
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRed-light photoswitching of indigos in polymer thin films†
Kim
Kuntze
 a,
Jani
Viljakka
a,
Matti
Virkki
a,
Chung-Yang (Dennis)
Huang
*b,
Stefan
Hecht
*cd and
Arri
Priimagi
*a
a,
Jani
Viljakka
a,
Matti
Virkki
a,
Chung-Yang (Dennis)
Huang
*b,
Stefan
Hecht
*cd and
Arri
Priimagi
*a
aSmart Photonic Materials, Faculty of Engineering and Natural Sciences, Tampere University, Fi-33101 Tampere, Finland. E-mail: arri.priimagi@tuni.fi
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: dcyhuang@icredd.hokudai.ac.jp
cDepartment of Chemistry & IRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Strasse 2, 12489 Berlin, Germany. E-mail: sh@hu-berlin.de
dDWI – Leibniz Institute for Interactive Materials, Forckenbeckstrasse 50, 52074 Aachen, Germany
First published on 22nd February 2023
Abstract
Through simple synthetic derivatisation, the parent indigo dye becomes a red-light E–Z photoswitch exhibiting negative photochromism and tuneable thermal isomerisation kinetics. These attributes make indigo derivatives extremely attractive for applications related to materials and living systems. However, there is a lack of knowledge in translating indigo photoswitching dynamics from solution to solid state – the environment crucial for most applications. Herein, we study the photoswitching performance of six structurally distinct indigo derivatives in five polymers of varying rigidity. Three key strategies are identified to enable efficient photoswitching under red (660 nm) light: (i) choosing a soft polymer matrix to minimise its resistance toward the isomerisation, (ii) creating free volume around the indigo molecules through synthetic modifications, and (iii) applying low dye loading (<1% w/w) to inhibit aggregation. These strategies are shown to improve both photostationary state distributions and the thermal stability of the Z isomer. When all three strategies are implemented, the isomerisation performance (>80% Z form in the photostationary state) is nearly identical to that in solution. These findings thus pave the way for designing new red-light photochromic materials based on indigos.
Introduction
The focus of materials research is steadily shifting towards interactive, “intelligent” materials that adapt to environmental changes and can be controlled with (multiple) external stimuli.1,2 Light is arguably the most attractive external stimulus for this purpose as it is non-invasive, waste-free, and can be applied in a highly controllable fashion with superior temporal, spatial and spectral resolution. Thus, systems responding to light are increasingly investigated,3,4 with applications emerging in a wide range of research fields from optics5,6 and (opto)electronics7,8 to photobiology9–13 and photomechanics.14,15 Among other strategies, photoresponsive materials can be designed by incorporating photochromic molecules (photoswitches) onto the surface of a metal particle16 or into the bulk of a macroscopic material. Photoswitches are organic compounds that upon excitation with light of a suitable wavelength interconvert reversibly between two or more isomeric ground state species, inducing changes to the macroscopic properties of a material when the physicochemical characteristics of the involved isomers differ.4 A multitude of photoswitches has been developed over the last few decades, mostly based on E–Z isomerisation around C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,17,18 CN19–21 and NN22,23 double bonds or cyclisation reactions.24–27
C,17,18 CN19–21 and NN22,23 double bonds or cyclisation reactions.24–27
The wide array of photoswitches with their unique properties has so far provided chemists with flexible options when designing photoresponsive systems with the desired functions. Beyond solution studies, the performance of various photoswitches has been investigated in different solid-like matrices,28,29 especially in thin polymer and hydrogel films, where the photochromic molecules can either be covalently bound or simply blended with the polymer matrix.30 Azobenzene thin films, in particular, are increasingly used in the development of smart materials that are reversibly deformed or patterned thanks to the E–Z switching of photoisomerisable units.4–6
On the other hand, most photoswitches isomerise upon excitation in the ultraviolet region. UV irradiation is absorbed by various materials, causing damage to many polymers and especially to living tissue in addition to suffering from low penetration depth.31–33 Hence, extensive research has been carried out to red-shift the isomerisation wavelengths, ultimately aiming for the therapeutic window above 650 nm.9,12,34,35 To this end, progress has been made by (i) changing the electronic properties of conventional photoswitches such as azobenzenes through structural changes,36–41 (ii) designing new photoswitchable scaffolds and structures that intrinsically absorb low-energy light,25,42–44 and (iii) exploiting indirect isomerisation using photocatalysts.45–47 Yet, the vast majority of these investigations has been restricted to solution, and switching with red or near-infrared light in solid matrices has remained largely unexplored.48,49
With its long history in providing blue colour, indigo is nowadays one of the most widely used dyes (Scheme 1). Structurally, it is composed of two indole-type subunits connected by a central CC bond. This central double bond conjugates two push–pull pairs of nitrogen/carbonyl groups, and as a result, indigo intrinsically absorbs in the red-light region. Although photoisomerisation of the parent indigo is inhibited by fast excited-state proton transfer,50 it is possible to enable photoswitching through N-functionalisation. Upon N-arylation and/or N-alkylation of the parent indigo, efficient isomerisation from the stable E isomer to the metastable Z form can be realised by red light.44 The rate of thermal Z → E isomerisation can be tuned from seconds to hours, being the slowest for compounds with electron-poor N-aryl substituents.44 Perhaps most importantly, indigos are negative photochromic compounds: the prominent visible light absorption band is blue-shifted upon E → Z isomerisation, which enables an increasing amount of red light to penetrate the sample as the content of the photo-generated Z isomer increases. These features combined make indigo derivatives attractive candidates for the design of smart materials.
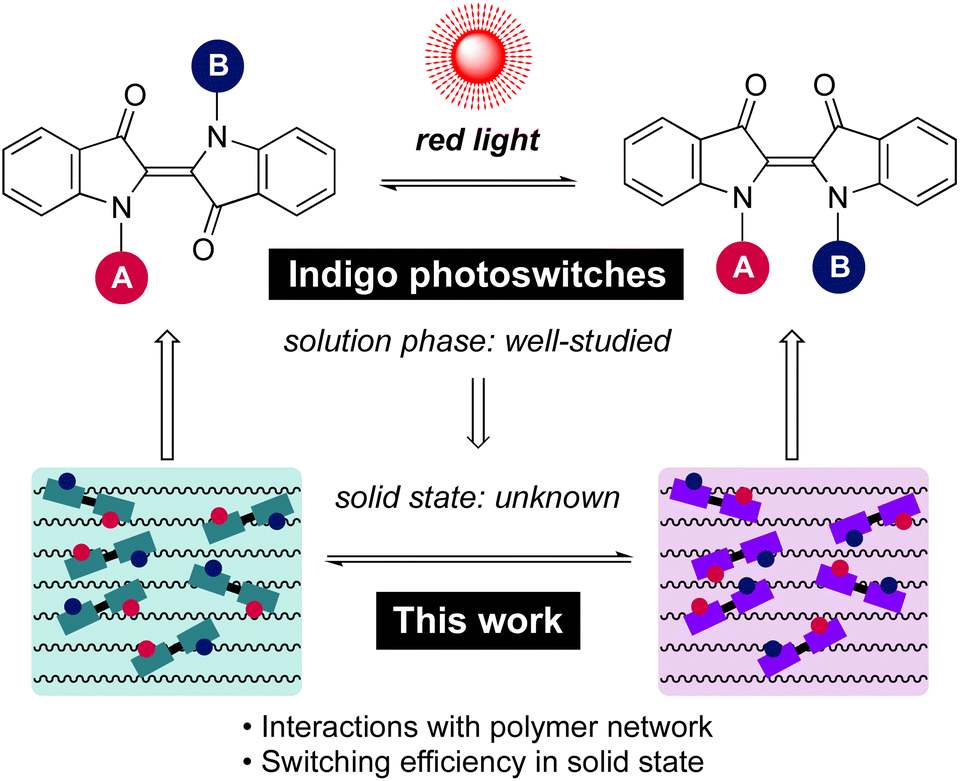 | ||
| Scheme 1 E–Z isomerisation of indigo photoswitches and the focus of this work. | ||
Although the isomerisation of indigo derivatives has been studied extensively in solution,51–54 only scattered observations have been made regarding their isomerisation in the solid state.55,56 Importantly, no information is available on their photoswitching in organic polymers, which represent a ubiquitous chemical host in materials science. At the outset, it is unclear whether photoisomerisation of indigos can be successfully translated from solution to polymers. One potential hurdle is that the E ↔ Z isomerisation of indigos brings about a greater spatial displacement as compared to other steric switches such as azobenzenes. While it is a desirable trait in the view of applications where the molecular-level geometrical changes are utilised to trigger macroscopic transformations in a bulk material, the isomerisation process in a rigid environment may also be more hindered, rendering switching of the indigos in the solid state more challenging than for other classes of photoswitches. Thus, there is a need for a systematic investigation of the solid-state switching behaviour of indigo derivatives to determine criteria enabling their good performance and successful application.
Herein, we present the first example of efficient photoswitching of indigo derivatives in polymer matrices under red light. The effects of N-substituents as well as supporting polymer network on the photoswitching dynamics are elucidated. Moreover, we reveal a strong dependence between indigo concentration and the switching properties, critical for the range of applications these switches can best be exploited in. Based on these findings, we demonstrate a strategy by which the photoswitching performance in the solid can be enhanced dramatically – to reach the same level as in solution.
Results and discussion
We chose to study the indigo derivatives Al-Me, Ar-Me and As-Me (Scheme 2a) that represent the three principal substitution patterns yielding functional red-light photoswitches.44 The ester moieties have a dual purpose: they function as electron-withdrawing substituents that stabilise the metastable Z isomer through an inductive effect, while also providing a linkage for further functionalisation with, e.g., polymerisable groups. The compounds were prepared in one or two steps in moderate to good yields from unsubstituted indigo by utilising the previously published mild nucleophilic substitution reaction44 for Al-Me and a copper-catalysed coupling57 for Ar-Me. The non-symmetric derivative As-Me was prepared through a selective mono-arylation followed by an alkylation reaction. Detailed synthetic protocols are given in the ESI.†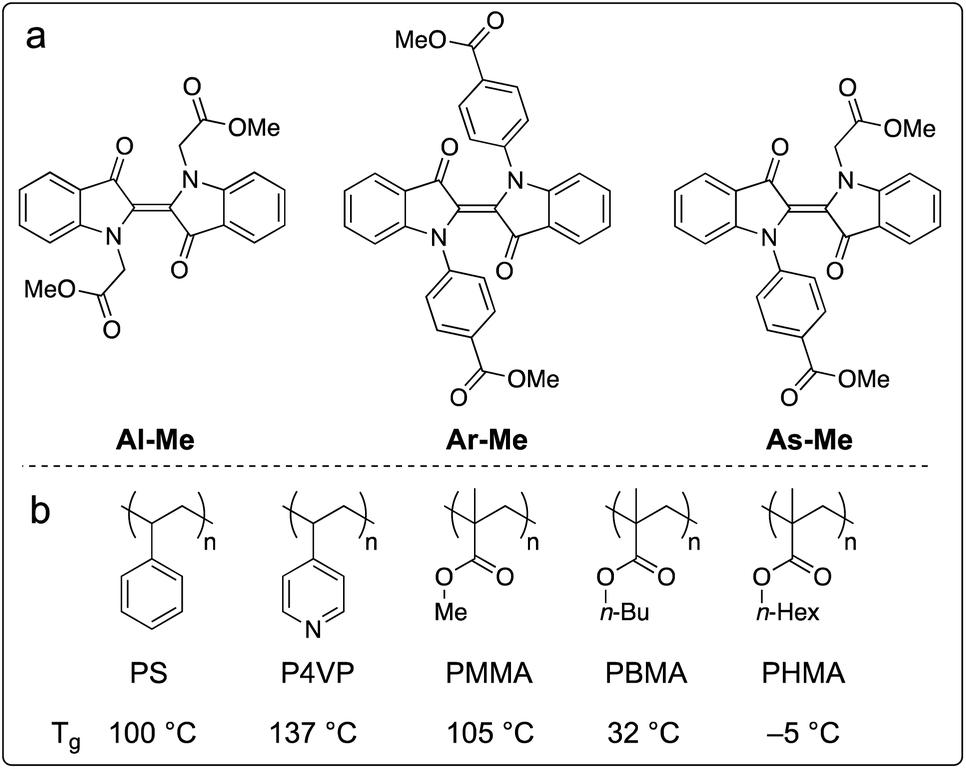 | ||
| Scheme 2 (a) Studied indigo derivatives. (b) Studied polymer environments and their glass transition temperatures (Tg). | ||
To confirm that the compounds have similar photochemical properties to the electronically analogous derivatives reported before, we first studied their photoswitching in dilute acetonitrile solutions. As expected, the compounds form photostationary states (PSS) with 75–88% and 3–11% Z upon excitation with 660 nm (250 mW cm−2, ≤30 s) and 500 nm light (85 mW cm−2, ≤2 min), respectively. Their Z half-lives range from 1.4 min (Al-Me) to 41 min (Ar-Me) (Fig. 1, Table 1). Whereas Al-Me and As-Me exist exclusively as their E isomers in the dark, Ar-Me reached an equilibrium with 22% Z in darkness, irrespective of whether starting from E- or Z-enriched solutions. The same phenomenon has been reported for other indigos functionalised with two N-aryl substituents, originating from the fact that this substitution pattern renders the ground state energy difference between the E and Z isomers down to ca. 1 kcal mol−1.44 Overall, all three compounds exhibit very similar photochemical properties to their previously reported analogues.51 The switching is relatively robust: 82% of As-Me and 90% of Ar-Me were conserved after 1000 cycles and more than 16 hours of continuous irradiation with alternating 660 and 500 nm light pulses (Fig. S1†) under ambient conditions. Al-Me seems, however, less fatigue resistant, with 46% of the original concentration remaining after an identical experiment (Fig. 1d).
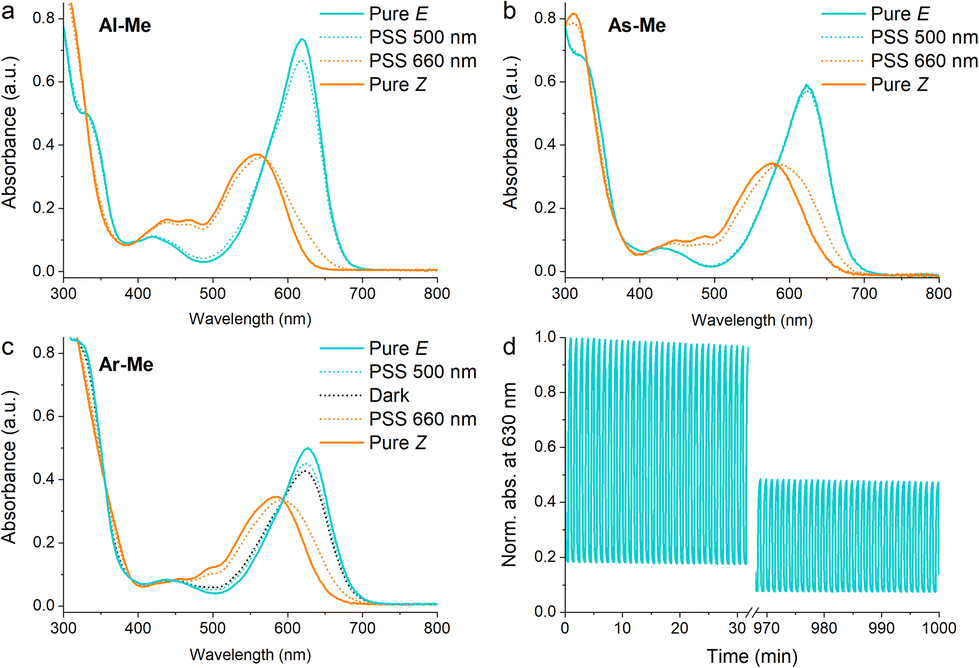 | ||
| Fig. 1 Absorption spectra of (a) Al-Me, (b) As-Me and (c) Ar-Me in solution (50 μM in acetonitrile, 25 °C). (d) 1000 switching cycles with alternating 660 and 500 nm light pulses for Al-Me. | ||
| λ E (nm) | λ Z (nm) | PSS660 nm (% Z) | PSS500 nm (% E) | Dark (% E) | t 1/2 (min) | |
|---|---|---|---|---|---|---|
| Al-Me | 619 | 559 | 88 | 90 | 100 | 1.4 |
| As-Me | 623 | 577 | 75 | 97 | 100 | 3.8 |
| Ar-Me | 627 | 584 | 75 | 87 | 78 | 41 |
Next, we proceeded to studying whether the compounds switch when doped into polymer matrices. We focused our studies on five amorphous polymers: polystyrene (PS), poly(4-vinylpyridine) (P4VP) and polyacrylates with methyl (PMMA), n-butyl (PBMA), and n-hexyl (PHMA) side chains (Scheme 1b). PS, P4VP and PMMA are rigid (Tg values between +100 and +137 °C) matrices often utilised as scaffolds for photoswitches,58–61 differing in polarity and aromaticity. PBMA and PHMA (Tg values +32 and −5 °C) were chosen to give information on the photoswitching behaviour in a softer polymeric environment. We began our studies by preparing thin polymer films containing 2% (w/w) of the indigo derivatives. After optimising the spin-casting procedure for each polymer, we were able to create uniform and sufficiently thick (5–20 μm) polymer films from viscous solutions of different polymer–indigo combinations (see the ESI† for details). Each sample was studied with UV-visible absorption spectroscopy. The Z-fraction in the photostationary state was estimated by comparing the absorbance values of the dark and PSS spectra (see the ESI† for details). The half-life of thermal isomerisation was determined by recording the absorbance values at a chosen wavelength over time after ceasing the excitation, using a stretched exponential function62 to account for the continuum of different microenvironments inside the polymer matrix. Since indigos are known to emit light,51 we also recorded the fluorescence spectra of the samples.
The polymer environment induced only minute changes in the absorption spectra of the studied compounds (Fig. S2†) with the exception of P4VP in which the major absorption band of all three switches was red-shifted by ca. 10 nm, presumably due to the more polar environment.51 Their photoswitching behaviour, however, proved to be highly polymer-dependent. In the rigid PS and P4VP matrices, only 15–20% of the switches isomerised from E to Z upon irradiation with 660 nm light (Fig. 2a). Similarly inefficient switching in rigid matrices has been observed for other switches that undergo a large geometric change upon isomerisation.28 In less rigid environments the PSS distribution improved notably, up to 55% Z for Al-Me in PBMA (Fig. 2b). The Z-fraction was typically largest for Al-Me and smallest for As-Me, the difference being most pronounced in PMMA (Fig. 2c).
 | ||
| Fig. 2 Absorption spectra of Al-Me at room temperature in (a) PS and (b) PBMA, showing the difference between inefficient and efficient switching. (c) Z-Fractions in the photostationary state (illumination with 660 nm light) and (d) Z-half-lives for all indigo–polymer combinations. The half-life of Ar-Me in P4VP is calculated with a biexponential fit due to large error margins with the stretched exponential function (see the ESI†). (e) 250 switching cycles for Al-Me in PHMA. (f) Photographs of PHMA films with Ar-Me upon irradiation with 500 and 660 nm light. Indigo content: 2% (w/w) except 0.5% in (e) and (f). Excitation intensities: 670 mW cm−2 (660 nm), 220 mW cm−2 (500 nm). | ||
Surprisingly, photodegradation leading to a yellow-coloured decomposition product was observed in PMMA and PBMA (visible in Fig. 2b as a missing isosbestic point) but was almost negligible in the other three polymers unless repeated switching cycles were carried out. Furthermore, even though bleaching did eventually take place in all surroundings, in the case of PS, P4VP and PHMA the films turned colourless without any yellow hue. To gain insight into the degradation mechanisms, an LC-MS (liquid chromatography coupled to high-resolution mass spectrometry) analysis was carried out after prolonged irradiation of the indigo switches in solution, in a PHMA film, and in a PBMA film (see the ESI† for details). In solution and in PHMA, the irradiation yielded exclusively a product with a mass of M + 16 (Fig. S3†) and no absorption in the visible range, most probably the well-known oxygen adduct.63 In PBMA, we also observed a species absorbing at around 400 nm with a mass corresponding to M − 2 (Fig. S3†), hinting towards a cyclisation product. Thus, an additional degradation pathway exists in PBMA and PMMA, which we believe to originate from the end groups or initiator residues present in PMMA and PBMA that were purchased from a different vendor than PHMA. However, the accurate determination of the degradation pathways and how they could be inhibited would require additional experimental and computational studies. The effect of degradation was taken into account when estimating the PSS distributions.
Interestingly, the half-life of the thermal Z → E isomerisation remained on the timescale of minutes for both Al-Me and As-Me in all studied polymers (Fig. 2d, Table S3†), even though thermal isomerisation of E–Z switches is often more than one magnitude faster in solid matrices than in solution.28 The thermal isomerisation of Ar-Me was significantly slower in the soft PBMA compared to the more rigid PS, P4VP and PMMA. It is possible that in a rigid matrix the E → Z isomerisation induces considerable mechanical strain which relaxes upon Z → E isomerisation, speeding up the kinetics compared to a softer environment. Somewhat surprisingly, the Z half-lives were not longest in PHMA, the least rigid polymer. This observation, as well as the slightly diminished PSS distributions (Fig. 2c), can be explained by aggregation that is most pronounced in this matrix (vide infra).
Having established that all switches can be successfully isomerised inside a polymer matrix, we investigated the effect of loading by preparing PHMA films with 0.25–10% (w/w) of Al-Me. This polymer was chosen as it exhibited the best combination of efficient and clean isomerisation. The isomerisation of azobenzenes in polymeric environments is typically unaffected by the azobenzene concentration, at least up to 10%.64 Thus, we were surprised by the drastic effect indigo loading had on the photochemical properties. Absorption spectra were identical up to 2% after which the band broadened and red-shifted (by 10 nm between 2% and 10%), whereas in the fluorescence spectra a shift was visible already between 1% and 2% (Fig. 3a and c). This result indicates the formation of aggregates upon higher loadings. We hypothesized that by increasing the steric bulk of the indigo molecules, their aggregation could be circumvented or at least alleviated, as a similar strategy has been successfully used to minimise diarylethene aggregation in polymer blends of thin film transistors.65 To this end, we synthesised Al-tBu (Scheme 3) with bulky tert-butyl groups to inhibit the aggregation, yet exhibiting similar electronic properties (Fig. S4 and Table S1†). Indeed, the absorption band of Al-tBu did not show any bathochromic shift from 0.5 to 10%, although a slight 5 nm shift could still be observed in the fluorescence spectra (Fig. 3b and c). Either some degree of agglomeration takes place even with the new molecular design or the mere changes in the dielectric constant of the surrounding medium are enough to affect the orbital energy levels. Similar trends were also observed in the fluorescence spectra of As-Me and Ar-Me, but their absorption spectra showed no red-shift, probably because the aryl groups (perpendicular to the indigo core44) inhibit aggregation (Fig. S5†). No shifts were observed in the absorption spectra of Al-Me in PS (Fig. 3c and S6†) and in PBMA (Fig. S7†), although a shift was visible in the fluorescence spectra. While it is conceivable that the π, π interactions between PS and the indigos could hamper aggregation, the origin of the difference between the spectral changes in PHMA and PBMA is less obvious. One possibility is that the longer alkyl chains in PHMA could lead to lower miscibility of the indigo. Alternatively, the different end groups in PBMA (vide supra) may play a role in breaking the aggregation.
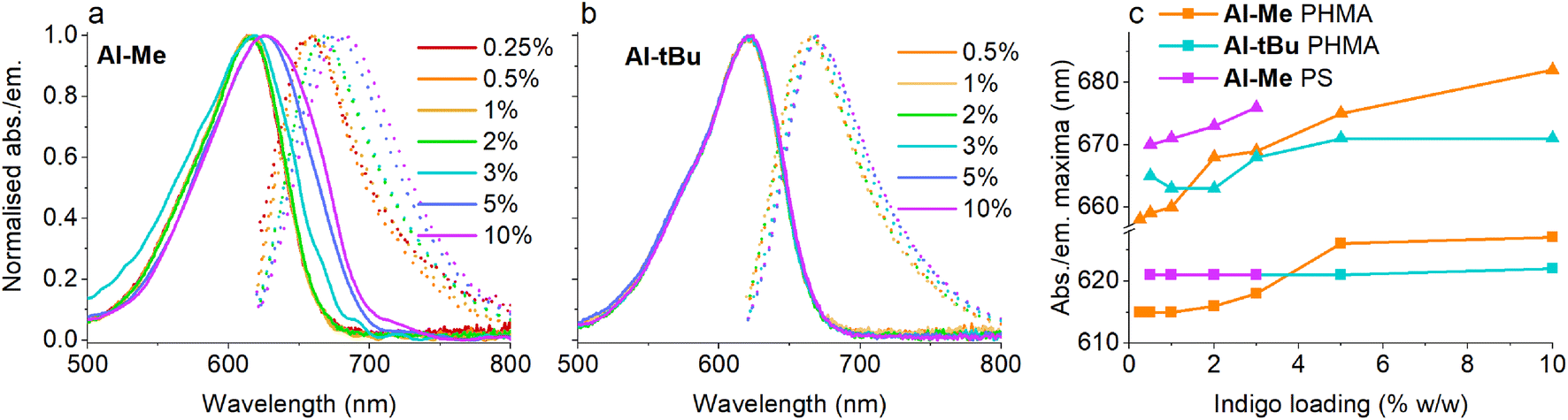 | ||
| Fig. 3 Absorption and emission spectra of (a) Al-Me and (b) Al-tBu at different dye loadings in PHMA. (c) Absorption (squares) and emission (triangles) maxima of Al-Me and Al-tBu in PHMA and Al-Me in PS at different dye loadings. | ||
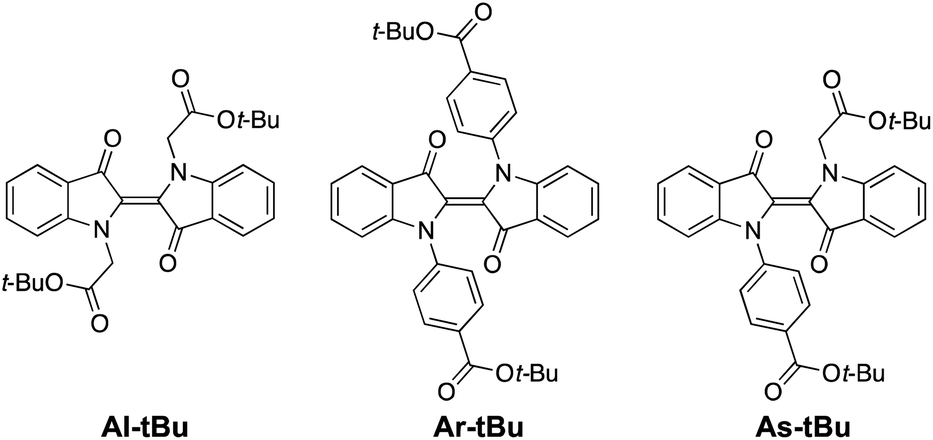 | ||
| Scheme 3 Studied tert-butyl-substituted indigo derivatives. | ||
Even more importantly, we found that dye concentration plays a major role in determining the photoswitching dynamics of indigos in a solid environment. At an Al-Me loading of 10% (in PHMA), the Z-content in PSS660 is 10% and the Z half-life only 0.14 seconds. At a loading of 0.5%, the respective values are 67% and 39 s (Fig. 4a). In terms of thermal energy barriers, the difference (ΔΔG‡) is approximately 1.8 kcal mol−1. This could easily be interpreted as a result of aggregation; also the exponential factor b in the stretched exponential fit decreased as the dye loading increased, pointing towards reduced homogeneity (Table S3†). However, even though the bulky tert-butyl groups prevent or decrease the aggregation of the indigos, the same trend is seen for Al-tBu. We note that although these two parameters correlate, there is no causality between them. Apart from perhaps the highest Al-Me loadings, thermal isomerisation is not fast enough to affect the PSS distribution with our experimental setup. Most likely the changes in spectra, PSS distribution, and thermal isomerisation kinetics are caused by additional intermolecular interactions between the switched molecules. Similar dependence between concentration and the isomerisation efficiency was observed for all other indigo–polymer combinations for which concentration series were studied: Ar-Me and As-Me in PHMA (Fig. 4b) as well as for Al-Me in PBMA and PS (Fig. S8†).
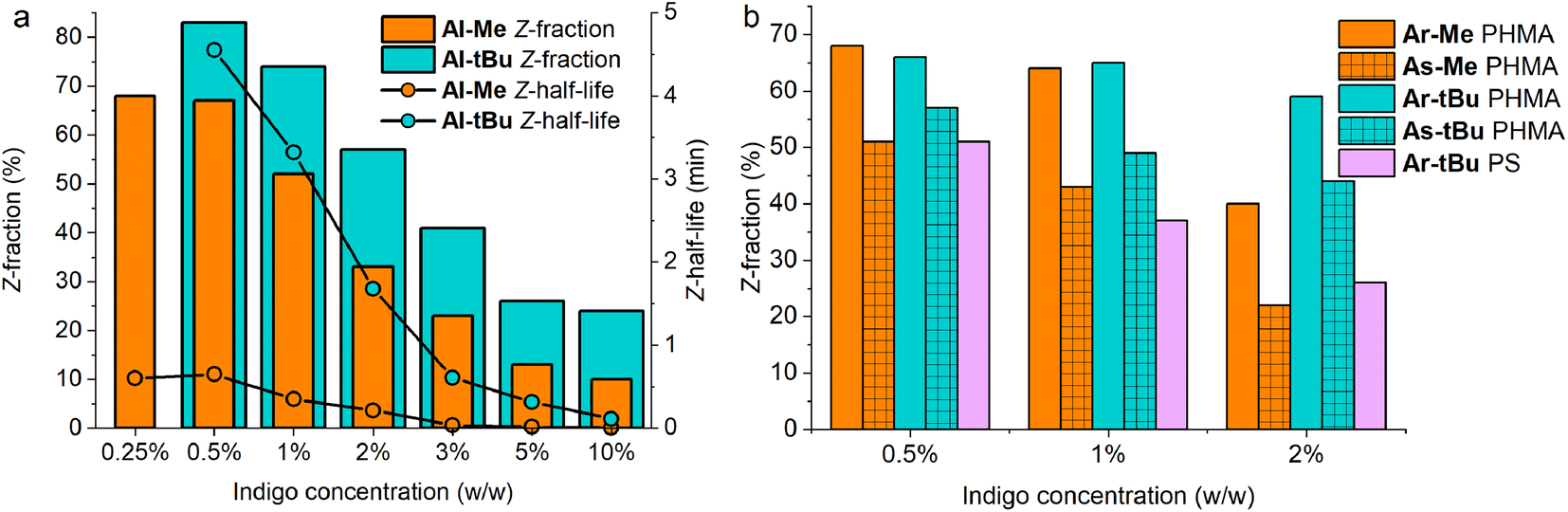 | ||
| Fig. 4 (a) Z-Fractions upon excitation with 660 nm light and thermal Z half-lives of Al-Me and Al-tBu, and (b) Z-fractions of Ar-Me, As-Me, Ar-tBu and As-tBu at different dye loadings in PHMA and Ar-tBu in PS. | ||
Gratifyingly, our strategy to enhance the solid-state switchability by bulking the indigo molecules is effective: Al-tBu exhibits generally better PSS distributions and longer Z half-lives than the electronically similar Al-Me, reaching a Z-content of 83% in the PSS and a 4.6 min thermal Z half-life at a loading of 0.5% (Fig. 4a). This is logical, as the addition of bulky groups is known to improve the photoswitching parameters of other E–Z photoswitches in a constrained environment by increasing the free space these molecules can operate in.65,66 Much to our delight, this approach is also applicable to other derivatives. We synthesised Ar-tBu and As-tBu and studied their isomerisation in PHMA at loadings from 0.5 to 2%, and the Z-fraction in PSS increased for both switches when compared to the respective methyl-capped compounds (Fig. 4b), especially at a loading of 2%. Using this strategy, switching was relatively efficient even in PS, a film containing 0.5% of Ar-tBu reaching a PSS with 51% Z. In this case, thermal Z-to-E isomerisation was very slow, exhibiting a half-life of 9.7 hours. This extended stability comes with a markedly greater value than the 26 minutes measured in solution, corresponding to a difference of approximately 1.9 kcal mol−1 in the thermal barrier. For applications operating on the timescale of minutes, the system is virtually bistable (Fig. S9†).
Conclusions
In just one or two synthetic steps, the familiar indigo dye yields photoswitches that operate with red light and exhibit negative photochromism, two key characteristics particularly intriguing for a range of applications from optics to photobiology. Although the synthetic pathways to indigo photoswitches and their photochemistry in solution have been studied in recent years, it has not been established whether their favourable properties can be transferred to solid matrices – which is crucial for real-world applications. Herein, we have for the first time studied indigo photoswitching in amorphous polymer films made of PS, P4VP, PMMA, PBMA and PHMA. We have doped the polymer matrices with six indigo switches that share favourable photochemical properties but differ in their N-substituents and thermal isomerisation dynamics. Every switch is decorated with two ester moieties which, in addition to improving the photochemical properties, serve as synthetic handles for potential further functionalisation. In all of the polymers, the switches can be reversibly isomerised between their E and Z forms with 660 and 500 nm light, respectively, conserving their spectral qualities with the thermal isomerisation half-lives in the range of minutes. Initial trial showed that the switched percentage is low in rigid environments (PS, P4VP, PMMA), where the Z-content remains mainly below 20% under red-light excitation. This inefficiency is presumably due to the constraints applied on the steric switching by the matrix.We propose that the distribution can be improved by reducing the amount of mechanical strain the E-to-Z isomerisation creates with one of the two means: (i) choosing a softer polymer matrix such as PBMA or PHMA, or (ii) incorporating bulky tert-butyl groups in the molecular design to increase the free volume surrounding the switch. Both strategies are proven effective for all studied indigos, and the switched fractions are greatly improved. Furthermore, as the most intriguing finding, indigo photoswitching is shown to be highly concentration-dependent in a solid environment, even at low loadings (e.g., from 0.5 to 1%): both PSS distribution and the thermal stability of the Z isomer increase when indigo concentration is decreased. The behaviour is common for all studied derivatives and can be exploited as a third strategy for enabling efficient solid state photoswitching. When these strategies work in concert (i.e., low loadings of a tert-butyl-decorated indigo in a soft polymer), isomerisation in the solid state is as efficient as in solution, reaching up to 83% Z for Al-tBu in PHMA. Simultaneously, the thermal stability of the Z isomer can be improved even beyond the stability in solution. Thus, we conclude that indigo photoswitches show great potential as red-light-responsive molecular tools in material applications. By judicious choice of molecular structures and the solid-state surrounding, photo-isomerisation can occur efficiently in polymer environments. Our current effort is thus focused on translating the indigo photoswitching from the molecular level to the macroscopic level, where the material properties can be modulated by red light.
Data availability
The data are available upon request.Author contributions
K. K. carried out the synthesis and characterisation of the compounds and preparation and photochemical studies of the polymer films with the help of J. V. and M. V., and wrote the original manuscript. C.-Y. H., S. H. and A. P. initiated and supervised the work and edited the draft. The final manuscript was written by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. K. is grateful for the funding from the Tampere University Graduate School. C.-Y. H. is indebted to the financial supports by Institute for Chemical Reaction Design and Discovery (WPI-ICReDD) and Hokkaido University. A. P. acknowledges the financial support of the Academy of Finland (Center of Excellence LIBER, No. 346107, Flagship Programme PREIN, No. 320165). S. H. thanks the German Research Foundation (DFG via SFB 1349, project ID: 387284271) for support. The authors acknowledge Dr Jussi Isokuortti for his help with the fluorescence measurements.Notes and references
- C. Kaspar, B. J. Ravoo, W. G. van der Wiel, S. V Wegner and W. H. P. Pernice, Nature, 2021, 594, 345–355 CrossRef CAS.
- A. Walther, Adv. Mater., 2020, 32, 1905111 CrossRef CAS.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS.
- A. Priimagi and A. Shevchenko, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 163–182 CrossRef CAS.
- S. L. Oscurato, F. Reda, M. Salvatore, F. Borbone, P. Maddalena and A. Ambrosio, Laser Photonics Rev., 2022, 16, 2100514 CrossRef.
- X. Huang and T. Li, J. Mater. Chem. C, 2020, 8, 821–848 RSC.
- C. Fedele, T.-P. Ruoko, K. Kuntze, M. Virkki and A. Priimagi, Photochem. Photobiol. Sci., 2022, 21, 1719–1734 CrossRef CAS.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS.
- Z. Mahimwalla, K. G. Yager, J. Mamiya, A. Shishido, A. Priimagi and C. J. Barrett, Polym. Bull., 2012, 69, 967–1006 CrossRef CAS.
- Y. Chen, J. Yang, X. Zhang, Y. Feng, H. Zeng, L. Wang and W. Feng, Mater. Horiz., 2021, 8, 728–757 RSC.
- T. Bian, Z. Chu and R. Klajn, Adv. Mater., 2020, 32, 1905866 CrossRef CAS.
- C. Petermayer and H. Dube, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef.
- I. Aprahamian, Chem. Commun., 2017, 53, 6674–6684 RSC.
- M. W. H. Hoorens, M. Medved', A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. Jan Buma and W. Szymanski, Nat. Commun., 2019, 10, 2390 CrossRef.
- S. Crespi, N. A. Simeth, M. Di Donato, S. Doria, C. N. Stindt, M. F. Hilbers, F. L. Kiss, R. Toyoda, S. Wesseling, W. J. Buma, B. L. Feringa and W. Szymański, Angew. Chem., Int. Ed., 2021, 60, 25290–25295 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- M. Quant, A. E. Hillers-Bendtsen, S. Ghasemi, M. Erdelyi, Z. Wang, L. M. Muhammad, N. Kann, K. V Mikkelsen and K. Moth-Poulsen, Chem. Sci., 2022, 13, 834–841 RSC.
- A. Gonzalez, E. S. Kengmana, M. V Fonseca and G. G. D. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- D. Mutruc, A. Goulet-Hanssens, S. Fairman, S. Wahl, A. Zimathies, C. Knie and S. Hecht, Angew. Chem., Int. Ed., 2019, 58, 12862–12867 CrossRef CAS PubMed.
- J. Boelke and S. Hecht, Adv. Opt. Mater., 2019, 7, 1900404 CrossRef.
- E. Yousif and R. Haddad, Springerplus, 2013, 2, 398 CrossRef PubMed.
- M. M. Becker and Z. Wang, J. Mol. Biol., 1989, 210, 429–438 CrossRef CAS PubMed.
- W. F. Cheong, S. A. Prahl and A. J. Welch, IEEE J. Quantum Electron., 1990, 26, 2166–2185 CrossRef.
- V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud’homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS.
- Z. Zhang, W. Wang, M. O'Hagan, J. Dai, J. Zhang and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202205758 CAS.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem.–Eur. J., 2014, 20, 16492–16501 CrossRef CAS.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS.
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS.
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547 CrossRef CAS.
- K. Kuntze, J. Viljakka, E. Titov, Z. Ahmed, E. Kalenius, P. Saalfrank and A. Priimagi, Photochem. Photobiol. Sci., 2022, 21, 159–173 CrossRef CAS.
- A. Kerckhoffs, K. E. Christensen and M. J. Langton, Chem. Sci., 2022, 13, 11551–11559 RSC.
- S. Wiedbrauk and H. Dube, Tetrahedron Lett., 2015, 56, 4266–4274 CrossRef CAS.
- C. Petermayer, S. Thumser, F. Kink, P. Mayer and H. Dube, J. Am. Chem. Soc., 2017, 139, 15060–15067 CrossRef CAS.
- C.-Y. Huang, A. Bonasera, L. Hristov, Y. Garmshausen, B. M. Schmidt, D. Jacquemin and S. Hecht, J. Am. Chem. Soc., 2017, 139, 15205–15211 CrossRef CAS.
- A. Goulet-Hanssens, C. Rietze, E. Titov, L. Abdullahu, L. Grubert, P. Saalfrank and S. Hecht, Chem, 2018, 4, 1740–1755 CAS.
- J. Isokuortti, K. Kuntze, M. Virkki, Z. Ahmed, E. Vuorimaa-Laukkanen, M. A. Filatov, A. Turshatov, T. Laaksonen, A. Priimagi and N. A. Durandin, Chem. Sci., 2021, 12, 7504–7509 RSC.
- K. Kuntze, J. Isokuortti, A. Siiskonen, N. Durandin, T. Laaksonen and A. Priimagi, J. Phys. Chem. B, 2021, 125, 12568–12573 CrossRef CAS.
- A.-L. Leistner and Z. L. Pianowski, Eur. J. Org. Chem., 2022, 2022, e202101271 CAS.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS.
- S. Yamazaki, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2011, 13, 1618–1628 RSC.
- Š. Budzák, J. Jovaišaitė, C.-Y. Huang, P. Baronas, K. Tulaitė, S. Juršėnas, D. Jacquemin and S. Hecht, Chem.–Eur. J., 2022, 28, e202200496 CrossRef.
- D. Pinheiro, M. Pineiro, A. M. Galvão and J. S. Seixas de Melo, Chem. Sci., 2021, 12, 303–313 RSC.
- D. C. Nobre, C. Cunha, A. Porciello, F. Valentini, A. Marrocchi, L. Vaccaro, A. M. Galvão and J. S. S. de Melo, Dyes Pigm., 2020, 176, 108197 CrossRef CAS.
- D. Farka, M. Scharber, E. D. Głowacki and N. S. Sariciftci, J. Phys. Chem. A, 2015, 119, 3563–3568 CrossRef CAS.
- S. Ganapathy, R. G. Zimmermann and R. G. Weiss, J. Org. Chem., 1986, 51, 2529–2535 CrossRef CAS.
- J. H. Porada, J.-M. Neudörfl and D. Blunk, New J. Chem., 2015, 39, 8291–8301 RSC.
- Y. Matsumoto and H. Tanaka, Heterocycles, 2003, 60, 1805–1810 CrossRef CAS.
- M. Poutanen, Z. Ahmed, L. Rautkari, O. Ikkala and A. Priimagi, ACS Macro Lett., 2018, 7, 381–386 CrossRef CAS.
- S. Hisham, N. Muhamad Sarih, H. A. Tajuddin, Z. H. Zainal Abidin and Z. Abdullah, RSC Adv., 2021, 11, 15428–15437 RSC.
- C. Pakula, C. Hanisch, V. Zaporojtchenko, T. Strunskus, C. Bornholdt, D. Zargarani, R. Herges and F. Faupel, J. Mater. Sci., 2011, 46, 2488–2494 CrossRef CAS.
- G. Clavier, F. Ilhan and V. M. Rotello, Macromolecules, 2000, 33, 9173–9175 CrossRef CAS.
- R. Kohlrausch, Ann. Phys., 1854, 167, 56–82 CrossRef.
- B. D. Smith, D. E. Alonso, J. T. Bien, E. C. Metzler, M. Shang and J. M. I. I. Roosenberg, J. Org. Chem., 1994, 59, 8011–8014 CrossRef CAS.
- C. Barrett, A. Natansohn and P. Rochon, Chem. Mater., 1995, 7, 899–903 CrossRef CAS.
- M. El Gemayel, K. Börjesson, M. Herder, D. T. Duong, J. A. Hutchison, C. Ruzié, G. Schweicher, A. Salleo, Y. Geerts, S. Hecht, E. Orgiu and P. Samorì, Nat. Commun., 2015, 6, 6330 CrossRef CAS.
- E. N. Cho, D. Zhitomirsky, G. G. D. Han, Y. Liu and J. C. Grossman, ACS Appl. Mater. Interfaces, 2017, 9, 8679–8687 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting results, synthetic details, 1H NMR, 13C NMR and UV-vis spectra of synthesised compounds, sample preparation details, UV-vis spectra and thermal isomerisation graphs of the studied indigo–polymer combinations. See DOI: https://doi.org/10.1039/d2sc06790k |
| This journal is © The Royal Society of Chemistry 2023 |