DOI:
10.1039/D2SC06234H
(Edge Article)
Chem. Sci., 2023,
14, 827-832
Inherently chiral calixarenes by a catalytic enantioselective desymmetrizing cross-dehydrogenative coupling†
Received
11th November 2022
, Accepted 5th December 2022
First published on 6th December 2022
Abstract
Under the catalysis of PdBr2 and a chiral phosphoramidite ligand, the upper-rim mono (2-bromoaroyl)-substituted calix[4]arene derivatives underwent a facile enantioselective desymmetrization reaction to afford 9H-fluorene-embedded inherently chiral calixarenes in good yields with excellent enantioselectivities. The transannular dehydrogenative arene–arene coupling reaction proceeded most probably through an oxidative addition of the Caryl–Br bond to a ligated palladium catalyst followed by a sequence of an enantioselective 1,5-palladium migration and an intramolecular C–H arylation sequence. This new family of inherently chiral calixarenes possesses unique chiroptical properties thanks to their highly rigid structure induced by the 9H-fluorene segment.
Introduction
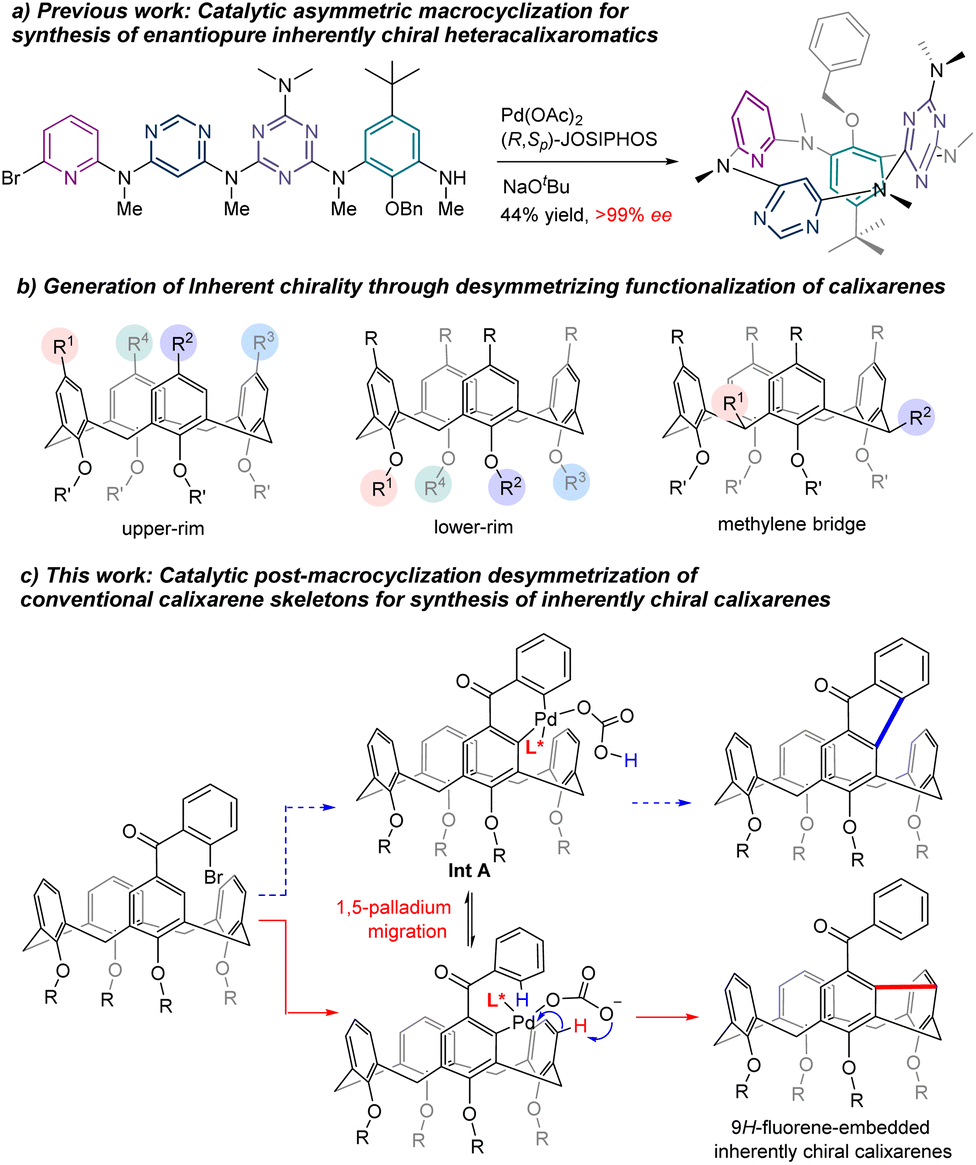
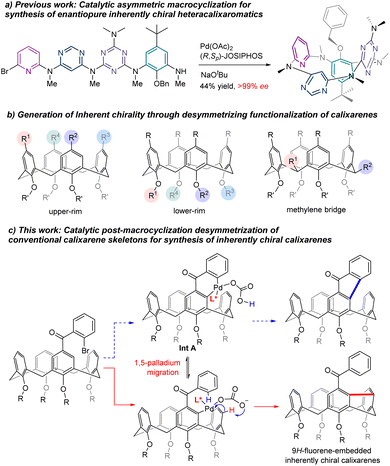
Inherently chiral macrocycles, coined by Böhmer in 1994, are a unique type of chiral chemical entity.1 Different from chiral molecules bearing a central, an axial and a planar chirality and helicity, the inherent chirality results from the three-dimensional curvature architecture of macrocycles.2–4 Calix[n]arenes are prototypical inherently chiral molecules from which the term originated. With their unique three-dimensional bowl-shaped structures, controllable cavity sizes and shapes, easy preparation and post-chemical modifications, calix[n]arenes and their aza- as well as oxa-analogues have become privileged scaffolds in catalysis, molecular recognition, sensing, nanotechnology, biotechnology, etc.5–7 The inherently chiral calixarenes and analogues have not been the subject of intensive investigation due mainly to the inaccessibility of enantioenriched chiral calix[n]arenes. Indeed, most of the enantioenriched calixarenes and resorcinarenes documented in the literature are synthesized by separation of enantiomers using chiral HPLC columns.2–4,8–12 Kinetic resolution of racemates has been reported to give chiral calixarenes with either low efficiency or modest enantioselectivity while diastereoselective synthesis using chiral auxiliary requires inevitably multiple reaction steps and sometimes tedious separation of diastereomers.13–18 Catalytic enantioselective synthesis of an intrinsically chiral tetraazacalix[4]arene was attempted by Tsue in 2009.19 However, under their optimized conditions, Pd-catalyzed Caryl–N bond-forming macrocyclization afforded the product with only 35% ee and only one example was documented in this paper. Very recently, we reported a general synthesis of highly enantioenriched ABCD-type heteracalixaromatics via a Pd-catalyzed intramolecular Buchwald–Hartwig reaction.20 Linear precursors containing benzene, pyridine, pyrimidine and triazine rings underwent efficient cyclization to furnish nitrogen-bridged calixarenes with ee up to >99% (Scheme 1a). These chiral macrocycles exhibit excellent and intriguing proton-triggered switchable CPL properties.
 |
| | Scheme 1 Strategies for the construction of inherently chiral macrocycles. | |
In view of the easy accessibility of calixarenes, desymmetrization is, without doubt, an obvious option to pursue in order to access enantioenriched calixarenes. In principle, selective functionalization of the upper rim,3 the lower rim11 and the methylene bridge21 could break the symmetry of calixarenes, generating therefore the inherent chirality (Scheme 1b). All these three routes have indeed been exploited using a chiral auxiliary approach with limited success. On the other hand, catalytic enantioselective desymmetrization of calixarenes remains, to the best of our knowledge, unknown. Aiming at creating new inherently chiral macrocyclic systems and searching for novel chiroptical organic molecules,22 we set out to investigate the catalytic enantioselective desymmetrization of conventional calix[4]arenes. Our initial design was to construct a 9H-fluoren-9-one moiety within the skeleton of calix[4]arenes taking advantage of the Pd-catalyzed intramolecular C–H arylation process (Scheme 1c). We hypothesized that, in the presence of an appropriate chiral ligand, the ArPdXL* species generated in situ via oxidative addition would be able to discriminate the two enantiotopic ortho C–H bonds leading, after reductive elimination of the 6-membered palladacycle Int A (Scheme 1c), to chiral calix[4]arene.23,24 Surprisingly, a more complex reaction sequence occurred affording chiral meta–meta bridged calix[4]arenes in good yields with high enantioselectivities (Scheme 1c). Although one example of this type of calix[4]arenes is reported in the literature,25,26 no enantioselective synthesis is known to date. We disclose herein the results of our study.
Results and discussion
Optimization of reaction conditions
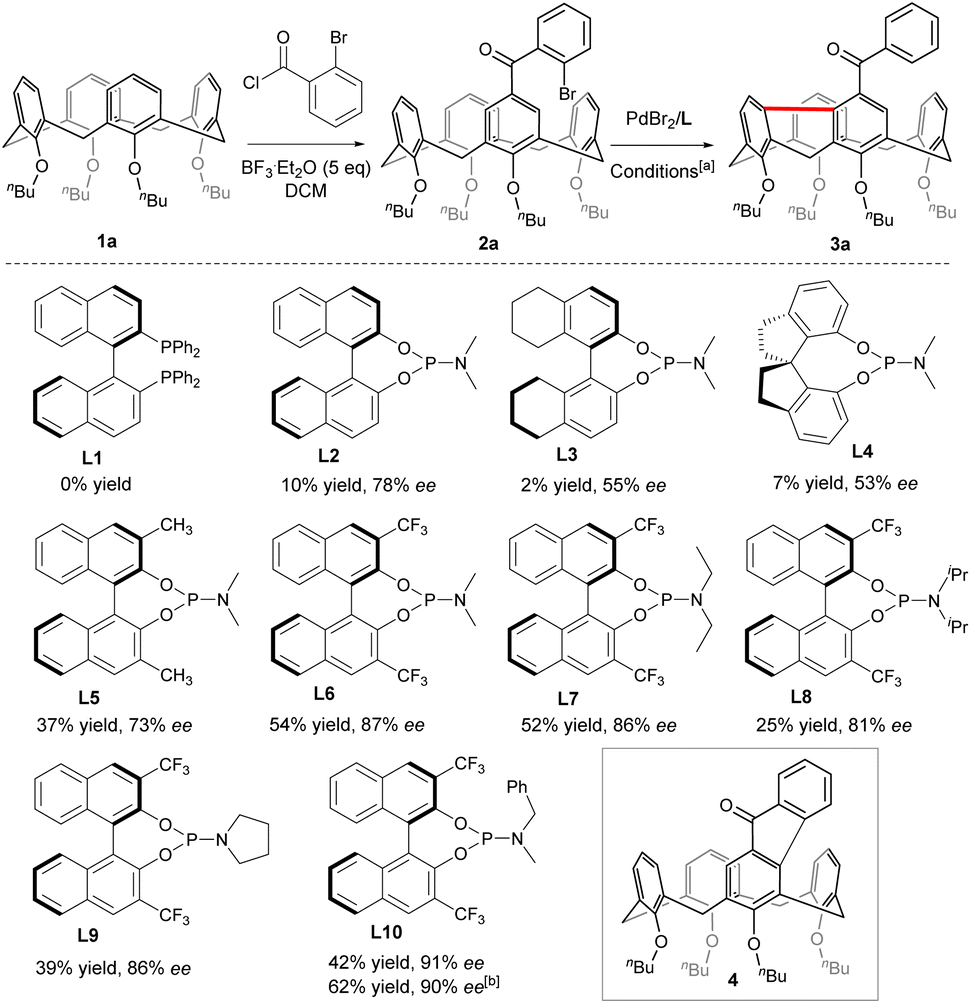
To begin with, Friedel–Crafts acylation between symmetric calix[4]arene 1a and 2-bromobenzoyl chloride afforded our starting reagent 2a (Scheme 2). The catalytic enantioselective desymmetrizing reaction of 2a was initially investigated by varying the Pd-sauces, the ligands, the solvents, and the temperature (ESI†). PdBr2 stood out as the best Pd source. The nature of chiral ligands was found to impact not only the enantioselectivity of the reaction but also the product yield (Scheme 2). While a bidentate phosphine ligand such as R-BINAP (L1) failed to promote the reaction, chiral phosphoramidite derived from R-BINOL (L2) was able to affect the enantioselective transformation of 2a to afford 3a with 78% ee, albeit in a very low yield. Since either the enantioselectivity or the efficiency of the reaction was not improved when phosphoramidites prepared from R-[H8]BINOL (L3) and S-2,2′,3,3′-tetrahydro-1,1′-spirobi[indene]-7,7′-diol (L4) were employed as ligands, we focused on the chiral BINOL scaffold by introducing substituents on 3,3′-positions. In contrast to 3,3′-diaryl-substituted phosphoramidites which did not perform well due to most probably the steric hindrance (ESI†), 3,3′-dimethylated ligand L5 was able to increase the yield considerably while maintaining the same enantioselectivity. Both the chemical yield and enantiomeric excess value of 3a were improved remarkably when 3,3′-bis(trifluoromethyl)-substituted phosphoramidite (L6) was used as a ligand in combination with Cs2CO3 (54% yield, 87% ee). The amino part of the ligand was subsequently varied and phosphoramidite bearing an N-benzyl-N-methylamino group (L10) gave the best results. Finally, replacing CsCO3 with Rb2CO3 further increased the reaction efficiency. Overall, under optimized conditions [PdBr2 (10 mol%), L10 (20 mol%), Rb2CO3 (3.0 equiv.), THF, 110 °C], compound 2a was converted to 3a in 62% yield with 90% ee. A sequence of oxidative addition, enantioselective C–H activation/1,5-palladium migration27,28 followed by the second C–H activation/reductive elimination could account for the reaction outcome. The exclusive formation of 3a at the expense of 4 indicated that the 1,5-palladium migration and subsequent transannular arylation proceed much faster than the Csp2–Csp2 bond-forming reductive elimination from intermediate Int A (Scheme 1c). The facile transannular Caryl–H and Caryl–H cross-coupling reaction is most likely facilitated by the preferential cone conformation of the calixarene backbone.
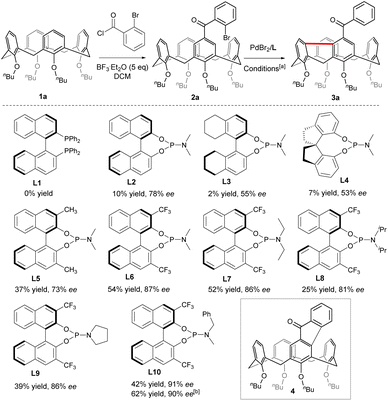 |
| | Scheme 2 Structures of ligands. [a] Sealed tube, inert atmosphere, 2a (0.05 mmol), PdBr2 (0.1 eq.), L (0.2 eq.), Cs2CO3 (2 eq.), THF (1.5 mL), 110 °C; [b] PdBr2 (0.1 eq.), L10 (0.2 eq.), Rb2CO3 (3 eq.), under otherwise standard conditions. | |
Scope of the reaction
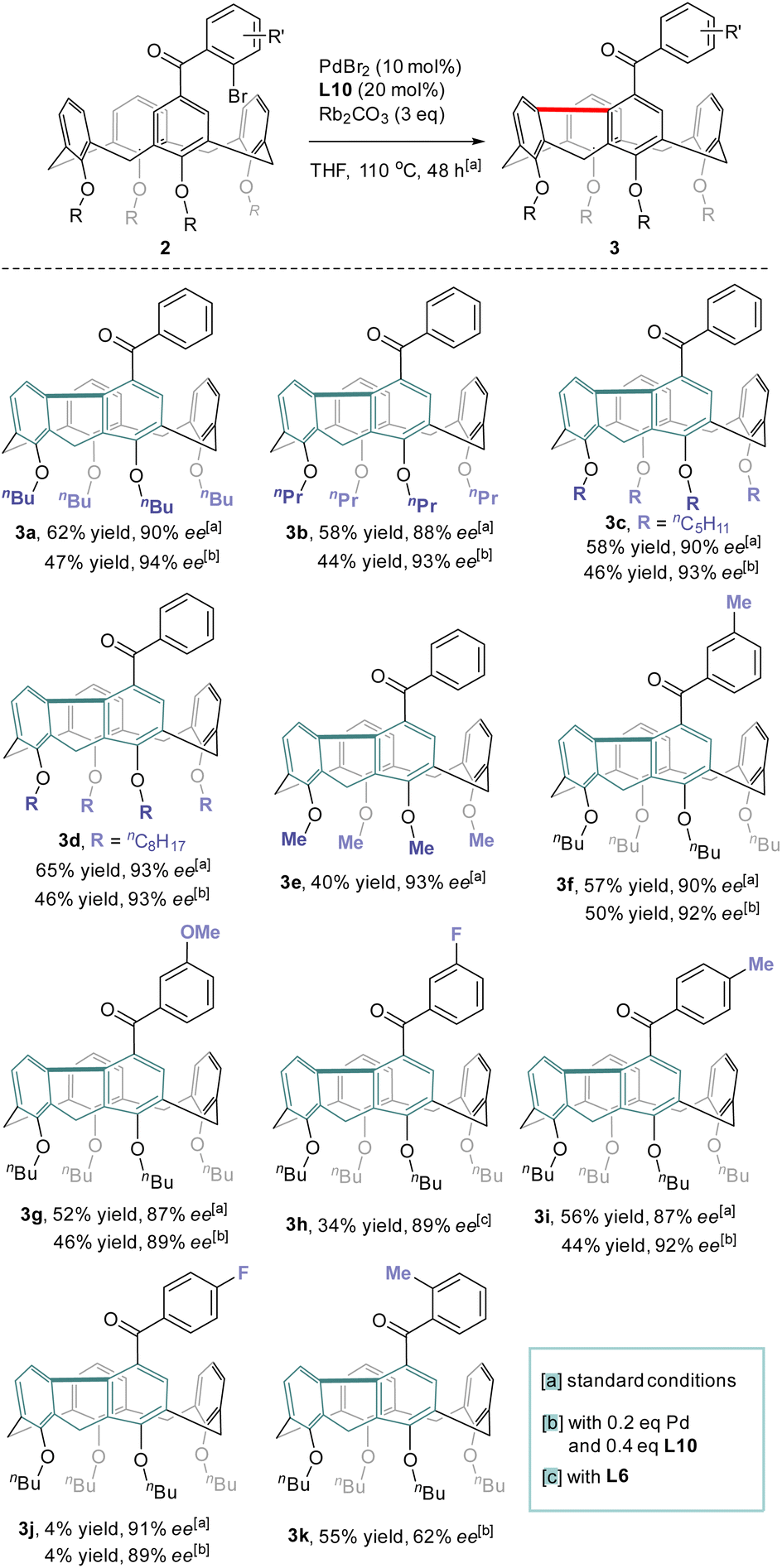
With the optimized conditions in hand, the reaction scope with regard to the structure of the lower-rim substituents of calix[4]arene was first examined (Scheme 3). Satisfyingly, substrates that contain alkoxy groups with different lengths of the alkyl chain such as n-Pr (3b), n-amyl (3c) and n-octyl (3d) were well accepted affording the desired products in good yields and enantiopurities. Noteworthily, when the alkyl substituent on each phenolic oxygen was replaced by methyl, the inherently chiral macrocycle 3e was obtained similarly with 93% ee. No racemization took place at an elevated temperature such as 150 °C (ESI†). Since an O-alkyl group larger than ethyl is generally required to stabilize the conformational structure of tetraethers of conventional calix[4]arenes,29 the isolation of highly enantioenriched compound 3e which contains only methoxy substituents at the lower-rim suggests that the rigidification of the macrocycle stemmed from the chemical bonding between proximal benzene rings. The formation of the 9H-fluorene unit also led to the contraction of the macrocyclic ring size which could prevent the methoxybenzene moieties from rotating or flipping around the macrocyclic annulus, the processes accounting for racemization. Substituents on 2-bromobenzoyl and substitution patterns subtly affected the outcomes of catalytic enantioselective desymmetrization. Reactants 2f–2h which bear respectively methyl, methoxy and fluorine groups on the para-position of bromine of 2-bromoaroyl acted as good substrates and their conversion to 3f–3h proceeded with the same high level of efficiency and enantiocontrol. Excellent enantioselectivity along with 44% yield was achieved for the transformation of 2i which has a methyl moiety para to the carbonyl. Surprisingly, substrate 2j having a fluorine para to the carbonyl appeared virtually inert to chiral catalysis with a large amount of starting material remaining intact. Moving methyl to the ortho position of the carbonyl, however, had a detrimental effect on enantioselectivity as the ee value of 3k dropped to 62%. A similarly high level of enantiocontrol of 1,5-Pd migration observed for most of the substrates 2a–2j except ortho-methyl-bearing reactant 2k implies the steric interaction between substrates and chiral catalyst may be crucial in chirality expression from a chiral catalyst to inherently chiral macrocycles.
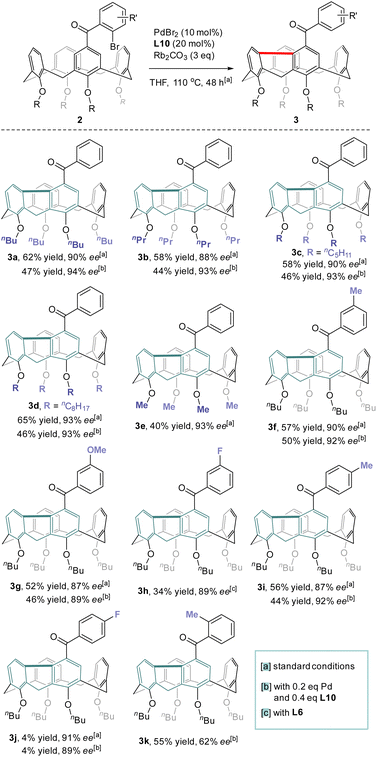 |
| | Scheme 3 Scope of the reaction. | |
Structural elucidation
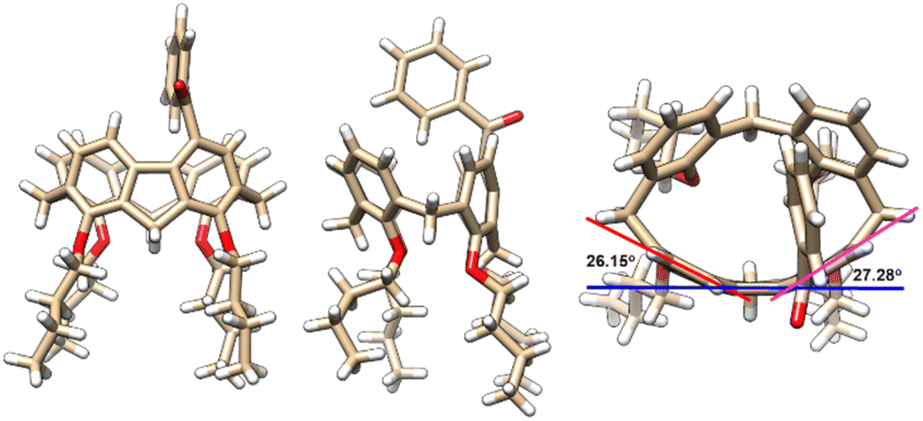
Molecular structures and their absolute configuration were determined unambiguously by crystallographic analysis of P-3a (Fig. 1). Similar to its calix[4]arene precursor, compound 3a adopts a cone conformation, indicating the stability of the conformational structure of the macrocycle during the course of palladation, 1,5-metal migration and transannular arylation. Noticeably, the newly formed 9H-fluorene fragment deviates severely from planarity.30 The dihedral angles between the five-membered ring and fused benzene rings are in the range of 26.15° and 27.38°, reflecting the ring strain of the macrocycle. The curved structure of 9H-fluorene results in the generation of an oval-shaped cavity.
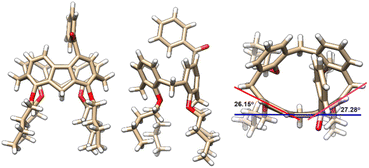 |
| | Fig. 1 X-ray molecular structures of P-3a with side (left and middle) and top (right) views. | |
Derivatization of inherently chiral calixarenes
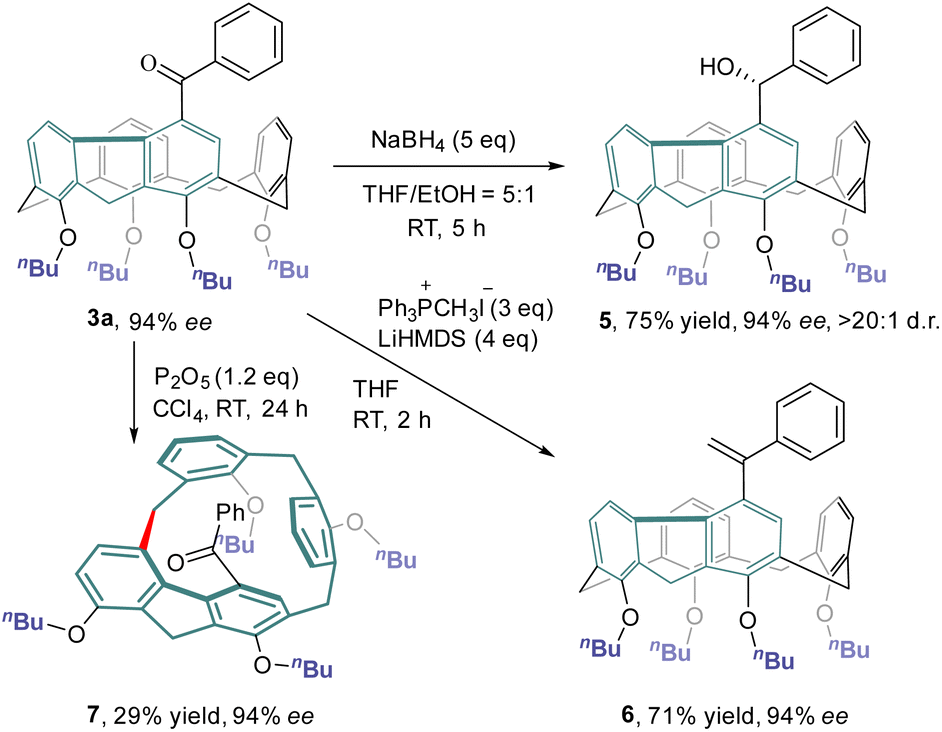
The chemical transformations of 3a taking advantage of the carbonyl group are depicted in Scheme 4. Treatment of 3a with an excess amount of NaBH4 at ambient temperature in a mixture of THF and ethanol produced alcohol 5 in 75% yield with a diastereomeric ratio of >20:1. The Wittig reaction of 3a with methylenetriphenylphosphorane provided olefin 6 in 71% yield. Both hydroxy and olefin functionalities would provide versatile handles for further elaboration of this new class of inherently chiral calixarenes. Interestingly, simply stirring a CCl4 solution of 3a at room temperature in the presence of P2O5 furnished inherently chiral macrocycle 7.31 No erosion of enantiopurity was observed in this transformation, indicating a probable concerted mechanism of macrocycle-to-macrocycle rearrangement.25b,c
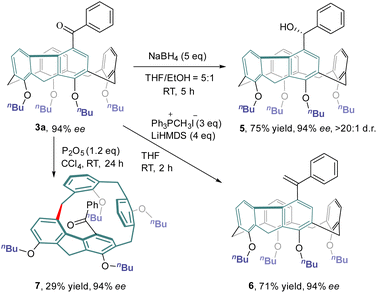 |
| | Scheme 4 Synthetic transformation of inherently chiral calixarene 3a. | |
Chiroptical properties
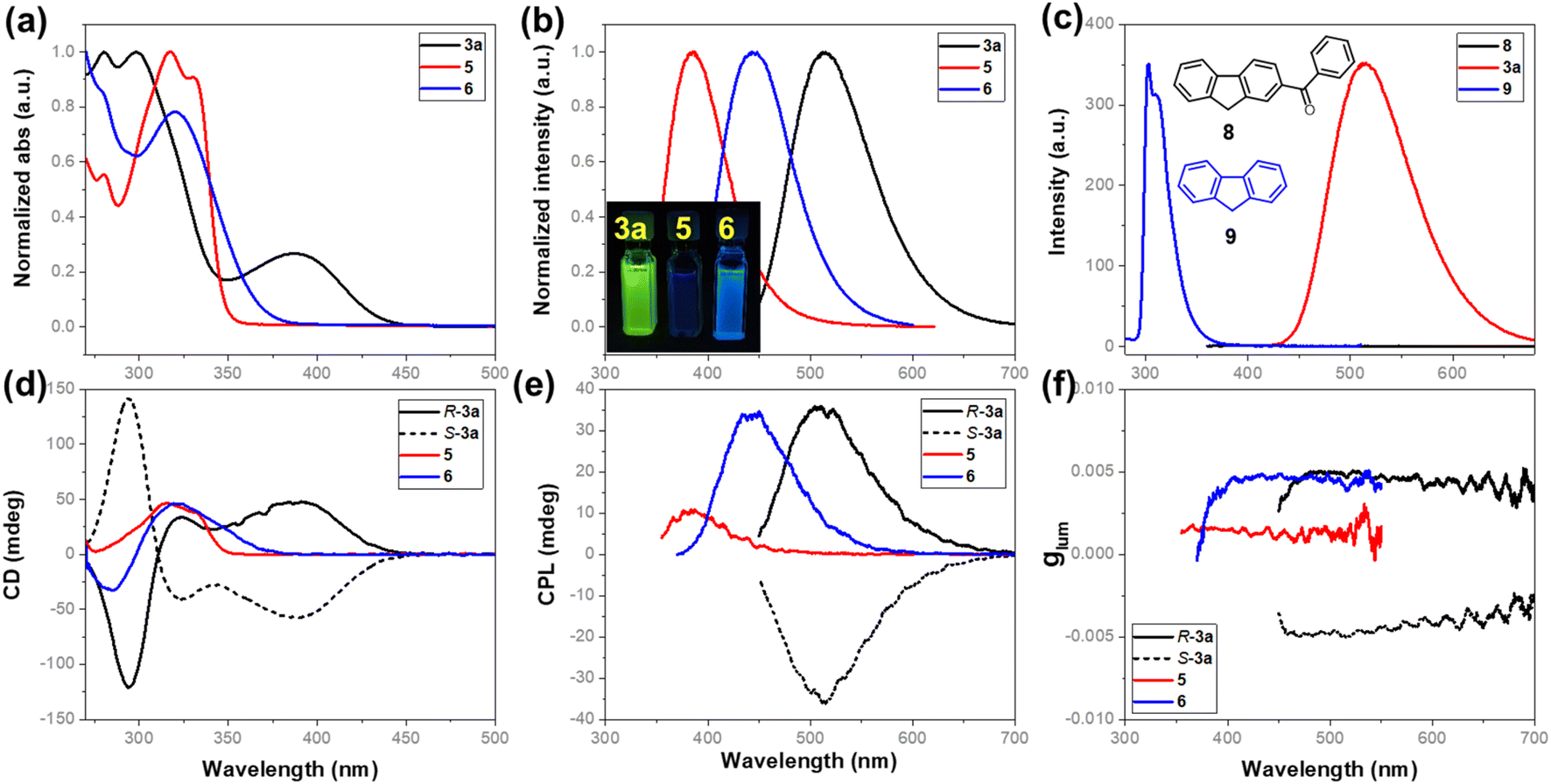
The acquired enantioenriched inherently chiral macrocycles 3 and their derivatives 5 and 6 exhibited promising chiroptical properties (Fig. 2 and ESI†). The UV–vis spectra of 3a and its analogues 3b–3k in acetonitrile show the lowest-energy absorption maximum at ca. 390 nm. When the carbonyl group was converted into hydroxyl (5) or a vinyl group (6), the absorption bands blue-shifted to 330 nm and 321 nm, respectively, indicating the fine-tuning of the properties by the substituents on the inherently chiral macrocycle skeleton. Relating to the UV–vis spectrum was the ECD spectrum which shows two positive Cotton effects at 391 and 325 nm and one negative effect at 285 nm for P-3a. Enantiomer M-3a gives the expected mirror-imaged ECD spectrum. For compounds 5 and 6, the positive Cotton effects at 390 nm disappear, while the other two (one positive and one negative) remain constant (Fig. 2d). Compound 3a was found to be fluorescent, giving a broad emission band (ϕF = 13.0%) centered at 514 nm upon excitation at 388 nm. Similarly, the hypsochromic shift of the emission band to 383 nm for 5 and 443 nm for 6 was observed (Fig. 2b). In addition, 3a showed pronounced positive solvatochromism in polar solvent (in toluene, λem = 476 nm; in CH3CN, λem = 514 nm, ESI†), indicating an excited state intramolecular charge transfer (ESICT) process. Notably, P-3a and M-3a appear strongly CPL active, giving emission maxima consistent with those observed in their fluorescence spectra. Under UV irradiation at 388 nm, for instance, P-3a and M-3a in acetonitrile gave complementary CPL spectra with large luminescence dissymmetry values of |glum| = 5 × 10−3 at 515 nm. Interestingly, despite having a different conjugation system, the vinyl-bearing compound 6 has the same luminescence dissymmetry factor (|glum| = 4.9 × 10−3) as ketone compound 3a. However, in the case of 5 in which conjugation was interrupted due to the reduction of carbonyl, the luminescence dissymmetry factor decreased sharply (|glum| = 1.7 × 10−3) with the concomitant hypsochromic shift from 515 nm to 479 nm (Fig. 2e and f). These indicated that the post-modification on the inherently chiral calixarene skeleton, especially the tuning of conjugation of chromophores, could effectively regulate the chiroptical properties. It is worth addressing that benzoyl-substituted 9H-fluorene 8 and fluorene 9 show strikingly different photophysical properties. The former compound is not fluorescent at all due to probably a very fast intercrossing process while the latter emits fluorescence at 303 nm. The photophysical properties of compounds 3, 5 and 6 therefore resulted from the deformed 9H-fluorene chromophore, suggesting that the regulation of conformation of chromophores by means of forming strained macrocycles would provide a new method to fine-tune the photophysical properties of conventional aromatic chromophores.
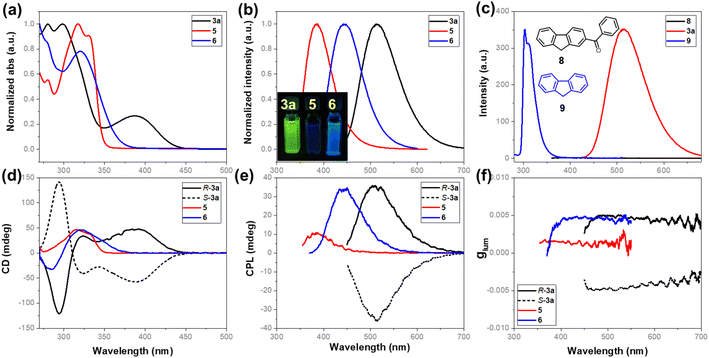 |
| | Fig. 2 (a) UV/vis and (b) fluorescence spectra of 3a, 5, and 6 in CH3CN; (c) fluorescence spectra of 3a (2 × 10−5 M), 8 (2 × 10−5 M), and 9 (5 × 10−6 M) in CH3CN; (d) ECD and (e) CPL spectra of P-3a, M-3a, 5, and 6 in CH3CN (5 × 10−5 M); (f) luminescence dissymmetry factors glum of P-3a, M-3a, 5, and 6. | |
Conclusions
In conclusion, we have developed a new type of inherently chiral calixarene by the catalytic enantioselective transannular arene–arene dehydrogenative coupling reaction of calixarene derivatives. We have demonstrated that the 9H-fluorene-embedded inherently chiral calixarenes are useful scaffolds for the fabrication of CPL materials. The outcomes opened new opportunities for the design and synthesis of novel CPL materials based on the calixarene skeleton. The acknowledged versatile molecular recognition properties and the above-mentioned post-modification nature of calixarenes would render the resulting inherently chiral macrocycles a unique and lively chiroptical system.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and the ESI,† as well as from the authors upon request.
Author contributions
X. Z., S. T., and M. X. W. conceived and designed the experiments. X. Z. carried out the experiments. S. T., J. Z., and M. X. W. interpreted the results and co-wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (21920102001 and 22171160) for financial support.
Notes and references
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- A. Dalla Cort, L. Mandolini, C. Pasquini and L. Schiaffino, New J. Chem., 2004, 28, 1198–1199 RSC.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed.
-
C. D. Gutsche, in Calixarenes, Royal Society of Chemistry, Cambridge, U.K., 1989 Search PubMed.
-
P. Neri, J. L. Sessler and M.-X. Wang, in Calixarenes and Beyond, Springer International Publishing, Cham, 2016 Search PubMed.
- For selected reviews, see:
(a) R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed;
(b) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed;
(c) S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC;
(d) D.-S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925–1934 CrossRef CAS PubMed.
-
(a) V. Böhmer, L. Merkel and U. Kunz, J. Chem. Soc., Chem. Commun., 1987, 1987, 896–897 RSC;
(b) L. Zetta, A. Wolff, W. Vogt, K.-L. Platt and V. Böhmer, Tetrahedron, 1991, 47, 1911–1924 CrossRef CAS.
- J.-T. Li, L.-X. Wang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2014, 79, 2178–2188 CrossRef CAS PubMed.
-
(a) R. Arnecke, V. Böhmer, E. F. Paulus and W. Vogt, J. Am. Chem. Soc., 1995, 117, 3286–3287 CrossRef CAS;
(b) M. T. El Gihani, H. Heaney and A. M. Z. Slawin, Tetrahedron Lett., 1995, 36, 4905–4908 CrossRef CAS;
(c) R. Arnecke, V. Böhmer, S. Friebe, S. Gebauer, G. Krauss, I. Thondorf and W. Vogt, Tetrahedron Lett., 1995, 36, 6221–6224 CrossRef CAS.
- S.-Y. Li, Y.-W. Xu, J.-M. Liu and C.-Y. Su, Int. J. Mol. Sci., 2011, 12, 429–455 CrossRef CAS PubMed.
- M. J. McIldowie and M. I. Ogden, Supramol. Chem., 2010, 22, 13–39 CrossRef CAS.
- Z.-X. Xu, C. Zhang, Z.-T. Huang and C.-F. Chen, Org. Lett., 2008, 10, 477–479 CrossRef CAS PubMed.
- K. Iwamoto, H. Shimizu, K. Araki and S. Shinkai, J. Am. Chem. Soc., 1993, 115, 3997–4006 CrossRef CAS.
-
(a) S. A. Herbert and G. E. Arnott, Org. Lett., 2009, 11, 4986–4989 CrossRef CAS PubMed;
(b) S. A. Herbert and G. E. Arnott, Org. Lett., 2010, 12, 4600–4603 CrossRef CAS PubMed.
- P. C. B. Page, H. Heaney and E. P. Sampler, J. Am. Chem. Soc., 1991, 121, 6751–6752 CrossRef.
- L. Hodson, K. J. Visagie, M.-P. Smith, L. Loots, D. Kuter, T. M. Snayer and G. E. Arnott, Chem. Commun., 2021, 57, 11045–11048 RSC.
-
(a) W.-Z. Zhang, H. Ma, G.-Y. Xiang, J. Luo and W.-S. Chung, ChemistrySelect, 2016, 1, 2486–2491 CrossRef CAS;
(b) W.-Z. Zhang, K. Yang, S.-Z. Li, H. Ma, J. Luo, K.-P. Wang and W.-S. Chung, Eur. J. Org. Chem., 2015, 765–774 CrossRef CAS.
- K. Ishibashi, H. Tsue, H. Takahashi and R. Tamura, Tetrahedron: Asymmetry, 2009, 20, 375–380 CrossRef CAS.
- S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed.
-
(a) O. Middel, Z. Greff, N. J. Taylor, W. Verboom, D. N. Reinhoudt and V. Snieckus, J. Org. Chem., 2000, 65, 667–675 CrossRef CAS;
(b) F. J. An, W.-Q. Xu, S. Zheng, S.-K. Ma, S.-Y. Li, R.-L. Wang and J.-M. Liu, Eur. J. Org. Chem., 2016, 5, 1012–1016 CrossRef;
(c) W.-Q. Xu, W.-S. Liu, J.-X. Yan, S.-K. Ma, J. Guo, J.-M. Liu, R.-L. Wang and S.-Y. Li, J. Org. Chem., 2016, 81, 10683–10687 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2011, 115, 7304–7397 CrossRef PubMed.
- For a recent review, see: O. Vyhivskyi, A. Kudashev, T. Miyakoshi and O. Baudoin, Chem.–Eur. J., 2021, 27, 1231–1257 CrossRef CAS PubMed , and the references cited therein..
- For selected related examples, see:
(a) D.-W. Gao, Q. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2014, 136, 4841–4844 CrossRef CAS PubMed;
(b) R. Deng, Y. Huang, X. Ma, G. Li, R. Zhu, B. Wang, Y.-B. Kang and Z. Gu, J. Am. Chem. Soc., 2014, 136, 4472–4475 CrossRef CAS PubMed.
-
(a) J. Holub, V. Eigner, L. Vrzal, H. Dvořáková and P. Lhoták, Chem. Commun., 2013, 49, 2798–2800 RSC;
(b) P. Slavík, M. Kohout, S. Böhm, V. Eigner and P. Lhoták, Chem. Commun., 2016, 52, 2366–2369 RSC;
(c) P. Slavík, M. Krupička, V. Eigner, L. Vrzal, H. Dvořáková and P. Lhoták, J. Org. Chem., 2019, 84, 4229–4235 CrossRef PubMed.
- For meta-functionalization of calixarenes, see:
(a)
O. Kundrat and P. Lhotkák, in Calixarens and Beyond, Springer International Pulishing, Cham, 2016, pp. 43–73 Search PubMed;
(b) P. Lhotkák, Org. Biomol. Chem., 2022, 20, 7377–7390 RSC.
- Reactions involving 1,4-palladium migration have been extensively investigated, for selected reviews, see:
(a) S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS PubMed;
(b) A. Rahim, J. Feng and Z. Gu, Chin. J. Chem., 2019, 37, 929–945 CrossRef CAS;
(c) Selected examples of dehydrogenative coupling induced with in situ generated Pd(II) species, see: Q. Huang, A. Fazio, G. Dai, M. A. Campo and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 7460–7461 CrossRef PubMed;
(d) J. Zhao and R. C. Larock, Org. Lett., 2005, 7, 701–704 CrossRef CAS PubMed;
(e) Z.-Y. Gu, C.-G. Liu, S.-Y. Wang and S.-J. Ji, Org. Lett., 2016, 18, 2379–2382 CrossRef CAS PubMed;
(f) T. Piou, A. Bunescu, Q. Wang, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12385–12389 CrossRef CAS PubMed;
(g) A. Bunescu, T. Piou, Q. Wang and J. Zhu, Org. Lett., 2015, 17, 334–337 CrossRef CAS PubMed.
- The asymmetric version of 1,5-palladium migration is scarce, see:
(a) J.-L. Han, Y. Qin, C.-W. Ju and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 6555–6560 CrossRef PubMed;
(b) Y. Sato, C. Takagi, R. Shintani and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 9211–9216 CrossRef CAS PubMed.
- K. Iwamoto, K. Araki and S. Shinkai, J. Org. Chem., 1991, 56, 4955–4962 CrossRef CAS.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed.
- Molecular structure of rearrangement product 7 was confirmed by NMR spectroscopy and single crystal X-ray analysis of rac-7.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Jieping
Zhu
*a,
Jieping
Zhu