
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-free photochemical C–F activation for the preparation of difluorinated-oxindole derivatives†
Bianca
Matsuo‡
 a,
Jadab
Majhi‡
a,
Albert
Granados
a,
Mohammed
Sharique
a,
Robert T.
Martin
b,
Osvaldo
Gutierrez
*bc and
Gary A.
Molander
*a
a,
Jadab
Majhi‡
a,
Albert
Granados
a,
Mohammed
Sharique
a,
Robert T.
Martin
b,
Osvaldo
Gutierrez
*bc and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
bDepartment of Chemistry and Biochemistry, University of Maryland, 8051 Regents Drive, College Park, Maryland 20742, USA
cDepartment of Chemistry, Texas A&M University, 580 Ross St., College Station, Texas 77843, USA. E-mail: og.labs@tamu.edu
First published on 9th February 2023
Abstract
The development of strategies for single and selective C–F bond activation represents an important avenue to overcome limitations in the synthesis of valuable fluorine-containing compounds. The synthetic and medicinal research communities would benefit from new routes that access such relevant molecules in a simple manner. Herein we disclose a straightforward and mechanistically distinct pathway to generate gem-difluoromethyl radicals and their installation onto N-arylmethacrylamides for the preparation of valuable difluorinated oxindole derivatives. To achieve operational simplicity, the use of a readily available benzenethiol as a photocatalyst under open-to-air conditions was developed, demonstrating the facile multigram preparation of the targeted fluorinated molecules. Additionally, dispersion-corrected density functional theory (DFT) and empirical investigations provide a new basis to support the proposed reaction pathway, indicating that arene thiolate is an efficient organophotocatalyst for this transformation.
Introduction
Challenging synthetic organic transformations have driven the development of new protocols to diversify and broaden existing methods. New technologies and activation modes have dramatically changed how chemical space is being explored. Among the various challenges in the field, the selective activation of a single C–F bond in polyfluorinated substrates has become a desirable transformation.1–3 The inherent interest in accessing C–F bond-containing molecules is attributed to the fact that these bonds are prevalent in biologically active compounds. Fluorinated molecules have proved to be key in drug discovery programs because they can dramatically change the properties of drug candidates by improving stability, permeability, and, consequently, the efficacy of molecules discovered therein.4,5 It is not a coincidence that fluorinated molecules are highly represented in many currently marketed drugs. Consequently, continuous efforts have been made to prepare and derivatize such molecules. A central challenge in this synthetic field remains the high bond dissociation energy of C–F bonds, which imposes limitations on selective bond cleavage and further functionalization in polyfluorinated substrates.Considering modern approaches to this challenge, electrochemical and photochemical methods have emerged as extremely useful technologies, allowing selective C–F bond activation in various different classes of compounds,3,6 including fluoroarenes,7–13 trifluoromethylated arenes14–17 and -alkenes,18–23gem-difluoroalkenes,24–29 and others, such as trifluoromethylated esters, -amides,30–33 and -ketones34 under mild reaction conditions.
In this arena, photoinduced metal-free protocols represent a highly attractive approach to C–F activation, providing valuable structures without trace metals; a feature that is especially relevant in the industrial sector. Small organic conjugated molecules have been shown to exhibit interesting properties as photocatalytic species, successfully allowing C–F bond activation. Some examples include the use of pyrenes35 and phenoxazines15 in catalytic hydrodefluorination of polyfluoroarenes and trifluoromethylarenes, respectively; the exploration of phosphinophenolates32 in defluoroalkylation and hydrodefluorination of trifluoromethylated substrates [e.g., esters, acetamides, and (hetereo)arenes]; in addition to the well-known applications of dicyanobenzenes as potent organophotocatalysts.16,33,36
In 2021, our group disclosed a metal-free and mechanistically novel approach to install gem-difluoromethylene groups from trifluoroacetates and -acetamides onto (un)activated alkenes.30 The method was designed to be promoted by a hydrogen atom transfer (HAT) step via combination of diaryl ketone, thiol, and sodium formate. Continuing with our interest in the synthesis of fluorinated compounds via photoredox catalysis,19,21,22,37–39 we focused on developing a metal-free preparation of difluorinated oxindole derivatives via a cascade reaction between gem-difluoromethylene radicals and N-arylmethacrylamides, exploring the photoreactive nature of thiols as photocatalysts.
Aryl acrylamides are useful precursors to build more complex structures containing the oxindole moiety, known to exhibit extraordinary activities as drugs as well as being present in many active natural products.40–42 They have been explored in cascade-type transformations where the sequence of addition to the alkene moiety followed by cyclization and rearomatization leads to interesting and valuable compounds from a medicinal point of view. Our approach would represent a complementary and alternative route to access these privileged motifs under milder conditions, extending the chemical space covered by previous advances in the field, which have demonstrated the use of trifluoromethylated arenes17 and -ketones34 as gem-difluoromethylene radical sources. In these examples, activation of the C–F bond was achieved by employing a combination of Ir-catalysts and Lewis acids to facilitate the fluoride elimination (Fig. 1a).
 | ||
| Fig. 1 Previous approaches to preparing difluoroalkylated compounds. | ||
Previous approaches to installing gem-difluoroacetates for the preparation of valuable difluorinated oxindoles make use of harsh conditions in the presence of metal-based catalysts, oxidants, and/or high temperatures, and are restricted regarding the structural diversification of the fluorinated moiety (Fig. 1b).43–46 Recently, a seminal Giese addition protocol using trifluoromethylated compounds was also reported by the Shang group, supporting the application of thiolate species as reductants and HAT catalysts. An efficient hydrodefluorination of trifluoroacetates, -arenes, and -amides and the coupling with a range of alkenes was reported (Fig. 1c).31 In another vein, Chiba and co-workers explored the powerful ability of photoexcited thiol anions to promote the single-electron reduction of aryl halides with reduction potentials as low as −2.4 V vs. SCE,47,48 while Hamashima explored thiobenzoic acid as a single-electron reducing agent and HAT agent to promote regioselectivie Cα–H arylation reactions.49 All of these precedents demonstrate the rich applicability of sulfur anions as potential photocatalysts.50
Seeking alternative methods that would provide easy access to gem-difluoromethyl radicals under metal-free and milder conditions, we recognized that benzenethiols play a key role as photocatalysts under blue and purple light irradiation and, in the presence of sodium formate,30–32,36,51,52 directly lead to selective mono C–F bond cleavage in trifluoroacetates. Herein, we report a transition metal-free, mild, and operationally simple method to overcome the limitations associated with the preparation of fluorinated oxindole molecules by single C–F bond activation (Fig. 1d).
Discussion
To initiate our studies, we chose 4-methoxybenzenethiol as a catalytic species. The screening of the reaction conditions for the defluorinative alkylation protocol between the N-arylmethacrylamide 1 and the trifluoroacetate 2 demonstrated that the use of 20 mol% of the organophotocatalyst, in combination with sodium formate and KHCO3, provides access to the corresponding difluorinated oxindole 3 in 95% yield under 390 nm wavelength LED irradiation (Table 1, entry 1). Lowering the catalyst loading led to erosion of the yield (Table 1, entry 2). The use of excess trifluoroacetate provided high yields, however the utilization of fewer equivalents also gave the product in moderate yields (Table 1, entry 3) (see Table S1 for the complete optimization, ESI†). Because this source of gem-difluoromethylene radical is a commercially available, bench-stable, and cost-accessible material in comparison to bromo/iodo-difluoroacetates, we decided to proceed with the use of 10 equivalents for the study of this transformation.|
|
||
|---|---|---|
| Entry | Deviation from std condition | Yieldb (%) |
| a Reaction conditions: 1 (0.1 mmol, 1 equiv.), 2 (10 equiv.), ArSH (0.20 equiv.), HCO2Na (3.0 equiv.) and KHCO3 (1.0 equiv.) in DMSO (0.1 M) under 390 nm wavelength Kessil light irradiation and open-to-air. b Yields determined by 19F NMR analysis using 4-bromo-2-fluoro-1-iodobenzene as internal standard. c Reaction performed under heating at 60 °C. ArSH = 4-methoxybenzenethiol. | ||
| 1 | None | 95 |
| 2 | 0.10 equiv. of ArSH | 50 |
| 3 | 7.0 equiv. of 2 | 64 |
| 4 | No base | 56 |
| 5 | 1.0 equiv. of sodium formate | 10 |
| 6 | Violet Kessil 427 nm | 10 |
| 7c | No Kessil lamp | nr |
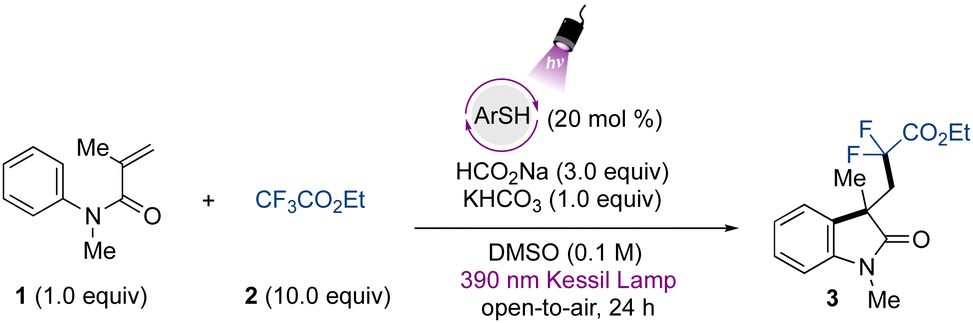
Notably, the reaction successfully takes place under an oxygen atmosphere, and no degassing of the reaction is required. These results indicate the operational simplicity of the developed protocol, and the open-to-air conditions still lead to excellent results even though oxygen is known to oxidize thiols.53 Preliminary studies were carried out in DMSO, and in the presence of other polar aprotic solvents, no product formation was observed. Other alternative sulfoxide and sulfone-based solvents, such as tetramethylene sulfoxide and sulfolane, did not lead to formation of the desired product (see ESI†). The use of base was also important to achieve high yields (Table 1, entry 4), and sodium formate was demonstrated to be a crucial component for the reaction to take place (Table 1, entry 5). Regarding the irradiation source, the photocatalyst showed poor activity under 427 nm wavelength Kessil irradiation (Table 1, entry 6). As control experiments, no transformation occurred in the absence of irradiation (Table 1, entry 7) or using TEMPO as a radical scavenger, indicating the radical nature of the transformation.
To demonstrate the applicability of the developed metal-free protocol, we prepared a library of oxindole derivatives containing diverse functional groups attached to the aromatic ring of the N-arylmethacrylamide component, as well as other diverse substructural motifs (Fig. 2). As expected, electron-donating groups were amenable to this photochemical difluoroalkylation protocol, providing compounds 4–6 with good yields. Halogen atoms were also successfully tolerated, with excellent compatibility with a para-fluoro substituent in 7. Other halogen moieties such as chloride and bromide substituents in the para-, meta-, and ortho positions on the aromatic ring (8–11) also showed good reactivity. A naphthyl aromatic ring was also included, providing the corresponding difluorinated oxindole 12 with lower yield, which can be attributed to a competitive hydroarylation reaction leading to the formation of the benzodihydroquinolinone product.54 The gem-difluoromethyl radical was also smoothly added to a 1,1-disubstituted olefin to provide compound 13 with 80% yield and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Given the potential of the method, we further applied it to the preparation of more complex fused tricyclic compounds. The oxindole derivative derived from tetrahydroisoquinoline (14) was successfully obtained with moderate yield. Further examples to explore the scope included modification of the alkene moiety of the N-arylmethacrylamide. Spirocyclic compounds possess recognized relevance in biological chemistry and are considered challenging targets in organic synthesis.41,55 By using internal alkenes in five- and six-membered rings, spirocyclic compounds 15 and 16 were readily prepared in excellent yields. Finally, the benzyl- and phenyl protecting groups (17 and 18, respectively) were also tolerated under the developed protocol, while the unprotected or Boc-protected acrylamides or phenyl methacrylate precursors did not lead to the desired products.
:1 dr. Given the potential of the method, we further applied it to the preparation of more complex fused tricyclic compounds. The oxindole derivative derived from tetrahydroisoquinoline (14) was successfully obtained with moderate yield. Further examples to explore the scope included modification of the alkene moiety of the N-arylmethacrylamide. Spirocyclic compounds possess recognized relevance in biological chemistry and are considered challenging targets in organic synthesis.41,55 By using internal alkenes in five- and six-membered rings, spirocyclic compounds 15 and 16 were readily prepared in excellent yields. Finally, the benzyl- and phenyl protecting groups (17 and 18, respectively) were also tolerated under the developed protocol, while the unprotected or Boc-protected acrylamides or phenyl methacrylate precursors did not lead to the desired products.
 | ||
| Fig. 2 Scope of N-aryl acrylamides in the cascade addition/cyclization reaction for the synthesis of difluorinated oxindoles. All values indicate the yield of the isolated product. Reaction conditions: N-arylmethacrylamide (0.5 mmol, 1 equiv.), 2 (10 equiv.), ArSH (0.20 equiv.), HCO2Na (3.0 equiv.) and KHCO3 (1.0 equiv.) in DMSO (0.1 M) under 390 nm wavelength Kessil light irradiation and open-to-air. aObtained as a mixture of isomers. ArSH = 4-methoxybenzenethiol. | ||
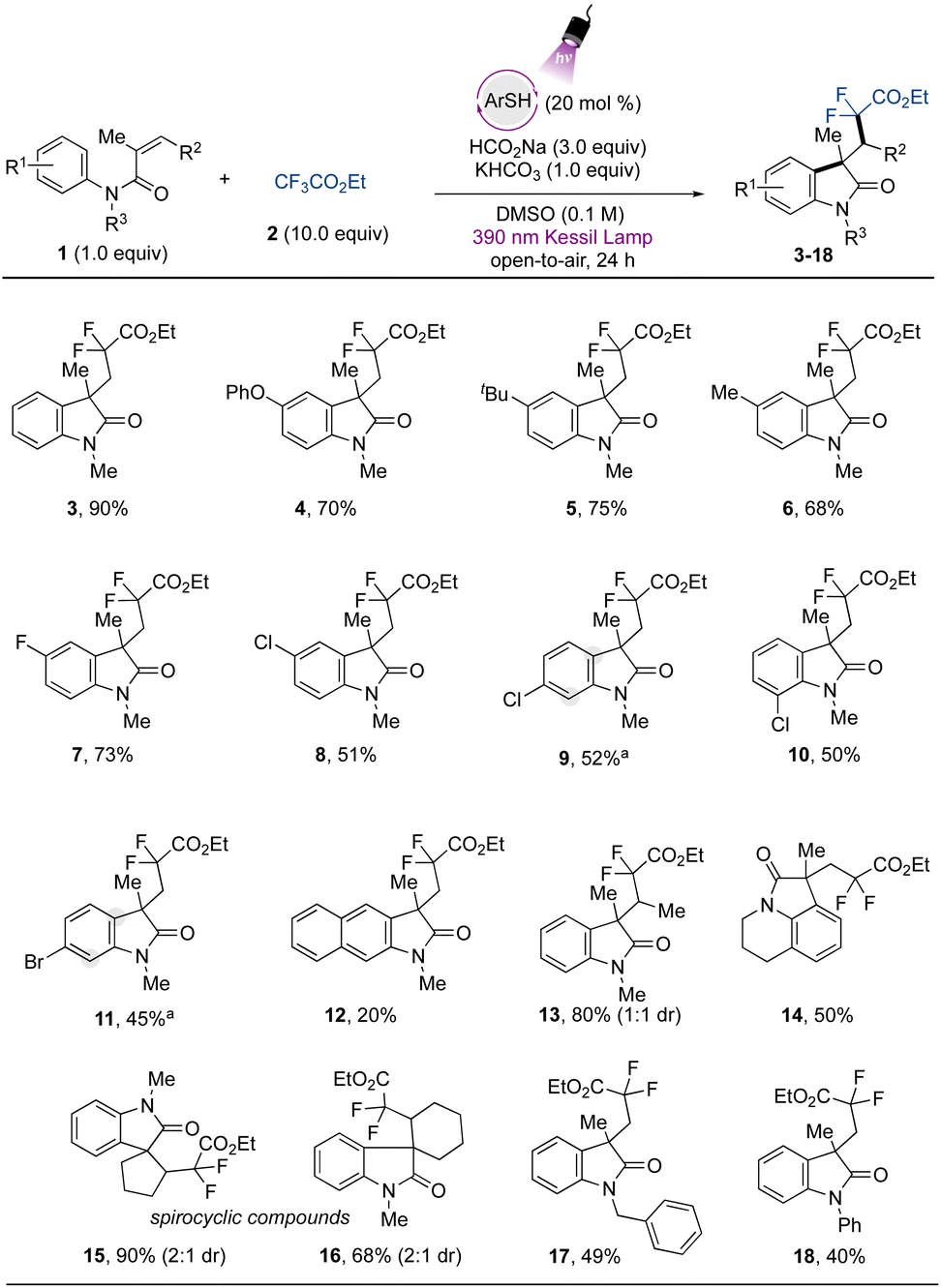
Attention was next turned to verifying the fluoroalkylation of other trifluoromethylated compounds via the developed metal-free strategy. Extending the scope using trifluoroacetates, selective C–F bond cleavage was found to occur smoothly when other derivatives were employed (Fig. 3). For example, aliphatic ester-containing alkyl- and aromatic groups (as in phenethyl- (19) and 2-cyclohexylethyl- (20) trifluoroacetates) underwent the desired transformation with good to excellent yields. Other α,α-difluorinated aliphatic carboxylate esters containing secondary aliphatic groups were accessed, such as those in the isopropyl- (21), and cyclohexyl (22) derivatives, and the tertiary example containing the sterically demanding adamantyl group (23). Other challenging CF3-containing substrates were engaged in the cascade transformation. Ethyl pentafluoropropionate (Ered = −2.6 V vs. SCE)30 smoothly underwent regioselective single C–F bond activation to provide the corresponding difluorooxindole derivative 24 containing an intact CF3 group in 78% yield. Additionally, 25, derived from ethyl difluoroacetate, which presents an even more challenging reduction potential than other substrates (Ered = −2.9 V vs. SCE),30 was also successfully accessed, although in lower yield.
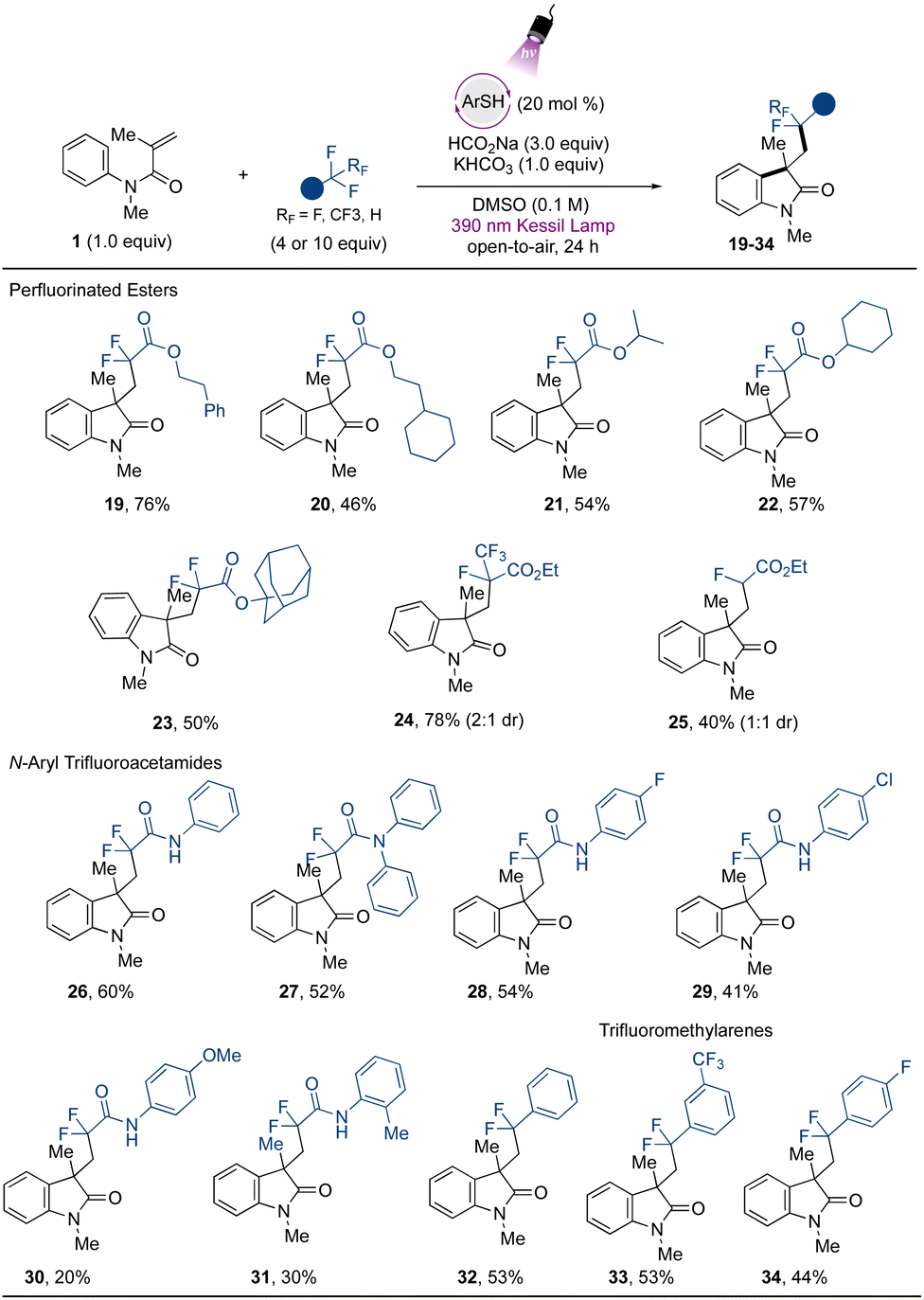 | ||
| Fig. 3 Scope of trifluoromethylated compounds in the cascade addition/cyclization reaction for the synthesis of difluorinated oxindoles. All values indicate the yield of the isolated product. Reaction conditions: N-arylacrylamide 1 (0.5 mmol, 1 equiv.), trifluoromethylated compound (10 equiv. for the scope of trifluoroacetates and CF3-arenes, and 4 equiv. for the scope of acetamides), ArSH (0.20 equiv.), HCO2Na (3.0 equiv.) and KHCO3 (1.0 equiv.) in DMSO (0.1 M) under purple Kessil light irradiation and open-to-air. ArSH = 4-methoxybenzenethiol. | ||
The protocol was further extended to trifluoromethylated amide precursors. Amide-containing compounds are valued structures from a medicinal and synthetic point of view, given the intrinsic properties associated with this functional group, such as their polarity, stability, and H-bonding capability.56 Difluorinated oxindole derivatives containing pendant amide-groups (26–29) were thus generated in modest to good yields. The trifluoroacetamide (Ered = −2.2 V vs. SCE) gave direct access to the desired compound 26 from the defluorinative-cascade process in 60% yield, and the diphenyl derivative 27 was also accessed in modest yield. Interestingly from a synthetic point of view, the acetamides containing halogen atoms at the para position on the aromatic ring underwent the transformation in moderate to good yield (28 and 29). The thiolate excited-state presents a highly reducing power (Ered = −3.3 V vs. SCE)31 capable of promoting a competitive homolytic C–X bond cleavage, which may be associated with the yields observed for halogenated compounds. Electron-rich acetamides provided poor results, as in 30 and 31. Limitations were found regarding the use of acetamides containing electron-withdrawing groups, and the derivatives substituted with cyano- and di-CF3 groups did not react under the developed conditions (see ESI for more information†). Although the method still presents opportunities to be explored to extend the scope regarding the use of structurally diverse amides, the method avoids the use of Lewis acids as additives, which are usually employed to lower the reduction potential of these species.30 Finally, exploring the further potential of the developed method, the use of trifluoromethyltoluene provides access to the arylated difluoroindole derivative 32 in 53% yield, and a similar yield was found when 1,3-(bis)trifluoromethyltoluene was evaluated (33). The scope also included the p-fluorotoluene derivative, which gave rise to 34 in 44% yield. For other halogenated arenes (X = Br, I), the homolytic C–X bond cleavage was observed instead, indicating the powerful reducing properties of the organophotocatalyst employed in the study. These results demonstrate the applicability of the method via another mechanistic approach, complementing the protocols disclosed recently introducing difluoromethyl ketones and -arenes under photocatalytic conditions.17,34
Given the importance of the scalability of new synthetic methods aiming at industrial applications, we successfully achieved the preparation of 1.9 grams (10 mmol scale) of compound 3 in 64% yield under the standard reaction conditions. For this demonstration, we employed 4 equivalents of trifluoroacetate 2, and the yield was comparable to the small-scale reaction (0.1 mmol). As noted in Fig. 4, the large-scale experiment was easily performed simply by using a large round-bottom flask irradiated with the 390 nm light source. The scope regarding the trifluoroacetates nicely complements and extends other reported protocols, allowing for the first time the installation of these motifs onto N-arylmethacrylamides via C–F bond activation.
 | ||
| Fig. 4 Scalability of the defluorinative alkylation protocol for the large-scale preparation of difluorinated oxindoles. aYield for the 0.1 mmol scale reaction using 4.0 equiv. of 2. ArSH = 4-methoxybenzenethiol. | ||
Finally, to gain insights into the mechanism of this transformation, we employed dispersion-corrected density functional theory (DFT) computations (Fig. 5 and ESI†). As shown in Fig. 5, the proposed mechanism initiates with the formation of the thiolate species II from deprotonation of the thiol I in the presence of base. The thiolate II is then promoted to the excited anion (III) under purple light irradiation. Given the strong ability of the latter to act as a reductant (Ered = −3.3 V vs. SCE),31 the initial defluorinative step to form the radical anion species A˙− should take place via single electron reduction of the ethyl trifluoroacetate 2 (Ered = −2.0 V vs. SCE), as supported by quenching experiments.30 In the thiolate catalytic cycle, the thiyl radical IV formed is prone to undergo dimerization, and it is in equilibrium with the disulfide V.
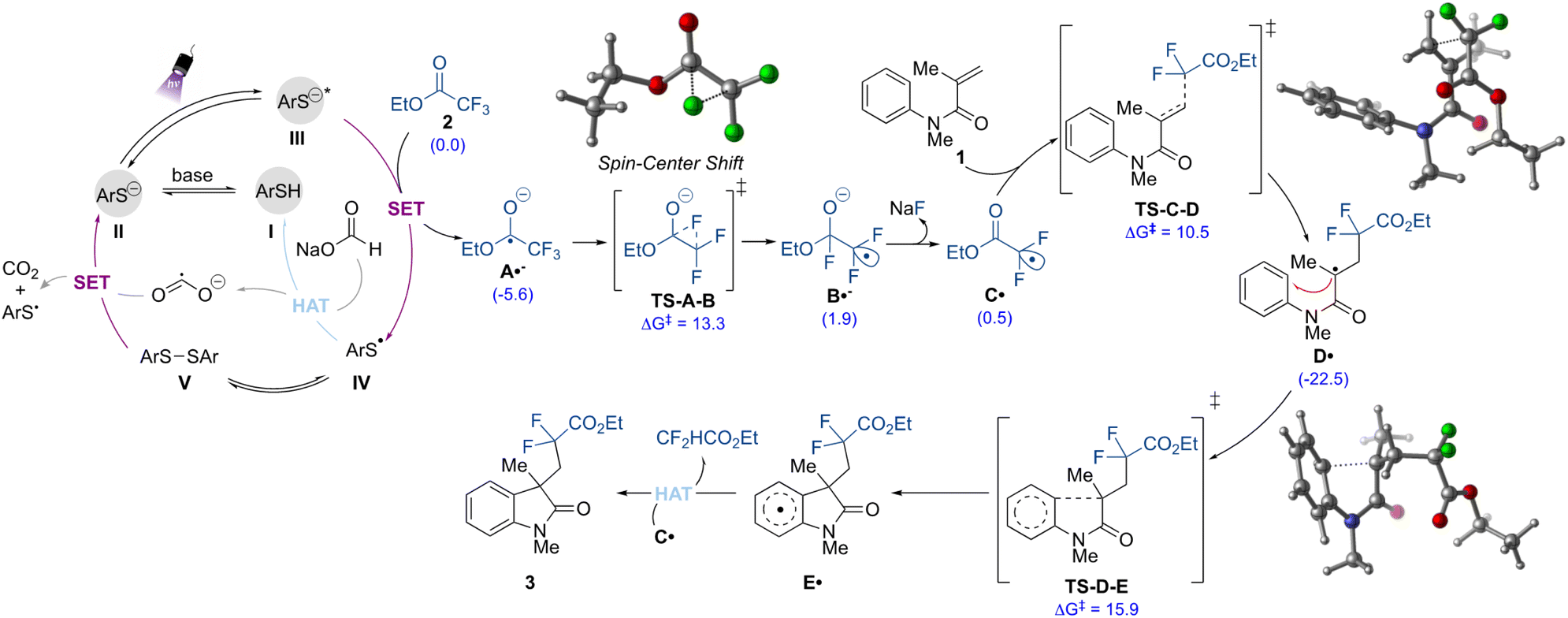 | ||
| Fig. 5 Proposed mechanism for the metal-free defluorinative alkylation strategy. Free energies of relevant species reported in kcal mol−1 at UωB97XD/def2-SVP-CPCM(DMSO)//UB3LYP-D3/def2-SVP-CPCM(DMSO) level of theory. | ||
This species also demonstrated the ability to play a role as the pre-catalyst, providing product 3 with a 95% yield when used instead of I (Table S2, ESI†). Regeneration of the precatalyst I can then take place via HAT between IV and sodium formate. In this step, the reductant CO2˙− is formed, which can also contribute to the generation of the active thiolate species II by promoting the reductive cleavage of the S–S bond in V. Each of these processes is energetically feasible (see ESI for full computational details†). Additionally, the quantum yield (Φ) value for the transformation was found to be 0.35, which suggests that a chain process is possibly not operating. Thus, the formate radical anion is unlikely to be contributing to the reduction of 2.30,33,36,57
From this point, the formed C-centered radical anion species A˙− undergoes a spin-center shift (SCS)58 to form the electrophilic gem-difluoromethyl radical intermediate C˙, which was also observed as a TEMPO adduct in the control experiments. DFT computations suggest that the SCS proceeds through a stepwise 1,2-F migration pathway (TS-A-B), wherein the fluorine on the terminal carbon shifts to the carbonyl carbon. This process is energetically feasible (ΔG‡ = 13.3 kcal mol−1). Following the 1,2-F migration, the fluoride is released as an anion (via coordination with sodium from sodium formate) which forms the carbon-centered radical C˙.59 In turn, alkyl radical C˙ then undergoes regioselective and irreversible Giese addition to the olefin moiety of the N-arylmethacrylamide (1) to form the radical intermediate D˙. Finally, a facile radical cyclization followed by rearomatization would take place to furnish the difluorinated oxindole 3. The DFT calculation indicated that this latter is likely to be proceeding via a HAT mechanism, where the hydrogen can be abstracted by the radical intermediate carbon centered radical C˙, which is being formed in excess in the reaction medium. Other radical intermediates, such as A˙− and CO2˙−, could be operative in this mechanism as well, however, the pathway viaC˙ hydrogen abstraction is more energetically feasible. A second reaction mechanism for the rearomatization step would involve a single electron oxidation of E˙ by the thiyl radical IV to provide the corresponding carbocation intermediate, which after proton loss would furnish the final difluorinated compound 3. Although this pathway (Fig. S11, ESI†) is slightly less energetically favorable than the radical HAT pathways described, the reaction is still energetically feasible and the rearomatization process is irreversible.
Conclusions
The photoinduced protocol developed herein represents a mechanistically distinct pathway to install fluorinated motifs easily onto N-arylmethacrylamides via a single, chemoselective C–F bond activation of trifluoroacetates, -acetamides, and -arenes. The preparation of a library of valuable oxindole derivatives was accomplished under mild reaction conditions using the thiolate excited state as an organophotocatalyst. DFT computational results supporting the proposed reaction pathway complement the experimental studies and shed light on the reactivity of these readily available and powerful catalytic species. Taking advantage of the operational simplicity of the method, the reaction was performed under open-to-air conditions, allowing an easy scale-up for the multigram preparation of a target molecule. This protocol complements the synthetic tools available to install gem-difluoromethyl radicals via C–F activation, avoiding the use of rare metal-based catalysts and the use of expensive halogen-difluoroacetates as radical sources. Given the relevance and challenges found in this synthetic approach, the practical and accessible procedure described herein contributes to the advancement of the field of selective C–F bond activation and extends chemical space of important fluorinated motifs, unlocking new opportunities to explore the powerful reduction potential of excited thiolates in further transformations under photocatalytic conditions.Data availability
The ESI† includes all experimental details, including quantum yield measurements, mechanistics studies, DFT calculation, synthesis and characterization of all products reported in this study. NMR spectra of all products are included as well.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support (RG2020) provided by Merck KGaA, Darmstadt, Germany, and NIGMS (R35 GM 131680 to G. A. M.). The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. O. G. gratefully acknowledges the NIGMS NIH (R35GM137797) and Camille and Henry Dreyfus Foundation for funding and Texas A&M University HPRC resources (https://hprc.tamu.edu), UMD Deepthought2, MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053) for computational resources. The authors thank Dr Venkatesh Yarra (UPenn) for helping in the quenching experiments.Notes and references
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed
.
- A. Das and N. Chatani, ACS Catal., 2021, 11, 12915–12930 CrossRef CAS
- S. Li and W. Shu, Chem. Commun., 2022, 58, 1066–1077 RSC
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC
- K. Reichenbacher, H. I. Suss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC
- Z. H. Wang, Y. Sun, L. Y. Shen, W. C. Yang, F. Meng and P. H. Li, Org. Chem. Front., 2022, 9, 853–873 RSC
- P. J. Xia, Z. P. Ye, Y. Z. Hu, J. A. Xiao, K. Chen, H. Y. Xiang, X. Q. Chen and H. Yang, Org. Lett., 2020, 22, 1742–1747 CrossRef CAS PubMed
- J. Wang, B. Huang, Y. Gao, C. Yang and W. Xia, J. Org. Chem., 2019, 84, 6895–6903 CrossRef CAS PubMed
- X. Sun and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 10557–10562 CrossRef CAS PubMed
- A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2015, 6, 7206–7212 RSC
- A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796–6802 RSC
- S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed
- S. Senaweera and J. D. Weaver, J. Am. Chem. Soc., 2016, 138, 2520–2523 CrossRef CAS PubMed
- H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed
- D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Medebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noel, C. W. Am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS PubMed
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed
- T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908–7916 CrossRef CAS PubMed
- S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS PubMed
- L. Li, T. Xiao, H. Chen and L. Zhou, Chem. - Eur. J., 2017, 23, 2249–2254 CrossRef CAS PubMed
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed
- R. J. Wiles, J. P. Phelan and G. A. Molander, Chem. Commun., 2019, 55, 7599–7602 RSC
- Y. Q. Guo, R. Wang, H. Song, Y. Liu and Q. Wang, Org. Lett., 2020, 22, 709–713 CrossRef CAS PubMed
- L. H. Wu, J. K. Cheng, L. Shen, Z. L. Shen and T.
P. Loh, Adv. Synth. Catal., 2018, 360, 3894–3899 CrossRef CAS
- H. Tian, Q. Xia, Q. Wang, J. Dong, Y. Liu and Q. Wang, Org. Lett., 2019, 21, 4585–4589 CrossRef CAS PubMed
- J. L. Wang, B. B. Huang, C. Yang and W. J. Xia, Chem. Commun., 2019, 55, 11103–11106 RSC
- C. Zhu, Y. F. Zhang, Z. Y. Liu, L. Zhou, H. D. Liu and C. Feng, Chem. Sci., 2019, 10, 6721–6726 RSC
- S. L. Xie, X. T. Gao, H. H. Wu, F. Zhou and J. Zhou, Org. Lett., 2020, 22, 8424–8429 CrossRef CAS PubMed
- Y. Li, X. Li, X. Li and D. Shi, Chem. Commun., 2021, 57, 2152–2155 RSC
- M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed
- C. Liu, K. Li and R. Shang, ACS Catal., 2022, 12, 4103–4109 CrossRef CAS
- C. Liu, N. Shen and R. Shang, Nat. Commun., 2022, 13, 354 CrossRef CAS PubMed
- J. H. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115456 CAS
- S. Ghosh, Z. W. Qu, S. Pradhan, A. Ghosh, S. Grimme and I. Chatterjee, Angew. Chem., Int. Ed., 2022, 61, e202115272 CAS
- J. Lu, N. S. Khetrapal, J. A. Johnson, X. C. Zeng and J. Zhang, J. Am. Chem. Soc., 2016, 138, 15805–15808 CrossRef CAS PubMed
- S. S. Yan, S. H. Liu, L. Chen, Z. Y. Bo, K. Jing, T. Y. Gao, B. Yu, Y. Lan, S. P. Luo and D. G. Yu, Chem, 2021, 7, 3099–3113 CAS
- R. K. Dhungana, A. Granados, M. Sharique, J. Majhi and G. A. Molander, Chem. Commun., 2022, 58, 9556–9559 RSC
- A. Granados, R. K. Dhungana, M. Sharique, J. Majhi and G. A. Molander, Org. Lett., 2022, 24, 4750–4755 CrossRef CAS PubMed
- G. Levitre, A. Granados, M. J. Cabrera-Afonso and G. A. Molander, Org. Lett., 2022, 24, 3194–3198 CrossRef CAS PubMed
- G. Cerchiaro and A. M. D. Ferreira, J. Braz. Chem. Soc., 2006, 17, 1473–1485 CrossRef CAS
- C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209–2219 CrossRef CAS
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed
- W. J. Fu, M. Zhu, G. L. Zou, C. Xu and Z. Q. Wang, Asian J. Org. Chem., 2014, 3, 1273–1276 CrossRef CAS
- X. L. Wang, W. Wan, Y. R. Chen, J. L. Li, H. Z. Jiang, Y. Wang, H. M. Deng and J. Hao, Eur. J. Org. Chem., 2016, 3773–3779 CrossRef CAS
- D. Liu, S. B. Zhuang, X. Chen, L. Yu, Y. Q. Yu, L. Hu and Z. Tan, Tetrahedron Lett., 2018, 59, 612–616 CrossRef CAS
- S. D. Ma, P. S. Zhou, X. Fan, D. J. Li and J. H. Yang, Tetrahedron Lett., 2022, 102, 153933 CrossRef CAS
- H. Y. Li, X. X. Tang, J. H. Pang, X. Y. Wu, E. K. L. Yeow, J. Wu and S. Chiba, J. Am. Chem. Soc., 2021, 143, 481–487 CrossRef CAS PubMed
- H. Li, Y. Liu and S. Chiba, Chem. Commun., 2021, 57, 6264–6267 RSC
- F. Kobayashi, M. Fujita, T. Ide, Y. Ito, K. Yamashita, H. Egami and Y. Hamashima, ACS Catal., 2021, 11, 82–87 CrossRef CAS
- S. Wang, H. Wang and B. König, J. Am. Chem. Soc., 2021, 143, 15530–15537 CrossRef CAS PubMed
- P. Xu, X. Y. Wang, Z. Wang, J. Zhao, X. D. Cao, X. C. Xiong, Y. C. Yuan, S. Zhu, D. Guo and X. Zhu, Org. Lett., 2022, 24, 4075–4080 CrossRef CAS PubMed
- J. H. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115456 CAS
- C. M. Q. Le, F. Morlet-Savary and A. Chemtob, Polym. Chem., 2021, 12, 6594–6605 RSC
- Z. S. Liu, S. Zhong, X. C. Ji, G. J. Deng and H. W. Huang, ACS Catal., 2021, 11, 4422–4429 CrossRef CAS
- P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS
- B. T. Matsuo, P. H. R. Oliveira, E. F. Pissinati, K. B. Vega, I. S. de Jesus, J. T. M. Correia and M. Paixão, Chem. Commun., 2022, 58, 8322–8339 RSC
- C. M. Hendy, G. C. Smith, Z. H. Xu, T. Q. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed
- B. Matsuo, A. Granados, J. Majhi, M. Sharique, G. Levitre and G. A. Molander, ACS Org. Inorg. Au, 2022, 6, 435–454 CrossRef PubMed
- Y. J. Yu, F. L. Zhang, T. Y. Peng, C. L. Wang, J. Cheng, C. Chen, K. N. Houk and Y. F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06179a |
| ‡ B. M. and J. M. contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |