
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of SARS-CoV-2 3CL protease mutations on nirmatrelvir inhibitory efficiency. Computational insights into potential resistance mechanisms†
Carlos A.
Ramos-Guzmán
 ab,
Milorad
Andjelkovic
a,
Kirill
Zinovjev
a,
J. Javier
Ruiz-Pernía
*a and
Iñaki
Tuñón
*a
ab,
Milorad
Andjelkovic
a,
Kirill
Zinovjev
a,
J. Javier
Ruiz-Pernía
*a and
Iñaki
Tuñón
*a
aDepartamento de Química Física, Universidad de Valencia, 46100 Burjassot, Spain. E-mail: ignacio.tunon@uv.es; j.javier.ruiz@uv.es
bInstituto de Materiales Avanzados, Universidad Jaume I, 12071 Castelló, Spain
First published on 14th February 2023
Abstract
The use of antiviral drugs can promote the appearance of mutations in the target protein that increase the resistance of the virus to the treatment. This is also the case of nirmatrelvir, a covalent inhibitor of the 3CL protease, or main protease, of SARS-CoV-2. In this work we show how the by-residue decomposition of noncovalent interactions established between the drug and the enzyme, in combination with an analysis of naturally occurring mutations, can be used to detect potential mutations in the 3CL protease conferring resistance to nirmatrelvir. We also investigate the consequences of these mutations on the reaction mechanism to form the covalent enzyme-inhibitor complex using QM/MM methods. In particular, we show that the E166V variant of the protease displays smaller binding affinity to nirmatrelvir and larger activation free energy for the formation of the covalent complex, both factors contributing to the observed resistance to the treatment with this drug. The conclusions derived from our work can be used to anticipate the consequences of the introduction of nirmatrelvir in the fitness landscape of the virus and to design new inhibitors adapted to some of the possible resistance mechanisms.
Introduction
Since the end of 2019, the fight against COVID-19 has been one of the priorities in research. Many efforts have been made towards the development of vaccines1 and antiviral drugs2 that can be used to prevent or treat this disease. However, the ability of viruses to mutate offers them possible ways to escape the action of drugs and vaccines, triggering an evolution battle between therapeutical tools and the virus. New SARS-CoV-2 variants, some of them showing partial evasion of vaccines, have already emerged. The Omicron variant, for example, has been responsible for most of the latest infection waves across the world.3 Typically, new virus strains display frequent mutations in the spike protein which is the target of current vaccines.4Regarding antiviral drugs, three of them have been already authorized for the treatment of COVID-19: remdesivir,5 molnupiravir6 and nirmatrelvir.2 The first two are directed against the RNA replication process, while the target of the third drug is one of the two proteases of the virus: the main or 3CL protease (3CLpro). This dimeric enzyme is key for the replication cycle of the virus because it is in charge of cutting the long polyproteins produced after the translation of the genomic material in 11 positions, recognizing a glutamine residue at the P1 site (just before the peptide bond to be hydrolysed).7 Due to the fact that none of the known human proteases use this recognition sequence, 3CLpro has attracted considerable attention as a target for the development of inhibitors that can act as antivirals with reduced side effects. This has motivated continuous efforts to look for adequate inhibitors of the protease to be used as potential drugs, using both experimental8–14 and computational approaches.15–19 Many of these compounds are covalent inhibitors that contain an electrophilic site able to react with the catalytic cysteine of 3CLpro, Cys145, mimicking the behavior of the peptidic substrate. Covalent inhibitors first bind into the active site of the protease forming a non-covalent complex (EI) and then react with Cys145 to form a covalent complex (E–I):
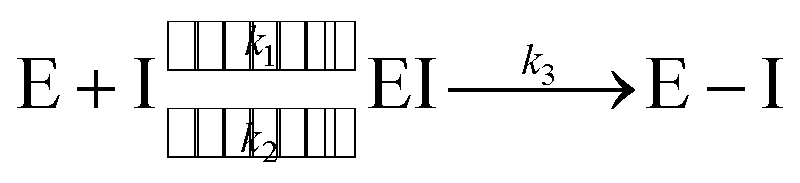 | (1) |
Nirmatrelvir, see Scheme 1, contains a nitrile group as a warhead that forms a thioimidate after reaction with the thiol group of the catalytic cysteine.20 Nirmatrelvir is associated with ritonavir, a compound that boosts its activity, under the commercial name of Paxlovid. This drug, developed by Pfizer, is administered orally and received authorization for emergency use in December 2021. Paxlovid drastically reduces hospitalization and death in patients with risk factors21 although it recently failed to show statistically significant protection for standard patients.22
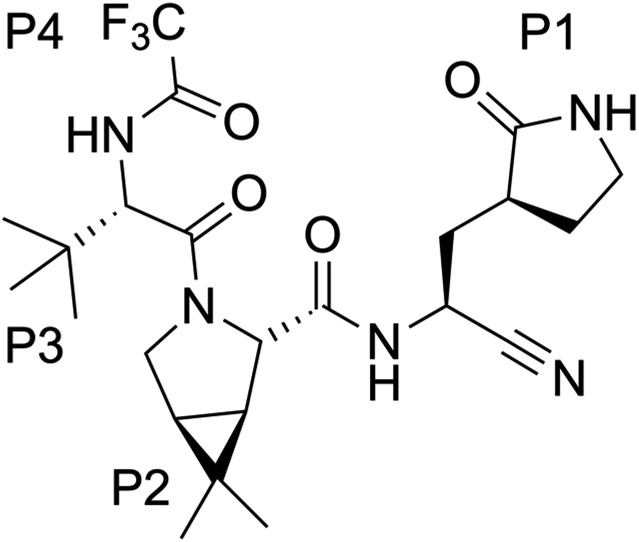 | ||
| Scheme 1 Chemical structure of nirmatrelvir. | ||
The generalized use of nirmatrelvir or other inhibitors of the 3CL protease could be threatened by the development of antiviral resistance, acquired after mutations in this enzyme. In principle, the comparison of the sequences of the main proteases of SARS-CoV-2 and other viruses belonging to the same sarbecovirus lineage of the β-CoV genus leads to the conclusion that this enzyme is highly conserved. For example, SARS-CoV and SARS-CoV-2 main proteases share 96% of the amino acid sequence and the main residues involved in catalysis are fully conserved.23 This would give some credit to the hypothesis that the 3CL protease is a safe target in terms of evolutive strategies of the virus to escape. However, a recent study showed that this conclusion is not maintained when the comparison is extended to viruses belonging to other lineages of the β-CoV genus; although the 3CL protease is significantly more conserved than the spike protein, the comparison of sequences coming from different coronaviruses indicates that this enzyme is also highly mutable.24 The conclusion from this study is that the use of nirmatrelvir will probably lead to new virus variants with mutations in the 3CL protease conferring resistance to the treatment. The analysis of the virus genome sequences from COVID-19 patients also indicates a high variability in some positions of this protein, according to mutation summary data from the Global Initiative on Sharing Avian Influenza Data (GISAID) CoVsurver app.25,70
A study by Pfizer showed that nirmatrelvir remained effective against several virus variants, including Omicron that contains a single mutation in 3CLpro (P132H).26 More recent studies have directly addressed the question of the evolutive pressure caused by the use of nirmatrelvir. One study found that after several rounds of nirmatrelvir treatment, the 3CL protease accumulated up to three mutations, at positions L50, E166 and L167, which conferred increased resistance to the drug.27 Single mutations increased the half-maximal inhibitory concentration (IC50) of the drug by a factor between 4 and 10, while the three combined mutations increased 72-fold this value.27 Another study also found that combined mutations L50F and E166V resulted in 80-fold resistance.28 The E166V mutant was also identified as a mutation conferring resistance to nirmatrelvir in a yeast-based mutational scanning approach.29 Interestingly, the position E166 is shown to be strictly conserved in the comparison among the sequences of different coronaviruses.24 While the mutation of this position could, in principle, reduce viral fitness decreasing the enzymatic activity versus natural substrates, additional mutations, for example at the L50 position, could compensate for the fitness cost of the E166 mutation.28,30 The position E166 was also identified in an analysis of nirmatrelvir resistance among naturally occurring mutations of 3CLpro.31 This study identifies S144, M165, E166, H172 and Q192 as hot spots for nirmatrelvir tolerance, because some mutations at these positions are likely to keep the enzymatic activity while increasing drug resistance. In a recent study using a chimeric virus the 3CLpro mutants Y54C, G138S, L167F, Q192R, A194S and F305L have been shown to present resistance against nirmatrelvir and GC376 inhibitors.32 A combined computational and experimental screening has identified five additional variants (N142L, E166M, Q189E, Q189I and Q192T) with reduced susceptibility to nirmatrelvir in vitro.33 Finally, a recent study has identified transmissible variants of SARS-CoV-2 with two mutations (P168 deletion, or ΔP168, and A173V) that when present together increase the resistance of 3CLpro to nirmatrelvir by 51-fold (versus 5- or 11-fold for each single mutant, respectively).34
In this study, we use simulation tools to study the binding and reactivity of nirmatrelvir in 3CLpro. We show that the analysis of the contributions of different residues to the formation of the noncovalent complex between the enzyme and the inhibitor is useful to identify some positions susceptible to mutations that confer resistance to the drug. We further analyse the consequences of one of these mutations on the reaction mechanism for the formation of the covalent complex, to rationalize strategies for the development of new and more efficient 3CLpro covalent inhibitors. All in all, our study shows that computational simulations are an important tool to be added to the arsenal to fight COVID-19 and to rationalize the origin of resistance mechanisms to the treatment with 3CLpro inhibitors.
Methods
Molecular mechanics simulations
Michaelis complexes of the wild type 3CLpro protease of SARS-CoV-2 with a peptidic substrate and with nirmatrelvir were obtained from our previous simulations.35,36 Briefly, these structures were prepared starting from the crystal structure of the 3CL protease of SARS-CoV-2 with PDB code 6Y2F.37 The peptide substrate was built using the Maestro tool38 with the sequence Ac-Ser-Ala-Val-Leu-Gln-Ser-GlyPhe-NMe and placed in both the active sites, in agreement with X-ray structures. The same procedure was used to prepare the Michaelis complex with nirmatrelvir. Michaelis complexes of the E166V variant, with either the peptide substrate or nirmatrelvir in the active sites, were prepared introducing mutations with Maestro38 in wild type structures with the corresponding ligand.The tleap tool of AmberTools18 (ref. 39) was employed to prepare all simulation systems. The Michaelis complexes described before were solvated in a box with a buffer region of at least 12 Å from any protein/substrate atom to the limits of the simulation box. Na+ atoms were added to neutralize the charges and PROPKA3.0 (ref. 40) was used to determine the most likely protonation state of every residue at pH 7.4. The catalytic dyad was simulated in the neutral state, in agreement with experimental41 and computational evidence.35,42 ll classical MD simulations have been run using Amber18.39 The protein was described by means of the ff14SB43 forcefield, while water molecules were described using TIP3P44 potential. Nirmatrelvir was parametrized using the Antechamber program45 available in the AmberTools package. Atomic charges were calculated using the restrained electrostatic potential (RESP)46 method at the HF/6-31G* level of theory. All long-range electrostatic interactions were described using the particle mesh Ewald method (PME),47 and the cut-off radius was set to 10 Å. SHAKE48 was used to freeze the bonds involving hydrogen atoms, allowing the use of a 2 fs time step during simulations. For all classical molecular dynamic simulations, the Amber18 GPU version of pmemd49,50 was employed. The details of the minimization and equilibration procedure are described elsewhere.35,36 After thermalization at 300 K, three replicas 1 μs each in the NVT ensemble were run for each system in order to guarantee enough sampling.
Molecular mechanics free energy calculations
The impact of a possible mutation on the formation of the noncovalent complex between the 3CL protease and nirmatrelvir can be formally quantified through an effect on the half-maximal inhibitory concentration (IC50). This magnitude depends on the values of the inhibitory (Ki) and Michaelis (Km) constants and substrate concentration ([S]):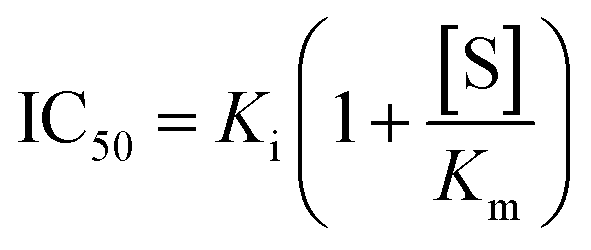 | (2) |
An increased value in the mutant (IC′50 > IC50) indicates a larger resistance to the drug. This depends on the effect of the mutation on both constants:
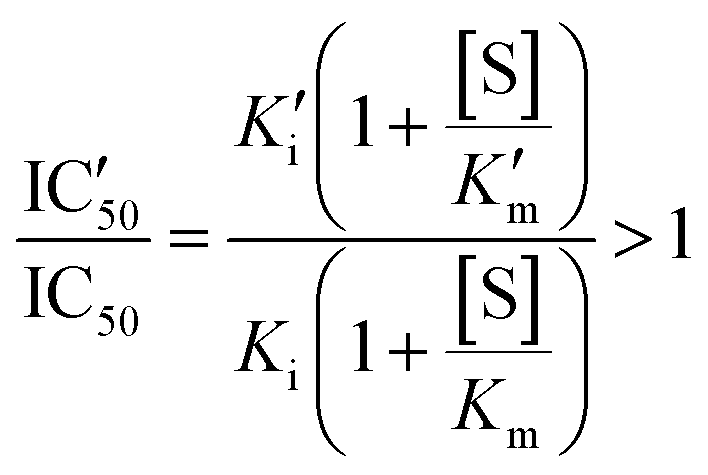 | (3) |
For small substrate concentration ([S] ≪ Km), the increase is solely determined by the change in the inhibitory constant and then by the effect on the inhibitor binding free energy.
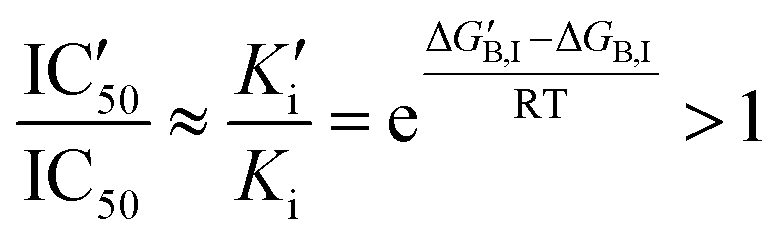 | (4) |
If gi,I is the impact of a mutation in position i to the binding free energy of the inhibitor 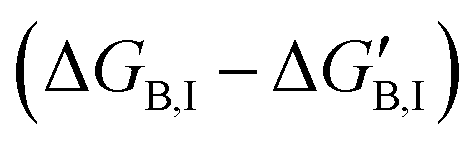 , then an increase in the half-maximal inhibitory concentration is due to those positions whose mutation makes a negative impact on the binding free energy:
, then an increase in the half-maximal inhibitory concentration is due to those positions whose mutation makes a negative impact on the binding free energy:
| gi,I < 0 | (5) |
For larger substrate concentrations ([S] >> Km), the change in the half-maximal inhibitory concentration depends on the relative effect of the mutation on the Michaelis and the inhibitory constants and then on the impact on the inhibitor and substrate binding free energies:
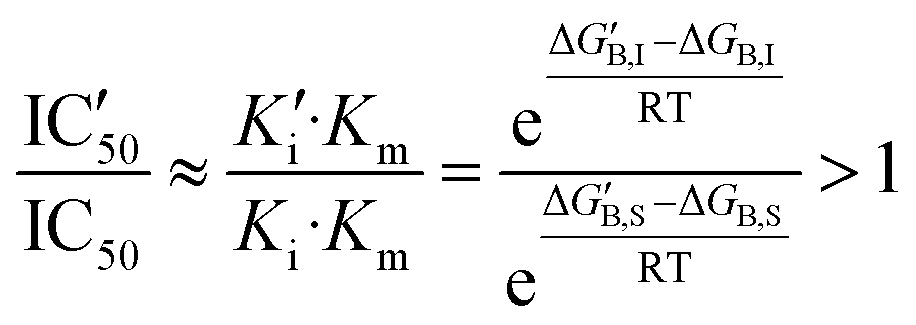 | (6) |
Introducing also the impact of the mutation on position i to the binding free energy of the substrate (gi,S), now the condition for a mutation to increase IC50 would be:
| gi,S − gi,I > 0 or gi,I − gi,S < 0 | (7) |
In this work we approximate the values of gi by the contribution of residue i to the binding free energy of the inhibitor or the substrate. Molecular Mechanics Generalized Born Surface Area (MMGBSA) calculations were run using the MMPBSA.py script51 available in AmberTools18. The Ante-MMGBSA.py51 utility was used to create compatible complex, receptor and ligand topology files and GB solvation radii were set to mbondi2. The default Generalized Born method was applied and the salt concentration was set to 0.1 M. In order to calculate the contribution of each residue to the binding free energy, full electrostatic and van der Waals contributions were considered, instead of using the standard MMPBSA.py decomposition scheme that attributes half of the interaction energy to the ligand and the other half to the corresponding residue.52 Both decompositions, per residue and side-chain options, were applied. MMGBSA calculations were carried out for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 frames evenly extracted from a total of 200 trajectories of 50 ns (a total of 10 μs) ran for the wild type 3CLpro enzyme in the complex with the peptide substrate and with nirmatrelvir. Finally, to estimate the statistical uncertainty of each residue's contribution to the free energy, we computed the standard deviation of 10 averages, each corresponding to 1000 frames covering 1 μs of simulation each.
000 frames evenly extracted from a total of 200 trajectories of 50 ns (a total of 10 μs) ran for the wild type 3CLpro enzyme in the complex with the peptide substrate and with nirmatrelvir. Finally, to estimate the statistical uncertainty of each residue's contribution to the free energy, we computed the standard deviation of 10 averages, each corresponding to 1000 frames covering 1 μs of simulation each.
QM/MM simulations
Hybrid Quantum Mechanics/Molecular Mechanics (QM/MM)53,54 simulations were used to describe the chemical transformation from the noncovalent complex to the covalent one (see eqn (1)). QM/MM calculations were performed using a modified version of Amber18, coupled to Gaussian16 (ref. 55) for density functional theory calculations. We selected the M06-2X functional56 with the 6-31+G* basis set and D3 dispersion corrections57 to describe the QM region. This included the side chains of the catalytic dyad (His41 and Cys145), a water molecule involved in the reaction mechanism and the backbone atoms of residue P1 and the warhead of nirmatrelvir. The cutoff radius for the QM/MM interactions was set equal to 15 Å. As discussed below, this M062X-D3/MM combination provides results in excellent agreement with our previous B3LYPD3/MM calculations, which supports the reliability of our QM/MM simulations.36In order to explore the free energy landscape associated with the chemical reaction, the Adaptive String Method (ASM)58,59 developed in our research group was employed. According to this method, N replicas of the system, that correspond to the nodes of the string, are evolved following the mean force acting on each node while being kept equidistant. In this way the nodes converge to the minimal free energy path (MFEP) in the space spanned by the selected collective variables (CVs). For the reaction under study we selected as CVs the distances of all the bonds being broken or formed during the transformation (see Scheme 2). Once the string converged, a single path-CV (s) is defined to drive the system along the MFEP. Along this path-CV, the free energy profiles are obtained using umbrella sampling (US)60 and later integrated using WHAM.61 For the reaction of nirmatrelvir with the wild type and E166V variants of the SARS-CoV-2 3CLpro enzyme, each string was composed of 96 nodes. The simulations for each node were propagated using a time step of 1 fs until the RMSD of the string nodes dropped to 0.1 amu1/2·Å for at least 2 ps. US simulations were then carried out around each converged node of the string accumulating 10 ps of data collection for each window. The force constants and positions of the nodes, employed to bias the ASM and US simulations, were determined on the fly to ensure a probability density distribution of the reaction coordinate that was as homogeneous as possible.58 Replica exchange between neighboring string nodes was attempted every 50 steps to improve the convergence.
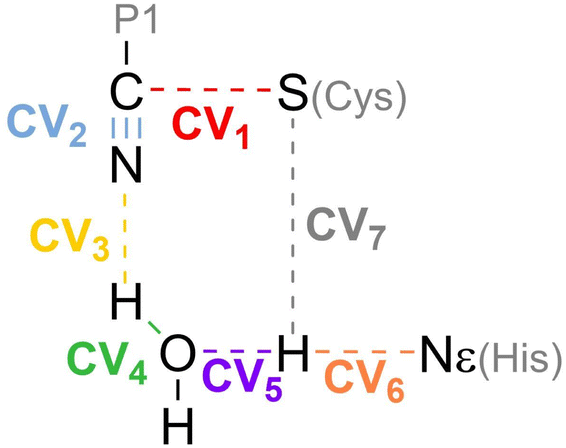 | ||
| Scheme 2 Collective Variables (CVs) employed to study the reaction mechanism of nirmatrelvir with 3CLpro. | ||
Results
Analysis of the noncovalent complex
As explained in the methodological section, we used the MMGBSA scheme to calculate the contributions of each residue of the 3CLpro enzyme to the binding of a peptidic substrate (gi,S) and of nirmatrelvir (gi,I). Fig. 1 displays the average values of the contribution of each residue to the total MMGBSA binding free energy of the inhibitor and the difference between substrate and inhibitor contributions, as obtained after a total of 10 μs of simulations. Only positions with gi,I smaller than −0.5 kcal mol−1 are displayed. The complete list of values and their statistical errors are given in Table S1,† while those represented in Fig. 1 are given in Table 1.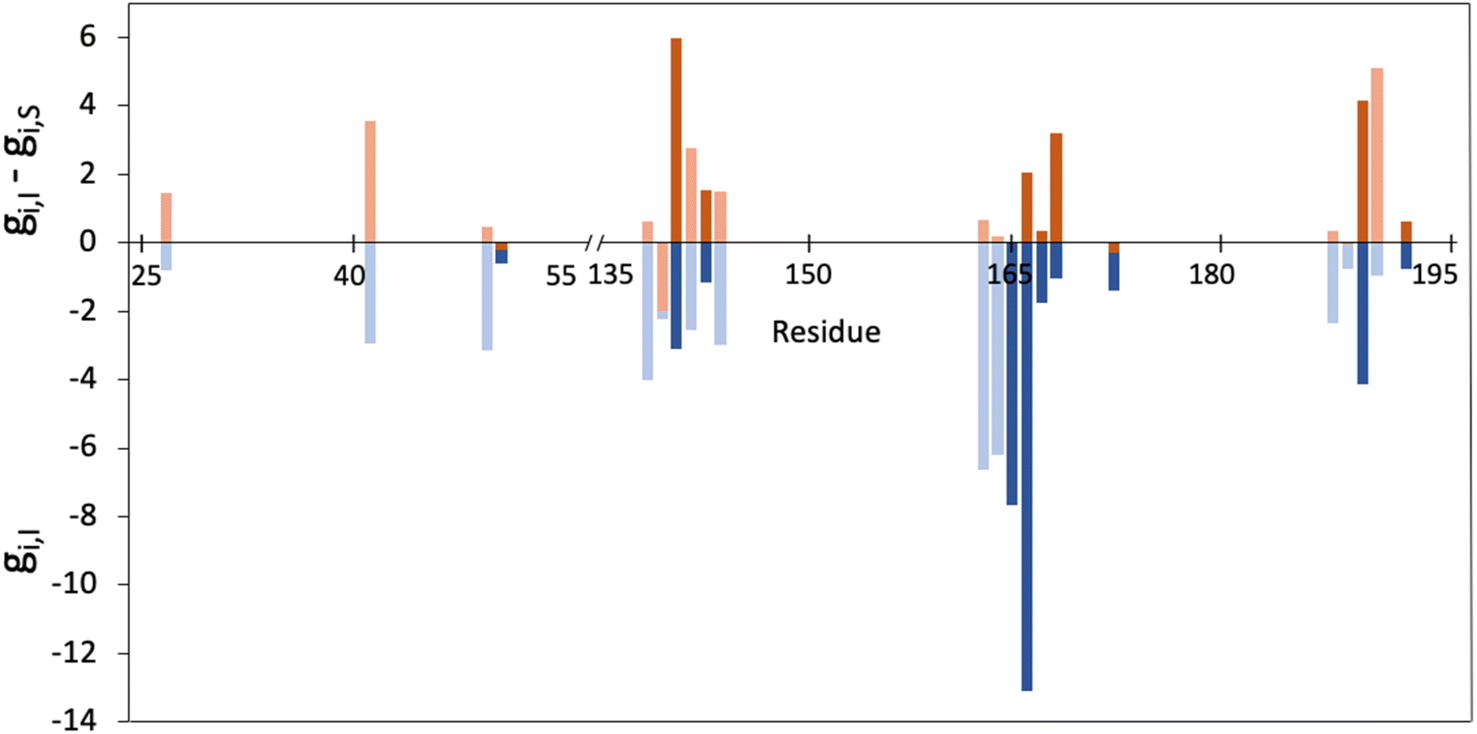 | ||
| Fig. 1 Representation of the contribution of each residue of the 3CLpro enzyme to the binding free energy of nirmatrelvir as determined from MMGBSA calculations (gi,I, in blue) and the differential contribution to the binding of nirmatrelvir and a peptide substrate (gi,I − gi,S, in orange). Only those residues with gi,I < 0.5 kcal mol−1 are shown. Free energies are given in kcal mol−1. Positions whose mutation was experimentally found to increase resistance are highlighted in darker colours. | ||
| Residue | g I | g I,sc | g S | g S,sc |
|---|---|---|---|---|
| L27 | −0.79 | −0.64 | −2.26 | −1.67 |
| H41 | −2.96 | −2.92 | −6.50 | −6.29 |
| M49 | −3.13 | −2.68 | −3.61 | −3.04 |
| L50 | −0.59 | −0.44 | −0.40 | −0.26 |
| F140 | −4.02 | −1.36 | −4.66 | −1.24 |
| L141 | - 2.25 | −0.93 | −0.25 | −0.76 |
| N142 | −3.09 | −2.17 | −9.08 | −6.00 |
| G143 | −2.54 | −0.15 | −5.31 | −0.79 |
| S144 | −1.15 | −0.64 | −2.67 | −0.84 |
| C145 | −2.97 | −1.74 | −4.47 | −2.29 |
| H163 | −6.65 | −6.51 | −7.32 | −7.16 |
| H164 | −6.20 | −1.15 | −6.38 | −1.39 |
| M165 | −7.66 | −4.69 | −7.70 | −4.78 |
| E166 | −13.10 | −8.42 | −15.14 | −7.17 |
| L167 | −1.75 | −1.14 | −2.10 | −1.50 |
| P168 | −1.04 | −0.66 | −4.26 | −3.28 |
| H172 | −1.39 | −0.97 | −1.10 | −0.81 |
| N187 | −2.37 | −1.71 | −2.72 | −2.15 |
| R188 | −0.76 | −0.82 | −0.72 | −0.68 |
| Q189 | −4.13 | −2.84 | −8.30 | −6.56 |
| T190 | −0.97 | −0.38 | −6.09 | −0.84 |
| Q192 | −0.76 | −0.73 | −1.39 | −0.47 |
Interestingly, up to ten of the positions that have been experimentally characterized as contributing to increase in resistance to nirmatrelvir (50, 142, 144, 165, 166, 167, 168, 172, 189 and 192)27,28,31–34 appear in the list of 22 residues that fulfil the condition gi,I < −0.5 kcal mol−1. In Fig. 1 these positions are highlighted with darker colours. This suggests that this criterion (eqn (5)) could be useful to predict potential mutations of 3CLpro that could confer SARS-CoV-2 resistance to treatments with nirmatrelvir. Instead, only two of the positions identified experimentally (50 and 172) fulfil the condition expressed in eqn (7). We will now discuss the positions appearing in this list.
H41, C145 and H163 are essential residues for the activity of this enzyme. These three residues are strictly conserved among CoVs,24 although, as discussed before, this criterion alone is not enough to discard possible mutations. Of these three residues, the first two form the catalytic dyad responsible for the protease activity, while the third one is part of the S1 site that recognizes the presence of glutamine in the peptidic substrate.35 The analysis of naturally occurring mutations shows that these three residues are needed for the enzymatic activity and that mutations in these position are not frequent because they led to inactive enzymes or to enzymes with decreased activity.31
The ten positions of Table 1 that have been experimentally characterized as possible spots for mutations conferring resistance are L50, N142, and S144 (strictly conserved among β-CoVs, but not in all CoVs), M165 and E166 (strictly conserved among CoVs), L167, P168, and H172 (strictly conserved among CoVs), Q189 (strictly conserved among β-CoVs, but not in all CoVs) and Q192 (also strictly conserved among CoVs).They all appear in the list of residues contributing to the binding free energy of nirmatrelvir with a free energy contribution below −0.5 kcal mol−1. For these residues the side chain contribution is a significant fraction of the free energy term, more than 50% in all cases (see Table 1). N142, E166, S144, P168 and Q192 contribute significantly more to the binding of the substrate than to nirmatrelvir (see Fig. 1 and Table 1). E166 is the residue contributing most to the binding of both the peptidic substrate and nirmatrelvir. This contribution is mostly due to three hydrogen bond interactions established with the substrate, two through the backbone (both the NH and CO moieties of this residue form hydrogen bonds with the substrate) and one between the carboxylate group of the side chain and the substrate P1 group.35 In this sense, E166A and E166V mutations, found after growing the virus in the presence of nirmatrelvir, increase the virus resistance to the treatment. The E166A variant shows a 10-fold increase in IC50, while the enzymatic activity is reduced approximately by the same factor.27 The E166V variant showed up to 260-fold nirmatrelvir resistance, as measure by the viral EC50.28 This mutation also reduces the enzymatic activity with the natural substrate.30 In the E166V and E166A mutants, the side chain carboxylate group at position 166 is substituted by an alkyl group, losing one hydrogen bond interaction with the P1 group of the substrate (see Fig. 2). According to our MMGBSA calculations (see Table 1), the side chain of residue E166 contributes −8.42 ± 0.13 kcal mol−1 to the binding of nirmatrelvir, a quantity larger, in absolute terms, than the contribution to the binding of the peptide substrate, −7.17 ± 0.16 kcal mol−1. This would explain the impact of its mutation on the increased resistance and the decreased natural activity observed experimentally. One should take into account that this quantity is probably overestimated due to the limitations of the MMGBSA approach: (i) specific solvent interactions of the carboxylate group are accounted by means of continuum model estimations; (ii) the geometry of the active site is not allowed to relax during the unbinding process. In fact, our simulations of the E166V mutant show that the loss of the hydrogen bond interaction between the side chain of residue 166 and the γ-lactam ring (P1 group) of nirmatrelvir is not the only effect contributing to the differences between the wild type and the mutant variants. The E166 residue in the wild type 3CLpro keeps a persistent salt-bridge interaction with the N-terminal group of the other protomer. In 3CLpro the N-finger of one protomer is known to play an active role pre-organizing the active site of the other protomer for catalysis.62 This interaction contributes to the integrity of the active site and to the stability of the dimer, the active form of the enzyme. In the E166V variant this salt-bridge is lost and the N-finger moves aways from the enzymatic S1 site, as observed in Fig. 2. The distributions of distances between the centers of mass of residue 166 and the N-terminal group obtained from MD simulations of wild type and E166V variants are provided in Fig. S1.†
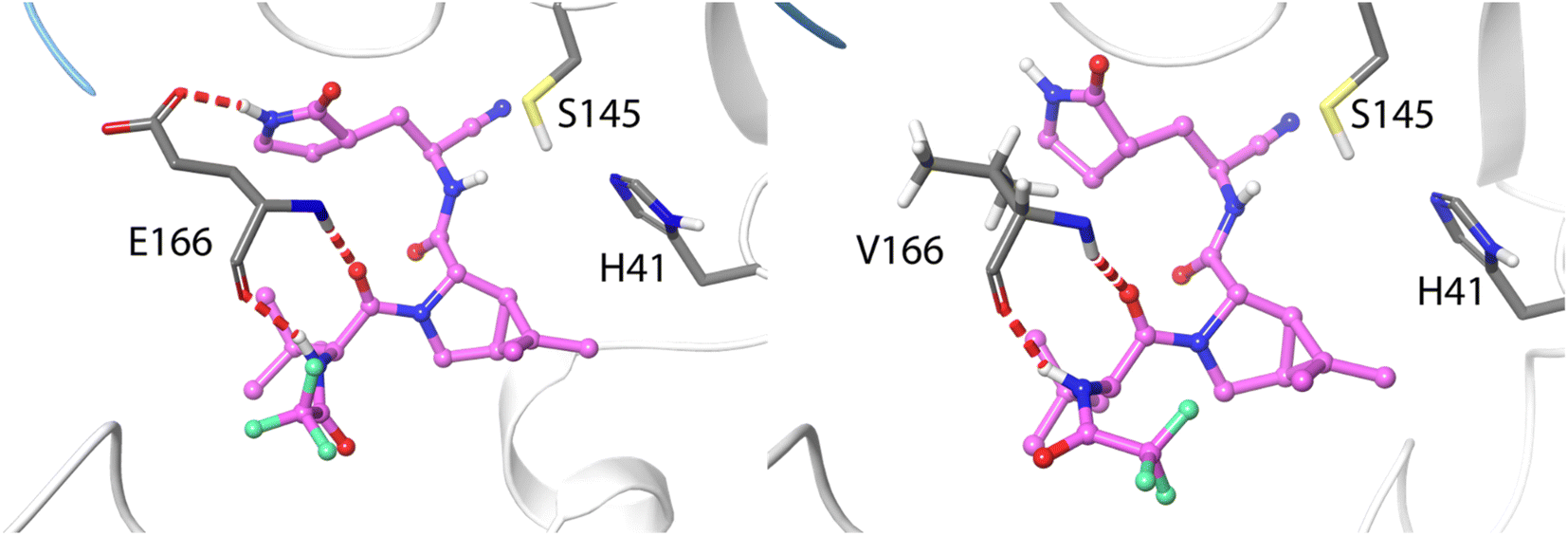 | ||
| Fig. 2 Hydrogen bond interactions formed between residue 166 and nirmatrelvir in wild type 3CLpro (left) and in the E166V variant (right). Nirmatrelvir is represented with carbon atoms in pink. The N-finger of the other protomer is depicted in blue. | ||
In general, experimental studies on resistance conferring mutations and our simulations agree on the importance of the residues found in the segment of the 3CLpro sequence comprised between positions 163 and 173. Up to five mutations experimentally found to increase resistance to nirmatrelvir are found in this fragment, while our free energy calculations demonstrate that these residues contribute significantly to the binding of the drug. The key role played by this group of residues in substrate binding, together with those forming the oxyanion hole (see below), was also confirmed by the analysis of molecular dynamics simulations of 3CLpro complexed with different peptides.42 We have performed a NCI analysis63,64 of nirmatrelvir-3CLpro intermolecular interactions, which confirms the relevant role of these residues establishing favourable interactions with the drug in the noncovalent complex. The result of this analysis is shown in Fig. S2.†
Regarding S144, this residue is strictly conserved among β-CoVs, but not in all CoVs.24 This residue makes part of the oxyanion hole, together with G143 and C145, that stabilizes the negative charge being developed on the carbonyl oxygen atom of the target peptide bond during the formation of the acylenzyme complex.35 A recent preprint reported a simulation study using nonequilibrium molecular dynamics to measure the response of the protein to substrate removal that highlights the dynamical behaviour of the oxyanion hole in the binding process.65 The analysis of the 15 most frequent naturally mutations at position 144 shows that ten of them present a significant reduction in enzymatic activity and only five still had comparable activity to the wild type enzyme. These five mutants also showed increased drug resistance to nirmatrelvir.31
From the remaining positions displaying the largest contributions to nirmatrelvir binding, one can select additional candidates for potential mutations that could confer drug resistance to the virus. L141 could be an interesting candidate because it contributes more to the binding free energy to nirmatrelvir than to the peptidic substrate, so, in principle, its mutation could confer resistance to nirmatrelvir without significantly affecting the enzymatic activity with the natural substrate. This residue is part of the P1 site and then used to recognize the substrate. However, this residue is not strictly conserved and it can be found substituted by I, V or M in other CoVs.24 The GISAID data also indicate that its deletion is the most frequent spontaneous mutation, although, in general, this position showed a small number of naturally occurring mutations until now.25,70 It must be taken into account that baseline mutations might appear in the databases just due to errors in the sequencing process.
L27 and R188 are two residues contributing similarly to the binding of nirmatrelvir and to the peptidic substrate. L27 is strictly conserved among all CoVs and contributes to the binding interaction with the P1 group of nirmatrelvir and also with the P1′ group of the peptidic substrate.36 R188 seems a better candidate for mutations because this residue varies significantly among CoVs: its position can be found occupied by other positively charged residues (K), neutral (Q or A) and even negatively charged (E). Naturally occurring mutations reported by the GISAID web page indicate that the most frequent change in this position is its deletion. This residue makes a significant contribution to binding through its side chain, presenting similar contributions to the binding of the substrate and the drug. Thus, mutations at this position could improve the viral resistance to nirmatrelvir, but probably also affecting its natural activity.
Other possible hot spots for mutations conferring nirmatrelvir resistance are M49 and H164, which interact significantly with the drug through their side chains. However, the most frequent natural mutations of these residues have been shown not to present significantly improved resistance to nirmatrelvir, even if some of them have a slightly improved enzymatic activity compared to the wild type variant.31
Finally, D187, F140 and G143 are less likely to present mutations that simultaneously confer resistance and keep the enzymatic activity. These residues establish key interactions to keep the integrity of the enzymatic structure (such as the salt-bridge between D187 and R40) or to bind the natural substrate at different stages of the reaction (F140 at the reactants state and G143 at the transition state).35 None of these positions is prone to frequent mutations according to GISAID data, (the most frequent is D187 deletion appearing in less than 0.008% of the sequences). However, a recent computational high-throughput protein design study identified F140I and G143D as potential mutations conferring resistance to nirmatrelvir.66
Formation of the covalent complex
We also analyse the impact of potential mutations on the formation of the covalent complex due to the reaction of nirmatrelvir with the catalytic cysteine, C145. We compare the reaction free energy profile obtained for the wild type enzyme to that obtained for the E166V mutant, selected because of its large impact on the binding of the drug, as discussed above. This mutation also displays a significant increase in the resistance of SARS-CoV-2 to the treatment with nirmatrelvir. While the impact of this mutation on the binding free energy associated with the formation of the noncovalent complex has been discussed, the possible impact on the reaction mechanism for the formation of the covalent complex remains to be clarified.Fig. 3 shows the behavior of relevant distances for the chemical reaction between nirmatrelvir and 3CLpro, both for the wild type and E166V variants. These include the distances between the C145-Sγ atom and the H41-Nε atom (Fig. 3a), which is associated with the proton transfer needed to activate the catalytic dyad forming an Ion Pair (IP), and the distance between the C145-Sγ atom and the nitrogen atom of the nirmatrelvir warhead (Fig. 3b), which is associated with the nucleophilic attack leading to the thioimidate complex. In both cases, we observe a bimodal distribution that can be explained considering that the side chain of C145 can be found in two different conformations (trans and gauche). These two possible conformations were already confirmed to be populated in the X-ray structure of the 3CLpro enzyme of the SARS-CoV virus62 and were also reported in the simulations of the SARS-CoV-2 variant in the complex with its natural substrate.35 Interestingly, our simulations show a slight decrease in the population of reactive conformers, those showing a shorter distance between the nitrogen atom of the nirmatrelvir warhead and C145-Sγ atoms, in the E166V mutant. The loss of a hydrogen bond between the E166 side chain and the P1 group of the substrate in the mutant also mismatches other interactions established between the drug and the enzyme. Fig. 3c shows the distribution of distances found between the NH amide moiety of the P1 group of nirmatrelvir and the C145-Sγ atom, in the wild type and E166V variants of SARS-CoV-2 3CLpro. In the mutant, the distances found in the simulations are longer, reflecting a slightly worse binding pose of the substrate for catalysis. As discussed below, this has consequences on the reaction free energy profile, because the S⋯HN interaction contributes to the stabilization of the negative charge developed on the sulphur atom when the IP state is reached.
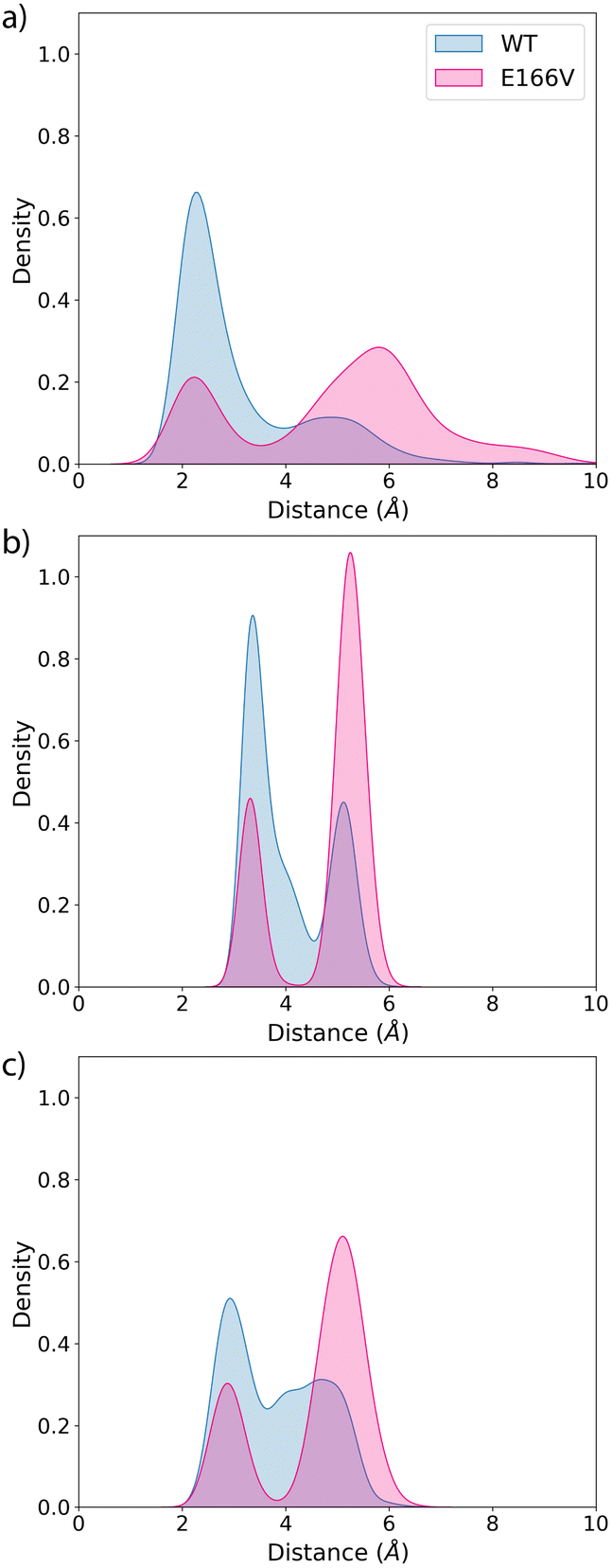 | ||
| Fig. 3 Probability distributions of the distances between: (a) the Sγ-H atom of C145 and the Nε atom of H41 (wild type and E166V variants in green and blue colors); (b) the Sγ atom of C145 and the electrophilic carbon atom of the inhibitor and (c) the Sγ atom of C145 and the amide N–H moiety of the P1 group of nirmatrelvir. | ||
Regarding the formation of the covalent complex, we computed the free energy profiles corresponding to the reaction of nirmatrelvir with the wild type and E166V 3CLpro variants using the Adaptive String Method, as described in the methodological section. The M06-2XD3/MM free energy profiles are shown in Fig. 4a and d together with the evolution of the relevant bond distances that change significantly during the chemical process (Fig. 4b and e) and a representation of the corresponding Transition States (TSs), Fig. 4c and f. Regarding the results obtained for the wild type enzyme, both the energetic and structural descriptions of the process are close to that previously obtained by us at the B3LYPD3/MM level.36 We have shown that the inhibition reaction in wild type enzymes with nirmatrelvir involves two consecutive stages.36 The first one is the activation of the catalytic dyad after the proton transfer from C145 to H41 to form the IP, a metastable species. Then the reaction continues with a proton transfer from H41 to the nitrogen atom of the nitrile warhead and the nucleophilic attack of the activated C145 on the electrophile carbon atom of the warhead to form a thioimidate. A water molecule is recruited to act as a proton shuttle between H41 and the nitrile warhead. This water molecule occupies the same position as the amide nitrogen atom of the targeted peptide bond in the natural substrate of the protease (see Fig. S3†). The same stages are now observed in the evolution of the bond distances obtained along the MFEPs in the wild type enzyme at the M06-2XD3 level (see Fig. 4b). The only difference between the B3LYPD3 and M06-2XD3 results is that the water mediated proton transfer is slightly more asynchronous in the second case, the transfer from H41 to the water molecule anticipating the proton transfer from the water to the nitrogen atom of the nitrile group. The free energy barriers obtained at both DFT levels are also quite similar (16.3 and 14.6 kcal mol−1 using B3LYP and M06-2X functionals, respectively). The B3LYP value is in better agreement with the activation free energy derived from the experimental kcat measured at 310 K and 18.2 kcal mol−1, which provides an upper limit to the barrier of the chemical step.67 The activation free energy of the chemical reaction is expected to be reduced at lower temperatures, because of the ion pair formation and water participation that result in negative activation entropy. A preprint reporting a QM/MM study of the potential energy surface for the reaction of nirmatrelvir suggests that the proton transfer from H41 to the substrate could take place through a complex proton wire involving three water molecules and two residues.68 A recent study using ONIOM calculations confirmed our proposal of a water mediated proton transfer from H41 to the nitrile warhead.69
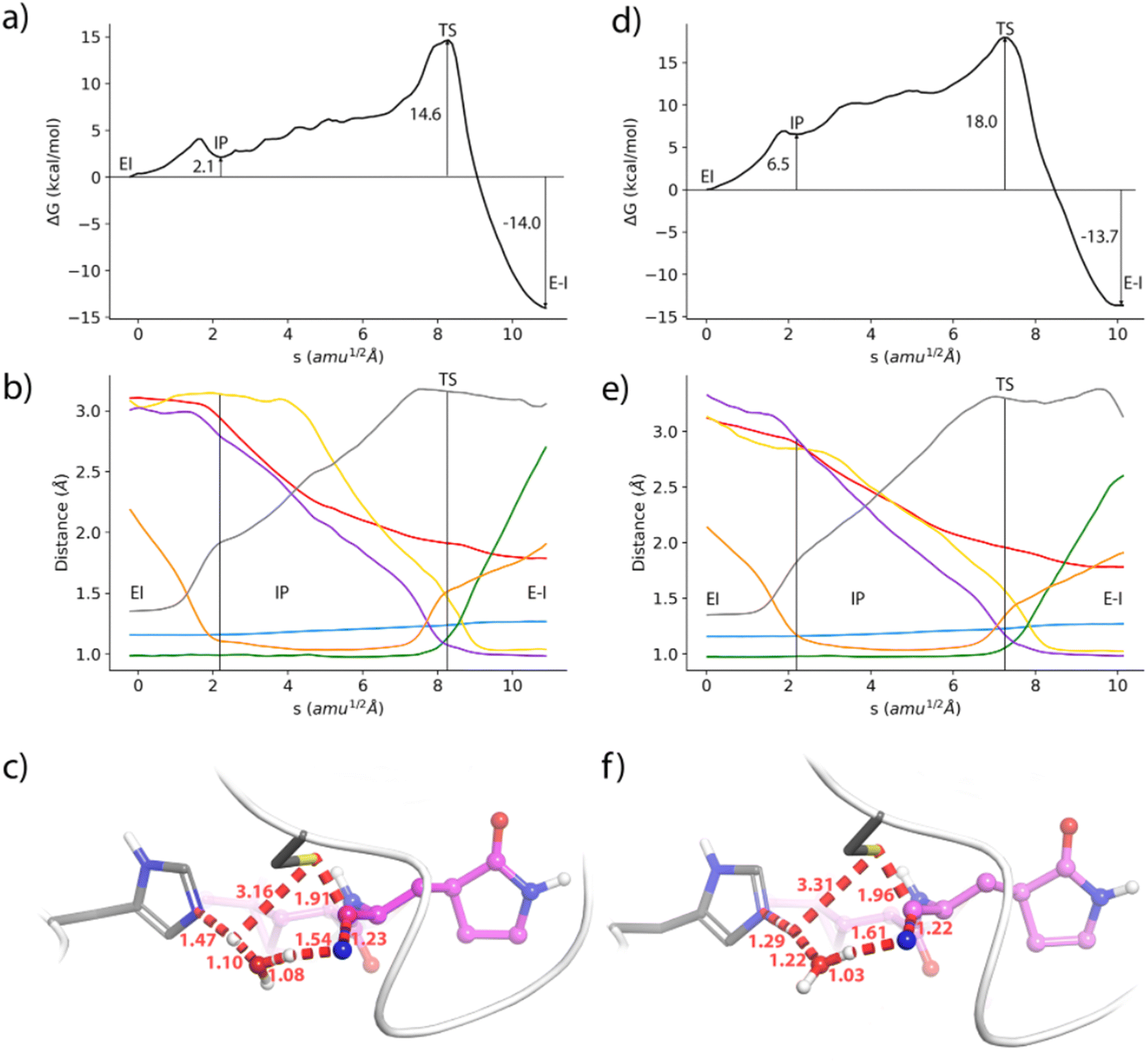 | ||
| Fig. 4 Formation of the thioimidate covalent product of nirmatrelvir with the 3CLpro enzyme. (a) M062X-D3/6-31+G*/MM free energy profile along the path-CV for the formation of the covalent E–I complex from the noncovalent (EI) one in the wild type enzyme. (b) Evolution of the selected CVs along the MFEP. Blue: C–N, orange: H-Nε, green: Hw-Ow, black: Sγ-H, yellow: N-Hw, red: Sγ-C, and purple: Ow-H. (c) Representation of the TS in the wild type enzyme. The values of the distances correspond (in Å) to the coordinates of the TS along MFEP. (d) M062X-D3/6-31+G*/MM free energy profile in the E166V mutant. (e) Evolution of the selected CVs along the MFEP in the E166V mutant. (f) Representation of the TS in the E166V mutant. The values of the distances correspond (in Å) to the coordinates of the TS along MFEP. | ||
The geometrical evolution of the reaction process obtained for the E166V variant is also very similar, with no significant differences with respect to the wild type results (compare Fig. 4b and e). This confirms that the reaction mechanism remains unaltered upon mutation. The free energy profiles obtained along the path collective variable are qualitatively similar in both cases (Fig. 4a and d) but the E166V variant presents a larger free energy barrier (18.0 kcal mol−1 in the mutant versus 14.6 kcal mol−1 in the wild type). An increase of almost 4 kcal mol−1 in the free energy cost is already observed in the formation of the IP, which has a relative free energy with respect to the reactants of 2.1 and 6.5 kcal mol−1 in the wild type and mutant versions, respectively. Similar differences in the free energy profiles are observed at the B3LYPD3/MM level (see Fig. S4†). This increased free energy cost is attributed to a slightly modified binding pose of nirmatrelvir in the active site of the mutant, due to the loss of the hydrogen bond interaction between the P1 γ-lactam ring and the side chain of residue 166 in the mutant, and the leaving of the N-finger. As shown above, the mutation slightly changes the position of the amide NH moiety of the substrate's P1 group in the active site, in such a way that establishes a weaker interaction with the thiol group of C145. Finally, the reaction free energy is similar in both variants of the enzyme, corresponding to a clearly exergonic process (−14.0 and −13.7 kcal mol−1, respectively). Clearly, the mismatch of the binding pose caused by the mutation plays a more important role in the intermediate stages of the reaction, when a larger negative charge is developed on the sulphur atom, than in the product state.
The average geometries of the wild type and mutant TSs are also similar (see Fig. 4c and f). These structures correspond to a water mediated proton transfer from H41 to the nitrile group, coupled to the nucleophilic attack of the sulphur atom to the nitrile carbon atom. In the wild type 3CLpro the TS is slightly more advanced along the path collective variable than in the mutant, showing a more advanced proton transfer from H41 to the water molecule, while the proton transfer from this water to the nitrile group has still not progressed significantly. In the mutant enzyme, the TS displays less advanced proton transfers from H41 to the water molecule and from this molecule to the nitrile group of nirmatrelvir. The distance of the nucleophilic Sγ atom to the electrophilic carbon is also slightly longer in the mutant than in the wild type TS (1.96 versus 1.91 Å).
These results show that mutations have an impact on the kinetics of the covalent inhibition process. Mismatching drug–protein interactions can have a significant impact not only on the binding process but also on the activation free energy barrier for the formation of a covalent bond between the 3CLpro enzyme and its inhibitors. Increasing the activation free energy reduces the inactivation rate constant, which in turn decreases the efficiency of the covalent inhibitor. This is a resistance strategy that should be taken into account when analyzing the consequences of mutations on the inhibition process.
Conclusions
Mutations may endow viruses with a mechanism of resistance to treatments developed to combat their infections. In the case of SARS-CoV-2, one of the antiviral treatments contains an active principle, nirmatrelvir, targeted against the main or 3CL protease of the virus. Nirmatrelvir has been designed as a covalent inhibitor, forming a covalent bond between its nitrile warhead and the catalytic cysteine of the enzyme. Several experimental studies have already identified several positions in the 3CLpro enzyme whose mutation increases the tolerance of the virus to the presence of this covalent inhibitor. However, the detailed mechanisms by which these mutations increase viral resistance are still unknown, in particular the consequences for the formation of the covalent complex. In this work, we have used molecular dynamics simulations and free energy methods to investigate the impact of possible mutations on the resistance mechanism of SARS-CoV-2 to nirmatrelvir, analyzing the impact on binding and on the chemical reaction between the enzyme and the drug.Using a per residue MMGBSA free energy decomposition scheme, we found that most of the enzymatic positions whose mutations were experimentally found to confer resistance to nirmatrelvir contribute significantly to the protein-ligand binding free energy. This suggests that this strategy may be a valid criterion to identify hot spots for potential mutations increasing the resistance of SARS-CoV-2 to the treatment with nirmatrelvir. In particular, the E166 position contributes to the binding of the drug and of the natural substrate, establishing up to three hydrogen bond interactions, two through the backbone and one between the carboxylate group of the side chain and the P1 group of the substrate. Its mutation to valine results in an increased resistance to the drug but also in a decreased enzymatic efficiency towards its natural function, efficiency that could be recovered after a second mutation. MMGBSA results show that the E166 residue contributes significantly to the binding free energy of nirmatrelvir to form a noncovalent complex with the enzyme. In addition, molecular dynamics simulations show that the E166V mutation can also reduce the stability of the active dimer, because of the loss of the interaction between residue 166 and the N-terminal group of the other protomer. The so-called N-finger is known to participate in the S1 pocket of the accompanying protomer. Our MMGBSA calculations have also allowed us to identify other positions for possible mutations conferring resistance to nirmatrelvir. In particular, L141, R188, F140 and G143 seem to be good candidates to suffer mutations improving the viral fitness to the pressure due to the use of nirmatrelvir.
We have also investigated the effect of the E166V mutation on the formation of the covalent complex between nirmatrelvir and the 3CLpro enzyme. According to our QM/MM simulations, the reaction mechanism remains essentially unaltered after mutation, but the kinetics of the process are slowed down. The loss of the hydrogen bond between residue 166 and the P1 group of the drug slightly changes the binding pose of nirmatrelvir in the active site, increasing then the free energy cost to reach the IP and the reaction TS. Instead, the reaction free energy is not substantially altered.
In this work we have analyzed the effect of single mutations on the two stages of the covalent inhibition process, the binding free energy for the formation of the noncovalent complex and the activation free energy for the reaction to form the covalent complex. The information provided by these simulations is useful to understand the impact of mutations and to assist in the development of new inhibitors that could help fight against new variants of the virus that could escape the action of nirmatrelvir. One should keep in mind that many residues play a similar role in the natural proteolytic activity of the enzyme and in the formation of the enzyme-inhibitor covalent complex. Then, single mutations that increase resistance may also probably reduce the enzymatic activity, resulting in non-transmissible viruses. However, the natural substrate is a polyprotein chain, much bulkier than nirmatrelvir or other drugs, which opens the possibility that multiple mutations, taking place beyond the active site, could have a different impact on the binding of the substrate and of the drug and then could restore the natural enzymatic activity while keeping an enhanced resistance to the drug. This scenario has been already observed experimentally and should also be considered from a computational perspective in order to provide additional tools to fight COVID-19.
Data availability
Data for this paper, trajectories of the Transition States described in the manuscript, are available from a link given in the ESI† of this article.Author contributions
C. A. R-G., K. Z., J. J. R-P. and I. T were responsible for the design of the work. C. A. R-G. and M. A. performed the calculations and all the authors participated in the discussion. I. T. wrote the original draft, while all the authors participated in manuscript revision and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from grant PID2021-123332OB-C22 funded by MCIN/AEI/10.13039/501100011033/and by “ERDF A way of making Europe” and also from grant PROMETEO CIPROM/2021/079 of Generalitat Valenciana. KZ acknowledges a Maria Zambrano fellowship from the Ministerio de Universidades (Spain). MA acknowledges the support of a fellowship (fellowship code LCF/BQ/DR21/11880026) from the “la Caixa” Foundation (ID 100010434). Computational resources were provided by the Barcelona Supercomputing Center and Servei d’Informàtica, Universitat de València (Tirant supercomputer). We gratefully acknowledge all data contributors, i.e., the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is partly based.References
- B. M. Bennet, J. Wolf, R. Laureano and R. S. Sellers, Toxicol. Pathol., 2020, 48, 800–809 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke and P. R. Verhoest, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- X. Zhang, S. Wu, B. Wu, Q. Yang, A. Chen, Y. Li, Y. Zhang, T. Pan, H. Zhang and X. He, Signal Transduction Targeted Ther., 2021, 6, 10–12 CrossRef PubMed.
- J. Zhang, T. Xiao, Y. Cai, C. L. Lavine, H. Peng, H. Zhu, K. Anand, P. Tong, A. Gautam, M. L. Mayer, R. M. Walsh, S. Rits-Volloch, D. R. Wesemann, W. Yang, M. S. Seaman, J. Lu and B. Chen, Science, 2021, 374, 1353–1360 CrossRef CAS PubMed.
- A. Frediansyah, F. Nainu, K. Dhama, M. Mudatsir and H. Harapan, Clin. Epidemiol. Global Health, 2021, 9, 123–127 CrossRef CAS PubMed.
- S. M. R. Hashemian, M. H. Pourhanifeh, M. R. Hamblin, M. K. Shahrzad and H. Mirzaei, Biomed. Pharmacother., 2022, 146, 112517 CrossRef CAS PubMed.
- R. Hilgenfeld, FEBS J., 2014, 281, 4085–4096 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X. M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L. K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum, M. A. Joyce, H. S. Young, D. L. Tyrrell, J. C. Vederas and M. J. Lemieux, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell, B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu, T. Suto, K. Kouki, A. Nakahashi, S. Kawashima, T. Sanaki, S. Toba, K. Uemura, T. Mizutare, S. Ando, M. Sasaki, Y. Orba, H. Sawa, A. Sato, T. Sato, T. Kato and Y. Tachibana, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed.
- P. Sahoo, D. R. Lenka, M. Batabyal, P. K. Pain, S. Kumar, D. Manna and A. Kumar, ACS Pharmacol. Transl. Sci., 2023, 6(1), 171–180 CrossRef CAS PubMed.
- W. Yu, Y. Zhao, H. Ye, N. Wu, Y. Liao, N. Chen, Z. Li, N. Wan, H. Hao, H. Yan, Y. Xiao and M. Lai, J. Med. Chem., 2022, 65, 16252–16267 CrossRef CAS PubMed.
- K. Gao, R. Wang, J. Chen, J. J. Tepe, F. Huang and G.-W. Wei, J. Med. Chem., 2021, 64, 16922–16955 CrossRef CAS PubMed.
- C. H. Zhang, E. A. Stone, M. Deshmukh, J. A. Ippolito, M. M. Ghahremanpour, J. Tirado-Rives, K. A. Spasov, S. Zhang, Y. Takeo, S. N. Kudalkar, Z. Liang, F. Isaacs, B. Lindenbach, S. J. Miller, K. S. Anderson and W. L. Jorgensen, ACS Cent. Sci., 2021, 7, 467–475 CrossRef CAS PubMed.
- L. E. Khoury, Z. Jing, A. Cuzzolin, A. Deplano, D. Loco, B. Sattarov, F. Hédin, S. Wendeborn, C. Ho, D. E. Ahdab, T. J. Inizan, M. Sturlese, A. Sosic, M. Volpiana, A. Lugato, M. Barone, B. Gatto, M. L. Macchia, M. Bellanda, R. Battistutta, C. Salata, I. Kondratov, R. Iminov, A. Khairulin, Y. Mykhalonok, A. Pochepko, V. Chashka-Ratushnyi, I. Kos, S. Moro, M. Montes, P. Ren, J. W. Ponder, L. Lagardère, J.-P. Piquemal and D. Sabbadin, Chem. Sci., 2022, 13, 3674–3687 RSC.
- C. A. Ramos-Guzmán, J. J. Ruiz-Pernía and I. Tuñón, Angew. Chem., Int. Ed., 2021, 60, 25933–25941 CrossRef PubMed.
- R. S. Tumskiy, A. V. Tumskaia, I. N. Klochkova and R. J. Richardson, Comput. Biol. Med., 2023, 153, 106449 CrossRef CAS PubMed.
- S. Martí, K. Arafet, A. Lodola, A. J. Mulholland, K. Świderek and V. Moliner, ACS Catal., 2022, 12, 698–708 CrossRef PubMed.
- R. Löser, K. Schilling, E. Dimmig and M. Gütschow, J. Med. Chem., 2005, 48, 7688–7707 CrossRef PubMed.
- J. Hammond, H. Leister-Tebbe, A. Gardner, P. Abreu, W. Bao, W. Wisemandle, M. Baniecki, V. M. Hendrick, B. Damle, A. Simón-Campos, R. Pypstra and J. M. Rusnak, N. Engl. J. Med., 2022, 386, 1397–1408 CrossRef CAS PubMed.
- Pfizer Press Release, https://www.pfizer.com/News/Press-Release/Press-Release-Detail/Pfizer-Reports-Additional-Data-Paxlovidtm-Supporting/, accessed November 7, 2022 Search PubMed.
- S. Chakraborti, S. Bheemireddy and N. Srinivasan, Molecular, 2020, 16, 474–491 CAS.
- K. S. Yang, S. Z. Leeuwon, S. Xu and W. R. Liu, J. Med. Chem., 2022, 65, 8686–8698 CrossRef CAS PubMed.
- GISAID – CoVsurver mutations App, https://gisaid.org/database-features/covsurver-mutations-app/, accessed November 7, 2022 Search PubMed.
- S. E. Greasley, S. Noell, O. Plotnikova, R. A. Ferre, W. Liu, B. Bolanos, K. Fennell, J. Nicki, T. Craig, Y. Zhu, A. E. Stewart and C. M. Steppan, J. Biol. Chem., 2022, 298, 101972 CrossRef CAS PubMed.
- D. Jochmans, C. Liu, K. Donckers, A. Stoycheva, S. Boland, S. K. Stevens, C. D. Vita, B. Vanmechelen, P. Maes, B. Trüeb, N. Ebert, V. Thiel, S. D. Jonghe, L. Vangeel, D. Bardiot, A. Jekle, L. M. Blatt, L. Beigelman, J. A. Symons, P. Raboisson, P. Chaltin, A. Marchand, J. Neyts, J. Deval and K. Vandyck, bioRxiv, 2019, preprint, DOI: DOI:10.1101/2022.06.07.495116.
- Y. Zhou, K. A. Gammeltoft, L. A. Ryberg, L. V. Pham, H. D. Tjørnelund, A. Binderup, C. R. Duarte Hernandez, C. Fernandez-Antunez, A. Offersgaard, U. Fahnøe, G. H. J. Peters, S. Ramirez, J. Bukh and J. M. Gottwein, Sci. Adv., 2022, 8(51), eadd7197, DOI:10.1126/sciadv.add7197.
- S. Iketani, S. J. Hong, J. Sheng, F. Bahari, B. Culbertson, F. F. Atanaki, A. K. Aditham, A. F. Kratz, M. I. Luck, R. Tian, S. P. Goff, H. Montazeri, Y. Sabo, D. D. Ho and A. Chavez, bioRxiv, 2022, preprint, DOI: DOI:10.1101/2022.06.23.497404.
- S. Iketani, H. Mohri, B. Culbertson, S. J. Hong, Y. Duan, M. I. Luck, M. K. Annavajhala, Y. Guo, Z. Sheng, A.-C. Uhlemann, S. P. Goff, Y. Sabo, H. Yang, A. Chavez and D. D. Ho, Nature, 2022, 1–7 CAS.
- Y. Hu, E. M. Lewandowski, H. Tan, X. Zhang, R. T. Morgan, X. Zhang, L. M. C. Jacobs, S. G. Butler, M. V. Gongora, J. Choy, X. Deng, Y. Chen and J. Wang, bioRxiv, 2022, preprint, DOI: DOI:10.1101/2022.06.23.497404.
- E. Heilmann, F. Costacurta, S. A. Moghadasi, C. Ye, M. Pavan, D. Bassani, A. Volland, C. Ascher, A. K. H. Weiss, D. Bante, R. S. Harris, S. Moro, B. Rupp, L. Martinez-Sobrido and D. von Laer, Sci. Transl. Med., 2022, 15(678), eabq7360 CrossRef PubMed.
- V. M. Sasi, S. Ullrich, J. Ton, S. E. Fry, J. Johansen-Leete, R. J. Payne, C. Nitsche and C. J. Jackson, Biochemistry, 2022, 61, 2495–2505 CrossRef CAS PubMed.
- S. A. Moghadasi, E. Heilmann, S. N. Moraes, F. L. Kearns, D. von Laer, R. E. Amaro and R. S. Harris, bioRxiv, 2022, preprint, DOI: DOI:10.1101/2022.08.07.503099.
- C. A. Ramos-Guzmán, J. J. Ruiz-Pernía and I. Tuñón, ACS Catal., 2020, 10, 12544–12554 CrossRef PubMed.
- C. A. Ramos-Guzmán, J. J. Ruiz-Pernía and I. Tuñón, Chem. Commun., 2021, 57, 9096–9099 RSC.
- L. Zhang, L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- Schrödinger Release 2023-1: Maestro, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, H. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, S. Sagui, J. Schott-Verdugo, C. L. Shen, J. Simmerling, R. Smith, J. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER, University of California, San Francisco, 2018 Search PubMed.
- M. H. M. Olsson, C. R. SØndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- K. A. Adem, J. C. Ferreira, S. Fadl and W. M. Rabeh, J. Biol. Chem., 2023, 299(2), 102790 CrossRef PubMed.
- H. T. H. Chan, M. A. Moesser, R. K. Walters, T. R. Malla, R. M. Twidale, T. John, H. M. Deeks, T. Johnston-Wood, V. Mikhailov, R. B. Sessions, W. Dawson, E. Salah, P. Lukacik, C. Strain-Damerell, C. D. Owen, T. Nakajima, K. Świderek, A. Lodola, V. Moliner, D. R. Glowacki, J. Spencer, M. A. Walsh, C. J. Schofield, L. Genovese, D. K. Shoemark, A. J. Mulholland, F. Duarte and G. M. Morris, Chem. Sci., 2021, 12, 13686–13703 RSC.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics, 2006, 25, 247–260 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- A. W. Götz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef PubMed.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- B. R. Miller, T. D. Mcgee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
- H. Gohlke, C. Kiel and D. A. Case, J. Mol. Biol., 2003, 330, 891–913 CrossRef CAS PubMed.
- M. W. V. D. Kamp and A. J. Mulholland, Biochemistry, 2013, 52, 2708–2728 CrossRef PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (version Revision C.01), Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104(1)–154104(19) CrossRef PubMed.
- K. Zinovjev and I. Tuñón, J. Phys. Chem. A, 2017, 121, 9764–9772 CrossRef CAS PubMed.
- K. Zinovjev, String-Amber, https://github.com/kzinovjev/string-amber, accessed June 22, 2022 Search PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld and Z. Rao, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190–13195 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- H. T. H. Chan, A. S. F. Oliveira, C. J. Schofield, A. J. Mulholland and F. Duarte, bioRxiv, 2022, preprint, DOI: DOI:10.1101/2022.12.10.519730.
- A. K. Padhi and T. Tripathi, Biochem. Biophys. Res. Commun., 2022, 629, 54–60 CrossRef CAS PubMed.
- S. Ullrich, K. B. Ekanayake, G. Otting and C. Nitsche, Bioorg. Med. Chem., 2022, 62, 128629 CrossRef CAS PubMed.
- B. L. Grigorenko, I. V. Polyakov, G. Giudetti, S. Faraji, A. I. Krylov and A. V. Nemukhin, ChemRxiv, 2022, preprint, DOI: DOI:10.26434/chemrxiv-2022-8lr3v.
- S. T. Ngo, T. H. Nguyen, N. T. Tung and B. K. Mai, RSC Adv., 2022, 12, 3729–3737 RSC.
- S. Khare, C. Gurry, L. Freitas, M. B. Schultz, G. Bach, A. Diallo, N. Akite, J. Ho, R. T. C. Lee, W. Yeo, GISAID Core Curation Team and S. Maurer-Stroh, China CDC Weekly, 2021, 3(49), 1049–1051 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete table with residue contributions to the MMGBSA binding free energy of nirmatrelvir and the peptide substrate; distribution of distances between 166 and N-terminal residues in wild type and E166V variants of 3CLpro; distribution of distances between residues 166 and the N-terminal in wild type and E166V mutants; TS structures for the acylation of a peptide substrate and nirmatrelvir in the active site of 3CLpro; B3LYPD3/MM free energy profiles for the reaction of nirmatrelvir with the wild type and E166V variants of 3CLpro; link to the coordinates from the trajectories of the TS for the reaction of nirmatrelvir. See DOI: https://doi.org/10.1039/d2sc06584c |
| This journal is © The Royal Society of Chemistry 2023 |