
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct conversion of amino acids to oxetanol bioisosteres via photoredox catalysis†
Avelyn Mae V.
Delos Reyes
 ac,
Christopher S.
Nieves Escobar
bc,
Alberto
Muñoz
c,
Maya I.
Huffman
cd and
Derek S.
Tan
*abcde
ac,
Christopher S.
Nieves Escobar
bc,
Alberto
Muñoz
c,
Maya I.
Huffman
cd and
Derek S.
Tan
*abcde
aPharmacology Graduate Program, Weill Cornell Graduate School of Medical Sciences, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA. E-mail: tand@mskcc.org
bTri-Institutional PhD Program in Chemical Biology, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA
cChemical Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA
dTri-Institutional Chemical Biology Summer Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA
eTri-Institutional Research Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA
First published on 15th September 2023
Abstract
Carboxylic acids are an important structural feature in many drugs, but are associated with a number of unfavorable pharmacological properties. To address this problem, carboxylic acids can be replaced with bioisosteric mimics that interact similarly with biological targets but avoid these liabilities. Recently, 3-oxetanols have been identified as useful carboxylic acid bioisosteres that maintain similar hydrogen-bonding capacity while decreasing acidity and increasing lipophilicity. However, the installation of 3-oxetanols generally requires multistep de novo synthesis, presenting an obstacle to investigation of these promising bioisosteres. Herein, we report a new synthetic approach involving direct conversion of carboxylic acids to 3-oxetanols using a photoredox-catalyzed decarboxylative addition to 3-oxetanone. Two versions of the transformation have been developed, in the presence or absence of CrCl3 and TMSCl cocatalysts. The reactions are effective for a variety of N-aryl α-amino acids and have excellent functional group tolerance. The Cr-free conditions generally provide higher yields and avoid the use of chromium reagents. Further, the Cr-free conditions were extended to a series of N,N-dialkyl α-amino acid substrates. Mechanistic studies suggest that the Cr-mediated reaction proceeds predominantly via in situ formation of an alkyl-Cr intermediate while the Cr-free reaction proceeds largely via radical addition to a Brønsted acid-activated ketone. Chain propagation processes provide quantum yields of 5 and 10, respectively.
Introduction
The carboxylic acid moiety is an important structural feature found in many drugs and other bioactive compounds.1 However, it is also associated with several pharmacological liabilities, including limited permeability across biological membranes, high plasma protein binding, rapid renal clearance, and conversion to chemically reactive metabolites associated with toxicity.2–7 Indeed, small-molecule drugs containing carboxylic acid moieties have been withdrawn from the market at a much higher rate (39%)8 than their prevalence would predict (13%).1 One approach to circumvent undesired pharmacological properties associated with a given chemical group is to replace it with a bioisostere, a structural mimic that can induce a similar biological response.9 Several carboxylic acid bioisosteres have been reported, including hydroxamic acids, phosphonic acids, tetrazoles, and isothiazoles.5 Recently, 3-oxetanols have also been identified as promising carboxylic acid bioisosteres that can accommodate similar hydrogen-bonding interactions with biological targets while being less acidic, non-anionic under physiologic conditions, and more lipophilic to provide increased membrane permeability (Fig. 1a).10 While several synthetic approaches to 3-oxetanols have been reported,10–13 they require multistep de novo synthesis, presenting an obstacle to broad exploration of this promising class of bioisosteres. In contrast, a method for direct conversion of carboxylic acids to 3-oxetanols would provide expedient access to this motif, facilitating its investigation in medicinal chemistry campaigns. Herein, we report a new synthetic approach that enables direct conversion of α-amino acids to corresponding 3-oxetanols using visible light photoredox-catalyzed decarboxylative addition to 3-oxetanone. The reaction can be carried out in the presence or absence of CrCl3 and TMSCl cocatalysts, with the Cr-free conditions generally providing higher yields and avoiding the use of chromium reagents. The reactions provide broad substrate scope and functional group compatibility across N-aryl α-amino acid substrates. In addition, the Cr-free reaction was extended to a series of N,N-dialkyl α-amino acid substrates. Mechanistic studies suggest that the Cr-mediated reaction proceeds primarily via a Nozaki–Hiyama–Kishi reaction manifold, while the Cr-free reaction proceeds largely via 1,2-radical addition. The reactions have quantum yields of 5 and 10, respectively, indicative of chain propagation mechanisms.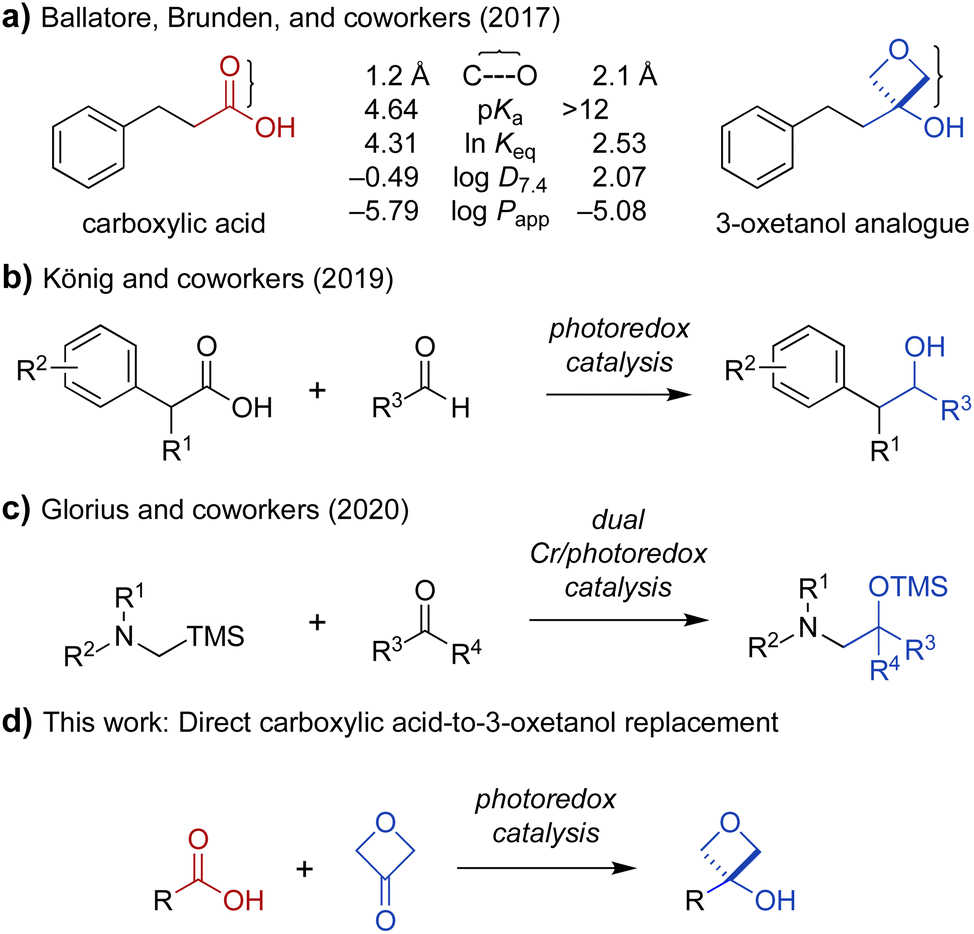 | ||
Fig. 1 (a) Physicochemical and pharmacological characteristics of carboxylic acids and 3-oxetanol bioisosteres (C⋯O = distance from carbonyl carbon to oxygen; ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Keq = hydrogen-bonding equilibrium constant determined by colorimetric assay for blue-shift of a fluorescent pyrazinone; logD7.4 = 1-octanol/water distribution coefficient, pH 7.4; logPapp = apparent permeability coefficient in parallel artificial membrane permeability assay).10,13 (b) Decarboxylative coupling of carboxylic acids and aldehydes under photoredox catalysis.14 (c) Addition of α-silyl amine-derived nucleophiles to aldehydes and ketones under photoredox catalysis.15 (d) Proposed direct transformation of carboxylic acids to 3-oxetanol bioisosteres. Keq = hydrogen-bonding equilibrium constant determined by colorimetric assay for blue-shift of a fluorescent pyrazinone; logD7.4 = 1-octanol/water distribution coefficient, pH 7.4; logPapp = apparent permeability coefficient in parallel artificial membrane permeability assay).10,13 (b) Decarboxylative coupling of carboxylic acids and aldehydes under photoredox catalysis.14 (c) Addition of α-silyl amine-derived nucleophiles to aldehydes and ketones under photoredox catalysis.15 (d) Proposed direct transformation of carboxylic acids to 3-oxetanol bioisosteres. | ||
Oxetanes have been investigated widely as bioisosteric replacements for gem-dimethyl groups,13,16–19 and have also attracted attention as carbonyl bioisosteres.13,17–23 Recently, the use of 3-oxetanols as carboxylic acid bioisosteres has been explored by Ballatore, Brunden, and coworkers.10 Comparison of the physicochemical properties of hydrocinnamic acid and its 3-oxetanol analogue, indicate that the latter is more lipophilic and membrane permeable (logD7.4: −0.49 vs. 2.07, logPapp: −5.79 vs. −5.08) (Fig. 1a).10 A 3-oxetanol analogue of ibuprofen was also evaluated and shown to have inhibitory activity against the cyclooxygenase (COX) pathway in a cell-based assay.10 This work provided important proof of concept for the use of 3-oxetanols as effective carboxylic acid bioisosteres. However, access to 3-oxetanols generally requires multistep de novo synthesis. Examples include addition of organometallic reagents to 3-oxetanone,10,16 Paternò–Büchi reaction of silyl enol ethers and aldehydes,11 and ring contraction of pentofuranose sugars.12 This lack of direct synthetic access from carboxylic acid substrates presents an obstacle to the broad exploration of 3-oxetanols as carboxylic acid bioisosteres. To address this problem, we sought to develop a method for direct conversion of carboxylic acids to the corresponding 3-oxetanol analogues.
Photoredox catalysis has emerged as an indispensable tool in synthetic organic chemistry. This mode of catalysis relies on photosensitive catalysts that convert light into chemical energy through single-electron transfer (SET) events with organic substrates, generating reactive radical intermediates under mild conditions, which can then engage in a variety of chemical transformations.24,25 With this in mind, we noted recent work by König and coworkers demonstrating photocatalytic decarboxylative activation of phenylacetic acids using the organic dye 4CzIPN (1,2,3,5-tetrakis(carbazole-9-yl)-4,6-dicyanobenzene, 2,4,5,6-tetrakis(9H-carbazol-9-yl) isophthalonitrile) for benzylation of aldehydes (Fig. 1b).14 More recently, Glorius and coworkers reported a dual Cr/photoredox catalytic system to convert trimethylsilylmethylamines to α-amino carbanion equivalents for addition to aldehydes and ketones (Fig. 1c).15 Inspired by these reports, we envisioned that carboxylic acids could be activated under photoredox catalysis for Nozaki–Hiyama–Kishi-type addition26 to 3-oxetanone to form the corresponding 3-oxetanol analogues (Fig. 1d), facilitating access to these understudied bioisosteres.
Results and discussion
Development of photoredox-catalyzed reaction for direct conversion of N-aryl α-amino acids to 3-oxetanol analogues
Photon-induced oxidative decarboxylation of α-amino acids is well known27,28 and the synthetic utility of the resulting α-amino radicals has been demonstrated.29–32 Rueping and coworkers have reported Ir photoredox-mediated decarboxylative couplings of N-aryl amino acids with enones33 and Zeng, Zhong, and coworkers,34 and Peng and coworkers35 have separately reported related couplings with aldehydes and ketones. With this in mind, we selected N-phenyl glycine (1a) as an initial substrate because it is readily oxidized (E1/2 = +0.42 V versus standard calomel electrode [SCE] in CH3CN)33,36 and commercially available. Unfortunately, treatment of N-phenyl glycine (1a) and 3-oxetanone (2) under conditions similar to those reported by Glorius for dual Cr/photoredox catalysis with 4CzIPN, did not afford any of the 3-oxetanol product 3a (Table 1, entry 1).15 However, addition of CsOAc, a base commonly used in decarboxylative photoredox platforms,37 resulted in a 7% yield of the desired product (entry 2). Previous studies have used TMSCl as an oxophilic additive to facilitate release of Cr back into the catalytic cycle,28,38 and inclusion of TMSCl resulted in an increased yield of 22% (entry 3). Carrying out the reaction in DMF instead of DMA slightly increased the yield to 25% (entry 4).|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 (equiv.) | Base | Additive | Solvent | Yielda (%) |
| a Yields based on 1H-NMR analysis of crude reaction product in the presence of an internal standard, relative to N-phenyl glycine (theoretical maximum 50% for entries 1–11). b Photocatalyst: Ir-A = [Ir{dF(CF3)2ppy}2(bpy)]PF6 = [2,2′-bipyridine-N1,N1′]bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate. c Photocatalyst: Ir-B = [Ir{dF(CF3)ppy}2(dtbpy)]PF6 = [4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate. d In absence of CrCl3. e In absence of blue LED light. 4CzIPN = 1,2,3,5-tetrakis(carbazole-9-yl)-4,6-dicyanobenzene, 2,4,5,6-tetrakis(9H-carbazol-9-yl) isophthalonitrile; DMA = N,N-dimethyl acetamide, DMF = N,N-dimethyl formamide; TES = triethylsilyl; THF = tetrahydrofuran; TMS = trimethylsilyl. | |||||
| 1 | 0.5 | — | — | DMA | 0 |
| 2 | 0.5 | CsOAc | — | DMA | 7 |
| 3 | 0.5 | CsOAc | TMSCl | DMA | 22 |
| 4 | 0.5 | CsOAc | TMSCl | DMF | 25 |
| 5b | 0.5 | CsOAc | TMSCl | DMF | 24 |
| 6c | 0.5 | CsOAc | TMSCl | DMF | 17 |
| 7 | 0.5 | KHCO3 | TMSCl | DMF | 24 |
| 8 | 0.5 | CsOPiv | TMSCl | DMF | 32 |
| 9 | 0.5 | CsOPiv | TMSCl | THF | 38 |
| 10 | 0.5 | CsOPiv | TMSCl | CH3CN | 40 |
| 11 | 0.5 | CsOPiv | TESCl | CH3CN | 37 |
| 12 | 1.0 | CsOPiv | TMSCl | CH3CN | 60 |
| 13 | 2.0 | CsOPiv | TMSCl | CH3CN | 55 |
| 14d | 1.0 | CsOPiv | TMSCl | CH3CN | 50 |
| 15d | 1.0 | CsOPiv | — | CH3CN | 47 |
| 16e | 1.0 | CsOPiv | TMSCl | CH3CN | 0 |

We also evaluated alternative photocatalysts Ir-A and Ir-B,24 but these reactions provided somewhat lower yields (Table 1, entries 5 and 6). We then tested other bases (entries 7 and 8), solvents (entries 8–10), and silyl chlorides (entries 10 and 11) (see ESI Table S1† for complete details).
In these initial experiments, we used 3-oxetanone as the limiting substrate, by analogy to the conditions reported by Glorius.15 Next, we investigated alternative stoichiometric ratios (entries 11–13 and ESI Table S1†), and found that the reaction was most effective with equimolar amounts of the two substrates, providing a serviceable 60% yield (entry 12). Interestingly, the reaction also proceeded in the absence of CrCl3 (Table 1, entry 14), as well as in the absence of both CrCl3 and TMSCl (entry 15), albeit in lower yields; the mechanistic implications of this finding are discussed below. In contrast, control reactions performed in the absence of light (entry 15) or photocatalyst (see ESI Table S1†) did not afford any of the desired product.
Substrate scope and functional group tolerance of the Cr-mediated carboxylic acid-to-3-oxetanol transformation
Next, we investigated the scope of the Cr-mediated reaction using other N-aryl α-amino acid substrates (Fig. 2). A variety of other substrates were tolerated, including systems derived from alanine (3b), leucine (3c), phenylalanine (3d), tryptophan (3e), valine (3f) and isoleucine (3g). The reaction was also effective in a proline-derived system containing a tertiary amine (3h), as well as a corresponding acyclic N-methyl alanine-derived system (3i) and an indoline-derived system (3j). Notably, the reaction also proceeded efficiently with an α,α-dimethylglycine-derived substrate to form 3-oxetanol 3k having two adjacent quaternary carbons.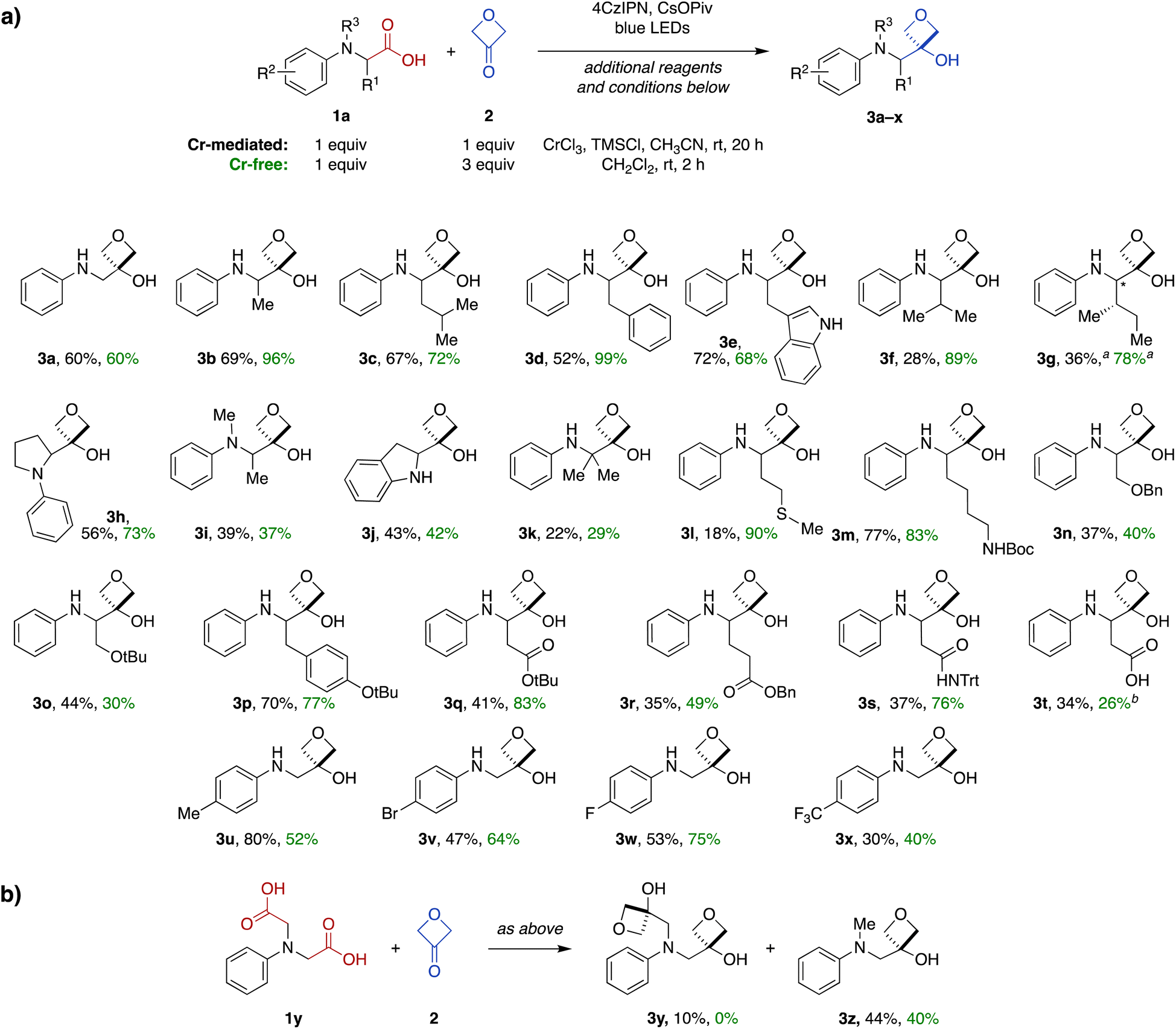 | ||
| Fig. 2 (a) Scope of the carboxylic acid-to-3-oxetanol transformation for N-aryl α-amino acid substrates under Cr-mediated (black yields) and Cr-free reaction conditions (green yields). (b) Conversion of diacid 1y to mono- (3y) and di-oxetanol (3z) products. Cr-mediated reaction conditions: 1 mol% 4CzIPN, 5 mol% CrCl3, 0.5 equiv. TMSCl, 1.2 equiv. CsOPiv, 0.8 M in CH3CN based on amino acid substrate 1, blue LED light, rt, 20 h. Cr-free reaction conditions: 2 mol% 4CzIPN, 1.2 equiv. CsOPiv, 0.5 M in CH2Cl2 based on amino acid substrate 1, blue LED light, rt, 2 h. adiastereomeric ratio = 1:1.4. bReaction carried out in isopropanol instead of CH2Cl2. | ||
A wide range of functional groups were tolerated in other substrates, including a methionine thioether (3l), Boc-protected lysine side chain (3m), serine benzyl and t-butyl ethers (3n and 3o), a tyrosine aryl ether (3p), protected aspartate and glutamate esters (3q and 3r), and an asparagine N-trityl amide (3s). Notably, a free carboxylic acid was also tolerated in an aspartate-derive system (3t), with transformation to the 3-oxetanol occurring regiospecifically at the main-chain carboxylate (34%).
We also investigated the influence of electronics of the aromatic ring using a variety of electron-donating and -withdrawing substituents (3u–x), but no clear reactivity trends were apparent across this series.
Finally, reaction of a symmetrical diacid substrate 1y was evaluated in the presence of 2 equiv. 3-oxetanone (2) (Fig. 2b). The major product was mono-oxetanol 3z with protodecarboxylation observed at the second site, while only 10% of the di-oxetanol 3y was recovered.
Development of Cr-free carboxylic acid-to-3-oxetanol transformation
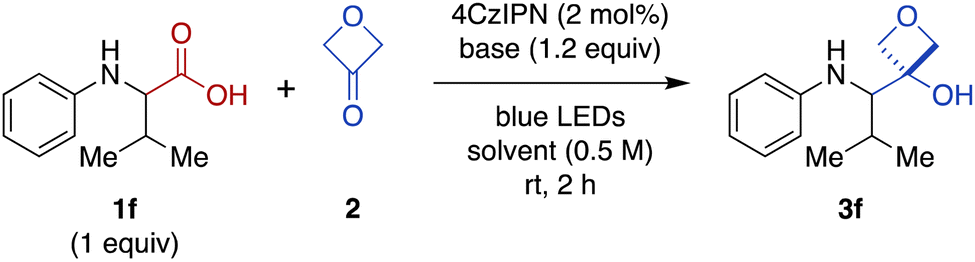
We were intrigued by the discovery above that the photoredox-catalyzed decarboxylative addition reaction also proceeded effectively in the absence of CrCl3 and TMSCl (Table 1, entry 14). Thus, we investigated further optimization of this Cr-free reaction using N-phenyl valine (1f), because the Cr-mediated reaction provided a low yield for this substrate (Figure 2, 28%). Omitting CrCl3 and TMSCl from the Cr-mediated reaction conditions afforded only a 9% of the desired product (Table 2, entry 1; see ESI Table S2† for complete details). We noted that there was little difference in yield when the reaction time was shortened from 20 h to 2 h (entry 2), facilitating further evaluation of reaction conditions. Investigation of various stoichiometric ratios (entries 2–5) showed that increasing 4CzIPN catalyst loading to 2 mol% and 3-oxetanone to 3 equiv. provided a modest increase in yield to 13%. Evaluation of other solvents (entries 5–8) afforded a dramatic increase in yield in CH2Cl2 (89%). Bases other than CsOPiv (entries 9 and 10) and photocatalysts other than 4CzIPN (entries 11 and 12) were far less effective. The reaction also remained dependent upon light (entry 13). We note that König and coworkers have previously reported photocatalyzed decarboxylative additions of arylacetic acids to aldehydes under similar conditions.14 However, application of the literature conditions (4CzIPN, Cs2CO3, DMA, LED, rt, 16 h) to our substrate 1f did not afford any of the desired 3-oxetanol product 3f.|
|
||||
|---|---|---|---|---|
| Entry | 2 (equiv.) | Base | Solvent | Yielda (%) |
| a Yields based on 1H-NMR analysis of crude reaction product in the presence of an internal standard, relative to N-phenyl valine (1f). b 1 mol% 4CzIPN. c 20 h reaction time. d Photocatalyst: Ir-A = [Ir{dF(CF3)2ppy}2(bpy)]PF6 = [2,2′-bipyridine-N1,N1′]bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate. e Photocatalyst: Ir-B = [Ir{dF(CF3)ppy}2(dtbpy)]PF6 = [4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate. f In absence of blue LED light. DCE = 1,2-dichloroethane; DIPEA = N,N-diisopropylethylamine. | ||||
| 1b,c | 1.0 | CsOPiv | CH3CN | 9 |
| 2b | 1.0 | CsOPiv | CH3CN | 8 |
| 3b | 2.0 | CsOPiv | CH3CN | 10 |
| 4b | 3.0 | CsOPiv | CH3CN | 11 |
| 5 | 3.0 | CsOPiv | CH3CN | 13 |
| 6 | 3.0 | CsOPiv | DCE | 57 |
| 7 | 3.0 | CsOPiv | CH2Cl2 | 89 |
| 8 | 3.0 | CsOPiv | i-PrOH | 78 |
| 9 | 3.0 | Na2CO3 | CH2Cl2 | 24 |
| 10 | 3.0 | DIPEA | CH2Cl2 | 21 |
| 11d | 3.0 | CsOPiv | CH2Cl2 | 43 |
| 12e | 3.0 | CsOPiv | CH2Cl2 | 30 |
| 13f | 3.0 | CsOPiv | CH2Cl2 | 0 |
Next, we investigated the substrate scope of the Cr-free reaction across the panel of N-aryl α-amino acid substrates (Fig. 2). In most cases, the Cr-free reaction provided higher yields of the 3-oxetanol products compared to those observed with the Cr-mediated reaction, in some cases dramatically so (e.g., 3d, 3f, 3g, 3l, 3q, 3s). Across the entire panel (3a–x), the average yield was 64% for the Cr-free reaction compared to 48% for the Cr-mediated reaction. In the case of the diacid substrate 1y, the Cr-free reaction provided the mono-oxetanol 3z exclusively. Overall, the Cr-free reaction provides significant advantages over the original Cr-mediated reaction with respect to efficiency (time, yield) and elimination of toxic and reactive reagents (CrCl3, TMSCl).
To expand the scope of this transformation beyond N-aryl α-amino acid substrates, we investigated Cr-free reactions of other amino acids. In preliminary experiments, we found that exposure of primary (phenylalanine), secondary (N-trityl glycine), and N-acylated (N-Boc-glycine, N-Cbz-proline, N-phthaloylglycine) α-amino acids to the reaction conditions did not afford any of the desired 3-oxetanol products (not shown). However, morpholine acetic acid was converted to the desired product, albeit with some bis and tris modification observed by MS, presumably at the ring carbons α to the amine. Selectivity for monofunctionalization was improved by decreasing 3-oxetanone stoichiometry from 3 equiv. to 1 equiv. With other slight modifications (changing solvent from CH2Cl2 to i-PrOH to improve solubility; increasing reaction time to 20 h), the desired 3-oxetanol 5a was obtained in 30% yield (Fig. 3). The reaction was also effective for systems containing N-methylpiperazine (5b), N-Boc-piperazine (5c), piperidine (5d), and an acyclic tertiary amine (5e), demonstrating tolerance of heteroatoms, protecting groups, and both cyclic and acyclic substrates.
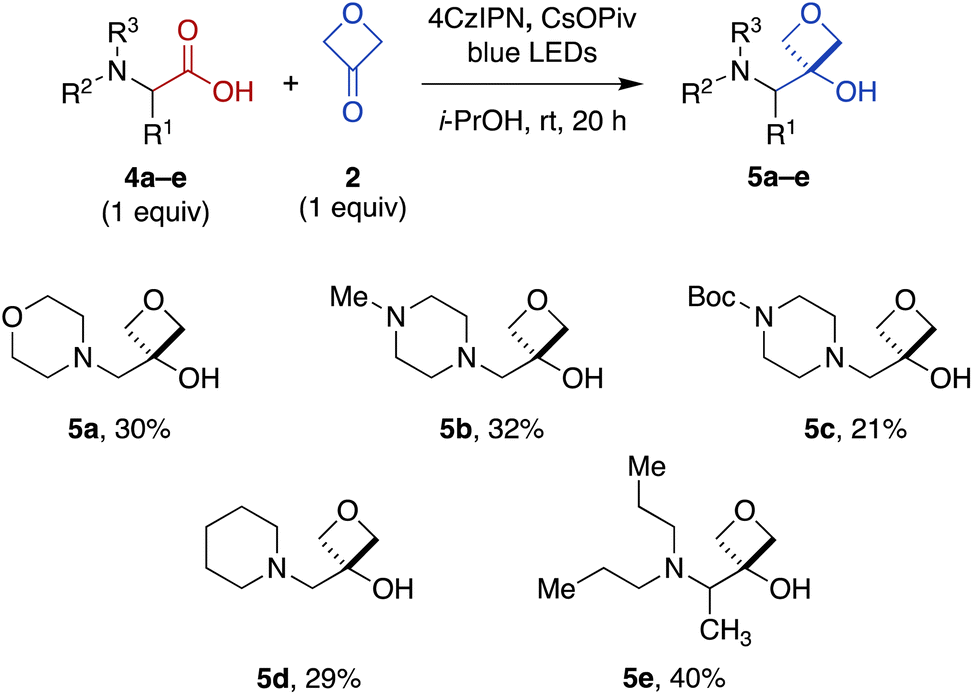 | ||
| Fig. 3 Direct carboxylic acid-to-3-oxetanol transformation for cyclic and acyclic N,N-dialkyl α-amino substrates under modified Cr-free reaction conditions. | ||
Mechanistic investigations
Next, we probed the mechanisms of the Cr-mediated and Cr-free transformations. We considered three possible mechanisms a priori: (1) addition of an α-amino carbanion (or Nozaki–Hiyama–Kishi alkyl-Cr intermediate) to 3-oxetanone,15,26 (2) addition of an α-amino radical to 3-oxetanone,39 or (3) radical–radical recombination of an α-amino radical and 3-oxetanone-derived radical.34First, to assess the reactivity of each of the substrates and reagents to photoactivated 4CzIPN, we conducted fluorescence quenching studies with N-phenylglycine (cesium salt) (1a), 3-oxetanone (2), CsOPiv, TMSCl, and CrCl3.40 Stern–Volmer analysis revealed that the quenching constant of the carboxylate 1a was substantially greater than that of the other reagents in both CH3CN and CH2Cl2.40 This supports a pathway in which the carboxylate substrate 1a reacts with the excited photocatalyst to undergo oxidative decarboxylation, forming an α-amino radical intermediate 6 (Fig. 4). Consistent with this mechanism, no product formation was observed when either transformation was carried out in the presence of TEMPO (1 equiv.).
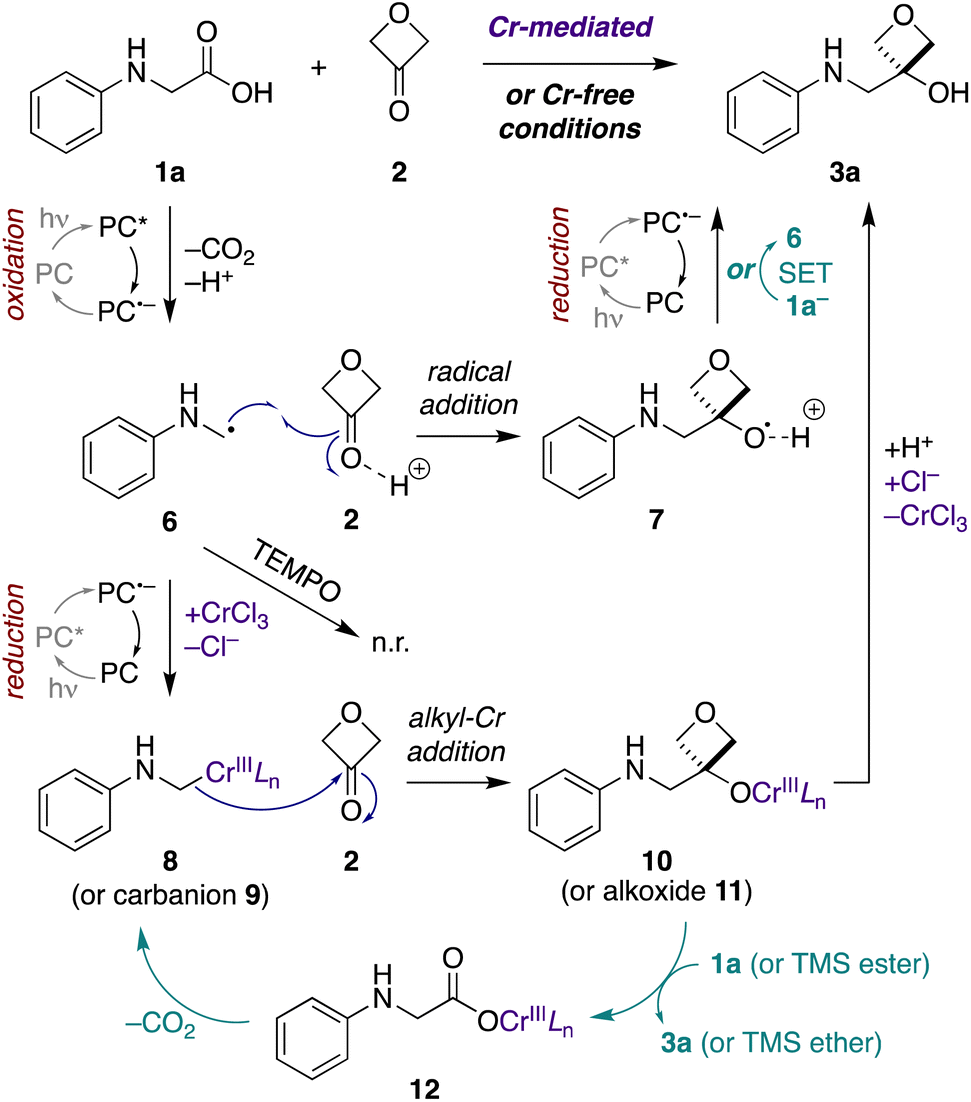 | ||
| Fig. 4 Possible mechanisms for Cr-mediated and Cr-free photoredox-catalyzed decarboxylative addition to 3-oxetanone (2). Initial photocatalytic oxidative decarboxylation of α-amino acid carboxylate 1a− forms α-amino radical 6. Under Cr-mediated conditions (purple), reduction to alkyl-Cr species 8 predominates, with nucleophilic addition to 3-oxetanone (2) forming Cr alkoxide 10. Chain propagation (teal) may occur by conversion of Cr-alkoxide 10 to Cr-carboxylate 12via either direct proton–Cr exchange or σ-bond metathesis of the corresponding TMS ester of 1a, followed by decarboxylation to regenerate alkyl-Cr species 8. Under Cr-free conditions, direct radical addition of α-amino radical 6 to Brønsted-acid activated 3-oxetanone (2) predominates, forming radical cation 7. The reaction may terminate by photocatalyzed reduction of 7 to the product 3a. Alternatively, chain propagation (teal) may occur through an SET event between 7 and carboxylate 1a− to furnish the product 3a and regenerate α-amino radical 6 (teal). A minor pathway in the Cr-mediated reaction may involve this same radical addition (6 + 2 → 7), while a minor pathway in the Cr-free reaction may involve a free carbanion/alkoxide mechanism (9 + 2 → 11). L = ligand, n.r. = no reaction, PC = photocatalyst, PC* = excited state, PC˙− = radical anion state. | ||
Next, to assess the possibility of a radical–radical recombination pathway (not shown), we measured the standard reduction potential (E1/2) of 3-oxetanone (2) using differential pulse voltammetry (DPV).40 We determined an E1/2 value of −2.51 V vs. SCE in CH3CN. In contrast, the redox potentials of 4CzIPN (E1/2[P˙+/P*] = −1.04 V; E1/2 [P/P˙−] = −1.21 V vs. SCE in CH3CN)41 are too small to drive reduction of 3-oxetanone (2) to the corresponding ketyl radical. Accordingly, radical–radical recombination pathways were ruled out for both reaction conditions.
In contrast, in the Cr-mediated reaction, the reduction potentials of 4CzIPN˙− are sufficient to reduce CrIIILn to CrIILn (E1/2 [CrIII/CrII] = −0.51 V vs. SCE in DMF).42 This reduced CrIILn can then intercept the α-amino radical 6 to generate alkyl-Cr intermediate 8, a step that has been extensively investigated in Nozaki–Hiyama–Kishi reaction manifolds,43 which may then add to 3-oxetanone (2) to form Cr alkoxide 10. The reaction may then terminate by protonation to form oxetanol 3a. Alternatively, it is possible that α-amino radical 6 may undergo direct addition to Brønsted acid-activated 3-oxetanone (2) to form radical cation 7, and there is precedent for such 1,2-additions.39,44 Subsequently, photocatalyzed reduction of the radical cation 7 would form oxetanol 3a, also completing the photocatalytic cycle.
Thus, to investigate these two possibilities, we carried out deuterium quenching experiments using the parent substrate N-phenyl glycine (1a) with 3-oxetanone (2) and/or methanol-d (CH3OD) (Table 3). We anticipated that, in the presence of methanol, the alkyl-Cr intermediate 8, but not the corresponding α-amino radical species 6, would be quenched to form the proto(deutero)decarboxylation products 13 (ESI Figure S1†). Under the standard Cr-mediated reaction conditions, we observed 60% of the 3-oxetanol product 3a and 14% protodecarboxylation product 13a (Table 3, entry 1). When 3-oxetanone was omitted and replaced by methanol-d, the yield of the proto/deuterodecarboxylation products 13a, b increased to 59% (combined), with 80% deuterium incorporation (entry 2), consistent with the alkyl-Cr addition pathway. Interestingly, when both 3-oxetanone (2) and methanol-d were included in the reaction, yields of both the 3-oxetanol product 3a and the protodecarboxylation products 13a, b were decreased (entry 3), suggesting that additional undesired reaction pathways become active under these conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditionsa | Electrophile | Quencher | 3a (%) | 13a + 13b (%) |
| a Cr-mediated reaction conditions: 1 mol% 4CzIPN, 5 mol% CrCl3, 0.5 equiv. TMSCl, 1.2 equiv. CsOPiv, 0.8 M in CH3CN based on amino acid substrate 1a, blue LED light, rt, 20 h. Cr-free reaction conditions: 2 mol% 4CzIPN, 1.2 equiv. CsOPiv, 0.5 M in CH2Cl2 based on amino acid substrate 1a, blue LED light, rt, 2 h. b Yields based on 1H-NMR analysis of crude reaction product in the presence of an internal standard, relative to N-phenyl glycine (1a). c Amino acid substrate 1a was deuterium exchanged with CH3OD prior to the reaction. d Percent deuterium incorporation (13b: R = D) shown in parentheses. | |||||
| 1 | Cr-mediated | 2 (1 equiv.) | — | 60 | 14 |
| 2 | Cr-mediatedc | — | CH3OD | — | 59 (80)d |
| 3 | Cr-mediatedc | 2 (1 equiv.) | CH3OD | 48 | 6 (57)d |
| 4 | Cr-free | 2 (3 equiv.) | — | 60 | 5 |
| 5 | Cr-freec | — | CH3OD | — | 47 (55)d |
| 6 | Cr-freec | 2 (3 equiv.) | CH3OD | 100 | — |

In the Cr-free reaction, quenching with methanol-d also resulted in formation of the proto/deuterodecarboxylation products 13a, b (entry 5), consistent with formation of an α-amino carbanion intermediate 9 (Fig. 4 and ESI Figure S1†). In contrast, when both 3-oxetanone (2) and methanol-d were included in the reaction, the yield of the 3-oxetanol product 3a increased to 100% (entry 6). This is contrary to expectation if the standard Cr-free reaction proceeds solely via a carbanion intermediate. Notably, Glorius and coworkers have proposed that photoredox-initiated intermolecular radical trapping by ketones and aldehydes may be promoted by Brønsted-acid activation of the carbonyl compound.39 Thus, the increased yield observed under these conditions (entries 3 and 6) may be attributed to such activation of 3-oxetanone by methanol. Unfortunately, the improved yield observed in Cr-free reaction in the presence of methanol did not prove generalizable to other N-aryl α-amino acid substrates (not shown).
The contrasting results in these competition experiments, in which the reaction conditions are significantly perturbed by omission of the electrophile or addition of a cosolvent, make it difficult to draw definitive conclusions regarding the predominant pathways under the standard Cr-mediated and Cr-free reaction conditions, and suggest that both are possible.
Lastly, we investigated the quantum yields of these transformations. Photon flux of the light source was determined using standard ferrioxalate actinometry.40 The quantum yield was then calculated by determining the amount of product formed in 3 min under the standard reaction conditions, and dividing by the photon flux. We observed quantum yields of 5.2 for the Cr-mediated reaction and 10.3 for the Cr-free reaction, indicative of chain propagation mechanisms under both conditions.
In the context of the Cr-mediated reaction, the reduction potentials of carboxylic acid 1a (E1/2 [1a+/1a] = +0.42 V vs. SCE in CH3CN)36 and CrIIILn (E1/2[CrIII/CrII] = −0.51 V vs. SCE in DMF)42 indicate that direct oxidative decarboxylation of 1a by Cr(III) would be thermodynamically unfavorable, making chain propagation via a redox mechanism unlikely.
An alternative possibility is that the alkyl-Cr species 8 is regenerated via a cycle in which the Cr-alkoxide intermediate 10 reacts with a new equivalent of the carboxylic acid substrate 1a to form Cr-carboxylate 12, which then undergoes metal-mediated decarboxylation to form alkyl-Cr species 8.45,46 Formation of Cr-carboxylate 12 could occur either via direct proton–Cr exchange with carboxylic acid 1a, or via σ-bond metathesis with the corresponding TMS ester, as postulated by Glorius and coworkers in related propagation reactions with trimethylsilylmethylamines,15 with subsequent desilylation of the resultant TMS ether to the product 3a. Consistent with the latter hypothesis, when TMSCl was omitted from the reaction, the quantum yield dropped to 1.6, indicating an important role in the propagation cycle.
In the Cr-free reaction, chain propagation may occur via SET between radical cation 7 and carboxylate 1a− (E1/2 [1a˙/1a−] = +0.42 V vs. SCE in CH3CN)36 to regenerate α-amino radical 6 and furnish 3-oxetanol product 3a. This electron transfer event should be thermodynamically favorable, based on the computationally determined redox potential of an alkoxy radical cation-to-alcohol conversion by Glorius and coworkers.39
Taken together, these results suggest that Cr-mediated reaction proceeds predominantly via the alkyl-Cr addition pathway (8 + 2 → 10), because omission of TMSCl results in a large decrease in quantum yield (5.2 to 1.6), indicating the importance of the Cr-based chain propagation cycle (10 → 12 → 8) compared to the SET chain propagation cycle (7 → 6) (ESI Fig. S2†). In contrast, the Cr-free reaction cannot involve the Cr-based chain propagation cycle (and the free carboxylate analogue of 12 would not decarboxylate spontaneously to form carbanion 9). Thus, the high quantum yield in that reaction (10.3) must be attributed to the SET propagation cycle, which can only arise from the radical addition pathway (6 + 2 → 7). Thus, while both reaction manifolds may be operative to some extent under both conditions, it appears that the Cr-mediated reaction proceeds mainly via the alkyl-Cr pathway and the Cr-free reaction proceeds mainly via the radical addition pathway.
Conclusions
In summary, by leveraging photoredox catalysis, we have successfully developed a method for direct conversion of α-amino acids to bioisosteric 3-oxetanols, thus avoiding the lengthy de novo synthesis approaches that have been used previously to access such motifs. Mechanistic investigations support a pathway involving initial oxidative decarboxylation to an α-amino radical species, which can then undergo direct radical addition to 3-oxetanone, or intermediate reduction to an α-amino alkyl-Cr or carbanion species followed by nucleophilic addition to 3-oxetanone, with the dominant reaction manifold dictated by the presence or absence of Cr. Notably, in both cases, chain propagation provides quantum yields >5. This methodology is applicable to a wide range of N-aryl α-amino acids, a motif which has been reported to have a variety of potential therapeutic applications in infectious disease, inflammation, neurodegeneration, and metabolic and gastrointestinal diseases.47–49 The substrate scope of the Cr-free reaction also includes N,N-dialkyl α-amino acid substrates. Efforts to expand the substrate scope further to other carboxylic acids are under active investigation in our lab. This direct conversion of carboxylic acids to 3-oxetanols should facilitate further investigation of these attractive bioisosteres in medicinal chemistry.Author contributions
A. M. V. D. R., C. S. N. E., A. M., and D. S. T. conceptualized the experiments; A. M. V. D. R. and C. S. N. E. performed the experiments with assistance from M. I. H.; A. M. V. D. R. and D. S. T. prepared the manuscript; A. M. V. D. R., C. S. N. E., A. M., and D. S. T. edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Tomislav Rovis (Columbia University), Prof. Tehshik Yoon and Dr Wesley Swords (University of Wisconsin–Madison), and Prof. Uttam Tambar (University of Texas, Southwestern Medical Center) for helpful discussions, Dr George Sukenick and Rong Wang (MSK Analytical NMR Core Facility) for expert NMR and mass spectral support, and Prof. Jonathan Goldberg (MSK) and Prof. Jason Lewis (MSK) for generous assistance with instrumentation. Financial support from the NIH (T32 CA062948-Gudas to A. M. V. D. R., T32 GM136640-Tan to C. S. N. E., and CCSG P30 CA008748 to S. M. Vickers) and MSK Basic Research Initiative (to A. M. V. D. R, A. M., and D. S. T.) is gratefully acknowledged.Notes and references
- F. Mao, W. Ni, X. Xu, H. Wang, J. Wang, M. Ji and J. Li, Molecules, 2016, 21, 1–18 Search PubMed
.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed
- C. Skonberg, J. Olsen, K. G. Madsen, S. H. Hansen and M. P. Grillo, Expert Opin. Drug Metab. Toxicol., 2008, 4, 425–438 CrossRef CAS PubMed
- A. Kalgutkar and J. Daniels, RSC Drug Discovery Ser., 2010, 1, 99–167 CAS
- C. Ballatore, D. M. Huryn and A. B. Smith III, ChemMedChem, 2013, 8, 385–395 CrossRef CAS PubMed
- P. S. Charifson and W. P. Walters, J. Med. Chem., 2014, 57, 9701–9717 CrossRef CAS PubMed
- M. V. Varma, B. Feng, R. S. Obach, M. D. Troutman, J. Chupka, H. R. Miller and A. El-Kattan, J. Med. Chem., 2009, 52, 4844–4852 CrossRef CAS PubMed
- Z. P. Qureshi, E. Seoane-Vazquez, R. Rodriguez-Monguio, K. B. Stevenson and S. L. Szeinbach, Pharmacoepidemiol. Drug Saf., 2011, 20, 772–777 CrossRef PubMed
-
N. Brown, in Bioisosteres in Medicinal Chemistry, 2012, pp. 1–14 Search PubMed
- P. Lassalas, K. Oukoloff, V. Makani, M. James, V. Tran, Y. Yao, L. Huang, K. Vijayendran, L. Monti, J. Q. Trojanowski, V. M. Y. Lee, M. C. Kozlowski, A. B. Smith, K. R. Brunden and C. Ballatore, Med. Chem. Lett., 2017, 8, 864–868 CAS
- T. Bach, K. Jödicke, K. Kather and R. Fröhlich, J. Am. Chem. Soc., 1997, 119, 2437–2445 CrossRef CAS
- D. R. Witty, G. W. J. Fleet, K. Vogt, F. X. Wilson, Y. Wang, R. Storer, P. L. Myers and C. J. Wallis, Tetrahedron Lett., 1990, 31, 4787–4790 CrossRef CAS
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed
- K. Donabauer, M. Maity, A. L. Berger, G. S. Huff, S. Crespi and B. König, Chem. Sci., 2019, 10, 5162–5166 RSC
- J. L. Schwarz, R. Kleinmans, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2020, 142, 2168–2174 CrossRef CAS PubMed
- G. Wuitschik, M. Rogers-Evans, K. Muller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 7736–7739 CrossRef CAS PubMed
- G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Muller, J. Med. Chem., 2010, 53, 3227–3246 CrossRef CAS PubMed
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Muller and E. M. Carreira, Angew Chem. Int. Ed. Engl., 2010, 49, 9052–9067 CrossRef CAS PubMed
- M. Rogers-Evans, H. Knust, J.-M. Plancher, E. M. Carreira, G. Wuitschik, J. Burkhard, D. B. Li and C. Guérot, Chimia, 2014, 68, 492–499 CrossRef CAS PubMed
- M. McLaughlin, R. Yazaki, T. C. Fessard and E. M. Carreira, Org. Lett., 2014, 16, 4070–4073 CrossRef CAS PubMed
- G. P. Möller, S. Müller, B. T. Wolfstädter, S. Wolfrum, D. Schepmann, B. Wünsch and E. M. Carreira, Org. Lett., 2017, 19, 2510–2513 CrossRef PubMed
- J. A. Burkhard, G. Wuitschik, J.-M. Plancher, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2013, 15, 4312–4315 CrossRef CAS PubMed
- S. Roesner, J. D. Beadle, L. K. B. Tam, I. Wilkening, G. J. Clarkson, P. Raubo and M. Shipman, Org. Biomol. Chem., 2020, 18, 5400–5405 RSC
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS PubMed
- A. Gil, F. Albericio and M. Álvarez, Chem. Rev., 2017, 117, 8420–8446 CrossRef CAS PubMed
- D. R. G. Brimage, R. S. Davidson and P. R. Steiner, J. Chem. Soc., Perkin Trans., 1973, 1, 526–529 RSC
- Z. Su, P. S. Mariano, D. E. Falvey, U. C. Yoon and S. W. Oh, J. Am. Chem. Soc., 1998, 120, 10676–10686 CrossRef CAS
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed
- Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed
- A. Millet, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464–13468 CrossRef CAS PubMed
- S. Pan, M. Jiang, J. Hu, R. Xu, X. Zeng and G. Zhong, Green Chem., 2020, 22, 336–341 RSC
- H. Wen, R. Ge, Y. Qu, J. Sun, X. Shi, W. Cui, H. Yan, Q. Zhang, Y. An, W. Su, H. Yang, L. Kuai, A. L. Satz and X. Peng, Org. Lett., 2020, 22, 9484–9489 CrossRef CAS PubMed
- H. Peng, S. Bi, M. Ni, X. Xie, Y. Liao, X. Zhou, Z. Xue, J. Zhu, Y. Wei, C. N. Bowman and Y.-W. Mai, J. Am. Chem. Soc., 2014, 136, 8855–8858 CrossRef CAS PubMed
- A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed
- A. Fürstner and N. Shi, J. Am. Chem. Soc., 1996, 118, 2533–2534 CrossRef
- L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS PubMed
- See ESI† for complete details.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS
-
K. Izutsu, in Electrochemistry in Nonaqueous Solutions, Wiley-VCH, 2009, pp. 89–110 Search PubMed
- Y. Katayama, H. Mitsunuma and M. Kanai, Synthesis, 2022, 54, 1684–1694 CrossRef CAS
- H.-M. Huang, P. Bellotti and F. Glorius, Acc. Chem. Res., 2022, 55, 1135–1147 CrossRef CAS PubMed
- T. Galcera, X. Jouan and M. Bolte, J. Photochem. Photobiol., A, 1988, 45, 249–259 CrossRef CAS
-
G. B. Deacon, S. J. Faulks and G. N. Pain, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1986, vol. 25, pp. 237–276 Search PubMed
- N. B. Tien, N. P. Buu-HoÏ and N. D. Xuong, J. Org. Chem., 1958, 23, 186–188 CrossRef CAS
- D. Schuster, M. Zederbauer, T. Langer, A. Kubin and P. G. Furtmüller, J. Enzyme Inhib. Med. Chem., 2018, 33, 1529–1536 CrossRef CAS PubMed
- N. Tanaka, T. Tamai, H. Mukaiyama, A. Hirabayashi, H. Muranaka, S. Akahane, H. Miyata and M. Akahane, J. Enzyme Inhib. Med. Chem., 2001, 44, 1436–1445 CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data for all new compounds. See DOI: https://doi.org/10.1039/d3sc00936j |
| This journal is © The Royal Society of Chemistry 2023 |