappeared and no heteroleptic macrocycles could be detected, indicating correction and dissociation processes were taking place (see Fig. S41†).
 | ||
| Fig. 4 (a) Component self-sorting in the competitive formation of two macrocycles pPh2(NON)2 and BiPh2(NON)2. (b) Evolution of the partial 1H NMR spectra (500 MHz, CDCl3, 23 °C) of the reaction 2pPh + 2BiPh + 4NON ([pPh]0 = [BiPh]0 = 3.6 mM; [NON]0 = 7.2 mM) over 9505 min. The bottom trace corresponds to the dialdehyde components, pPh and BiPh. (c) 1H NMR monitoring of dialdehydes pPh, BiPh and macrocycles pPh2(NON)2 and BiPh2(NON)2 over 9505 min. The distribution (%) have been obtained by the integration of the of the aromatic regions and aldehyde CHO proton signals in the 500 MHz 1H NMR spectra. Error in 1H-NMR signal integration: ±5%. The corresponding t1/2 values are given in the ESI.† | ||
HRMS was also used to identify some intermediates and to follow their evolution as a function of time (see in ESI, Fig. S44 and S45†). The intermediates [pPh + NON], [BiPh + NON] and [pPh + 2NON] were very rapidly formed after mixing the solutions of the three components. They reached their highest abundance after 180 min, and then were smoothly transformed into the desired homoleptic macrocycles. The intermediates [pPh + BiPh + NON] and [pPh + BiPh + 2NON] as well as macrocycle (pPh)(BiPh)(NON)2 were detected in the course of the reaction and disappeared latter on. No signals corresponding to a heteroleptic macrocycle could be observed at the end of the reaction. A plausible self-sorting process may be suggested: in an initial step, the diamine NON reacted reacts with the aldehydes pPh or BiPh to form [1 + 1] intermediates; the [1 + 1] intermediates then convert into their homoleptic macrocycles, pPh2(NON)2 and BiPh2(NON)2, via both homoleptic cyclization following three possible pathways (Scheme 2) and self-correction pathways (Scheme 4). Due to the competition between pPh and BiPh containing species, two heteroleptic intermediates, namely [pPh + BiPh + NON] and [pPh + BiPh + 2NON], may be formed and lead to the final homoleptic products by both dissociation and self-correction processes (Scheme 4).13
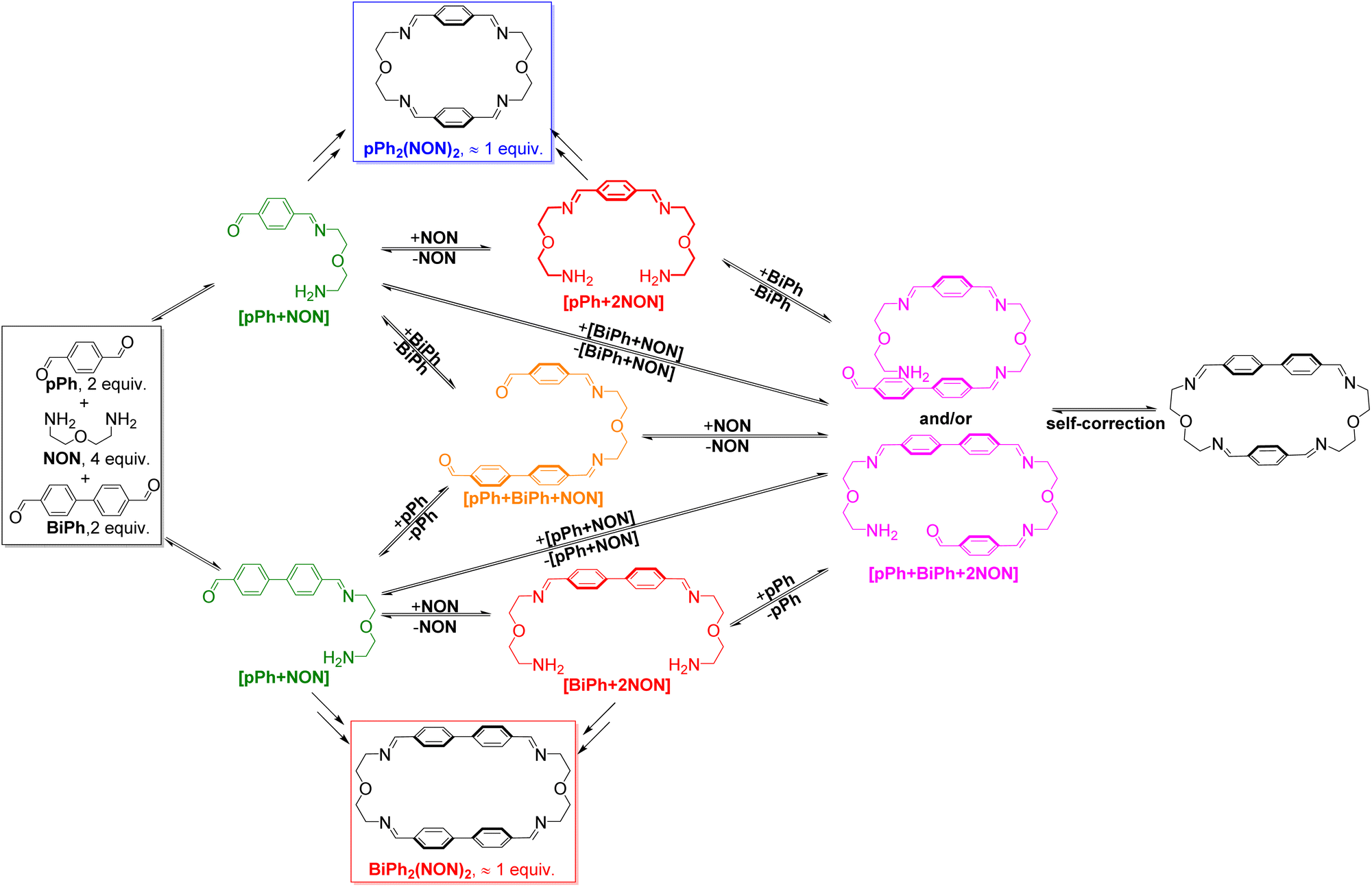 | ||
| Scheme 4 Self-correction and dissociation processes showing the conversion of heteroleptic intermediates on the way to self-sorting of the imine-based dynamic covalent macrocycles pPh2(NON)2 and BiPh2(NON)2. | ||
Self-correction in the self-sorting processes leading to the formation of homoleptic macrobicyclic cages
A three-component homo-self-sorting experiment resulting in the parallel formation of two cages was set up using two different bridging components pPh:BiPh:T in a 3:3:4 ratio. Fig. 5 shows that after mixing the components, the concentration of pPh rapidly decreased. Simultaneously broad new signals appeared which progressively disappeared as the reaction proceeded, indicating that they were due to intermediate species. After about 60 min, new small peaks appeared, the signals of the intermediates previously formed were attenuated and an intense imine peak corresponding to the small cage pPh3T2 arose at 8.17 ppm. Shortly after, the imine signal of the larger cage BiPh3T2 appeared, while both pPh and BiPh were progressively consumed. The aldehyde pPh was nearly fully consumed after 720 min, whereas 9% of BiPh remained unreacted. The consumption sequence was in accordance with their corresponding cage formation rates. Full conversion into 51% of pPh3T2 and 46% of BiPh3T2 as well as 2% of unreacted BiPh was attained after about 4157 min (69 h).§ The final spectrum indicated the high-fidelity of self-sorting and confirmed the absence of any heteroleptic structures. Comparing these observations to the results obtained for separate formation, both the consumption of BiPh and formation of BiPh3T2 were accelerated in the self-sorting experiment with respect to their separate formation, presumably because of the higher initial concentration of T, analogous to the observations in the macrocycle self-sorting experiment. In contrast the rate of formation of pPh3T2 remained the same compared as in the case of separate formation, indicating the probable involvement of correction and dissociation processes of heteroleptic intermediates (for more kinetic data see ESI Fig. S46†).
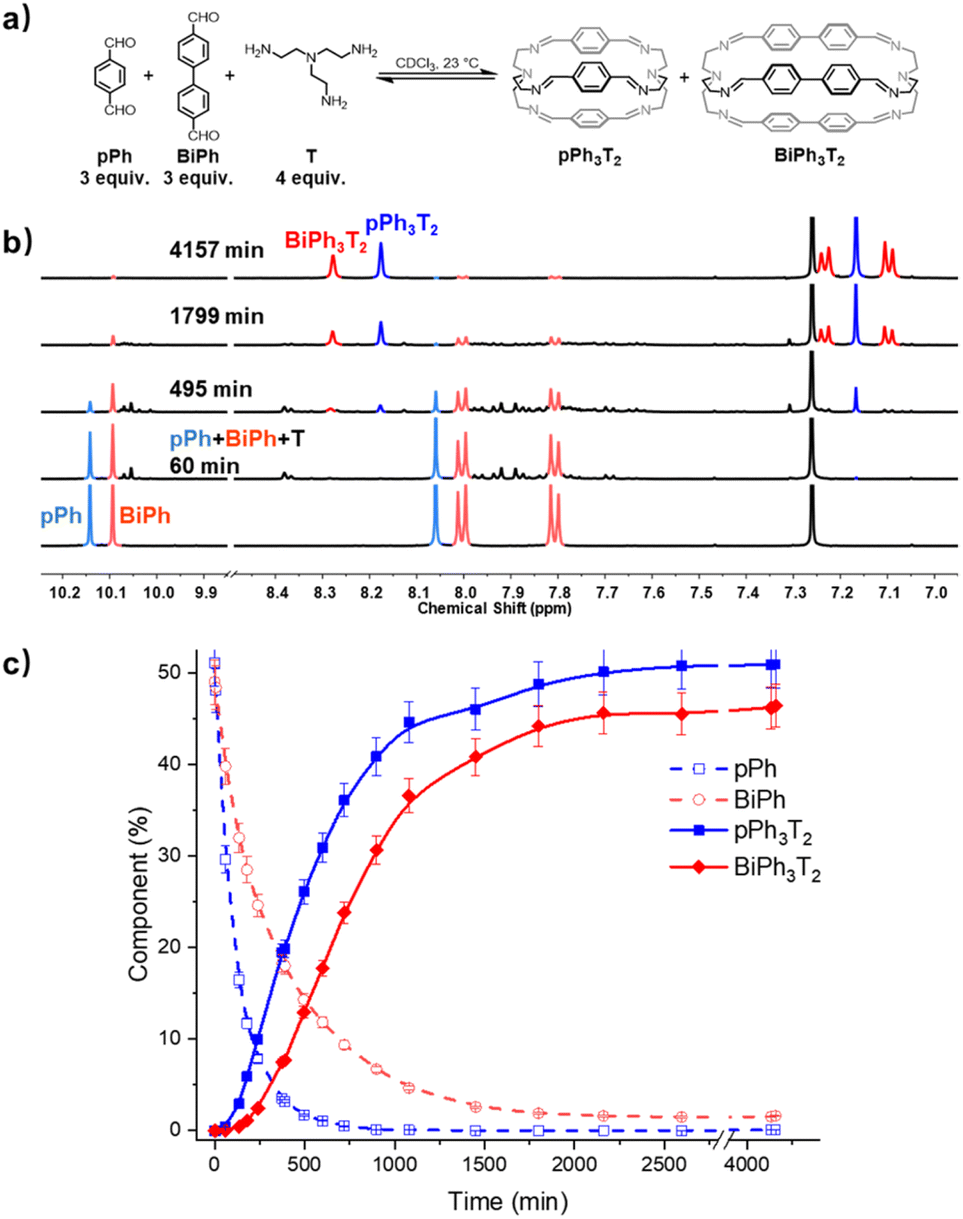 | ||
| Fig. 5 (a) Component self-sorting in the competitive formation of two macrobicyclic cages pPh3T2, BiPh3T2. (b) Evolution of the partial 1H NMR spectra (500 MHz, CDCl3, 23 °C) of the reaction 3pPh + 3Biph + 4T ([pPh]0 = [BiPh]0 = 3.6 mM, [T]0 = 4.8 mM) over 4157 min. The bottom trace corresponds to the dialdehyde components, pPh and BiPh. (c) 1H NMR monitoring of dialdehydes pPh, BiPh and cages pPh3T2, BiPh3T2 over 4157 min. The distribution (%) have been obtained by the integration of the of the aromatic regions and aldehyde CHO proton signals in the 500 MHz 1H NMR spectra. Error in 1H-NMR signal integration: ±5%. The corresponding t1/2 values are given in the ESI.† | ||
In order to gain some more information about the mechanism of the macrobicycle self-sorting processes, the key intermediates formed during the reaction were identified by their mass from the full HRMS spectra (Fig. S49 and S50†). Table S6† lists all identified intermediates and cages observed from time dependent HRMS experiments. The full evolution of the spectra over time suggests qualitative trends for these species. The reaction of pPh:BiPh with T started by the rapid appearance of [pPh + T] and [BiPh + T] intermediates. Thereafter several heteroleptic intermediates formed. All these intermediates progressively disappeared resulting in the simultaneous formation of the homoleptic cages pPh3T2 and BiPh3T2. Close inspection of the HRMS plot (under high amplification, Fig. 6b) revealed a decrease in concentration of the heteroleptic cage (pPh)(Biph)2T2 after its formation, indicating the operation of a self-correction process. Support for the general occurrence of such self-correction was obtained by investigation of a sequential cage formation experiment.
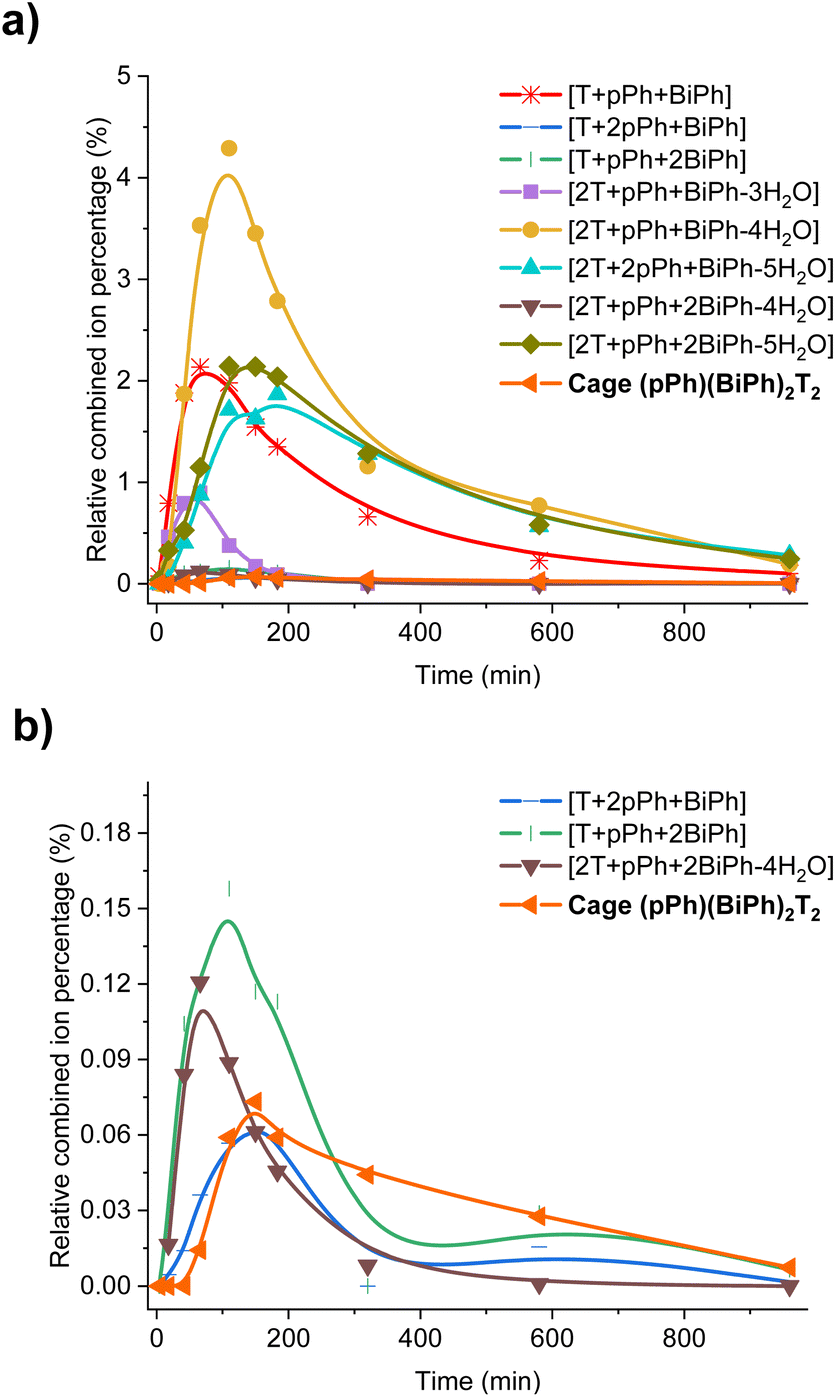 | ||
| Fig. 6 Evolution of the HRMS spectra displaying the self-correction process in the course of the self-assembly. (a) HRMS-ESI monitoring of the evolution of the heteroleptic intermediates generated from the 3pPh + 3BiPh + 4T ([pPh]0 = [BiPh]0 = 2 mM, T = 2.7 mM) library in the course of the homo-self-sorting process leading to the homoleptic structures pPh3T2, BiPh3T2. (b) Amplified view along the y axis. NB: the relative combined ion percentage is obtained by the ratio of combined ion count of each species to the sum of combined ion counts for all species at each time point. These data do not provide quantitative information about the relative amounts of each species identified by its mass, but, taken separately, they display the evolution of a given identified species during the course of the reaction. See Fig. S50† for the evolution of all species generated during the reaction course. | ||
A 1H-NMR study indicated that, in a solution (with hexamethyldisilane as internal standard) prepared by mixing 3 equiv. BiPh and 4 equiv. T, 36% of the initial T was converted into the cage BiPh3T2 after 48 h. As no CHO signal of free BiPh remained, the rest of BiPh appeared to have formed [BiPh + 2T] (10% with respect to T) and [2BiPh + 2T] (15% with respect to T), accompanied by the remaining free T (40%). This composition did not change over 3 more days. Then, on addition of 3 equiv. pPh to this solution, all intermediates and free T were converted into >47% (with respect to the internal reference) of each of the two macrobicycles pPh3T2 and BiPh3T2 (see ESI, Fig. S51†). These results indicated that the reaction between (i) the added pPh, (ii) the [BiPh + 2T] and [2BiPh + 2T] intermediates, and (iii) the remaining free T gave the pPh3T2 cage together with the additional BiPh3T2 cage. Taken together, they confirm that self-correction did indeed take place by component recombination driven by the formation of the pPh3T2 cage.
These data define a progressive evolution towards the final homoleptic structures along a sequence of steps (Scheme 5) which may involve up to 38 intermediates (see ESI Fig. S52†): the [1 + 1] intermediate forms initially in high abundance, followed by the three-component intermediates [1 + 2] and [2 + 1] which first increase and then slowly decrease to a plateau; thereafter, the four-component [2 + 2], [1 + 3] and the five-component [2 + 3] larger intermediates increase in abundance before declining to give the final homoleptic cage structures. The eventual formation of transient oligomeric intermediates has not been taken into account. The results indicate that self-correction of the heteroleptic species must take place at the different steps along the reaction sequence up to the final processing into the pure homoleptic macrobicyclic structures pPh3T2 and BiPh3T2.
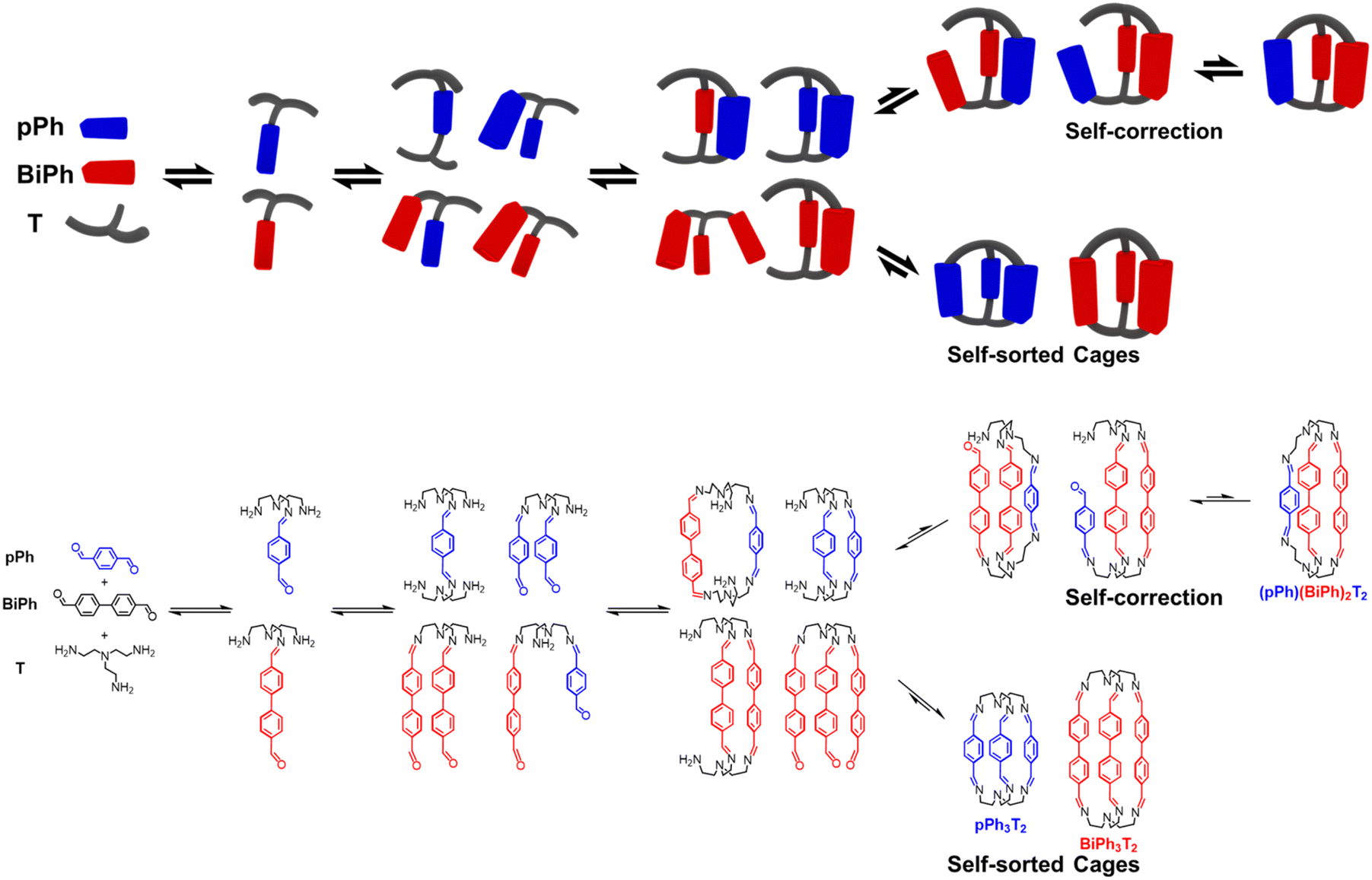 | ||
| Scheme 5 Schematic (top) and molecular (bottom) representations of the stepwise self-sorting process showing the most abundant intermediates on the way to the formation of the imine-based dynamic covalent homoleptic macrobicyclic structures pPh3T2 and BiPh3T2. | ||
DFT calculations elucidating the homo-self-sorting driving forces in macrocyclic and macrobicyclic systems
DFT calculations for the homoleptic and heteroleptic species of the two-cyclic order systems were performed to shed light on the contributions of the forces leading to the observed homo-self-sorting. Firstly, the macrocyclic structures were considered. The most stable conformations calculated corroborate the expected D2h symmetry for both pPh2(NON)2 and BiPh(NON)2 (Fig. 7, see also Fig. S53†). On the other hand, the heteroleptic macrocycle (pPh)(BiPh)(NON)2 presents a C2-symmetric structure. This lower symmetry (number of microstates: 2 for C2vs. 4 for D2h; Fig. S53†) seems to be decreasing the thermodynamic stability of (pPh)(BiPh)(NON)2 due to entropic implications.14 Moreover, the cyclic structure of the heteroleptic species adopts a quite arched conformation that could further destabilize this constituent, as evidenced by the higher energy calculated in comparison with the ones for the homoleptic macrocycles (+2.5 kcal mol−1 for (pPh)(BiPh)(NON)2, Fig. 7a). A similar scenario was found for the macrobicyclic system. Interestingly, in this case, the difference in symmetry numbers between the homoleptic and heteroleptic species is even more pronounced, as pPh3T2 and BiPh3T2 present a D3h symmetry (number of microstates: 12) but the heteroleptic cages (pPh)(BiPh)2T2 and (pPh)2(BiPh)T2 are C2-symmetric species (number of microstates: 2) (Fig. S54†). One might thus consider that the macrobicyclic system should undergo a more efficient homo-self-sorting considering the energy difference (heteroleptic–homoleptic) calculated for the two systems (Fig. 7 and S55†), with values increasing following the order: (pPh)(BiPh)(NON)2 < (pPh)(BiPh)2T2 ≪ (pPh)2(BiPh)T2. This hypothesis was corroborated by HRMS analyses. Whilst the heteroleptic macrocycle (pPh)(BiPh)(NON)2 was formed in ca. 1% relative abundance after 500 min, only minor traces of the heteroleptic molecular cage (pPh)(BiPh)2T2 were observed (max. abundance of ca. 0.1% after 180 min), with cage (pPh)2(BiPh)T2 not being detected during the analyses (Fig. 6b and S50 in ESI†).¶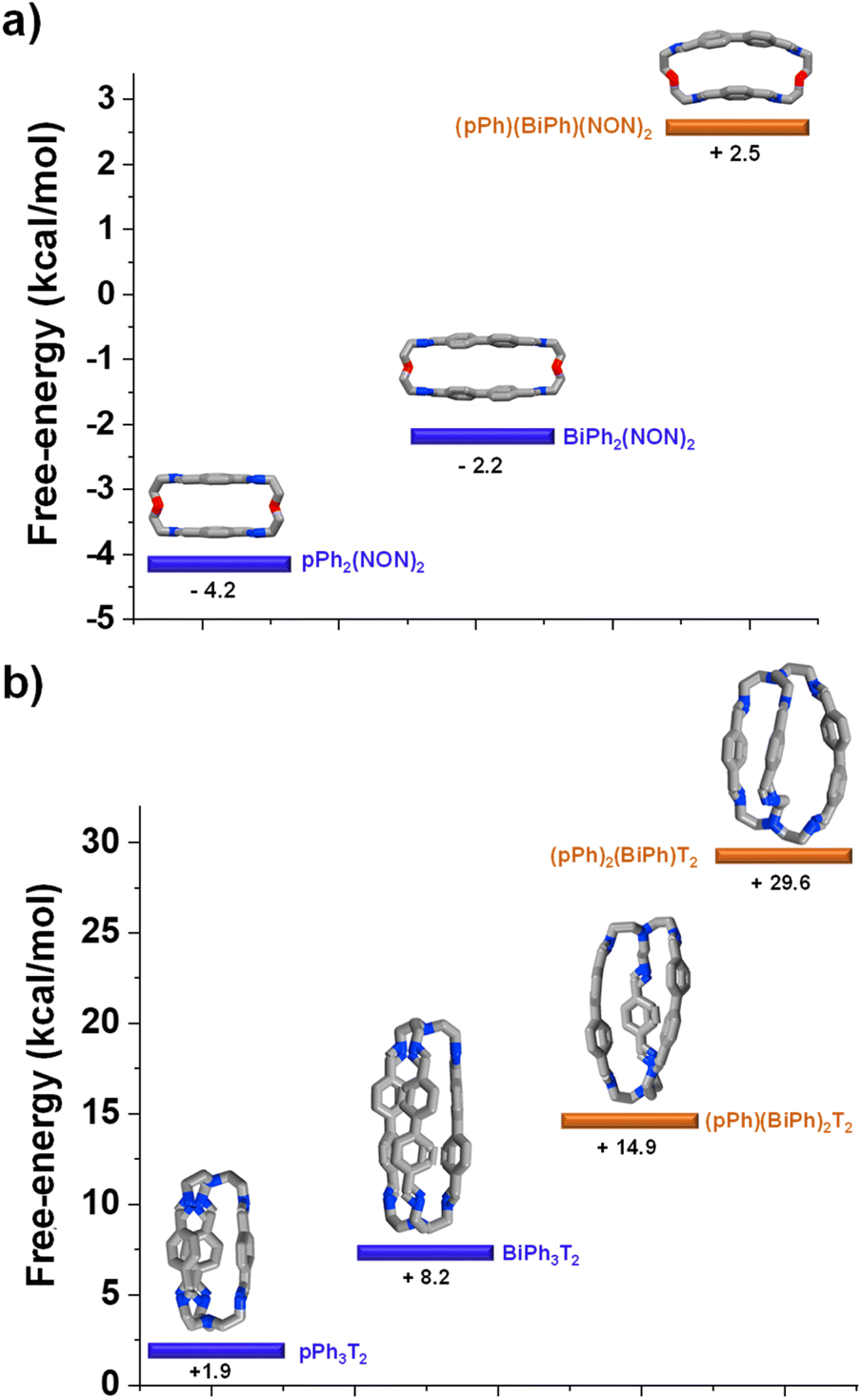 | ||
| Fig. 7 Most stable conformation (DFT, B3LYP, 6-311+G(d,p), Gaussian 09) for the homoleptic and heteroleptic (a) macrocycles and (b) cryptands. Free energies (kcal mol−1) have been calculated from the formation energies of the reaction components. See DFT calculations section (Section 7.4) in ESI† for more details. | ||
Homo-self sorting from [3 × 1] four-component DCovLs
A four-component DCovL composed of pPh, BiPh, TriPh and NON in 2:2:2:6 ratio was investigated in CDCl3 (Fig. 8a and b). The 1H NMR monitoring indicated that the reaction equilibrium was reached after 5510 min of heating at 40 °C. After the addition of NON to the mixture of dialdehydes, the concentration of the four components declined as that of the macrocycles rose to be the only species present at equilibrium. The generation of macrocycles followed the rate sequence pPh2(NON)2 > BiPh2(NON)2 ≥ TriPh2(NON)2. The composition of the final equilibrium solution was 36% pPh2(NON)2, 32% BiPh2(NON)2 and 32% TriPh2(NON)2.§ The ultimate formation of the three macrocycles was also confirmed by HRMS (ESI, Fig. S57†). The sum of all the constituents present in the equilibrium state agreed well with a high-fidelity self-sorting of the three competing macrocycles.
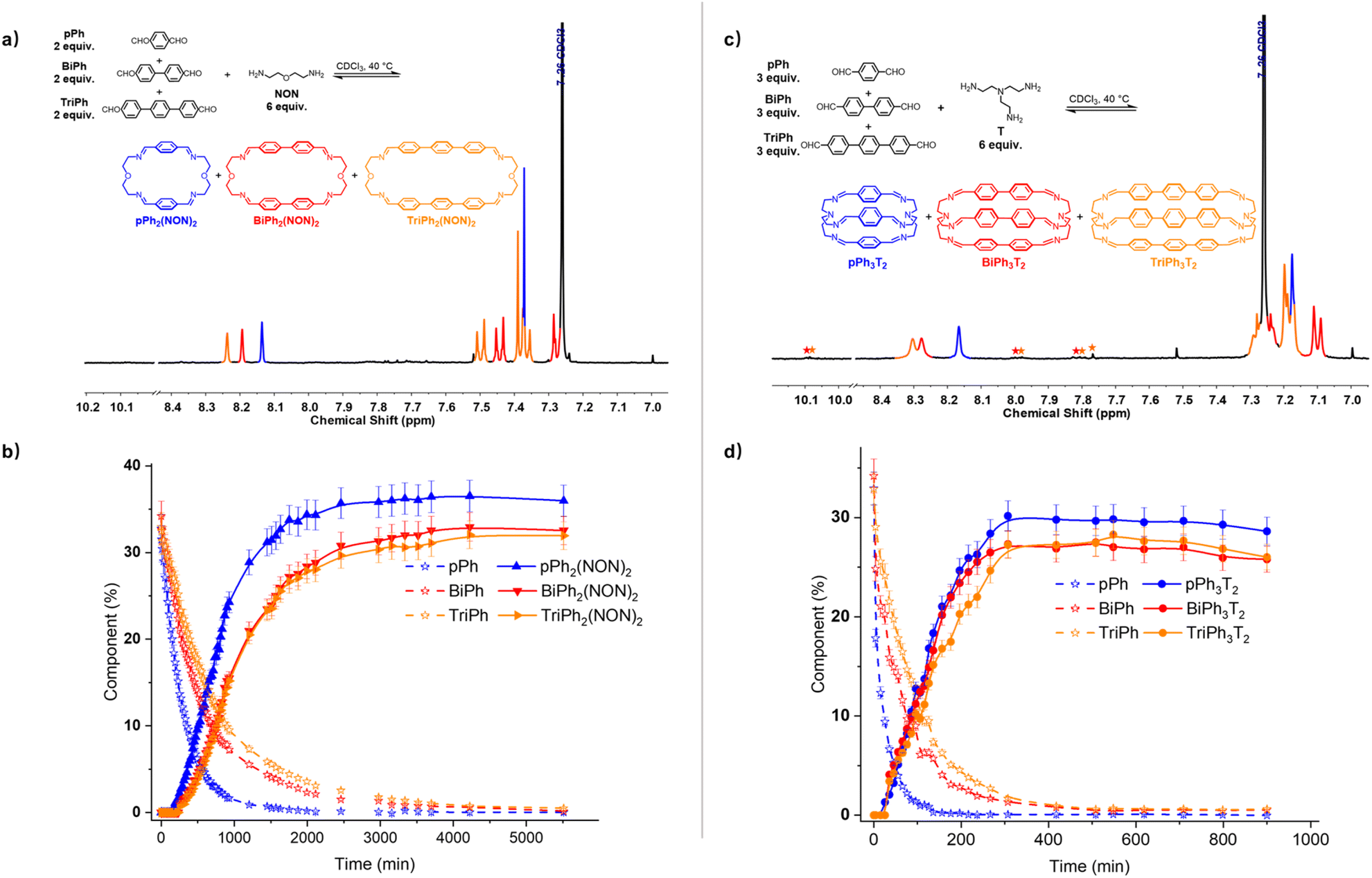 | ||
| Fig. 8 (a) Partial 1H NMR spectrum (400 MHz, CDCl3) of the parallel formation of a mixture of macrocycles pPh2(NON)2, BiPh2(NON)2 and TriPh2(NON)2 through self-sorting of 2pPh + 2BiPh + 2TriPh + 6NON after 5510 min at 40 °C. (b) 1H NMR monitoring of the formation of the macrocycles pPh2(NON)2, BiPh2(NON)2 and TriPh2(NON)2 over 5510 min. The initial concentration of each dialdehyde was 1.0 mM. (c) Partial 1H NMR spectrum (400 MHz, CDCl3) of the parallel formation of macrobicyclic cages pPh3T2, BiPh3T2 and TriPh3T2 though self-sorting of 3pPh + 3BiPh + 3TriPh + 6T after 900 min at 40 °C. (d) 1H NMR monitoring of the formation of the cages pPh3T2, BiPh3T2 and TriPh3T2 over 900 min. The initial concentration of each dialdehyde was 1.0 mM. Error in 1H-NMR signal integration: ±5%. The spectra and kinetic curves are coloured in accordance with the macrocycles and cages. The signals of free dialdehydes BiPh and TriPh are indicated by red and orange stars, respectively. | ||
In another case, a four-component DCovL composed of pPh, BiPh, TriPh and T in a 3:3:3:6 ratio was investigated in CDCl3 (Fig. 8c and d). The evolution was monitored by 1H NMR at 40 °C. Again, the reactant concentrations decreased uniformly with time as signals belonging to cages pPh3T2, BiPh3T2 and TriPh3T2 progressively rose following the sequence of relative rates pPh3T2 ≥ BiPh3T2 ≥ TriPh3T2. After 900 min, signals corresponding to macrobicyclic cages pPh3T2, BiPh3T2 and TriPh3T2 were clearly apparent (as also confirmed by HRMS, ESI, Fig. S59†) and the distribution was 29% pPh3T2, 26% BiPh3T2, 26% TriPh3T2 as well as <1% unreacted BiPh and <1% unreacted TriPh.§ Despite the poor solubility of cage TriPh3T2, the system was still able to self-sort towards the homoleptic macrocycles, but the final molar balance was of ca. 83%, suggesting that very small amounts of insoluble species might have been formed.
These results, taken together, indicate that self-sorting systems of higher diversity can be set up by appropriate choice of precursor components.
Homo-self sorting from [3 × 2] five-component and [4 × 2] six-component DCovLs and networks
To evaluate the self-sorting behaviour on increasing the complexity and diversity two systems containing a larger number of components were investigated. A first system [3 × 2] involved five components (three dialdehydes of increasing lengths, Py, BiPh and TriPh) together with the two polyamines of linear NON and branched T types with stoichiometries 3:3:3:6:2 for the [3 × 2] (Fig. 9). The resulting mixture was heated at 40 °C for up to 2340 min and the evolution of its composition was followed by 1H NMR spectroscopy. The affinity of Py for T was high enough to allow for selective formation of cage Py3T2. Indeed, after 90 min, all of the Py component had transformed into 23% of the anticipated cage Py3T2 and 2% of macrocycle Py2(NON)2. Meanwhile, approximately 33% of BiPh and 31% of TriPh (or a little less as some intermediates also contain –CHO groups whose signals might overlap with –CHO signals in BiPh and TriPh) remained unreacted, and no cages and macrocycles belonging to BiPh and TriPh were detected. After 720 min, no Py could be detected and the composition of the mixture was, BiPh (<5%), TriPh (<7%), Py3T2 (30%), Py2(NON)2 (6%), BiPh3T2 (2%), BiPh2(NON)2 (12%), TriPh3T2 (<6%), and TriPh2(NON)2 (10%), the remainder being in the form of unassigned intermediates. After 2340 min, the distribution was BiPh (<1%), TriPh (<1%), Py3T2 (29%), Py2(NON)2 (6%), BiPh3T2 (<1%), BiPh2(NON)2 (30%), TriPh3T2 (<1%), TriPh2(NON)2 (28%).§ The final distribution showed an almost completely sorted outcome (molar sum of constituents: 96%) with the cage Py3T2 and its agonistic macrocycles BiPh2(NON)2 and TriPh2(NON)2 as predominant compounds in the mixture.
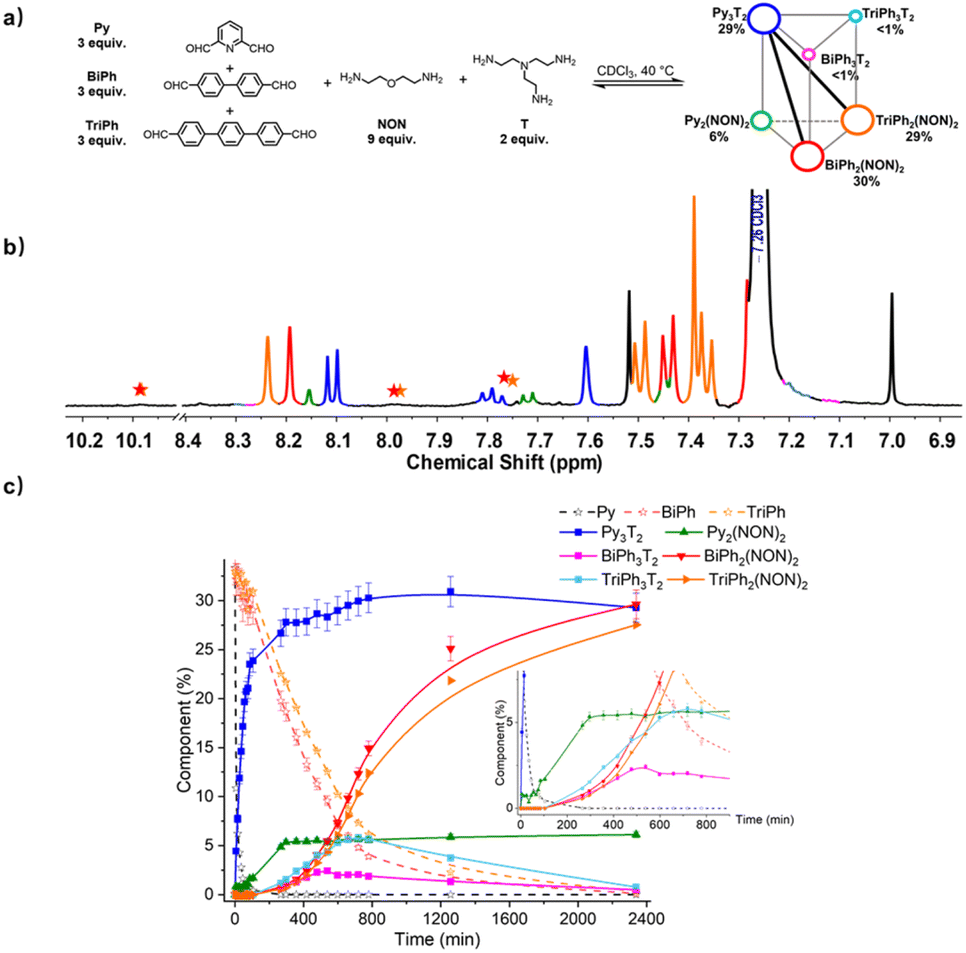 | ||
| Fig. 9 (a) [3 × 2] trigonal prismatic DCovL generated from 3Py + 3BiPh + 3TriPh + 2T + 6NON; (b) partial 1H NMR spectrum (400 MHz, CDCl3) of the equilibrated reaction mixture; (c) 1H NMR monitoring of macrocycles and macrocyclic cages over 2340 min. The initial concentration of each dialdehyde was 1.0 mM. Error in 1H-NMR signal integration: ±5%. The signals of free dialdehydes BiPh and TriPh are indicated by red and orange stars, respectively. The spectra and kinetic curves are coloured in accordance with the molecular structures. | ||
The second system concerned the self-sorting behaviour of [4 × 2] six-component DCovL built up from four dialdehydes Py, pPh, BiPh and TriPh and two diamines NON and T in 3:3:3:3:9:2 ratio. It was studied by NMR and HRMS at 40 °C for 3 days (Fig. 9). The equilibrated solution gave BiPh (1%), TriPh (2%), Py3T2 (21%), Py2(NON)2 (3%), pPh3T2 (<1%), pPh2(NON)2 (17%), BiPh3T2 (1%), BiPh2(NON)2 (21%), TriPh3T2 (3%), and TriPh2(NON)2 (18%).§ These products contained approximately 88% of the initial reactants. The final NMR spectrum inferred a less efficient self-sorting in this more complex DCovL, as evidenced by the unassigned signals in Fig. 10b. Such undesired products could be acting as kinetic traps that preclude an entirely sorted outcome. One might note, however, starting with six components generated only four dominant self-sorted architectures out of the eight possible homoleptic products. These results further corroborated the proposed important role of structural features towards achieving self-sorting of DCovLs. Nonetheless, it should be noted that the system is biased by the fast and dominant formation of the Py3T2 cage which precludes the formation of the other two cages as there is no T left (see Fig. S14 and S33† for kinetic profiles of formation of Py2NON2 and Py3T2 alone, and Fig. S35† for the competition between these three components). It again verifies the conclusion that, especially on introduction of a prevalent competitor, an increased compositional complexity may lead to a simplified output through a process of competitive self-sorting.15
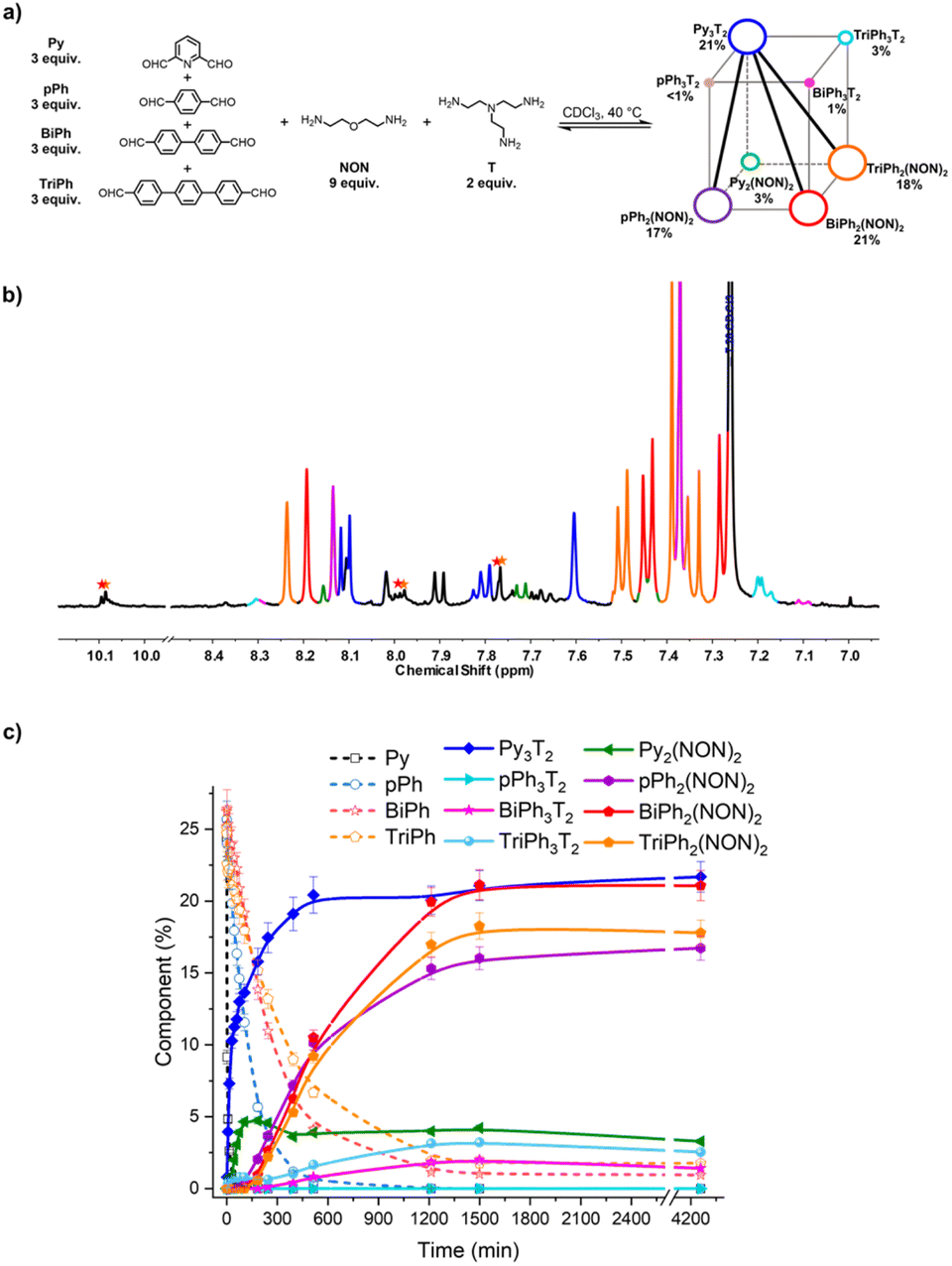 | ||
| Fig. 10 (a) [4 × 2] square prismatic DCovL generated from 3Py + 3pPh + 3BiPh + 3TriPh + 2T + 9NON; (b) partial 1H NMR spectrum (400 MHz, CDCl3, 40 °C) of the equilibrated reaction mixture; (c) 1H NMR monitoring of macrocycles and macrocyclic cages over 4260 min. The initial concentration of each dialdehyde was 2.0 mM. The signals of free dialdehydes BiPh and TriPh are indicated by red and orange stars, respectively. The larger size and the bold diagonal lines indicate the selective cogeneration of agonistic constituents. The spectra and kinetic curves are coloured in accordance with the molecular structures. | ||
Finally, the two DCovLs [3 × 2] and [4 × 2] can be represented by constitutional dynamic networks (CDNs)16 forming respectively a trigonal prism (Fig. 9a) and a square prism (Fig. 10a). The edges link antagonistic constituents that share a component and the diagonals share agonistic constituents not sharing a component.
Conclusions
Delineating the reaction pathways followed in multiple component DCovLs is crucial for the rational design of self-sorting systems of high diversity and complexity capable of generating several outputs side-by-side. The present study of the temporal evolution of imine-based dynamic covalent libraries and of the corresponding self-sorting processes involved both the separate and the concomitant formation of two types of representative structures of different cyclic order,1 macrocycles and macrobicyclic cages. The results suggest a rather complicated, multistep and multibranched pathway through consecutive reactions and numerous intermediates. Upon mixing the components, kinetically determined assembly first occurs to form a wide range of intermediates, some of which are then transformed into metastable heteroleptic structures. These metastable species then undergo dissociation and reassociation through component exchange to give homoleptic intermediates. The latter slowly converted to the final macrocycles and macrocyclic cages through pathways involving self-correction processes, while a small number of intermediates remained in the final product. The results highlight the important role played by structural features in controlling both the kinetic and thermodynamic parameters to balance a sorted versus a scrambled output. Besides, the DFT and HRMS outcomes infer the role of entropy in the homo-self-sorting attained. One may anticipate that they will contribute to the rational design of higher order self-sorting systems of increased complexity and diversity as well as inspire chemists towards the generation of novel dynamic covalent architectures and the investigation of the dynamic networks underlying self-sorting processes.Data availability
The ESI contains experimental details, characterization, spectral data, computational data and cartesian coordinates. CCDC 2006765, 2006766 and 2018494 contain the supplementary crystallographic data for this paper.†Author contributions
Z. Y. and J.-M. L. conceptualized the project; Z. Y., and C. A. performed the NMR and HRMS experiments; Z. Y. analysed the data; F. E. performed the DFT calculations; Z. Y. and F. E. wrote the original draft; all authors revised the manuscript; J.-M. L. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the ERC (Advanced Research Grant SUPRADAPT 290585), as well as the University of Strasbourg Institute for Advanced Study (USIAS) for financial support. This work was also supported by the NSFC (National Natural Science Foundation of China, 21720102007) and the Lehn Institute of Functional Materials (LIFM). Dr Z. Y. thanks Dr Jean-Louis Schmitt for helpful discussions of NMR and HRMS data, Corinne Bailly and Dr Lydia Karmazin for the X-ray crystallographic structure determinations. He also thanks Prof. Jack Harrowfield, Dr Artem Osypenko and Dr Youssef Atoini for extensive helpful discussions, as well as Stanislav Osypenko for drawing the schematic representation of cages.Notes and references
- (a) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS; (b) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (c) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (d) J. W. Sadownik and R. V. Ulijn, Curr. Opin. Biotechnol., 2010, 21, 401–411 CrossRef CAS PubMed; (e) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (f) Constitutional Dynamic Chemistry, Topics in Current Chemistry, ed. M. Barboiu, Springer Berlin, Heidelberg, 2012, vol. 322 Search PubMed; (g) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC; (h) Dynamic Covalent Chemistry: Principles, Reactions, and Applications, ed., W. Zhang and Y. Jin, Wiley-VCH, 2017 Search PubMed.
- (a) J. Jazwinski, J.-M. Lehn, D. Lilienbaum, R. Ziessel, J. Guilhem and C. Pascard, J. Chem. Soc., Chem. Commun., 1987, 1691–1694 RSC; (b) D. MacDowell and J. Nelson, Tetrahedron Lett., 1988, 29, 385–386 CrossRef CAS; (c) J. De Mendoza, E. Mesa, J. C. Rodríguez-Ubis, P. Vázquez, F. Vögtl, P. M. Windscheif, K. Rissanen, J. M. Lehn, D. Lilienbaum and R. Ziessel, Angew. Chem., Int. Ed., 1991, 30, 1331–1333 CrossRef; (d) R. Abidi, F. Arnaud-Neu, M. G. B. Drew, S. Lahély, D. Marrs, J. Nelson and M.-J. Schwing-Weil, J. Chem. Soc., Perkin Trans. 2, 1996, 2747–2755 RSC; (e) V. McKee, J. Nelson and R. M. Town, Chem. Soc. Rev., 2003, 32, 309–325 RSC; (f) P. A. Vigato, S. Tamburini and L. Bertolo, Coord. Chem. Rev., 2007, 251, 1311–1492 CrossRef CAS; (g) L. Fabbrizzi, J. Org. Chem., 2020, 85, 12212–12226 CrossRef CAS PubMed.
- (a) J.-M. Lehn, Science, 1985, 227, 849–856 CrossRef CAS PubMed; (b) J.-M. Lehn, R. Méric, J.-P. Vigneron, I. Bkouche-Waksman and C. Pascard, J. Chem. Soc., Chem. Commun., 1991, 2, 62–64 RSC; (c) V. Amendola, G. Bergamaschi, A. Buttafava, L. Fabbrizzi and E. Monzani, J. Am. Chem. Soc., 2010, 132, 147–156 CrossRef CAS PubMed; (d) M. Brutschy, M. W. Schneider, M. Mastalerz and S. R. Waldvogel, Chem. Commun., 2013, 49, 8398–8400 RSC; (e) K. Acharyya, A. Chowdhury, B. Mondal, S. Chakraborty and P. S. Mukherjee, Chem.–Eur. J., 2017, 23, 8482–8490 CrossRef CAS PubMed.
- (a) M. W. Hosseini, J.-M. Lehn and M. P. Mertes, Helv. Chim. Acta, 1983, 66, 2454–2466 CrossRef CAS; (b) T. Lu, X. Zhuang, Y. Li and S. Chen, J. Am. Chem. Soc., 2004, 126, 4760–4761 CrossRef CAS PubMed; (c) B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS PubMed; (d) Y. Zhang, Y. Xiong, J. Ge, R. Lin, C. Chen, Q. Peng, D. Wang and Y. Li, Chem. Commun., 2018, 54, 2796–2799 RSC; (e) L. Qiu, R. McCaffrey, Y. Jin, Y. Gong, Y. Hu, H. Sun, W. Park and W. Zhang, Chem. Sci., 2018, 9, 676–680 RSC; (f) D.-C. Zhong and T.-B. Lu, Chem. Commun., 2016, 52, 10322–10337 RSC.
- For selected general resources on self-sorting, see (a) R. Krämer and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 CrossRef PubMed; (b) S. J. Rowan, D. G. Hamilton, P. A. Brady and J. K. M. Sanders, J. Am. Chem. Soc., 1997, 119, 2578–2579 CrossRef CAS; (c) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; (d) Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC; (e) S. Ghosh and L. Isaacs, Dynamic Combinatorial Chemistry: In Drug Discovery, Bioorganic Chemistry, and Materials Science, ed. B. L. Miller, Wiley-VCH, Hoboken, 2010, pp. 118–154 Search PubMed; (f) H. Jędrzejewska and A. Szumna, Chem. Rev., 2017, 117, 4863–4899 CrossRef PubMed; (g) K. Acharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed; (h) C. Li, Y. Zuo, Y.-Q. Zhao and S. Zhang, Chem. Lett., 2020, 49, 28–53 CrossRef.
- (a) K. Acharyya, S. Mukherjee and P. S. Mukherjee, J. Am. Chem. Soc., 2013, 135, 554–557 CrossRef CAS PubMed; (b) K. Acharyya and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 1646–1657 CrossRef CAS PubMed; (c) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Angew. Chem., Int. Ed., 2014, 53, 7823–7827 CrossRef CAS PubMed; (d) X. Wang, P. Peng, W. Xuan, Y. Wang, Y. Zhuang, Z. Tian and X. Cao, Org. Biomol. Chem., 2018, 16, 34–37 RSC; (e) Z. Yang and J.-M. Lehn, J. Am. Chem. Soc., 2020, 142, 15137–15145 CrossRef CAS PubMed; (f) R. Gu and J.-M. Lehn, J. Am. Chem. Soc., 2021, 143, 14136–14146 CrossRef CAS PubMed; (g) P. Li, Z. Sun, J. Chen, Y. Zuo, C. Yu, X. Liu, Z. Yang, L. Chen, E. Fu, W. Wang, Z. Liu, J. Hu and S. Zhang, J. Am. Chem. Soc., 2022, 144, 1342–1350 CrossRef CAS PubMed; (h) M. Ovalle, M. Kathan, R. Toyoda, C. N. Stindt, S. Crespi and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202214495 CrossRef CAS PubMed.
- (a) S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed; (b) D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed; (c) R. L. Greenaway, V. Santolini, F. T. Szczypiński, M. J. Bennison, M. A. Little, A. Marsh, K. E. Jelfs and A. I. Cooper, Chem.–Eur. J., 2020, 26, 3718–3722 CrossRef CAS PubMed; (d) V. Abet, F. T. Szczypiński, M. A. Little, V. Santolini, C. D. Jones, R. Evans, C. Wilson, X. Wu, M. F. Thorne, M. J. Bennison, P. Cui, A. I. Cooper, K. E. Jelfs and A. G. Slater, Angew. Chem., Int. Ed., 2020, 59, 16755–16763 CrossRef CAS PubMed; (e) P. Wagner, F. Rominger, W.-S. Zhang, J. H. Gross, S. M. Elbert, R. R. Schröder and M. Mastalerz, Angew. Chem., Int. Ed., 2021, 60, 8896–8904 CrossRef CAS PubMed.
- (a) S. Jiang, J. T. A. Jones, T. Hasell, C. E. Blythe, D. J. Adams, A. Trewin and A. I. Cooper, Nat. Commun., 2011, 2, 207 CrossRef PubMed; (b) Q. Wang, C. Zhang, B. C. Noll, H. Long, Y. Jin and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10663–10667 CrossRef CAS PubMed; (c) S. Lee, A. Yang, T. P. Moneypenny and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 2182–2185 CrossRef CAS PubMed; (d) R. L. Greenaway, D. Holden, E. G. B. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James and A. I. Cooper, Chem. Sci., 2017, 8, 2640–2651 RSC.
- (a) M. Kołodziejski, A. R. Stefankiewicz and J.-M. Lehn, Chem. Sci., 2019, 10, 1836–1843 RSC; (b) F. Esteve, B. Altava, E. García-Verdugo, S. V. Luis and J.-M. Lehn, Chem, 2022, 8, 2023–2042 CrossRef CAS.
- V. H. Krässig and G. Greber, Makromol. Chem., 1955, 17, 131–153 CrossRef.
- (a) M. G. B. Drew, P. C. Yates, B. P. Murphy, J. Nelson and S. M. Nelson, Inorg. Chim. Acta, 1986, 118, 37–47 CrossRef CAS; (b) P. C. Haussmann, S. I. Khan and J. F. Stoddart, J. Org. Chem., 2007, 72, 6708–6713 CrossRef CAS PubMed.
- M. G. B. Drew, D. McDowell and J. Nelson, Polyhedron, 1988, 7, 2229–2232 CrossRef CAS.
- W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309–2320 CrossRef CAS PubMed.
- (a) S.-K. Lin, J. Chem. Inf. Comput. Sci., 1996, 36, 367–376 CrossRef CAS; (b) S. W. Siscoa and J. S. Moore, Chem. Sci., 2014, 5, 81–85 RSC; (c) J. Atcher, J. Bujons and I. Alfonso, Chem. Commun., 2017, 53, 4274–4277 RSC.
- (a) S. Dhers, J. Holub and J.-M. Lehn, Chem. Sci., 2017, 8, 2125–2130 RSC; (b) O. Š. Miljanić, Chem, 2017, 2, 502–524 CrossRef; (c) J.-F. Ayme, S. Dhers and J.-M. Lehn, Angew. Chem., Int. Ed., 2020, 59, 12484–12492 CrossRef CAS PubMed; (d) P. Adamski, M. Eleveld, A. Sood, Á. Kun, A. Szilágyi, T. Czárán, E. Szathmáry and S. Otto, Nat. Rev. Chem., 2020, 4, 386–403 CrossRef PubMed.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed; (b) G. Men and J.-M. Lehn, J. Am. Chem. Soc., 2017, 139, 2474–2483 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2006765, 2006766 and 2018494. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01174g |
| ‡ Crystals suitable for X-ray analyses were obtained for macrocycles pPh2(NON)2 and BiPh2(NON)2 and for the cage TriPh3T2. Both macrocyclic structures revealed a C2-symmetry enforced by intramolecular π⋯π interactions, with the shortest Car⋯Car distance of 3.840 Å and 3.585 Å, respectively. Despite the long distances between the oxygen atoms −dO⋯O = 10.584 and 15.116 Å for pPh2(NON)2 and BiPh2(NON)2 – no solvent molecules were observed within the cavity. A water molecule was found to be interacting with the lone pair of the N atoms (imino groups) in pPh2(NON)2, with a N⋯Owater distance of 2.896 Å. The TriPh3T2·molecular structure showed a distorted D3 symmetry and the aromatic bridging units were also interacting through π⋯π forces (shortest C⋯C distance = 3.425 Å). These intramolecular interactions were likely compressing the 3D-conformation of the cyclic species and thus reducing the cavity void, hampering the encapsulation of guest molecules. |
| § The composition (component %) of constituents in the self-sorting experiments is presented on the basis of the percentages of the dialdehyde components. |
| ¶ We have also calculated the relative intensities of the metastable heteroleptic macrocycles and macrobicycles in view of the analogous homoleptic compounds, as the HRMS comparison will be more representative due to similar response for each set of constituents. In this case, the maximum relative abundance for (pPh)(BiPh)(NON)2 was of ca. 7% and for (pPh)(BiPh)2T2 was of ca. 0.3% (Tables S6 and S8 in ESI†). |
| This journal is © The Royal Society of Chemistry 2023 |