
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriarylamines as catalytic donors in light-mediated electron donor–acceptor complexes†
Durbis J.
Castillo-Pazos‡
 ab,
Juan D.
Lasso‡
ab,
Ehsan
Hamzehpoor‡
a,
Jorge
Ramos-Sánchez
a,
Jan Michael
Salgado
ab,
Gonzalo
Cosa
ab,
Dmytro F.
Perepichka
a and
Chao-Jun
Li
*ab
ab,
Juan D.
Lasso‡
ab,
Ehsan
Hamzehpoor‡
a,
Jorge
Ramos-Sánchez
a,
Jan Michael
Salgado
ab,
Gonzalo
Cosa
ab,
Dmytro F.
Perepichka
a and
Chao-Jun
Li
*ab
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bFRQNT Centre for Green Chemistry and Catalysis, McGill University, Montreal, QC H3A 0B8, Canada
First published on 14th March 2023
Abstract
Recently, photochemistry of Electron Donor–Acceptor (EDA) complexes employing catalytic amounts of electron donors have become of interest as a new methodology in the catalysis field, allowing for decoupling of the electron transfer (ET) from the bond-forming event. However, examples of practical EDA systems in the catalytic regime remain scarce, and their mechanism is not yet well-understood. Herein, we report the discovery of an EDA complex between triarylamines and α-perfluorosulfonylpropiophenone reagents, catalyzing C–H perfluoroalkylation of arenes and heteroarenes under visible light irradiation in pH- and redox-neutral conditions. We elucidate the mechanism of this reaction using a detailed photophysical characterization of the EDA complex, the resulting triarylamine radical cation, and its turnover event.
1 Introduction
Modern photocatalysis explores myriad ways of accelerating chemical reactions by channeling energy from a light source. Currently, the paradigm for light-powered reactions in industry and academia is mostly represented by the use of transition metal complexes—for example, iridium and ruthenium photocatalysts—which pose challenges of elevated cost and contamination of the final product with often toxic metals.1 Therefore, it is highly desirable to find alternative photocatalytic approaches based on commercially available organic (metal-free) molecules. Complementary to the many advances in the field of organic photoredox catalysis in the last decade,2,3 the synthetic potential of EDA complexes has become of great recent interest through the pioneering studies by Kochi and later by the groups of Melchiorre and Chatani.4–6EDA complexes are formed by association of an electron donor molecule and an acceptor molecule in the ground state. They are characterized by a reduction of the HOMO–LUMO gap compared to individual components, and a new charge-transfer absorption band often appears in the UV-vis spectra. Irradiation of EDA complexes with visible light can lead to a single-electron transfer (SET), which upon the irreversible cleavage of a leaving group in either the donor or the acceptor, can be exploited to trigger further radical reactivity.7 This SET enables new chemical transformations under mild conditions with substrates that do not absorb visible light, and without the need for an exogenous photocatalyst. However, a caveat of traditional EDA-mediated synthetic platforms comprises the need for stoichiometric amounts of donor and acceptor, where both components end up in the product structure.8–10 This drawback can be overcome by a catalytic approach to EDA complex formation, in which either the donor or acceptor is added in a catalytic amount, decoupling the complexation/photoactivation steps from the substrate functionalization.11
In 2019, Fu et al. reported a modified EDA-mediated system consisting of an electron-deficient acceptor (N-(acyloxy)phthalimides) in association with a catalytic donor (triphenylphosphine) and sodium iodide as an additive, where a substrate of interest traps the resulting radical species, and the donor is later regenerated by a one-electron reduction in a turnover event (Fig. 1a).12 Using this catalytic EDA-system they promoted the photodecarboxylative alkylation of silyl enol ethers and heteroaromatics. Since this seminal work, a limited number of catalytic donors for EDA-mediated transformations have been reported (Fig. 1b), categorized into ionic and neutral EDA systems. Milestone examples of the former category include Melchiorre's decarboxylative Giese additions employing dithiocarboxylate anions,13 the use of tetramethylethylenediamine (TMEDA) and phosphonium iodide salts for the monofluoromethylation of alkenes,14 Stephenson's 2-methoxynaphthalene-mediated trifluoromethylation and alkylation of aromatics,15 and the synthesis of benzo[b]phosphine oxides by the photoredox catalyst Eosin Y.16 On the other hand, more scarce reports on neutral catalytic EDA systems include the quinuclidine-catalyzed aminodecarboxylation of tetrachlorophthalamide esters,17 and hydroquinones/organophosphines in the halogen bonding-mediated modification of arenes and heterocycles,18,19
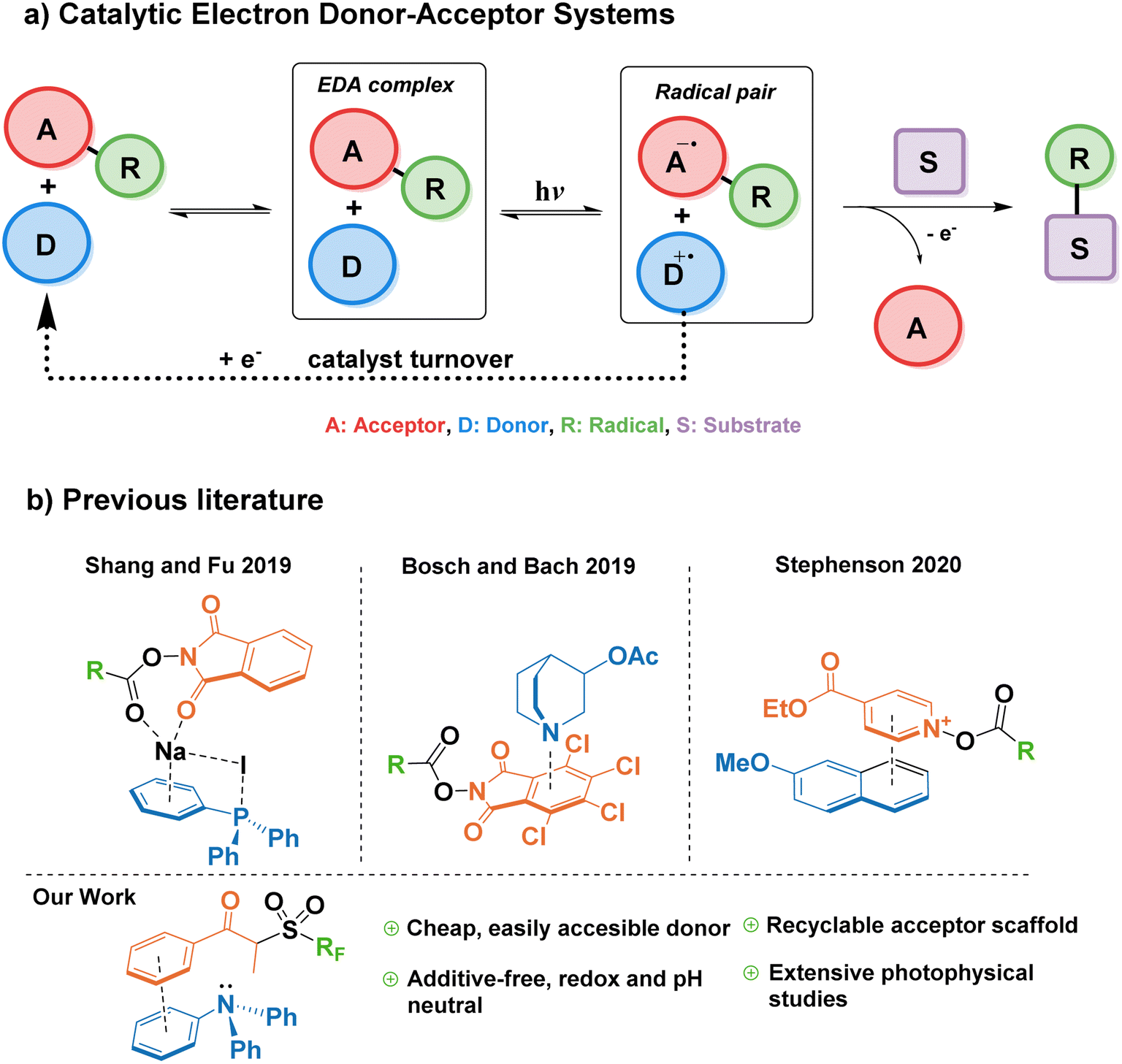 | ||
| Fig. 1 Overview of catalytic EDA complexes in synthetic chemistry. | ||
While all these reports showcase the expansion of the catalytic donor toolbox in the last three years, there still exist challenges that hamper their efficacy when compared to transition metal photocatalysts; for example, the instability of most donors in the catalytic cycle, the need for superstoichiometric amounts of additives (i.e. acids, bases, or inorganic salts), long reaction times (>24 h), or catalyst loadings above 10 mol%. Therefore, it is important to explore new functionalities capable of addressing such limitations.
Triarylamines are among the most studied electron donors for their versatility, stability, and the easy tunability of the electronic levels by permutations of the aryl groups synthesized in one step by well-established methodologies. The parent molecule of this family, triphenylamine (TPA), is widely used in materials chemistry as a redox-active scaffold, spanning numerous applications such as dyes, semiconductors, or electrochemical redox mediators.20–24 In its ground state, TPA is also one of the least basic organic amines, making it compatible with a wide range of functional groups, useable in pH-neutral reactions, and orthogonal to acid–base processes within the same system.25 However, until a recent report by Procter and co-workers26—published after the preparation of this manuscript—, triarylamines remained unexplored as catalytic donors in a general synthetic platform for radical generation and functionalization of arenes. In their work, a naphthyldiphenylamine donor proved successful in the alkylation and cyanation of arenes from their corresponding thianthrenium salts. Nevertheless, in addition to their elegant C–H activation for the formation of aryl radicals—challenging to form without the use of highly reducing photoredox catalysts such as 10-phenylphenothiazine27—, it would also be advantageous for the field to explore the capability of EDA catalytic triarylamine donors in the context of Minisci-type reactions onto aromatic substrates.28
For the reasons listed above, we were compelled to study triphenylamine—the simplest of triarylamines, until now synthetically relegated to cyclization and dimerization reactions—as a suitable catalytic donor that is not only readily available, economically accessible, and synthetically tunable; but also safe, efficient and obtainable from renewable feedstocks. In this work, we describe the design of an EDA system utilizing TPA as a catalytic donor, and study the mechanism behind its association with a propiophenone redox tag as a suitable acceptor for the first time. Additionally, we explore the catalytic turnover ability of triarylamines towards the innate perfluoroalkylation and trifluoromethylation of electron-rich arenes and biologically-relevant scaffolds as a benchmark reaction. Finally, we provide comprehensive photophysical studies that describe the catalytic cycle in detail, establishing a general reference framework for the design of future EDA-mediated catalytic donors and acceptors.
2 Results and discussion
2.1. Design of a catalytic system: triphenylamine in the context of β-cleavage reactions
The first step in the design of an acceptor complementary to TPA in EDA catalysis was to find a suitable “recognition element” pertaining to an electron-deficient moiety capable of interacting with the lone pair of the amine. Kannappan and coworkers studied the charge transfer complexes between arylamines and aryl ketones and showed that the thermodynamic stability of such complexes is aided by secondary interactions such as dipole–dipole forces or π-stacking (Fig. 2a).29 Reactions involving ketone β-cleavage have been studied previously under high intensities of ultraviolet light (450 W Xenon lamp) with the presence of a suitable leaving group such as a halogen, acetoxy group, or sulfones.30–33 Thus, we hypothesized that the addition of an electron (photoreduction) into such scaffolds would lead to the generation of a stabilized anion and a radical species (Fig. 2b), providing an opportunity to study the capability of triphenylamine as a catalytic electron donor in a general synthetic platform.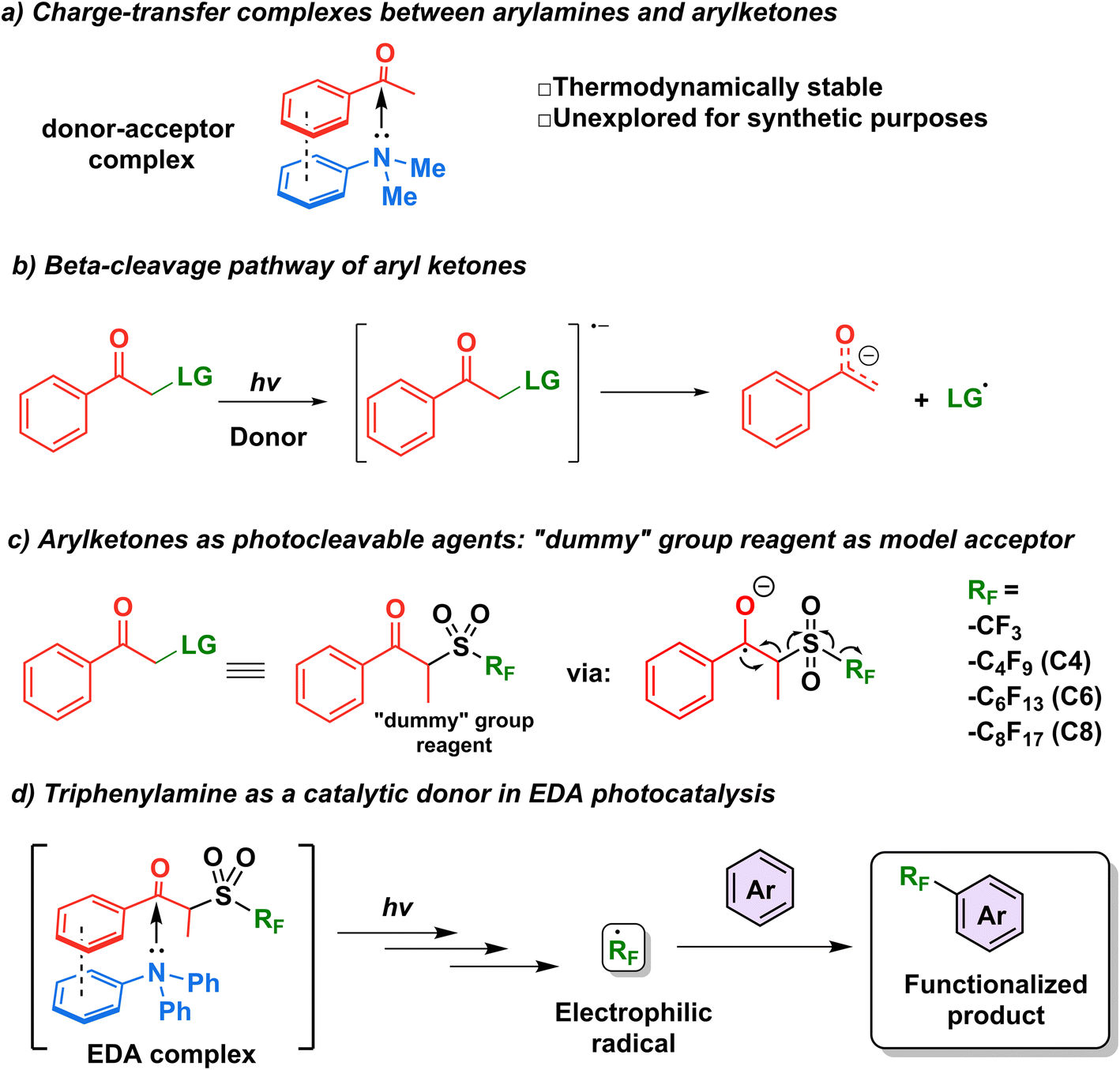 | ||
| Fig. 2 Catalytic EDA system design and application. | ||
Recently, the β-cleavage of ketones was included in the design of a series of alkylating reagents reported by our laboratory, exploiting propiophenone as a photocleavable fragment (Fig. 2c).34 In this work, inspired by García-Garibay's engineering of Norrish I reactions35,36 and the captodative effect,37,38 these reagents were capable of homolytic cleavage into three elements upon irradiation of light with a xenon arc lamp (>295 nm): a propiophenone radical—dubbed “dummy” due to its stability—, a molecule of SO2 gas, and the radical of interest, which is then trapped by the aromatic substrate to be functionalized innately.34 Such propiophenone-based reagents have several advantages versus the well-established perfluoroalkyl iodides: they are thermo- and photostable, and can be used under redox- and pH-neutral condition in late-stage functionalization. These “dummy group” reagents allow for easy installation of perfluoroalkyl (perfluorooctyl, perfluorohexyl, perfluorobutyl and trifluoromethyl) group and also alkyl radicals (such as isopropyl and n-hexyl) on electron-rich aromatics and various biologically relevant substrates.39 However, while a diverse set of perfluoroalkyl and alkyl radicals were successfully utilized in this approach, the scope of the substrate is limited to molecules with minimal absorptions overlap to that of the “dummy” group (<290 nm) to achieve high photoconversion yields. It is noteworthy that while the literature pertaining to perfluoroalkylation reactions is vast,40–44 methods for the metal-free C–H perfluoroalkylation of aromatic molecules through the use of EDA complexes in the catalytic regime remain limited.18,19,45
We thus hypothesized that these dummy group reagents constituted the ideal candidates as the benchmark reaction to test TPA's capability as a catalytic donor (Fig. 2d): firstly, they contain a benzoyl moiety that can be used as a recognition element, secondly, their structure is designed for a facile cleavage leading to the generation of the desired radicals, and thirdly, the formation of an EDA complex with TPA would allow for the photoactivation to be initiated under milder conditions with the use of blue LEDs rather than a high energy ultraviolet light source.
3 Mechanistic studies: exploring the EDA catalytic cycle
EDA complexes are evidenced by the appearance of a new absorption band typically within the visible range of light, beyond the absorption of pure donor or acceptor. Upon mixing solutions of TPA in MeCN with ketone C8 (or C6) −1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio, 0.02 M and 0.1 M, respectively—an immediate shift in the absorption with a new broad feature (attributed to charge transfer) was observed compared to the absorption profile of the separate components (Fig. 3a and c). Furthermore, the sequential dilution of a mixture of TPA (0.005 M) and C6 (0.02 M) showed a quadratic concentration dependency for the charge transfer band consistent with its bimolecular complexation origin (λmon = 427 nm; Fig. S2a and c†), whereas that of the π–π* band (λmon = 295 nm; Fig. S2b and d†), linearly decreased upon dilution as is to be expected based on Beer–Lambert law, further confirming the bimolecular nature of the new charge transfer band. Overlapping the emission profile of our 427 nm LED lamp suggests that this freshly mixed solution absorbs light from the photoreactor as a result of the EDA complex formation, having ca. 20% light absorption (absorbance of 0.097) at 427 nm (maximum of our photoreactor incident light) for the given concentrations. Furthermore, after irradiating the sample under blue light for ten minutes, the sample's absorption surged considerably by reaching 64%, and a new absorption band was formed at ∼660 nm. We attributed this new species to triphenylamine radical cation (TPA˙+), previously observed upon SET events.46,47
:5 ratio, 0.02 M and 0.1 M, respectively—an immediate shift in the absorption with a new broad feature (attributed to charge transfer) was observed compared to the absorption profile of the separate components (Fig. 3a and c). Furthermore, the sequential dilution of a mixture of TPA (0.005 M) and C6 (0.02 M) showed a quadratic concentration dependency for the charge transfer band consistent with its bimolecular complexation origin (λmon = 427 nm; Fig. S2a and c†), whereas that of the π–π* band (λmon = 295 nm; Fig. S2b and d†), linearly decreased upon dilution as is to be expected based on Beer–Lambert law, further confirming the bimolecular nature of the new charge transfer band. Overlapping the emission profile of our 427 nm LED lamp suggests that this freshly mixed solution absorbs light from the photoreactor as a result of the EDA complex formation, having ca. 20% light absorption (absorbance of 0.097) at 427 nm (maximum of our photoreactor incident light) for the given concentrations. Furthermore, after irradiating the sample under blue light for ten minutes, the sample's absorption surged considerably by reaching 64%, and a new absorption band was formed at ∼660 nm. We attributed this new species to triphenylamine radical cation (TPA˙+), previously observed upon SET events.46,47
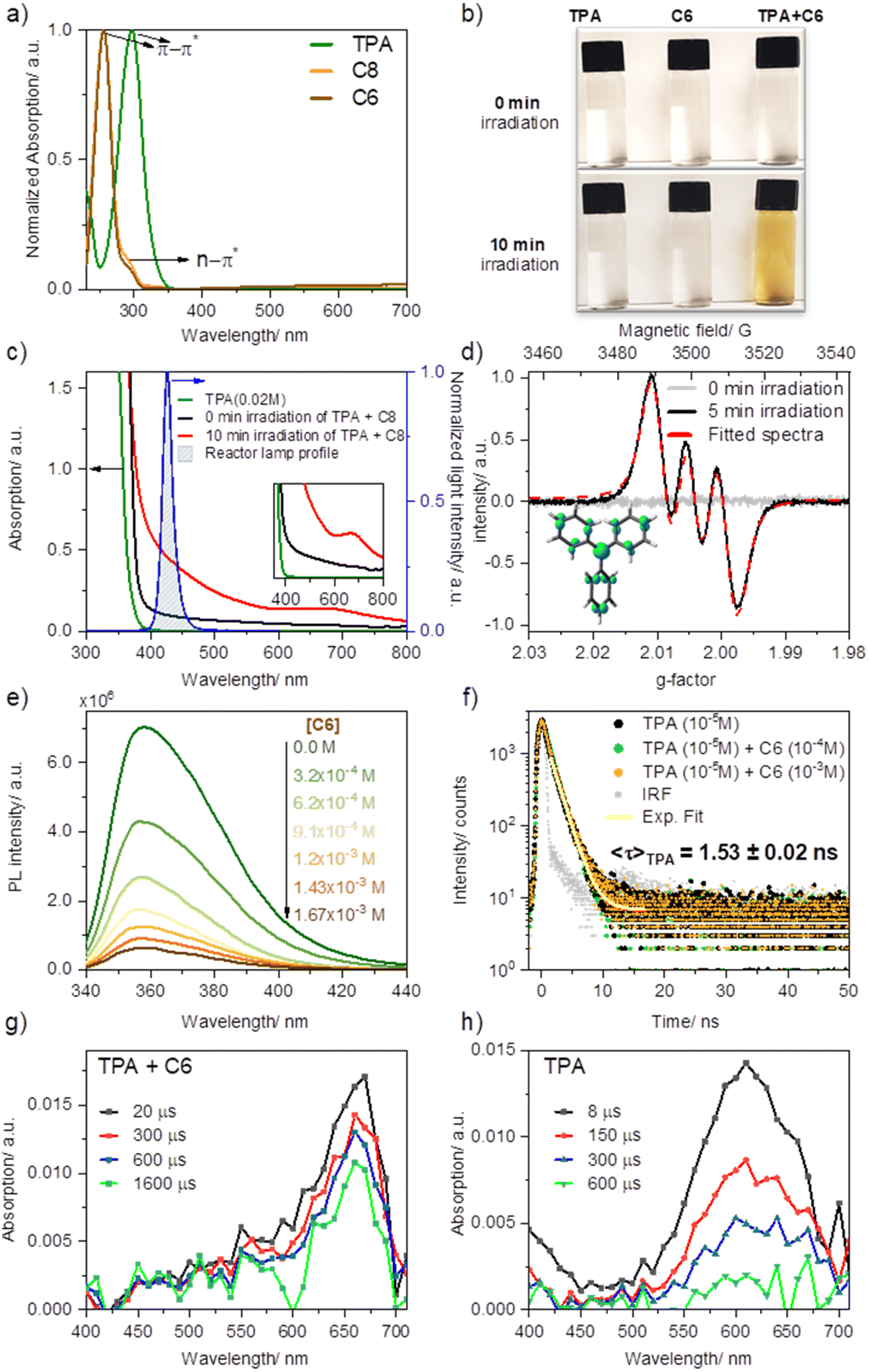 | ||
| Fig. 3 Mechanistic studies on triphenylamine as an EDA (a) UV-visible spectra of TPA (1 × 10−5 M) and the perfluoroalkylating reagents C8 and C6 (1 × 10−5 M) (b) Formation of color by the mixture of TPA (0.02 M) and C8 (0.1 M, 5 equiv.) solutions, freshly made (0 min irradiation) and after 10 min irradiation using blue light (10 min, 40 W Kessil lamp 427 nm). (c) Absorption evolution of the EDA complex formed between TPA (0.02 M) and C8 (0.1 M, 5 equiv.) and its absorption overlap with the employed light source. (d) EPR spectrum of a mixture of C8 with TPA at room temperature before and after shining light (5 min, 40 W Kessil lamp 427 nm). Below the spectra: spin distribution for TPA radical cation calculated by density functional theory (DFT; B3LYP 6-31g(d)) as a visual reference of spin delocalization. (e) Quenching of TPA (1 × 10−5 M) by forming an EDA complex with various concentrations of C6. (f) Time-correlated single-photon counting spectra of TPA in the absence of perfluoroalkylating agent (black), 10 equiv. C6 (green) and 100 equiv. C6 (orange) (1 × 10−4 M and 1 × 10−3 M, respectively); the decreasing emission intensity and the constant lifetime upon the addition of the quencher indicate a static quenching. (g) Transient absorption spectrum of TPA (0.005 M) and C6 (0.02 M) upon excitation with a 355 nm laser yielding the TPA radical cation. (h) Transient absorption of TPA (0.005 M) showing the formation of 4a,4b-dihydrocarbazole in the ground state upon excitation with a 355 nm laser. | ||
Consistent with TPA˙+ formation upon irradiation of the TPA and acceptor mixture in acetonitrile at room temperature (5 minutes of irradiation under blue light, 427 nm), a strong and long-lived electron paramagnetic resonance (EPR) signal was recorded confirming the presence of a radical species, Fig. 3d. Fitting of the observed EPR spectra suggests a hyperfine coupling (hfc) attributed to a single electron coupled with an N nucleus (S = 1, hfc ∼8.5 G) and nine H nuclei (3Hpara with hfcH(para) ∼1.9 G and 6Hortho, hfc ∼1.4 G), which is in line with the previous reports48–52 and the density functional theory calculations (Fig. 3e, Table S1†). Such observation not only confirmed the in situ formation of the TPA radical cation but also substantiated the formation of an EDA complex, as discussed in the work of Stamires and Turkevich.53 Importantly, no EPR signal was observed in a freshly mixed solution of TPA and the acceptor before irradiation, wherefrom a relatively weak charge transfer between the donor (TPA) and acceptor (perfluoroalkyl) may be inferred.53
Quenching of TPA emission in the presence of increasing amounts of acceptor (C6) provided further insights into the nature of their interaction, studied through steady-state and time-resolved fluorescence quenching experiments (Fig. 3e and f). Here we sought to reveal whether the TPA interaction with the dummy group existed a priori of their excitation (static quenching, no diffusion required) or rather, following photoexcitation of TPA, the electronically excited donor and the acceptor (C6) encounter in solution (dynamic quenching). The drop in intensity of TPA with increasing C6 (Fig. 3e) was analyzed using Stern–Volmer formula. A linear relationship was obtained with a Stern–Volmer quenching constant of KSV ∼3.1 × 103 M−1. Combining this value and the fluorescence decay lifetime of TPA in acetonitrile (1.53 ns, Fig. 3e), a bimolecular quenching rate constant kq = 2.1 × 1012 M−1 s−1 (Fig. S4a†) was estimated. The resulting kq far exceeds the diffusion-controlled rate constant value in acetonitrile, i.e. 2.0 × 1010 M−1 s−1,54 indicating that “dynamic quenching” is not a major pathway involved in TPA-C6 interaction. Consistent with a preformed complex, static emission quenching was confirmed in time-resolved emission studies by the lack of changes in the TPA emission decay lifetime, recorded from time-correlated single-photon counting (TCSPC) upon the addition of C6 up to 10−3 M concentrations. In the case of a dynamic process, both intensity and emission decay lifetime would vary proportionally to the increasing C6 concentration. Analysis of the quenching plot using static equilibrium conditions and assuming no emission from the EDA complex (see ESI Fig. S4, eqn S(2)†) suggests the formation of a 1:1 ratio complex with a binding constant of K = 1.04 ± 0.40 × 104 M−1, which is significantly higher than the previously studied charge transfer complexes between a similar system of N,N-dimethylaniline and acetophenone derivatives.29,55
To gain further mechanistic insight, we also conducted transient absorption studies via laser flash photolysis upon 355 nm excitation of TPA in Ar purged acetonitrile solutions, alone or in the presence of C6. Following excitation in the presence of C6 we observed the formation of a transient band with a peak at 660 nm consistent with that previously reported for TPA˙+(Fig. 3g).46,56 Formation of TPA˙+ was observed immediately within excitation; however, we noted that the high sample absorption at the excitation wavelength generated a thermal wave that prevented accurate spectral assignments within the first ∼1 μs upon excitation. Notably, excitation of TPA in the absence of C6 but under otherwise identical conditions, rendered the spectra of 4a,4b-dihydrocarbazole in the ground state (DHC0) with a characteristic absorption at 610 nm (Fig. 3h). This transient species is consistent with the previously reported photo-processes of TPA.57–59 In short, for the latter, upon excitation, rapid intersystem crossing (ISC) from the singlet excited state (1TPA*) generates TPA in the triplet excited state (3TPA*) with a quantum yield of 0.9. The 3TPA* in turn, cyclizes to quantitatively generate 3DHC*, which decays into its ground state upon intersystem crossing (DHC0). In the absence of oxygen, DHC0 converts back to TPA. Importantly, while we were unable to excite at a wavelength where only the EDA complex absorbs, our LFP results position SET as an active mechanism operating when TPA is in the presence of C6. Here TPA˙+ is unequivocally observed within the first few microseconds following excitation. Combined with the absorption and fluorescence studies that indicate the formation of an EDA complex and the radical formation at lower excitation energies (427 nm) measured by EPR, transient absorption studies altogether confirm that electron transfer originates from the complex and not from the excitation of TPA alone.
The observed high stability of the TPA radical cation in the solution (∼3 h in air, as confirmed by EPR analysis) is likely due to its low concentration, which suppresses the coupling of radical cations, as studied by Creason, Wheeler, and Nelson.60 Moreover, we see no evidence for the formation of N,N,N′,N′-tetraphenyl benzidine (TPB)—the product of dimerization between two TPA units—neither by GC-MS of reaction mixtures nor by fluorescence spectroscopy (Fig. S3a†),46,61,62 ruling out the possibility of TPB being a secondary catalyst formed in situ by photooxidation of TPA.
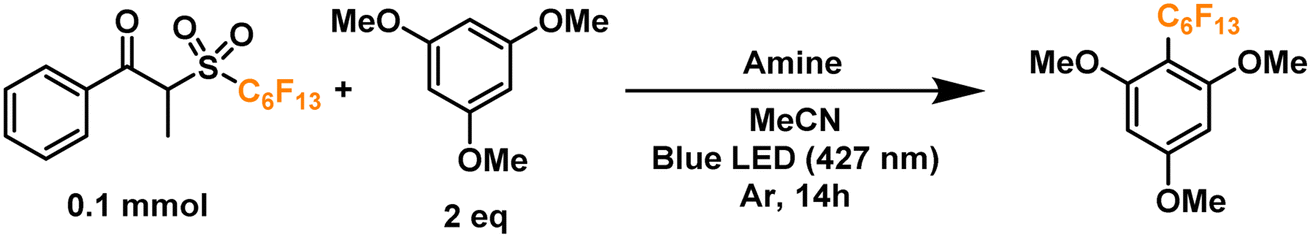
Finally, we proceeded to confirm the formation of the desired perfluoroalkyl radicals with a trapping experiment (Fig. 4). These radicals are electrophilic in nature, making them more prone to addition to electron-rich aromatics. The first trial using 1,3,5-trimethoxybenzene as the trapping substrate provided an NMR yield of 85% when irradiated overnight (14 h) in the presence of a 25 mol% load of TPA with respect to the C6 perfluoroalkylating reagent. The formation of propiophenone was confirmed by GCMS and NMR, corroborating the proposed cleavage pathway for the C6 reagent. Regarding controls for this system, light irradiation is essential for the formation of product given the yields obtained when running a dark reaction, both at room temperature and 40°C—none and traces, respectively. Most importantly, when a TPA-free reaction was run under light irradiation, less than 10% of the product was formed, indicating that the functionalization of the aromatic substrate is clearly driven by the addition of the donor. Lastly, we investigated the tolerance of the EDA catalytic cycle to oxygen. The reaction still proceeds in air, albeit with a 50% decrease in the yield, which we attribute to possible interference of oxygen in the radical cascade derived from the cleavage of the perfluoroalkylating reagent furthermore, since commercially available triphenylamines are usually synthesized through metal-mediated cross-coupling reactions (e.g. traditional Ullmann coupling), we wanted to rule out the possibility of trace metals such as Cu and Pd promoting the reaction. For this, an extra control was run with sublimated TPA, giving the same yield as the commercial-grade reagent (98% purity).
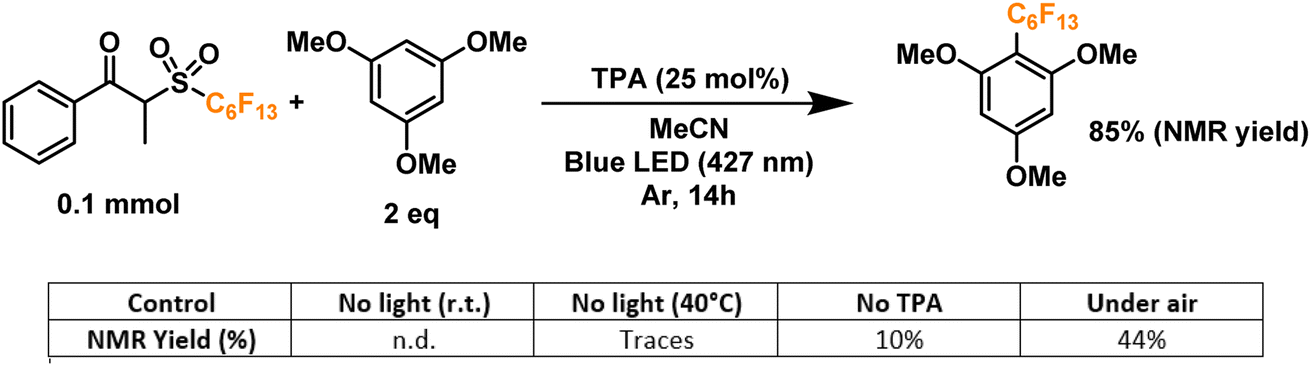 | ||
| Fig. 4 Radical trapping and control experiments. | ||
With this information in mind, a proposed mechanism is depicted in Fig. 5. When present in solution, our TPA catalyst I and the propiophenone-containing perfluoroalkylating reagent II form a charge transfer complex III (absorbing in the visible spectra). Upon excitation with blue light at 427 nm, SET creates a radical pair of the triphenylaminium radical cation and the radical anion of the perfluoroalkylating reagent. Due to a weakened C–S bond and driven by entropy, this radical anion breaks into three components: a resonance-stabilized enolate, an SO2 molecule, and the free perfluoroalkyl radical. The latter thereafter adds to the aromatic substrate V, forming the intermediate VI which is then oxidized by the triphenylaminium radical cation releasing the functionalized product and regenerating the TPA catalyst.
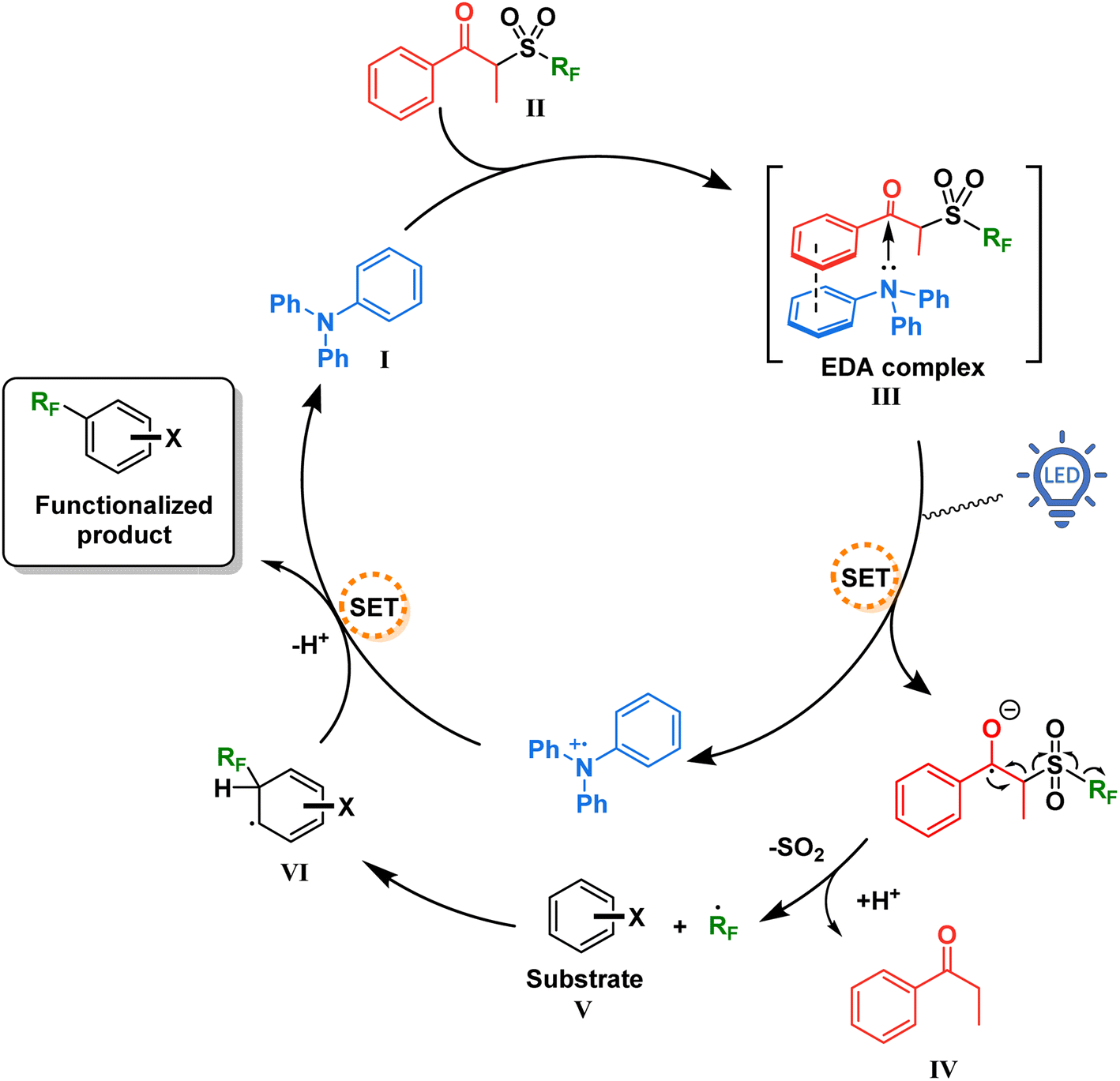 | ||
| Fig. 5 Proposed mechanism for the catalytic EDA complex formation between TPA and the perfluoroalkylating reagent. | ||
4 Optimization of conditions and expansion of scope: visible-light promoted perfluoroalkylation and trifluoromethylation of aromatics
Having placed the pieces together in our mechanistic puzzle, we proceeded to optimize the proposed conditions and study the scope, while using our previously reported UV-light promoted perfluoroalkylating methodology as a ref. 39. In the original report, 2 equivalents of the propiophenone-based perfluoroalkylating reagent were used to functionalize electron-rich aromatics under irradiation of a 300 W xenon arc lamp with a long-pass filter of >295 nm, reaching almost quantitative NMR yields for the perfluorohexylation of 1,3,5-trimethoxybenzene.39 Here we aimed to attain the same yields albeit with an electron donor—preferably in catalytic amounts—and using commercial blue LEDs.Firstly, taking the conditions shown in Fig. 4 as our starting point (Table 1, entry 1), we explored the use of other amines to study their efficiency and behavior compared to TPA (entries 2–6). Starting with N,N-dimethylaniline (N,N-DMA) as a reference, the yield of functionalization decreased to 37%, possibly due to both a lower affinity towards the C6 reagent and a lower turnover number. Also, despite its structural similarity with TPA, N-methyldiphenylamine (MeDPA) presented a lower yield at 52%. Such observation could be explained by the known reactivity of alkylamines in photocatalysis, where upon abstraction of one electron α-aminoalkylradicals are readily formed.63 Trials with amines bearing no aromatic substituents (e.g., Hünig's base) resulted in even lower yield of 25%, suggesting that, while the amine could reduce the reagent, it acts as a stoichiometric (sacrificial) donor. Additionally, when switching to another strong organic base such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)—traditionally known as a suitable electron donor in thermochemistry–the reaction was shut down, suggesting that the donating power of the amine may not depend on its basicity in the ground state. Finally, the highly reducing 10-phenylphenothiazine (PTH) photocatalyst showed lower selectivity towards the monoperfluorohexylated product with 21%, in addition to a 39% yield for the diperfluorohexylated product. These results confirm that the reactivity between TPA and propiophenone-based redox tags is significantly different from previous literature reports employing aryl amines, including the closely related phenothiazines.
|
|
||||
|---|---|---|---|---|
| Entry | Amine | Mol% amine | Solvent | Product yielda (%) |
| a NMR yields using dimethylsulfone as a standard. b Yields of mono- and di-functionalized products, respectively. c 24 h reaction. d Double concentration (0.4 M). e Inverted equivalents of substrate and reagent. f Perfluorooctylation. TPA: triphenylamine, N,N-DMA: N,N-dimethylaniline, MeDPA: N-methyldiphenylamine, DIPEA: N,N-diisopropylethylamine/Hünig's base, DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene, PTH: 10-phenylphenothiazine. | ||||
| 1 | TPA | 25 | MeCN | 85 |
| 2 | N,N-DMA | 25 | MeCN | 37 |
| 3 | MeDPA | 25 | MeCN | 52 |
| 4 | DIPEA | 25 | MecN | 25 |
| 5 | DBU | 25 | MeCN | Traces |
| 6 | PTH | 25 | MeCN | 21, 39b |
| 7 | TPA | 25 | MeCN | 80 |
| 8 | TPA | 25 | MeCN | 65d |
| 9 | TPA | 25 | IPA | 18 |
| 10 | TPA | 25 | EtOAc | 77 |
| 11 | TPA | 25 | MeOH | 48 |
| 12 | TPA | 25 | DMSO | 47 |
| 13 | TPA | 50 | MeCN | 59 |
| 14 | TPA | 75 | MeCN | 54 |
| 15 | TPA | 100 | MeCN | 47 |
| 16 | TPA | 5 | MeCN | 82 |
| 17 | TPA | 5 | MeCN | 87 |
| 18 | TPA | 5 | MeCN | 88 , |
|
||||
Further extension of the reaction time to 24 h resulted in no change in the yield (entry 7), prompting us to keep the initially proposed 14 hours as the optimal timeframe. Moreover, by doubling the crude concentration to around 0.4 M, a slight decrease in yield was noticed (entry 8), possibly due to decreased light penetration in the reaction mixture. Next, we moved on to analyze the influence of solvent in the reaction (entries 9–12). Trials to optimize the conditions selecting more polar solvents (higher chance of EDA complexation)8 such as isopropanol (IPA), ethyl acetate (EtOAc), methanol (MeOH) and dimethylsulfoxide (DMSO), showed lower yields compared to acetonitrile. Next, we proceeded to find the optimal amount of TPA in our system. Interestingly, increasing the amine's load proved counter-productive to the reaction, with yield dropping to ∼50% when using 50–100 mol% of TPA (entries 13–15). On the other hand, the use of 5 mol% of TPA allowed us to replicate a >80% yield observed in our initial radical trapping experiment, with just one fifth of the original catalyst loading (entry 16). Motivated by this result, we moved on to test whether an inversion of the equivalents between the aromatic substrate and the perfluoroalkylating reagent would have an effect on the yield, allowing us to apply these conditions when the substrate is available in very limited amounts for applications such as late-stage functionalization. By keeping a 5 mol% load of TPA with respect to the limiting reagent −2.5 mol% with respect to the perfluoroalkylating reagent (a very low loading of catalyst compared to the literature on EDA catalytic donors), we successfully obtained a yield of ∼87% (entries 17 and 18).
With these encouraging results we moved on to show the wide scope of our optimized conditions (Fig. 6). As already discussed above, perfluorooctylation and perfluorohexylation of 1,2,3-trimethoxybenzene resulted in good yields after purification, reaching 80% yield for product PF1 and 79% yield for product PF2. An additional test for perfluorobutylation of the same substrate yielded similar amounts by NMR, albeit a lower isolated yield for the perfluorobutylated PF3. When looking at less electron-rich aromatics such as 1,4- and 1,2-dimethoxybenzene we observed moderate yields of 49% (PF4) and 46% (PF5), respectively, with high regioselectivity towards the 4-position in the latter case. On the other hand, an inseparable mixture of monoperfluoroalkylated isomers with approximately equimolar distribution, as determined by GCMS, was obtained for 1,2,3-trimethoxybenzene (PF6) with a total yield of 20%.
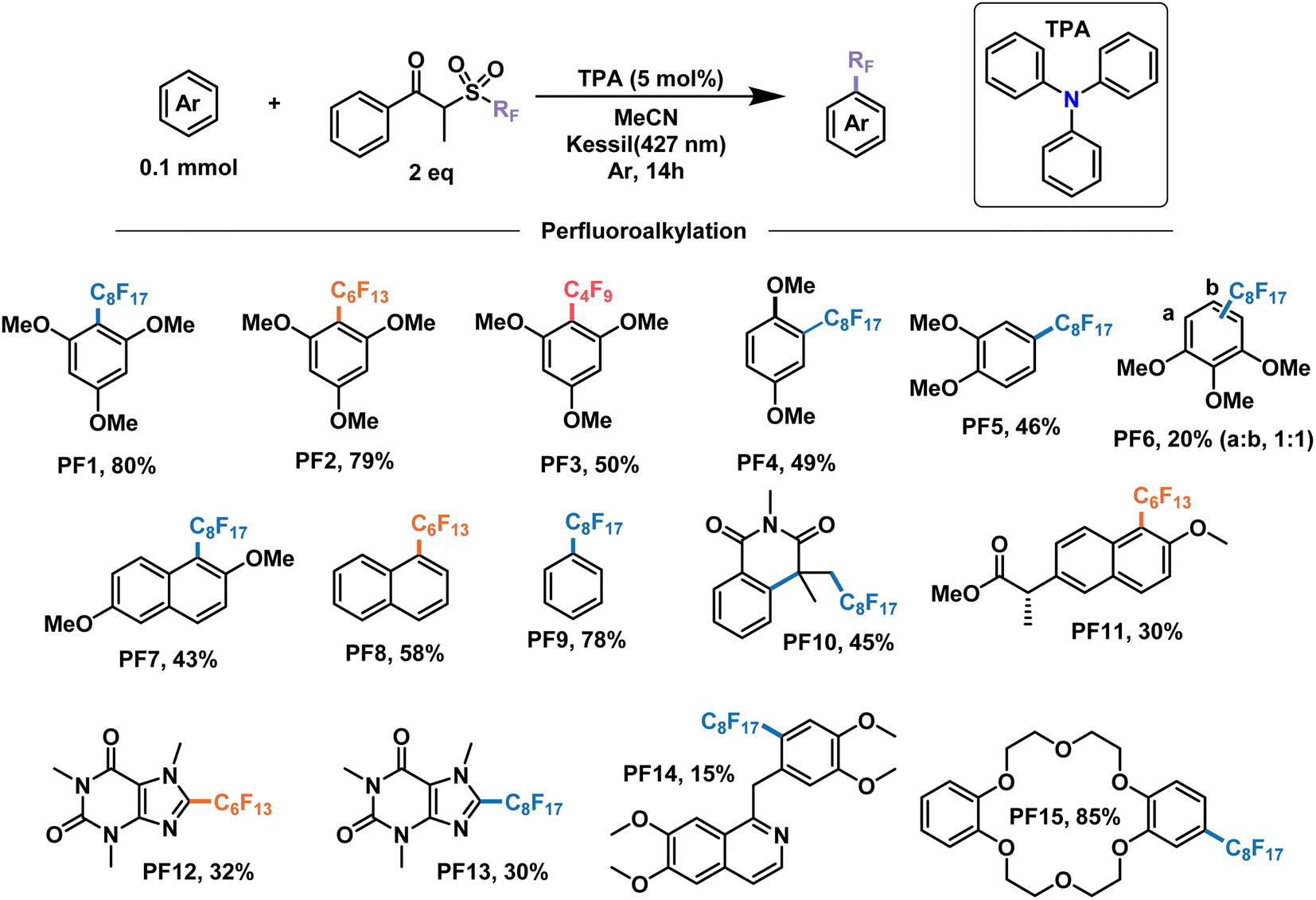 | ||
| Fig. 6 Demonstrative scope for the catalytic EDA-mediated perfluoroalkylation of electron-rich aromatics. Note: PF9 is done under modified conditions (25% benzene v/v as solvent). | ||
We then decided to test substrates that have proved challenging to perfluoroalkylate in the literature, such as those that absorb at wavelengths similar to the perfluoroalkylating reagent. For example, 2,6-dimethoxynapthalene is strongly absorbing at 295 nm, preventing the cleavage of the dummy group reagent and the subsequent generation of radicals in solution. However, when subject to the conditions of this work, perfluorooctylation was successful in the 1-position, giving the product PF7 with a yield of 43%. Unsubstituted aromatic rings constitute another challenging family of molecules in perfluoroalkylation reactions due to their less electron-rich character. Nevertheless, the proposed EDA catalysis proved useful for the perfluorohexylation of naphthalene and the perfluorooctylation of benzene in good yields giving products PF8 and PF9, respectively. Monofunctionalized products were observed for both substrates, with the latter having modified reaction conditions—25% v/v in solution. Lastly, UV light also prevented the application of these perfluoroalkylating reagents for the addition to olefins due to the generation of inseparable mixtures. When submitting N-methacryloyl-N-methylbenzamide to our conditions, the addition of the perfluorooctyl radical and the subsequent cascade cyclization with the benzene ring were observed, yielding product PF10 in 45% yield.
In addition to the electron-rich substrates presented here, our strategy proved applicable for the late-stage functionalization of some molecules of biological importance. For example, a methylated derivative of naproxen proved responsive towards perfluorohexylation in 30% yield to give product PF11. In addition, similar yields were observed for free caffeine to give the perflurorohexylated product PF12 and its perflurooctylated analogue PF13 under our conditions. Monosubstituted PF14 was also obtained with high selectivity albeit in low yield (15%) by perfluorooctylation of papaverine—an antispasmodic alkaloid—in its free base form. Finally, we tested our conditions on dibenzo-18-crown-6, a popular phase-transfer agent, obtaining the 4-perfluorooctylated product PF15 in excellent yield. We envision that the late-stage perfluoroalkylation of aromatic substrates shown in this work will find applications beyond this scope, such as the study of their physical–chemical properties for the modification of materials, the study of their interactomes in biological systems,64 their use as probes and the development of new analytical techniques for their binding and detection,65,66 among others.
The previously described results motivated us to turn next to a highly-sought reaction in medicinal chemistry: the radical late-stage trifluoromethylation of aromatic scaffolds.67,68 The introduction of the trifluoromethyl moiety allows for the control of fundamental properties in the drug development process such as potency, conformation, metabolism and membrane permeability, making it a valuable transformation for the pharmaceutical industry.69,70 Following the same principle as the previous section, we began to optimize the trifluoromethylation of 1,3,5-trimethoxybenzene in the presence of the CF3 reagent and TPA at 427 nm. Starting with similar conditions to the optimized perfluoroalkylation of aromatics, TPA reached a yield of only 15% towards the desired product (Table 2, entries 1 and 2). Such a decrease in the yield against a different reagent can be explained by the difference in bond dissociation energies between the carbon atom alpha to the ketone and the sulfone moiety, showing 174.9 kJ mol−1 and 185.0 kJ mol−1 for C6 and CF3, respectively (calculated using density functional theory, DFT- B3LYP 6-31g(d)). Nonetheless, this represented an opportunity to modify the triphenylamine scaffold without compromising the simplicity of the catalytic design. In our search for more electron-donating amines, tris(4-methoxyphenyl)amine (TMPA) appeared to be a viable commercially-available alternative to TPA. A reaction with a 5 mol% loading of TMPA showed a significant increase in yield at 64% under the same conditions (entries 3 and 4). Similar to our previous optimization, lower loadings of catalyst provided higher yields of up to 77% with 2.5 mol% of catalyst (entries 4–6); however, loadings of ≤1 mol% led to poor results (entry 7). On the other hand, using half the amount of acetonitrile brought the yield of the reaction up to 74% (entry 8), similar to the use of 2.5 equivalents of trifluoromethylating reagent (entry 10). A longer reaction time of up to 18 h further increased the yield to 89% (entry 9). Combining these factors together, a yield of 91% was achieved when running the reaction for 18 hours in 0.25 mL of acetonitrile with a 2.5 mol% loading of TMPA (entry 11). Additionally, we tested the efficiency of this system under a 440 nm light source (entries 12–14); however, a maximum yield of 50% was obtained even after 24 hours of irradiation.
|
|
||||
|---|---|---|---|---|
| Entry | Amine | mol% amine | Volume (mL) | Product yielda (%) |
| a NMR yields using dimethylsulfone as a standard. b 18 hours of irradiation. c Using 2.5 equivalents of CF3 reagent. d 440 nm light. e 24 hours of irradiation. | ||||
| 1 | TPA | 0 | 0.5 | Traces |
| 2 | TPA | 5 | 0.5 | 15 |
| 3 | TMPA | 0 | 0.5 | Traces |
| 4 | TMPA | 5 | 0.5 | 64 |
| 5 | TMPA | 10 | 0.5 | 63 |
| 6 | TMPA | 2.5 | 0.5 | 77 |
| 7 | TMPA | 1 | 0.5 | 13 |
| 8 | TMPA | 5 | 0.25 | 74 |
| 9 | TMPA | 5 | 0.5 | 89 |
| 10 | TMPA | 2.5 | 0.5 | 75c |
| 11 | TMPA | 2.5 | 0.25 | 91 |
| 12 | TMPA | 2.5 | 0.25 | 45b,d |
| 13 | TMPA | 2.5 | 0.25 | 45d,e |
| 14 | TMPA | 2.5 | 0.25 | 50c,d |
Next, we proceeded to explore the substrate scope for the trifluoromethylation reaction (Fig. 7). As with our perfluoroalkylation scope, the addition of trifluoromethyl radicals proceeds readily with electron-rich arenes such as trimethoxyarenes. After isolating our first trifluoromethylated product TF1 at 82%, we tested our conditions against 3,4,5-trimethoxytoluene (TF2) and methyl 3,4,5-trimethoxybenzoate (TF3) resulting in yields of 56% and 45%, respectively. Moreover, nitrogen-containing heterocycles proved successful with moderate yields: N-phenylpyrrole (TF4) at 46% and indoles such as 3-methylindole (TF5) and 2-phenylindole (TF6) with yields of 50% and 38%, respectively. Similarly, as with our perfluorooctylation of PF10, the trifluoromethylation of olefins was proven feasible with the cyclization of N-methyl-N-phenylcinnamamide (TF7) in 42% yield. Lastly, in the context of late-stage functionalization, trifluoromethylation of caffeine proved more efficient than its perfluorohexyl and perfluorooctyl analogues with a yield of 48%. Given this result, we proceeded to test our conditions on pentoxifylline (TF9)—a hemorheological agent used in the treatment of muscle pain—resulting in a 41% yield.
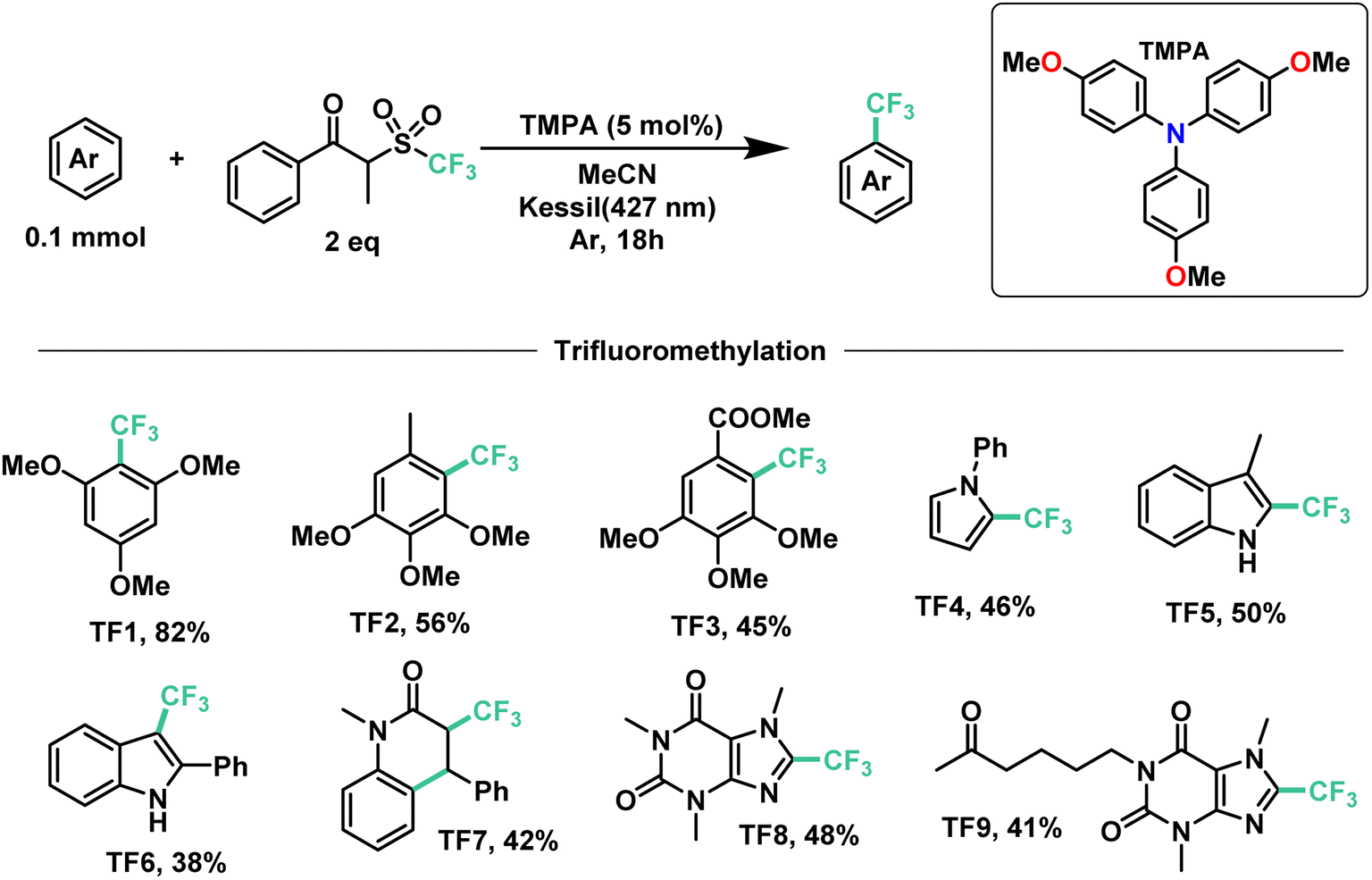 | ||
| Fig. 7 Demonstrative scope for the catalytic EDA-mediated trifluoromethylation of electron-rich aromatics. | ||
Conclusions
We have performed a comprehensive mechanistic study of perfluoroalkylation reactions using Electron Donor–Acceptor (EDA) complexes of triarylamines as photocatalytic donors and α-(perfluoroalkylsulfonyl)propiophenones as the model acceptor for the generation of perfluorooctyl, perfluorohexyl, perfluorobutyl, and trifluoromethyl radicals. We have confirmed the formation of an EDA complex via UV-vis absorption and fluorescence quenching experiments and studied the mechanism behind its successful turnover under blue light irradiation. We showed a great synthetic utility of this catalytic system at low catalytic loading (2.5 mol%) in the Minisci-type perfluoroalkylation and trifluoromethylation reactions for the diversification of aromatics, including the late-stage functionalization of a biologically-relevant scaffolds in moderate to good yields. This work opens the new applications of triarylamines in synthetic chemistry as photocatalysts. We also envisage that our results will open new avenues for general applications EDA-mediated systems in photocatalysis.Data availability
General experimental conditions, calculations related to the mechanism elucidation, an characterization of all compounds synthesized are available in the ESI.†Author contributions
DJCP, JL, and EH: conceptualization, methodology, and investigation. DJCP, JL, and CJL conceived the catalytic system. DJCP and JL conducted the experiments for optimization and scope. EH and JRS performed all spectroscopic studies. JMS helped validate the reproducibility of the experiments and purify the final products. DJCP wrote the manuscript and all authors and coauthors contributed to data analysis and revising manuscript. CJL, GC and DFP supervised the work, providing mentorship regarding the mechanistic studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the McGill Chemistry Characterization (MC2) Facility for their contribution to the compound characterization in this work, specifically, Robin Stein on the NMR and Nadim Saadé and Alexander Wahba on HRMS. We also thank Chia-Yu Huang for providing the starting materials for products PF10 and TF7. The authors acknowledge the Canada Research Chair (Tier I) foundation, the E. B. Eddy endowment fund, the CFI, NSERC, and FQRNT for support of our research. DJCP acknowledges the financial support granted by the Vanier Canada Graduate Scholarship, CONACYT Mexico, and the Richard H. Tomlinson Fellowship. JRS acknowledges the financial support granted by CONACYT Mexico.References
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- Y. Lee and M. S. Kwon, Eur. J. Org. Chem., 2020, 2020, 6028–6043 CrossRef CAS.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- M. Tobisu, T. Furukawa and N. Chatani, Chem. Lett., 2013, 42, 1203–1205 CrossRef CAS.
- E. K. Kim, T. M. Bockman and J. K. Kochi, J. Am. Chem. Soc., 1993, 115, 3091–3104 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- Y.-Q. Yuan, S. Majumder, M.-H. Yang and S.-R. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- T. Tasnim, M. J. Ayodele and S. P. Pitre, J. Org. Chem., 2022, 87, 10555–10563 CrossRef CAS PubMed.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed.
- Q. Liu, Y. Lu, H. Sheng, C.-S. Zhang, X.-D. Su, Z.-X. Wang and X.-Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 25477–25484 CrossRef CAS PubMed.
- E. J. McClain, T. M. Monos, M. Mori, J. W. Beatty and C. R. J. Stephenson, ACS Catal., 2020, 10, 12636–12641 CrossRef CAS.
- V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Lalevée, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–7441 CrossRef CAS PubMed.
- I. Bosque and T. Bach, ACS Catal., 2019, 9, 9103–9109 CrossRef CAS.
- T. Tasnim, C. Ryan, M. L. Christensen, C. J. Fennell and S. P. Pitre, Org. Lett., 2022, 24, 446–450 CrossRef CAS PubMed.
- H. Lu, D.-Y. Wang and A. Zhang, J. Org. Chem., 2020, 85, 942–951 CrossRef CAS PubMed.
- P. Blanchard, C. Malacrida, C. Cabanetos, J. Roncali and S. Ludwigs, Polym. Int., 2019, 68, 589–606 CrossRef CAS.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- P.-Y. Ho, Y. Wang, S.-C. Yiu, W.-H. Yu, C.-L. Ho and S. Huang, Org. Lett., 2017, 19, 1048–1051 CrossRef CAS PubMed.
- C. Dai, L. Zhong, X. Gong, L. Zeng, C. Xue, S. Li and B. Liu, Green Chem., 2019, 21, 6606–6610 RSC.
- K. Yuan Chiu, T. Xiang Su, J. Hong Li, T.-H. Lin, G.-S. Liou and S.-H. Cheng, J. Electroanal. Chem., 2005, 575, 95–101 CrossRef.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 2019, 6735–6748 CrossRef CAS.
- A. Dewanji, L. van Dalsen, J. A. Rossi-Ashton, E. Gasson, G. E. M. Crisenza and D. J. Procter, Nat. Chem., 2023, 15, 43–52 CrossRef CAS PubMed.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- R. Kumar, S. Jayakumar and V. Kannappan, Thermochim. Acta, 2012, 536, 15–23 CrossRef CAS.
- J. C. Sheehan, R. M. Wilson and A. W. Oxford, J. Am. Chem. Soc., 1971, 93, 7222–7228 CrossRef CAS.
- H. E. Zimmerman and T. W. Flechtner, J. Am. Chem. Soc., 1970, 92, 6931–6935 CrossRef CAS.
- H. E. Zimmerman, K. G. Hancock and G. C. Licke, J. Am. Chem. Soc., 1968, 90, 4892–4911 CrossRef CAS.
- J. C. Sheehan and R. M. Wilson, J. Am. Chem. Soc., 1964, 86, 5277–5281 CrossRef CAS.
- P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef CAS PubMed.
- L. M. Campos, H. Dang, D. Ng, Z. Yang, H. L. Martinez and M. A. Garcia-Garibay, J. Org. Chem., 2002, 67, 3749–3754 CrossRef CAS PubMed.
- Z. Yang and M. A. Garcia-Garibay, Org. Lett., 2000, 2, 1963–1965 CrossRef CAS PubMed.
- H. G. Viehe, R. Merényi, L. Stella and Z. Janousek, Angew. Chem., Int. Ed., 1979, 18, 917–932 CrossRef.
- H. G. Viehe, Z. Janousek, R. Merenyi and L. Stella, Acc. Chem. Res., 1985, 18, 148–154 CrossRef CAS.
- D. J. Castillo-Pazos, J. D. Lasso and C.-J. Li, Beilstein J. Org. Chem., 2022, 18, 788–795 CrossRef CAS PubMed.
- Z. Guan, H. Wang, Y. Huang, Y. Wang, S. Wang and A. Lei, Org. Lett., 2019, 21, 4619–4622 CrossRef CAS PubMed.
- L. Zhang, G. Zhang, P. Wang, Y. Li and A. Lei, Org. Lett., 2018, 20, 7396–7399 CrossRef CAS PubMed.
- K. Muralirajan, R. Kancherla, J. A. Bau, M. R. Taksande, M. Qureshi, K. Takanabe and M. Rueping, ACS Catal., 2021, 11, 14772–14780 CrossRef CAS.
- D. A. Nagib and D. W. C. Macmillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- W. J. Choi, S. Choi, K. Ohkubo, S. Fukuzumi, E. J. Cho and Y. You, Chem. Sci., 2015, 6, 1454–1464 RSC.
- D. J. Castillo-Pazos, J. D. Lasso and C.-J. Li, Org. Biomol. Chem., 2021, 19, 7116–7128 RSC.
- R. F. Nelson and R. H. Philp, J. Phys. Chem., 1979, 83, 713–716 CrossRef CAS.
- K. Sreenath, C. V. Suneesh, K. R. Gopidas and R. A. Flowers, J. Phys. Chem. A, 2009, 113, 6477–6483 CrossRef CAS PubMed.
- L. Mao, M. Zhou, X. Shi and H.-B. Yang, Chin. Chem. Lett., 2021, 32, 3331–3341 CrossRef CAS.
- M. R. Talipov, M. M. Hossain, A. Boddeda, K. Thakur and R. Rathore, Org. Biomol. Chem., 2016, 14, 2961–2968 RSC.
- T. A. Schaub, T. Mekelburg, P. O. Dral, M. Miehlich, F. Hampel, K. Meyer and M. Kivala, Chem. – Eur. J., 2020, 26, 3264–3269 CrossRef CAS PubMed.
- M. U. Munshi, G. Berden, J. Martens and J. Oomens, Phys. Chem. Chem. Phys., 2017, 19, 19881–19889 RSC.
- P. Cias, C. Slugovc and G. Gescheidt, J. Phys. Chem. A, 2011, 115, 14519–14525 CrossRef CAS PubMed.
- D. N. Stamires and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 2557–2561 CrossRef CAS.
- W. R. Ware, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS.
- V. Kannappan and N. I. Gandhi, Phys. Chem. Liq., 2008, 46, 510–521 CrossRef CAS.
- M. Oyama, T. Higuchi and S. Okazaki, Electrochem. Solid-State Lett., 2002, 5, E1 CrossRef CAS.
- E. W. Forster and K. H. Grellman, Chem. Phys. Lett., 1972, 14, 536–538 CrossRef.
- H. Görner, J. Phys. Chem. A, 2008, 112, 1245–1250 CrossRef PubMed.
- R. A. Sanmartín, M. L. Salum, S. Protti, M. Mella and S. M. Bonesi, ChemPhotoChem, 2022, 6, e202100247 CrossRef.
- S. C. Creason, J. Wheeler and R. F. Nelson, J. Org. Chem., 1972, 37, 4440–4446 CrossRef CAS.
- A. Vallat and E. Laviron, J. Electroanal. Chem., 1976, 74, 309–314 CrossRef CAS.
- O. Yurchenko, D. Freytag, L. Zur Borg, R. Zentel, J. Heinze and S. Ludwigs, J. Phys. Chem. B, 2012, 116, 30–39 CrossRef CAS PubMed.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed.
- Q. Zhang, X. Dong, J. Lu, J. Song and Y. Wang, Anal. Chem., 2021, 93, 9634–9639 CrossRef CAS PubMed.
- R. Khan, D. Andreescu, M. H. Hassan, J. Ye and S. Andreescu, Angew. Chem., Int. Ed., 2022, 61, e202209164 CAS.
- M. Kralj, L. Tušek-Božić and L. Frkanec, ChemMedChem, 2008, 3, 1478–1492 CrossRef CAS PubMed.
- J. D. Lasso, D. J. Castillo-Pazos and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10955–10982 RSC.
- H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc07078b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |